
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of crystalline layered coordination polymers that respond to light and heat stimuli
Kenichiro
Omoto
 *a and
Gwénaël
Rapenne
*bc
*a and
Gwénaël
Rapenne
*bc
aDivision of Chemistry and Materials Science, Graduate School of Integrated Science and Technology, Nagasaki University, 1-14, Bunkyo-machi, Nagasaki, 852-8521, Japan. E-mail: omoto@nagasaki-u.ac.jp
bDivision of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama-cho, Ikoma, 630-0192, Japan. E-mail: gwenael-rapenne@ms.naist.jp
cCEMES, Université de Toulouse, CNRS, 29 Rue Marvig, F-31055 Toulouse Cedex 4, France
First published on 14th April 2025
Abstract
Layered coordination polymers have attracted significant attention as a class of crystsalline materials characterized by the layer-by-layer stacking of rigid two-dimensional (2D) coordination networks. One of their remarkable features is the flexibility of their crystal structures, which allows for interlayer displacement, swelling, and exfoliation. Incorporating stimuli-responsive moieties into their structures is a promising strategy for the rational design of layered coordination polymers with targeted flexible properties and functions. Despite the challenges associated with crystal design, a variety of stimuli-responsive layered coordination polymers have been developed over the past two decades. This article provides an overview of representative examples of layered coordination polymers whose properties and functions can be modulated by photo- and thermal stimuli.
 Kenichiro Omoto | Kenichiro Omoto obtained his PhD in 2016 from the University of Tokyo (Japan) under the supervision of Prof. M. Shionoya working on host–guest chemistry related to metallo-macrocycles. After post-doc positions in Kyoto University (Prof. S. Kitagawa) and at the University of Tokyo (Prof. H. Nishihara), he obtained in 2019 a specially appointed assistant professor position in the laboratory of Prof. G. Rapenne (NAIST, Japan) working on molecular machines and supramolecular chemistry related to lipid assemblies and coordination polymers. In 2023, he moved in Nagasaki University as an assistant professor. |
 Gwénaël Rapenne | Gwénaël Rapenne obtained his PhD in 1998 from Louis Pasteur University (Strasbourg, France) under the supervision of Prof. J.-P. Sauvage working on the synthesis and resolution of double helices and knots. After spending one year as a Lavoisier postdoctoral fellow working on fullerenes with Prof. F. Diederich at ETH Zürich, he joined the NanoSciences Group (CEMES-CNRS) as a Maître de Conférences at the University Paul Sabatier (Toulouse) to work in the field of single molecular machines and motors. Promoted to Full Professor in 2011 and Distinguished Professor in 2019, he also obtained a Professor position in NAIST (Japan) in 2018 in the frame of a crossed-position. |
Introduction
Coordination polymers have garnered significant attention as solid-state materials with supramolecular frameworks precisely organized and constructed from metal ions and organic ligands.1 By combining diverse metal ions with various ligands, researchers have developed a wide range of coordination polymers with one- to three-dimensional periodic networks. These materials exhibit unique structure-dependent functionalities, including applications in catalysis,2 adsorption,3 and as magnetic4 or optical materials.5Layered coordination polymers constitute a subclass of coordination polymers characterized by rigid layers of two-dimensional (2D) coordination networks stacked through weak interlayer interactions (Fig. 1).6 One of their most intriguing features is the flexibility of their crystal structure. The rigid 2D layers can undergo displacement and swelling, allowing modular modifications of their stacking arrangements and interlayer distances while maintaining their in-plane periodic linkages. This flexibility imparts unique properties and functions, such as molecular adsorption and high processability.
 | ||
| Fig. 1 Schematic representation of the structure of layered coordination polymer and its flexible morphological changes involving interlayer displacement/swelling and exfoliation. | ||
For instance, layered coordination polymers exhibiting adsorption behaviors accompanied with distinctive structural transformations, like displacement and swelling, have been reported to influence the selectivity of guest molecule.7 Additionally, technologies to process layered coordination polymers into nanosheets have been developed by physically or chemically exfoliating the stacked 2D coordination networks.8 These nanosheets hold promise for applications such as gas separation membranes.
To rationally control the flexible properties and functions of layered coordination polymers, it is essential to design materials capable of modifying their stacking modes, structures, and dynamic behaviors in response to physical stimuli such as photoirradiation or temperature changes.9 However, research on the design and application of layered coordination polymers utilizing physical stimuli remains limited and underdeveloped.
A significant challenge lies in the inherent nature of crystalline materials, where molecular arrangements are difficult to predict, and the movement of chemical components is highly constrained. As a result, the synthesis of layered coordination polymers equipped with light- or heat-responsive sites has seen limited progress.10–18 Furthermore, examples of layered coordination polymers that exhibit significant structural transformations, such as interlayer displacement, swelling, or exfoliation, enabled by their unique flexibility in response to external stimuli are still scarce (Fig. 1). Addressing these challenges requires careful consideration of the arrangement, aggregation, and isolation of molecules within layered coordination polymers. By tackling these factors, it may become possible to expand the range of photo- and thermally responsive layered coordination polymers with tailored properties and dynamic structural behaviors.
This article highlights notable examples of layered coordination polymers developed over the past two decades that utilize external physical stimuli to regulate their structures and functions. The focus is specifically on structural transformation of crystal upon physical stimuli, such as photoirradiation and temperature changes, while structural transformation in response to chemical signals, such as those involving adsorbates as well as changes in electronic properties without significant structural transformation, such as spin crossover complexes, are beyond from the scope of this article.6,19
Design directions of stimuli-responsive layered coordination polymers
Layered coordination polymers that undergo structural transformations in response to physical stimuli can be designed by introducing stimulus-responsive functional groups into the organic ligands.6,20 Herein, two major approaches can be considered as design strategies for such structural transformations.The first approach is incorporation of stimulus-responsive functional groups into the linkers (organic ligands) that connect metal ions creating the main skeleton of the coordination network (Fig. 2a). In this case, external stimulus structural change of the functional group induces in-plane deformation of the coordination network, leading to changes in pore size and dimensionality.
