
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithiation mechanism of sulfur surfaces during discharge of Li–S batteries from quantum chemical calculations†
Jonas
Lührs
,
Daniel
Sebastiani
 and
Pouya
Partovi-Azar
*
and
Pouya
Partovi-Azar
*
Van-Danckelmann-Platz 4, Halle (Saale), Germany. E-mail: pouya.partovi-azar@chemie.uni-halle.de
First published on 25th April 2025
Abstract
We present a computational study based on quantum-chemical calculations to investigate the initial lithiation reactions on the (001) surface of α-sulfur. The study aims to explore the possible emerging structures during consecutive lithiation steps and to analyze their reaction enthalpies. Our results show that during the first lithiation reactions, S8 rings in the lower layers of the (001) surface are preferentially lithiated. In subsequent lithiation steps, we find that S8 rings on the upper layers, adjacent to previously lithiated molecules, may also undergo lithiation. Once Li2S8 dimers are formed, further reactions on the surface can proceed, leading to the formation of Li2S8 trimers in a lower/upper/lower layer arrangement or lower-order Li-polysulfides, such as Li2S6/Li2S2 and Li2S5/Li2S3. Notably, in contrast to sulfur reduction reactions in the electrolyte, the formation of Li2S4/Li2S4 does not occur on the (001) surface, likely due to the surface morphology, which prevents complete exposure of S8 rings to lithium ions. This suggests that surface lithiation predominantly leads to the formation of high-order polysulfides in the early stages of discharge, while the dissolution of these higher-order polysulfides into the electrolyte may facilitate their reduction to Li2S4, a process observed experimentally. Our study provides an atomistic mechanism for the discharge process of Li–S batteries with a crystalline α-sulfur cathode, contributing to a deeper understanding of both solid- and liquid-phase reactions during the early discharge stages.
1. Introduction
The ever-increasing global energy demand calls for more efficient energy-storage devices. Currently, lithium-ion batteries are the most commonly used in electronic devices such as mobile phones, tablets, and electric cars etc.1 However, the energy density of these batteries has only incrementally increased in the past decade approaching a saturated energy density of 250–400 W h kg−1.2,3 This has been made possible only by using rare metals, which increases the net weight of Li-ion batteries as well as their cost. As an alternative, lithium–sulfur (Li–S) batteries have the potential to replace Li-ion batteries due to their very high energy density of around 2600 W h kg−1.4,5 Moreover, sulfur is an abundant material, and therefore considerably cheaper6 compared to the transition metals required for Li-ion batteries. Nevertheless, Li–S batteries still face technical issues that prevent their everyday use. These issues are partially related to the formation of soluble lithium polysulfides which result in shuttle effect during the discharge process, which is believed to be the main reason for the poor cycle life of Li–S batteries.1,7–12Among the proposed cathode materials to mitigate the shuttle effect, sulfur/carbon copolymers13–23 and covalent organic frameworks24–30 have recently attracted much attention. Despite the promising performance of these materials to immobilize the lithium polysulfides (or even to facilitate sulfur reduction reactions), the overall electrochemical performance of the whole cathode, is determined to a great extent by that of the crystalline sulfur to which the copolymers or covalent organic frameworks are added. Therefore, to prevent the shuttle effect, it is important to gather an atomistic insight into the lithiation mechanism of crystalline sulfur. Although the main reactions during discharge are believed to occur in the solvent,10–12,31,32 the initial lithiation reactions occur primarily on the cathode surface, as octa-sulfur is nonpolar. Additionally, previous quantum-chemical studies have addressed lithiation reaction at sulfur/solvent interface including explicit solvent molecules using ab initio molecular dynamics (AIMD) which suggest the occurrence of consecutive lithiation reactions at the sulfur surfaces forming lower-order Li-polysulfides, before their dissolution into the electrolyte.33 Therefore, it is important to understand the morphological changes of the sulfur surfaces during early stages of discharge.
The purpose of this study is to investigate the lithiation mechanism of the (001) surface of the crystalline α-sulfur, determine the most stable products formed during this process, and study the structural changes of the surface during lithiation using quantum chemical calculations. Despite several studies to elucidate the lithiation mechanism of solvated S8 and on sulfur surfaces,33–35 an atomistic understanding of the underlying processes and structural changes remains incomplete due to the complex nature of the reactions.
