
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsocyanate-based multicomponent reactions
Clara Zavarise,
Jean-Christophe Cintrat,
Eugénie Romero* and
Antoine Sallustrau *
*
Université Paris Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), SCBM, Gif-sur-Yvette 91191, France. E-mail: antoine.sallustrau@cea.fr
First published on 12th December 2024
Abstract
Since their discovery, multicomponent reactions have attracted significant attention due to their versatility and efficiency. This review aims to explore the latest advancements in isocyanate-based multicomponent reactions and the sophisticated chemical opportunities they present for generating molecules of interest. The added value of the methodologies described, supported by mechanism schemes, as well as scopes of application, will be discussed. These developments will be organised as the main accessible chemical functions and sorted according to their type of MCR (3, 4 or 5-MCR).
1. Introduction
Since the pioneer work of Strecker1,2 in 1850, multicomponent reactions (MCRs) have garnered significant interest in synthetic organic chemistry due to their widespread applications in various fields such as drug discovery, medicinal chemistry, total synthesis, pharmaceuticals and agrochemical industry. These one-pot reactions incorporate more than two reactants into a single final product, providing access to complex molecular architectures, with high molecular diversity.3 Regarded as a convenient and effective approach, MCRs present several advantages over traditional one or two-component reactions or in one-pot stepwise synthesis. In one-pot stepwise synthesis, multiple reactions occur sequentially in a single vessel. In contrast, a multicomponent reaction involves the simultaneous combination of three or more reactants to form a product in a single reaction step. MCRs simplify synthetic routes by reducing the requirement for numerous sequential steps and the number of purification operations, leading frequently to better yields. Additionally to expand the chemical space, the atom-economical nature of MCRs and their alignment with green chemistry principles4 have highlighted their environment-friendly aspects.Hantzsch,5 Biginelli,6 Passerini7 and Ugi8 among notable contributors have paved the way for the development of MCRs. For decades, these reactions remained largely underexplored before being used in the synthesis of complex and biologically active compounds. Over time, the scope of these reactions expanded using a combination of serendipity and rational design. Several strategies have been developed for the rational design of new MCRs:9 (i) single reactant replacement (SRR), (ii) modular reaction sequences (MRS), (iii) adjusting conditions towards divergent MCRs. Some of these strategies have been used in tandem with innovative technologies such as flow chemistry,10 microwave chemistry,11 mechanochemistry12 and photochemistry13 to further enhance the applications of MCRs. A final strategy implemented by different research teams consists in combining multiple known MCRs to achieve higher-order MCRs that can involve six or more reactants.14 An example of the diversification of known-MCRs is the widely applied Ugi-deprotection–cyclization synthetic strategy15,16 or post-Ugi cyclization,17 which both largely contributed to the construction of diverse heterocyclic and drug-like compounds.
These developments have significantly impacted many domains and applications, such as material science for the preparation of covalent organic frameworks18 (COFs), combinatorial chemistry providing an efficient chemistry tool for the preparation of asymmetrical product libraries,19 or for the development of biocompatible reactions20,21 through tailored MCR methodologies. Polymer science has also benefited from MCRs, enabling efficient and cost-effective polymer construction and post-polymerization modifications.22 Due to their ability to serve as a starting point for diversity orientating synthesis and provide access to a myriad of diverse chemical scaffolds, MCRs have also gained considerable significance in the pharmaceutical industry, and more particularly in drug discovery.3 MCR-derived products exhibit high atom density, enhancing their interaction potential with biological targets. This often leads to a superior hit ratio compared to non-MCR compounds, particularly in the context of specific targets like protein–protein interactions.20 Moreover, they can generate libraries of complex small molecules that are easily screened in direct-to-biology approaches.23 Some notable applications in the pharmaceutical industry include their utilization for synthetic routes optimization, facilitating the transition to industrial scale production, and ultimately resulting in faster and more cost-effective synthesis processes. For instance, penicilin, telaprevir, nifedipine and praziquantel syntheses involve MCR steps within their industrial manufacturing processes.3
As one of the most widely recognized reagent in multicomponent reactions, isocyanides have been playing a crucial role in many fields, notably due to the high structural diversity it reaches. Carbonyl compounds and amines are fundamental components in Hantzsch and Biginelli reactions, among others MCRs. Other reagents have emerged as very useful building blocks in MCRs, such as sodium azide and isocyanates. However, even if recent reviews reported the use of different chemical class such as isocyanide24–26 or sodium azide27 in MCRs, none of them focused on the use of isocyanates. Reviews on isocyanate chemistry28,29 or their use in specific transformations of interest as amide bond formation30 or urethane/urea bonds formation31 have been reported. This review will discuss the interest of isocyanates in another prism: it will spotlight the potential of isocyanates as versatile building blocks in 3, 4, or 5-component reactions (MCRs) for the efficient synthesis of structurally diverse and complex molecules (Fig. 1). We delve into the various strategies employed by researchers, exploring the use of isocyanates as both pre-formed building blocks and those generated in situ within diverse synthetic transformations.
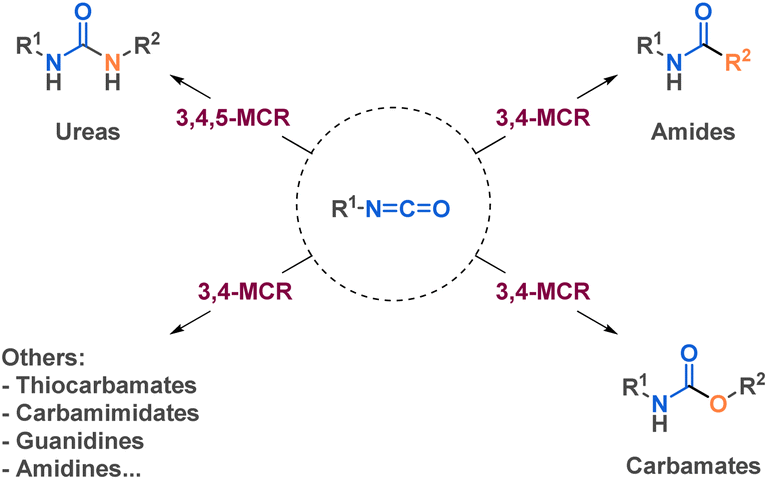 | ||
| Fig. 1 General representation of possible MCRs with isocyanates. | ||
2. Synthesis and reactivity of isocyanates
2.1. Discovery and synthesis of isocyanates
Isocyanates are very commonly used building blocks in many chemical applications since their discovery by Wurtz in 1848.32 They have emerged as remarkably useful for the synthesis of essential compounds across diverse fields, including pharmaceuticals, agrochemicals, materials science, and fine chemicals. The global market of isocyanates is increasing by 5% every year, mostly due to their valuable industrial use31 and to their highly efficient and cost-effective large scale production by phosgenation of amines,33 or by cleavage of urethanes. Besides these techniques, many lab-scale reactions have been developed including the Curtius rearrangement, but also Hofmann, Lossen and Schmidt rearrangements involving an isocyanate intermediate. These methods provide access to a diverse range of isocyanates, more or less sensitive to hydrolysis and prone to dimerization or trimerization. Other preparation methods have been described, such as the reaction of amides and amines with oxalyl chloride or the use of isocyanides with carboxylic derivatives. Recent advancements have introduced milder methods like the Staudinger-Aza Wittig (SAW) sequence. This approach utilizes an azide and CO2, resulting in a less toxic, more environmentally friendly reaction that exhibits high tolerance towards functional groups sensitive to phosgene (Fig. 2).34 The abundance of efficient synthetic methods, coupled with the development of in situ isocyanate generation, facilitates the utilization of a vast library of alkyl and aryl isocyanates in the construction of diverse scaffolds.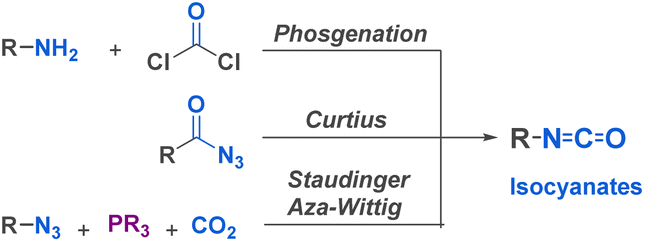 | ||
| Fig. 2 Several examples of reactions producing isocyanates. | ||
2.2. Fundamental mechanisms of reactions involving isocyanate
Isocyanates can be described as strained linear structures characterized by an electrophilic carbon surrounded by a nitrogen atom and an oxygen atom. The resonance structure of the isocyanate reinforces the electrophilicity of the carbon (Fig. 3).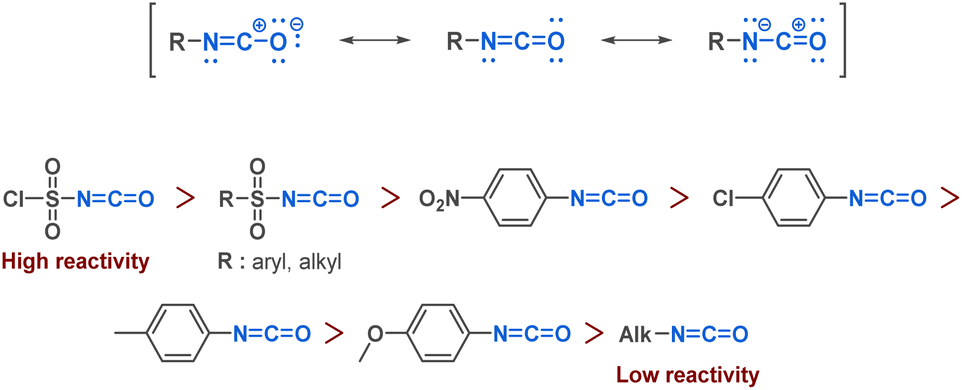 | ||
| Fig. 3 Reactivity of isocyanates. | ||
As with many functional groups, the reactivity of isocyanates depends on their substituents. An electron-withdrawing group increases carbon electrophilicity and enhances reactivity. Conversely, any electron-donating group lowers the reactivity of the isocyanate (Fig. 3). Due to the electrophilic carbon, isocyanates can undergo nucleophilic attacks from various nucleophiles such as amines, alcohols or thiols, where the commonly accepted mechanism involves a first step of nucleophilic attack followed by a proton transfer (Fig. 4).
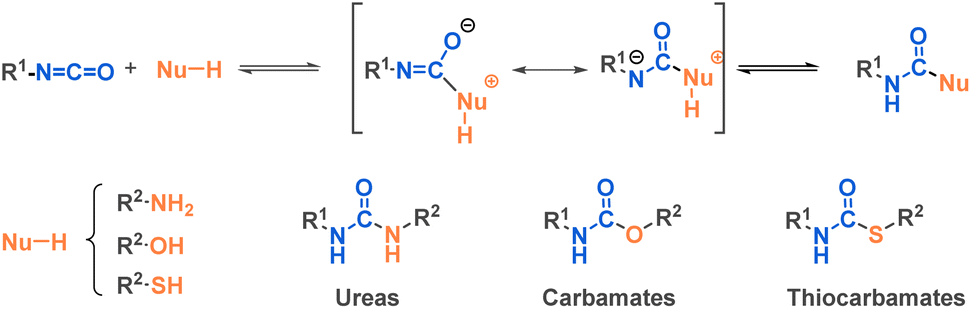 | ||
| Fig. 4 Main classes of products resulting from nucleophilic attack on isocyanates. | ||
2.3. Reactivity and stability of isocyanates
Isocyanates are known for their high reactivity but also for their inherent toxicity particularly since the 1984 Bhopal tragedy that led to the release of 40 tons of methyl isocyanates in the atmosphere killing thousands of citizens. Isocyanates toxicity come from its ability to react with nucleophilic biological molecules such as DNA bases or glutathione, leading to the production of carbamoyl metabolites that could react with other macromolecules in the surrounding tissues.35 In this context, driven by the goal of sustainable development and the reduction of hazardous chemical use, there has been a trend toward substituting certain substances, such as isocyanates. One area that has received significant attention is the development of non-isocyanate polyurethanes (PUs) and polyureas (PUas), like CO2-based PUs/PUas.36,37 This is particularly significant considering that PUs are currently ranked as the 6th most widely sold polymer worldwide.In certain cases, their high reactivity can also cause complications in handling and storage. As an example, isocyanates are known to be prone to polymerization when bearing a nucleophilic functional group. The blocked-isocyanate strategy elegantly bypasses this limitation by providing the in situ generation of an isocyanate from a precursor under activation. This controlled release allows for direct reaction with specific substrates. For instance, the research team led by Beauchemin38–42 devised several methodologies to generate isocyanates in situ. One of these strategies implies the functionalisation of the electrophilic carbon of the isocyanate by a leaving group, which is then cleaved upon microwave irradiation or heating, thus generating the corresponding isocyanate in situ. This innovative approach provides on-demand formation of isocyanates with precise control over their reactivity. Beauchemin successfully synthesized N-isocyanates, O-isocyanates, and isocyanates with nucleophilic functional groups that readily react with desired substrates to yield targeted products (Scheme 1).
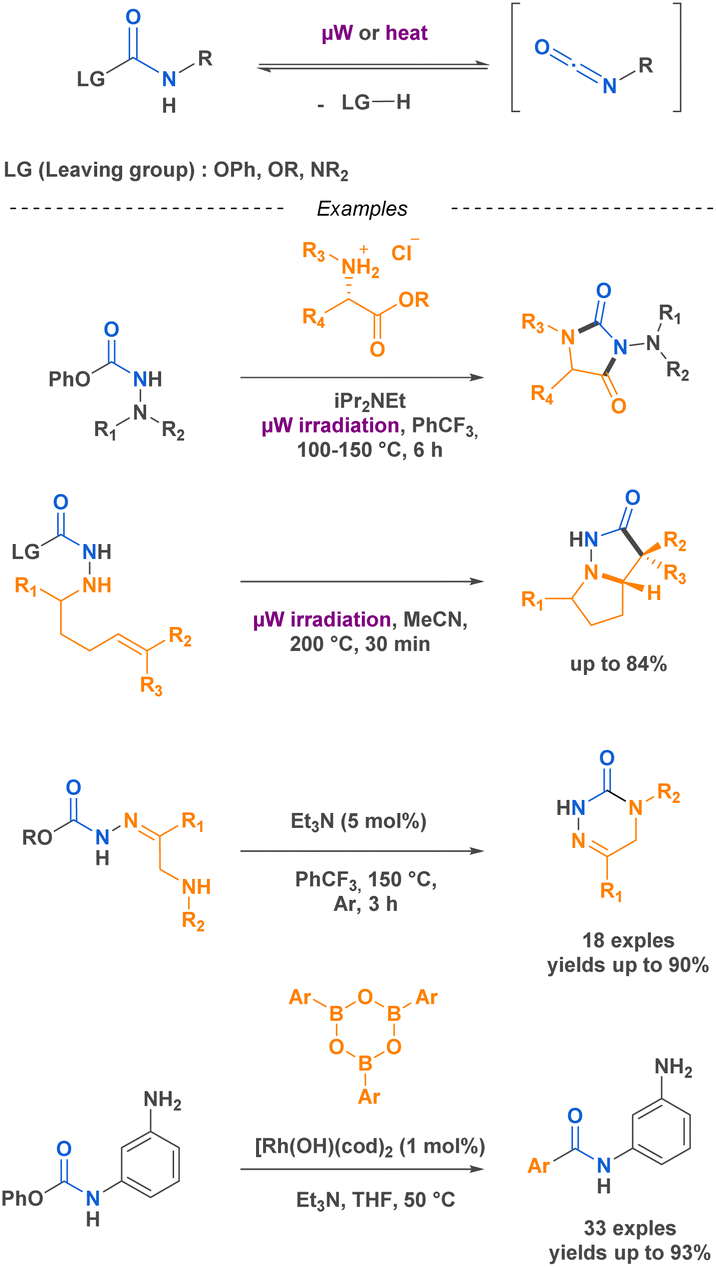 | ||
| Scheme 1 Examples of in situ generation of isocyanates by the team of Beauchemin. | ||
Due to their high reactivity and interesting properties, isocyanates have been used in multicomponent reactions as developed in the following sections.
3. Isocyanate-based multicomponent reactions
3.1. Urea derivatives
Ureas hold tremendous biological significance, sparking a wealth of research into their synthesis. This structural motif plays a vital role in numerous drugs used to treat a wide range of diseases, such as antitumoral and antiparkinsonian applications.43,44 The extensive biological applications of ureas notably stem from their ability to form stable hydrogen bonds with the target molecules, thereby improving ligand–receptor interactions. The substantial interest in ureas has pushed multiple scientific teams to dedicate significant efforts towards developing linear substituted ureas or cyclic ureas using diverse synthetic strategies, including multi-component reaction approaches.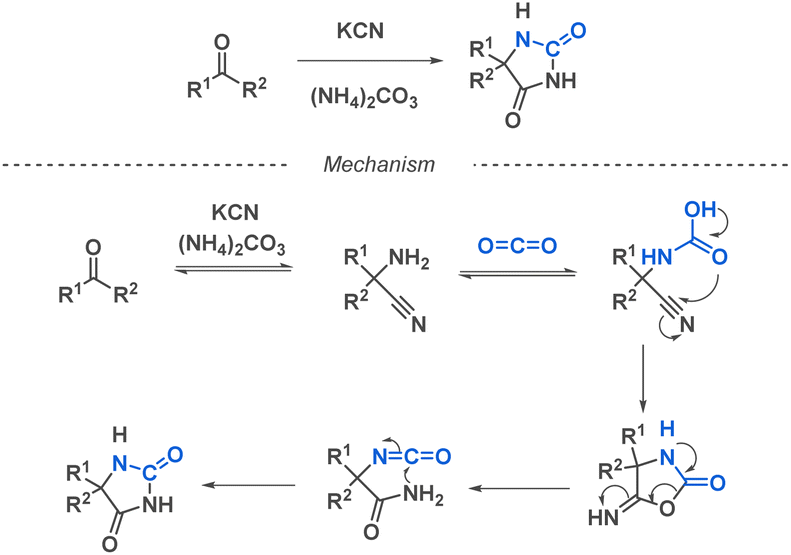 | ||
| Scheme 2 General scheme and a proposed mechanism of the Bucherer–Bergs reaction. | ||
One of the limitation of this reaction is that there is only one point of diversity based on the starting carbonyl, limiting the broadness of the scope. Another strategy to prepare hydantoins from isocyanates was proposed by Bellucci et al. using α-azido ester through a pseudo 3-MCR (Scheme 3a).46 Pseudo-MCRs are one-pot processes involving combinations of at least three reactants, in which two of them are identical.47 This had the advantage to provide another simple and efficient route to other type of hydantoins increasing also the diversity in their scaffold. Another example was reported by Alizadeh et al. where they successfully combined three compounds in the presence of triphenylphosphine: an amine, an arylsulfonyl isocyanate and an alkyl propiolate or a dialkyl acetylenedicarboxylate (Scheme 3b) providing complex hydantoin structures in very good yields but with a limited scope of 5 examples.48
 | ||
| Scheme 3 Synthesis of hydantoins by 3-MCR methodologies. | ||
Rigby et al. reported a dual-copper catalyst system that enables the reaction between aryl isocyanates and dimethoxycarbene for the preparation of hydantoin derivatives.49 In contrast to the previous methodologies, this methodology could be regarded as a pseudo-3-MCR given that two components are identical. Some of the isocyanates used for the scope were derived from a Curtius rearrangement (Scheme 4). Importantly, while activated aromatic rings leaded to excellent yield up to 84%, electron-donating groups on the aromatic ring resulted in moderated yield (range of 50%). Other multicomponent reactions involving more than three reactants have been developed to prepare hydantoin derivatives and will be described in the following sections.
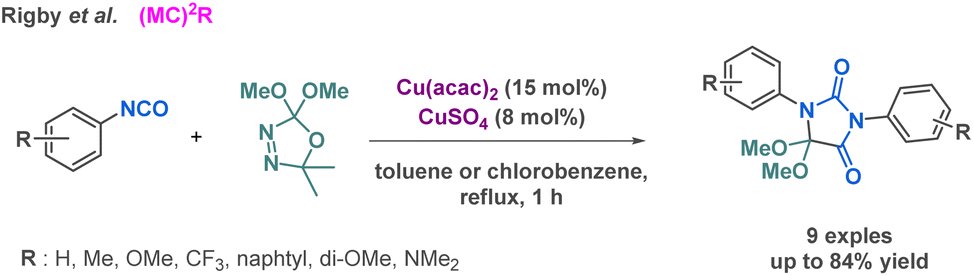 | ||
| Scheme 4 Synthesis of hydantoin derivatives by Rigby et al. | ||
Sharing a close structural relationship with the hydantoin core, uracils are heterocyclic molecules featuring a six-membered ring system, which are prevalent in numerous bioactive molecules, including uracil in RNA, and are found in various compounds exhibiting promising anticancer, antiviral, and antiparasitic properties. These molecules play crucial roles in biological processes and offer potential for therapeutic applications.50 Due to the attention paid to uracil in medicinal chemistry, different synthetic routes have been developed to prepare these compounds.
Similarly to the work of Rigby et al. on the synthesis of hydantoin derivatives via a pseudo-MCR methodology, two teams have independently developed innovative pseudo-MCR methods for the synthesis of uracil derivatives, including dihydrouracils. Fernández de Trocóniz et al. reported a pseudo 3-MCR using two isocyanates allowing the preparation of fluorinated triazinane-2,4-diones from two phenyl isocyanate precursors and an unsaturated imine at room temperature (Scheme 5a).51 At the moment of the report, it was the first synthesis of such fluorinated compounds. Morimoto et al. also developed a pseudo 3-MCR but employed a nickel-catalysed strategy to afford the uracil derivatives (Scheme 5b).52 Dekamin et al. capitalized on the high reactivity and versatility of isocyanates to provide an efficient cyclotrimerisation approach tailored for alkyl and aryl isocyanates.53,54 The innovative aspect of these approaches lie in the use of organocatalysts under mild and solvent free reaction conditions, either tetra-butylammonium phthalimide-N-oxyl or tetra-ethylammonium 2-(carbamoyl) benzoate or sulphate anions (Scheme 5c and d). These catalytic systems present the most popular and industrial route for the cyclotrimerisation of isocyanates to access isocyanurates, as other methods described remain laborious.
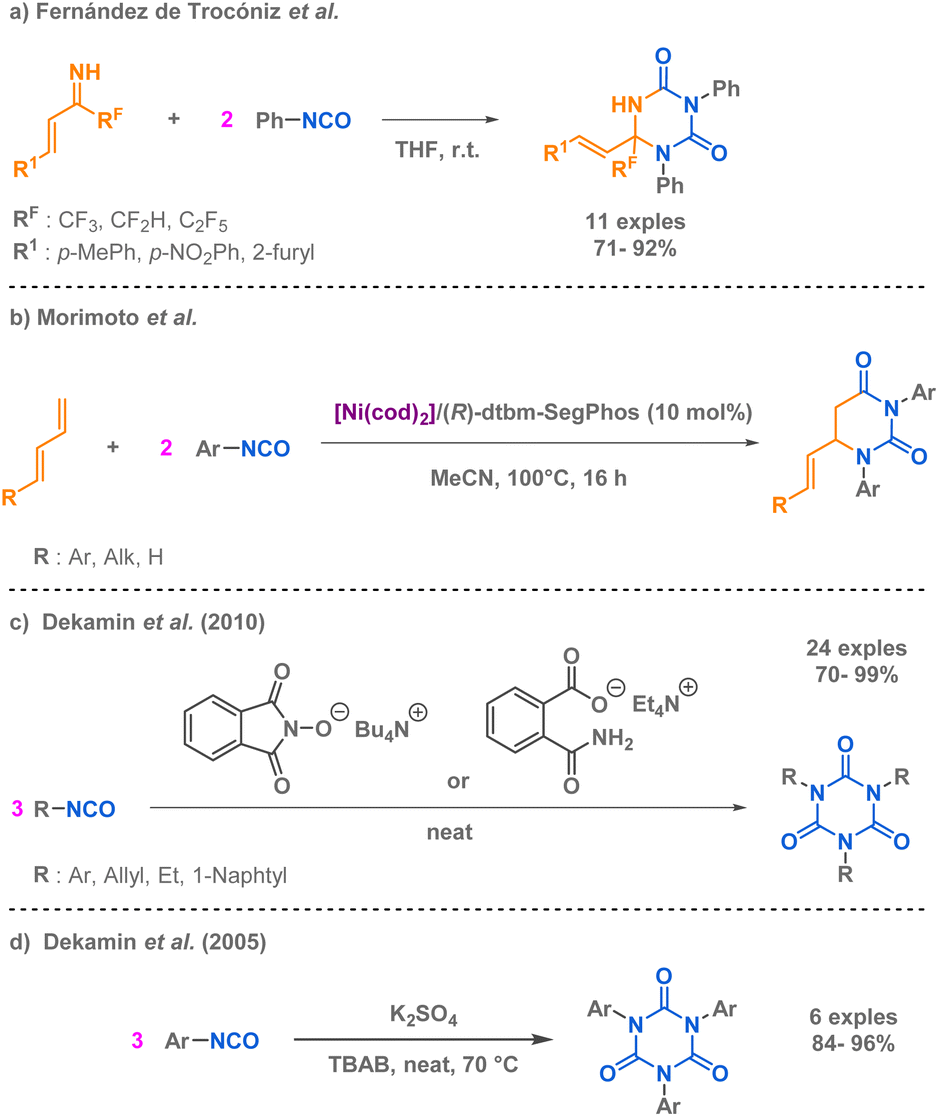 | ||
| Scheme 5 Synthesis of uracil and aza-uracil derivatives via pseudo-MCR methodologies. | ||
Beyond hydantoins and uracil derivatives, cyclic ureas are present in several FDA-approved drugs, including avibactam, trimetaphan, and azlocillin. They also have garnered significant attention due to their potent biological activities as selective neurokinin-1 receptor antagonists55 and HIV-1 protease inhibitors.56 Cyclic urea can usually be prepared by cycloaddition, condensation, Curtius rearrangement of acyl azide, phosgenation followed by cyclisation, or ring-closing metathesis. These applications pushed forward several research groups to develop other methodologies to provide cyclic ureas using MCR methodologies.
As a peculiar example, Pan et al. put forward an innovative approach using visible-light photoredox catalysis for the synthesis of spirocyclic quinazolin-2-ones in a single step, compounds for which a very limited number of methods are describe, compared to their analogues spirocyclic quinazolin-4-ones.57 Behind the advantages of being a new strategy to access these complex structures, this methodology proceeds under mild conditions with the assistance of metal-free catalysts, visible light as energy activation, and air serving as a natural oxidant, enhancing its overall sustainability. The synthesis of the desired products involves the use of isocyanates, α-anilinosilanes, and 2-aminoacetophenones (Scheme 6a). With a scope composed by more than 30 examples, it however has to be pointed out that it remains mostly applicable to aryl isocyanates (one example with benzyl isocyanate, no conversion with tert-butyl isocyanate). The proposed mechanism involves both a Brønsted acid mediated catalytic cycle using TFA and a photoredox catalytic cycle using a catalytic amount of mesityl-methylacridinium perchlorate to obtain the quinazolinone core. Just a year before, they investigated a similar multicomponent reaction using iron-III catalysis for the synthesis of spiroquinazolin-2-ones combining an isocyanate, a 2-aminoacetophenone, and a 2-amino-benzaldehyde (Scheme 6b).58 In terms of scope limitations, if most examples report aryl isocyanates, one desired spirocyclic quinazolin-2-one from a propyl isocyanate shows a decent yield of 59%. Importantly, the aminoacetophenone needs to be N-Me or N-Et functionalized, exclusively, to avoid rearomatization of the quinolone motif. The reported mechanism starts with a first iron catalysed [4 + 2] annulation between 2-aminoacetophenone and an isocyanate, followed by an intramolecular cyclization, affording 4-methylene-quinazolinone. Then, a second iron catalysed [4 + 2] annulation followed by a cyclization afforded the desired product. It is worth mentioning that the presence of a hydrogen atom at the R1 position leads to an additional aromatization of the spiroquinazolinone and the consequent formation of quinolinureas (Scheme 6c).
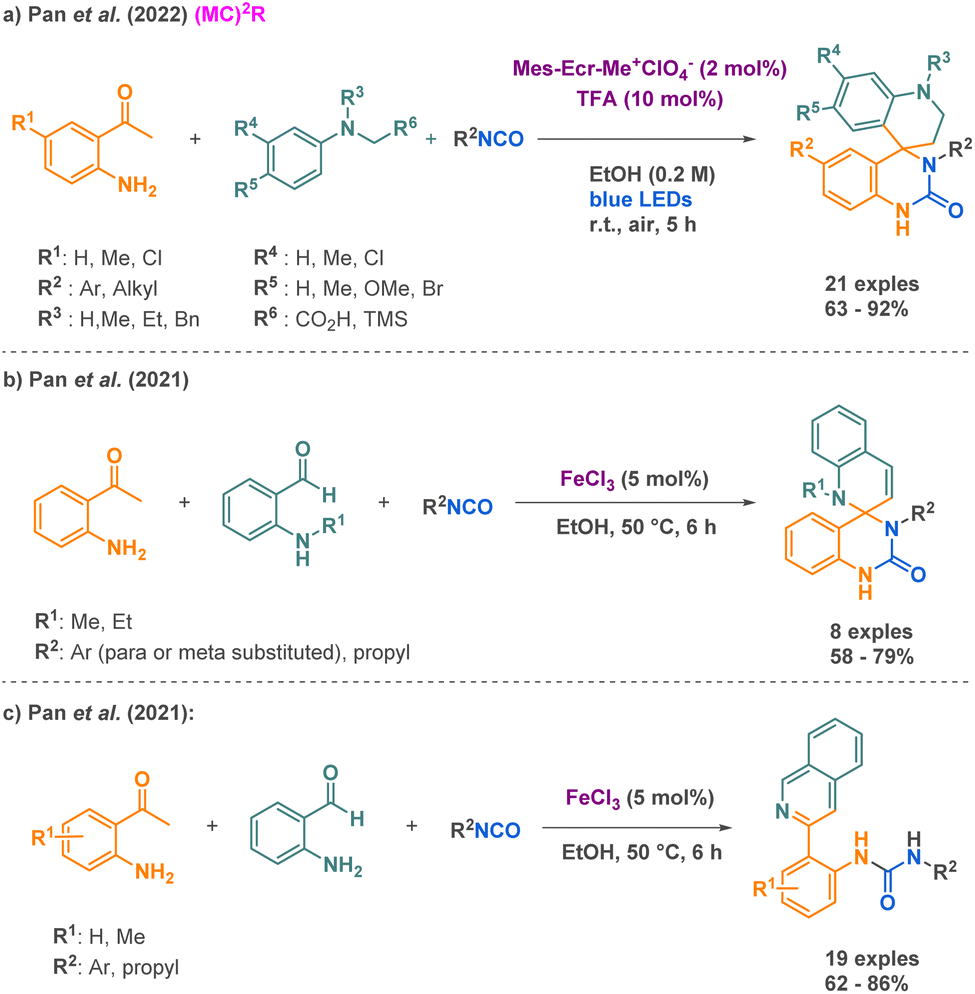 | ||
| Scheme 6 Synthesis of urea derivatives by Pan et al. | ||
More complex urea derivatives such as semicarbazides and semicarbazones represent an attractive category of compounds that are already present on the market, such as naftazone which is used for the treatment of varicose veins or haemorrhoids. Ongoing studies on the stimulation of the dopamine D4 receptor by APH199,59 implied in various psychiatric disorders further highlight the promising nature of these compounds for future applications. To meet the challenge of synthesizing these compounds, Mohammadi and Adib developed one of the few examples of microwave-assisted MCRs.60 In their work, the authors demonstrated a highly efficient method for the synthesis of 1,2,4-triazole-3-one derivatives. By combining aldehydes, phenylhydrazines, and phenyl isocyanates, they achieved excellent yields (around 90%) under their reaction conditions. This represents a significant improvement over conventional heating methods, which typically afford low yields for similar transformations. However, the substrate scope of this method was limited to aryl-substituted compounds (Scheme 7a). Du et al. successfully obtained similar products using a palladium-catalysed methodology involving hydrazonoyl chlorides, sodium azide and carbon monoxide surrogate as starting materials.61 The advantage of this method relies on the use of azide as substrate, leading to a free nitrogen in position N1 in the desired compounds. The proposed mechanism involves the carbonylation of hydrazonoyl chloride and the insertion of the azide under palladium catalysis. The acyl azide intermediate undergoes a Curtius rearrangement, resulting in a reactive isocyanate intermediate that facilitates intramolecular nucleophilic addition (Scheme 7b).
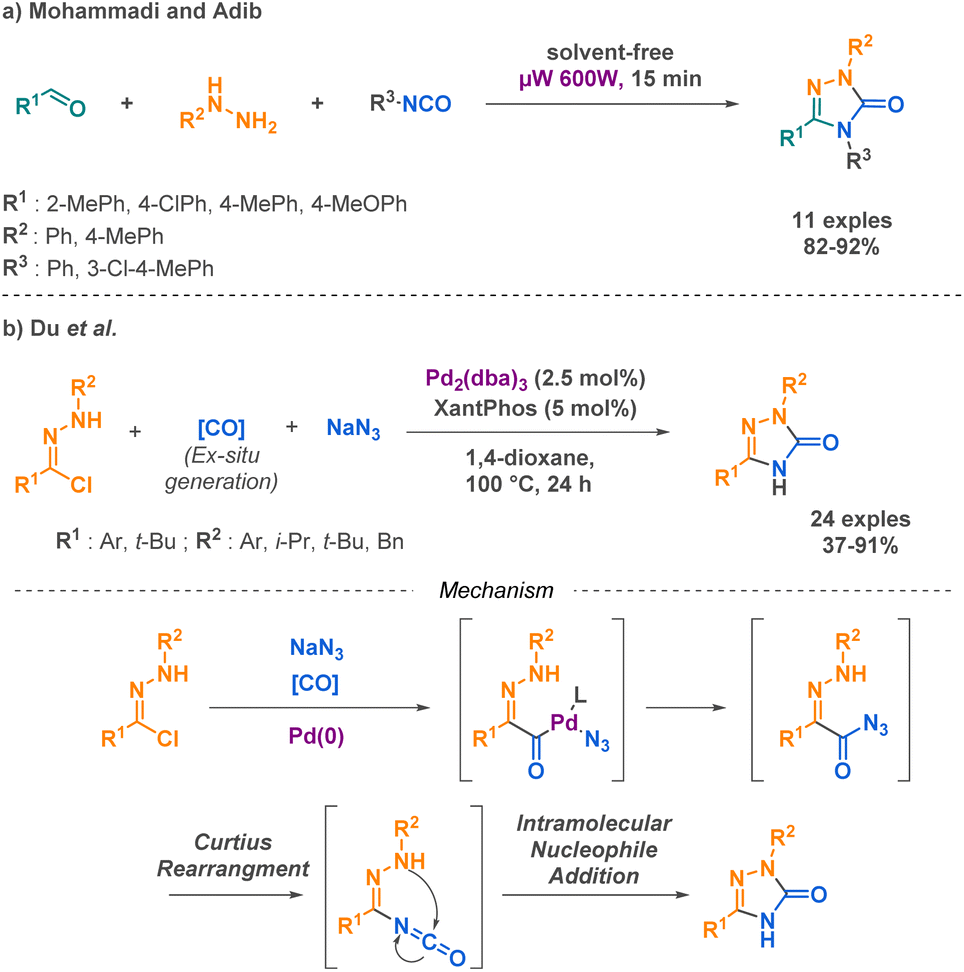 | ||
| Scheme 7 Synthesis of cyclic semicarbazones by Mohammadi and Adib and Du et al. | ||
Concerning the preparation of cyclic semicarbazides, a highly effective and practical method was developed by Soeta et al., directly inspired by the Ugi reaction.62 They substituted a carboxylic acid by an isocyanate to be reacted with a nitrile imine, an isocyanide and an isocyanate resulting in the formation of a complex 1,2,4-triazinedione scaffold (Scheme 8). The key challenge of this work was to replace carboxylic acids with isocyanates. Carboxylic acids play a critical role in the Ugi reaction mechanism by activating the imine intermediate, which enables the addition of an isocyanide and the formation of a nitrilium ion. This ultimately leads to the formation of the final product through an acyl group migration. However, to expand the chemical space of the reaction and access a wider range of products, it was necessary to explore alternative reaction partners such as isocyanates.
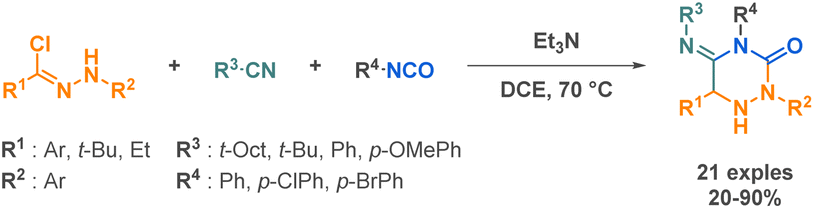 | ||
| Scheme 8 Ugi-type synthesis of cyclic semicarbazides by Soeta et al. | ||
Martinez-Ariza et al. also described the preparation of semicarbazones using MCRs. They showcased various multicomponent strategies for this purpose. Among them, two involve the use of an isocyanate. The most studied one consists in the combination of a ketone, an isocyanate and 2-hydrazinoazine to afford triazolo-carboxamide leading to fused 6,6-bicyclic pyrido[2,1-c][1,2,4]triazines with enhanced sp3 character over Groebke–Blackburn–Bienaymé derived scaffolds. It is the very first use of 2-hydrazinoazines in this particular MCR transformation, allowing access to a built-in H-bond donor–acceptor warhead property, highly advantageous for many biological targeting phenomena. Four examples of the scope of the reaction developed by the group involve an aldehyde, an isocyanate and a hydrazine instead of 2-hydrazinoazine, leading to the synthesis of indazole-carboxamides with correct yields around 50% (Scheme 9).63
 | ||
| Scheme 9 Synthesis of triazolo-carboxamides and indazole-carboxamides by Martinez-Ariza et al. | ||
Among more than 40 linear urea examples, Babin et al. described the preparation of several linear semicarbazides through a Staudinger-Aza-Wittig (SAW) procedure able to generate isotopically labelled isocyanates in situ directly from carbon dioxide (13C, 14C, 11C) and azides in the presence of a phosphine.64 This technique opens up new possibilities for radiolabeling, which holds a place of choice in pre-clinical studies. It is worth mentioning that the reaction conditions are influenced by the substituents on both the azide and the amine. However, a key advantage of this approach lies in its utilization of carbon dioxide in near-stoichiometric quantities (Scheme 10a). Another SAW-MCR was previously reported as a microwave assisted SAW procedure by Carnaroglio et al. in 2006 (Scheme 10b).65 Despite employing supported phosphines to streamline purification, their method consumed over ten times more CO2 than that developed by Babin et al. and exhibited a narrower scope of application.
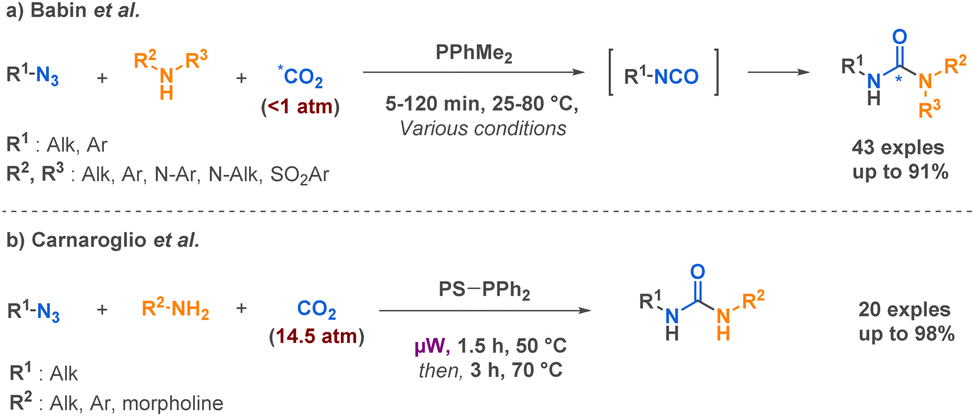 | ||
| Scheme 10 Synthesis of linear ureas derivatives by SAW procedures by Babin et al. and Carnaroglio et al. | ||
Other teams have developed MCR-methods for the synthesis of linear urea derivatives. For example, Yavari and Nematpour's work offers an innovative solution to this complex challenge through their copper-catalysis method (Scheme 11).66
 | ||
| Scheme 11 Synthesis of linear ureas derivatives by Yavari et al. | ||
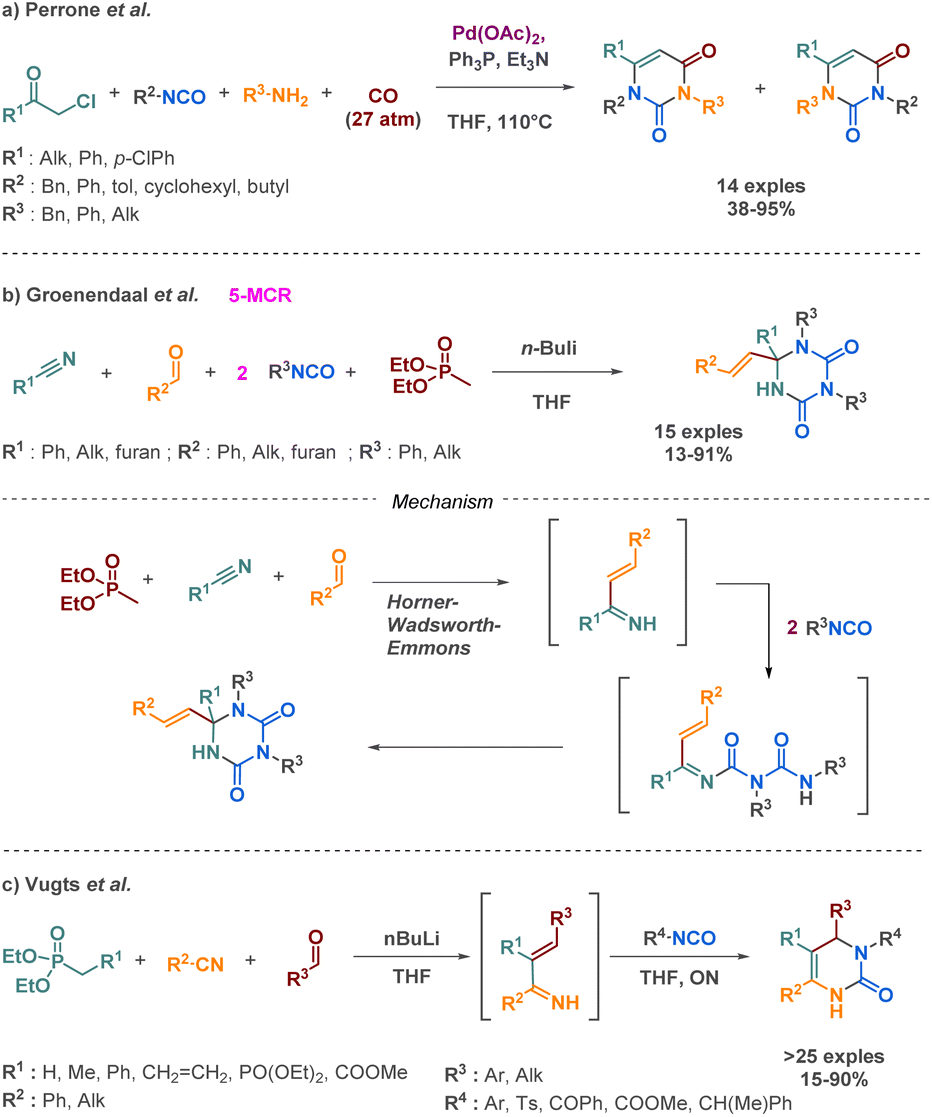 | ||
| Scheme 12 Synthesis of cyclic urea derivatives by Perrone et al., Groenendaal et al. and Vugts et al. | ||
The same team implemented a comparable approach two years before to obtain dihydropyrimidines. Their strategic approach also involves the use of a Horner–Wadsworth intermediate, but instead of engaging in reactions with two isocyanates, the intermediate subsequently reacts with one isocyanate in an aza-Diels–Alder reaction (Scheme 12c).69
The latest example of cyclic urea synthesis via a 4-MCR was reported by Messa et al., who described the palladium catalysed formation of a pyrimidine-dione from an α-chloroketone, an isocyanate, an aniline and carbon monoxide (Scheme 13).70 The authors proposed a concerted mechanism where urea formation occurs simultaneously with the α-chloroketone carbonylation. These intermediates then cyclize to provide the desired product in 73% yield.
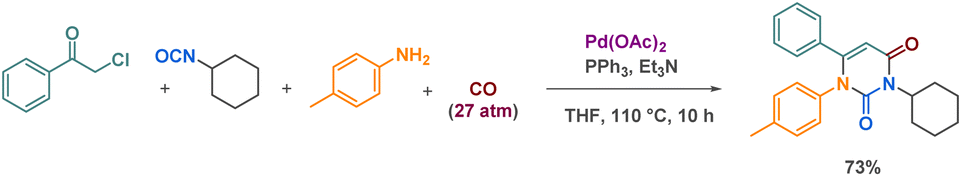 | ||
| Scheme 13 4-MCR synthesis of a cyclic urea example by Messa et al. | ||
Sulfonylureas a subclass of linear urea derivatives, are particularly attractive due to their diverse applications across various fields, notably, in the treatment of type 2 diabetes. Zhao et al. developed a palladium-catalysed reaction combining sulfonyl azides with carbon monoxide to generate an isocyanate intermediate that undergoes a nucleophilic attack from a primary or secondary amine (Scheme 14).71 They successfully extended their methodology on a scope of 37 compounds with yields higher than 90% and applied it for the gram scale synthesis of Glibenclamide, an antidiabetic drug.
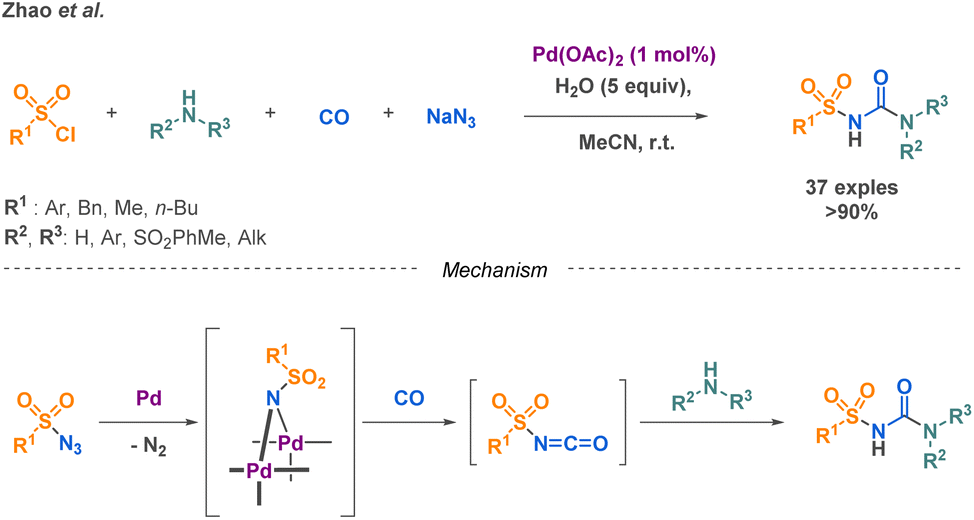 | ||
| Scheme 14 4-MCR for the preparation of sulfonylureas and the proposed mechanism by Zhao et al. | ||
Ghosh et al. reported two multicomponent reaction (MCR) approaches, a pseudo 4-MCR and a standard 4-MCR, for the synthesis of 5,6-dihydropyrrolo[2,1-a]isoquinolines. The pseudo 4-MCR method demonstrated a broader substrate scope, affording 22 diverse products with yields up to 92%. The 4-MCR approach, while offering increased molecular diversity, can sometimes lead to lower yields (up to 43%) and a narrower substrate scope (9 examples) due to potential side reactions and competing pathways. Both strategies are based on the first formation of the isocyanate from the indol-2,3-dione building block. After the addition of the tetrahydro-isoquinoline derivative and a [3 + 2] cycloaddition with the alkyne compound, the indol-2,3-dione core undergoes a C2–C3 bond cleavage leading to the isocyanate intermediate. Then, a primary or secondary amine realises a nucleophile attack on this reactive intermediate (Scheme 15).72 Although the reaction presents limitations in terms of scope application, it presents an efficient strategy to access a high molecular complexity via the access of 5,6-dihydropyrrolo[2,1-a]isoquinoline frameworks, relevant core in various bioactive compounds.
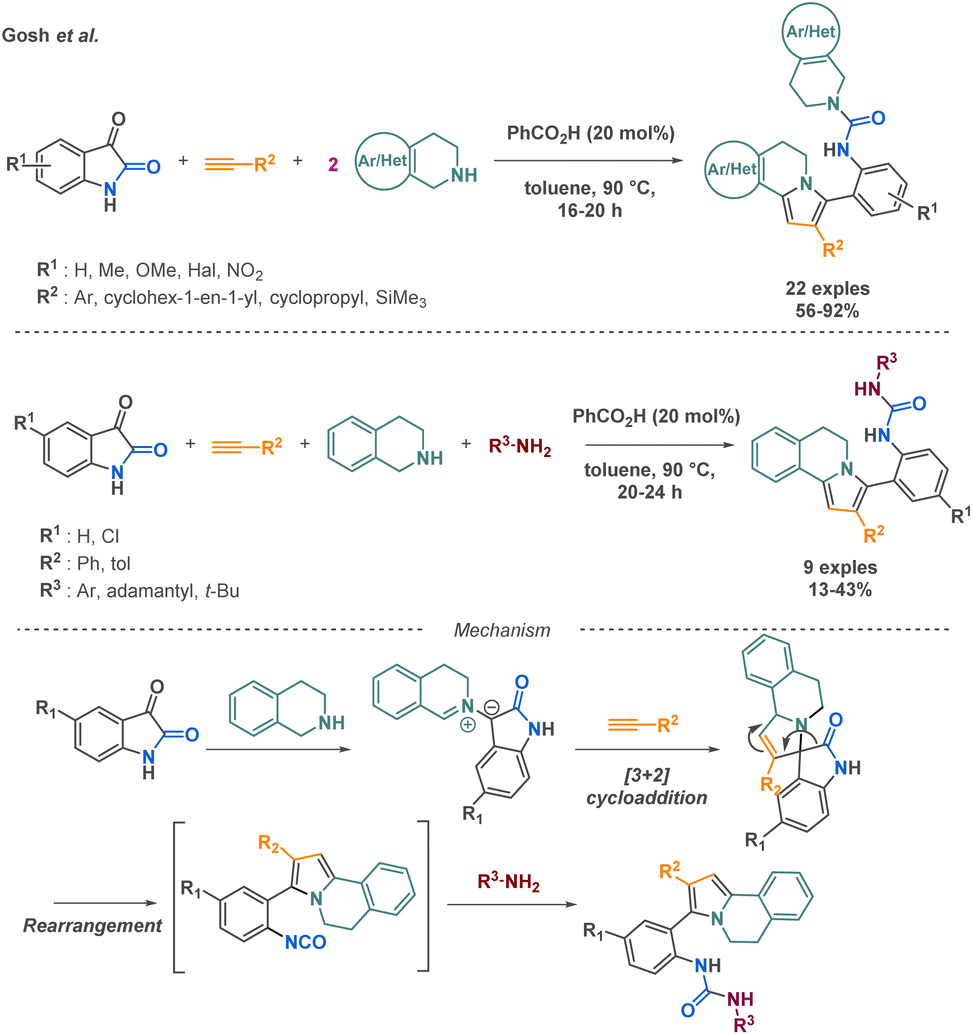 | ||
| Scheme 15 4-MCR Synthesis of 5,6-dihydropyrrolo[2,1-a]isoquinolines by Ghosh et al. | ||
3.2. Amide derivatives
Isocyanates offer the opportunity to react with diverse nucleophiles such as organometallic reagents to form amide bonds.73 Amide bonds hold significant interest not only for their natural abundance in numerous biological components but also for their widespread presence in numerous pharmaceutical and bioactive compounds. From the well-known techniques of peptidic coupling, less classical routes to amide bond formation have emerged including isocyanate-based MCR. | ||
| Scheme 16 Synthesis of succinimide derivatives by Esmaelli et al. and Alizadeh et al. | ||
Numerous studies on the transformation of aryl sulfonyl isocyanates have been reported for the preparation of amide derivatives.78 Alizadeh et al. described two methodologies involving the combination of amines, arylsulfonamides, and acetylenic compounds to yield arylsulfonamides or 1-(arylsulfonyl)-3-[(diethylamino)-methylene]-2,4-azetidinedione derivatives (Scheme 17a).79 In this work, the authors have unveiled the diverse reactivity profiles of intermediates, which can undergo proton transfer and other transformations depending on the type of starting materials employed. This inherent flexibility allows for the synthesis of a variety of molecular architectures through a single, mild reaction. Notably, the method avoids the need for pre-functionalized starting materials, streamlining the synthetic process and providing a compelling alternative to complex multi-step sequences. In a very similar approach, they investigated the preparation of 3-oxo-3,4-dihydroquinoxalines combining o-phenylenediamine, dialkyl acetylenedicarboxylates, and arylsulfonyl isocyanates through a 3-MCR (Scheme 17b).80 Building upon their previous work, the research group has introduced another straightforward and efficient synthetic method. The protocol operates under mild conditions, enabling the synthesis of complex molecules with high yields. Sulfonyl isocyanates have also been used by Huang et al. in combination with diazooxindoles and internal alkenes to generate complex scaffolds via MCR. Their innovative approach relies on the use of zinc catalysis, which facilitates [2 + 2 + 1] annulation that produces spirooxindole derivatives (Scheme 17c).81 This method employs an abundant and inexpensive catalyst to enable the efficient synthesis of spirooxindole derivatives with yields up to 96% across 38 examples. The broad substrate scope and high catalytic efficiency of this approach represent a significant improvement over previous methods, which typically exhibit limitations in terms of both substrate compatibility and catalytic activity. The proposed mechanism involves an initial step where the isocyanate group activated by the zinc, goes through a nucleophilic attack of the diazooxindole leading to cyclization and the subsequent elimination of nitrogen gas. Further coordination between the zinc, the resulting intermediate and the alkene leads to a [3 + 2] cycloaddition yielding the final product.
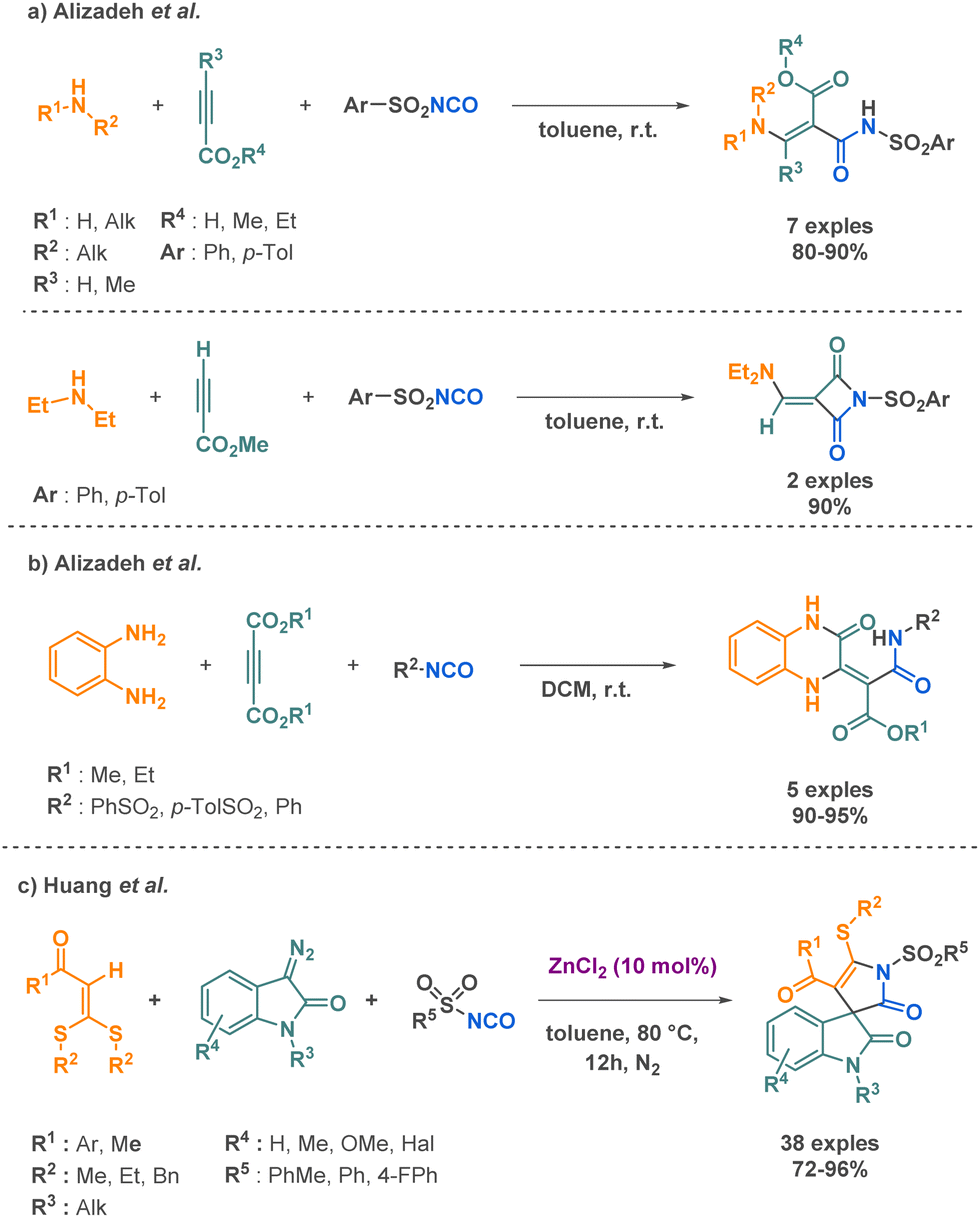 | ||
| Scheme 17 Synthesis of sulfonylamide derivatives by Alizadeh et al. and Huang et al. | ||
Imidazoles are nitrogen-containing heterocycles found in diverse biological contexts and found significant interest for their anticancer, anti-inflammatory, or antimicrobial properties.82 Three teams have proposed 3-MCR synthetic routes for their functionalisation with cyclic or linear amide moieties. First, Adib et al. reported a methodology involving imidazoles and isocyanates. This 3-MCR occurs with acetylenic esters as the third reactant. A mixture of the three compounds at room temperature in dichloromethane affords tetrahydroimidazo[1,2-a]-pyrimidines in one hour (Scheme 18a).83 Although the scope evaluation is limited (6 examples), this method is an efficient, fast and easy strategy to access such ring systems having hydrogen at bridgehead carbon atom. Shen et al. reported the C2-functionalisation of nitrogen-substituted imidazoles. This reaction involves acetylenic ketones and isocyanates in combination with imidazoles (Scheme 18b).84 The 3-MCR methodology, which operates under catalyst-free conditions, offers a straightforward approach to the synthesis of moderately complex molecules. The method exhibits excellent functional group tolerance and provides good to excellent yields (up to 98%) across a broad range of 21 substrates. The functionalisation at the C2 position can also be achieved using isocyanates and cyanophenylacetylene as reported by Belyaeva et al. (Scheme 18c) in a very similar approach.85 The transformation allows the formation of N-(Z)-alkenylimidazole-2-carboxamides at room temperature, without catalyst and without solvent and goes through the in situ formation of imidazole carboxamide zwitterions. A further advantage of this reaction is its ability to proceed with complete Z-stereoselectivity, which the authors explained by the authors by the simultaneous cleavage of the C–N bond and the formation of the bond between the Nitrogen of the anionic carboxamide and the corresponding carbon (Scheme 18b, mechanism).
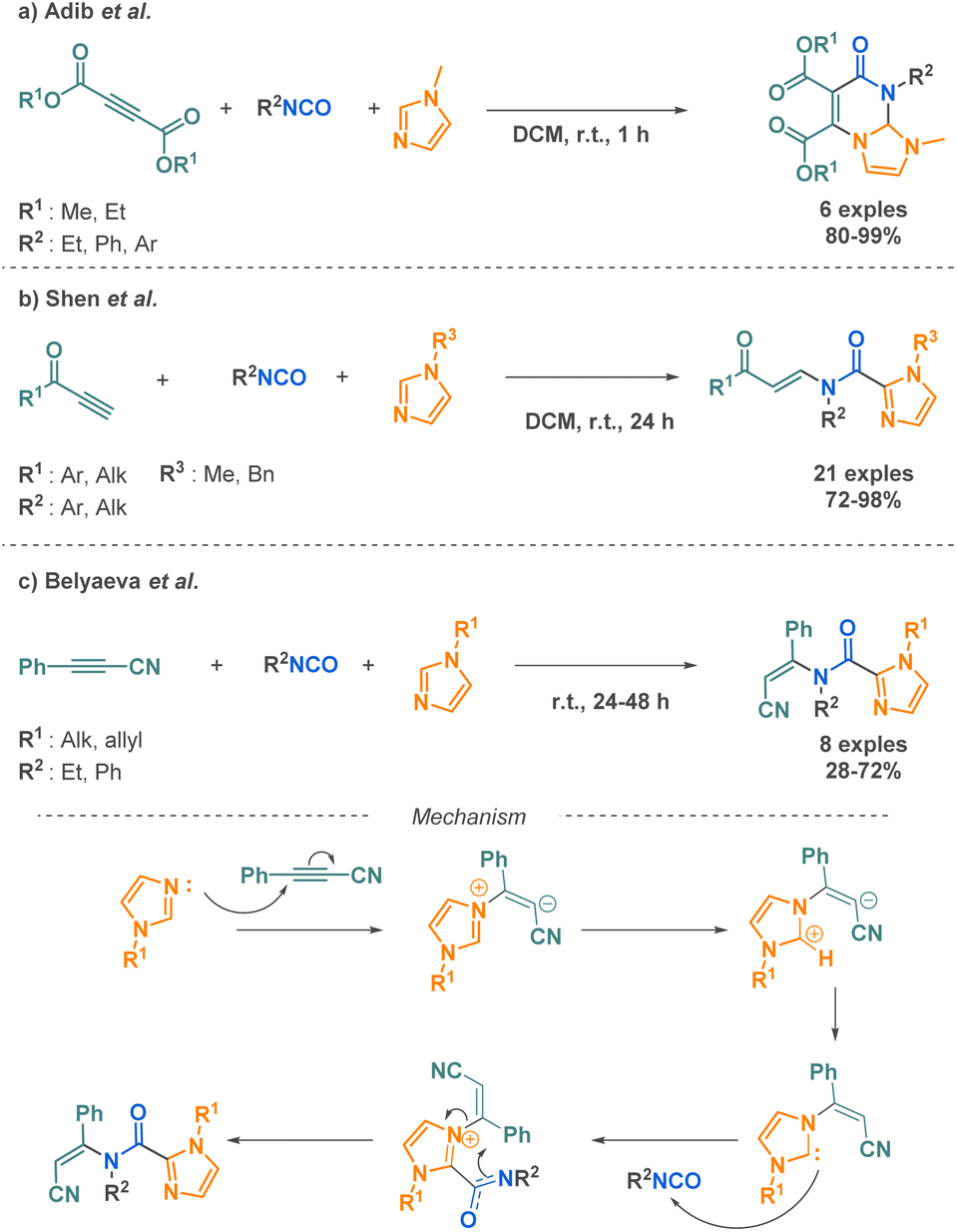 | ||
| Scheme 18 Synthesis of imidazole derivatives by 3-MCR by Adib et al., Shen et al. and Belyaeva et al. | ||
Since their development, several examples of MCR forming cyclic amide derivatives have been published. At the end of the 70's, Hong and Hoberg already investigated, respectively, cobalt and nickel catalysis via a pseudo 3-MCR involving isocyanates and 2 equivalents of alkyne to prepare pyridone.86–89 Later on, Oberg et al. employed rhodium catalysis to achieve cycloadditions between terminal alkynes and isocyanates via another pseudo 3-MCR since it involves two equivalents of the alkyne (Scheme 19a).90 This reaction came after the development of very similar rhodium catalysed pseudo 3-MCR in milder conditions by Tanaka et al. providing substituted 2-pyridones (Scheme 19b).91 Both studies have reported similar catalytic systems, pioneered by Tanaka et al. but Oberg et al. demonstrated the versatility of this approach by carefully selecting a ligand/metal complex that could direct the reaction toward different regioselectivities, depending on the nature of the starting material. Those two studies are highly complementary in regards of the synthesis of 2-pyridones by [2 + 2 + 2] cycloaddition.
 | ||
| Scheme 19 Synthesis of amide derivatives by Oberg et al. and Tanaka et al. | ||
In 2020, Beigi-Somar and co-workers prepared dihydropyridine-3-carboxylates from terminal alkynes, isocyanates and malonates (Scheme 20b).92
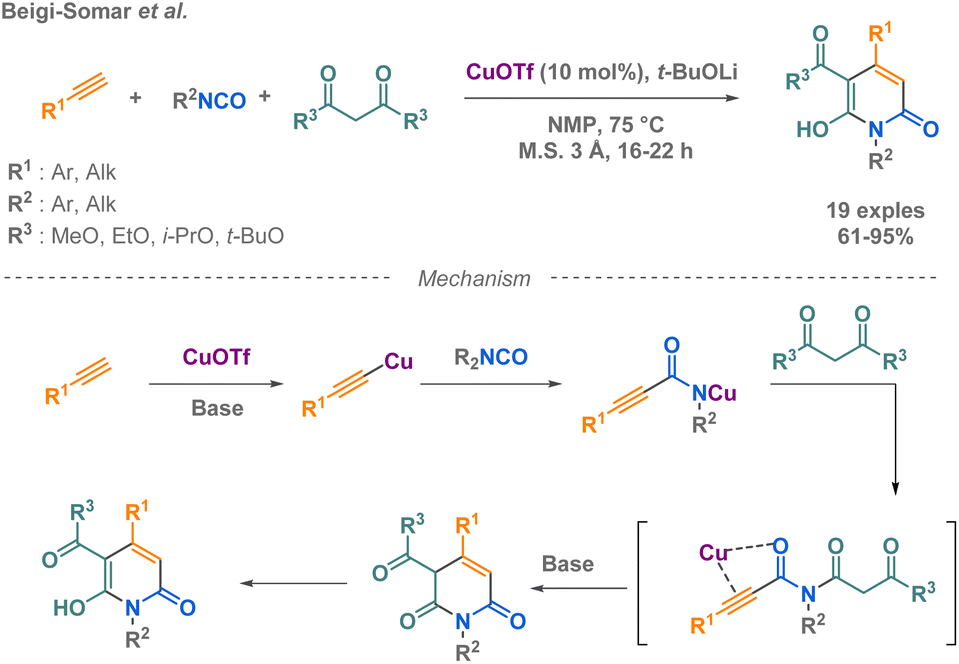 | ||
| Scheme 20 Synthesis of cyclic amide derivatives Beigi-Somar et al. | ||
Using a copper(I) catalysis facilitates the reaction by the formation of a copper acetylide in situ, which subsequently reacts with the electrophilic carbon of the isocyanate compound. The team hypothesized a mechanism and conducted experiments to test the regioselectivity of the reaction using an unsymmetrical malonate, the ethyl methyl malonate. The results showed that 57% of the product formed was ethyl carboxylate, while 31% was methyl carboxylate. Although the scope of malonate remains limited, the efficiency of the reaction with a large scope of isocyanates has been demonstrated (R = alkyl, aryl) with good to excellent yields.
Combined with isocyanates, acetylene dicarboxylates are useful building blocks to access amide derivatives as presented in the above-mentioned reactions. Another example of MCR employing this starting material was described by Lei et al., where they combined acetylene dicarboxylates with aryl isocyanates along with α,β-unsaturated imines to afford pyrimidone derivatives (Scheme 21).93 Although the scope presents the evaluation of 24 new structures with moderate to good yields, the variation of functional groups remains restricted, as for instance only aryl isocyanates and N-phenyl imines are tolerated.
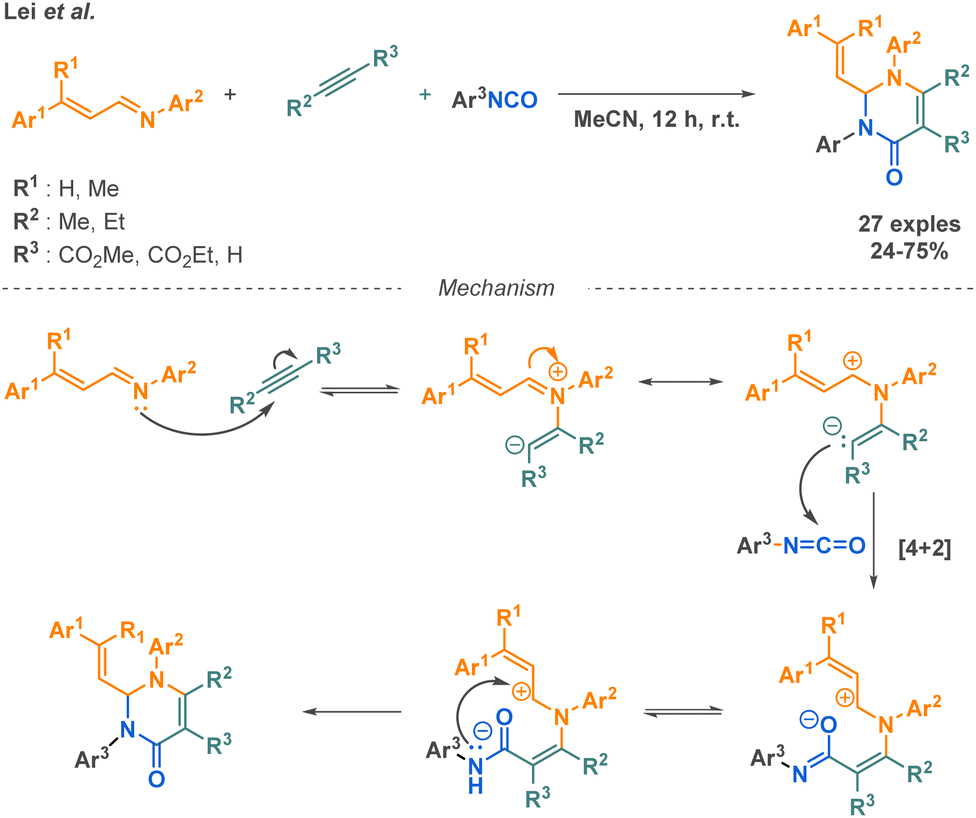 | ||
| Scheme 21 Synthesis of pyrimidine derivatives by Lei et al. | ||
3.3. Carbamate derivatives
Carbamates are found in FDA-approved drugs with antitumor or anticonvulsant properties.94,95 Additionally, they represent a class of commonly used pesticides and serve as starting materials in the synthesis of polyurethanes.96,97 Their widespread use in medicinal chemistry is attributed to their biological compatibility and functional stability. Carbamate groups, acting as bioisosteres of amide bonds, offer improved pharmacokinetic properties and enhanced bioavailability due to their stability against proteases. Consequently, carbamate derivatives are of interest in peptidomimetic synthesis. The nucleophilic attack of alcohols on isocyanates is a common route to carbamate formation, which has been employed by multiple research teams in multicomponent reaction methodologies to access various scaffolds containing cyclic or linear carbamate moieties.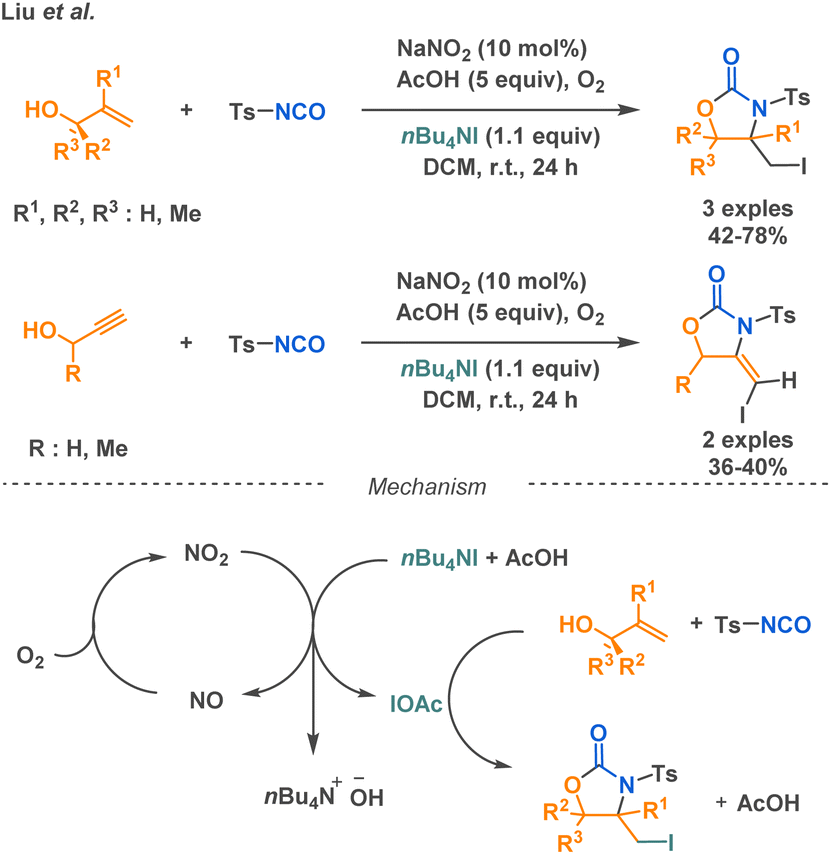 | ||
| Scheme 22 Synthesis of cyclic carbamate derivatives Liu et al. | ||
Church et al., also explored a catalytic 3-MCR process but this time using an aluminium complex. While investigating its ability to act as a catalyst in a reaction involving an isocyanate, carbon monoxide, and an epoxide, they demonstrated the selective production of 1,3-oxazinane-2,4-diones stereospecifically (Scheme 23).99 The harsh reaction conditions (55 atm. of CO) and the complex synthesis of the catalyst limit the practical applicability of this methodology. However, the reaction demonstrates a broad tolerance for various substituents on the epoxide component, with enhanced selectivity observed for electron-deficient isocyanates. Additionally, the use of chiral epoxides enables the synthesis of enantiopure products.
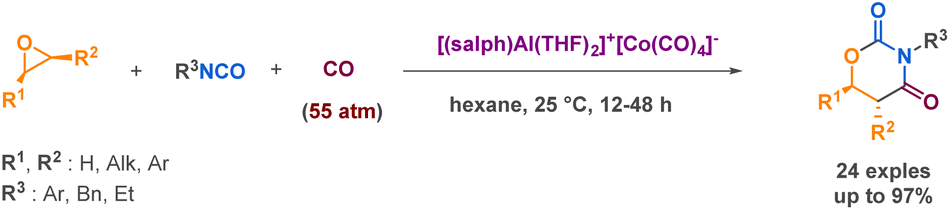 | ||
| Scheme 23 Synthesis of oxazinanedione derivatives by Church et al. | ||
Potuganti et al. explored the synthesis of cyclic carbamates via a metal-catalysed multicomponent reaction (MCR) approach. They developed a novel strategy to produce spiro oxazolidinones using copper catalysis. The interest of the methods among other strategies to access spiro oxazolidinones relies on the mild and inexpensive conditions. By combining carbonyl compounds, arylacetylenes, and isocyanates, they successfully synthesized 24 compounds with moderate to good yields (up to 88% yields) (Scheme 24).100
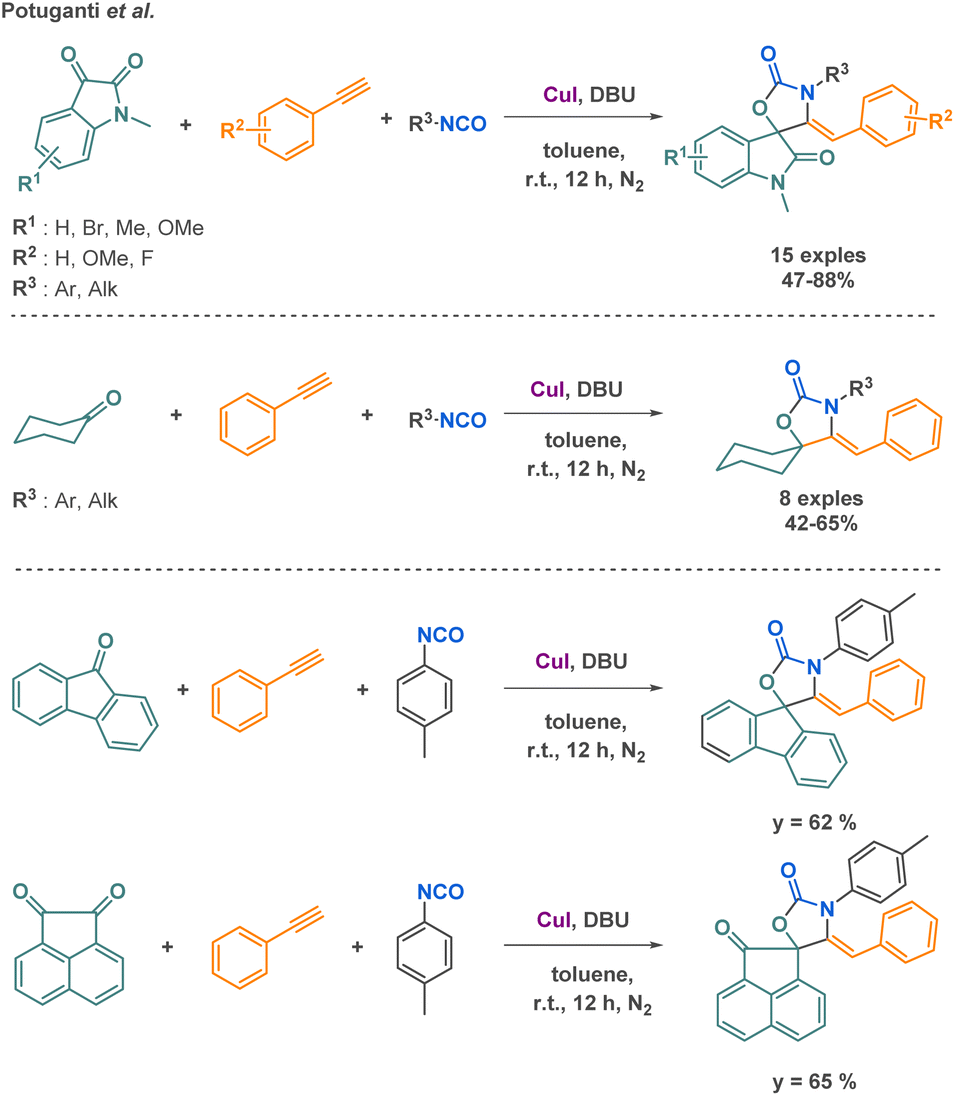 | ||
| Scheme 24 Synthesis of cyclic carbamate derivatives by Potuganti et al. | ||
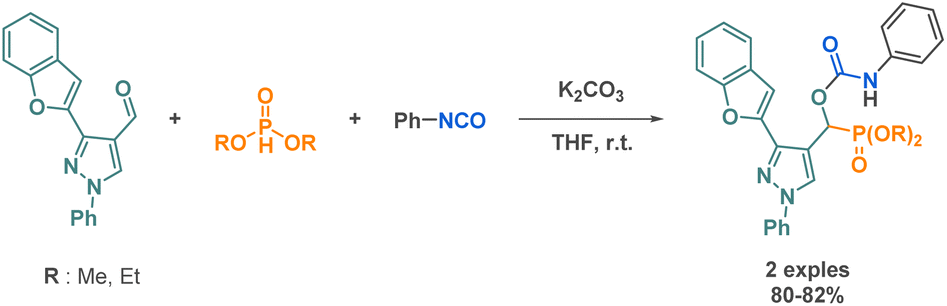 | ||
| Scheme 25 Synthesis of linear carbamate derivatives by Abdou et al. | ||
As previously described by Church et al. carbon monoxide could be used to prepare cyclic carbamates via MCR.99 On this base, Yin et al. combined aryl bromides and carbon monoxide to perform a 4-MCR palladium-catalysed carbonylation.102 Following this carbonylation, the potassium cyanate moiety coordinates to the palladium complex formed. Eventually, an alcohol performs a nucleophilic attack on the cyanate to provide a carbamate that couples with the carbonylated intermediate (Scheme 26). The advantage of this method relies on the milder conditions employed compared to Church et al. (1.5 equivalent of CO, catalytic amount of commercially accessible metal/ligands). Also, the use of CO allows the access to carbamate derivatives from aryl bromide as building blocks, a vast and easily accessible feedstock. This is demonstrated by the large scope investigated by the authors (34 examples up to 99% yield).
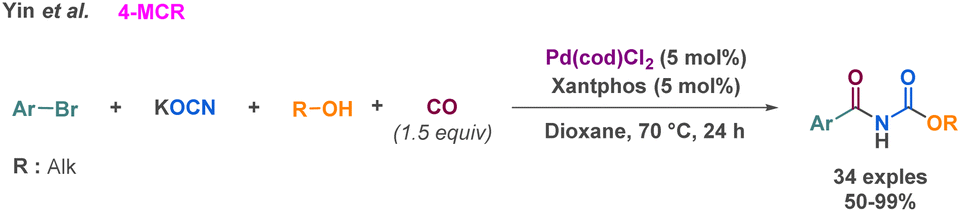 | ||
| Scheme 26 Synthesis of linear carbamate derivatives by 4-MCR by Yin et al. | ||
3.4. Miscellaneous
Thiocarbamates represent a class of compounds that can be prepared using an isocyanate and a thiol as nucleophilic agent. This strategy has been employed by Babin et al. in a 3-MCR. While they were investigating the robustness of the SAW reaction for linear urea synthesis they found that thiols directly competed with the amine, resulting in the predominant formation of thiocarbamate by-product. Building upon their in situ isocyanate generation method, the researchers further applied it to synthesize isotopically labeled thiocarbamates using thiols (Scheme 27).34 | ||
| Scheme 27 Synthesis of thiocarbamates using a 3-MCR by Babin et al. | ||
Carbamimidates is another class of compounds that can be accessed via isocyanate-based MCR. Among the three examples described in the literature, Campbell and Toste reported the synthesis of cyclic carbamimidates using a (MC)2R approach.103 Their technique combines imines, terminal alkynes, and p-toluenesulfonylisocyanate (p-TSNCO) under the influence of monophosphine gold catalysis. Using unsual ligands, i.e., trans-1-diarylphosphino-2-aminocycloalkanes, they were able to propose the sequence with high regio- and enantioselectivities (41–95% ee). The proposed mechanism involves a gold-catalysed addition reaction between an alkyne and an imine, producing a propargyl-amine, which is then trapped with p-TSNCO to afford acyclic ureas. Then, a 5-exo-dig cyclization occurs via a nucleophilic attack of the urea oxygen to afford the desired product. This strategy allows for the conservation of the stereocenter along the synthetic sequence, where traditional biaryl bisphosphine ligands were not competent in the cyclization step (Scheme 28a). Moreover, by changing the monophosphine for a sulfonyl urea ligand, they were able to form product A enantioselectively (Scheme 28b).
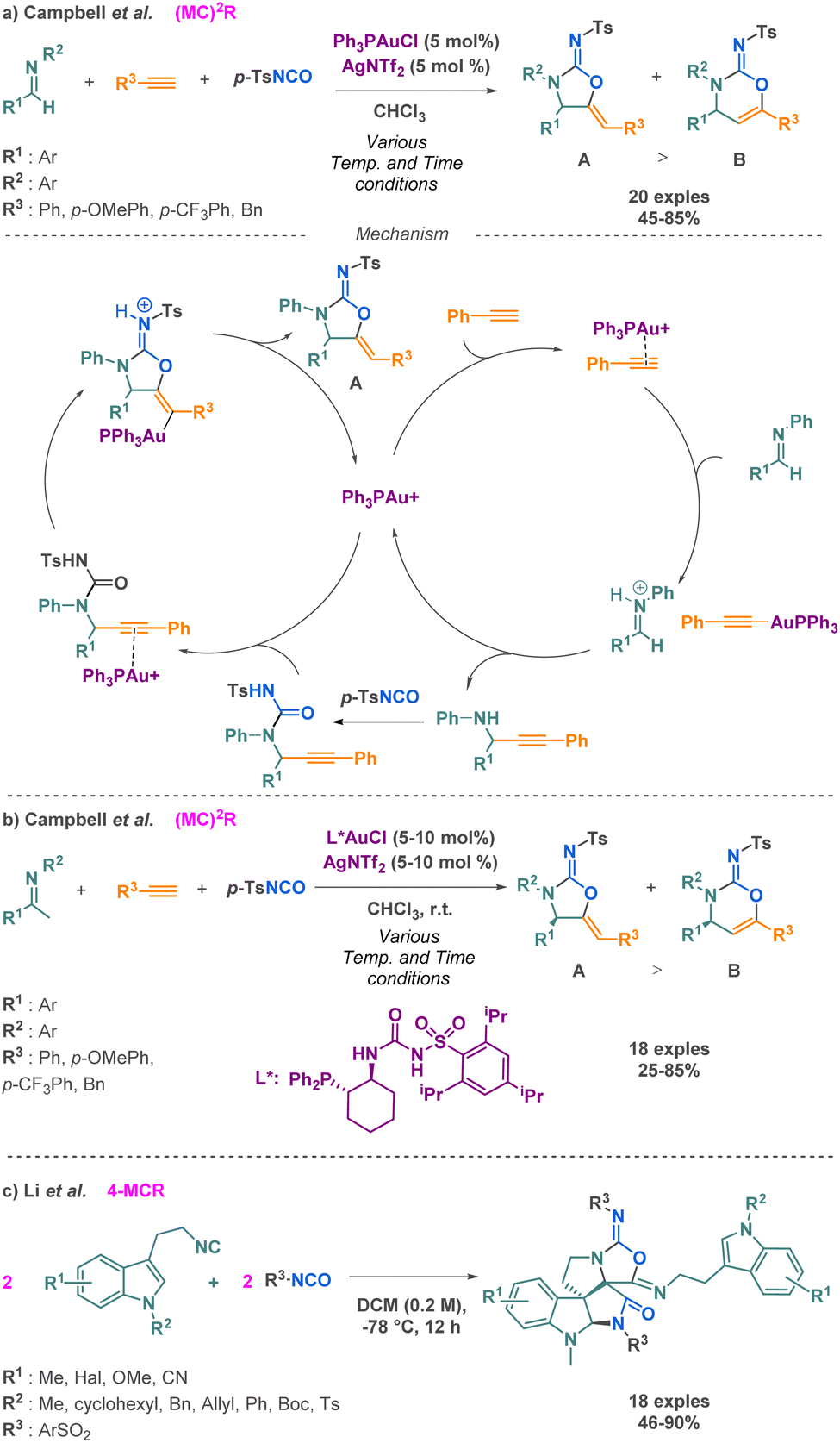 | ||
| Scheme 28 Synthesis of carbamimidate derivatives by Campbell et al. and Li et al. | ||
In 2018, Li et al. conducted a novel pseudo 4-MCR yielding fused pyrocyclic bispiroindolines. These compounds belong to a group of biologically significant molecules, including strychnine. This Ugi-type reaction occurred between two 2-isocyanoethylindoles and two benzylsulfonates isocyanates, leading to the construction of polycyclic bispiroindolines (Scheme 28c).104 This method presents the enormous advantage of a simple protocol to access large molecular complexity, rarely observed in a single-step synthesis.
In recent years, there have been several reports on Biginelli-like reactions incorporating isocyanates. Liang et al. showcased a one-pot synthesis of N-aryl-5-carboxyl-6-methyl cytimidine derivatives utilizing an aromatic isocyanate, ethyl acetoacetate, and urea derivatives. This innovative approach highlights the remarkable potential of isocyanates as versatile building blocks for the synthesis of diverse nitrogen heterocycles (Scheme 29a).105 Similarly, Elhefny et al. reported a 3-MCR affording amidine derivatives. This strategy was implemented to introduce the quinoxaline motif in biologically tested compounds. This scaffold holds significant interest due to its frequent presence in diverse antibiotic molecules (Scheme 29b).106
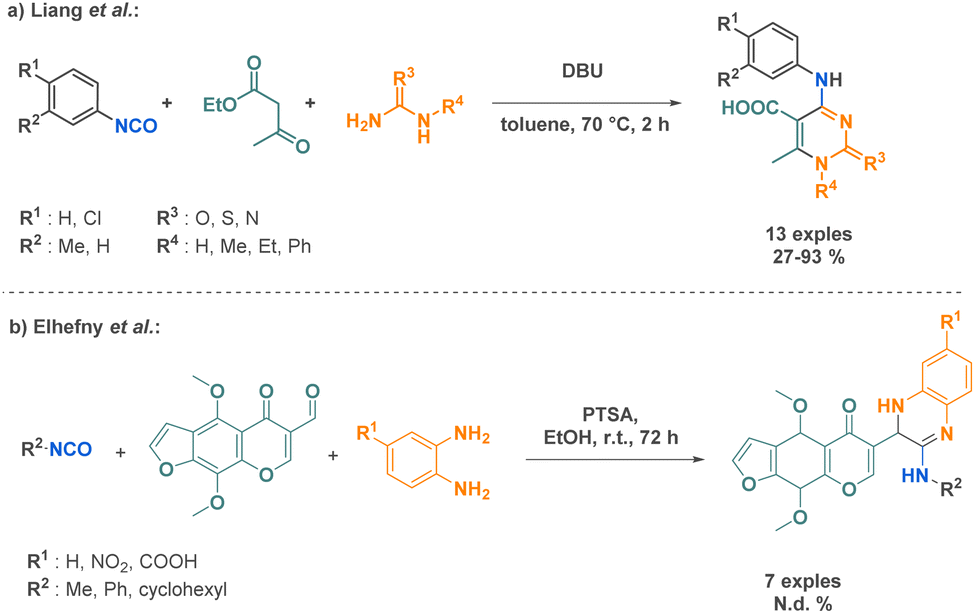 | ||
| Scheme 29 MCR reactions involving isocyanates by Liang et al. and Elhefny et al. | ||
To conclude this part on miscellaneous isocyanate-based MCR, one last example reported by Wang et al. in 2019 describes an interesting copper-catalyzed MCR to synthetize Boc-protected α-aminals from di-tert-butyl peroxide (DTBP), unactivated ethers and isocyanates (Scheme 30).107
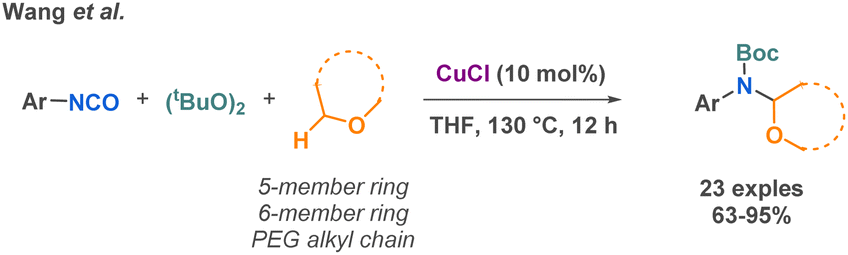 | ||
| Scheme 30 MCR reactions involving isocyanates by Wang et al. | ||
4. Conclusion
In conclusion, isocyanates have proven to be incredibly versatile building blocks in multicomponent reactions (MCRs), playing a key role in the efficient creation of complex and diverse molecules. Their ability to take part in 3-, 4-, and 5-component reactions has opened up exciting possibilities for designing compounds that are not only structurally varied but also biologically relevant. This makes isocyanates a valuable tool for developing new molecules with potential applications in fields like drug discovery and material science. Isocyanates, whether used as pre-formed reagents or generated in situ, contribute to the development of novel scaffolds with enhanced interaction potential for drug discovery and material science. The integration of isocyanates into MCRs, coupled with modern techniques like flow and microwave chemistry, has further expanded their applications. As MCR strategies evolve, isocyanates are poised to play an increasingly important role in accelerating the discovery of new functional compounds, particularly in pharmaceutical and agrochemical industries. Their use aligns well with green chemistry principles, making them an environmentally friendly and efficient choice for synthetic transformations.Data availability
No new data were created or analysed in this study. Data sharing is not applicable to this article.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Commissariat à l'Energie Atomique et aux Energies Alternatives (PhD fellowship of C. Zavarise).References
- A. Strecker, Ann. Chem. Pharm., 1850, 75, 27–45 CrossRef.
- A. Strecker, Ann. Chem. Pharm., 1854, 91, 349–351 CrossRef.
- A. Dömling and A. D. AlQahtani, in Multicomponent Reactions in Organic Synthesis, Wiley, 1st edn, 2014, pp. 1–12 Search PubMed.
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, 2000 Search PubMed.
- A. Hantzsch, Ann. Chem. Pharm., 1882, 215, 1–82 CrossRef.
- P. Biginelli, Gazz. Chim. Ital., 1893, 23, 360–416 Search PubMed.
- M. Passerini and L. Simone, Gazz. Chim. Ital., 1921, 51, 126–129 CAS.
- I. Ugi and C. Steinbrückner, Angew. Chem., 1960, 72, 267–268 CrossRef CAS.
- E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- C. Lambruschini, L. Moni and A. Basso, in Multicomponent Reactions towards Heterocycles, 1st edn, 2022, pp. 211–237 Search PubMed.
- J. Fairoosa, S. Saranya, S. Radhika and G. Anilkumar, ChemistrySelect, 2020, 5, 5180–5197 CrossRef CAS.
- M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042–2064 RSC.
- P. Renzi, J. Scarfiello, A. Lanfranco and A. Deagostino, Eur. J. Org. Chem., 2023, 26, 41 Search PubMed.
- S. E. Hooshmand, H. Yazdani and C. Hulme, Eur. J. Org. Chem., 2022, 2022, 34 Search PubMed.
- V. Tyagi and P. M. S. Chauhan, Synlett, 2013, 24, 1291–1297 CAS.
- C. R. B. Rhoden, D. G. Rivera, O. Kreye, A. K. Bauer, B. Westermann and L. A. Wessjohann, J. Comb. Chem., 2009, 11, 1078–1082 CrossRef CAS PubMed.
- J. Bariwal, R. Kaur, L. G. Voskressensky and E. V. Van Der Eycken, Front. Chem., 2018, 6, 557 CrossRef PubMed.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, J. Am. Chem. Soc., 2023, 145, 1475–1496 CAS.
- M. Carvalho, G. Amarante and P. De Castro, J. Braz. Chem. Soc., 2023, 34, 1041–1070 CAS.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef.
- G. Zhao, H. Wang, J. Luo, X. He, F. Xiong, Y. Li, G. Zhang and Y. Li, Org. Lett., 2023, 25, 665–670 CrossRef CAS.
- Z. Zhang, Y. You and C. Hong, Macromol. Rapid Commun., 2018, 39, 1800362–1800374 CrossRef PubMed.
- M. J. Buskes, A. Coffin, D. M. Troast, R. Stein and M.-J. Blanco, ACS Med. Chem. Lett., 2023, 14, 376–385 CrossRef CAS PubMed.
- C. Russo, F. Brunelli, G. Cesare Tron and M. Giustiniano, Chem.–Eur. J., 2023, 29, e202203150–e202203169 CAS.
- A. Váradi, T. Palmer, R. Notis Dardashti and S. Majumdar, Molecules, 2015, 21, 19 CrossRef PubMed.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 Search PubMed.
- Z. Wang, J. Li, N. Wang, H. Liu and K. Wang, Asian J. Org. Chem., 2023, 12, e202300105–e202300124 CAS.
- J. H. Saunders and R. J. Slocombe, Chem. Rev., 1948, 43, 203–218 CAS.
- R. G. Arnold, J. A. Nelson and J. J. Verbanc, Chem. Rev., 1957, 57, 47–76 CrossRef CAS.
- E. Serrano and R. Martin, Eur. J. Org. Chem., 2018, 3051–3064 CrossRef CAS.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- A. Wurtz, Comptes Rendus, 1848, 27, 241–243 Search PubMed.
- C. Six and F. Richter, in Ullmann's Encyclopedia of Industrial Chemistry, 1st edn, 2003 Search PubMed.
- V. Babin, A. Sallustrau, M. Molins, A. Labiche, A. Goudet, F. Taran and D. Audisio, Eur. J. Org. Chem., 2021, 57, 6680–6683 CAS.
- R. S. Obach and A. S. Kalgutkar, in Comprehensive Toxicology, 2010, pp. 309–347 Search PubMed.
- H. Li, H. Cheng and F. Zhao, Asian J. Org. Chem., 2022, 11, e202200338–e202200355 CAS.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CAS.
- J. F. Vincent-Rocan, C. Clavette, K. Leckett and A. M. Beauchemin, Chem.–Eur. J., 2014, 21, 3886–3890 CrossRef.
- R. A. Ivanovich, C. Clavette, J. F. Vincent-Rocan, J. G. Roveda, S. I. Gorelsky and A. M. Beauchemin, Chem.–Eur. J., 2016, 22, 7906–7916 CrossRef CAS.
- R. A. Ivanovich, D. E. Polat and A. M. Beauchemin, Adv. Synth. Catal., 2017, 359, 4289–4293 CrossRef CAS.
- J. S. Derasp and A. M. Beauchemin, ACS Catal., 2019, 9, 8104–8109 CrossRef CAS.
- F. Vuillermet, M. A. Dahab, D. Abdelhamid, D. E. Polat, M. A. Gill and A. M. Beauchemin, J. Org. Chem., 2023, 88, 2095–2102 CrossRef CAS PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2020, 63, 2751–2788 CrossRef CAS.
- R. Ronchetti, G. Moroni, A. Carotti, A. Gioiello and E. Camaioni, RSC Med. Chem., 2021, 12, 1046–1064 RSC.
- M. Kalník, P. Gabko, M. Bella and M. Koóš, Molecules, 2021, 26, 4024–4057 CrossRef PubMed.
- M. C. Bellucci, M. Frigerio, T. Marcelli and A. Volonterio, Synlett, 2013, 24, 727–732 CrossRef CAS.
- J. C. Flores-Reyes, V. D. C. Cotlame-Salinas, I. A. Ibarra, E. González-Zamora and A. Islas-Jácome, RSC Adv., 2023, 13, 16091–16125 RSC.
- A. Alizadeh and E. Sheikhi, Tetrahedron Lett., 2007, 48, 4887–4890 CrossRef CAS.
- J. H. Rigby and S. A. Brouet, Tetrahedron Lett., 2013, 54, 2542–2545 CrossRef CAS.
- A. Pałasz and D. Cież, Eur. J. Med. Chem., 2015, 97, 582–611 CrossRef.
- G. Fernández De Trocóniz, A. M. Ochoa De Retana, G. Rubiales and F. Palacios, J. Org. Chem., 2014, 79, 5173–5181 CrossRef.
- M. Morimoto, Y. Nishida, T. Miura and M. Murakami, Chem. Lett., 2013, 42, 550–552 CrossRef CAS.
- M. G. Dekamin, K. Varmira, M. Farahmand, S. Sagheb-Asl and Z. Karimi, Catal. Commun., 2010, 12, 226–230 CrossRef CAS.
- M. G. Dekamin, S. Mallakpour and M. Ghassemi, J. Chem. Res., 2005, 2005, 177–179 CrossRef.
- H.-J. Shue, X. Chen, N.-Y. Shih, D. J. Blythin, S. Paliwal, L. Lin, D. Gu, J. H. Schwerdt, S. Shah, G. A. Reichard, J. J. Piwinski, R. A. Duffy, J. E. Lachowicz, V. L. Coffin, F. Liu, A. A. Nomeir, C. A. Morgan and G. B. Varty, Bioorg. Med. Chem. Lett., 2005, 15, 3896–3899 CrossRef CAS.
- N. Hodge, P. E. Aldrich, L. T. Bacheler, C.-H. Chang, C. J. Eyermann, S. Garber, M. Grubb, D. A. Jackson, K. Jadhav, B. Korant, P. Y. Lam, M. B. Maurin, J. L. Meek, M. J. Otto, M. M. Rayner, C. Reid, T. R. Sharpe, L. Shum and S. Erickson-Viitanen, Chem. Biol., 1996, 3, 301–314 CrossRef PubMed.
- Z. Pan, X. Yang, B. Chen, S. Shi, T. Liu, X. Xiao, L. Shen, L. Lou and Y. Ma, J. Org. Chem., 2022, 87, 3596–3604 CrossRef CAS.
- Z. Pan, S. Shi, X. Yang, X. Xiao, W. Zhang, S. Wang and Y. Ma, Green Chem., 2021, 23, 2944–2949 RSC.
- D. Chestnykh, F. Graßl, C. Pfeifer, J. Dülk, C. Ebner, M. Walters, S. Von Hörsten, J. Kornhuber, L. S. Kalinichenko, M. Heinrich and C. P. Müller, Psychopharmacology, 2023, 240, 1011–1031 CrossRef CAS.
- B. Mohammadi and M. Adib, Chin. Chem. Lett., 2014, 25, 553–556 CrossRef CAS.
- S. Du, W.-F. Wang, Y. Song, Z. Chen and X.-F. Wu, Org. Lett., 2021, 23, 974–978 CrossRef CAS.
- T. Soeta, S. Takashita, Y. Sakata and Y. Ukaji, Asian J. Org. Chem., 2016, 5, 1041–1047 CrossRef CAS.
- G. Martinez-Ariza, M. Ayaz, F. Medda and C. Hulme, J. Org. Chem., 2014, 79, 5153–5162 CrossRef CAS.
- V. Babin, A. Sallustrau, O. Loreau, F. Caillé, A. Goudet, H. Cahuzac, A. D. Vecchio, F. Taran and D. Audisio, Chem. Commun., 2021, 57, 6680–6683 RSC.
- D. Carnaroglio, K. Martina, G. Palmisano, A. Penoni, C. Domini and G. Cravotto, Beilstein J. Org. Chem., 2013, 9, 2378–2386 CrossRef CAS.
- I. Yavari and M. Nematpour, Helv. Chim. Acta, 2014, 97, 1132–1135 CrossRef CAS.
- S. Perrone, M. Capua, A. Salomone and L. Troisi, J. Org. Chem., 2015, 80, 8189–8197 CrossRef CAS.
- B. Groenendaal, D. J. Vugts, R. F. Schmitz, F. J. J. De Kanter, E. Ruijter, M. B. Groen and R. V. A. Orru, J. Org. Chem., 2008, 73, 719–722 CrossRef CAS.
- D. J. Vugts, M. M. Koningstein, R. F. Schmitz, F. J. J. De Kanter, M. B. Groen and R. V. A. Orru, Chem.–Eur. J., 2006, 12, 7178–7189 CrossRef CAS.
- F. Messa, S. Perrone and A. Salomone, Molbank, 2023, 2023, M1611–M1616 CrossRef CAS.
- J. Zhao, Z. Li, S. Song, M.-A. Wang, B. Fu and Z. Zhang, Angew. Chem., 2016, 128, 5635–5639 CrossRef.
- A. Ghosh, S. Kolle, D. S. Barak, R. Kant and S. Batra, ACS Omega, 2019, 4, 20854–20867 CrossRef CAS PubMed.
- R. M. De Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS.
- Z. Zhao, J. Yue, X. Ji, M. Nian, K. Kang, H. Qiao and X. Zheng, Bioorganic Chem., 2021, 108, 104556–104585 CrossRef.
- A. A. Esmaeili, H. Khoddam-Mohammadi, A. Moradi, E. Davamdar, M. Izadyar, M. Khavani and M. R. Islami, RSC Adv., 2014, 4, 37900–37907 RSC.
- A. Alizadeh, F. Movahedi and A. A. Esmaili, Tetrahedron Lett., 2006, 47, 4469–4471 CrossRef CAS.
- A. Alizadeh, A. Rezvanian and L.-G. Zhu, Helv. Chim. Acta, 2007, 90, 2414–2420 CrossRef CAS.
- D. Huang and G. Yan, Org. Biomol. Chem., 2017, 15, 1753–1761 RSC.
- A. Alizadeh and A. Rezvanian, Synthesis, 2008, 1747–1752 CrossRef CAS.
- A. Alizadeh, M. Babaki, N. Zohreh and A. Rezvanian, Synthesis, 2008, 3793–3796 CrossRef CAS.
- Z. Huang, J. Lin, M. Li, Y.-G. Zhou and Z. Yu, Org. Lett., 2023, 25, 2328–2332 CrossRef CAS.
- N. Kerru, S. V. H. S. Bhaskaruni, L. Gummidi, S. N. Maddila, S. Maddila and S. B. Jonnalagadda, Synth. Commun., 2019, 49, 2437–2459 CAS.
- M. Adib, M. Mollahosseini, H. Yavari, M. H. Sayahi and H. R. Bijanzadeh, Synlett, 2004, 1086–1088 Search PubMed.
- Y. Shen, S. Cai, C. He, X. Lin, P. Lu and Y. Wang, Tetrahedron, 2011, 67, 8338–8342 CAS.
- K. V. Belyaeva, L. V. Andriyankova, L. P. Nikitina, A. G. Mal'Kina, A. V. Afonin and B. A. Trofimov, Tetrahedron Lett., 2012, 53, 7040–7043 CAS.
- P. Hong and H. Yamazaki, Synthesis, 1977, 1, 50–52 Search PubMed.
- P. Hong and H. Yamazaki, Tetrahedron Lett., 1977, 15, 1333–1336 Search PubMed.
- H. Hoberg and B. W. Oster, J. Organomet. Chem., 1982, 234, C35–C38 CrossRef CAS.
- H. Hoberg and B. W. Oster, J. Organomet. Chem., 1983, 252, 359–364 CrossRef CAS.
- K. M. Oberg, E. E. Lee and T. Rovis, Tetrahedron, 2009, 65, 5056–5061 CrossRef CAS PubMed.
- K. Tanaka, A. Wada and K. Noguchi, Org. Lett., 2005, 7, 4737–4739 CrossRef CAS PubMed.
- V. Beigi-Somar, S. S. Homami, M. Ghazanfarpour-Darjani and A. Monzavi, Monatsh. fur Chem., 2020, 151, 231–241 CrossRef CAS.
- M. Lei, W. Tian, W. Li, P. Lu and Y. Wang, Tetrahedron, 2014, 70, 3665–3674 CrossRef CAS.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- A. Matošević and A. Bosak, Arch. Ind. Hyg. Toxicol., 2020, 71, 285–299 Search PubMed.
- N. L. Mdeni, A. O. Adeniji, A. I. Okoh and O. O. Okoh, Molecules, 2022, 27, 618 CrossRef CAS.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- H. Liu, Y. Pan and C.-H. Tan, Tetrahedron Lett., 2008, 49, 4424–4426 CrossRef CAS.
- T. L. Church, C. M. Byrne, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 8156–8162 CrossRef CAS PubMed.
- G. R. Potuganti, D. R. Indukuri, J. B. Nanubolu and M. Alla, J. Org. Chem., 2018, 83, 15186–15194 CrossRef CAS PubMed.
- W. M. Abdou and M. S. Bekheit, Arab. J. Chem., 2018, 11, 1260–1269 CrossRef CAS.
- H. Yin, A. M. De Almeida, M. V. De Almeida, A. T. Lindhardt and T. Skrydstrup, Org. Lett., 2015, 17, 1248–1251 CrossRef CAS.
- M. J. Campbell and F. D. Toste, Chem. Sci., 2011, 2, 1369–1378 RSC.
- L. Li, J. Liu and M. Shi, Org. Lett., 2018, 20, 7076–7079 CrossRef CAS.
- C. Liang, H. Song, H. Jiang and Q. Yao, Synth. Commun., 2014, 44, 2930–2935 CrossRef CAS.
- E. A. Elhefny, S. M. Abu-Bakr, N. M. Fawzy, A. M. Nasef and M. S. Aly, Int. J. Pharm. Sci. Rev. Res., 2015, 32, 85–94 CAS.
- L. Wang, Q. Tian, C. Bin and G. Zhang, Tetrahedron Lett., 2019, 60, 2084–2087 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |