
Design and synthesis of pillared metal–organic frameworks featuring olefinic fragments†
Rachel Leigh
Mander
 a,
Ashley
Schmidt
b,
Michael
Ruf
b and
Maciej Damian
Korzyński
*a
a,
Ashley
Schmidt
b,
Michael
Ruf
b and
Maciej Damian
Korzyński
*a
aUniversity of Toronto Mississauga, 3359 Mississauga Road, Mississauga, ON L5L 1C6, Canada. E-mail: maciej.korzynski@utoronto.ca; Tel: +1-416-874-7440
bBruker AXS, 5465 E. Cheryl Parkway, Madison, WI 53711, USA
First published on 5th October 2024
Abstract
While metal–organic frameworks (MOFs) are known primarily for their well-defined crystalline porous structures that make them desirable for a myriad of applications, they also distinguish themselves with their chemical tunability. One strategy for chemical tailoring of MOF structures is post-synthetic modification (PSM) targeting moieties present in their organic building blocks (linkers). In this context, alkene (olefinic) fragments are underrepresented in the realm of MOFs despite their extremely well-established and versatile chemistry. With the majority of reported olefinic MOFs falling into the microporous regime, the PSM opportunities involving bulkier reagents are severely limited. Herein, we report a family of UofT (University of Toronto) pillared MOFs constructed around olefinic 1,4-bis(2-(pyridin-4-yl)vinyl)benzene (BPVB) and tetrakis(4-carboxyphenyl)porphyrin (TCPP) linkers. By utilizing a variety of M(II) [M = Zn, Ni, Co] precursors, three structurally distinct frameworks were synthesized and characterized. Most notably, the nickel-based framework represents the first reported example of a stable mesoporous olefinic pillared MOF. In addition to the de novo formation of a stable pillared MOF, Ni(II) is also used in a cation exchange process to structurally reinforce zinc-based frameworks.
Introduction
Metal–organic frameworks (MOFs) are a burgeoning class of materials that exhibit a unique combination of crystallinity and high porosity that makes them desirable for a myriad of applications such as gas storage and separation,1–5 catalysis,6,7 sensing8,9 and energy storage,1,10 to name a few. MOFs are typically assembled with inorganic secondary building units (SBUs) and organic linkers. While thousands of MOF structures have been realized to date by combining these building blocks, certain structural motifs are much more prevalent than others. In particular, linker design has relied heavily on two classes of unsaturated hydrocarbons, arenes and alkynes, due to their rigid geometries and relative ease with which these can be incorporated into more complex molecules.11 Conversely, the third general class of unsaturated hydrocarbons, alkenes (olefins), remains underrepresented in the realm of MOF structural chemistry.While the sp2-hybridized carbons introduce additional flexibility into the framework,12–14 the olefinic fragment provides great opportunities for post-synthetic modification. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond can be used as a versatile synthon for decoration of pores with more elaborate functional groups,15 to tune the framework flexibility and associated guest adsorption properties via reversible bromination,16 in reactive chlorine capture,17 and in MOF pore size engineering using the clip-off chemistry approach.18 Notwithstanding post-synthetic modification, MOFs featuring olefinic linkers have been recently explored in the context of water harvesting and luminescence applications.19–23 Additionally, other recent olefinic-based MOFs have shown exceptional gas separation capabilities of acetylene from carbon dioxide and ethylene.24–26 While these studies clearly highlight the prospects of olefinic MOFs, the portfolio of such frameworks remains limited to primarily microporous materials.18,24,27–34 Because of the small pore sizes and/or apertures, there is only a limited array of molecules that can be introduced into the framework. Consequently, post-synthetic modification techniques aimed at olefinic fragment elaboration cannot be used to their full potential.
C bond can be used as a versatile synthon for decoration of pores with more elaborate functional groups,15 to tune the framework flexibility and associated guest adsorption properties via reversible bromination,16 in reactive chlorine capture,17 and in MOF pore size engineering using the clip-off chemistry approach.18 Notwithstanding post-synthetic modification, MOFs featuring olefinic linkers have been recently explored in the context of water harvesting and luminescence applications.19–23 Additionally, other recent olefinic-based MOFs have shown exceptional gas separation capabilities of acetylene from carbon dioxide and ethylene.24–26 While these studies clearly highlight the prospects of olefinic MOFs, the portfolio of such frameworks remains limited to primarily microporous materials.18,24,27–34 Because of the small pore sizes and/or apertures, there is only a limited array of molecules that can be introduced into the framework. Consequently, post-synthetic modification techniques aimed at olefinic fragment elaboration cannot be used to their full potential.
Recognizing these limitations, our laboratory has recently focused on a search for new olefinic MOFs that feature large pores that could easily be accessed by bulkier reactants. In our synthetic efforts we targeted pillared MOFs (PMOFs), a class of MOFs in which two-dimensional layers composed of SBUs and anionic (primarily carboxylate) linkers are interconnected with neutral pillaring linkers to afford three-dimensional structures (Fig. 1).35 One of the main advantages of pillared MOFs is the ease with which one can predict the shape and size of the pore apertures. Furthermore, the pillaring linkers allow for a straightforward incorporation of diverse functionalities, including alkene fragments.20–23,36 Herein, we report the synthesis and characterization of a novel family of olefinic PMOFs assembled from paddlewheel SBUs, the tetrakis(4-carboxyphenyl)porphyrin (TCPP) interlayer linker and the 1,4-bis(2-(pyridin-4-yl)vinyl)benzene (BPVB) pillaring linker.
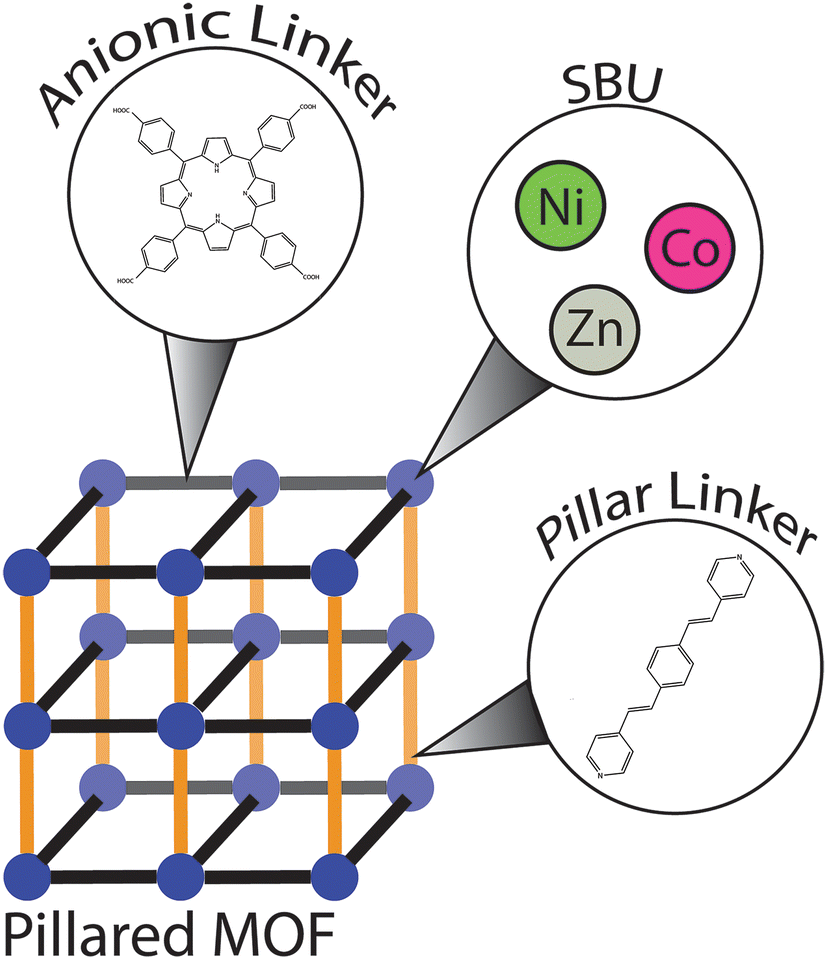 | ||
| Fig. 1 General representation of PMOFs highlighting the building blocks used in this work. | ||
Results and discussion
Synthesis and structural characterization of the as-synthesized MOFs
The solvothermal reaction of M(NO3)2·6H2O (M = Zn(II), Ni(II) and Co(II)) with varying amounts of TCPP and BPVB in N,N-dimethylformamide (DMF)/HNO3/ethanol mixtures (for experimental details, see the ESI†) afforded three novel crystalline materials denoted as UofT-1(Zn), UofT-2(Ni), and UofT-3(Co). Single crystal X-ray diffraction (SCXRD) measurements revealed that both UofT-1(Zn) and UofT-2(Ni) crystallize in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with the M3(TCPP)(BPVB) general formula, while UofT-3(Co) assumes the C2/m space group with Co1.5(TCPP)0.25(BPVB) stoichiometry (for other relevant metrics see Table S1†). In all reported frameworks, the metal cation used during the synthesis is coordinated to the central porphyrinic site in the TCPP linkers. Even though all three materials can be categorized as PMOFs based on the fact that they consist of paddle-wheel SBU-TCPP layers with BPVB pillaring linkers in between (Fig. 2 and Fig. S4–S6†), each of them exhibits unique structural features.
space group with the M3(TCPP)(BPVB) general formula, while UofT-3(Co) assumes the C2/m space group with Co1.5(TCPP)0.25(BPVB) stoichiometry (for other relevant metrics see Table S1†). In all reported frameworks, the metal cation used during the synthesis is coordinated to the central porphyrinic site in the TCPP linkers. Even though all three materials can be categorized as PMOFs based on the fact that they consist of paddle-wheel SBU-TCPP layers with BPVB pillaring linkers in between (Fig. 2 and Fig. S4–S6†), each of them exhibits unique structural features.
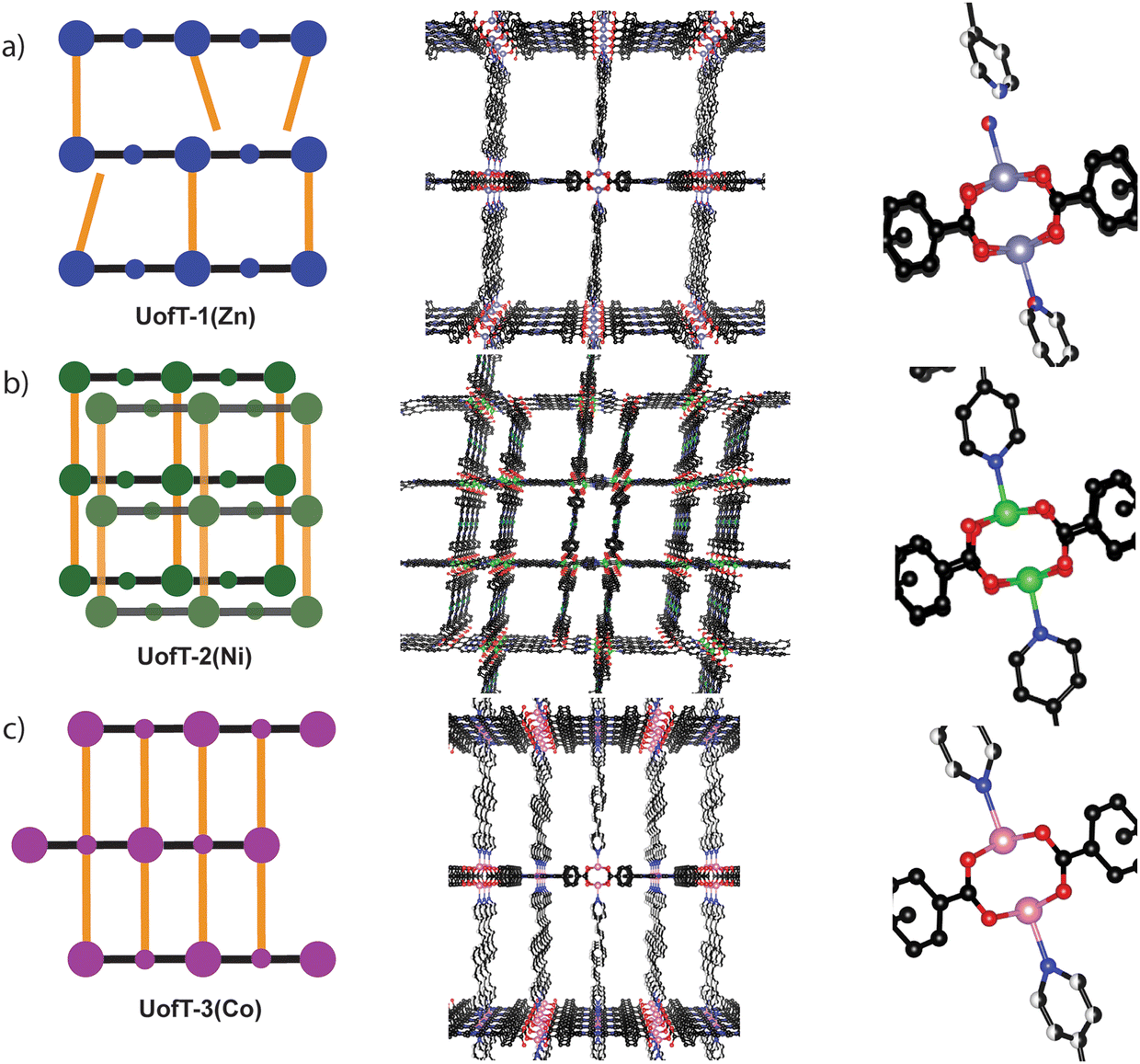 | ||
| Fig. 2 Cartoon representation (left), view of the largest pore (center), and view of SBU coordination (right) for (a) UofT-1(Zn), (b) UofT-2(Ni), and (c) UofT-3(Co). Hydrogen atoms have been omitted for clarity. Carbon (black), nitrogen (blue), oxygen (red), zinc (purple), nickel (green), cobalt (pink). | ||
In an idealized paddle-wheel PMOF SBU, two separate pillaring linkers are coordinated to the apical positions of the M2(COO)4 unit. However, in the case of UofT-1(Zn), the crystal structure refinement revealed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 disorder between coordinated BPVB and water molecules (Fig. 2). Notably, H2O coordination to the SBU causes the one end of the nearby BPVB linker to shift slightly towards the center of the pore. Based on the geometry of two residues and a relatively short distance between the H2O oxygen atom and the nearby BPVB pyridyl nitrogen atom (1.75 Å), it is unlikely that any hydrogen bonding interaction is present.37 A similar type of structural defect has been reported in another paddle-wheel zinc-based PMOF featuring 9,10-bis((E)-2-(pyridine-4-yl)vinyl)anthracene (BP4VA) and 1,1′-biphenyl-4,4′-dicarboxylic acid linkers, FDM-22.36 Using a combination of spectroscopic techniques, the authors demonstrated that ≈10% of BP4VA linkers in the as-synthesized FDM-22 are not coordinated to the SBU, leaving an open apical coordination site. Following thermal activation under vacuum, the ratio between coordinated and non-coordinated pillaring linker termini changes to 1:1, a value that is very close to that found in UofT-1(Zn) using SCXRD data. The high number of defects in FDM-22 manifests itself in scanning electron microscopy (SEM) images of the activated MOF, where prominent parallel striations in the crystals can be observed. Interestingly, we were able to observe similar striations in the as-synthesized UofT-1(Zn) (Fig. S8†). All of the obtained data suggest that the interlayer connectivity in UofT-1(Zn) is highly defective. Notwithstanding structural disorder, the solid-state structure of the as-synthesized UofT-1(Zn) consists of large pores with 23.0 by 17.2 Å apertures (Fig. 2) when viewed along the a crystallographic direction that account for 81% void space.
:1 disorder between coordinated BPVB and water molecules (Fig. 2). Notably, H2O coordination to the SBU causes the one end of the nearby BPVB linker to shift slightly towards the center of the pore. Based on the geometry of two residues and a relatively short distance between the H2O oxygen atom and the nearby BPVB pyridyl nitrogen atom (1.75 Å), it is unlikely that any hydrogen bonding interaction is present.37 A similar type of structural defect has been reported in another paddle-wheel zinc-based PMOF featuring 9,10-bis((E)-2-(pyridine-4-yl)vinyl)anthracene (BP4VA) and 1,1′-biphenyl-4,4′-dicarboxylic acid linkers, FDM-22.36 Using a combination of spectroscopic techniques, the authors demonstrated that ≈10% of BP4VA linkers in the as-synthesized FDM-22 are not coordinated to the SBU, leaving an open apical coordination site. Following thermal activation under vacuum, the ratio between coordinated and non-coordinated pillaring linker termini changes to 1:1, a value that is very close to that found in UofT-1(Zn) using SCXRD data. The high number of defects in FDM-22 manifests itself in scanning electron microscopy (SEM) images of the activated MOF, where prominent parallel striations in the crystals can be observed. Interestingly, we were able to observe similar striations in the as-synthesized UofT-1(Zn) (Fig. S8†). All of the obtained data suggest that the interlayer connectivity in UofT-1(Zn) is highly defective. Notwithstanding structural disorder, the solid-state structure of the as-synthesized UofT-1(Zn) consists of large pores with 23.0 by 17.2 Å apertures (Fig. 2) when viewed along the a crystallographic direction that account for 81% void space.
Despite having the same formulas and comparable unit cell dimensions within the same space group, UofT-1(Zn) and UofT-2(Ni) differ significantly in their solid-state structures. Firstly, UofT-2(Ni) is doubly interpenetrated, with one of the sub-lattices growing within the pores of the other one (Fig. 2). While interpenetration can drastically decrease the porosity of a material, the intergrowing sub-lattice is offset from the center of the other's pores. As a result, UofT-2(Ni) has two kinds of channels parallel along the b crystallographic direction—larger (14.1 by 18.7 Å) and smaller (8.8 by 17.7 Å). Unsurprisingly, the interpenetration leads to a lower percentage of vacant space (65%) in the MOF structure when compared to UofT-1(Zn). Secondly, inspection of the coordination environment around Ni(II) sites revealed that there is no disorder associated with the BPVB-SBU linkages (Fig. 2), suggesting a more robust interlayer connectivity in UofT-2(Ni) when compared to the Zn(II) framework. Correspondingly, the SEM images of the Ni(II) MOF do not show similar striations to those seen in UofT-1(Zn) (Fig. S9†).
In contrast to the archetypal SBU-to-SBU pillaring linker coordination seen in UofT-1(Zn) and UofT-2(Ni), the 3D structure of UofT-3(Co) is formed via the coordination of BPVB donor nitrogen atoms to the SBU and the Co(II) sites located in the middle of the TCPP linker forming an AB stacking pattern (Fig. 2). Such non-innocent (from the MOF assembly perspective) character of the linker-bound metal centers has been demonstrated before in porphyrin-based pillared MOFs.38,39 Despite the higher density of BPVB linkers in the interlayer space, the material still exhibits sizable pores (25.8 by 9.0 Å) with 74% vacant structure. Similar to UofT-2(Ni), no pillar coordination disorder could be observed in SCXRD data (Fig. 2). The SEM images show uniform crystals with no sign of striations or other defects (Fig. S10†).
Powder X-ray diffraction (PXRD) experiments were performed to verify the bulk phase purity of the synthesized materials. The experimental PXRD patterns for UofT-1(Zn) and UofT-3(Co) generally match up well in terms of peak positions with the patterns predicted from the respective SCXRD structures (Fig. 3 and Fig. S11, S13†). Comparison of the experimental and simulated PXRD patterns for UofT-2(Ni) in the 7.5°–9.5° region revealed the absence of certain features in the former (Fig. 3 and Fig. S12, S40†). We believe that these differences stem from enhanced dynamics of the framework under ambient conditions when compared to the patterns simulated from SCXRD data collected at 200 K. As a result, an averaging of distances between closely related crystallographic planes such as 110 and 1−10 can be observed (Fig. S40†). The 1H nuclear magnetic resonance (NMR) spectra of the MOFs digested in D2SO4 and DMSO-d6 show that both linkers are present in all materials (Fig. S24–S26†). Finally, we have also confirmed that the compositions of all synthesized MOFs reasonably match the theoretical values using elemental analysis (EA) and inductively coupled plasma optical emission spectroscopy (ICP-OES) (see the ESI† for details).
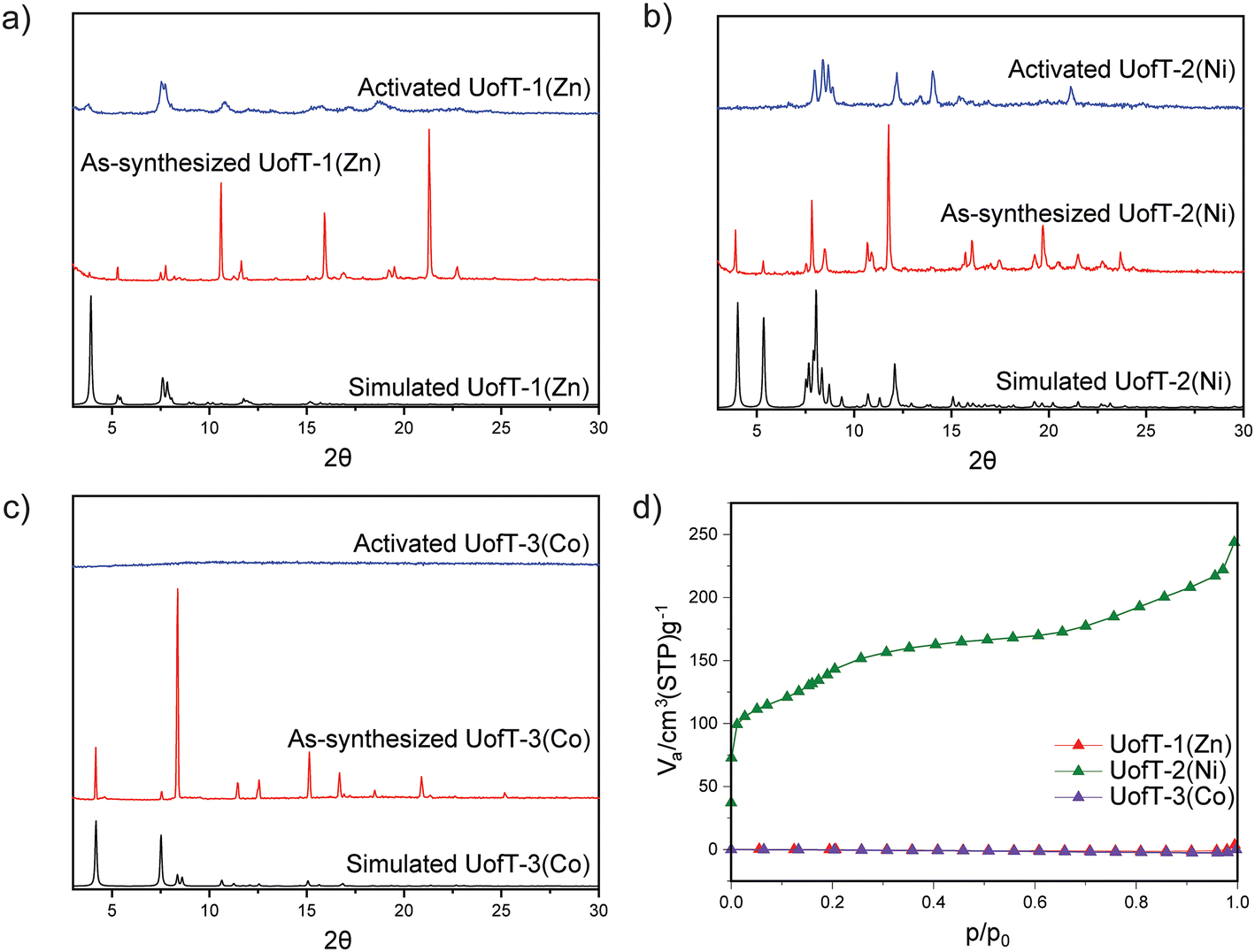 | ||
| Fig. 3 Comparison of the simulated, as-synthesized and activated PXRD patterns for (a) UofT-1(Zn), (b) UofT-2(Ni), and (c) UofT-3(Co); (d) comparison of 77 K nitrogen gas adsorption isotherms for all synthesized PMOFs. | ||
Activation and gas adsorption studies
In order to evaluate the stability of the synthesized PMOFs, we attempted thermal activation under vacuum at 150 °C following exchange of DMF for a more volatile and less coordinating solvent, acetone. The activation temperature was selected based on the thermogravimetric analysis data for the as-synthesized and activated samples (Fig. S14–S17†). In all synthesized MOFs, pronounced changes in PXRD patterns can be observed following the activation. In the case of UofT-1(Zn), a broadening and overall decrease in the definition of the features is evident in the diffraction data (Fig. 3). Additionally, some of the peaks present in the as-synthesized material (e.g., 5.3°) cannot be detected anymore. The addition of DMF to the activated material did not lead to the recovery of the as-synthesized UofT-1(Zn) PXRD pattern (Fig. S18†). Such behavior is likely indicative of pronounced and irreversible structural changes impacting the crystallinity of the material. SCXRD measurements performed on activated UofT-1(Zn) revealed a complete delamination of the material with TCPP-SBU layers intact and aqua ligands coordinated to all apical Zn(II) sites (Fig. S7†). It is worth noting that the SBU itself undergoes distortion from an idealized paddle-wheel to a distorted one with concomitant elongation of the Zn–Zn distance (2.73 to 2.92 Å). Curiously, while complete chemical delamination occurs in the material, the interlayer spacing remains similar to that of the as-synthesized material. Based on this observation and the fact that activated UofT-1(Zn) samples were exposed to ambient moisture before SCXRD measurements, we posit that the activated material does not inherently feature coordinated water molecules. Instead, the thermal treatment causes further dissociation of the pillaring linkers (similar to that described by Qi et al.36), which results in the formation of Zn(II) open metal sites prone to water coordination. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) data collected on activated UofT-1(Zn) corroborate our discussion since no spectral signatures of H2O expected above 3500 cm−1 can be observed (Fig. S21†). Furthermore, the liquid-phase 1H NMR spectra of the digested MOF (Fig. S24†) show that BPVB is still present in the sample. These pillars remain unbound to SBUs and occupy the interlayer space without exhibiting any long-range order.While the activation of UofT-2(Ni) also results in significant changes in the PXRD pattern, the peaks remain narrow and well-defined, similar to those of the as-synthesized MOF (Fig. 3). This is indicative of the structural change within the framework that does not disrupt the long-range order of the material. The disappearance of the lowest 2θ peak (3.9°) associated with the 001 plane family and the emergence of the additional peaks at higher angles suggest that UofT-2(Ni) exhibits structural flexibility that is often encountered in pillared MOFs.35,40,41 These structural changes are reversible and solvent dependent, as evidenced by the recovery of the as-synthesized UofT-2(Ni) PXRD pattern following the exposure of the activated material to DMF (Fig. S19†). Expectedly, the DRIFTS spectrum of activated UofT-2(Ni) is very similar to that of UofT-1(Zn) (Fig. S22†). As opposed to Zn(II) and Ni(II) MOFs, the activation of UofT-3(Co) leads to a complete loss of crystallinity, as evidenced by a featureless PXRD diffractogram (Fig. 3). The amorphization upon activation in cobalt-based paddle-wheel frameworks has been observed before, e.g., in the case of DUT-49(Co).42,43
In order to identify if any of the synthesized materials exhibit permanent porosity, we performed nitrogen adsorption measurements at 77 K. Consistent with the PXRD characterization, UofT-1(Zn) and UofT-3(Co) show minimal uptake of N2 and can be considered non-porous (Fig. 3). Conversely, the dinitrogen adsorption into UofT-2(Ni) is non-negligible and results in a type IV isotherm with two distinct steps at p/p0 values of 0.011 and 0.133 (Fig. 3), which could be related to the structural flexibility described above. Fitting of the adsorption data to the Brunauer–Emmett–Teller (BET) equation yields a surface area of 514 m2 g−1, the highest value reported for olefin-bearing pillared porphyrin-based MOFs to date. Consistent with the type IV isotherm, the Barrett–Joyner–Halenda (BJH) analysis indicated that the average pore size in UofT-2(Ni) is ≈2 nm (Fig. S30–S32†), making it the first reported permanently mesoporous olefinic PMOF.
Reinforcement of UofT-1(Zn)via Ni(II) cation exchange
Due to its accessible permanently porous structure, UofT-2(Ni) stands out as the most promising PMOF platform for further chemical elaboration targeting the olefinic bonds. However, the interpenetrated structure of this material does limit the vacant space available to reactants.44 Thus, we decided to explore the chemical reinforcement of the non-interpenetrated UofT-1(Zn) structure as a path to a permanently porous non-interpenetrated PMOF. To this end, we focused on the cation exchange of Ni(II) into UofT-1(Zn), a process that has been shown to increase the thermal stability of Zn(II) PMOFs due to the substitution of zinc(II) present in the SBU for nickel(II).45 Treatment of UofT-1(Zn) with either 1 or 10 molar equivalents (with respect to Zn(II)) of NiCl2·6H2O in DMF at 80 °C for 72 hours yielded two new materials UofT-1(Zn/Nilow) and UofT-1(Zn/Nihigh), respectively. According to the ICP-OES analysis of the digested MOFs, the Ni:Zn ratio in the former is approximately 1:1, while in the latter the same ratio is equal to 3.5:1. The higher than nominal loading of Ni(II) in UofT-1(Zn/Nilow) likely results from inaccuracies in weighing of the solvated MOF (for additional discussion, see the ESI†). The PXRD patterns of activated UofT-1(Zn/Nilow) and UofT-1(Zn/Nihigh) (Fig. 4) closely resemble those of UofT-1(Zn). In addition, as more Ni(II) is introduced into the parent Zn(II) MOF, a systematic shift of the peaks towards higher 2θ values can be observed (Fig. S33–S36†). This change can be attributed to a decrease of M–M and M–O distances upon substitution of zinc for nickel. Similar systematic shifts towards higher 2θ values can be readily observed when comparing the PXRD patterns of pristine UofT-1(Zn) and UofT-2(Ni) (where the M–M distance changes from 2.7 Å for Zn(II) to 2.6 Å for Ni(II)).
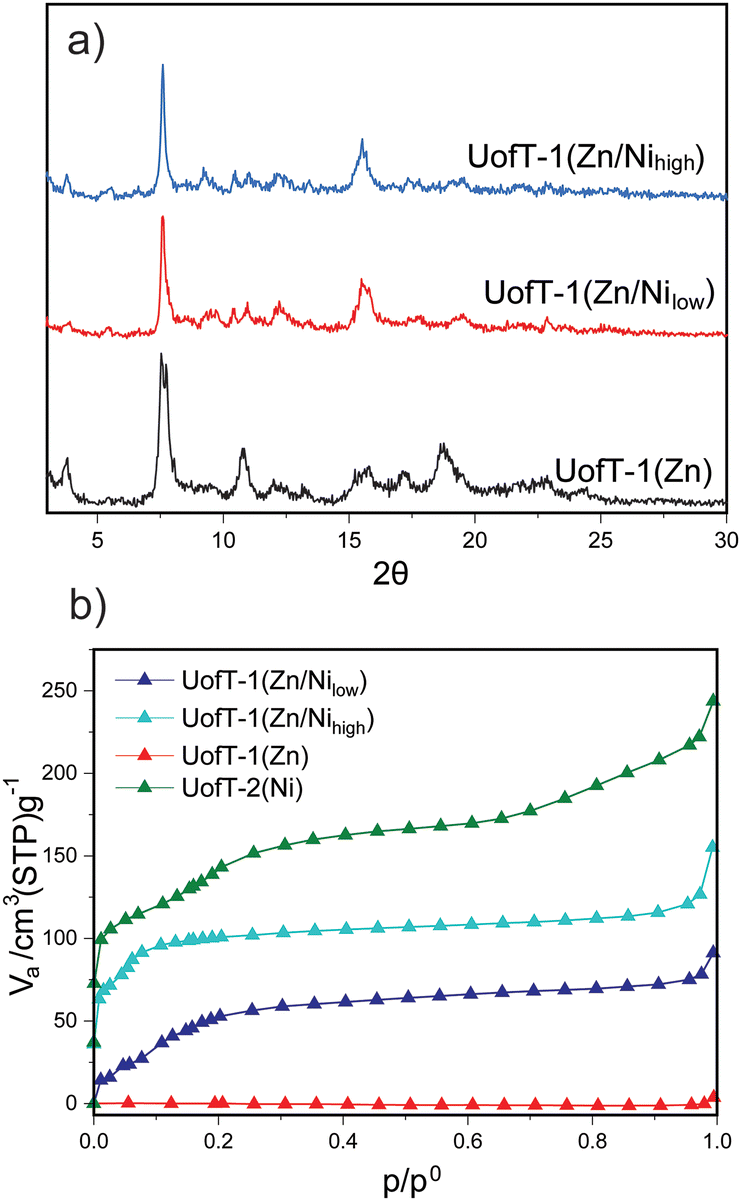 | ||
| Fig. 4 (a) Comparison of PXRD patterns for activated cation-exchanged PMOFs and UofT-1(Zn); (b) comparison of 77 K nitrogen gas adsorption isotherms for cation-exchanged PMOFs as well as UofT-1(Zn) and UofT-2(Ni). | ||
The most prominent difference between the parent MOF and Ni(II) exchanged samples can be observed in 77 K N2 adsorption data (Fig. 4). In contrast to UofT-1(Zn), both cation exchanged samples show positive gas uptake with BET surface areas of 153 m2 g−1 for UofT-1(Zn/Nilow) and 395 m2 g−1 for UofT-1(Zn/Nihigh). Accordingly, the BJH pore size analysis of UofT-1(Zn/Nihigh) showed an average value of 1.7 nm (Fig. S39†). While these porosity benchmarks fail to surpass those of UofT-2(Ni), the impact of Ni(II) exchange (even at lower concentrations) leads to a significant improvement in MOF stability and should be considered a viable strategy for structural reinforcement of Zn(II) PMOFs.
Conclusions
In this work, we introduced three new olefinic PMOFs based on Zn(II), Ni(II), and Co(II). Each of the frameworks exhibits unique structural features demonstrating the rich chemistry associated with this class of materials. Of the synthesized PMOFs, UofT-1(Zn) and UofT-3(Co) exhibit low stability in thermal activation and cannot be considered permanently porous. A significant improvement in the stability of UofT-1(Zn) can be achieved by modest doping with Ni(II) in a cation exchange process. Notably, the pristine Ni(II) framework, UofT-2(Ni), is the first example of an olefinic PMOF that is permanently mesoporous, making it an ideal platform for further post-synthetic modification targeting the alkene moiety.Author contributions
R. L. M.: investigation, data analysis and curation, validation, visualization, and writing – original draft; A. S. and M. R.: investigation, data analysis and curation, and writing – reviewing and editing; M. D. K.: data analysis, supervision, funding acquisition, project administration, conceptualization, writing – original draft, reviewing and editing.Data availability
The data supporting this article have been uploaded as part of the ESI.† Crystallographic data for UofT-1(Zn) (2378084), activated UofT-1(Zn) (2378083), UofT-2(Ni) (2378082) and UofT-3(Co) (2378081)† have been deposited at the Cambridge Crystallographic Data Center (CCDC).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of the Natural Sciences and Engineering Research Council of Canada (NSERC), Discovery grant RGPIN-2023-04211. We would like to thank Wendy Wu for assistance with the collection of SEM images.References
- B. M. Connolly, D. G. Madden, A. E. H. Wheatley and D. Fairen-Jimenez, J. Am. Chem. Soc., 2020, 142, 8541–8549 CrossRef.
- J. Tan, Y. Tao, X. Zhang, Q. Wang, T. Zeng, Z. Shi, K. E. Cordova, Y. Lee, H. Liu and Y.-B. Zhang, J. Mater. Chem. A, 2021, 9, 24857–24862 RSC.
- Z. Zhu, H. Tsai, S. T. Parker, J.-H. Lee, Y. Yabuuchi, H. Z. H. Jiang, Y. Wang, S. Xiong, A. C. Forse, B. Dinakar, A. Huang, C. Dun, P. J. Milner, A. Smith, P. G. Martins, K. R. Meihaus, J. J. Urban, J. A. Reimer, J. B. Neaton and J. R. Long, J. Am. Chem. Soc., 2024, 146, 6072–6083 CrossRef CAS.
- H. Wang, D. Luo, E. Velasco, L. Yu and J. Li, J. Mater. Chem. A, 2021, 9, 20874–20896 RSC.
- S. Hiraide, Y. Sakanaka, H. Kajiro, S. Kawaguchi, M. T. Miyahara and H. Tanaka, Nat. Commun., 2020, 11, 3867 CrossRef.
- P. M. Stanley, J. Haimerl, N. B. Shustova, R. A. Fischer and J. Warnan, Nat. Chem., 2022, 14, 1342–1356 CrossRef.
- Y. Wang, G. Ning and D. Li, Chem. Eur. J., 2024, 30, e202400360 CrossRef.
- H. Sohrabi, S. Ghasemzadeh, S. Shakib, M. R. Majidi, A. Razmjou, Y. Yoon and A. Khataee, Ind. Eng. Chem. Res., 2023, 62, 4611–4627 CrossRef.
- H. Yamagiwa, S. Sato, T. Fukawa, T. Ikehara, R. Maeda, T. Mihara and M. Kimura, Sci. Rep., 2014, 4, 6247 CrossRef.
- P. Peng, H. Z. H. Jiang, S. Collins, H. Furukawa, J. R. Long and H. Breunig, ACS Energy Lett., 2024, 9, 2727–2735 CrossRef.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- I. H. Hwang, H.-Y. Kim, M. M. Lee, Y. J. Na, J. H. Kim, H.-C. Kim, C. Kim, S. Huh, Y. Kim and S.-J. Kim, Cryst. Growth Des., 2013, 13, 4815–4823 CrossRef.
- H.-C. Kim, S. Huh, J. Y. Kim, H. R. Moon, D. N. Lee and Y. Kim, CrystEngComm, 2017, 19, 99–109 RSC.
- S.-S. Meng, M. Xu, H. Guan, C. Chen, P. Cai, B. Dong, W.-S. Tan, Y.-H. Gu, W.-Q. Tang, L.-G. Xie, S. Yuan, Y. Han, X. Kong and Z.-Y. Gu, Nat. Commun., 2023, 14, 5347 CrossRef.
- K. Hindelang, A. Kronast, S. I. Vagin and B. Rieger, Chem. Eur. J., 2013, 19, 8244–8252 CrossRef.
- X. Xu, F. Yang, S.-L. Chen, J. He, Y. Xu and W. Wei, Chem. Commun., 2017, 53, 3220–3223 RSC.
- T. J. Azbell, R. M. Mandel, J.-H. Lee and P. J. Milner, ACS Appl. Mater. Interfaces, 2022, 14, 53928–53935 CrossRef.
- V. Guillerm, H. Xu, J. Albalad, I. Imaz and D. Maspoch, J. Am. Chem. Soc., 2018, 140, 15022–15030 CrossRef.
- A. H. Alawadhi, S. Chheda, G. D. Stroscio, Z. Rong, D. Kurandina, H. L. Nguyen, N. Rampal, Z. Zheng, L. Gagliardi and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 2160–2166 CrossRef.
- B. B. Rath, D. Kottilil, W. Ji and J. J. Vittal, ACS Appl. Mater. Interfaces, 2023, 15, 26939–26945 CrossRef.
- M. Liu, H. S. Quah, S. Wen, Z. Yu, J. J. Vittal and W. Ji, Chem. Mater., 2016, 28, 3385–3390 CrossRef.
- H. S. Quah, W. Chen, M. K. Schreyer, H. Yang, M. W. Wong, W. Ji and J. J. Vittal, Nat. Commun., 2015, 6, 7954 CrossRef.
- H. S. Quah, V. Nalla, K. Zheng, C. A. Lee, X. Liu and J. J. Vittal, Chem. Mater., 2017, 29, 7424–7430 CrossRef.
- L. Wang, W. Sun, Y. Zhang, N. Xu, R. Krishna, J. Hu, Y. Jiang, Y. He and H. Xing, Angew. Chem. Int. Ed., 2021, 60, 22865–22870 CrossRef.
- W. Sun, J. Hu, S. Duttwyler, L. Wang, R. Krishna and Y. Zhang, Sep. Purif. Technol., 2022, 283, 120220 CrossRef.
- N. Xu, T. Yan, J. Li, L. Wang, D. Liu and Y. Zhang, Inorg. Chem. Front., 2023, 10, 522–528 RSC.
- B. Chen, S. Ma, F. Zapata, E. B. Lobkovsky and J. Yang, Inorg. Chem., 2006, 45, 5718–5720 CrossRef.
- B. Chen, S. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H.-C. Zhou, Inorg. Chem., 2007, 46, 1233–1236 CrossRef.
- C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136–7144 CrossRef.
- W. Lin, E. Ning, L. Yang, Y. Rao, S. Peng and Q. Li, Inorg. Chem., 2021, 60, 11756–11763 CrossRef.
- I.-H. Park, K. Kim, S. S. Lee and J. J. Vittal, Cryst. Growth Des., 2012, 12, 3397–3401 CrossRef.
- S. Sanda, S. Parshamoni, S. Biswas and S. Konar, Chem. Commun., 2015, 51, 6576–6579 RSC.
- N. Liu, Z. Chen, W. Fan, J. Su, T. Lin, S. Xiao, J. Meng, J. He, J. J. Vittal and J. Jiang, Angew. Chem. Int. Ed., 2022, 61, e202115205 CrossRef.
- Z. Zhu, Z. Wang, Q.-H. Li, Z. Ma, F. Wang and J. Zhang, Dalton Trans., 2023, 52, 4309–4314 RSC.
- F. ZareKarizi, M. Joharian and A. Morsali, J. Mater. Chem. A, 2018, 6, 19288–19329 RSC.
- Y. Qi, H. Xu, X. Li, B. Tu, Q. Pang, X. Lin, E. Ning and Q. Li, Chem. Mater., 2018, 30, 5478–5484 CrossRef.
- T. K. Harris and A. S. Mildvan, Proteins, 1999, 35, 275–282 CrossRef.
- P. M. Barron, H.-T. Son, C. Hu and W. Choe, Cryst. Growth Des., 2009, 9, 1960–1965 CrossRef.
- E.-Y. Choi, P. M. Barron, R. W. Novotny, H.-T. Son, C. Hu and W. Choe, Inorg. Chem., 2009, 48, 426–428 CrossRef.
- I. Senkovska, V. Bon, L. Abylgazina, M. Mendt, J. Berger, G. Kieslich, P. Petkov, J. L. Fiorio, J. Joswig, T. Heine, L. Schaper, C. Bachetzky, R. Schmid, R. A. Fischer, A. Pöppl, E. Brunner and S. Kaskel, Angew. Chem., Int. Ed, 2023, 62, e202218076 CrossRef.
- S. Henke, R. Schmid, J. Grunwaldt and R. A. Fischer, Chem. Eur. J., 2010, 16, 14296–14306 CrossRef.
- B. Garai, V. Bon, S. Krause, F. Schwotzer, M. Gerlach, I. Senkovska and S. Kaskel, Chem. Mater., 2020, 32, 889–896 CrossRef.
- S. Ehrling, M. Mendt, I. Senkovska, J. D. Evans, V. Bon, P. Petkov, C. Ehrling, F. Walenszus, A. Pöppl and S. Kaskel, Chem. Mater., 2020, 32, 5670–5681 CrossRef.
- A. B. Crom, J. L. Strozier, C. J. Tatebe, C. A. Carey, J. I. Feldblyum and D. T. Genna, Chem. Eur. J., 2023, 29, e202302856 CrossRef.
- O. Karagiaridi, W. Bury, D. Fairen-Jimenez, C. E. Wilmer, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Inorg. Chem., 2014, 53, 10432–10436 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General information and synthetic procedures; further SCXRD, PXRD, SEM, TGA, DRIFTS, 1H NMR, and nitrogen adsorption data. CCDC 2378081–2378084. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02397h |
| This journal is © The Royal Society of Chemistry 2024 |