2. Computational details
To represent the sulfur surface, certain steps were taken to create a suitable system for geometry optimizations using density functional theory (DFT) calculations.36 The calculations were performed on the orthorhombic α-sulfur, considering its (001) surface which, along with the (100) surface, has the lowest energy compared to other α-sulfur surfaces.33 The original unit cell structure was extracted from X-ray diffraction measurements.37 We employed periodic boundary conditions (PBC) along the x and y axes to represent crystallinity. In addition, a vacuum buffer along the z-axis was implemented to expose the (001) surface for lithiation reactions. To prevent the formation of radical electron structures, a modification was made to the top layer in the z-direction to ensure the absence of broken S8 rings. Detailed information can be found in Fig. 1 and Table 1. To study lithiation reactions, we used CP2K/QUICKSTEP package,38 incorporating dual plane-wave and Gaussian basis functions. A double-zeta plus polarization basis set optimized for molecular systems (DZVP-MOLOPT-SR-GTH)39 was used for the valence electrons together with Goedecket–Teter–Hutter-type pseudopotentials,40,41 and the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.42 To account for dispersion interactions, the DFT-D343 method was incorporated. Electronic structure calculations employed an SCF convergence criterion set at 10−6 and the atomic coordinate optimizations were conducted using the BFGS optimizer.44–47 Our calculations including an implicit solvent model, did not show significant structural differences in the product structures of lithiation reactions in electrolyte (see ESI†). Besides, the main concern here is to understand the deformation of the sulfur surface during lithiation reactions rather than the solubility or dissociation of the lithiated products. Therefore, here we did not include an implicit solvent model for the electrolyte. | ||
| Fig. 1 Unit cell with S8 slab. Perpendicular to the z axis are (001) surfaces, while PBC as used along x and y axes. | ||
| Number of atoms | 578 |
| Unit cell size [Å] | (a, b, c) = (31.39, 25.73, 60.00) |
| Angles [°] | (α, β, γ) = (90, 90, 90) |
The sulfur surface structure was prepared for the lithiation reaction by initially relaxing the sulfur slab in a vacuum. This involved relaxing the upper three layers while keeping the lower three layers fixed, as shown in Fig. 2. The relaxation of the upper layers represents the surface flexibility, which is important for the lithiation process. Meanwhile, the constraint of the lower layers mimics the crystallinity of the sulfur system. Fig. 2(a) shows the z-displacement of each layer during the relaxation process while (b) highlights the corresponding layers, encircled using the same color coding. The obtained relaxed structure is the starting point for the lithiation process. To study the product structures in each lithiation reaction, distinct positions for two lithium atoms were chosen on the sulfur surface. Always two lithium atoms with a distance of around 3 Å were added to different positions on the surface at each lithiation step to ensure that no radical systems are present in the process of the lithiation. For each position two lithium atoms were arranged once parallel (p) and once orthogonal (o) to the (001) surface. The product structures were then obtained by optimizing the entire system, including the two lithium atoms, while constraining the lower three ring layers. Consequently, for each subsequent lithiation step, the lowest-energy product from the previous step was used as the starting point to define new distinct positions for additional lithium atoms.
 | ||
| Fig. 2 (a) z-Displacement of the upper layers, (b) the final structure and the marked upper layers. | ||
The reaction energy for each lithiation step was calculated via
 | (1) |
| S8 + 2Li+ + 2e− → Li2S8, | (2) |
3. Results and discussion
In particular, we try to provide answers to the following questions:1. How does the surface topography affect the lithiation reactions?
2. How comparable are the product structures to the known gas/liquid-phase products at each lithiation step?
3. Is the lithiation of the surface a homogeneous process or happens through nucleation?
To provide answers to these questions, we focus on the first few lithiation reactions corresponding to the early stages of discharge.
3.1. First lithiation
The periodicity of the (001) surface along x and y axes reveals two distinct S8 rings on the surface [Fig. 3(a)]. Notably, in Fig. 3(b), one S8 ring resides in the upper layer, while the other is positioned in the lower layer. These layers alternate on the surface and are highlighted in Fig. 3(a) and (b) as “up” and “down”. The repeating unit of two S8 rings [Fig. 3(a)] results in the recurrence of distinct positions for the lithiation reactions [Fig. 3(c)]. These distinct positions serve as representative sites for two Li atoms in the reactant structures and are expected to break one S–S covalent bond in the S8 rings after the reaction. As such, S(1)/S(2) in Fig. 3(c) denote the upper S8 ring, while S(3)/S(4) represent the distinct positions on the lower S8 ring. | ||
| Fig. 3 First lithiation step: (a) repetitive unit of the S8 surface from top view, (b) upper and lower layers of the S8 surface from side view, (c) distinct positions as reaction centers. | ||
Fig. 4(a) presents the product structures of the first lithiation reaction (only the affected S8 ring is shown), while Fig. 4(b) shows the reaction energy distribution of all the possible products. In Fig. 4(b), for example, 4p refers to the S8(4) structure in (a) where the two Li atoms were initially placed in a parallel configuration with respect to the (001) surface. The difference in reaction energies between the product structures is significant, namely up to 0.7 eV per lithium atom. This is due to the fact that the molecular structure of each product is sizably different. The energy distributions of the subsequent lithiation steps (i.e. second and third) were not shown as the reaction energy ranges were found to be similar to the energy distribution in Fig. 4(b). Despite their similar configuration (Table 2), the 2p product structure on the upper ring corresponds to a lithiation reaction with a reaction energy ∼0.21 eV higher than that of the 3o product structure on the lower ring. Based on these results, it can be concluded that lower S8 rings are energetically preferred for lithiation compared to upper ones. This conclusion was additionally validated for the following lithiation steps with a broader sample size of initial geometries.
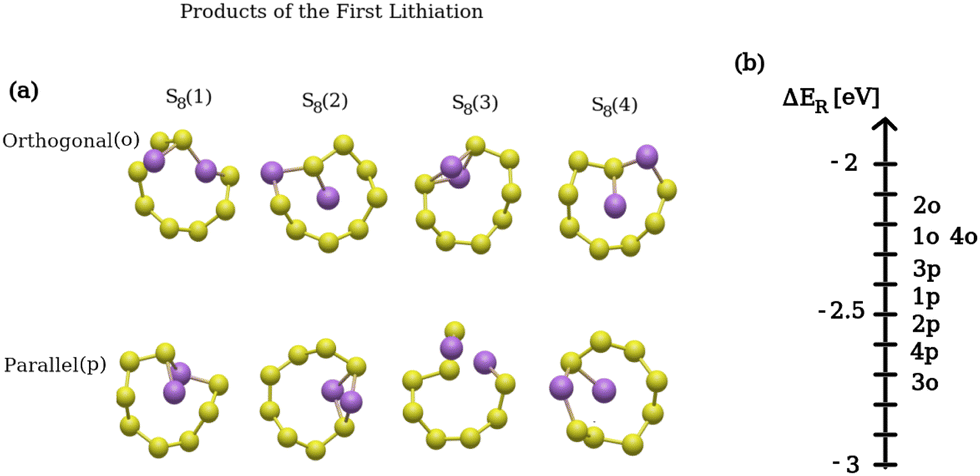 | ||
| Fig. 4 First lithiation step: (a) calculated product structures after the first lithiation (only the affected S8 ring is shown). (b) Distribution of the reaction energies. In (b), for example 4p refers to the S8(4) structure in (a) where the two Li atoms were initially positioned in parallel with respect to the (001) surface. | ||
| Structure | S(1)–Li(1) | S(2)–Li(1) | S(1)–Li(2) | S(2)–Li(2) |
|---|---|---|---|---|
| 2p | 2.35 | 2.38 | 2.45 | 2.38 |
| 3o | 2.40 | 2.41 | 2.38 | 2.41 |
| Ref. 48 | 2.44 | 2.44 | 2.44 | 2.44 |
Another important point to note is that the optimal structures closely resembled the Li2S8 structure in the gas/liquid phase, as shown in Table 2. The previous gas/liquid-phase results,48 were obtained on a ring-shaped structure where a mesomeric rhombus forms via two similar Li–S bonds after the lithiation. These observations also align with the 2p and 3o structures obtained here. Additionally, we calculated the product structures corresponding to the gas- and liquid-phase lithiation reactions and are reported in the ESI.† Our results also showed a diamond-shaped structure for Li2S8 with slightly different bond angles and distances compared to those reported in ref. 48. Besides, the structural differences between gas- and liquid-phase structures are marginal (see Fig. S5 and Table S2 in the ESI†). On the (001) surface, the Li2S8 structures of 2p and 3o also exhibit equidistant Li–S bonds and form a mesomeric rhombus structure. They also result in a ring-shaped product structure. Here, the 3o structure, corresponding to a lithiation reaction with the lowest reaction energy, was considered as the initial structure for the next lithiation step.
