
Two amino-functionalized metal–organic frameworks with different topologies for C2H2/C2H4 separation†
Yue
Li‡
a,
Mingming
Xu‡
a,
Hongyan
Liu
a,
Xiaokang
Wang
 *a,
Yutong
Wang
a,
Meng
Sun
a,
Weidong
Fan
*ab and
Daofeng
Sun
*ab
*a,
Yutong
Wang
a,
Meng
Sun
a,
Weidong
Fan
*ab and
Daofeng
Sun
*ab
aSchool of Materials Science and Engineering, China University of Petroleum (East China), Qingdao, Shandong 266580, China. E-mail: xiaokangwang0625@163.com; wdfan@upc.edu.cn; dfsun@upc.edu.cn
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China), Qingdao, Shandong 266580, China
First published on 6th November 2024
Abstract
The rational design of metal–organic framework adsorbents is crucial for target gas separation. Herein, we report two three-dimensional MOFs with different topologies by regulating metal ions with amino-functionalized V-type ligands. Adsorption isotherms and Grand Canonical Monte Carlo simulation reveal that UPC-122 with channel-cavity structure has the potential to separate C2H2/C2H4 at room temperature with a separation ratio of 2.35 (50/50).
Metal–organic frameworks (MOFs) are coordination compounds with potential porosity formed by the self-assembly of organic ligands and metal ions/clusters.1–3 As a new class of porous materials, MOFs have unique flexibility in framework design and regulation in addition to their large specific surface area and excellent porosity, and have attracted more and more attention in the field of gas adsorption and separation.4–6
Among them, the separation and purification of ethylene is one of the research hotspots in recent years. Ethylene is the primary raw material for many chemicals and polymers in the chemical industry, and mainly comes from naphtha or ethane cracking, in which acetylene is inevitably present as the main by-product. The separation of acetylene from ethylene/acetylene to produce high-purity ethylene is essential in the chemical industry.7 However, acetylene is difficult to be separated due to its similar molecular size and boiling point to ethylene. Acetylene is usually removed by catalytic hydrogenation or solvent extraction, and finally polymerized ethylene is obtained by low temperature distillation.8 Due to the high energy consumption of traditional methods, adsorptive separation technology based on porous materials has attracted people's attention due to their great energy-saving potential. As a novel porous material with various structures, MOFs show great potential in the separation of C2H2/C2H4.9–11
In recent years, researchers have focused on tuning the structure of MOFs to achieve better gas adsorption and separation performance.12,13 To further elucidate the effect of structure on the gas adsorption and separation performance, it is crucial to develop MOFs with novel structures. One of the motivations for designing and fabricating novel MOFs is their fascinating topology. The regulation of MOF frameworks can generally be carried out from the following aspects: ligand configuration,14–16 introduction of functional groups,17,18 types of solvents,19 types of metal ions,20,21 and insertion of auxiliary ligands.22 Among them, MOF materials with different topological structures can be constructed with the same ligand by changing the types of metal ions, which is conducive to discovering new topological structures.
As carboxylic acids have multiple coordination modes and can form strong coordination bonds with many metal ions, most of the early MOFs were based on various carboxylic acid ligands, such as MOF-5 and HKUST-1.23–25 Later, with the development of MOFs, the types of ligands became more and more diverse, and researchers began to use various nitrogen-containing heterocyclic ligands.26,27 Ligands with multiple coordinating atoms provide the possibility to synthesize novel MOFs. Therefore, ligands with both nitrogen-containing heterocyclic rings and carboxylic acids have attracted extensive attention of researchers due to their strong coordination patterns and diverse coordination modes.28–30
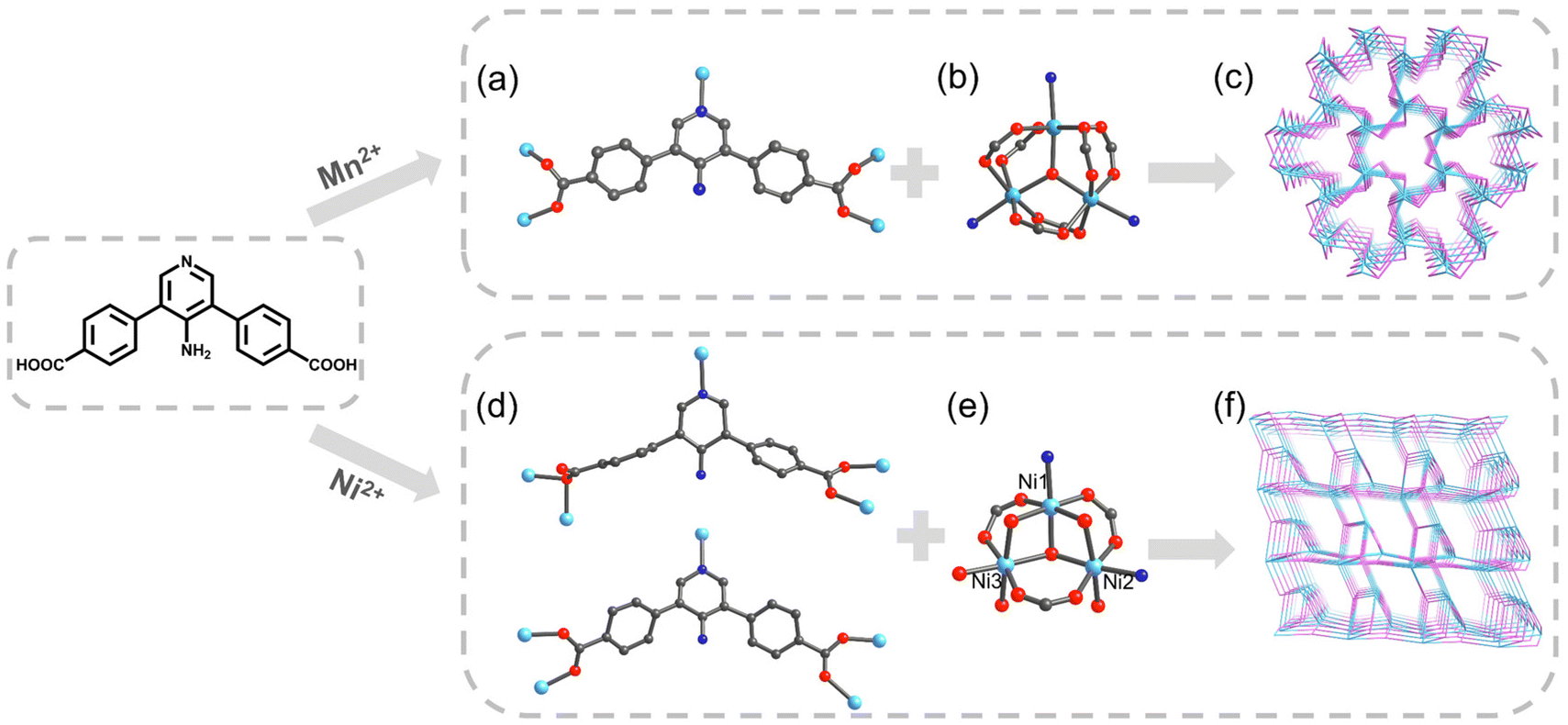
Based on the above considerations, we constructed and studied two MOFs composed of the same amino-functionalized V-shaped Ligand—H2APDA (4,4′-(4-aminopridine-3,5-diyl)dibenzoic acid) with different structures, namely UPC-121 and UPC-122 (UPC, China University of Petroleum), constructed from MnCl2·4H2O and Ni(NO3)2·6H2O, respectively. Details of the synthesis and its 1H NMR spectrum are shown in Scheme S1 and Fig. S3.† They have different trinuclear secondary building units (SBUs), and the ligands adopt different coordination modes, resulting in two completely different framework structures. Crystal data are summarized in Table S1.†
The single-crystal structure shows that UPC-121 crystallizes in the hexagonal system P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2c. The asymmetric unit contains half a deprotonated ligand and one Mn2+. The crystallographic C2 axis passes by the midpoint of the C1–C2v bond (symmetry code: (v) +X, +Y, −3/2 − Z). Mn2+ coordinates with four oxygen atoms and one pyridine N atom from the ligands, and one bridging oxygen atom. The three manganese ions in the same coordination environment form a classical [Mn3O(COO)6] cluster (Fig. 1b). The pyridine N atom and each carboxylic oxygen atom of the ligand are bridged with a manganese ion (Fig. 1a). The metal clusters and ligands are interconnected to form a three-dimensional framework with a channel diameter of 3.6 Å. The entire structure is determined by TOPOS Pro as the connected network of 3,9-c gfy topology with point symbol {412·615·89}{43}3 (Fig. 1c). The calculated molecular formula of UPC-121 is Mn(μ3-O)(APDA). The experimental powder X-ray diffraction (PXRD) pattern is consistent with the simulation results, indicating good phase purity (Fig. S1†). Thermogravimetric analysis (TGA) confirms that UPC-121 has good thermal stability, and the framework can be stabilized to 430 °C in a nitrogen atmosphere (Fig. S4†). Infrared (IR) spectrum shows the broad absorption peak at 3357 cm−1, corresponding to the hydroxyl stretching vibration peak of the coordinated water molecule. There is no absorption peak around 1740–1690 cm−1, indicating the deprotonation of carboxyl groups on the ligand (Fig. S6†).
2c. The asymmetric unit contains half a deprotonated ligand and one Mn2+. The crystallographic C2 axis passes by the midpoint of the C1–C2v bond (symmetry code: (v) +X, +Y, −3/2 − Z). Mn2+ coordinates with four oxygen atoms and one pyridine N atom from the ligands, and one bridging oxygen atom. The three manganese ions in the same coordination environment form a classical [Mn3O(COO)6] cluster (Fig. 1b). The pyridine N atom and each carboxylic oxygen atom of the ligand are bridged with a manganese ion (Fig. 1a). The metal clusters and ligands are interconnected to form a three-dimensional framework with a channel diameter of 3.6 Å. The entire structure is determined by TOPOS Pro as the connected network of 3,9-c gfy topology with point symbol {412·615·89}{43}3 (Fig. 1c). The calculated molecular formula of UPC-121 is Mn(μ3-O)(APDA). The experimental powder X-ray diffraction (PXRD) pattern is consistent with the simulation results, indicating good phase purity (Fig. S1†). Thermogravimetric analysis (TGA) confirms that UPC-121 has good thermal stability, and the framework can be stabilized to 430 °C in a nitrogen atmosphere (Fig. S4†). Infrared (IR) spectrum shows the broad absorption peak at 3357 cm−1, corresponding to the hydroxyl stretching vibration peak of the coordinated water molecule. There is no absorption peak around 1740–1690 cm−1, indicating the deprotonation of carboxyl groups on the ligand (Fig. S6†).
 | ||
| Fig. 1 (a) Coordination mode of the ligand of UPC-121. (b) Secondary building unit of UPC-121. (c) Simplified topological structure of UPC-121 (symmetry codes: (i) +X, +Y, −1/2 − Z; (ii) +Y − X, −1 − X, +Z; (iii) +Y − X, −1 − X, −1/2 − Z) (d) two coordination modes of the ligand of UPC-122. (e) Secondary building unit of UPC-122. (f) Simplified topological structure of UPC-122 (symmetry codes: (i) 1 − Y, 1 + X − Y, +Z; (ii) 5/3 − Y, 4/3 + X − Y, 1/3 + Z; (iii) 1/3 + X, 2/3 + Y, −1/3 + Z). | ||
UPC-122 crystallized in the trigonal system R3. The asymmetric unit contains two dicarboxylic acid ligands and three crystallographically independent Ni2+: Ni1, Ni2, and Ni3. Ni1 coordinates with two oxygen atoms from two ligands, three bridging oxygen atoms, and one pyridine N atom; Ni2 coordinates with two oxygen atoms from two ligands, two bridging oxygen atoms, one pyridine N, and one oxygen atom from water; Ni3 coordinates with two oxygen atoms from two ligands, two bridging oxygen atoms, and two oxygen atoms from water. Ni1, Ni2, and Ni3 form a three-nucleus SBU different from UPC-121 (Fig. 1e). The ligands have two different coordination modes, one of which is the same as that in UPC-121, while in the other mode, one of the four oxygen atoms do not participate in the coordination (Fig. 1d). The calculated molecular formula of UPC-122 is Ni3(μ3-O)O2(APDA)2(H2O)3. The phase purity of the product was confirmed by PXRD. As shown in Fig. S2,† the experimental PXRD pattern is consistent with the simulation results. TGA (Fig. S5†) confirms that UPC-122 has good thermal stability and the framework collapses at around 300 °C. The IR absorption peak at 3460 cm−1 corresponds to the hydroxyl stretching vibration peak of the coordinated water molecule. The deprotonation of carboxyl groups is proved by the missing stretching vibration absorption peak of carboxyl groups around 1740–1690 cm−1 (Fig. S7†).
The complex coordination patterns provide a completely new topology for UPC-122 (Fig. 1f). Topologically, each Ni3 cluster links six ligands, and each ligand connects three clusters, and thus they are simplified as 6- and 3-connected nodes, respectively (Fig. 2a and b). The entire structure is identified by TOPOS Pro as a new 3,6-c connected network with the point symbol: {42·6}2{44·66·85}. Notably, the pores of UPC-122 are not simple one-dimensional channels, but “pocket cavities” of approximately 7.7 Å in diameter are uniformly bulge from the approximately 5.3 Å diameter channels (Fig. 2c and d). Instead of a normal cage, the cavities are semi-exposed. The structure can provide more storage space for gas molecules than simple one-dimensional channels. After removing dioxane, DMA (N,N-dimethylacetamide), and H2O from the pores, the calculated porosity is 58.5%, which is much higher than the 14% of UPC-121.
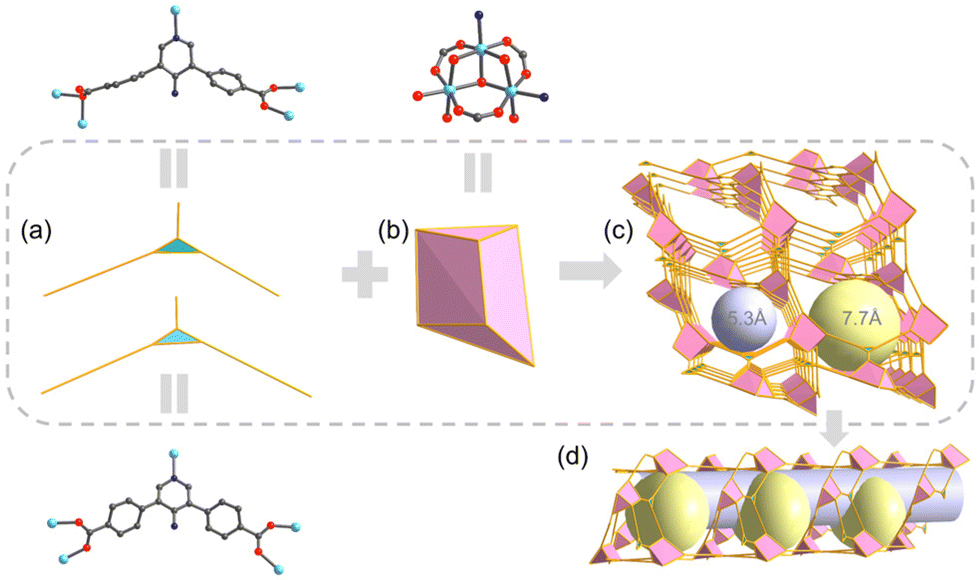 | ||
| Fig. 2 (a) Simplified 3-connected nodes for the two coordination modes of UPC-122. (b) Simplified 6-connected triangular prism for the secondary building units of UPC-122. (c) Three-dimensional stacking diagram of UPC-122. The purple sphere indicates one-dimensional channel with a diameter of about 5.3 Å. The yellow ball indicates “pocket-like cavities” with a diameter of about 7.7 Å. (d) A schematic diagram of the “pocket-like cavities” spaced below the one-dimensional channel. | ||
Due to the large porosity and novel structure of UPC-122, we intend to study the adsorption and separation performance of UPC-122 for acetylene and ethylene, and then compare it with UPC-121 constructed with the same ligand. First, duplicated UPC-121 and UPC-122 were activated. The MOF was washed three times with DMA and soaked in methanol for three days. The methanol solution in the upper layer was changed every day to replace the high-boiling solvent in the pores. After methanol was removed by supercritical carbon dioxide, the gas adsorption performance was measured. Fig. 3a shows the nitrogen adsorption isotherm of UPC-122 at 77 K, and the maximum adsorption capacity of N2 is 203.70 cm3 g−1 (9.09 mmol g−1), and the pore size distribution of 6.0 and 7.1 Å is consistent with the structure. According to the classification of IUPAC, the nitrogen adsorption isotherm of UPC-122 is type I, indicating that UPC-122 has a permanent microporous structure with a BET surface area of 557.93 m2 g−1 and a pore volume of 0.32 cm3 g−1.
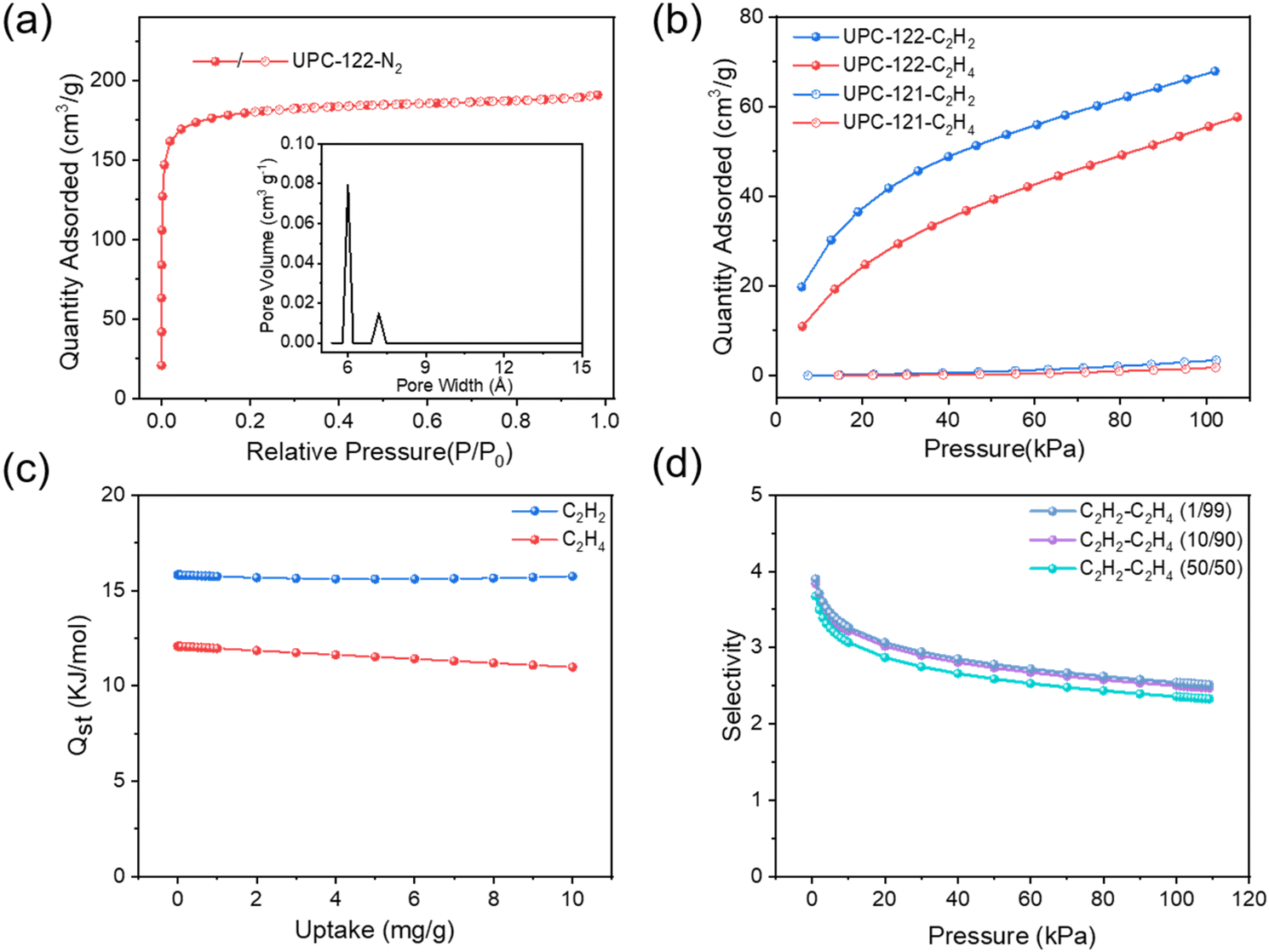 | ||
| Fig. 3 (a) N2 adsorption isotherms and pore size distribution of UPC-122 at 77 K. (b) C2H2 and C2H4 adsorption isotherms of UPC-121 and UPC-122 at 298 K. (c) Adsorption enthalpy (Qst) of C2H2 and C2H4 for UPC-122. (d) The separation ratios of C2H2 and C2H4 (1/99, 10/90, 50/50) at 298 K calculated with IAST. | ||
The C2H2 and C2H4 adsorption isotherms of UPC-121 and UPC-122 were collected at 298 K (Fig. 3b). The maximum adsorption capacities of UPC-122 for ethylene and acetylene were 57.60 cm3 g−1 (2.57 mmol g−1) and 67.82 cm3 g−1 (3.03 mmol g−1), respectively, which are much higher than that of UPC-121. The difference in the adsorption capacities of the two MOFs may be caused by the large difference in porosity and pore size. Using the ideal adsorption solution theory (IAST), the selectivity of UPC-122 to different proportions of ethylene and acetylene mixtures at room temperature was calculated, and the separation selectivities of C2H2/C2H4 (1/99, 10/90, 50/50) at 101 kPa are 2.54, 2.50, and 2.35, respectively (Fig. 3d). Its separation selectivity was higher than that of NOTT-300 (2.3 (50/50))5 and ZJNU-115 (2.19 (1/99)).31 The adsorption enthalpies (Qst) were calculated using the virial method to evaluate the interaction force of UPC-122 to C2H2 and C2H4 (Fig. 3c). The adsorption enthalpy of UPC-122 for C2H2 is 15.81 kJ mol−1, while that of C2H4 is only 12.07 kJ mol−1, indicating that the framework has a stronger force for C2H2, and its low adsorption enthalpy is more conducive to the activation and regeneration of the material.
To further explore the separation of ethylene and acetylene by UPC-122, Grand Canonical Monte Carlo (GCMC) simulation was carried out for the simulation study. According to the probability distribution of the simulated adsorbed gas molecules, we found that the binding of C2H2 and C2H4 to the framework is mainly due to the hydrogen bond and van der Waals interaction between the gas molecule and the framework (Fig. 4). In addition to hydrogen bonds with carboxylic acid oxygen, C2H2 form more hydrogen bonds with the nitrogen atoms of the amino group, while C2H4 does not interact directly with the amino group, indicating that the introduction of the amino group successfully increases the affinity of the framework for C2H2 rather than C2H4. The analysis shows that the hydrogen bonds (3.44–4.00 Å) and van der Waals interactions (3.46–3.98 Å) between C2H2 and amino group, carboxylic acid and framework are more and shorter than those (3.37–3.62 Å, 3.68–4.03 Å) of C2H4, indicating that the force between UPC-122 and C2H2 is stronger than that of C2H4, corresponding to the value of adsorption enthalpy. The stronger force between C2H2 and the framework makes UPC-122 have a larger adsorption capacity for C2H2.
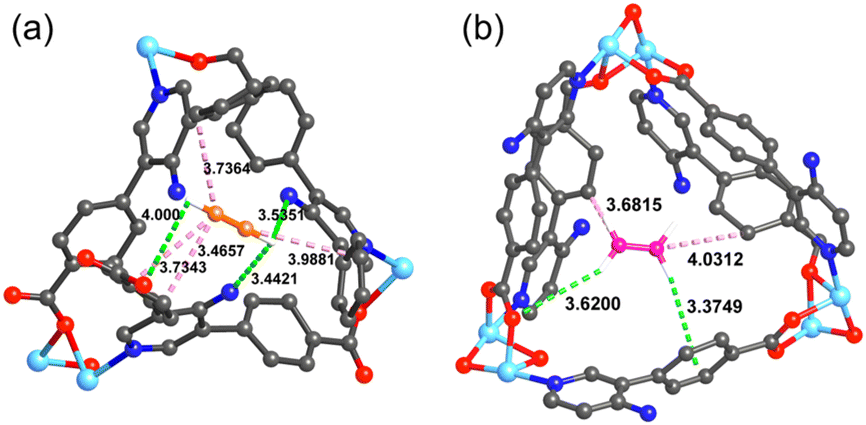 | ||
| Fig. 4 Simulated preferred adsorption sites for C2H2 and C2H4 in UPC-122. | ||
In conclusion, we constructed two structurally distinct MOFs, UPC-121 and UPC-122, using an amino-functionalized pyridine dicarboxylic acid ligand H2APDA, with Mn2+ and Ni2+, respectively. Among them, UPC-122 has a complex coordination mode, resulting in a new topology structure. Interestingly, its one-dimensional channels are evenly interspersed with pocket-like cavities. Compared with UPC-121, UPC-122 has higher porosity and shows the separation potential of ethylene and acetylene. This work can provide guidance for the design and synthesis of novel MOFs with specific application functions based on the principles of network chemistry. Our next step is to optimize its pore environment to further improve the gas adsorption capacity of the material and achieve better performance in C2H2/C2H4 separation.
Data availability
The data are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Basic Research Projects of Natural Science Foundation of Shandong Province (ZR2023ZD40), the Key Research and Development Projects of Shandong Province (2023CXGC010315), the National Natural Science Foundation of China (22275210, 22201305), the Outstanding Youth Science Fund Projects of Shandong Province (2022HWYQ-070), the Taishan Scholar Foundation of Shandong Province (tsqnz20221123), and the Fundamental Research Funds for the Central Universities (22CX06024A, 23CX04001A).References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef PubMed
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC
- S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef
- B. Li, Y. Zhang, R. Krishna, K. Yao, Y. Han, Z. Wu, D. Ma, Z. Shi, T. Pham, B. Space, J. Liu, P. K. Thallapally, J. Liu, M. Chrzanowski and S. Ma, J. Am. Chem. Soc., 2014, 136, 8654–8660 CrossRef
- L. Zhang, T. Xiao, X. Zeng, J. You, Z. He, C.-X. Chen, Q. Wang, A. Nafady, A. M. Al-Enizi and S. Ma, J. Am. Chem. Soc., 2024, 146, 7341–7351 CrossRef PubMed
- X. Feng, X. Wang, H. Yan, H. Liu, X. Liu, J. Guan, Y. Lu, W. Fan, Q. Yue and D. Sun, Angew. Chem., Int. Ed., 2024, 63, e202407240 CrossRef
- Y. Zhang, W. Sun, B. Luan, J. Li, D. Luo, Y. Jiang, L. Wang and B. Chen, Angew. Chem., Int. Ed., 2023, 62, e202309925 CrossRef PubMed
- J. You, H. Wang, T. Xiao, X. Wu, L. Zhang and C.-Z. Lu, Chem. Eng. J., 2023, 477, 147001 CrossRef CAS
- X.-W. Gu, E. Wu, J.-X. Wang, H.-M. Wen, B. Chen, B. Li and G. Qian, Sci. Adv., 2023, 9, eadh0135 CrossRef CAS
- L. Zhang, F. Li, J. You, N. Hua, Q. Wang, J. Si, W. Chen, W. Wang, X. Wu, W. Yang, D. Yuan, C. Lu, Y. Liu, A. M. Al-Enizi, A. Nafady and S. Ma, Chem. Sci., 2021, 12, 5767–5773 RSC
- W. Fan, S. Yuan, W. Wang, L. Feng, X. Liu, X. Zhang, X. Wang, Z. Kang, F. Dai, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 8728–8737 CrossRef
- L. Yang, P. Cai, L. Zhang, X. Xu, A. A. Yakovenko, Q. Wang, J. Pang, S. Yuan, X. Zou, N. Huang, Z. Huang and H.-C. Zhou, J. Am. Chem. Soc., 2021, 143, 12129–12137 CrossRef CAS PubMed
- Y. Suzuki, M. Gutiérrez, S. Tanaka, E. Gomez, N. Tohnai, N. Yasuda, N. Matubayasi, A. Douhal and I. Hisaki, Chem. Sci., 2021, 12, 9607–9618 RSC
- S. Wu, D. Ren, K. Zhou, H.-L. Xia, X.-Y. Liu, X. Wang and J. Li, J. Am. Chem. Soc., 2021, 143, 10547–10552 CrossRef CAS
- Y. Wang, C. Hao, W. Fan, M. Fu, X. Wang, Z. Wang, L. Zhu, Y. Li, X. Lu, F. Dai, Z. Kang, R. Wang, W. Guo, S. Hu and D. Sun, Angew. Chem., Int. Ed., 2021, 60, 11350–11358 CrossRef CAS PubMed
- S. Xing, J. Liang, P. Brandt, F. Schäfer, A. Nuhnen, T. Heinen, I. Boldog, J. Mollmer, M. Lange, O. Weingart and C. Janiak, Angew. Chem., Int. Ed., 2021, 60, 17998–18005 CrossRef CAS
- W. Wang, K. Su, E. M. El-Sayed, M. Yang and D. Yuan, ACS Appl. Mater. Interfaces, 2021, 13, 24042–24050 CrossRef CAS
- H. Yang, F. Peng, D. E. Schier, S. A. Markotic, X. Zhao, A. N. Hong, Y. Wang, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2021, 60, 11148–11152 CrossRef CAS PubMed
- M. R. Mian, H. Chen, R. Cao, K. O. Kirlikovali, R. Q. Snurr, T. Islamoglu and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 9893–9900 CrossRef CAS PubMed
- K. Su, W. Wang, S. Du, C. Ji and D. Yuan, Nat. Commun., 2021, 12, 3703 CrossRef CAS
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS
- N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 51–54 CrossRef CAS PubMed
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS
- P.-Q. Liao, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 8697 CrossRef
- Q.-L. Qian, X.-W. Gu, J. Pei, H.-M. Wen, H. Wu, W. Zhou, B. Li and G. Qian, J. Mater. Chem. A, 2021, 9, 9248–9255 RSC
- Y. A. Belousov, A. A. Drozdov, I. V. Taydakov, F. Marchetti, R. Pettinari and C. Pettinari, Coord. Chem. Rev., 2021, 445, 214084 CrossRef CAS
- C.-R. Ye, J. Zheng, M. Li and X.-C. Huang, Chem. Commun., 2018, 54, 8769–8772 RSC
- Y. Li, X. Wang, X. Yang, H. Liu, X. Chai, Y. Wang, W. Fan and D. Sun, Inorg. Chem., 2023, 62, 3722–3726 CrossRef PubMed
- L. Fan, P. Zhou, X. Wang, L. Yue, L. Li and Y. He, Inorg. Chem., 2021, 60, 10819–10829 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal data, PXRD, TGA curves, and IR pattern. CCDC 2141832 and 2141833. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02592j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |