
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReducing non-radiative voltage losses in organic solar cells using molecular encapsulation†
Anirudh
Sharma‡
 a,
Lisa
Sharma‡
b,
Jules
Bertrandie
a,
Diego R.
Villalva
a,
Yajun
Gao
a,
Catherine S. P.
De Castro
a,
Joel
Troughton
a,
Julien
Gorenflot
a,
Frederic
Laquai
a,
Hugo
Bronstein
*b and
Derya
Baran
*a
a,
Lisa
Sharma‡
b,
Jules
Bertrandie
a,
Diego R.
Villalva
a,
Yajun
Gao
a,
Catherine S. P.
De Castro
a,
Joel
Troughton
a,
Julien
Gorenflot
a,
Frederic
Laquai
a,
Hugo
Bronstein
*b and
Derya
Baran
*a
aKAUST Solar Center (KSC), Physical Sciences and Engineering Division (PSE), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955, Kingdom of Saudi Arabia. E-mail: derya.baran@kaust.edu.sa
bDepartment of Chemistry and Physics, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: hab60@cam.ac.uk
First published on 3rd January 2023
Abstract
Non-radiative voltage loss is one of the significant bottlenecks limiting the performance of organic solar cells (OSCs). This work presents a material design approach for conjugated polymers isolating the polymer chains with molecular encapsulation, enabling better control over the donor–acceptor spacing. Bulk-heterojunction OSCs based on encapsulated polymers as electron donors show a remarkable increase of ∼50 mV in the Voc. Significant improvement in the Voc is attributed to reduced non-radiative voltage losses as evidenced by enhanced electroluminescence as a result of suppressed intermolecular aggregation and an increase in structural disorder. Importantly, molecular encapsulation resulted in notable improvement in the operational stability of OSCs with no burn-in loss in Voc, presenting a promising strategy to simultaneously enhance the performance and operational stability of OSCs.
Introduction
Organic solar cells (OSCs) witnessed tremendous progress in the last two decades,1 now achieving power conversion efficiencies (PCEs) of up to 19% for single-junction devices.2,3 The PCE of OSCs is limited mainly by the open-circuit voltage (Voc), often lower than the optical bandgap (Eg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) opt), mainly due to non-radiative charge recombination. As per the Shockley–Queisser model, an ideal solar cell should also be an ideal light-emitting diode (LED), having only radiative recombination (100% electroluminescence external quantum efficiency EQEEL).4,5 However, in actual devices, non-radiative recombination simultaneously takes place, which is relatively more predominant in OSCs than inorganic counterparts.6
opt), mainly due to non-radiative charge recombination. As per the Shockley–Queisser model, an ideal solar cell should also be an ideal light-emitting diode (LED), having only radiative recombination (100% electroluminescence external quantum efficiency EQEEL).4,5 However, in actual devices, non-radiative recombination simultaneously takes place, which is relatively more predominant in OSCs than inorganic counterparts.6
In most OSCs, EQEEL is very low typically on the order of ∼10−6–10−4, resulting in total energy loss ΔEloss (Egopt − qVoc) of ∼500–600 meV, with non-radiative voltage loss (ΔVocnr) being the key loss mechanism,7–9 and is expressed as:
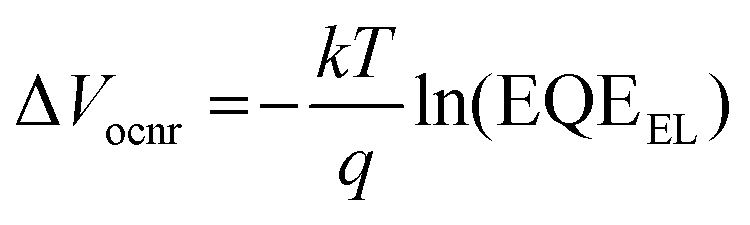 | (1) |
So far, the rapid progress in the development of OSCs can be attributed mainly to the design and synthesis of new materials, primarily focusing on minimizing the voltage losses,10,12 and tuning the optical bandgap and frontier molecular orbital energies.13–16 Nevertheless, besides optimizing the energetic offsets, material design strategies are still needed to intrinsically limit the (ΔVocnr) that occur due to the recombination of the photo-generated charges.
Conjugated push–pull polymers based on electron-deficient lactam core of diketopyrrolopyrrole (DPP) have found a wide range of applications for optoelectronic devices,17 owing to their good electronic properties, photochemical stability, and industrial relevance. However, due to a planar backbone, DPP polymers have been found to have a strong tendency to aggregate, which can limit the performance of OSCs via geminate charge recombination.18 Furthermore, polymer aggregates, for example, are known to result in the formation of non-emissive charge transfer states,19 limiting the device performance. Wang et al. have recently demonstrated that by increasing the donor–acceptor (DA) spacing, Voc of OSCs can be improved via reduced non-radiative decay of charge carriers.20
Covalently attached molecular encapsulation previously reported by Sugiyasu,21 Pan,22 and others has been shown to reduce π–π stacking interactions in conjugated polymers by isolating the polymer chains, significantly improving the emissivity of the polymers.23 However, due to the reduction/elimination of π–π interactions, which would likely result in reduced charge-carrier mobility, encapsulated conjugated polymers have rarely been successfully implemented into optoelectronic devices.
In this study, we use DPP-based polymers as the model system to demonstrate the reduction of (ΔVocnr) in OSCs. This is achieved by tuning the morphological and optoelectronic properties of polymers utilizing molecular encapsulation.23 We utilize a half-encapsulated approach to modulate the properties of thiophene-flanked DPP-based conjugated polymer. By only protecting a single face of a monomer unit and additionally introducing it at a low concentration into a non-encapsulated host polymer, we aim to maintain sufficient bulk charge carrier mobility whilst improving the luminescence efficiency of a BHJ OSC and hence reduce the (ΔVocnr). We selected narrow bandgap polymer PDPP-TT, first synthesized by Li et al.,24 due to its good performance in OSCs and high solubility. Here, new analogs of this polymer were synthesized by systematically incorporating varying amounts of half-encapsulated DPP monomer units into the polymer backbone. Encapsulation of the DPP core resulted in a notable enhancement of Voc by 50 mV via reduced(ΔVocnr). Molecular encapsulation reduced polymer aggregation, enhancing the electroluminescence in polymer:PC71BM OSCs. Importantly, no changes were observed in the ionization energy of the polymers as a result of molecular encapsulation, presenting a promising molecular strategy to reduce (ΔVocnr), without the need to design completely new molecules. This promising molecular design strategy can be implemented to a variety of existing π-conjugated systems and be used with alternative non-fullerene acceptors.
Results and discussion
The synthesis of the half-encapsulated monomer is shown in Scheme 1, and its detailed procedure is described in the ESI.† The synthesis of the encapsulated DPP monomer began with the di-alkylation of 1 (DPP) using 1,10-dibromodecane in DMF and K2CO3 as the base. Compound 1 (DPP) was prepared via a method previously reported in the literature.25 In order to minimize the production of the competing side products, including mono- and di-alkylated DPP, a solution of the alkyl chain in DMF underwent slow dropwise addition to a largely diluted basic solution of 1. Upon workup, the crude material was purified via column chromatography, followed by sonication in hexane. The pure product was then collected via filtration to give 2 as a bright pink solid (16%). Finally, 2 underwent di-bromination with NBS at 50 °C to afford 3 as a dark purple solid (79%) (Scheme 1). The structure of compound 3, was determined by X-ray crystallographic analysis (single crystals grown from CHCl3: MeOH, Fig. 1),26 confirming the successful encapsulation of the DPP core. The X-ray structure of 2 was also obtained and can be found in the ESI† (Fig. S1). The successful half-encapsulation can clearly be seen, and the molecule retains a co-planar conformation. However, slight bending of the DPP core can be seen, suggesting some strain may have been introduced through encapsulation.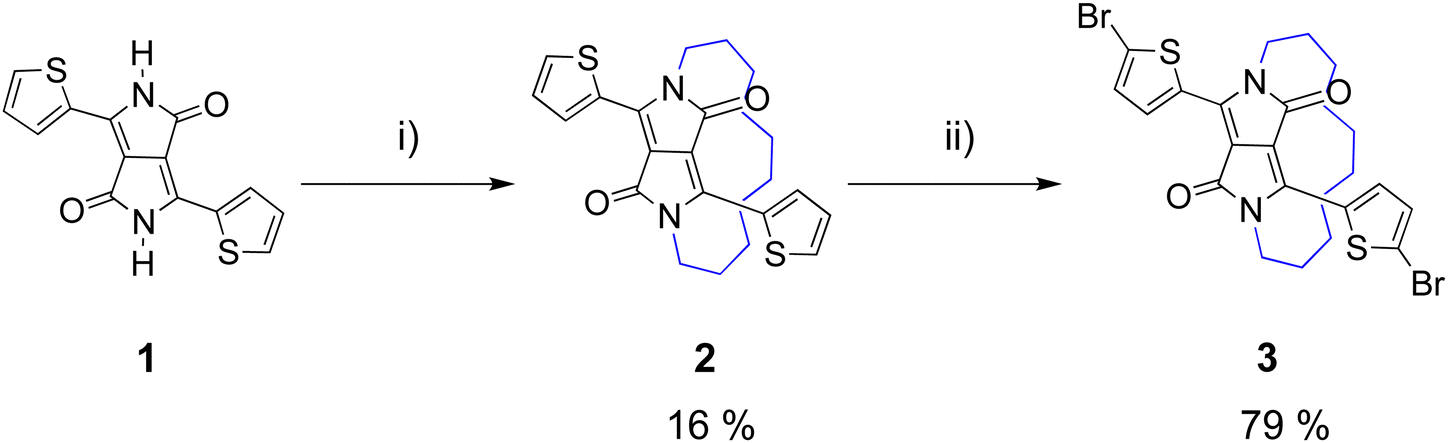 | ||
| Scheme 1 Synthesis of encapsulated DPP monomer 3 (i) 1,10-dibromodecane, K2CO3, 18-crown-6, DMF, 120 °C, 18 h; (ii) NBS, CHCl3, 50 °C, 5 h. | ||
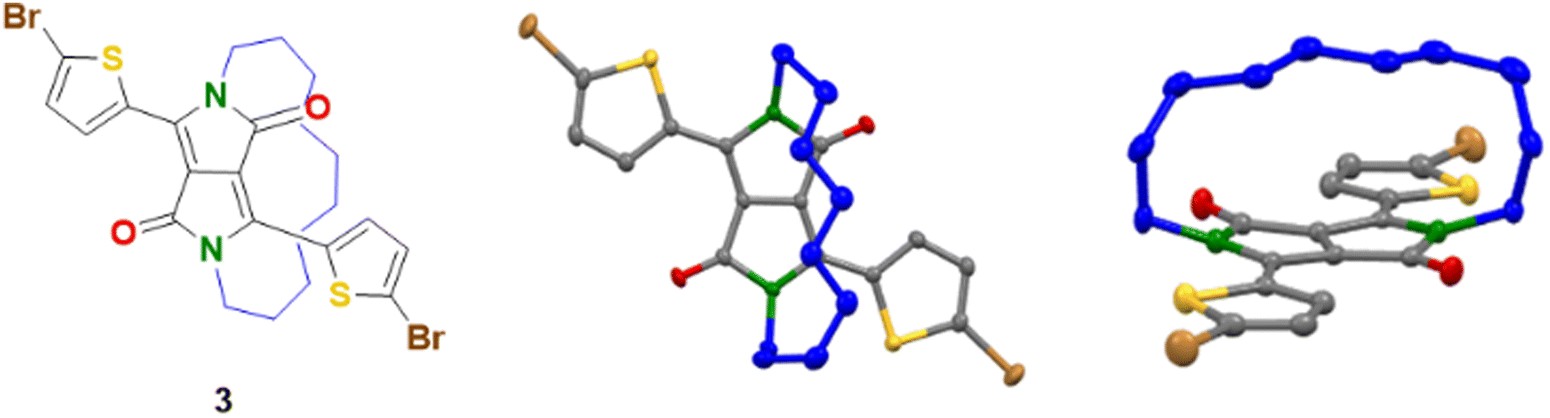 | ||
| Fig. 1 Crystal structure of encapsulated monomer 3 (forward and side perspective view). Crystals grown from CHCl3:MeOH. | ||
Monomer 4 was synthesized based on previously reported literature.27 The reference polymer, PDPP-TTref, (which contains no encapsulated monomer) was synthesized via a Stille cross-coupling between monomer 4 and 2,5-bis(trimethylstannyl)-thieno[3,2-b]thiophene, based on a procedure by Heeger et al.27 (Scheme 2). The polymer then underwent Soxhlet extraction using acetone, hexane and dichloromethane to afford PDPP-TTref at a 77% yield. The new encapsulated polymers, PDPP-TT5%, PDPP-TT10%, and PDPP-TT20%, were then synthesized by incorporating an increasing ratio of monomer 3 in the place of monomer 4 (Table 1), using the same procedure used to synthesize PDPP-TTref, where the term x% reflects the feed ratio of the encapsulated monomer in the polymer. The polymers were then purified via Soxhlet extraction as before and isolated in either dichloromethane (DCM), chlorobenzene (CB), or chloroform (CHCl3). The monomer ratios, molecular weights, and polydispersity index (PDI) of the polymers in the study are summarized in Table 1.
 | ||
| Scheme 2 Synthesis of encapsulated DPP polymers. | ||
| Polymer | Monomer ratio | Molecular weighta | ||||
|---|---|---|---|---|---|---|
| 4 | 3 | TT | M n (g mol−1) | M w (g mol−1) | PDI | |
| a Determined by gel permeation chromatography (GPC) in CB against polystyrene (PS) standard. | ||||||
| PDPP-TTref | 1 | 0 | 1 | 26.3 K | 102.7 K | 3.9 |
| PDPP-TT5% | 0.95 | 0.05 | 1 | 24.4 K | 79.1 K | 3.2 |
| PDPP-TT10% | 0.90 | 0.1 | 1 | 78.4 K | 221.2 K | 2.8 |
| PDPP-TT20% | 0.80 | 0.2 | 1 | 14.0 K | 76.4 K | 5.5 |
The thin-film absorption spectra of PDPP-TTref, as well as of its analogs with varying amounts of encapsulated units, are shown in Fig. 2a. For PDPP-TTref, the absorption peak was found to be at 821 nm, and blue-shifted when increasing the fraction of encapsulated units in the polymer backbone. For all polymer analogs, a blue shift in the absorption was also observed in the solution (Fig. 2b), suggesting reduced aggregation with the introduction of encapsulated units in the polymer backbone. Alternatively, the slight bending of the DPP chromophore as observed in the X-ray structure may have resulted in a slight loss of conjugation along the polymer backbone.
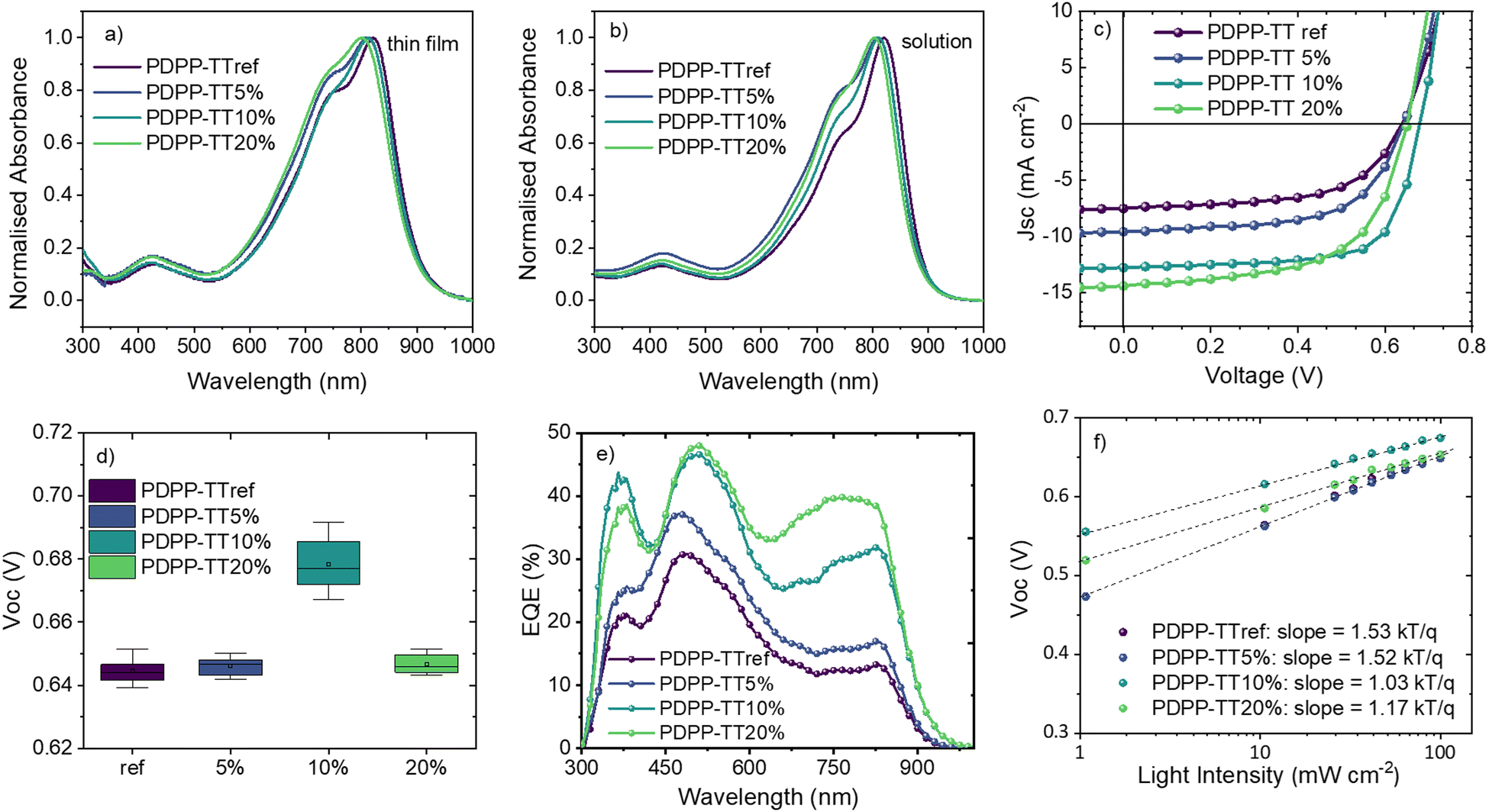 | ||
| Fig. 2 Normalized UV-Vis absorption spectra of different PDPP-TT polymer analogs in (a) thin films (spin-coated from 5 mg mL−1 in CB) and (b) solutions (CB). (c) J–V characteristics, (d) box-plot depicting the statistical distribution of Voc, (e) EQE, and (f) intensity-dependent Voc measurements for OSCs with different PDPP-TT donor polymer analogs. | ||
The photovoltaic properties of OSCs fabricated with an active layer based on different polymer analogs and PC71BM BHJ blends are shown in Fig. 2 and summarized in Table 2. OSCs based on PDPP-TTref demonstrated a modest PCE of 2.8% with a Voc of 0.64 V, which remained unchanged for PDPP-TT5% devices. However, devices based on PDPP-TT10% demonstrated a significantly enhanced Voc of 0.69 V, resulting in the highest PCE of 6.1%. It is worth noting that in the case of PDPP-TT20% devices, a drop of 40 mV was observed in the Voc, compared to the PDPP-TT10% devices. The statistical distribution of device parameters can be found in Fig. S17 (ESI†). Though all devices exhibited a broad spectral response, the EQE of the PDPP-TT10% and PDPP-TT20% devices were found to be significantly higher between ∼650–850 nm resulting in enhanced Jsc, as compared to the devices based on the PDPP-TTref and PDPP-TT5% donor polymers (Fig. 2e). This is potentially due to the increase in the donor–acceptor intermixing owing to the expected reduction in molecular packing in the polymer phase, leading to better extraction of the photo-generated charges.
opt, Urbach energy (Eu), Voc loss of OSCs fabricated using donor polymers based on PDPP-TT analogs with varying fraction of the encapsulated units. The average PCE values are calculated over six devices
| PDPP-TT | J sc (mA cm−2) | V oc (V) | FF (%) | PCEavg (%) | PCEmax (%) |
E
gopt (eV) |
E u (meV) | eΔVoc (eV) | V ocrad (V) | ΔVocnr (V) |
|---|---|---|---|---|---|---|---|---|---|---|
| Ref | 7.5 | 0.64 | 58 | 2.7 | 2.8 | 1.43 | 34.11 ± 0.61 | 0.79 | 1.05 | 0.41 |
| 5% | 9.6 | 0.64 | 61 | 3.6 | 3.8 | 1.43 | 40.33 ± 0.44 | 0.79 | 1.05 | 0.41 |
| 10% | 12.8 | 0.69 | 69 | 6.0 | 6.1 | 1.42 | 38.90 ± 0.22 | 0.73 | 1.04 | 0.35 |
| 20% | 14.4 | 0.65 | 59 | 5.5 | 5.6 | 1.42 | 40.91 ± 0.26 | 0.77 | 1.03 | 0.38 |
To verify the enhancement of Voc with molecular encapsulation in the polymer backbone, analogs of another DPP-based polymer, PDPP-P, were also synthesized.18 OSCs based on PDPP-P:PC71BM BHJ blends also showed an improvement of ∼30 mV in the Voc when encapsulated units were incorporated into the polymer backbone (Fig. S18 and Table S4, ESI†).
To understand the improvement in the Voc upon molecular encapsulation of the donor polymer, we first studied possible changes in the ionization energy (IE) and electron affinity (EA) of DPP-TT polymer analogs using ultraviolet photoelectron spectroscopy (UPS) and low-energy inverse photoelectron spectroscopy (LE-IPES).14 For PDPP-TTref, IE and EA were measured to be 5.16 eV and 3.38 eV (Fig. 3), respectively, corresponding to a transport gap of 1.78 eV. Though the IE of all-polymer analogs was found to be similar within the experimental uncertainties (∼0.05 eV) (PDPP-TT5% – 5.12 eV, PDPP-TT10% – 5.14 eV, PDPP-TT20% – 5.17 eV), the EA of PDPP-TT5%, PDPP-TT10%, and PDPP-TT20% were measured to be 3.48 eV, 3.46 eV, and 3.29 eV, respectively (Fig. 3c). From the frontier molecular orbital energies measured using UPS and LE-IPES, a similar photovoltaic gap (ΔEpv) between the highest occupied molecular orbital (HOMO) of all polymer analogs and the lowest occupied molecular orbital (LUMO) of the electron acceptor material in this case PC71BM, is expected. Thus, the observed enhancement in the device Voc upon molecular encapsulation of the polymer is independent of the material's energetics.
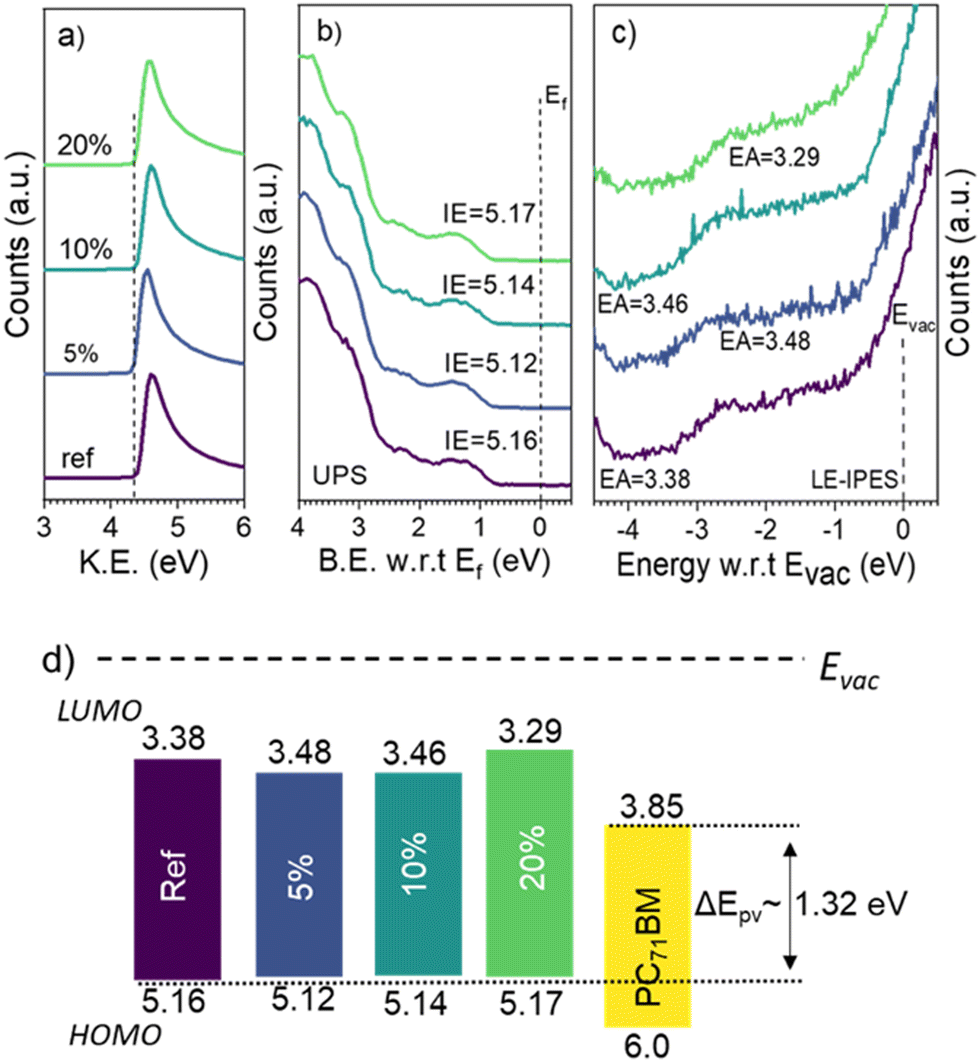 | ||
| Fig. 3 (a and b) Ultraviolet photoelectron spectroscopy (UPS), (c) low-energy inverse photoelectron spectroscopy (LE-IPES) spectra of PDPP-TT polymer analogs, and (d) schematic depicting the measured frontier molecular orbital energies of PDPP-TT polymer analogs and the resultant photovoltaic gap (ΔEpv). | ||
Detailed electrical and photo-physical characterization of PDPP-TT: PC71BM devices were further performed for an insightful understanding of the Voc trend. First, the charge recombination mechanisms were probed by measuring J–V characteristics at different light intensities. Jsc and Voc plotted against the natural logarithm of the light intensity provide information about the bimolecular and trap-assisted recombination, respectively.28 Light intensity-dependent Voc measurements show that when 10% encapsulated units are introduced within the polymer backbone, trap-assisted recombination is significantly reduced, as evident from the calculated slope of 1.53 kTq−1 and 1.03 kTq−1 for PDPP-TTref and PDPP-TT10%, respectively (Fig. 2f). For PDPP-TT20%, the slope slightly increased to 1.17 kTq−1 compared to PDPP-TT10%. The above results suggest that a 10% fraction of encapsulated units within the polymer backbone results in an optimum intra and inter-molecular order, thus facilitating efficient charge generation and transport. This results in the least trap-assisted charge recombination and explains the higher Voc of 0.69 V and FF of 69% for the PDPP-TT10% devices (Table 2). The relatively higher trap-assisted recombination in the case of PDPP-TT20% devices suggests that even though the relatively higher amount of encapsulated fraction in PDPP-TT20% increases the donor–acceptor intermixing (via reduced aggregation in the polymer chains), in this case, the further increased encapsulation has a detrimental impact on the device performance, as reflected by the reduced FF.29 On the other hand, the calculated slope of Jscvs. the natural logarithm of light intensity for various polymer analogs saw a non-significant increase when encapsulated, suggesting little change in the bimolecular recombination (Fig. S17, ESI†).
In order to probe the disorder in the BHJ films when molecular encapsulation is introduced into the polymer backbone, sub-bandgap absorption was measured using photothermal deflection spectroscopy (PDS) (Fig. 4a and Table 2). Below the bandgap, a characteristic single exponential Urbach tail was observed,30 which can be fitted according to eqn (2), where α is the material's absorption coefficient, E, the photon energy, Eu the Urbach energy, and Ec and A are material constants:
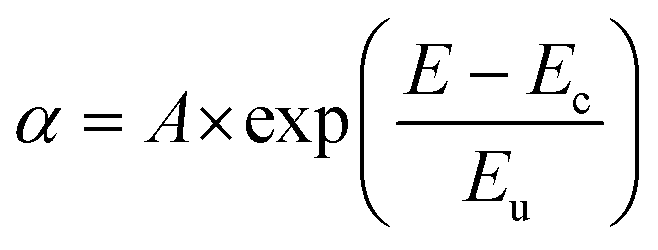 | (2) |
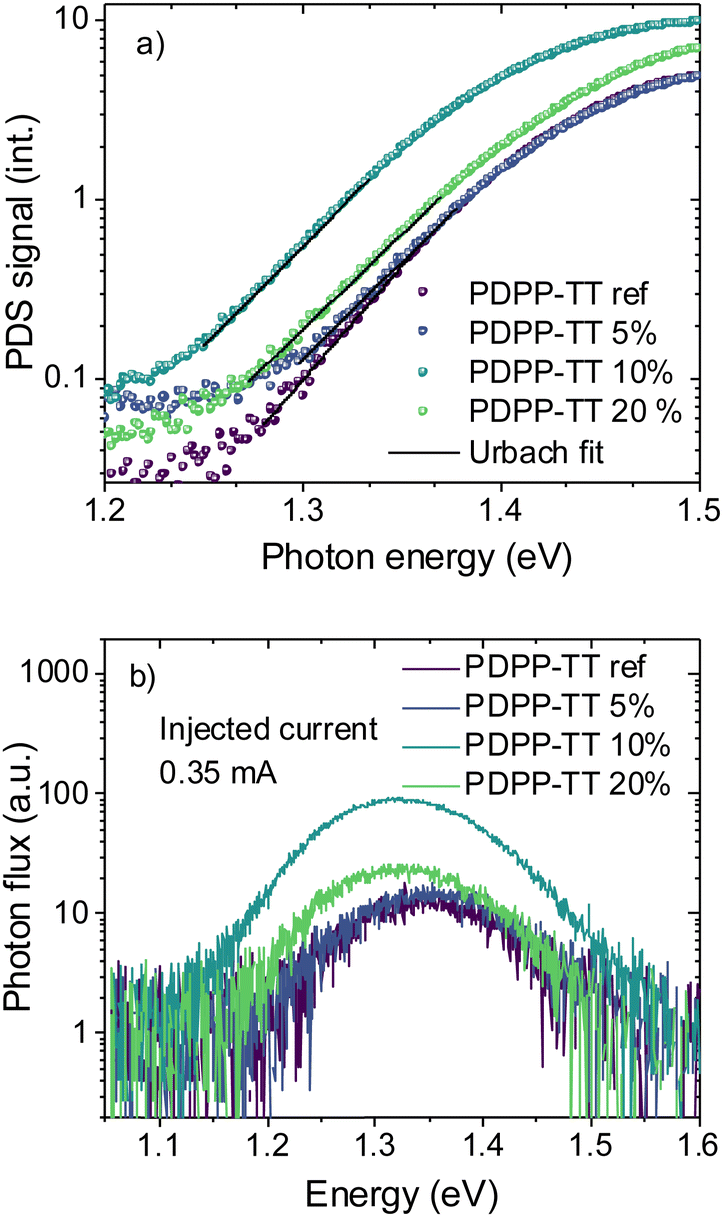 | ||
| Fig. 4 (a) Photothermal deflection spectroscopy (PDS) spectra of polymer-PC71BM BHJ blend films, (b) electroluminescence (EL) spectra (uncorrected for detector sensitivity) measured at the same exposure time, of OSCs based on BHJ blends having various PDPP-TT polymer analogs with varying amounts of encapsulated units in the polymer backbone. | ||
This interpretation is further supported by electroluminescence (EL) measurements performed to study the changes in the emissivity of polymers when encapsulated. Fig. 4b shows the spectra of devices measured at a fixed injection current of 0.35 mA with different PDPP-TT polymer analogs. The EL intensity of the device based on PDPP-TT10% was found to be about an order of magnitude higher at a fixed injection current density of 3.5 mA cm−2, as compared to that in the case of PDPP-TTref devices. This demonstrates that the molecular encapsulation in the polymer backbone leads to higher emissivity by limiting the formation of non-radiative quenching sites, and recombination in such devices is more radiative in nature. Interestingly, when encapsulation was further increased to 20%, the EL intensity was reduced again, identifying PDPP-TT10% devices to be the most emissive. This is in agreement with the photovoltaic parameters of devices, which show that PDPP-TT10% results in OSCs with the highest Voc, and a relatively lower Voc is observed in the case of PDPP-TT20% devices.
To obtain further insight into the Voc enhancement with molecular encapsulation in the polymer backbone, we evaluated the radiative and non-radiative energy loss. The Egopt of the PDPP-TTref and PDPP-TT5% devices, determined as per the reported procedure,32 from the derivative of EQE spectra of the fully functional devices was found to be 1.43 eV. For PDPP-TT10% and PDPP-TT20% devices, an Egopt of 1.42 eV was measured. This agrees with the nearly unchanged IE of the PDPP-TT polymer analogs with varying amounts of encapsulated units (Fig. 3b). Utilizing the measured Egopt and the Voc of devices, ΔEloss was calculated to be 0.79 V for PDPP-TTref and PDPP-TT5% devices, whereas it was 0.73 V for PDPP-TT10% and 0.77 V for the PDPP-TT20% devices, respectively.
The radiative limit (ΔVocrad) and the (ΔVocnr) was calculated by using the relation expressed in eqn (3)–(5),33 where the reconstructed EQEpv from blackbody (ΦBB) corrected EL spectrum was combined with the EQE spectrum (Fig. S19b, ESI†) to enhance the resolution of the measurements.4 In eqn (3), J0,rad represents the current density simulating the blackbody radiation of the device at room temperature, kB is the Boltzmann's constant, E is the photon energy, and T is 300 K.
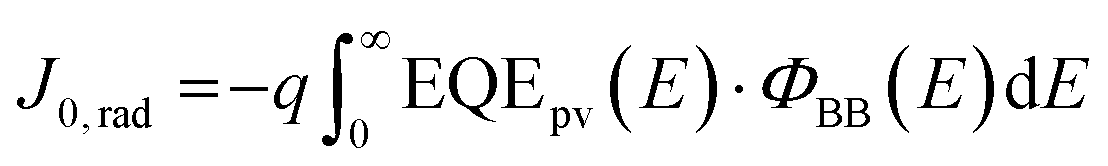 | (3) |
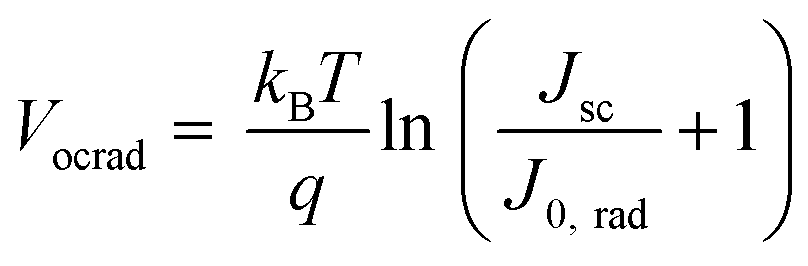 | (4) |
| ΔVocnr = Vocrad − Voc | (5) |
(ΔVocnr) for the PDPP-TTref and PDPP-TT5% devices were calculated to be 0.41 V (Table 2). In the case of PDPP-TT10%, (ΔVocnr) of 0.35 V, ∼ 60 mV lower than the reference device was measured. For PDPP-TT20%, (ΔVocnr) was evaluated to be 0.38 V, 30 mV higher than that in PDPP-TT10% devices, and is in agreement with the higher Voc and enhanced emissivity observed with the PDPP-TT10% devices. This demonstrates that the introduction of molecular encapsulation in the polymer backbone is an effective strategy to reduce(ΔVocnr) in devices by making encapsulated polymers intrinsically more emissive, at the expense of increased structural disorder.
Lastly, morphology characterizations were performed to directly probe the morphological changes upon molecular encapsulation. The surface topographical images of polymer thin films from atomic force microscopy (AFM) show domains with fibrillar structures of polymer chains for PDPP-TTref (Fig. S21a, ESI†). Upon introducing the encapsulated units within the polymer backbone, the fibrillar structures were slightly suppressed for PDPP-TT5%. The fibrillar structures were not evident within the experimental resolution for the PDPP-TT10% and PDPP-TT20%, which show lobular domains. The thin film surface topography of all four polymer analogs, when mixed with [6,6]phenylC71-butric acid methyl ester (PC71BM) in a BHJ blend, was largely consistent with that found for neat polymers. PDPP-TTref and PDPP-TT5% blends with PC71BM displayed molecular aggregates with fibrillar structures, which were subsequently suppressed for BHJ blends of PDPP-TT10% and PDPP-TT20% (Fig. S21b, ESI†).
Grazing-incidence X-ray diffraction (GIXRD) analysis (Fig. 5) of the BHJ blend films shows prominent 100 and 200 peaks for the PDPP-TTref:PC71BM BHJ, owed to the lamellar stacking of the PDPP-TTref polymer, along with isotropic rings corresponding to the packing of PC71BM (Fig. 5). The introduction of the encapsulation (PDPP-TT5%) led to a reduction of the d-spacing of the lamellar stacking of the 100 and 200 reflections (Table S5, ESI†). Additionally, the area of both peaks reduces, suggesting a lower crystallinity of this DPP polymer. Finally, both polymers PDPP-TT10%, and PDPP-TT20% show no prominent lamellar reflections (Fig. 5c and d, respectively), further confirming the suppression of the molecular packing with the introduction of encapsulation into the polymer backbone. The reduced molecular order in the BHJ films upon encapsulation agrees with the observed enhancement in Voc.34
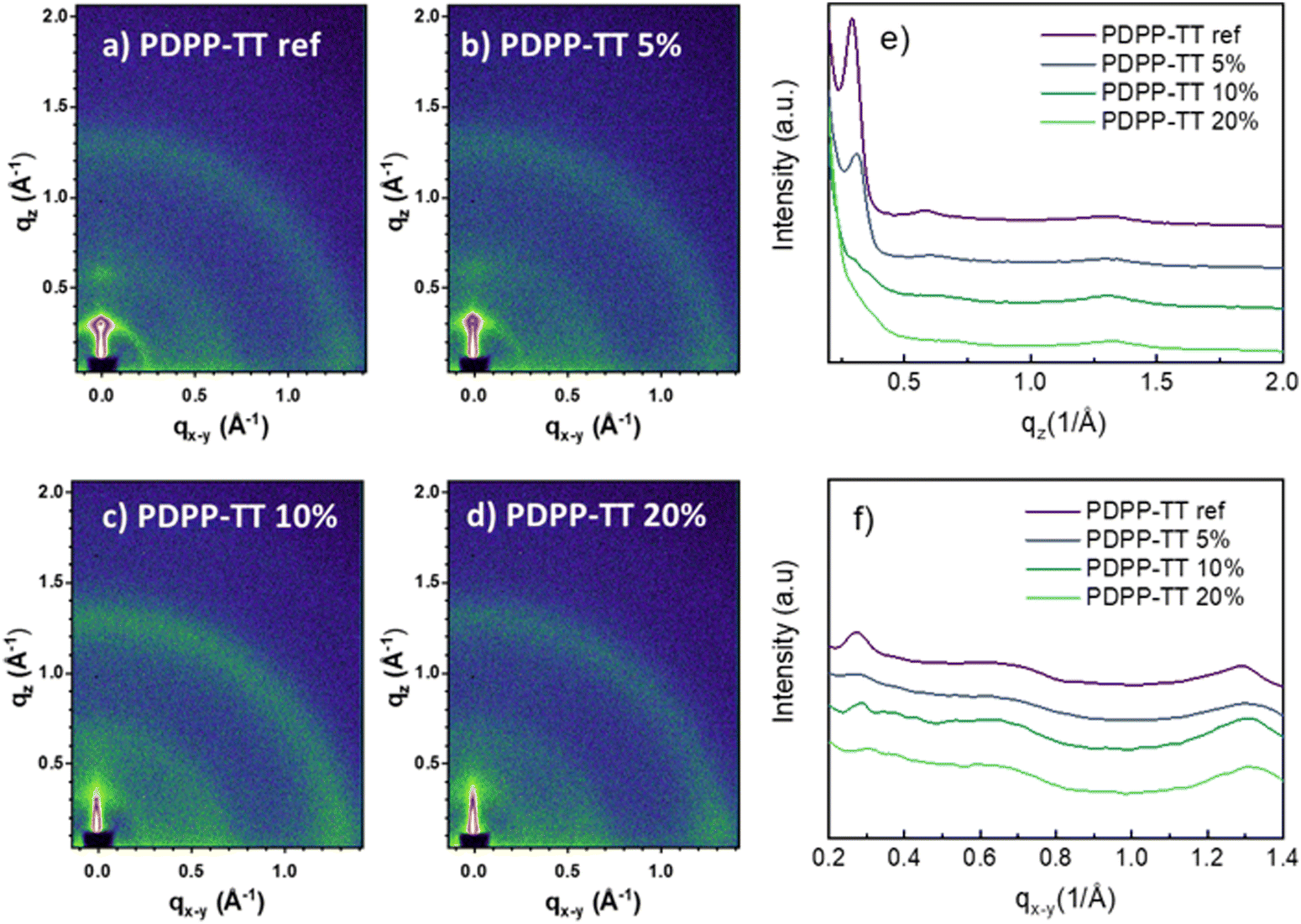 | ||
| Fig. 5 2D-GIXRD profiles of polymer-PC71BM BHJ films with DPP polymer analogs (a) PDPP-TTref, (b) PDPP-TT5%, PDPP-TT10%, and PDPP-TT20%, (e) and (f) show the respective line-cuts. | ||
Changes in the morphology and crystallinity or molecular ordering in polymer thin films often correlate with changes in the thermal35,36 and carrier transport characteristics.37 The charge carrier mobility of all PDPP-TT polymer analogs was thus measured using an organic field-effect transistor (OFET) with a bottom contact-top gate configuration. The OFET demonstrated dominant hole transport characteristics with high linear mobility (μlin) of 0.5 cm2 V−1 S−1 for PDPP-TTref (Fig. S22, ESI†), comparable to that reported earlier.21 However, with molecular encapsulation in the polymer backbone, μlin was found to systematically decrease with an increasing number of encapsulated units in the backbone, resulting in μlin of 0.35 cm2 V−1 S−1, 0.18 cm2 V−1 S−1 and 0.03 cm2 V−1 S−1 for PDPP-TT5%, PDPP-TT10%, and PDPP-TT20%, respectively. A similar trend was observed for the saturation mobility (μsat), where increasing the ratio of encapsulated units in the polymer backbone results in a systematic decrease of μsat. Lower charge mobility with molecular encapsulation in the polymer backbone confirms that the introduction of encapsulated units in the polymer backbone decreases the molecular packing between adjacent polymer chains, resulting in reduced molecular order in thin polymer films. Interestingly, the threshold voltage (Vth) is also reduced in the case of encapsulated polymer analogs. PDPP-TTref shows Vth = 49.8 V, which is decreased to 26.7 V for PDPP-TT10%, suggesting a reduction in the trap density.38 Finally, PDPP-TT20% shows Vth of 54.3 V, suggesting a higher trap density. These results are in good agreement with the intensity-dependent Voc measurements, where PDPP-TT10% shows the lowest trap-assisted recombination. Even though FET-measured mobility is not directly relevant for understanding the properties of solar cells as FET characterizes the charge transport parallel to the substrate, it provides useful insights into the morphology of polymer films after encapsulation. Nevertheless, the observed changes in mobility and trap-assisted recombination in OSCs are in agreement.
Finally, the photostability of OSCs based on different PDPP-TT polymer analogs was studied. Devices based on the PDPP-TTref and PDPP-TT5% polymers had significant burn-in losses, losing almost 80% of PCE in the first 25 h of operation with all device parameters undergoing severe degradation (Fig. 6). On the contrary, the PDPP-TT10% and PDPP-TT20% devices were relatively more stable, retaining almost 50% of the initial PCE after up to 200 h of operation. The significant enhancement in photostability of PDPP-TT10% and PDPP-TT20% devices is largely due to the negligible change in the Voc during the initial burn-in period of up to 25 h. This suggests that the increased intermixing of the polymer and PC71BM phases in the case of PDPP-TT10% and PDPP-TT20% devices result in more stable BHJ morphology, ensuring more resilient devices under operation. This strategy can be extended further to other π-conjugated systems and utilized as a new strategy to extend the lifetime of OPV devices.
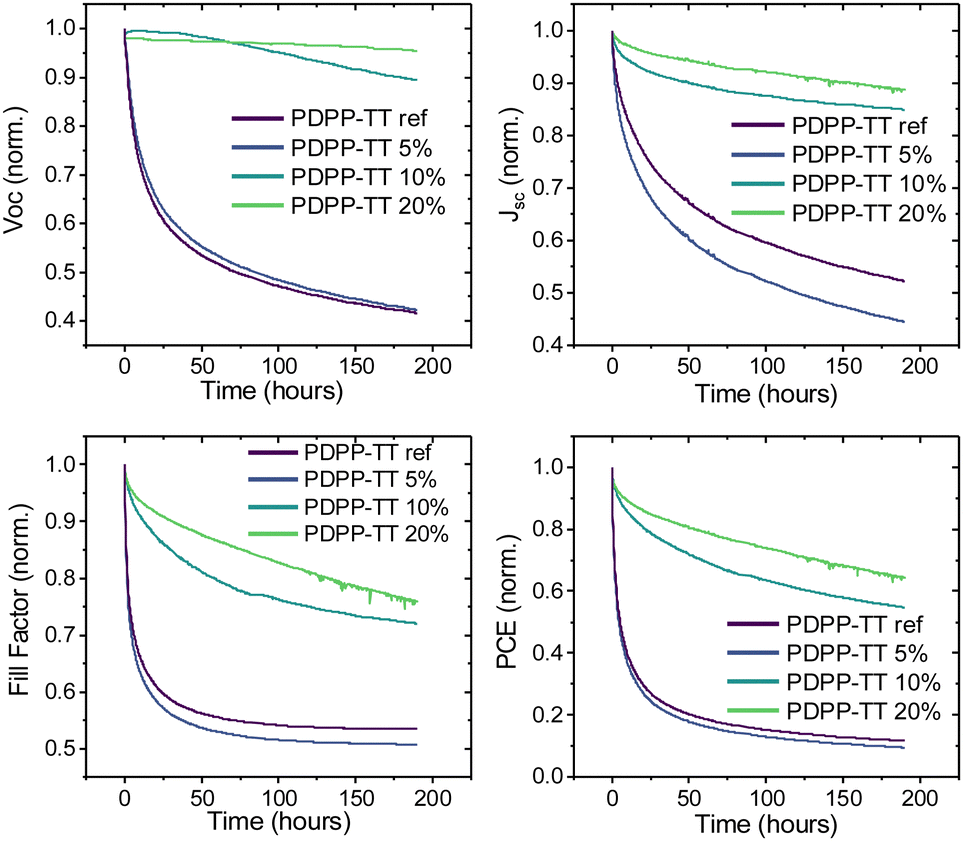 | ||
| Fig. 6 Photostability of polymer-PC71BM BHJ OSCs incorporating different PDPP-TT polymer analogs with varying amounts of molecular encapsulation as donor material. | ||
Conclusion
In conclusion, a series of low-bandgap encapsulated DPP-based polymers were synthesized using molecular encapsulation. The encapsulated material design enhanced the electroluminescence in OSCs via suppressed intramolecular aggregation, resulting in a notable reduction of non-radiative voltage losses. OSCs based on the encapsulated polymers demonstrated reduced trap-assisted charge recombination and improved Voc (∼50 mV). Moreover, molecular encapsulation led to a significant enhancement in the OSC photo-stability via better control over the D–A morphology. The implications of the molecular encapsulation strategy presented in this work are highly relevant for designing high-performing π-conjugated materials and solar cells with high operational stability.Experimental
UV-visible absorption spectroscopy
UV-vis absorption spectra of encapsulated PDPP-TT polymer analogs were measured either in solution with chlorobenzene (CB) as the solvent or on thin films deposited from CB solution (5 mg mL−1) on a glass substrate.OSC Fabrication and Testing
OSCs were fabricated with the structure ITO/PEDOT:PSS (20 nm)/BHJ active layer (∼100 nm)/DPO (∼6 nm)/Ag (80 nm). ITO (10 Ω sq−1) was cleaned by ultrasonication in a 2% aqueous solution of glass detergent (Hellmanex TM III) for 10 minutes before rinsing in DI water. ITO substrates were subsequently ultrasonicated in DI water, acetone, and isopropyl alcohol (IPA) for 10 minutes each, before cleaning in oxygen plasma for 10 minutes. PEDOT:PSS (CLEVIOS P VP AI 4083) was then spin-coated at 4000 rpm for 40 seconds, and the substrates were then annealed at 160 °C for 10 minutes in the air before transferring the substrates to the glove box. The active layer was deposited from a polymer: PC71BM (1:3) BHJ solution in chlorobenzene with 3% diphenyl ether as the solvent additive. DPO (0.5 mg mL−1 in IPA) was spin-coated at 2000 rpm for 20 seconds to result in a ∼6 nm ETL before thermally evaporating a 100 nm silver electrode. The active area of all devices, as defined by the electrode geometry, was 0.1 cm−2. Average PCE values were calculated from an average of at least six devices.
The photovoltaic characteristics of devices were measured in a nitrogen-filled glove box with a Keithly 2400 source meter and an Orial Sol3A Class AAA solar simulator with an AM1.5 filter and calibrated to 1 sun using a Newport certified KG-5 silicon reference cell. External quantum efficiency (EQE) measurements were performed using an integrated system with a xenon arch lamp from Enlitech, Taiwan.
Ultraviolet photoelectron spectroscopy (UPS) and low-energy inverse photoelectron spectroscopy (LE-IPES)
The ionization energy (IE) and electron affinity (EA) of polymers were measured using UPS and LE-IPES, as described elsewhere.39 Briefly, the polymer thin films were deposited on Au-coated Si substrates by spin-coating polymer solution (2 mg mL−1 in chlorobenzene) at 2500 rpm. UPS measurements were performed in an ultrahigh vacuum chamber with a base pressure of 10–10 mbar, equipped with a 7-channel hemispherical analyzer (Sphera II EAC 125), calibrated with the Fermi edge of polycrystalline silver. The measurements were performed at a pass energy of 10 eV using He 1 line with excitation energy of 21.22 eV. The estimated precision and spectral reproducibility in UPS measurements was ±0.05 eV. LE-IPES measurements were performed in isochromatic mode (base pressure 10−9 mbar) using a PMT detector (Hamamatsu R585) mounted with a 280 nm filter (10 nm wavelength window) and an in-house build setup. The onset energies of the unoccupied molecular orbitals were estimated by deconvoluting the spectra using a Gaussian function and a Tougaard background, with an estimated uncertainty of ±0.1 eV.Electroluminescence (EL)
EL spectra were measured on best devices inside a nitrogen-filled glove box either by applying a DC voltage to the device equal to the Voc at one sun illumination or at a fixed injection current of 0.35 mA (3.5 mA cm−2). The emission collected by a collimator reached the spectrograph (Princeton Instruments SP-2300) through an optical fiber. The Pix100BRX Si detector was wavelength calibrated using a Ne/Ar light source (Princeton Instruments Intellical). The EL spectra measured using the Si detector were corrected for detector sensitivity to calculate the (ΔVocnr).Sensitive EQE (sEQE)
sEQE measurements were performed at short-circuit under focused illumination from xenon arch lamp. The light beam was modulated by an optical chopper (275 Hz). The photocurrent was measured as a function of incident photon energy using a lock-in amplifier (Stanford Instruments SR830, and the lamp intensity was calibrated using a Si photodiode. The EL spectra (corrected for detector sensitivity) were used to reconstruct the EQEpv spectra at low photon energies according to the reciprocity relation.4Photothermal deflection spectroscopy (PDS)
PDS measurements were performed on thin polymer: PC71BM (1:3) blend films (20 mg mL−1 in chlorobenzene with 3% diphenyl ether as solvent additive) spin-coated on Quartz using a home-built PDS setup. The light from a 250 W quartz tungsten-halogen lamp (Newport 66996-250Q-R1) was sent through a monochromator (LOT MSH-300) and used as a pump allowing excitation across the UV-to-near-infrared spectral region. The pump light was modulated by a chopper operating at a constant frequency of a few hertz (4 Hz for the present case) and focused on the sample, which was immersed in a chemically inert liquid [perfluorohexane (C6F14); Sigma-Aldrich] during the measurement. A small fraction of the monochromatic pump light was split off as intensity reference and measured by lock-in detection (Stanford Research Systems SR830 lock-in amplifier) using a pyroelectric detector (Newport DET-L-PYC5-R-P). Thin-film samples for PDS were prepared on cleaned quartz substrates by spin coating the polymer: PC71BM BHJ solution (20 mg mL−1 in chlorobenzene). A stabilized continuous-wave laser (Thorlabs HRS015 HeNe, 633 nm) was used as a probe beam source focused closely on the sample surface. The deviation of the probe beam was detected by a Si quadrant detector (Thorlabs PDP90A) using lock-in detection (Stanford Research Systems SR830). The entire setup was controlled by a home-built LabVIEW-based data acquisition and device control code. Further details can be found in the ESI.†
Author contributions
A. S.: conceptualization, data curation and analysis, methodology, investigation, project management, and writing the original draft. L. S.: polymer synthesis and characterization, writing – review & editing. J. B.: investigation, D. R. V.: mobility and morphology characterization; Y. G.: PDS measurements; C. D. C. and J. G.: writing – review & editing; J. T.: stability measurements; F. L.: resources, writing – review & editing; H. B. and D. B.: resources, conceptualization, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research under award no. OSR-CARF/CCF-3079, OSR-CRG2018-3737, OSR-CRG2018-3746, and OSR-CRG2019-4025. HB acknowledges the support from Engineering and Physical Sciences Research Council (EPSRC) under award no. EP/S003126/1.References
- O. Inganas, Organic Photovoltaics over Three Decades, Adv. Mater., 2018, 30, e1800388 CrossRef PubMed.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Non-fullerene acceptors with branched side chains and improved molecular packing to exceed 18% efficiency in organic solar cells, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Single-Junction Organic Photovoltaic Cell with 19% Efficiency, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- U. Rau, Reciprocity relation between photovoltaic quantum efficiency and electroluminescent emission of solar cells, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 085303 CrossRef.
- W. Shockley and H. J. Queisser, Detailed Balance Limit of Efficiency of p–n Junction Solar Cells, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- M. A. Green, Radiative efficiency of state-of-the-art photovoltaic cells, Prog. Photovoltaics, 2012, 20, 472–476 CAS.
- K. D. Rosenthal, M. P. Hughes, B. R. Luginbuhl, N. A. Ran, A. Karki, S.-J. Ko, H. Hu, M. Wang, H. Ade and T.-Q. Nguyen, Quantifying and Understanding Voltage Losses Due to Nonradiative Recombination in Bulk Heterojunction Organic Solar Cells with Low Energetic Offsets, Adv. Energy Mater., 2019, 9, 1901077 CrossRef.
- X. Liu, X. Du, J. Wang, C. Duan, X. Tang, T. Heumueller, G. Liu, Y. Li, Z. Wang, J. Wang, F. Liu, N. Li, C. J. Brabec, F. Huang and Y. Cao, Efficient Organic Solar Cells with Extremely High Open-Circuit Voltages and Low Voltage Losses by Suppressing Nonradiative Recombination Losses, Adv. Energy Mater., 2018, 8, 1801699 CrossRef.
- X.-K. Chen and J.-L. Brédas, Voltage Losses in Organic Solar Cells: Understanding the Contributions of Intramolecular Vibrations to Nonradiative Recombinations, Adv. Energy Mater., 2018, 8, 1702227 CrossRef.
- D. Qian, Z. Zheng, H. Yao, W. Tress, T. R. Hopper, S. Chen, S. Li, J. Liu, S. Chen, J. Zhang, X. K. Liu, B. Gao, L. Ouyang, Y. Jin, G. Pozina, I. A. Buyanova, W. M. Chen, O. Inganas, V. Coropceanu, J. L. Bredas, H. Yan, J. Hou, F. Zhang, A. A. Bakulin and F. Gao, Design rules for minimizing voltage losses in high-efficiency organic solar cells, Nat. Mater., 2018, 17, 703–709 CrossRef CAS PubMed.
- O. D. Miller, E. Yablonovitch and S. R. Kurtz, Strong Internal and External Luminescence as Solar Cells Approach the Shockley-Queisser Limit, IEEE J. Photovolt., 2012, 2, 303–311 Search PubMed.
- X. K. Chen, D. P. Qian, Y. M. Wang, T. Kirchartz, W. Tress, H. F. Yao, J. Yuan, M. Hulsbeck, M. J. Zhang, Y. P. Zou, Y. M. Sun, Y. F. Li, J. H. Hou, O. Inganas, V. Coropceanu, J. L. Bredas and F. Gao, A unified description of non-radiative voltage losses in organic solar cells, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, Fluorinated Copolymer PCPDTBT with Enhanced Open-Circuit Voltage and Reduced Recombination for Highly Efficient Polymer Solar Cells, J. Am. Chem. Soc., 2012, 134, 14932–14944 CrossRef CAS PubMed.
- J. Bertrandie, J. Han, C. S. P. De Castro, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, The Energy Level Conundrum of Organic Semiconductors in Solar Cells, Adv. Mater., 2022, 34, e2202575 CrossRef PubMed.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. D. Peña, W. Liu, R.-Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Intrinsic efficiency limits in low-bandgap non-fullerene acceptor organic solar cells, Nat. Mater., 2021, 20, 378–384 CrossRef CAS PubMed.
- A. Classen, C. L. Chochos, L. Lüer, V. G. Gregoriou, J. Wortmann, A. Osvet, K. Forberich, I. McCulloch, T. Heumüller and C. J. Brabec, The role of exciton lifetime for charge generation in organic solar cells at negligible energy-level offsets, Nat. Energy, 2020, 5, 711–719 CrossRef CAS.
- Q. Liu, S. E. Bottle and P. Sonar, Developments of Diketopyrrolopyrrole-Dye-Based Organic Semiconductors for a Wide Range of Applications in Electronics, Adv. Mater., 2020, 32, 1903882 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, W. S. C. Roelofs, S. C. J. Meskers, M. M. Wienk and R. A. J. Janssen, Effect of the Fibrillar Microstructure on the Efficiency of High Molecular Weight Diketopyrrolopyrrole-Based Polymer Solar Cells, Adv. Mater., 2014, 26, 1565–1570 CrossRef CAS PubMed.
- Z. Hu, A. P. Willard, R. J. Ono, C. W. Bielawski, P. J. Rossky and D. A. Vanden Bout, An insight into non-emissive excited states in conjugated polymers, Nat. Commun., 2015, 6, 8246 CrossRef PubMed.
- J. Wang, X. Jiang, H. Wu, G. Feng, H. Wu, J. Li, Y. Yi, X. Feng, Z. Ma, W. Li, K. Vandewal and Z. Tang, Increasing donor-acceptor spacing for reduced voltage loss in organic solar cells, Nat. Commun., 2021, 12, 6679 CrossRef CAS PubMed.
- K. Sugiyasu, Y. Honsho, R. M. Harrison, A. Sato, T. Yasuda, S. Seki and M. Takeuchi, A Self-Threading Polythiophene: Defect-Free Insulated Molecular Wires Endowed with Long Effective Conjugation Length, J. Am. Chem. Soc., 2010, 132, 14754–14756 CrossRef CAS PubMed.
- C. Pan, K. Sugiyasu, Y. Wakayama, A. Sato and M. Takeuchi, Thermoplastic Fluorescent Conjugated Polymers: Benefits of Preventing π–π Stacking, Angew. Chem., Int. Ed., 2013, 52, 10775–10779 CrossRef CAS PubMed.
- A. Leventis, J. Royakkers, A. G. Rapidis, N. Goodeal, M. K. Corpinot, J. M. Frost, D.-K. Bučar, M. O. Blunt, F. Cacialli and H. Bronstein, Highly Luminescent Encapsulated Narrow Bandgap Polymers Based on Diketopyrrolopyrrole, J. Am. Chem. Soc., 2018, 140, 1622–1626 CrossRef CAS PubMed.
- W. W. Li, K. H. Hendriks, W. S. C. Roelofs, Y. Kim, M. M. Wienk and R. A. J. Janssen, Efficient Small Bandgap Polymer Solar Cells with High Fill Factors for 300 nm Thick Films, Adv. Mater., 2013, 25, 3182–3186 CrossRef CAS PubMed.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. Shakya Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, Thieno[3,2-b]thiophene−Diketopyrrolopyrrole-Containing Polymers for High-Performance Organic Field-Effect Transistors and Organic Photovoltaic Devices, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- Deposition numbers CCDC 2164409 (for 2), and CCDC 2177219 (for 3) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- H. Choi, S.-J. Ko, T. Kim, P.-O. Morin, B. Walker, B. H. Lee, M. Leclerc, J. Y. Kim and A. J. Heeger, Small-Bandgap Polymer Solar Cells with Unprecedented Short-Circuit Current Density and High Fill Factor, Adv. Mater., 2015, 27, 3318–3324 CrossRef CAS.
- P. Schilinsky, C. Waldauf and C. J. Brabec, Recombination and loss analysis in polythiophene based bulk heterojunction photodetectors, Appl. Phys. Lett., 2002, 81, 3885–3887 CrossRef CAS.
- K. Vandewal, J. Widmer, T. Heumueller, C. J. Brabec, M. D. McGehee, K. Leo, M. Riede and A. Salleo, Increased Open-Circuit Voltage of Organic Solar Cells by Reduced Donor-Acceptor Interface Area, Adv. Mater., 2014, 26, 3839–3843 CrossRef CAS PubMed.
- Y. Gao, K. Wang, M. Wang, J. I. Khan, A. H. Balawi, W. Liu, S. De Wolf and F. Laquai, Impact of Cesium/Rubidium Incorporation on the Photophysics of Multiple-Cation Lead Halide Perovskites, Solar RRL, 2020, 4, 2000072 CrossRef CAS.
- C. Kaiser, O. J. Sandberg, N. Zarrabi, W. Li, P. Meredith and A. Armin, A universal Urbach rule for disordered organic semiconductors, Nat. Commun., 2021, 12, 3988 CrossRef CAS PubMed.
- U. Rau, B. Blank, T. C. M. Muller and T. Kirchartz, Efficiency Potential of Photovoltaic Materials and Devices Unveiled by Detailed-Balance Analysis, Phys. Rev. Appl., 2017, 7, 044016 CrossRef.
- M. Azzouzi, J. Yan, T. Kirchartz, K. Liu, J. Wang, H. Wu and J. Nelson, Nonradiative Energy Losses in Bulk-Heterojunction Organic Photovoltaics, Phys. Rev. X, 2018, 8, 031055 CAS.
- F. Piersimoni, S. Chambon, K. Vandewal, R. Mens, T. Boonen, A. Gadisa, M. Izquierdo, S. Filippone, B. Ruttens, J. D'Haen, N. Martin, L. Lutsen, D. Vanderzande, P. Adriaensens and J. V. Manca, Influence of Fullerene Ordering on the Energy of the Charge-Transfer State and Open-Circuit Voltage in Polymer:Fullerene Solar Cells, J. Phys. Chem. C, 2011, 115, 10873–10880 CrossRef CAS.
- A. Sharma, X. Pan, J. A. Campbell, M. R. Andersson and D. A. Lewis, Unravelling the Thermomechanical Properties of Bulk Heterojunction Blends in Polymer Solar Cells, Macromolecules, 2017, 50, 3347–3354 CrossRef CAS.
- A. Sharma, X. Pan, J. M. Bjuggren, D. Gedefaw, X. Xu, R. Kroon, E. Wang, J. A. Campbell, D. A. Lewis and M. R. Andersson, Probing the Relationship between Molecular Structures, Thermal Transitions, and Morphology in Polymer Semiconductors Using a Woven Glass-Mesh-Based DMTA Technique, Chem. Mater., 2019, 31, 6740–6749 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, A general relationship between disorder, aggregation and charge transport in conjugated polymers, Nat. Mater., 2013, 12, 1038 CrossRef CAS PubMed.
- S. Olthof, S. Singh, S. K. Mohapatra, S. Barlow, S. R. Marder, B. Kippelen and A. Kahn, Passivation of trap states in unpurified and purified C60 and the influence on organic field-effect transistor performance, Appl. Phys. Lett., 2012, 101, 253303 CrossRef.
- A. Sharma, S. Singh, X. Song, D. Rosas Villalva, J. Troughton, D. Corzo, L. Toppare, G. Gunbas, B. C. Schroeder and D. Baran, A Nonionic Alcohol Soluble Polymer Cathode Interlayer Enables Efficient Organic and Perovskite Solar Cells, Chem. Mater., 2021, 33, 8602–8611 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2164409 and 2177219. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qm01044e |
| ‡ A.S. and L.S. contributed equally |
| This journal is © the Partner Organisations 2023 |