3.2. Second lithiation
For the second reaction, two possibilities can be considered: (i) a neighboring ring undergoes a lithiation; or (ii) the Li2S8 becomes further lithiated to two lower-order polysulfides. These correspond to the following two reactions,| S8 + 2Li+ + 2e− → Li2S8 |
| Li2S8 + 2Li+ + 2e− → Li2S8−x/Li2Sx, | (3) |
For the second lithiation, two different classes of distinct positions were selected. Firstly, positions surrounding the previously lithiated Li2S8 ring, and secondly, positions around intact S8 rings. The positions surrounding the Li2S8 molecule were selected in such a way that the new Li atoms are allowed to attack S–S bonds of the Li2S8 molecule which are accessible on the surface. Due to its asymmetrical geometry, the accessible bonds on both sides of every Li2S8 molecule were considered. For the intact S8 molecules, positions were chosen to ensure that the rings are not adjacent to the lithiated ones. The chosen positions are identical to those used in the previous lithiation step, with the exception that an additional position was considered closest to the already lithiated center. Moreover, positions on both sides of each S–S bond were considered to account for all lithiation possibilities. Two S8 molecules in the lower layer (L) and two S8 rings in the upper layer (U) were chosen [Fig. 5(b)]. It is worth mentioning that, in the second lithiation, the use of ‘U’ for upper and ‘L’ for lower sulfur layers in Fig. 5 refers only to the initial structures. This implies that the position of the two Li atoms on an upper layer could result in a polysulfide product structure formed on the lower layer and vice versa. Although such an occurrence was observed to happen rarely, it was taken into account for the average energy calculation of upper- and lower-layer conformations, as well as for comparing structures.
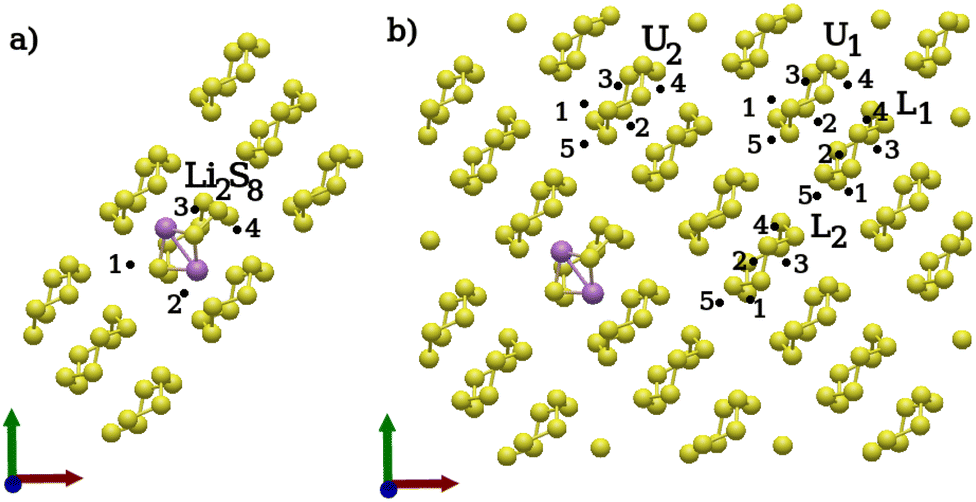 | ||
| Fig. 5 Second lithiation step: distinct positions considered for the second lithiation. (a) Distinct positions around an Li2S8 molecule. (b) Distinct positions around an intact S8. | ||
Fig. 6 shows the lowest-reaction energy structures formed on the upper and lower sulfur layers. We observe that the two Li atoms around the Li2S8 ring do react with the upper neighboring rings but not with the already lithiated one. While the direct subsequent lithiation of the already lithiated ring is in principle possible, it appears to be less likely. The underlying reason could be that the lithiated ring is strongly protected by the surrounding pristine S8 rings of the surface, and the only exposed site is where the two lithium atoms from the first lithiation step are located. Hence, the reactivity of the Li2S8 ring in a second lithiation step is low, and the second lithiation step is more likely to occur at neighboring S8 rings. Li2S8 1p describes the product of sulfur ring in an upper layer [Fig. 6] interacting with the adjacent Li2S8 ring from the lower layer. Therefore, Li2S8 emerges as the only stable product of the second lithiation step, as the lithiation around an Li2S8 molecule from the first lithiation step leads to the formation of a Li2S8 dimer. As such, only the first reaction in (3) seems to be thermodynamically likely.
 | ||
| Fig. 6 Second lithiation step: comparison of the lowest-reaction energy structures formed on the lower (left) and the upper (right) sulfur layers. Li2S8 1p refers to the product of the lithiation reaction on an adjacent sulfur ring to the already lithiated one. The others are isolated rings. | ||
Table 3 represents Li–S distances in these product structures, namely L1 1p, L2 3o, and Li2S8 1p, for comparison with a Li2S8 molecule in the gas phase. All products have comparable Li–S bond distances, with an average bond length of ∼2.40 Å. Their atomic configurations are also similar to an Li2S8 molecule in the gas phase. The reaction energies of all structures are also similar, with the product structures L2 3o and L1 1p being around 50 meV lower in energy than Li2S8 1p obtained through lithiation around Li2S8. A closer look reveals that structures L2 3o and L1 1p (two Li2S8 rings far away from each other) are the products of lithiation of two distant S8 rings both in lower layers. We also observe that the L2 5p product structure consisting of two Li2S8 rings both on lower layers shows about 200 meV higher reaction energy than L2 3o, L1 1p, and Li2S8 1p. One possible reason for this is that in the Li2S8 1p product structure, the lithium atoms can interact strongly with the lone-pair electrons on the terminal sulfur atoms from the neighboring Li2S8 ring [Fig. 6, top-right panel].
| Structure | S(1)–Li(1) | S(2)–Li(1) | S(1)–Li(2) | S(2)–Li(2) |
|---|---|---|---|---|
| Li2S8 1p | 2.43 | 2.42 | 2.37 | 2.35 |
| L2 3o | 2.42 | 2.39 | 2.38 | 2.43 |
| L1 1p | 2.39 | 2.42 | 2.38 | 2.38 |
| Ref. 48 | 2.44 | 2.44 | 2.44 | 2.44 |
Compared to the first lithiation, the reaction energies of L1 1p and L2 3o are similar to that of the product of the first lithiation. This is expected since they occur on isolated S8 rings. For the next lithiation step, the product structure Li2S8 1p was selected, because even though the other two structures (L1 1p and L2 3o) correspond to a slightly lower reaction energies, they would only represent a similar starting structure as for the second lithiation.
3.3. Third lithiation
Here, all possible reaction paths for a third lithiation step are considered as following| S8 + 2Li+ + 2e− → Li2S8 |
| Li2S8 + 2Li+ + 2e− → Li2S7/Li2S |
| Li2S8 + 2Li+ + 2e− → Li2S6/Li2S2 |
| Li2S8 + 2Li+ + 2e− → Li2S5/Li2S3 |
| Li2S8 + 2Li+ + 2e− → Li2S4/Li2S4 | (4) |
As before, several S8 rings and prelithiated Li2S8 structures were considered as potential reaction sites. The positions for the two additional Li atoms around the preexisting Li2S8 are shown in Fig. 7(a). The structures of the two neighboring Li2S8 rings are sizably different from a single Li2S8 molecule in the gas phase. Therefore, different positions for the two Li atoms are evenly distributed around the Li2S8 rings. Here, the positions 1–4, 11–12 are around the upper Li2S8 ring and 5–9 around the lower one [Fig. 7(a)]. For the distinct positions around an intact S8 in Fig. 7(b), lower layer rings (L1, L4) adjacent to the two Li2S8 molecules and those farther away (L2, L3) from them are considered. The same procedure was applied for the upper S8 rings in Fig. 7(c). The lithiation process on isolated rings (L2, L3 and U2, U3) only result in Li2S8 molecules similar to the previous lithiation steps. However, a third lithiation reaction adjacent and on the two Li2S8 rings (L1, L4 and U1, U4, Li2S8) results in more complex structures, such as Li2S7/Li2S, Li2S6/Li2S2 and Li2S5/Li2S3 clusters and bridged structures like Li2S8/Li2S8. Atomic coordinate optimizations of the reactant structures mostly result in Li2S7/Li2S and Li2S6/Li2S2 complexes while clusters of Li2S4/Li2S4 are not observed. One explanation for the absence of Li2S4/Li2S4 clusters is that the S–S bonds are not accessible to the two Li atoms due to their location beneath the reachable surface. This is in stark contrast to the gas/liquid-phase reactions. Without exception, the structures of the isolated or non-interacting Li2S8 molecules with the lowest reaction energies are those that are most similar to Li2S8 in the gas phase [Fig. 8]. This is consistent with the first and second lithiation steps.
 | ||
| Fig. 7 Third lithiation step: distinct positions on the (001) α-sulfur surface considered for the third lithiation reaction, (a) around Li2S8 dimer obtained in the previous lithiation step, (b) on the lower layers, and (c) on the upper layers around the dimer. | ||
 | ||
| Fig. 8 Third lithiation step: comparison of the structures with the lowest reaction energies formed on the lower (left) and the upper (right) layers of the (001) surface. | ||
We observe that in cases where an adjacent S8 ring is lithiated, the atomic configuration of the emerging Li2S8 cluster is very different from the monomers in the gas phase. Therefore, the comparison of the lowest-reaction energy structures to the Li2S8 molecules in the gas phase is only possible when the Li2S8 molecule is not directly interacting with other Li-polysulfides. This is illustrated in Fig. 9, where a distorted structure called Li2S8 7p is shown corresponding to a lower reaction energy than a similar gas-phase structure. The formation of Li2S8 7p might be an indicator that it is thermodynamically preferred that the lithiated structure forms on a lower adjacent S8 ring where the two Li atoms are exposed to the terminal S atoms of the neighboring Li2S8 ring [Fig. 9].
 | ||
| Fig. 9 Third lithiation step: comparison of two Li2S8 adjacent structures formed during the third lithiation reaction. | ||
Quite comparable in reaction energies to the product structures such as Li2S8 7p and L4 3p [Fig. 9] are the cluster/bridged structures shown in Fig. 10.
 | ||
| Fig. 10 Third lithiation step: mesomeric structures. | ||
Of all the cluster and bridged structures, only those with the lowest reaction energies are presented in Fig. 10. For example, the Li2S7/Li2S family of product structures [see reactions (4)], which exhibit considerably higher reaction energies, are not shown. All of these structures display a specific mesomeric pattern, as indicated by the dashed lines in Fig. 10. The results highlight two distinct types of lithiation products: those involving two lithiated Li2S8 rings (L4 2o) and those where an already lithiated Li2S8 molecule undergoes further lithiation (Li2S8 8p and L4 1p). Additionally, some cluster structures, such as Li2S6/Li2S2 (Fig. S1 in the ESI†), exhibit similar mesomerism but have higher reaction energies, likely due to strong electrostatic repulsion between closely positioned lithium atoms.
Despite these observations, the third lithiation step exhibits limited predictive insights onto the preferred conversion pathway due to comparable reaction energies across multiple product configurations. While mesomeric structures and lower adjacent Li2S8 products are thermodynamically favored, Li2S7/Li2S formation is energetically unfavorable on the sulfur surface. Additionally, Li2S4 dimerization is precluded by steric hindrance, as simultaneous Li+ diffusion beneath Li2S8 structures to access buried S–S bonds is geometrically restricted. Reactivity trends further indicate that lower adjacent S8 rings exhibit a higher lithiation propensity compared to isolated or upper-layer rings. This is likely due to reduced Li+ migration barriers and cooperative interactions with neighboring intermediates. While definitive reaction pathways remain unresolved, these insights help narrow plausible mechanisms by eliminating improbable reactions (e.g., Li2S7 formation) and identifying preferred structural motifs (e.g., adjacent Li2S8 interactions). Ultimately, our findings suggest that lower-lying Li2S8 rings interacting with already lithiated S8 molecules, as well as Li-polysulfide cluster-type structures such as Li2S6/Li2S2 or Li2S5/Li2S3, are the most favorable products of the third lithiation step.
4. Conclusions
Our study provides a detailed atomistic insight into the initial lithiation process on the (001) surface of -sulfur, offering a fundamental understanding of the early-stage reactions in Li–S batteries. The findings suggest that lithiation preferentially begins with S8 rings on the lower layer of the surface, leading to the formation of Li2S8 molecules. These surface-stabilized Li2S8 molecules exhibit strong similarities to their counterparts in gas/liquid-phase lithiation, indicating that early surface lithiation plays a critical role in determining subsequent reaction pathways. As lithiation progresses, further reactions around Li2S8 dimers lead to the formation of Li-polysulfides, including Li2S6, Li2S5, and lower-order species like Li2S3 and Li2S2. Notably, the formation of Li2S4 is sterically hindered on the surface, which may explain its preferential formation in the liquid phase. The dissolution of higher-order polysulfides into the electrolyte could further drive their reduction, reinforcing the experimentally observed discharge mechanism. These insights contribute to a deeper understanding of solid–liquid interactions during Li–S battery discharge and highlight the significance of surface lithiation in shaping polysulfide evolution.Therefore, the current study presents a detailed atomistic picture of the first lithiation reactions on the (001) surface of α-sulfur and suggests a discharge mechanism of Li–S batteries with crystalline sulfur cathode which can contribute to a better understanding of such a complex process involving both solid- and liquid-phase reactions.
Author contributions
J. L. contributed to formal analysis, investigation, methodology, visualization, and writing (original draft, review & editing); D. S. contributed to supervision, validation, and writing (review & editing); P. P.-A. contributed to conceptualization, funding acquisition, methodology, supervision, validation, and writing (review & editing).Data availability
All data and code supporting this study are publicly available at https://github.com/jolu75/Data. The data were curated and maintained by J. Lührs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the RTG BEAM (project number 329482734). The authors gratefully acknowledge DFG funding via projects PA3141/3 (project number 420536636), PA3141/5 (project number 446879138). The authors also acknowledge the computing time made available on the high-performance computer at the NHR Center of TU Dresden via the project ‘p_oligothiophenes’. This center is jointly supported by the Federal Ministry of Education and Research and the state governments participating in the NHR (https://www.nhr-verein.de/unsere-partner). P. P.-A. also gratefully acknowledges Rafael Müller for fruitful discussions.Notes and references
- M. Wild and G. Offer, Lithium–Sulfur Batteries, Wiley, London UK, 1st edn, 2019 Search PubMed.
- P. Stenzel, M. Baumann, J. Fleer, B. Zimmermann and M. Weil, 2014 IEEE International Energy Conference (ENERGYCON), 2014.
- G. E. Blomgren, J. Electrochem. Soc., 2016, 164, A5019–A5025 CrossRef.
- K. Amine, R. Kanno and Y. Tzeng, MRS Bull., 2014, 39, 395–401 CrossRef CAS.
- Y. Jin, K. Liu, J. Lang, X. Jiang, Z. Zheng, Q. Su, Z. Huang, Y. Long, C.-A. Wang, H. Wu and Y. Cui, Joule, 2020, 4, 262–274 CrossRef CAS.
- A. Rosenman, E. Markevich, G. Salitra, D. Aurbach, A. Garsuch and F. F. Chesneau, Adv. Energy Mater., 2015, 5, 1500212 CrossRef.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969 CrossRef CAS.
- Y. Huang, L. Lin, C. Zhang, L. Liu, Y. Li, Z. Qiao, J. Lin, Q. Wei, L. Wang, Q. Xie and D. Peng, Adv. Sci., 2022, 9, 2106004 CrossRef CAS PubMed.
- R. Xu, I. Belharouak, X. Zhang, R. Chamoun, C. Yu, Y. Ren, A. Nie, R. Shahbazian-Yassar, J. Lu, J. C. Li and K. Amine, ACS Appl. Mater. Interfaces, 2014, 6, 21938–21945 CrossRef CAS PubMed.
- T. D. Pham, et al. , Small, 2024, 20, 202307951 Search PubMed.
- Y. Lin, J. Zheng, C. Wang and Y. Qi, Nano Energy, 2020, 75, 104915 CrossRef CAS.
- J. L. D. Salvo, G. L. Luque and G. D. Luca, Mol. Syst. Des. Eng., 2022, 7, 364–373 RSC.
- P. Partovi-Azar, S. Panahian Jand and P. Kaghazchi, Phys. Rev. Appl., 2018, 9, 014012 CrossRef CAS.
- J. Shen, Y. Feng, P. Wang, G. Qiu, L. Zhang, L. Lu, H. Wang, R. Wang, V. Linkov and S. Ji, ACS Sustainable Chem. Eng., 2020, 8, 10389–10401 CrossRef CAS.
- C.-J. Huang, J.-H. Cheng, W.-N. Su, P. Partovi-Azar, L.-Y. Kuo, M.-C. Tsai, M.-H. Lin, S. P. Jand, T.-S. Chan and N.-L. Wu, et al. , J. Power Sources, 2021, 492, 229508 CrossRef CAS.
- Q. Zhang, Q. Huang, S.-M. Hao, S. Deng, Q. He, Z. Lin and Y. Yang, Adv. Sci., 2022, 9, 2103798 CrossRef CAS PubMed.
- P. Partovi-Azar, Phys. Rev. Appl., 2022, 18, 044072 CrossRef CAS.
- R. Kiani, D. Sebastiani and P. Partovi-Azar, ChemPhysChem, 2022, 23, e202100519 CrossRef CAS PubMed.
- S. de Kock, K. Skudler, R. Matsidik, M. Sommer, M. Müller and M. Walter, Phys. Chem. Chem. Phys., 2023, 25, 20395–20404 RSC.
- H. Raza, S. Bai, J. Cheng, S. Majumder, H. Zhu, Q. Liu, G. Zheng, X. Li and G. Chen, Electrochem. Energy Rev., 2023, 6, 29 CrossRef CAS.
- R. Kiani, M. Steimecke, M. Alqaisi, M. Bron, D. Sebastiani and P. Partovi-Azar, RSC Adv., 2023, 13, 27756–27763 RSC.
- Y. Schütze, D. Gayen, K. Palczynski, R. de Oliveira Silva, Y. Lu, M. Tovar, P. Partovi-Azar, A. Bande and J. Dzubiella, ACS Nano, 2023, 17, 7889–7900 CrossRef PubMed.
- R. Kiani, H. Sheng, T. Held, O. Löhmann, S. Risse, D. Sebastiani and P. Partovi-Azar, ChemPhysChem, 2024, e202400681 Search PubMed.
- H. Liao, H. Ding, B. Li, X. Ai and C. Wang, J. Mater. Chem. A, 2014, 2, 8854–8858 RSC.
- Z.-J. Zheng, H. Ye and Z.-P. Guo, Energy Environ. Sci., 2021, 14, 1835–1853 RSC.
- Z. Wang, F. Pan, Q. Zhao, M. Lv and B. Zhang, Front. Chem., 2022, 10, 1055649 CrossRef CAS.
- R. Yan, B. Mishra, M. Traxler, J. Roeser, N. Chaoui, B. Kumbhakar, J. Schmidt, S. Li, A. Thomas and P. Pachfule, Angew. Chem., Int. Ed., 2023, 62, e202302276 CrossRef CAS PubMed.
- B. Hu, J. Xu, Z. Fan, C. Xu, S. Han, J. Zhang, L. Ma, B. Ding, Z. Zhuang and Q. Kang, et al. , Adv. Energy Mater., 2023, 13, 2203540 CrossRef CAS.
- S. Lv, X. Ma, S. Ke, Y. Wang, T. Ma, S. Yuan, Z. Jin and J.-L. Zuo, J. Am. Chem. Soc., 2024, 146, 9385–9394 CrossRef CAS PubMed.
- Z. Wang, X. Wu, S. Wei, Y. Xie and C.-Z. Lu, Chem. Mater., 2024, 36, 2412–2419 CrossRef CAS.
- P. P. R. M. L. Harks, C. B. Robledo, T. W. Verhallen, P. H. L. Notten and F. M. Mulder, Adv. Energy Mater., 2016, 7, 1601635 CrossRef.
- T. Ma, et al. , Angew. Chem., Int. Ed., 2023, 62, e202310761 CrossRef CAS PubMed.
- C. Arneson, Z. D. Wawrzyniakowski, J. T. Postlewaite and Y. Ma, J. Phys. Chem. C, 2018, 122, 8769–8779 CrossRef CAS.
- L. Wang, T. Zhang, S. Yang, F. Cheng, J. Liang and J. Chen, J. Energy Chem., 2013, 22, 72–77 CrossRef CAS.
- R. Xu, J. Lu and K. Amine, Adv. Energy Mater., 2015, 5, 1500408 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- S. C. Abrahams, Acta Crystallogr., 1955, 8, 661–671 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. VandeVondele and J. Hutter, J. Phys. Chem., 2007, 127, 114105 CrossRef.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Phys. Chem., 2010, 132, 154104 CrossRef PubMed.
- C. G. Broyden, IMA J. Appl. Math., 1970, 6, 76–90 CrossRef.
- R. Fletcher, Comput. J., 1970, 13, 317–322 CrossRef.
- D. Goldfarb, Math. Comput., 1970, 24, 23–26 CrossRef.
- D. F. Shanno, Math. Comput., 1970, 24, 647–656 CrossRef.
- M. Cheviri and S. Lakshmipathi, Comput. Theor. Chem., 2021, 1202, 113323 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ya00050e |
| This journal is © The Royal Society of Chemistry 2025 |