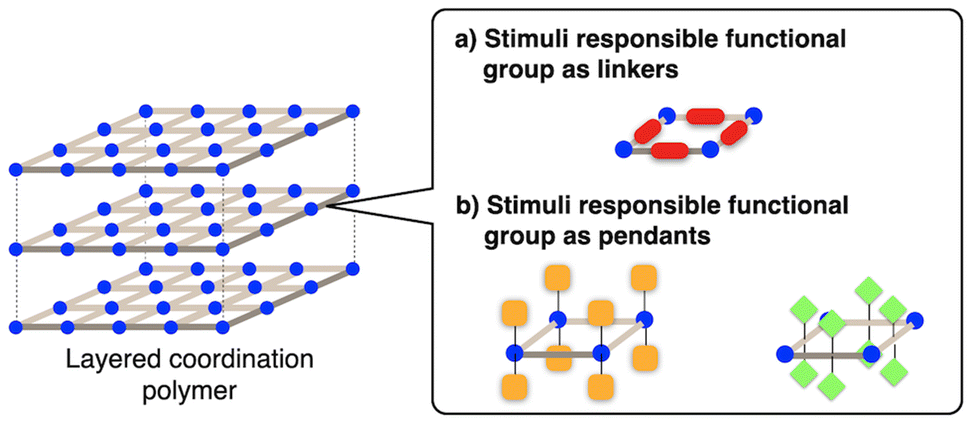 | ||
| Fig. 2 Schematic image of the stimulus-responsive functional groups incorporated (a) as linkers and (b) as pendants of the 2D coordination networks of layered coordination polymers. | ||
The second approach is incorporation of stimulus-responsive functional groups at the surface of the 2D coordination network (Fig. 2b). These moieties can be incorporated as pendant functional groups grafted to linkers or pendant ligands attached to nodes (metal ions), governing the interaction modes between layers by contacting or interpenetrating with adjacent stacked layers. In this case, the structural transformation of the functional groups can modify interlayer interactions. This would allow for the modulation of the stacking mode (swelling/expansion) and morphological changes (exfoliation) specific to layered coordination polymers in response to external stimuli. These features offer potential applications in molecular adsorption control, as well as technologies in crystal processing.
As mentioned above, movement of molecules in a crystal are highly constrained owing to the intermolecular interaction. Therefore, structural transformations based on in-plane deformation of 2D layer (the first approach), which is restricted by robust coordination network, are less commonly reported compared to transformations proceeding at the interlayer interface (the second approach), where weaker interactions govern the stacking. Most of the examples introduced in this paper are related to the latter.
Photo-responsive layered coordination polymers
Light is a widely used energy for controlling the structure and function of crystalline materials including layered coordination polymers.21,22 Key advantages of photons lie in its ability to provide high-energy stimuli to target coordination polymer crystals without direct contact. Furthermore, its versatility in terms of wavelength and irradiation angle enables precise control over the material's functions.Various photoactive layered coordination polymers have been reported by incorporating organic ligands with different photoactive functional groups as building blocks. One of the most well-studied examples involve the [2 + 2] photocycloaddition reaction of stilbene analogues.23 This reaction, induced by UV light, triggers the coupling of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in stilbene analogues to form a cyclobutadiene ring. This reaction is known to proceed topotactically in the crystalline state, with the distance between the reactive olefinic CC double bonds needing to be less than ca. 4.2 Å, as described by Schmidt's criteria.24 By utilizing organic ligand containing CC double bonds, various photoreactive layered coordination polymers capable of undergoing crystal-to-crystal structural transformation have been reported.
C double bonds in stilbene analogues to form a cyclobutadiene ring. This reaction is known to proceed topotactically in the crystalline state, with the distance between the reactive olefinic CC double bonds needing to be less than ca. 4.2 Å, as described by Schmidt's criteria.24 By utilizing organic ligand containing CC double bonds, various photoreactive layered coordination polymers capable of undergoing crystal-to-crystal structural transformation have been reported.
The earliest example of a photoreactive layered coordination polymer is the Zn-based compound [Zn(MeOip)(bpe)(DMF)], which demonstrates a single-crystal-to-single-crystal (SCSC) structural transformation via intralayer [2 + 2] photocycloaddition, enabling modulation of its porous functionality.10 In this crystals, 1D double-chain coordination polymers, composed of 5-methoxyisophtalate (MeOip) and Zn(II) with a distorted octahedral coordination geometry, are bridged by 1,2-bis(4-pyridyl)ethene (bpe) to form porous 2D layers. These layers are stacked along the [100] direction, creating 1D channels with a solvent accessible volume of 27.0% of the total cell volume (Fig. 3). Within the layers, two CC bonds of adjacent bpe molecules are arranged in face-to-face manner separated by 3.71 Å, sufficiently close to undergo a [2 + 2] cycloaddition reaction. Upon UV irradiation at −180 °C, the intralayer [2 + 2] cycloaddition reaction proceeded quantitatively, resulted in the in-plane structural change: widening of channel path and an increase in solvent-accessible volume to 28.1%. Remarkably, this intralayer [2 + 2] cycloaddition induces a dimensional change in the nanochannels, thereby modulating the CO2 absorption properties of the material.
