
Direct O2 mediated oxidation of a Ni(II)N3O structural model complex for the active site of nickel acireductone dioxygenase (Ni-ARD): characterization, biomimetic reactivity, and enzymatic implications†
Kelsey E.
Kirsch
 a,
Mary E.
Little
b,
Thomas R.
Cundari
c,
Emily
El-Shaer
b,
Georgia
Barone
b,
Vincent M.
Lynch
d and
Santiago A.
Toledo
*a
a,
Mary E.
Little
b,
Thomas R.
Cundari
c,
Emily
El-Shaer
b,
Georgia
Barone
b,
Vincent M.
Lynch
d and
Santiago A.
Toledo
*a
aDepartment of Chemistry, American University, 4400 Massachusetts Ave NW, Washington, DC 20016, USA. E-mail: stoledo@american.edu
bDepartment of Chemistry, St Edward's University, 3001 South Congress Ave, Austin, Texas 78704, USA
cDepartment of Chemistry, University of North Texas, 1155 Union Cir, Denton, Texas 76203, USA
dDepartment of Chemistry, The University of Texas at Austin, 120 Inner Campus Dr Stop G2500, Austin, Texas 78712, USA
First published on 11th October 2024
Abstract
A new biomimetic model complex of the active site of acireductone dioxygenase (ARD) was synthesized and crystallographically characterized ([Ni(II)(N-(ethyl-N′Me2)(Py)(2-t-ButPhOH))(OTf)]-1). 1 displays carbon–carbon oxidative cleavage activity in the presence of O2 towards the substrate 2-hydroxyacetophenone. This reactivity was monitored via UV-Visible and NMR spectroscopy. We postulate that the reactivity of 1 with O2 leads to the formation of a putative Ni(III)-superoxo transient species resulting from the direct activation of O2via the nickel center during the oxidative reaction. This proposed intermediate and reaction mechanism were studied in detail using DFT calculations. 1 and its substrate bound derivatives display reactivity toward mild outer sphere oxidants, suggesting ease of access to high valent Ni coordination complexes, consistent with our calculations. If confirmed, the direct activation of O2 at a nickel center could have implications for the mechanism of action of ARD and other nickel-based dioxygenases and their respective non-traditional, enzymatic moonlighting functions, as well as contribute to a general understanding of direct oxidation of nickel(II) coordination complexes by O2.
Introduction
The exquisite natural specificity of metalloenzymes is a result of the protein's structural enforcement of entatic states that optimize reactivity and selectivity of metal–substrate binding.1 Structural changes and/or metal promiscuity of metalloenzymes can lead to alternative reactivity pathways resulting in moonlighting functions.2,3 Moonlighting, aberrant functions of metalloenzymes, has been shown to play a role in the development of disease.3 Atomistic understanding of the underlying mechanistic operations of these aberrant behaviors is key to elucidating the role of metalloenzymes in disease development and ultimately the design of effective therapeutics.The metalloenzyme acireductone dioxygenase (ARD) plays a key role in the ubiquitous methionine salvage pathway (MSP) in animals, plants, and bacteria.4 The MSP makes S-adenosyl methionine (SAM) as a precursor to methylthioadenosine (MTA), and it is essential for polyamine synthesis in animals and ethylene synthesis in plants. It is known that the inhibition of MTA halts DNA replication and elevated amounts of polyamines are associated with tumor formation.5 ARD catalyzes the penultimate step in the MSP, the oxidative cleavage of the substrate acireductone (1,2-dihydroxo-3-keto-5-(thiomethyl)pent-1-ene) into 2-keto-4-(thiomethyl) butyrate (KMBT-methionine precursor) and formic acid (Fig. 1). This reaction occurs when iron is bound to the active site of the enzyme and is known as the “on-pathway” route. In contrast, when nickel (bacterial systems) is bound to the active site, an “off-pathway” shunt route follows with the formation of 3-(methylthio)propionate (MTP), formate, and carbon monoxide (Fig. 1).6–8 The cellular role of the “off-pathway” reaction remains unknown. ARD is a unique metalloenzyme in that it is the only known example of a metalloenzyme displaying a dual function solely based on the identity of the metal ion cofactor at the active site. In vitro binding studies showed that mammalian ARD (MmARD) and human ARD (HsARD) display identical “off-pathway” chemistry when bound to cobalt(II) or manganese(II).8–10 CO, the off-pathway product, is a known anti-apoptotic signaling molecule and neurotransmitter in mammals.11 Additionally, the dysregulation of ARD's Hs-ADI1 gene has been linked to a variety of cancer types.12–15 Most recently, a metabolomic study of uterine serous carcinoma (SC) proposed ARD as a potential therapeutic target, specifically for the most aggressive forms of this cancer.12 ARD has a known structural moonlighting function ascribed to its interaction with matrix metalloproteinase (MT1-MMP) through its cytoplasmic tail leading to inhibition of MT1-MMP function.14,16,17 MT1-MMP is overexpressed in a variety of cancer cell lines and is a known promoter of cancer cell metastasis.13,18,19
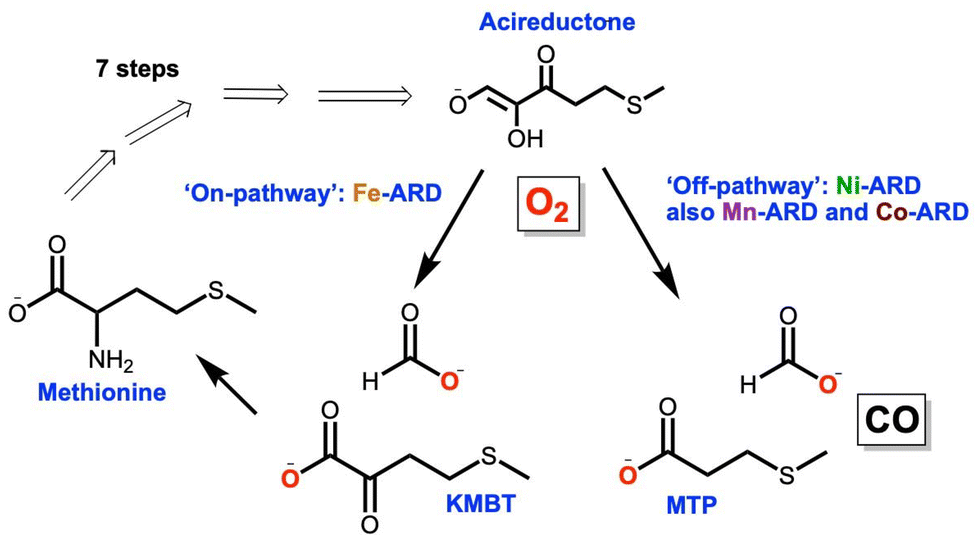 | ||
| Fig. 1 Essential features of the methionine salvage pathway (MSP). “On and off” acireductone dioxygenase (ARD) pathways display distinct product formation based solely on the identity of the metal center. | ||
It is still unknown if ARD remains enzymatically active during these interactions, or if the structural interactions enforce one pathway over another. It is a wide-open question whether there is a direct mechanistic, enzymatic involvement for ARD in cancer development and progression. Most relevant to this work, the reasons behind the metal mediated regioselective mechanism of ARD are still subject of debate in the literature8,10,20,21 with a variety of hypothesis ranging from the nature of substrate binding (chelate hypothesis),7,9 the role of water accessibility to the active site,21 and the possible redox activity of the metal cofactor.10,22 Our work provides needed empirical, biomimetic evidence to further contribute to these ideas. Prior to this work, only one functional (albeit not structural) model for ARD was published.23 This model system was instrumental in understanding the role that water might play in the regioselective oxidation mediated by ARD. More recently, we published a family of active site models of ARD where we attempted to better mimic the electronic environment of ARDs active site (Fig. 2b).24 This family of compounds used O2 to oxidatively cleave the C–C bond of a beta-diketonate substrate in dioxygenase fashion. Through computational work we ascribed this reactivity with a relatively inactive substrate, to the presence of an oxygen donating moiety in the coordinating ligand of the model complexes.
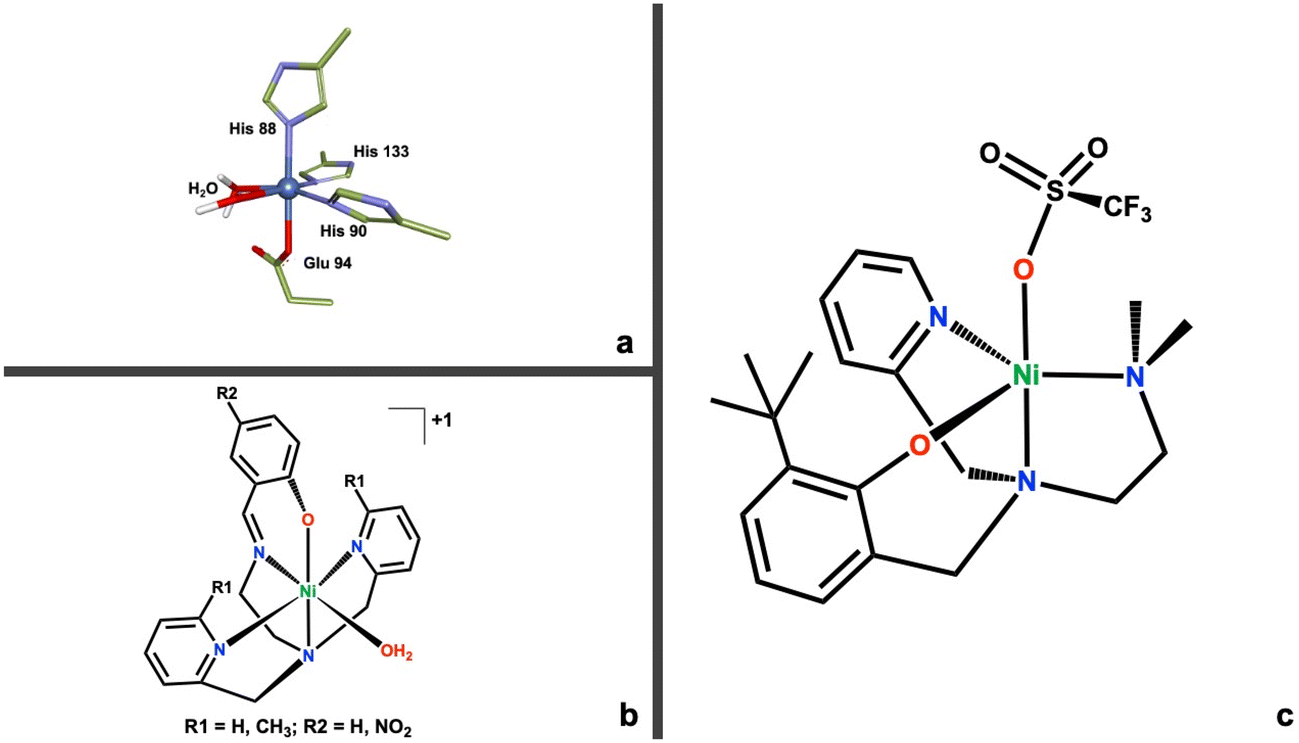 | ||
| Fig. 2 (a) First coordination sphere for the active site of ARD (PDB ID: 1VR3).25 (b) Structure of the first family of model complexes for ARD.24 (c) Structure of the new second generation Ni(II)N3O ARD model complex (1). | ||
In this work we introduce a new N3O chelating ligand and its corresponding nickel(II) complex (Fig. 2c), that more accurately mimics the His3–Glu1 coordination motif of ARD's active site (Fig. 2a). This second-generation model system is capable of biomimetic dioxygenase activity towards a C1–H substrate, 2-hydroxyacetophenone (2HAP-H). Furthermore, this Ni(II)N3O (1) complex can access a high-valent Ni(III) state via direct oxidation by O2. This reactivity is unprecedented in non-organometallic coordination complexes. In organometallic compounds, the Anderson lab reported the generation of a dimeric Ni(III)2μ-1,2-peroxo complex derived from O2, built on a tris-imidazolylborate ligand.26 The Mirica lab has done extensive work in the generation of high valent Ni organometallic complexes using a tacn-cycloneophyl ligands.27 Recent work in their lab demonstrated the chemoselective C–C and C–O bond formation mediated by a high valent nickel complex using O2 as the primary oxidant.28 Similarly, the McDonald lab has extensive work on the oxidative chemistry of high valent nickel using pyridinecarboxamidate ligands, albeit generated through chemical oxidation.29 We propose the formation of a Ni(III)-superoxo species as the active oxidant during the oxidative cleavage reaction. We provide both experimental and computational evidence for this reactivity which could be relevant and provide further clues for the regioselective mechanism of ARD. More generally, a proposed high-valent Ni(III) derived from the reaction with O2 warrants consideration as a relevant intermediate in the normal and/or aberrant function of other dioxygenases and their involvement in disease; and opens up the door for further inquiry on high valent nickel complexes, which have generated interest in recent years as versatile oxidants.27,30–32
Experimental
Materials and methods
The preparation and manipulation of air-sensitive compounds were performed using standard Schlenk techniques under an N2 atmosphere. Reagents and solvents were purchased from commercial suppliers of the highest available purity and used without further purification unless otherwise noted. Solvents were purchased ultra-dried and further dried using 4 Å molecular sieves (butanone and MeOH). For acetonitrile, dichloroethane, and diethyl ether the solvents were purified via a drying column on a PPTechnologies solvent purification system. All solvents including deuterated solvents were degassed via freeze–pump–thaw cycles. 1H NMR spectra were recorded on a Varian 300 MHz spectrometer at room temperature and referenced to a residual deuterated solvent; as well as a Bruker 400 MHz spectrometer at room temperature. UV-vis spectra were recorded in a 1 cm cuvette on a Varian Cary 60 spectrometer. Cyclic voltammograms were recorded inside an N2 atmosphere glove box using a Pine Research Wavenow wireless galvanostat/potentiostat system using a 2.0 mm glassy carbon working electrode and an Ag/AgNO3 reference electrode, and a platinum auxiliary electrode. Elemental analyses were performed by Galbraith Atlantic Microlabs, Norcross, GA. Synthetic procedures and characterization details are presented in the ESI.†Computational techniques
Density Functional Theory simulations employed the Gaussian 16 software package.33 As in an earlier study of ARD biomimetics, the BP86 functional and 6-31+G(d) basis sets were used for geometry optimizations and to confirm that stationary points had the appropriate number of imaginary frequencies. For more accurate energetics, single point calculations were performed at all stationary points at the BP86+GD3BJ/6-311++G(d,p)/SMD-methanol level of theory. Enthalpic and entropic corrections to electronic energies were derived from the unscaled BP86/6-31+G(d) frequencies. All reasonable spin states, tautomer's, and conformers were evaluated as needed. Thermodynamic quantities assume standard state conditions.Synthesis of ligand N,N-(2-pyridil)(2-hydroxy-3-tbutylphenol)-N′-dimethylethane ((N-(ethyl-N′Me2)-(Py)-(2-t-ButPhOH))-L1)
The ligand L1, was synthesized via an in situ reductive amination reaction. N,N-Dimethylethylenediamine (140 μL, 1.30 mmol), and 3-tert-butyl-2-hydroxybenzaldehyde (197 μL, 1.10 mmol), were added to a Schlenk flask containing 4 mL of dry 1,2-dichloroethane and 4 Å sieves. These reacted at 40 °C under an N2 atmosphere. The condensation reaction was monitored via thin layer chromatography using ninhydrin stain to test for any unreacted amine (∼1 h). Following, sodium triacetoxyborohydride (NaBH4(OAc)3 (STAB)) (0.308 g, 1.40 mmol) was added to the reaction mixture and allowed to stir for 24 hours at which time 2-pyridine carboxaldehyde (110 μL, 1.15 mmol) was added and the reaction was allowed to stir for 1 hour prior to the second addition of the reducing agent, NaBH4(OAc)3 (0.308 g, 1.40 mmol). The final mixture reacted for 24 hours. The reaction was quenched with 10 mL of 2 M sodium hydroxide and extracted with dichloromethane and washed with saturated sodium chloride solution, followed by drying over anhydrous sodium sulfate. The solvent was removed under reduced pressure, and the resulting oil was further dried under vacuum and isolated as a light-yellow oil. The product was further purified by dissolving it in 2 mL of n-hexane, that was precooled in a flask in liquid nitrogen. The resulting solution was then frozen in liquid nitrogen and allowed to thaw. As it thawed, a dark precipitate and a light-yellow liquid formed. The yellow liquid was collected, and the solvent was removed under vacuum, resulting in light-yellow crystals. Yield: 0.243 g (61%).1H NMR CDCl3 (300.1 MHz, CDCl3, 298 K) of L1δ 8.42 (d, 1H), 7.53 (t, 1H), 7.25 (d, 1H), 7.11 (t, 1H), 7.06 (d, 1H), 6.82 (d, 1H), 6.63 (t, 1H), 3.68 (s, 4H), 2.58 (t, 2H), 2.41 (t, 2H), 1.95 (s, 6H), 1.37 (s, 9H).
Synthesis of complex [Ni(II)(N-(ethyl-N′Me2)(Py)(2-t-ButPhOH))(OTf)] (1)
Complex 1 was synthesized in an anaerobic environment inside the glovebox. L1 (0.243 g, 0.70 mmol) was dissolved in 3 mL of dry acetonitrile in a Schlenk flask. One equivalent of triethylamine (Et3N) (99 μL, 0.70 mmol) was added and reacted for five minutes, followed by the addition of 1.1 equivalents of nickel(II) trifluoromethanesulfonate (0.253 g, 0.71 mmol). The reaction was left to stir for 24 hours at room temperature. A white-yellow precipitate forms and this is filtered resulting in a dark brown mother liquor. The solvent was removed under vacuum and the resulting residue was washed with diethyl ether (3× ∼5 mL) resulting in a bright green solid after drying. The powder was further layered with butanone and diethyl ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio) and left to sit overnight to yield a semi-crystalline bright green powder (59% yield). Crystals of 1 were obtained from the slow vapor diffusion of diethyl ether into a concentrated solution of 1 in butanone.
:3 ratio) and left to sit overnight to yield a semi-crystalline bright green powder (59% yield). Crystals of 1 were obtained from the slow vapor diffusion of diethyl ether into a concentrated solution of 1 in butanone.
Yield = 59% (0.174 g, 0.32 mmol). Elemental analysis for NiN3O4C22H30SF3 calcd: C, 48.198%; H, 5.516%; N, 7.665%. Found: C, 48.45%; H, 5.62%; N, 7.76%.
Electronic absorption spectrum (MeOH): λ (nm) (ε (M−1 cm−1)): 252 (18100), 302 (7089), 395 (758). HRMS (ESI-MS, m/z): calc. for [NiC21H30N3O]+: 398.1744, calculated: 398.1737.
O2 reactivity studies
For a typical UV-visible oxygen reactivity experiment, a stock solution of 1 (1.11 mM) dissolved in methanol was prepared. A 750 μL aliquot (0.005 mmol) of 1 was added to a cuvette with a septum cap inside of the glovebox. 3 mL of MeOH were added to the cuvette. Separately, 20 mol equivalents of 2-hydroxyacetophenone (2HAP-H model substrate) were dissolved in 200 μL of MeOH and placed into a gastight syringe. A stock solution of Et3N (700 mM) dissolved in methanol was prepared. A 16 μL aliquot (2 eq., 0.011 mmol) of Et3N was added to a gastight syringe. The cuvette and syringes were cycled out of the glovebox. After initial scanning of 1 (200 nm to 1100 nm), the substrate was injected into the cuvette and monitored for 5 minutes. Then, Et3N was injected, and the reaction was monitored for 15 more minutes. The cuvette was then bubbled with oxygen from a needle fitted to tubing attached to an oxygen tank. The oxygen bubbled for 40 minutes at room temperature, before the reaction was monitored every 5 minutes for 2 hours, and then every 30 minutes until complete disappearance of the intermediates are observed.For a typical 1H-NMR oxygen reactivity experiment 15 mg of 1 was added to 750 μL of degassed deuterated methanol (20.7 mM with respect to 1) and transferred directly into an NMR tube for in situ monitoring via1H NMR. Subsequently, stoichiometric, or excess (∼1,3,5 or 20 mol equivalents with respect to 1) of model substrate dissolved in 500 μL of MeOD and 2 mol eq. of Et3N were added to the NMR tube and a spectrum was collected to monitor the changes. To the reaction mixture O2 gas was bubbled via a needle for 120 minutes total, and then monitored for 24 hours.
GC-MS product detection
The sample for GCMS analysis was prepped from the products resulting from the reaction of 1 (15 mg, 0.027 mmol) with 20 mol equivalents of 2-hydroxyacetophenone (2HAP-H) and 2 eq. of Et3N, dissolved in 5 mL of dried, degassed MeOH in a Schlenk reaction flask. This was left to react at room temperature under a dynamic atmosphere of oxygen gas via a needle fitted to a Schlenk manifold for 40 minutes. The sample was then quenched with 10 mL of 2 M hydrochloric acid and extracted 3× with 10 ml of dichloromethane (DCM). The solvent was removed under vacuum. The sample was diluted to 5 ml with chloroform (CHCl3). This was then further diluted by taking a 10 μL aliquot and adding 490 μL of CHCl3. A final dilution was performed by taking 50 μL of the previous stock and adding 100 μL of CHCl3 and 50 μL of the derivatizing agent, N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA). The sample was then heated in a water bath at 60 °C for 30 minutes and finally diluted to 1 mL of CHCl3 to give the sample for analysis.34In situ oxidation studies with outer sphere oxidants
A stock solution of 1 (5.5 mM) was prepped in degassed methanol and a 110 μL (6.21 × 10−4 mmol) aliquot was added to a cuvette outfitted with a septum cap inside of the glovebox along with 3 mL of MeOH. The reaction was run without and with the substrate. For those reactions containing substrate, a stock solution of 2-hydroxyacetophenone (110 mM) was prepped in methanol and a 50 μL aliquot (20 eq., 0.005 mmol) was syringed into a gastight syringe and cycled out of the glovebox along with the cuvette. A stock solution of the oxidant (tris(4-bromophenyl) ammoniumyl hexachloroantimonate (magic blue) or ammonium cerium(IV) nitrate) was prepared in acetonitrile (3.2 mM). A 250 μL aliquot (1 eq.) of the oxidant was transferred out of the glovebox in a gas tight syringe. After adding 2HAP-H to the cuvette containing 1, the mixture reacted for an incubation period of 5 minutes. The oxidant aliquot was then injected into the cuvette and the reaction was monitored over several hours via UV-Visible spectroscopy.Results and discussion
Synthesis and characterization of [Ni(II)(N-(ethyl-N′Me2)(Py)(2-t-ButPhOH))(OTf)] (1)
The asymmetric, tripodal N3O chelating ligand (N-(ethyl-N′Me2)-(Py)-(2-t-ButPhOH)) (L1) was synthesized via the in situ reductive amination between N,N-dimethylethelenediamine and the corresponding aldehydes (Scheme 1 top). The resulting oil was purified via a wash with cold n-hexane to yield the desired ligand in 61% yield. L1 was used for the synthesis of the complex [NiII(N-(ethyl-N′Me2)(Py)(2-t-ButPhOH))(OTf)] (1). Complex 1 was synthesized via the addition of one equivalent of triethylamine to an acetonitrile solution of L1, followed by the addition of 1.1 equivalent of nickel(II) trifluoromethanesulfonate at room temperature, and allowed to stir for 24 hours (Scheme 1 bottom). The resulting turbid mixture was filtered to remove a pale-yellow precipitate, and the mother liquor was dried under vacuum, washed with diethyl ether (3×), and layered with butanone and diethyl ether to result in a ‘bright green powder in good yields (59%)’. The green powder was crystallized via slow vapor diffusion of diethyl ether into a concentrated solution of 1 in butanone to yield green crystals (Fig. 3).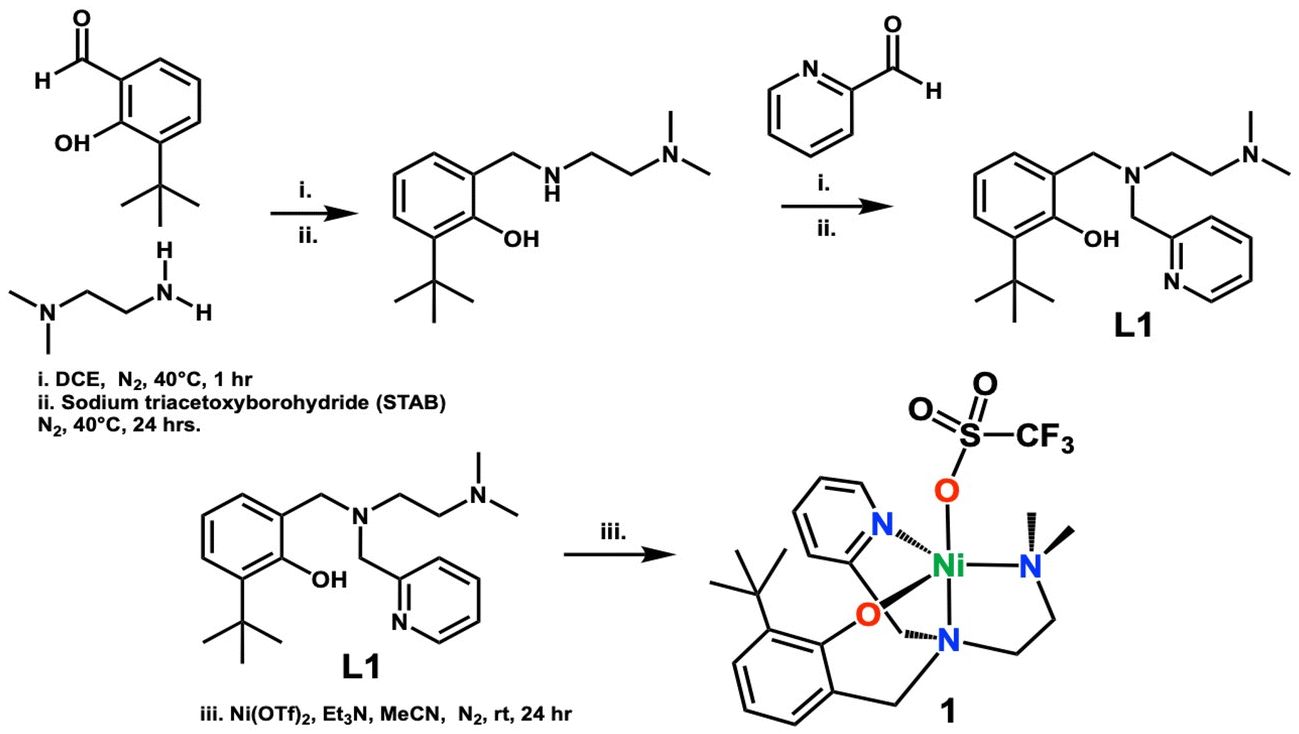 | ||
| Scheme 1 Synthetic procedures for making ligand L1 and biomimetic metal complex 1. | ||
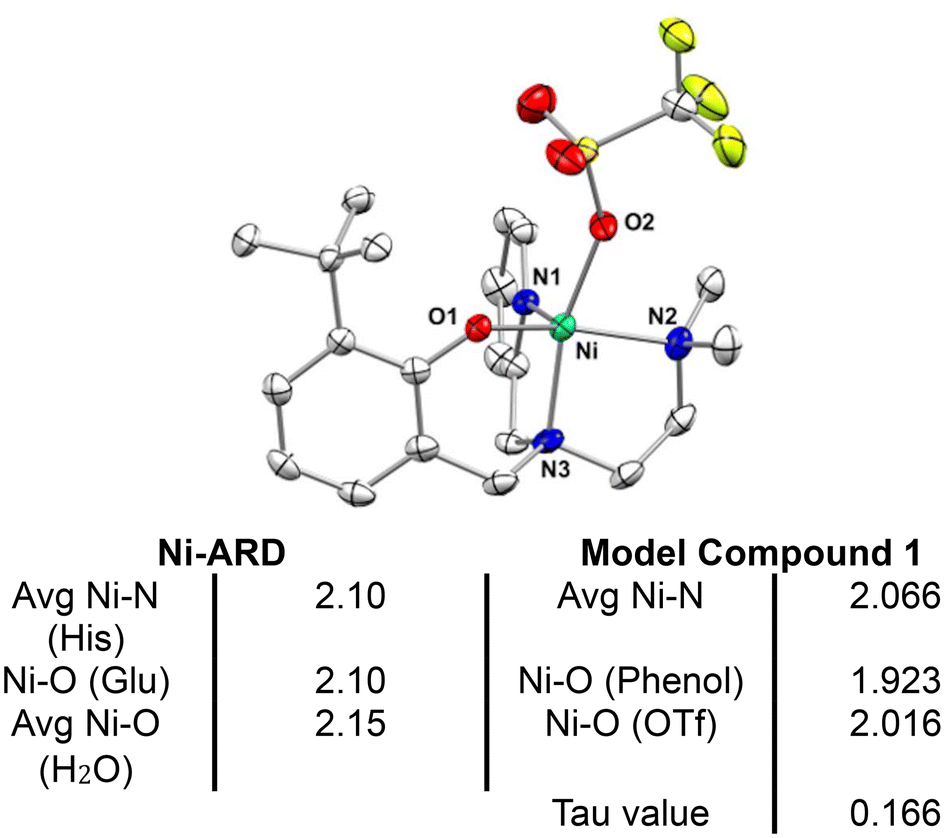 | ||
| Fig. 3 Top – Crystal structure of [NiII (N-(ethyl-N′Me2)(Py)(2-t-ButPhOH))(OTf)] (1) with ellipsoids drawn at the 50% probability level. H atoms omitted for clarity. Bottom – Table of selected bond lengths in Å for Ni-ARD (PDB ID: IVR3)25 and for complex 1. | ||
Crystallographic analysis shows that 1 is a nickel(II), pentacoordinate, pseudo-square pyramidal complex (Tau value = 0.166), coordinated to a triflate counterion (Fig. 3). 1 mimics the structural features of ARD's active site. The shorter bond lengths for 1 in comparison to those of Ni-ARD can be attributed to the stronger donating character of the tertiary amine (N3) and the more basic phenolate donor (instead of carboxylate of glutamate-O1). The labile triflate ligand gives accessibility to two open sites for substrate binding in analogous way to ARD. This ligand environment is an improvement over previous N4 and N4O models of ARD (Fig. 2b).
The 1HNMR of complex 1 in deuterated methanol displays paramagnetically shifted signals between 170 and 0.25 ppm (Fig. S5†) consistent with a Ni(II) S = 1 complex. The UV-Visible absorption spectrum of 1 in methanol (Fig. S4†) displays bands that absorb intensely in the ultra-violet region: 250 nm (ε = 8100), and 302 nm (ε = 7089). Additionally, there is a weakly absorbing broad band around 395 nm (ε = 758). The UV bands can be attributed to ligand-based charge transfer transitions while the weak visible band are characteristic of other nickel(II) complexes of similar geometry.24,35,36
The cyclic voltammogram of complex 1 in acetonitrile (2.2 mM) was obtained using a Ag/AgNO3 reference electrode (10 mM in MeCN) with 0.1 M tetrabutylammonium phosphate (TBAP) as supporting electrolyte. All values are reported vs. an internal Fc/Fc+ standard. 1 displays a main irreversible peak in the cathodic region at Epa = 159 mV (Fig. S6†). Additionally, and more anodically shifted, we observe two much smaller irreversible peaks at Epc = −159 mV and −409 mV. We posit that the wave at +159 mV corresponds to an irreversible Ni2+/Ni3+ couple, an assignment supported by the observance of a quasi-reversible wave for the iron(II) analogous complex (Fig. S9†),37 which displays an E1/2 = 48 mV (ΔEp = 95.8 mV). Based on the relative ease of oxidation of Fe(II) vs. more redox inactive Ni(II) complexes, it is reasonable to assume that the corresponding +2/+3 wave for 1 should be cathodically shifted, as we have observed. Additionally, this does not appear to be a ligand centered process, based on the absence of this peak for the iron(II) complex, which would be expected to possess a similar ligand-based oxidation wave. The other two very minor peaks at −159 mV and −409 mV cannot be assigned at this moment although they could correspond to a chemically irreversible reduction of a Ni(III) species generated electrochemically, and/or a Ni1+/Ni2+. These three peaks appear to be connected to the nickel sample analyzed, since stoichiometric titration of 1 in the CV results in current growth for all three peaks, with the peak at 159 mV still the dominant species (Fig. S7†). This peak's potential also changes due to the addition of substrate (2HAP-H 144 mV Fig. S7†) and base (Et3N 69 mV Fig. S7†) consistent with a bidentate binding of protonated and deprotonated substrate respectively (anodic shifts). Based on NMR spectra of the same batch of complex used for CV, we note there were no other ligand containing metal complexes found in solution (Fig. S5†). When compared to the previously reported family of N4O based Ni(II) complexes that served as models for ARD (Fig. 2b), 1 is the most anodically shifted molecule amongst the group (1 159 mV vs. 570 mV irrev. for the parent Ni(II)N4O complex) consistent with the stronger electron donating nature of the L1.24 The redox behavior of 1 suggests the likely accessibility of a possible Ni(III) state in the presence of mild oxidants (vide infra).
Biomimetic oxidative C–C bond cleaving reactivity studies
ARD catalyzes the regioselective, oxidative, carbon–carbon bond cleavage of acireductone using O2 as the oxidant (Fig. 1). The enzyme requires substrate binding prior to oxidation with O2, displaying no reactivity when O2 is added first.8 This suggests a mechanism that requires substrate binding prior to involvement of O2. Whether O2 attacks the substrate–enzyme complex, or if it is directly activated by the metal is unknown. However, the current working hypotheses emphasize the limited role of the metal in activating dioxygen.4 Most dioxygenases activate O2 into a more active oxidant (i.e. superoxide or peroxide) prior to substrate oxidation.38–40 This is often mediated by the redox activity at the metal center. In ARD's case it is argued that the metals (on and off pathway) do not engage in redox activity during the oxidative cleavage of acireductone.41 However, computational studies, suggest that the on-pathway metal, iron, can access a charge transfer, transient redox state (Fe3+) in the catalytic cycle.10,22 The authors of this work argue nickel is not capable of this, but that the off-pathway metals (Co or Mn) may be able to access these changes in redox state. These results highlight that more work needs to be done to understand how the metal redox state influences the oxidation of substrate.In our recent work we showed that a family of Ni(II) N4O mimics of ARD were capable of oxidatively cleaving the C2–C3 bond in acetylacetonate (acac).24 Through computational work we argued that the ability of these complexes to activate this relatively inactive substrate, was due to the presence of the O donating moiety in the ligand. We showed that these complexes activated acac without any direct activation of O2 at the metal center, and that instead the reactivity was mainly driven by the Lewis acidity of the metal activating the substrate, priming it for reactivity with O2. This was likely due to the inaccessibility of a second site available for O2 activation in the coordinatively saturated N4O ligand environment.
For this research we chose to use the substrate 2-hydroxyacetophenone (2HAP-H) for our biomimetic studies (Scheme 2). 2HAP-H is a C1–H substrate analogous to acireductone and an analogue substrate of 2,4′-dihydroxyacetophenone dioxygenase (DAD).42 Chavez et al. recently showed that an iron-based model complex was capable of oxidatively cleaving the C1–C2 bond of 2HAP-H in the presence of dioxygen under mild conditions.43
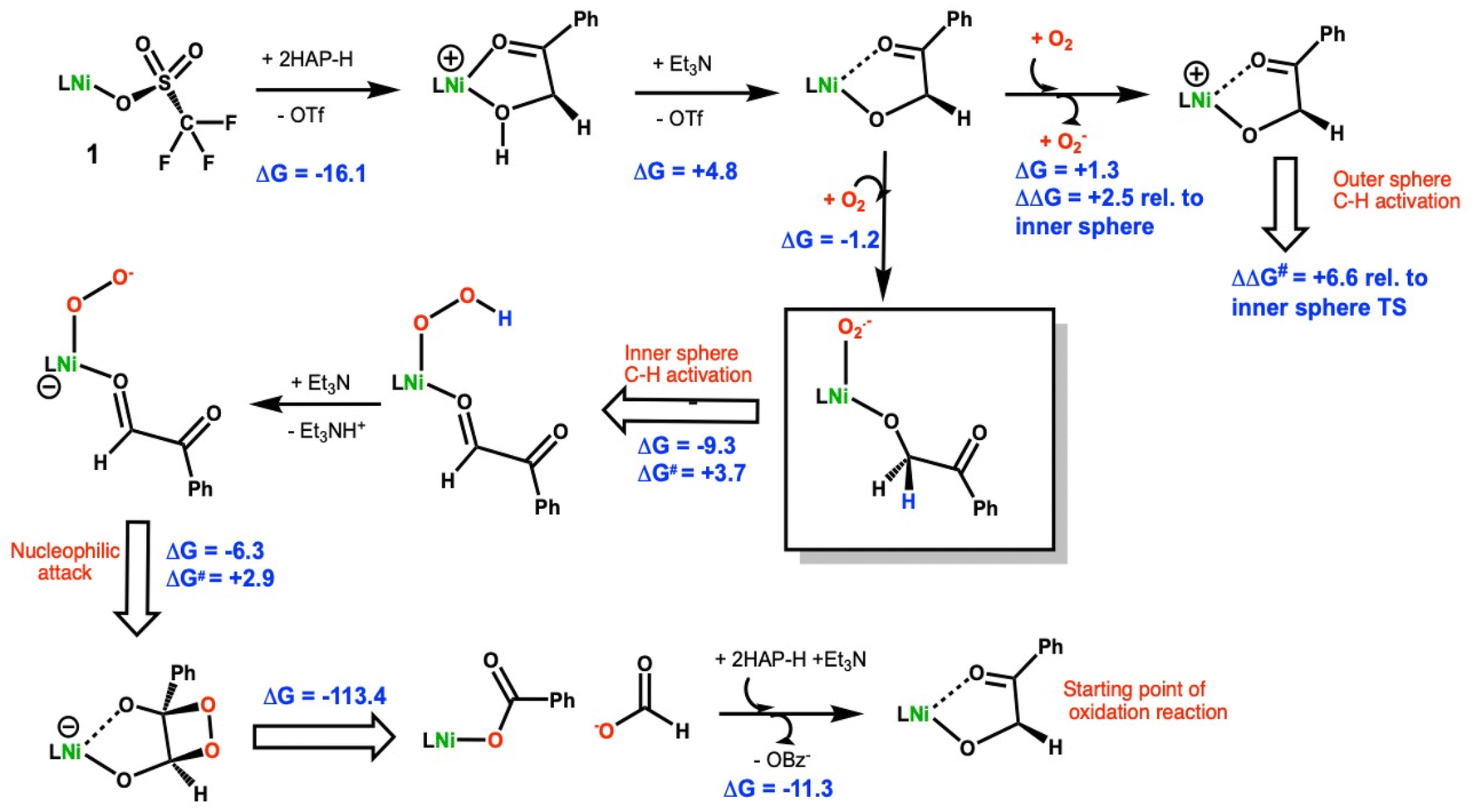 | ||
| Scheme 2 Proposed reaction mechanism and relevant free energies (kcal mol−1) for the O2 mediated oxidation of 2-hydroxyacetophenone (2-HAP-H) by 1. All nickel species are calculated to be ground state triplets with the exception of the Ni(III) complexes resulting from O2 oxidation, which are predicted to be doublets. The remaining species are best described as Ni(II). Geometries optimized at the BP86/6-31+G(d) level of theory. At those geometries, single point BP86/6-311++G(d,p) calculations are run with dispersion (GD3BJ) and solvent (SMD-methanol) corrections. | ||
Addition of an excess (20 equivalents) of 2HAP-H to a methanolic solution of 1 in the presence of 2 eq. of triethylamine (Et3N) results in the growth of a weak visible d–d band at 596 nm and one in the near-IR region at 950 nm (Fig. S11†). We attribute these changes to the spectrum of 1 to the binding of deprotonated 2HAP-H (2HAP) to 1. 2HAP is likely binding to Ni(II) in a bidentate fashion, because we have been able to isolate and crystallographically characterize the analogous acac bound Ni(II) derivative of 1 (Fig. S10†). This compound displays the substrate coordinated in bidentate fashion to the Ni(II) center resulting in a pseudo-octahedral complex. Acac bound 1 displays similar absorption bands at 608 nm and 950 nm respectively (Fig. S9†). The resulting compound, 1-2HAP, is oxygen sensitive. Attempts to crystallize the 1-2HAP complex were unsuccessful and only yielded the crystallization of oxidative cleavage products (1-benzoate, Fig. S13†), highlighting the sensitivity of this compound towards reacting with O2.
The 1H-NMR of 1-2HAP displays distinctly different paramagnetically shifted signals compared to those of 1, indicative of changes to the coordination environment of the nickel center (Fig. S12†). These results suggest a binding event in solution consistent with the UV-Vis results, and analogous to the binding of acac to 1.
When dry oxygen is bubbled into a methanolic solution of 1-2HAP for 40 minutes, we see changes to the UV-Visible spectrum (Fig. 4). This reaction results in the growth of several intense bands in the visible region (400 nm (sh), 530 nm, 690 nm, 774 nm, 878 nm – Fig. 4). The latter three bands grow first and rapidly over the course of ∼20 minutes after O2 addition. Some growth is observed for the 400 and 530 nm bands in this period. These two bands maximize in ∼105 min with concomitant decay of the other three original peaks. The rates of growth and decay of these two sets of peaks suggest that they likely belong to at least two separate transient species. The 400 (sh) and 530 nm peaks go away completely in ∼10 h. These dramatic changes to the visible region of the complex are likely due to the O2 mediated oxidation of the Ni(II) complex to a putative transient Ni(III) species. Ni(III)–N(amidate) ligated complexes from the literature display characteristic intense bands in the visible region.31,44,45 Furthermore, reactivity studies of 1 in the presence of non-coordinating external oxidants, tris(4-bromophenyl) ammoniumyl hexachloroantimonate (magic blue) and ammonium cerium(IV) nitrate, result in the formation of similar, but not identical, absorption spectrum patterns in the visible region also displaying transient behavior (Fig. S18–21†). DFT thermodynamic parameters (vide infra) and TD-DFT studies for the various Ni(III) cationic complexes (Ni(III)-OTf, Ni(III)-2HAP, and Ni(III)-benzoate, Fig. S22–27†) support our designation, and attribute these absorption bands to charge transfer transitions from metal orbitals to phenolate-ligand based unoccupied orbitals. These complexes were calculated as low spin d7 Ni(III) cationic complexes with very little discernable ligand based redox activity (see Computational section for details).
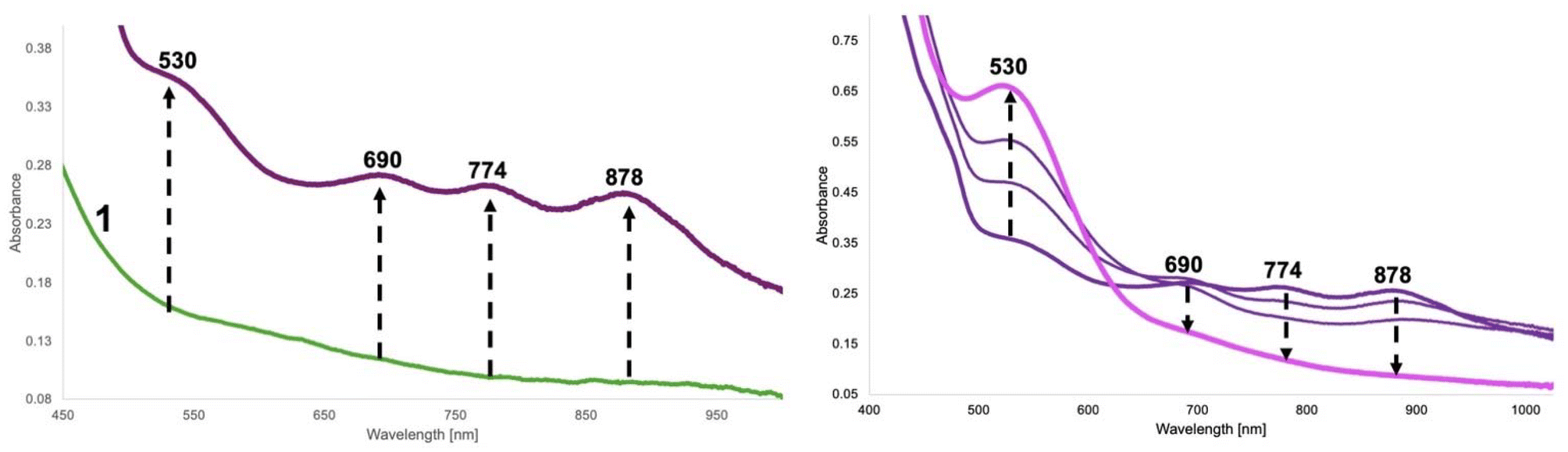 | ||
| Fig. 4 Left – UV-Visible absorption spectrum monitoring the growth of the reaction between 1, 2HAP-H (20 eq.) + Et3N (2 eq.) and excess O2 (40 min bubbling) in MeOH at room temperature. Right – Second phase growth and decay of transient species during the reaction. | ||
We can follow the O2 mediated reaction via1H-NMR spectroscopy (Fig. 5). For the NMR experiment we used 1 eq. of substrate (2HAP-H), 2 eq. base (Et3N) with respect to 1 (20.7 mM). After a short incubation time with the substrate and base, initial dioxygen bubbling is done directly to the NMR tube for ∼40 minutes. We monitored the reaction by the changes in the aromatic region over the course of 3 hours to track the disappearance of signals associated with the substrate 2HAP-H (7.95 ppm, 7.63 ppm, 7.51 ppm), with the concomitant appearance of signals corresponding to benzoic acid (8.00 ppm, 7.58 ppm, 7.46 ppm), the proposed C1–C2 oxidative cleavage product of 2HAP-H (Scheme 2). Over the course of this reaction the complex appears to cycle between at least two different species as evidenced by the color changes of the reaction in the NMR tube during consumption of O2 (ESI-Video 1†). Additionally, we ran this reaction under conditions where we titrated an excess of substrate (1, 3, and 5 eq.) and over the course of 24 hours and 120 min of O2 we noticed the disappearance of most of the 2HAP-H added (Fig. S14†). Under large excess of substrate ∼20 eq., we have observed disappearance of greater than 50% of the substrate added (Fig. S15†). These observations point to possible catalytic activity of the model complex towards substrate in the presence of excess O2.
 | ||
| Fig. 5 1HNMR timelapse spectra (CD3OD) of the aromatic region showing reactivity between complex 1 with 2HAP-H (1 eq., green trace), Et3N (2 eq., brown trace), excess O2 (40 min bubbling, purple trace), and final scan 180 minutes after the first addition of O2 (teal trace) showing the complete disappearance of the starting substrate. | ||
Large scale oxidation reactions of 1 were performed and the products from these reactions were analyzed by GC-MS after an acidic workup of the reaction mixture to remove the metal. These results confirm the formation of benzoic acid, as a product of oxidation, consistent with the oxidative C1–C2 cleavage of 2HAP-H (Fig. S17†).
Computational analysis for the mechanism of the O2 mediated oxidation of 2-hydroxyacetophenone (2HAP-H) by 1
DFT calculations using methods previously validated on a related Ni(II)–N4O ARD model24 were employed to understand the oxidative C–C cleavage of 2HAP-H by 1. TD-DFT calculations used the level of theory put forth by Chavez et al.43 in their recent study of related iron dihydroxyacetophenone dioxygenase biomimics. Particular attention was focused on the early steps in the reaction scheme given the growing interest in biomimetic oxidation by Earth-abundant 3d metals using dioxygen and the fact that the C–C cleavage steps are computed to be highly exergonic.Based on the combination of these experimental results, the mechanism in Scheme 2 is proposed for the oxidative C–C bond cleavage of 2HAP-H by 1.
The energetics of 2-hydoxy-acetophenone (2HAP-H) deprotonation in SMD continuum methanol solvent were calculated. The free energy to displace triflate from 1 by 2HAP-H is exergonic by 16.1 kcal mol−1, Scheme 2. Based on this, we assume that the dissociation of the anionic triflate by neutral 2HAP-H is likely a multi-step process with a reasonable barrier. Using triethylamine (Et3N) as a model base, ΔGdeprot is computed to be +12.0 kcal mol−1 to remove the hydroxyl proton of 2HAP-H. Upon coordination of 2HAP-H, ΔGdeprot by Et3N is reduced by ∼7 kcal mol−1 to +4.8 kcal mol−1. Coordination of the neutral substrate (k2-O,O′) to the nickel center thus greatly enhances its acidity. Interestingly, the optimized structure of the neutral ligand ligated to the Ni(II) center shows a hydrogen bonding interaction with the phenolate moiety of the N3O ligand (Fig. S28†). The Mulliken atomic charge on the hydroxyl proton is +0.28 vs. +0.33e− for free 2HAP-H at the same level of theory. The charges thus infer that the enhanced acidity of the metal-bound substrate primarily arises from stabilization of the anionic ligand upon deprotonation, more so than enhanced Lewis acidity of the hydroxyl proton from sigma-donation to the central metal ion.
The DFT calculations also suggest that coordination of the substrate to the Ni(II) center makes it easier to oxidize relative to the Ni(II)-triflate starting material (1). To model an outer-sphere electron transfer (ET) pathway, the free energy of electron transfer to O2 to make superoxide was computed. For the neutral starting material, 1, we found a ΔGET = +17.3 kcal mol−1 to form superoxide and the cationic Ni(III) triflate complex. For the neutral Ni(II) 1-2HAP complex the corresponding ET reaction to make superoxide is essentially thermoneutral, ΔGET = +1.3 kcal mol−1. There is little change in the geometry of the 1-2HAP complex upon oxidation, RMSD ∼0.29 Å. Comparing the HOMO (highest occupied Kohn–Sham molecular orbital) energies for 1 and 1-2HAP indicates the latter orbital energy is higher, thus the 1-2HAP complex is predicted to be more easily oxidized by ∼0.6 eV, consistent with the calculated energy difference in the modeled electron transfer reactions (ΔΔGET = 17.3–1.3 = 16.0 kcal mol−1 = 0.69 eV). This is consistent with our experimental observations that 1 is air stable indefinitely in solution but 1-2HAP is very O2 sensitive.
The same model reaction was used to assess the relative proclivity toward oxidation of neutral, Ni(II) complexes, viz. complex + O2 = O2− + cation. For the triflate starting complex (1) as a baseline, the referenced oxidation is ΔG = +17.3 kcal mol−1. For the corresponding 1-2HAP complex, oxidation is much more facile as noted earlier. For the benzoate adduct, oxidation of the neutral complex is calculated to lie in between the triflate and 2HAP complexes, ΔG = +11.9 kcal mol−1. Thus, among these three complexes, 1-2HAP is clearly the most prone to oxidation, and indeed even close to thermoneutral, ΔG = +1.3 kcal mol−1, with respect to oxidation by dioxygen. Furthermore, all the cations are predicted to have low-spin (doublet) ground states. For the cationic [1-2HAP]+ complex, 0.67 unpaired e− is calculated to reside on the nickel ion, commensurate with that calculated (via Mulliken population analyses) for the cationic benzoate and triflate congeners, 0.74 and 0.62 unpaired e− on Ni, respectively. The remainder of the spin density is on the ligating atoms. Hence, the DFT data suggests low-spin d7 Ni(III) complex for the cations, which no evidence of significant ligand noninnocence in the redox event.
An inner-sphere oxidation pathway was also considered and the free energy for O2 coordination was computed to be mildly exergonic, ΔG = −1.2 kcal mol−1. In coordinating to the Ni(II) center in 1, the O2 displaces the carbonyl arm of the erstwhile k2-O,O′-2HAP. The optimized O–O bond length is 1.303 Å, which compares with 1.369 Å and 1.231 Å for free superoxide anion and triplet dioxygen, respectively, at the same level of theory (Fig. 6). The electronic structure of the triplet Ni(III)-2HAP-O2 adduct is particularly interesting, showing ∼1e− of total spin density on the two oxygen atoms of the O2 ligand, with slightly more on the terminal oxygen and ca. 1e− on the remainder of the complex; the latter spin density is largely situated on the nickel with a modest amount of spin density on the ligating atoms of the N3O supporting ligand (Fig. 6). Considering the calculated spin density and O–O bond lengths, this complex is viewed primarily as a Ni(III)-superoxide with an admixture of Ni(II)-dioxygen character.
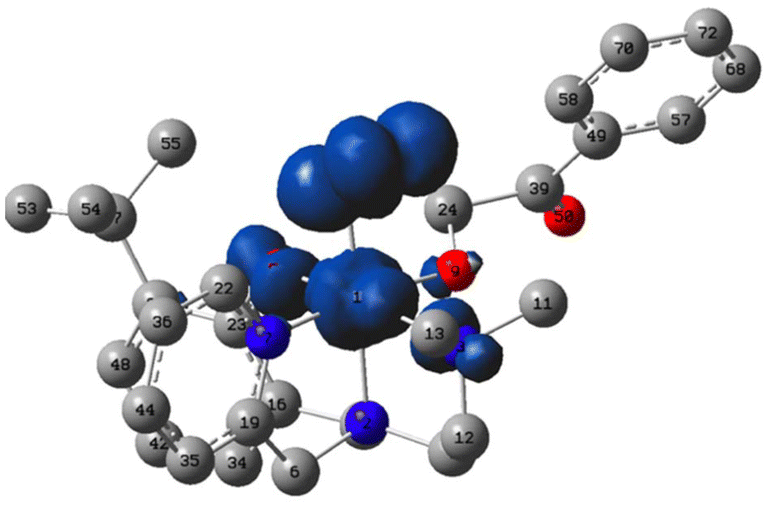 | ||
| Fig. 6 Plot of spin density (IsoValue = 0.004 a.u.) of the triplet ground state of the Ni(III)-superoxide complex. Spheres: O = red, C = gray, N = blue, Ni = light blue; hydrogen atoms are omitted from the figure for clarity. Optimized bond lengths (Å): NiOo2 = 1.87, O–O = 1.30, NiOalkoxide = 1.88, NiNamine (trans to phenolate) = 2.50, NiNpy = 1.97, NiOphenolate = 2.05, NiNamine (trans to O2) = 2.05. Ligand–metal–ligand angles = 90 ± 10°. | ||
For the Ni(III)-superoxide, there is a transition state (Fig. S29†) with a small barrier to C–H activate the C1–H bond of the 2-HAP substrate by the coordinated superoxide, ΔG‡ = 3.7 kcal mol−1, leading to a Ni(II)-hydroperoxide (Scheme 2). The H-atom transfer process is computed to be exergonic, ΔGHAT = −9.3 kcal mol−1. An isomeric outer-sphere C1–H bond activation TS was also isolated and computed to be 6.6 kcal mol−1higher than the inner sphere TS just mentioned (Fig. S29†). In conjunction with its calculated thermodynamic advantage (vide supra), ΔΔGET = 2.5 kcal mol−1, the computations suggest both a kinetic and thermodynamic advantage for an inner-sphere oxidation pathway. The resulting product is a Ni(II)–OOH also bound to the resulting oxidation product of 2HAP, phenylglyoxal. A ligated organic (2HAP based) hydroperoxyl isomeric to the Ni(II)–OOH intermediate was calculated and found to be downhill by 9.8 kcal mol−1 (Fig. S30†). Calculations suggest that the Ni–OOH intermediate is easier to deprotonate – again using Et3N as a model base – than the organic R–OOH intermediate, ΔG = +11.4 vs. +5.7 kcal mol−1, respectively. Hence, we propose that after the HAT step to make Ni–OOH this complex is deprotonated to give anionic NiOO−, which nucleophilically attacks phenylglyoxal at the carbonyl C1 position closing the ring via attack to C2, to make a dioxetane intermediate, ΔG‡ = +2.9 kcal mol−1, ΔG = −4.9 kcal mol−1. This intermediate is common in the literature for C–C cleaving reactions in biomimetic and enzymatic systems relevant to quercetin dioxygenase.38 We were unable to isolate the TS for decomposition of the dioxetane intermediate, but this reaction is extremely exergonic, ΔG = −113.4 kcal mol−1, and is computed to favor the observed products, i.e., Ni(II)-benzoate + formate anion by ∼11 kcal mol−1 relative to the alternative formulation of Ni(II)-formate + benzoate anion. We calculated the displacement of the benzoate bound 1 product by the conjugate base of 2HAP-H to initiate another round of catalysis to be exergonic by 11.3 kcal mol−1. This is consistent with the catalytic behavior of 1 observed via1H-NMR. In our attempts to isolate a substrate bound 1-2HAP species, we only have been able to crystallize the product of oxidation, a Ni(II)-benzoic acid bound complex (Fig. S13†).
Time-dependent density functional theory calculations of proposed intermediates
Using the TD-DFT conditions described by Chavez et al.,43 the electronic spectra were computed for several of the intermediates in the proposed 2-HAP oxidation catalytic cycle. These spectra are plotted on the same scale in the 400–900 nm range (Fig. S31†). The oxidation of 1-2HAP to the corresponding Ni(III)-2HAP cation shows not only a shift in the computed UV-Vis bands, but also a roughly 5× increase in the intensity of the most intense bands consistent with our experimental oxidation studies with intense charge transfer bands, in good agreement with our calculations. Similar TD-DFT calculations on neutral and cationic Ni-triflate complexes also show an increase in intensity upon oxidation of the former to the latter (Fig. S22–27†). For the Ni(III)-superoxide new bands at ∼480, 600 and 800 nm are predicted, and an increase in intensity of the UV-Vis bands relative to neutral 1-2HAP. For the hydroperoxide intermediate, three new bands at ∼500, 690 and 880 nm are seen, Fig. S31,† also in reasonable agreement with our UV-visible O2 reactivity studies depicting a mixture of species (Fig. 4).Implications of reactivity
Complex 1 is a new structural and functional model of ARD. It displays biomimetic dioxygenase-like, oxidative, C–C bond cleaving reactivity towards the C1–H substrate 2HAP-H using O2 as the oxidant. Experimental and computational data suggests that the Ni(II) center in 1 can access a high-valent Ni(III) state via direct oxidation by O2, only when substrate is bound to 1. Computational data suggests that the anionic 2HAP substrate modulates the redox potential of the metal center, making it more susceptible to oxidation. We propose the formation of a transient Ni(III)-superoxo complex derived directly from O2 activation at the nickel center. This species begins the oxidation process by activating the C1–H bond of the substrate followed by a series of steps with low activation barriers consistent with our mild reactivity conditions. In ARD, substrate must bind to the active site before substrate oxidation,6,46 following an enzyme–substrate + O2 mechanism without an initial O2 activation step. Initial metal centered activation of O2 is more typical of other non-nickel-based dioxygenases. It remains unclear whether the metal at the active site of ARD is capable of undergoing oxidation or if it can activate dioxygen once substrate is bound. The results from the reactivity of 1 in this work suggest that there could be a pathway by which the substrate primes the active site for O2 activation and perhaps even transient oxidation of the nickel center. This proposed metal oxidation and activation of O2 is a more traditional reactivity pathway for dioxygenases and should not be ruled out for ARDs off pathway or as a possible moonlighting reaction. This reactivity might also be present for ARD bound to metals that more readily undergo oxidation chemistry, such as the on-pathway iron, and off-pathway cobalt and manganese. Preliminary work in our laboratory with derivatives of 1 with these metals (Fig. S8†)37 suggest this might be the case. It is worth hypothesizing that an O2 activation mechanism might be operable in the enzyme, and we are currently exploring this via computational studies on the enzyme. The insights of this work shed light on possible mechanisms that might be operable in the less common N3O (3His–1Glu) motif of other dioxygenases such as quercetin dioxygenase, where direct O2 binding to the metal is observed.Conclusions
A second generation structural and functional biomimetic model for the active site of acireductone dioxygenase was synthesized and characterized. This complex displays catalytic, biomimetic, dioxygenase-like reactivity towards 2-hydroxyacetophenone, resulting in the C1–C2 oxidative bond cleavage of the substrate. This reactivity was studied via1H-NMR, UV-Vis, and DFT studies. A computationally derived reaction mechanism suggests the formation of a transient Ni(III)-superoxo species, directly derived from O2 oxidation of the Ni(II) center. The propensity of Ni(II) to form a high valent Ni(III) coordination compound is supported by the strong donating qualities of the N3O ligand. Furthermore, complex 1 only reacts with O2 once the substrate coordinates to the metal in analogous way to ARD. This type of reactivity might have implications on the mechanism of action in ARD and that of other dioxygenases, opening questions about non-traditional/moonlighting reactivity pathways for other nickel dioxygenases. Understanding the factors driving this reactivity will be essential for future studies. Additionally, this work suggests the possibility of accessing high valent nickel coordination complexes via direct O2 oxidation, which could be utilized as versatile oxidants.Future work will focus on understanding the substrate oxidation scope, kinetic, and thermodynamic parameters of this reaction, and the characterization of the dioxygen derived intermediate. Additionally, we are currently synthesizing the mammalian relevant analogues using Fe, Mn, and Co, to better understand the metal mediated, regioselective bifurcating reactivity of ARD. This, coupled to our ongoing computational biochemical studies, will help to provide further evidence for the mechanism of action occurring in ARD. With the current call for ARD to serve as a therapeutic target for treatment of certain cancers, it is imperative that the mechanistic, electronic, and structural parameters driving the dual chemistry of ARD are understood.
Author contributions
Kirsch, K. E.: conceptualization, data curation, formal analysis, investigation, supervision, validation, visualization, writing – original draft, review and editing. Little M. E.: data curation, formal analysis, investigation, supervision, validation. Cundari, T. R.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, resources, software, validation, visualization, writing – review and editing. El-Shaer, E.: data curation, formal analysis, investigation. Barone, G.: data curation, formal analysis, investigation. Lynch, V. M.: data curation, investigation. Toledo S. A.: lead scientist; conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing – original draft, review and editing.Data availability
The data supporting this article, and accompanying figures, images, video, and crystallographic data have been included as part of the ESI.†Crystallographic data for compounds 1, 1-acac (2), 1-benzoic acid (3), have been deposited with the CCDC under deposition numbers 2375944 (1), 237724 (2), and 2375818 (3).†
Data for this article, including NMR, CV, X-ray diffraction, UV-Visible and computational are available at the American University Research Archive (AURA) at https://doi.org/10.57912/c.7419103.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Liviu Mirica and his post-doctoral student Dr Daniel Hu for essential discussions on high valent systems and experimental consultations. We thank Adam Fadul and Colleen O'Dowd for ligand synthesis optimization and reactivity discussions. We thank Dr Matthew Hartings for GC-MS consultations and Dr Alexander Zestos for useful cyclic voltammetry discussions.National Institute of General Medical Sciences of the National Institutes of Health under Award Number SC2GM130438. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. American University College of Arts and Sciences Faculty start-up fund, NASA District of Columbia Space Grant Consortium (NNX15AT64H), College of Arts & Sciences Summer Undergraduate Fellowship and the Chemistry Department at American University for student stipend support, Departmental Welch Foundation grant (BH-0018-20151106) for support of students at St Edward's University. T. R. C. acknowledges the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences Division, Catalysis Science Program, for partial support of this research through grant DE-FG02-03ER15387.
References
- A. Das, C. Hessin, Y. Ren and M. D.-E. Murr, Chem. Soc. Rev., 2020, 49, 8840–8867 RSC.
- P. Singla and R. D. Bhardwaj, Biocatal. Biotransform., 2020, 38, 81–92 CrossRef CAS.
- H. Eom and W. J. Song, J. Biol. Inorg. Chem., 2019, 24, 517–531 CrossRef CAS PubMed.
- A. R. Deshpande, T. C. Pochapsky and D. Ringe, Chem. Rev., 2017, 117, 10474–10501 CrossRef CAS.
- A. E. Pegg, Cancer Res., 1988, 48, 759–774 CAS.
- T. C. Pochapsky, T. Ju, M. Dang, R. Beaulieu, G. M. Pagani and B. OuYang, in Nickel and Its Surprising Impact in Nature, ed. A. Sigel, H. Sigel and R. K. O. Sigel, John Wiley & Sons, Ltd, Chichester, UK, 2007, pp. 473–500 Search PubMed.
- S. C. Chai, T. Ju, M. Dang, R. B. Goldsmith, M. J. Maroney and T. C. Pochapsky, Biochemistry, 2008, 47, 2428–2438 CrossRef CAS PubMed.
- A. R. Deshpande, T. C. Pochapsky, G. A. Petsko and D. Ringe, Protein Eng., Des. Sel., 2017, 30, 109–206 Search PubMed.
- A. R. Deshpande, K. Wagenpfeil, T. C. Pochapsky, G. A. Petsko and D. Ringe, Biochemistry, 2016, 55, 1398–1407 CrossRef CAS.
- A. Miłaczewska, E. Kot, J. A. Amaya, T. M. Makris, M. Zając, J. Korecki, A. Chumakov, B. Trzewik, S. Kędracka-Krok, W. Minor, M. Chruszcz and T. Borowski, Chem. – Eur. J., 2018, 24, 5225–5237 CrossRef.
- A. S. Almeida, C. Figueiredo-Pereira and H. L. A. Vieira, Front. Physiol., 2015, 6, 33 Search PubMed.
- S. Gatius, M. Jove, C. Megino-Luque, M. Albertí-Valls, A. Yeramian, N. Bonifaci, M. Piñol, M. Santacana, I. Pradas, D. Llobet-Navas, R. Pamplona, X. Matías-Guiu and N. Eritja, Cancers, 2022, 14, 2842 CrossRef CAS.
- A. M. Knapinska and G. B. Fields, Pharmaceuticals, 2019, 12, 77 CrossRef CAS.
- S. Pahwa, M. J. Stawikowski and G. B. Fields, Cancers, 2014, 6, 416–435 CrossRef.
- L. A. Field, B. Love, B. Deyarmin, J. A. Hooke, C. D. Shriver and R. E. Ellsworth, Cancer, 2012, 118, 1334–1344 CrossRef CAS PubMed.
- D. Yang, L. Su, X. Li, C. Xie and Y. Zhang, Exp. Neurol., 2023, 365, 114410 CrossRef CAS PubMed.
- W. Hirano, I. Gotoh, T. Uekita and M. Seiki, Genes Cells, 2005, 10, 565–574 CrossRef CAS.
- L. Qiang, H. Cao, J. Chen, S. G. Weller, E. W. Krueger, L. Zhang, G. L. Razidlo and M. A. McNiven, J. Cell Biol., 2019, 218, 317–332 CrossRef CAS.
- V. S. Golubkov, S. Boyd, A. Y. Savinov, A. V. Chekanov, A. L. Osterman, A. Remacle, D. V. Rozanov, S. J. Doxsey and A. Y. Strongin, J. Biol. Chem., 2005, 280, 25079–25086 CrossRef CAS PubMed.
- X. Liu and T. C. Pochapsky, Inorganics, 2019, 7, 101 CrossRef CAS.
- C. J. Allpress, K. Grubel, E. Szajna-Fuller, A. M. Arif and L. M. Berreau, J. Am. Chem. Soc., 2013, 135, 659–668 CrossRef CAS.
- C. E. Valdez, N. M. Gallup and A. N. Alexandrova, Chem. Phys. Lett., 2014, 604, 77–82 CrossRef CAS.
- E. Szajna, A. M. Arif and L. M. Berreau, J. Am. Chem. Soc., 2005, 127, 17186–17187 CrossRef CAS PubMed.
- G. A. Blade, R. Parveen, J. L. Jaimes, W. Ilustre, D. Saldaña, D. A. Ivan, V. M. Lynch, T. R. Cundari and S. Toledo, J. Inorg. Biochem., 2020, 212, 111253 CrossRef CAS.
- Q. Xu, R. Schwarzenbacher, S. Sri Krishna, D. McMullan, S. Agarwalla, K. Quijano, P. Abdubek, E. Ambing, H. Axelrod, T. Biorac, J. M. Canaves, H.-J. Chiu, M.-A. Elsliger, C. Grittini, S. K. Grzechnik, M. DiDonato, J. Hale, E. Hampton, G. W. Han, J. Haugen, M. Hornsby, L. Jaroszewski, H. E. Klock, M. W. Knuth, E. Koesema, A. Kreusch, P. Kuhn, M. D. Miller, K. Moy, E. Nigoghossian, J. Paulsen, R. Reyes, C. Rife, G. Spraggon, R. C. Stevens, H. van den Bedem, J. Velasquez, A. White, G. Wolf, K. O. Hodgson, J. Wooley, A. M. Deacon, A. Godzik, S. A. Lesley and I. A. Wilson, Proteins: Struct., Funct., Bioinf., 2006, 64, 808–813 CrossRef CAS.
- N. Zhao, A. S. Filatov, J. Xie, E. A. Hill, A. Yu. Rogachev and J. S. Anderson, J. Am. Chem. Soc., 2020, 142, 21634–21639 CrossRef CAS.
- C. Magallón, L. Griego, C. H. Hu, A. Company, X. Ribas and L. M. Mirica, Inorg. Chem. Front., 2022, 9, 1016–1022 RSC.
- C.-H. Hu, S.-T. Kim, M.-H. Baik and L. M. Mirica, J. Am. Chem. Soc., 2023, 145, 11161–11172 CrossRef CAS PubMed.
- P. Pirovano, B. Twamley and A. R. McDonald, Chem. – Eur. J., 2018, 24, 5238–5245 CrossRef CAS PubMed.
- I. M. DiMucci, C. J. Titus, D. Nordlund, J. R. Bour, E. Chong, D. P. Grigas, C.-H. Hu, M. D. Kosobokov, C. D. Martin, L. M. Mirica, N. Nebra, D. A. Vicic, L. L. Yorks, S. Yruegas, S. N. MacMillan, J. Shearer and K. M. Lancaster, Chem. Sci., 2023, 14, 6915–6929 RSC.
- P. Pirovano, E. R. Farquhar, M. Swart and A. R. McDonald, J. Am. Chem. Soc., 2016, 138, 14362–14370 CrossRef CAS PubMed.
- P. Heim, R. Gericke, G. Spedalotto, M. Lovisari, E. R. Farquhar and A. R. McDonald, Dalton Trans., 2023, 52, 2663–2671 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gausian 16 (version C.01), Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- K. Zhang and Y. Zuo, J. Agric. Food Chem., 2004, 52, 222–227 CrossRef CAS.
- T. Nagataki, K. Ishii, Y. Tachi and S. Itoh, Dalton Trans., 2007, 1120–1128 RSC.
- C. J. Allpress and L. M. Berreau, Eur. J. Inorg. Chem., 2014, 2014, 4642–4649 CrossRef CAS.
- A. Fadul, J. A. Bertkey and S. A. Toledo, An iron complex with broad dioxygenase reactivity and substrate scope: Expanding our understanding of Fe-N3O active sites in dioxygenases, manuscript in preparation.
- X. Yan, H. Xiao, J. Song and C. Li, Molecules, 2023, 28, 6238 CrossRef CAS PubMed.
- A. J. Jasniewski and L. Que, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- X.-P. Zhang, A. Chandra, Y.-M. Lee, R. Cao, K. Ray and W. Nam, Chem. Soc. Rev., 2021, 50, 4804–4811 RSC.
- M. Sparta, C. E. Valdez and A. N. Alexandrova, J. Mol. Biol., 2013, 425, 3007–3018 CrossRef CAS PubMed.
- K. M. Roberts, G. C. Connor, C. H. Cave, G. T. Rowe and C. A. Page, Arch. Biochem. Biophys., 2020, 691, 108441 CrossRef CAS.
- A. Banerjee, J. Li, M. A. Molenda, A. A. Opalade, A. Adhikary, W. W. Brennessel, A. Y. S. Malkhasian, T. A. Jackson and F. A. Chavez, Inorg. Chem., 2021, 60, 7168–7179 CrossRef CAS.
- J. Zhang, Y.-M. Lee, M. S. Seo, M. Nilajakar, S. Fukuzumi and W. Nam, Inorg. Chem., 2022, 61, 19735–19747 CrossRef CAS.
- P. Heim, G. Spedalotto, M. Lovisari, R. Gericke, J. O'Brien, E. R. Farquhar and A. R. McDonald, Chem. – Eur. J., 2023, 29, e202203840 CrossRef CAS.
- Y. Dai, T. C. Pochapsky and R. H. Abeles, Biochemistry, 2001, 40, 6379–6387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2375818, 2375944 and 2377248. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02538e |
| This journal is © The Royal Society of Chemistry 2024 |