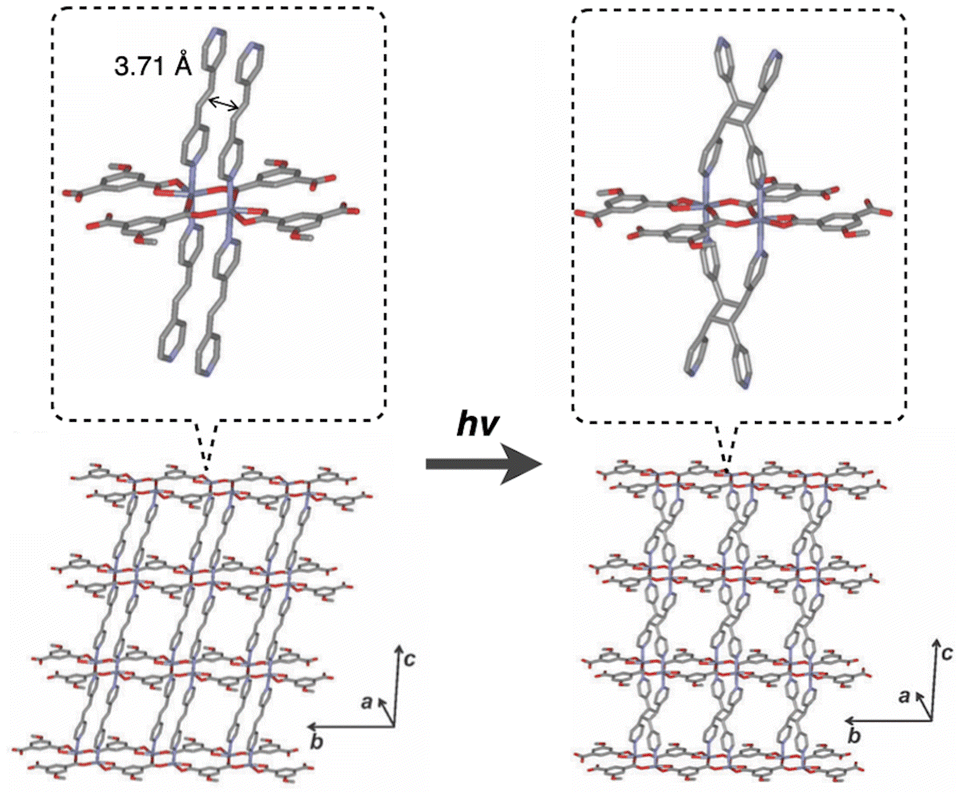 | ||
| Fig. 3 Structural conversion of [Zn(MeOip)(bpe)(DMF)] by photoinduced [2 + 2] cyclization. Adapted with permission from ref. 10. Copyright 2012 Royal Society of Chemistry. | ||
Inspired by the above pioneering research, examples of layered coordination polymers incorporating stilbene analogues as pendant ligands have been reported, [2 + 2] cycloaddition among which realize photoinduced interlayer coupling.11 This structural transformation enables modification of the coordination network's dimensionality (from 2D to 3D) and allows for the control of its flexibility.
J. J. Vittal et al. reported a layered coordination polymer [Zn(1,4-bdc)(2F-4spy)] composed of interdigitated stacks of 2D grid structure formed by Zn(II) and 1,4-benzenedicarboxylate (1,4-bdc).11b The surfaces of these layers are functionalized with a coordinating 4-(2-fluorostyryl)pyridine (2F-4spy) as pendants. In the crystal structure (Fig. 4), the olefinic CC bonds of 2F-4spy in adjacent layers are oriented in a crisscross manner, with a centroid distance of 4.12 Å, satisfying Schmidt's criteria. Upon UV irradiation, interlayer [2 + 2] cycloaddition occurs, realizing a photoconversion from the 2D layered structure to a 3D coordination network.
 | ||
| Fig. 4 Schematic representation of the structural conversion of [Zn(1,4-bdc)(2F-4spy)] by photoinduced interlayer [2 + 2] cycloaddition. Adapted with permission from ref. 11b. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KgaA. | ||
Similar structural changes have been leveraged to modify the physical and mechanical properties of crystals. L.-F. Wang et al. demonstrated that photoinduced [2 + 2] interlayer coupling in a Hoffmann-type 2D coordination polymer functionalized with 4-styrylpyridine (4spy) as pendants, [Fe(4spy)2{Ag(CN)2}2], leads to the formation of 3D coordination networks (Fig. 5).11c The centroid distance between the olefinic CC bonds of 4spy in adjacent layers is 3.84 Å, consistent with Schmidt's criteria. This photoinduced interlayer [2 + 2] coupling causes distortion in the coordination geometry of the Fe(II) center, altering its ligand field and consequently modifying the spin-crossover behavior of the crystals.
 | ||
| Fig. 5 Structural conversion of [Fe(4spy)2{Ag(CN)2}2] by photoinduced interlayer [2 + 2] cycloaddition. Adapted with permission from ref. 11c. Copyright 2019 Royal Society of Chemistry. | ||
The interlayer stacking mode (interlayer distance and displacement) of 2D coordination networks is known to depend on the adsorbed guest molecules. Examples linking such structural flexibility and photoreactivity have been reported.11e A layered coordination polymer, [Zn(bpdc)(4spy)] adopts distinct interlayer stacking modes depending on the adsorbed guest species. This variation modulates the distance between the nearest CC bonds, resulting in corresponding variations in photoreactivity under UV irradiation. In the absence of a guest molecule, the distance between the nearest CC bonds is small (3.9 Å), enabling a [2 + 2] cycloaddition reaction (Fig. 6a). In contrast, upon adsorption of DMF, the interlayer distance expands, increasing the nearest CC bond distance to 4.4 Å, which favors olefin/phenyl cyclization (Fig. 6b). Furthermore, when a bulkier guest molecule such as NMP is adsorbed, the nearest CC bond distance increases even further (10 Å), thereby preventing photochemical coupling between 4spy moieties. Furthermore, photoinduced interlayer coupling alters the gas adsorption behavior of the layered coordination polymer. Before coupling, a stepwise gas adsorption, accompanied with expansion in interlayer distance, is observed. However, after coupling, a typical Langmuir-type isotherm without remarkable structural transformation in the coordination polymer is observed due to the reduction of flexibility of the structure by interlayer coupling. This study exemplifies the correlation between the structural flexibility of layered coordination polymers and their stimulus-responsive behavior.
 | ||
| Fig. 6 Schematic representation of the photoreaction of 4spy within [Zn(bpdc)(4spy)] in the (a) absence and (b) presence of guest molecules. Adapted with permission from ref. 11e. Copyright 2019 American Chemical Society. | ||
Y.-X. Shi reported the utilization of an interlayer [2 + 2] coupling reaction in the layered coordination polymer [Zn(bdc)(3-F-spy)] (3-F-spy: 4-(3-fluorostyryl)pyridine) to achieve actuation in its single crystals.11d Upon photoirratiation from the [0−10] direction, the single crystal undergoes a shape transformation (Fig. 7). In the crystal structure, the olefinic CC groups are aligned with a distance of 3.76 Å making them well-suited for [2 + 2] cycloaddition. Detailed structural analysis revealed that the photoinduced interlayer [2 + 2] cycloaddition leads to changes in the dimensions of the unit cell, resulting in macroscopic distortions within the crystal. These distortions manifest as bending and, subsequently, a twisting motion that transforms the structure into a helical shape (Fig. 7b). Notably, the anisotropic nature of the photoreaction in crystals is effectively exploited. The reaction initiates from the irradiated surface and propagates through the crystal, creating a macroscopic gradient that drives the distortion.
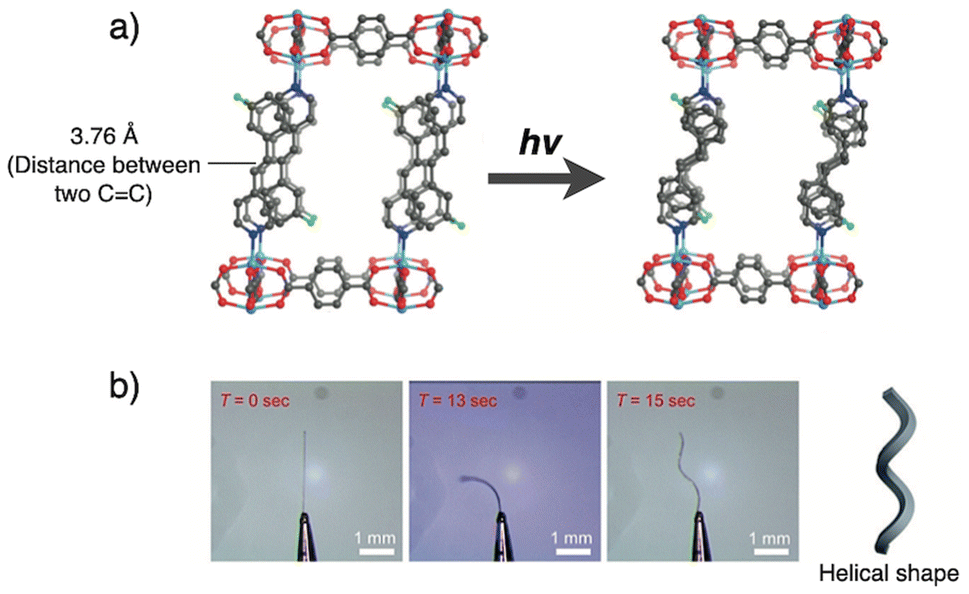 | ||
| Fig. 7 (a) Structural conversion of [Zn(bdc)(3-F-spy)] by photoinduced interlayer [2 + 2] cyclization and (b) optical microscope images of photo mechanical deformations of the crystal induced by photo irradiation. The crystal was fixed to a pin sample holder and photo irradiation was conducted from the left side on the image. Adapted with permission from ref. 11d. Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The [4 + 4] photocyclization reaction of anthracene derivatives, which leads to the formation of dianthracene, has also been utilized in the design of layered coordination polymers that undergo structural transformation upon photoirradiation.12 A notable characteristic of this reaction is its reversibility under UV irradiation or heating. From this perspective, anthracene and dianthracene can also be categorized as thermally responsive functional moiety discussed in the next section. Moreover, this transformation is accompanied by changes in emission color, realizing its potential for use as a luminescent material. For instance, L.-M. Zheng et al. constructed a layered coordination polymer, [Dy(NO3)3(depma2)1.5]·(depma2)0.5, composed of Dy(III) and depma2 (a dimer of 9-diethylphosphonomethylanthracene) as the skeleton of the 2D coordination network.12a Upon heating (140 °C), part of depma2 in crystal undergoes de-dimerization into dempa (9-diethylphosphonomethylanthracene), leading to in-plane structural changes from 2D coordination network to 1D chains, while UV irradiation at 365 nm induces re-dimerization, restoring the original structure (Fig. 8). The authors have also succeeded in photo-lithography, providing heterocrystals made of crystals with different emission colors by spatially irradiating the crystal with UV light.
 | ||
| Fig. 8 Photo and thermally induced dimerization and de-dimerization of anthracene derivatives in layered coordination polymers accompanied with change in the luminescent color. Adapted with permission from ref. 12a. Copyright 2023 Royal Society of Chemistry. | ||
In layered coordination polymer crystals, the mobility of constituent chemical species and their interactions or collisions with other molecules are precisely limited by the packing arrangements and isolation effects inherent to the crystal structure. These structural constraints enable precise control over chemical reactivity, including the stabilization and manipulation of highly unstable photochemical species within the crystals. H. Sato and S. Kitagawa et al. reported a layered coordination polymer (CID-N3) composed of interdigitated stacks of 2D nanosheets of [Zn(N3-ipa)(bpy)] (N3-ipa: 5-azidoisophthalate) with surfaces functionalized by photolabile azide (N3) moieties.13 Upon UV irradiation, photolysis of the azide groups, generating highly reactive nitrene (N), which is stabilized by the isolation effects of the coordination network (Fig. 9). Remarkably, this layered coordination polymer exhibits efficient gas absorption under UV irradiation in O2 or CO atmospheres. The generated nitrene reacts with the gas molecules to form NO or NCO species, as confirmed by SXRD analysis of the resulting crystals. This study represents the first example of photoinduced gas sorption realized through the specific reactivity of photolabile moieties confined within a layered coordination polymer.
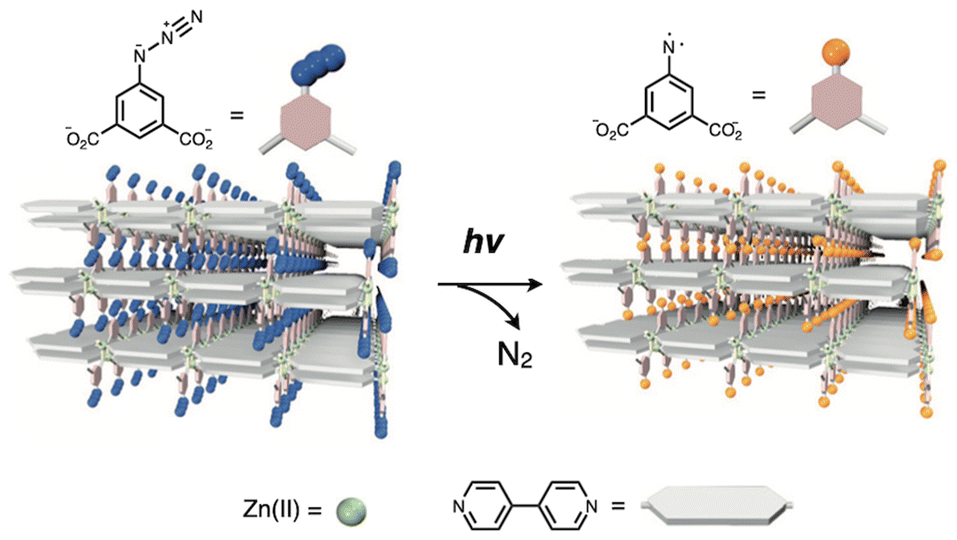 | ||
| Fig. 9 Schematic representation of the conversion of azide (N3) into nitren (N) by photoirradiation of [Zn(N3-ipa)(bpy)]. Adapted with permission from ref. 13. Copyright 2010 Springer. | ||
As discussed earlier, the photoresponsiveness of layered coordination polymers predominantly relies on photoreactive functional moieties that undergo covalent bond formation or cleavage upon photoexcitation. However, photo-responsive behavior arising from photoinduced electron transfer between neighbouring components in layered coordination polymers has also been reported.14 These photoreactions have been predominantly studied for chromic changes in crystal, while there have also been reports of their application in inducing structural and morphological transformations. J. Xie and S. Wang et al. identified a luminescent layered coordination polymer consisting of stacked anionic porous coordination networks [(UO2)2(ox)3]2− (ox: oxalate) intercalated with protonated 1,10-phenanthroline ([Hphen]+) (Fig. 10a and b).14d Upon photoexcitation, charge transfer between [(UO2)2(ox)3]2− and [Hphen]+ efficiently proceeded, generating radicals, which leads to quenching of the material's luminescence.
 | ||
| Fig. 10 Crystal structures of [Hphen]2[(UO2)2(ox)3]. Views from (a) above and (b) side of the 2D coordination network. (c) Photographs of the crystal after continuous UV (365 nm) irradiation. Adapted with permission from ref. 14d. Copyright 2019 Royal Society of Chemistry. | ||
Single-crystal structural analysis revealed that photoirradiation of the crystal induced an expansion in the distance between [Hphen]+ and [(UO2)2(ox)3]2− accompanied by intralayer contraction of the coordination network. Notably, continuous irradiation led to exfoliation of the crystal along the layers of the 2D coordination network, resulting in the formation of nanosheets (Fig. 10c). This study represents a pioneering example of harnessing the photo-responsive properties of the constituent components to facilitate nanosheet formation.
These examples of crystal structures with precise arrangement of photo-functional species realized development of photo-responsive coordination polymers, but modulation by heat is also a powerful strategy that could modify the stacking mode and actions of chemicals within the layers.
Thermally-responsive layered coordination polymers
Thermal energy which is closely associated with the vibrational and rotational motion of molecules, holds significant potential as an energy source for tuning the function of various solid-state materials, including layered coordination polymers.22,25 However, reports focusing on the use of such thermal rotation or vibration of components to influence the functionality of layered coordination polymers remain limited. This is likely due to the lack of clear design strategies compared to photoresponsive systems. Here, we present several examples where thermally-responsive properties are utilized for guest adsorption/permeation15,16 or for the control of crystal morphology.17,18S. Ma and H. C. Zhou et al. reported the thermally dependent gas adsorption properties of layered coordination polymer MAMS-1 [Ni8(5-bbdc)6(μ3-OH)4] (5-bbdc: 5-tert-butyl-1,3-benzenedicarboxylate), which is composed of a 2D coordination network formed by intrication of [Ni8(μ3-OH)4] clusters and 5-bbdc ligands (Fig. 11).15 MAMS-1 features two distinct types of nanopores: hydrophilic nanochannels located at the interface between 2D coordination networks and hydrophobic chambers that run across the layers. Access to the hydrophobic chambers is regulated by gates at the hydrophobic/hydrophilic interface, where two 5-bbdc ligands orient their tert-butyl groups toward each other.
 | ||
| Fig. 11 (a) Crystal structure of MAMS-1. Two pack of trilayers along the c axis form the hydrophobic chambers. (b) Top and (c) side views of the hydrophobic chamber. (d) Schematic representation of the mechanism of the temperature dependent gating effect in MAMS-1. Adapted with permission from ref. 15. Copyright 2007 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
These gates are stabilized by van der Waals interactions, effectively controlling access to the hydrophobic chambers. The hydrophilic nanochannels in MAMS-1 serve as pathways for gas molecules to access the hydrophobic nanochambers, with temperature playing a critical role in regulating this accessibility (Fig. 11d). At low temperature, MAMS-1 demonstrates high selectivity for small gas molecules, such as H2, since the gate permits access only to small-sized gas molecules. In contrast, at higher temperatures, the rotation and vibrational motion of the tert-butyl groups widens the gate, allowing larger gas molecules to access the hydrophobic pores. This thermoresponsive behavior enables MAMS-1 to function as a size-adjustable molecular sieve, capable of tuning its size selectivity for gas molecules based on temperature. Notably, the thermoresponsive gas adsorption properties of MAMS-1 arise from the synergistic effect of two key factors: (1) anisotropic and efficient gas diffusion along the in-plane direction of the layered coordination polymer, and (2) interlayer gas diffusion controlled by the temperature-dependent rotational and vibrational dynamics of the framework.
A notable feature of layered coordination polymers is their ability to be exfoliated into nanosheets, making them ideal for applications such as gas separation membranes. X. Wang and D. Zhao successfully developed thermoresponsive gas separation membranes using the same layered coordination polymer (MAMS-1).16 MAMS-1 nanosheets were prepared by exfoliating the crystals from a freeze–thaw method in a solvent. These nanosheets were then aligned on a porous aluminum oxide substrate to form the membrane, which exhibited unique temperature-dependent gas permeation selectivity (Fig. 12).
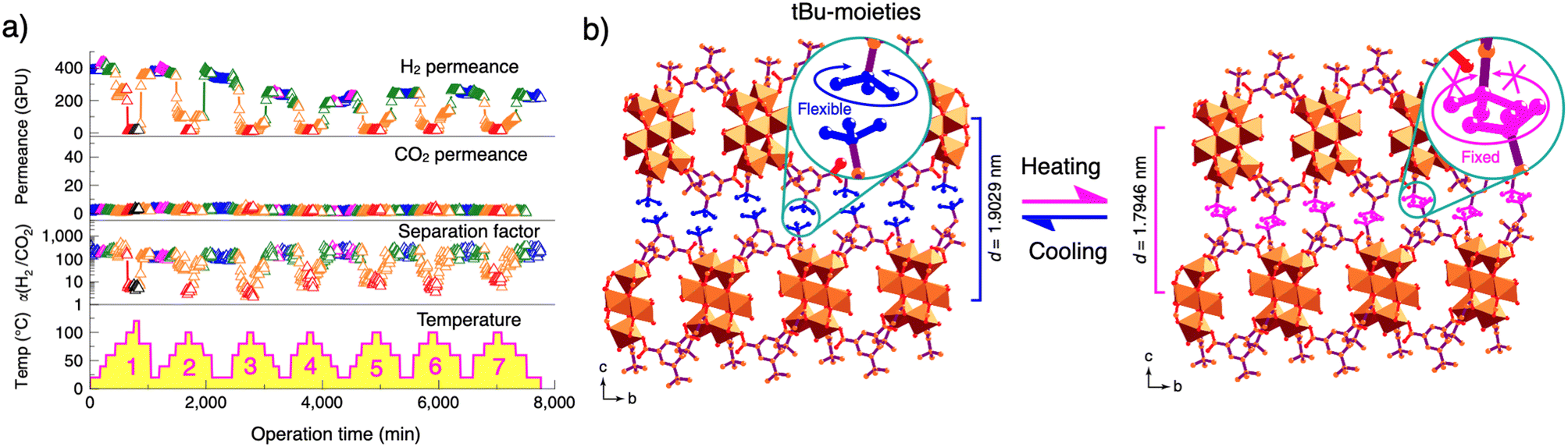 | ||
| Fig. 12 (a) Gas permeance and H2/CO2 separation factors of the MAMS-1 membrane under seven heating/cooling cycles. Different colours represent various temperatures: blue, 20 °C; magenta, 40 °C; green, 60 °C; orange, 80 °C; red, 100 °C and black, 120 °C. (b) Illustration of the shrinkage (during heating) and expansion (during cooling) of interlayer distance in MAMS-1. The freely rotated tert-butyl groups are highlighted in blue and the frozen ones are highlighted in magenta. Adapted with permission from ref. 16. Copyright 2017 Springer. | ||
Around 20 °C, the nanosheets demonstrate sixfold higher permeability for H2 compared to CO2. However, at elevated temperatures (>100 °C), the H2 permeability decreases to a level comparable to CO2. The gas permeability can be reversibly tuned by temperature changes (Fig. 12a). Variable-temperature PXRD analysis revealed that the distance between the tert-butyl moieties in adjacent layers decreased with increasing temperature, resulting in a reduction in their free rotational motion due to the shrinking interlayer distance (Fig. 12b).
This structural change caused by the restricted rotation of the tert-butyl groups hinders the interlayer diffusion of H2 molecules through the hydrophobic chamber of MAMS-1, leading to a reduction in H2 permeability at high temperature.
The strategy of incorporating flexible functional groups into ligands has gained significant attention for the development of crystalline materials capable of undergoing structural changes.22 Long-chain alkyl compounds, such as lipids and liquid crystals, are known to self-assemble into flexible and dynamic structures in condensed phases, driven by weak intermolecular interactions, solvophobic effects, and excluded volume effects.26 These assemblies exhibit unique phase transition behaviors originating from the conformational disordering/ordering (i.e. melting and crystallisation) of alkyl chains, which significantly affect molecular mobility, flexibility, and fluidity in soft materials. While the thermal responsiveness of long alkyl chains has been extensively explored in soft materials, their potential as thermally responsive domains in layered coordination polymers has also been investigated.
S.-H. Lee and co-workers synthesized a series of layered coordination polymers composed of Zn(II) and terephthalate modified with long alkyl chains.17 By optimizing the reaction conditions, the authors developed several crystalline coordination polymer phases. Among them, they demonstrated the fusible properties of the coordination polymer ZnC14-L2 which consists of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of Zn(II) and telephthalate with two tetradecyloxy groups on its aromatic ring (Fig. 13). Although the lack of SXRD data prevented conclusive confirmation of a two-dimensional coordination network, PXRD and IR measurements suggested a stacked structure, with soft alkyl segments intercalated between rigid coordination polymer layers.
:1 stoichiometry of Zn(II) and telephthalate with two tetradecyloxy groups on its aromatic ring (Fig. 13). Although the lack of SXRD data prevented conclusive confirmation of a two-dimensional coordination network, PXRD and IR measurements suggested a stacked structure, with soft alkyl segments intercalated between rigid coordination polymer layers.
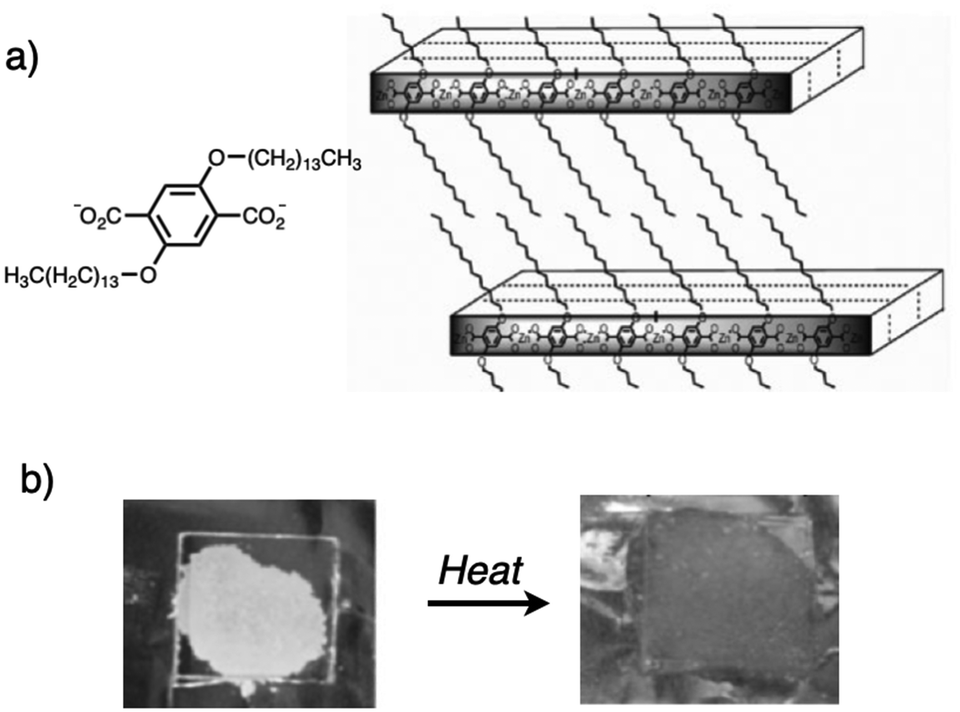 | ||
| Fig. 13 (a) Proposed structure for ZnC14-L2 phases and (b) its melting process on hot plate (130 °C). Adapted with permission from ref. 17b. Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Long-chain alkyl groups act as thermoresponsive domains, providing fusible properties to the coordination polymers. Upon heating ZnC14-L2 to 130 °C, the colorless crystalline polymer melts into a transparent liquid, which can be re-solidified upon cooling while retaining its layered nanostructure. This fusible character enables the material to be processed into fibers, demonstrating a method for controlling the processability of layered coordination polymers through the incorporation of thermally responsive domains.
Recently, our group synthesized a paraffinic layered coordination polymer (ZnC16) by complexation of Zn(OAc)2·H2O with isophthalate functionalised with a hexadecyloxy chain (C16) on its 5 position (Fig. 14a).18 This coordination polymer features a layer-by-layer structure, consisting of a rigid two-dimensional coordination network and an assembly of interdigitated alkyl chains that serve as thermally responsive moiety. Single crystals of ZnC16 exhibit a reversible crystal-to-crystal thermal phase transition (Fig. 14b–d), with a transition temperature around 150–160 °C. This phase transition is dominated by melting/crystallization of the long alkyl chains, as confirmed by SXRD, DSC, and Raman spectroscopy. Notably, variable temperature single crystal structural analysis revealed that the two-dimensional periodic structure of the coordination network remains intact during the phase transition, while the interlayer distance expands by up to 18%. Such anisotropic expansion/contraction of the crystal structure can be attributed to the combination of contrasting characteristics: the rigidity of the coordination polymer network and the flexibility of the long alkyl chain assemblies. Moreover, this crystal phase transition is accompanied by macroscopic morphological changes, including elongation and delamination into sheet-like structures, indicating significant crystal shape transformations (Fig. 14e). These thermally responsive coordination polymers, combining flexible alkyl chains with rigid coordination frameworks, hold great promise for thermally induced control over processability, molecular diffusivity within the structure, and related functionalities, all while preserving the intrinsic properties of the periodic coordination network.
 | ||
| Fig. 14 (a) Molecular structure of C16 and (b–d) crystal structures of the coordination polymer ZnC16. (b) Structure of the 2D coordination polymer network (20 °C, low temperature phase). Alkyl chains are omitted for clarity. Packing structures viewed along the a-axis of (c) the low temperature phase at 20 °C and (d) high temperature phase at 177 °C, respectively. Atoms of the n-hexadecyloxy chains of each C16 in high temperature phase (d) are disordered over two sites with 50% occupancy. (e) Microscopic images on a variable-temperature stage illustrating the crystal-to-crystal deformation of ZnC16. Heating from 140 °C to 160 °C and cooling from 160 °C to 140 °C at 20 °C min−1. Adapted with permission from ref. 18. Copyright 2022 Royal Society of Chemistry. | ||
As mentioned earlier, despite the use of long alkyl chains, rationally designing the vibrational and rotational motions of molecules in crystals remains challenging, as well as the design of thermoresponsive functional groups is still underdeveloped. On the other hand, heat serves as a universal and versatile external stimulus that can be applied to all chemical species. Properties of layered coordination polymers, such as gas adsorption and magnetism, are significantly influenced by temperature. Therefore, considering heat as a stimulus to control the functions of layered coordination polymers could open up new avenues for developing advanced functionalities. From this perspective, a detailed investigation of thermal responsiveness in existing layered coordination polymers may yield new concepts and drive further expansion of their applications in the future.
Conclusions
The development of layered coordination polymers with tunable structures and functions responsive to external stimuli has been realized through the integration of diverse metal ions and ligands. The incorporation of stimulus-responsive sites is particularly crucial for modulating their functions. In particular, as seen in most of the recent examples presented in this paper, incorporating stimulus-responsive moieties at the interfaces of 2D coordination networks, which govern interlayer interactions, is the most effective approach for imparting stimulus-responsive functions by leveraging the inherent flexibility and crystal processability of layered coordination polymers. However, the rational synthesis of soft crystals incorporating stimulus-responsive moieties remains a challenge, due to the difficulty in predicting or designing crystal structures and the suppression of molecular motion within the crystalline environments. Despite these challenges, innovative design strategies rooted in coordination chemistry, combined with the inherent flexibility of layered coordination polymers, offer promising solutions.Looking ahead, advancing crystal design methodologies to enable rational structural changes of stimulus-responsive moieties, even in densely packed crystals, represents an essential future direction. Specifically, the integration of stimuli-responsive 2D aggregates, such as soft alkyl chains, into 2D coordination polymers offers a powerful approach for fabricating functional, stimuli-responsive layered coordination polymers. These advances are expected to open-up new possibilities and expand the applications of this versatile class of materials.
Author contributions
Kenichiro Omoto – conceptualization, funding acquisition, writing original draft, review and editing. Gwénaël Rapenne – conceptualization, funding acquisition, project administration, validation, supervision, review and editing.Data availability
No primary research results have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Scientific Research (A) (22H00325; G. R.), Early-Career Scientists (21K14478; K. O.), and a Grant-in-Aid for Scientific Research (C) (23K04768; K. O.). The CNRS is acknowledged for its support through the International Research Project POEMES (2024–2028). The University of Toulouse and NAIST are also warmly thanked for providing a crossed position to G. R.References
-
(a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed
; (b) W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688–764 CrossRef CAS PubMed
-
(a) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 26, 2334–2375 CrossRef PubMed
-
(a) M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed
- G. Chakraborty, I.-H. Park, R. Medishetty and J. J. Vittal, Chem. Rev., 2021, 121, 3751–3891 CrossRef CAS PubMed
-
(a) K. Biradha, Y. Hongo and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3395–3398 CrossRef CAS
-
(a) Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed
- Z. Liu, L. Zhang and D. Sun, Chem. Commun., 2020, 56, 9416–9431 RSC
- H. Sato, R. Matsuda, M. H. Mir and S. Kitagawa, Chem. Commun., 2012, 48, 7919–7921 RSC
-
(a) R. Medishetty, L. L. Koh, G. K. Kole and J. J. Vittal, Angew. Chem., Int. Ed., 2011, 50, 10949–10952 CrossRef CAS PubMed
-
(a) X.-D. Huang, B.-K. Hong, G.-H. Wen, S.-H. Li and L.-M. Zheng, Chem. Sci., 2023, 14, 1852–1860 RSC
- H. Sato, R. Matsuda, K. Sugimoto, M. Takata and S. Kitagawa, Nat. Mater., 2010, 9, 661–666 CrossRef CAS PubMed
-
(a) X.-D. Yang, C. Chen, Y.-J. Zhang, L.-X. Cai and J. Zhang, Inorg. Chem. Commun., 2015, 60, 122–125 CrossRef CAS
- S. Ma, D. Sun, X.-S. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2007, 46, 2458–2462 CrossRef CAS PubMed
- X. Wang, C. Chi, K. Zhang, Y. Qian, K. M. Gupta, Z. Kang, J. Jiang and D. Zhao, Nat. Commun., 2017, 8, 14460 CrossRef CAS PubMed
-
(a) E.-Y. Choi, C. Gao, H.-J. Lee, O.-P. Kwon and S.-H. Lee, Chem. Commun., 2009, 7563–7565 RSC
- K. Omoto, S. Aoyama, T. Galica, E. Nishibori, S. Katao, K. Yasuhara and G. Rapenne, Dalton Trans., 2022, 51, 17967–17972 RSC
-
(a) N. F. Sciortino and S. M. Neville, Aust. J. Chem., 2014, 67, 1553–1562 CrossRef CAS
-
(a) G. A. Leith, C. R. Martin, A. Mathur, P. Kittikhunnatham, K. C. Park and N. B. Shustova, Adv. Energy Mater., 2022, 12, 2100441 CrossRef CAS
- V. Ramamurthy and K. Venkatesan, Chem. Rev., 1987, 87, 433–481 CrossRef CAS
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS
- S. K. Park and Y. Diao, Chem. Soc. Rev., 2020, 49, 8287–8314 RSC
- M. Sorai and K. Saito, Chem. Rec., 2003, 3, 29–39 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |