
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of aromatic to quinoidal transformation on the degradation kinetics of imine-based semiconducting polymers†
Naoya
Nozaki‡
a,
Azalea
Uva‡
 b,
Takashi
Iwahashi
a,
Hidetoshi
Matsumoto
a,
Helen
Tran
*bcd and
Minoru
Ashizawa
*a
b,
Takashi
Iwahashi
a,
Hidetoshi
Matsumoto
a,
Helen
Tran
*bcd and
Minoru
Ashizawa
*a
aDepartment of Materials Science and Engineering, Institute of Science Tokyo, Meguro-ku, Tokyo, 152-8552, Japan. E-mail: ashizawa.m.aa@m.titech.ac.jp
bDepartment of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada. E-mail: tran@utoronto.ca
cDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada
dAcceleration Consortium, University of Toronto, Toronto, Ontario M5S 3H6, Canada
First published on 13th December 2024
Abstract
Degradable semiconducting polymers featuring acid-labile imine bonds are often investigated for use in transient electronics. However, the structure–property relationship of these polymers, particularly regarding degradation kinetics, remains underexplored. Herein, we designed and synthesized two imine-based semiconducting polymers which undergo an aromatic to quinoidal transformation upon acidification, leading to slower degradation rates compared to previously reported imine-based polymers. By utilizing a thieno[3,2-b]thiophene (TT)-inserted thienoisoindigo (TII)-dimer unit (TT-(TII-CHO)2) and two diamines, p-phenylenediamine (PD) and 2,6-naphthalenediamine (2,6ND), we generated polymers p(TT-TII-PD) and p(TT-TII-2,6ND). The insertion of the TT unit between TII units results in high lying HOMO and low lying LUMO levels, facilitating a shift from an aromatic to quinoidal structure in the polymer backbone. Using ultraviolet-visible-near infrared (UV-vis-NIR) spectroscopy, infrared (IR) spectroscopy, and density functional theory (DFT) calculations, we investigated the influence of the quinoidal form on the degradation properties of these polymers. Notably, complete degradation of p(TT-TII-2,6ND) required over 30 days, indicating enhanced stability towards acid compared to previously reported TII-based polymers without the TT unit. Additionally, the protonated polymers demonstrated improved electrical properties compared to the pristine polymers, with field-effect transistor mobilities in the order of 10−2 cm2 V−1 s−1. These findings highlight the importance of quinoidal stability in modulating lifetimes and improving charge carrier transport in imine-based semiconducting polymers.
Introduction
Understanding structure–property relationships in polymers is paramount for imparting features such as flexibility, durability or thermal stability into a given material. In the context of electron-conducting organic polymers, structural modifications can be made to introduce electronic, biocompatible or degradable properties, opening avenues for these materials to be used in a wide range of applications. For example, the synthesis of degradable semiconducting polymers with various molecular architectures have garnered much attention in recent years due to their potential to be used in transient electronics.1–4 These materials incorporate π-conjugated, chemically labile bonds such as imines,5–9 1,2,4-oxadiazoles10 or imidazoles11–13 into polymer backbones to achieve degradability under predetermined conditions. However, few reports exist on how structural modifications influence the rate at which these semiconducting polymers degrade – a knowledge gap that is imperative for the future integration of these materials into stimuli-responsive applications.Imine-based semiconducting polymers are one class of degradable organic polymers that are often studied due to their modest charge carrier mobilities and triggerable degradation under acidic conditions.8,14–16 With a multitude of imine-based polymers synthesized in the last few decades, including donor–acceptor systems,8,9,17 researchers have now shifted their attention to understanding how this class of polymers degrade under acidic conditions,15 with a greater focus on structure–property relationships. For example, Bao and coworkers examined the influence of side chain modification on degradation kinetics14 as well as the effect of monomer structure on the concentration of acid required to degrade imine-based polymers.18 Analogously, Collier and coworkers reported a comprehensive degradation study on their poly(azomethine) system,19 noting the difference in degradation kinetics based on solvent polarity and acid strength.20 However, to the best our knowledge, one factor that is rarely studied is the impact of aromatic to quinoidal transformations on the rate of imine bond hydrolysis. For π-conjugated polymers, the quinoid structure is advantageous for charge-carrier mobility as it extends π-electron delocalization and promotes planarity in the polymer backbone.21–24 Coupling high charge-carrier mobility with predictable degradability has been a long withstanding challenge, with the stabilization of the quinoidal structure possibly being the key to achieving both. Therefore, we chose to focus our efforts on selecting a monomer that favors the quinoidal form, notably the thienoisoindigo (TII) moiety,25–28 a thiophene analogue of isoindigo (IIG). Previously, we reported two TII-based chemically recyclable semiconducting polymers, which utilized a dialdehyde TII monomer that could be recovered for repolymerization.29 Upon acid hydrolysis, these polymers demonstrated a bathochromic shift in their absorption spectrum during degradation, for which we attributed to a quinoidal intermediate forming upon protonation of the imine bond. As such, we chose to further investigate this quinoidal intermediate by creating a thieno[3,2-b]thiophene (TT)-inserted TII-dimer (TT-(TII-CHO)2), for which we hypothesized could extend the lifetime of the quinoidal intermediate (Fig. 1). The inserted TT unit not only favors the quinoid structure but is also beneficial for improving charge carrier transport in donor–acceptor polymers.30–32
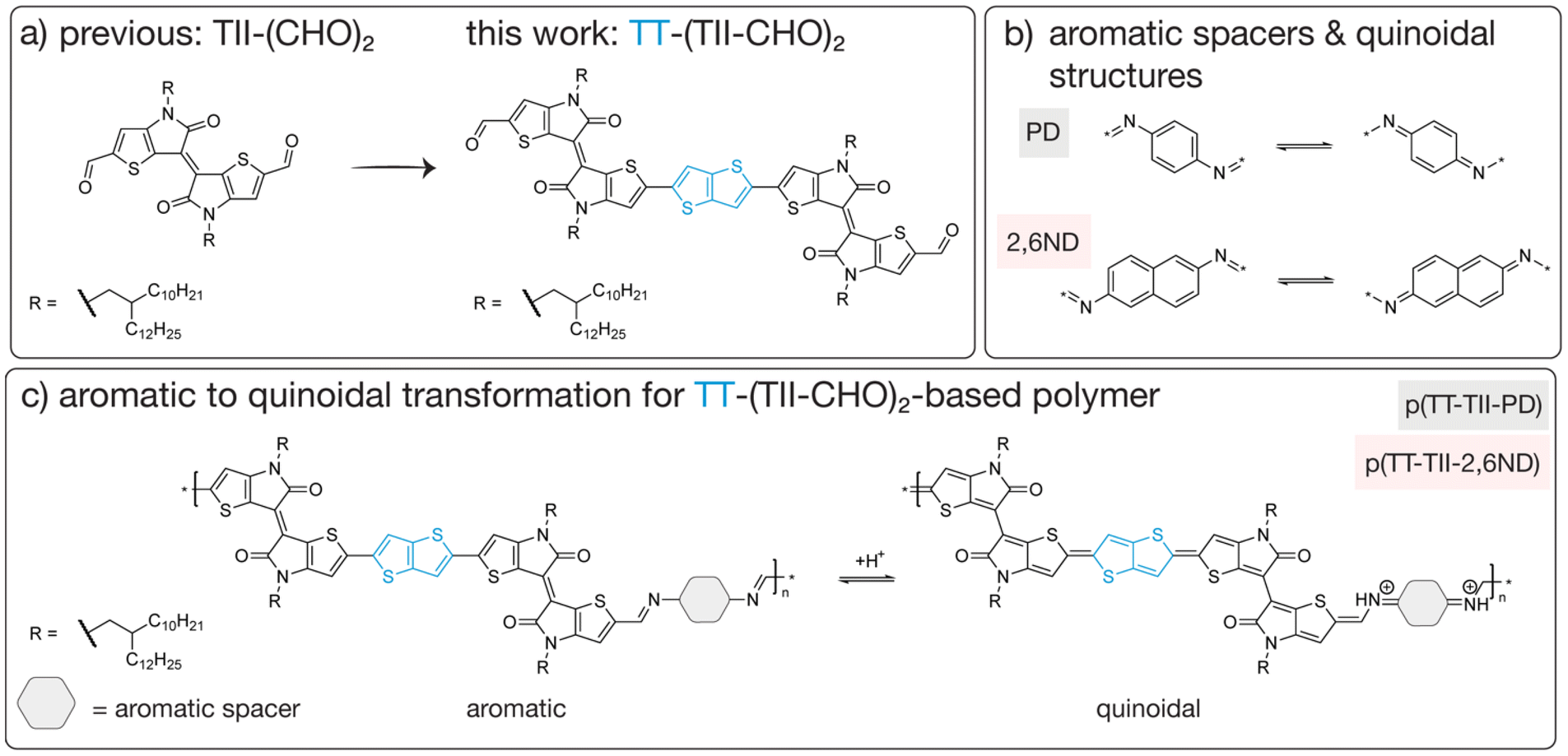 | ||
| Fig. 1 (a) Overview of previous work vs. this work focusing on the TT unit inserted TII dimer structure bearing aldehyde groups. (b) Aromatic spacers PD and 2,6ND used in this work and their quinoidal structures. (c) Aromatic to quinoidal transformation via acid-mediated protonation for polymers p(TT-TII-PD) and p(TT-TII-2,6ND). | ||
In this study, we designed and synthesized two degradable π-conjugated polymers, p(TT-TII-PD) and p(TT-TII-2,6ND), to investigate the effect of aromatic to quinoidal transformations on degradation lifetimes and charge carrier mobilities (Fig. 1). We report the chemical, thermal, and photophysical properties of these polymers as well as the charge carrier transport through field-effect transistor (FET) fabrication. Furthermore, we examined the degradation mechanism of these polymers under acidic conditions using ultraviolet-visible-near infrared (UV-vis-NIR) spectroscopy, infrared (IR) absorption spectroscopy, and density functional theory (DFT) calculations. Our results indicate the formation of quinoidal structures for both polymers upon acidification, with significantly slower degradation rates compared to our previously reported polymers without the TT unit. We attribute the delayed degradation to the TT insertion, which is known to facilitate the aromatic to quinoidal transformation,33 resulting in a rigid quinoidal structure capable of stabilizing positively charged protonated species along the polymer backbone. Further, we demonstrate that these new polymers exhibit comparable charge carrier mobilities to our previously reported TII-based polymers29 and could be improved by one order of magnitude through doping of polymer thin films with p-toluenesulfonic acid (PTSA). These findings not only provide new structure–property relationship guidelines for customizing degradation rates, but also support acid-mediated doping to improve charge carrier mobility in semiconducting polymers. Our approach of utilizing aromatic to quinoidal transformations to tailor degradation rates holds substantial promise in broadening the applicability of imine-based semiconducting polymers for transient electronics.
Results and discussion
Synthesis and polymer characterization
The synthesis of TT-(TII-CHO)2 and two TT-TII imine-based polymers is illustrated in Fig. 2, with experimental details and complete characterizations described in the ESI (Fig. S1–S8†). To improve the solubility of the polymers for solution processing of thin films,7,34 2-decyl-1-tetradecyl alkyl chains were installed on the TII unit used in this study. To synthesize the TT-(TII-CHO)2 unit, first monoaldehyde-modified TII-CHO was prepared via a Vilsmeier–Haack reaction using oxalyl chloride. Next, TII-CHO was selectively brominated at the α-position of the other thiophene ring using N-bromosuccinimide to obtain Br-TII-CHO. Finally, a Stille cross-coupling26 between Br-TII-CHO and bistrimethylstannylated TT unit was performed to yield the final monomer, TT-(TII-CHO)2.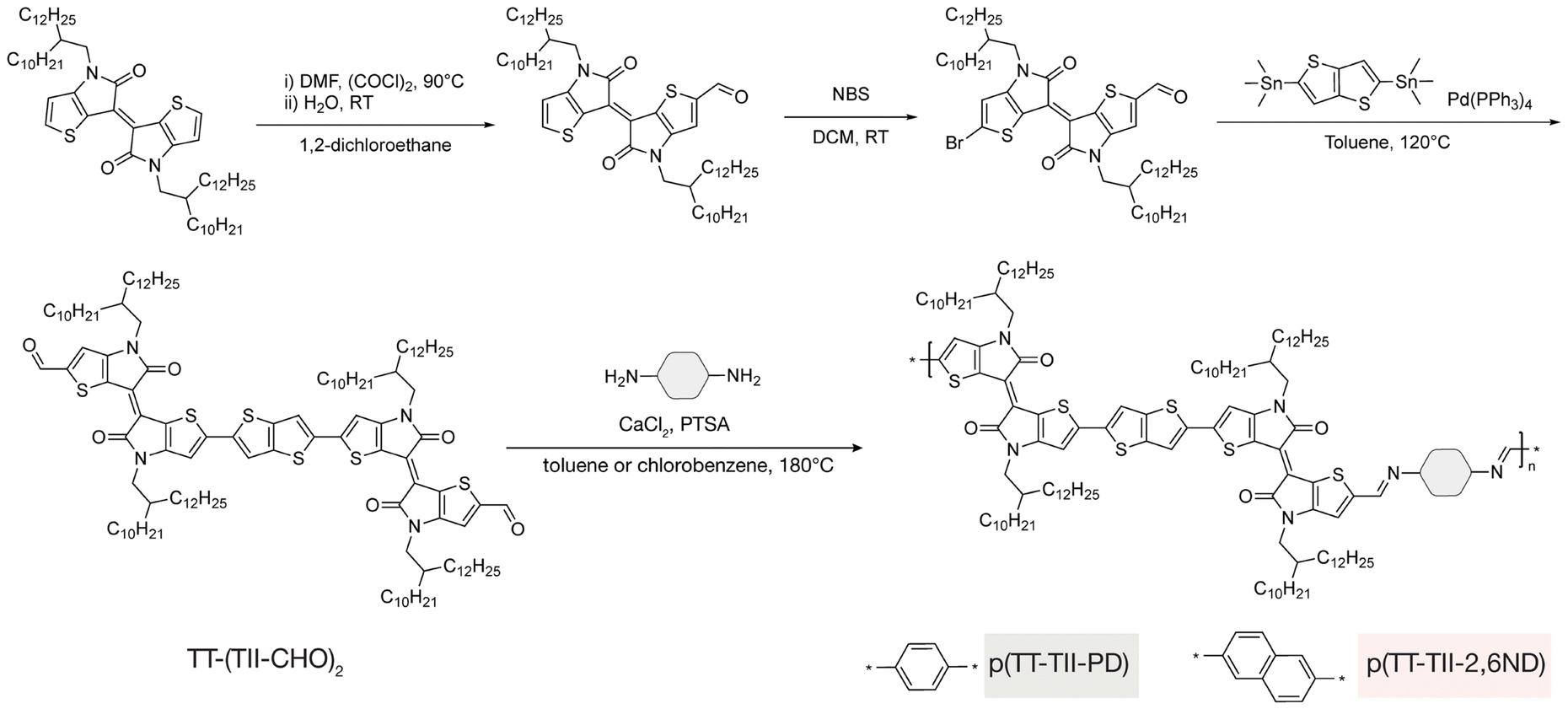 | ||
| Fig. 2 Synthetic scheme of TT-(TII-CHO)2 and target polymers p(TT-TII-PD) and p(TT-TII-2,6ND). | ||
The polymers were synthesized via an imine polycondensation reaction using p-toluenesulfonic acid (PTSA) as the catalyst and an excess of calcium chloride (CaCl2) as the drying agent.7,29 Taking into consideration the solubilities of diamines PD and 2,6ND, toluene was used as the polymerization solvent for p(TT-TII-PD) and chlorobenzene for p(TT-TII-2,6ND). The polymers were precipitated in methanol, followed by a Soxhlet extraction to remove impurities and dimers. The molecular weight (Mn) and dispersity (Đ) of the polymers were determined using high-temperature gel permeation chromatography (HT-GPC) in 1,2,4-trichlorobenezene at 135 °C against polystyrene standards (Table 1). The Mn values for p(TT-TII-PD) and for p(TT-TII-2,6ND) were both 7.6 kDa, with Đ values of 1.4 and 1.6, respectively, with notably small dispersity values compared to the typical range for poly(azomethine)s35 prepared via imine polycondensation reactions as a result of the small degrees of polymerization (DP) obtained for both polymers. The results of thermogravimetric analysis (TGA) (Fig. S9a† and Table 1) indicate reliable thermal stability for both polymers with thermal decomposition temperatures (5% weight loss) above 370 °C, which is greater than our previously reported TII-based polymers.29 We attribute the greater thermal decomposition temperature to the enlarged π-conjugation of the monomer due to the TT unit insertion. A glass transition temperature of 91.7 °C was found via differential scanning calorimetry (DSC) for polymer p(TT-TII-PD) (Fig. S9b†); with additional thermal transitions observed around 200–250 °C for both polymers, for which we hypothesize are related to melting transitions.
| M n (kDa) | M w (kDa) | Đ | DPa | T g (°C) |
T
d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (°C) b (°C) |
|
|---|---|---|---|---|---|---|
| a The degree of polymerization (DP) was calculated from Mn results from high-temperature gel permeation chromatography (HT-GPC). b T d was determined from 5 wt% loss temperature in the TG curve. | ||||||
| p(TT-TII-PD) | 7.6 | 10.5 | 1.4 | 4 | 91.7 | 384 |
| p(TT-TII-2,6ND) | 7.6 | 11.9 | 1.6 | 3 | N/A | 381 |
Optical and electrochemical properties
The optical and redox properties for both polymers were examined by UV-Vis-NIR absorption spectroscopy and cyclic voltammetry (CV), with the results summarized in Table 2 and Fig. S10.† In solution, p(TT-TII-PD) and p(TT-TII-2,6ND) exhibited similar absorption profiles, including a weak absorption band from 300 nm to 500 nm attributed to a n → π* transition from the imine bond and a strong absorption band from 600 nm to 1100 nm, attributed to the elongated π-conjugated backbone.36 In the solid state, a planarized polymer backbone yields red-shifted absorption profiles of both polymers, suggesting enhanced intermolecular interactions. The optical energy gaps of p(TT-TII-PD) and p(TT-TII-2,6ND) determined from the onset of absorption were 1.17 eV and 1.14 eV, respectively (Table 2). This small energy band gap is typically observed for TII-based polymers.26| Solution | Thin film | ||||||
|---|---|---|---|---|---|---|---|
| λ solmax (nm) | λ solonset (nm) |
E
solga (eV) |
λ filmonset (nm) |
E
filmgb (eV) |
E
HOMOc (eV) |
E
LUMOd (eV) |
|
| a E solg was calculated from λsolonset. b E filmg was calculated from λfilmonset. c E HOMO was determined using the onset curve of CV. d E LUMO was calculated using ELUMO = EHOMO + Efilmg. | |||||||
| p(TT-TII-PD) | 860.6 | 999.8 | 1.24 | 1055.9 | 1.17 | −5.13 | −3.96 |
| p(TT-TII2,6ND) | 860.8 | 993.7 | 1.25 | 1086.1 | 1.14 | −4.95 | −3.81 |
In the CV curves, both polymers exhibited an irreversible oxidation step, and a small irreversible reduction step, similar to our previously reported polymers.29 Hence, EHOMO for each polymer was estimated from the onset of the oxidation step in the CV curves, and ELUMO was calculated using the difference between EHOMO and the optical energy gaps for each polymer thin film. From the oxidation onset potentials referenced to the Fc/Fc+ couple, EHOMO and ELUMO were estimated to be −5.13 eV and −3.96 eV for p(TT-TII-PD) and −4.95 eV and −3.81 eV for p(TT-TII-2,6ND), respectively (Table 2). Compared to our previously reported polymers,29 these results indicate that the TT unit insertion elevates the HOMO level and lowers the LUMO level, resulting in a smaller HOMO–LUMO energy gap. When considering the work function is about −5.1 eV for Au electrodes utilized in FET devices,37 both polymers are expected to show p-channel performance since HOMO levels are close to −5 eV with reducing charge carrier injection barrier.
Degradation studies
In general, the formation of an imine bond is reversible in the presence of acid, with the hemiaminal intermediate being unstable. Therefore, it is necessary to consider the stability of a protonated imine bond when studying poly(azomethine) degradation rates. To determine the types of chemical bond present during protonation of polymer samples, IR spectroscopy using the KBr pellet method was employed (Fig. 3a and b). To prepare the protonated polymer samples, 2 mg of pristine polymers were dissolved into 1 mL of PTSA/chloroform solution (14.7 mmol L−1) and casted on a glass substrate. Following the evaporation of chloroform, the polymer solids were peeled off and used to make KBr pellets. Since both polymers and TT-(TII-CHO)2 monomer showed a moderate peak around 1690–1670 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration on the cyclic amide of TII, all spectra were normalized utilizing this peak. Both polymers exhibited a peak around 1600–1550 cm−1 corresponding to CN stretching vibration of the imine moiety,38,39 resulting from the polycondensation between the aldehyde and amine groups. In the profiles of protonated p(TT-TII-PD) and protonated p(TT-TII-2,6ND), new peaks between 1660–1600 cm−1 appeared (1627 cm−1 and 1635 cm−1 for protonated p(TT-TII-PD) and protonated p(TT-TII-2,6ND), respectively), which were not observed in pristine polymers nor in TT-(TII-CHO)2. These newly appeared peaks are attributed to the structural changes derived from the protonation of polymers corresponding to CNH+ stretching vibrations. The differences in peak shifts for protonated p(TT-TII-PD) and protonated p(TT-TII-2,6ND) suggest that p(TT-TII-2,6ND) forms a stronger protonated imine bond, leading to a delayed degradation rate.
O stretching vibration on the cyclic amide of TII, all spectra were normalized utilizing this peak. Both polymers exhibited a peak around 1600–1550 cm−1 corresponding to CN stretching vibration of the imine moiety,38,39 resulting from the polycondensation between the aldehyde and amine groups. In the profiles of protonated p(TT-TII-PD) and protonated p(TT-TII-2,6ND), new peaks between 1660–1600 cm−1 appeared (1627 cm−1 and 1635 cm−1 for protonated p(TT-TII-PD) and protonated p(TT-TII-2,6ND), respectively), which were not observed in pristine polymers nor in TT-(TII-CHO)2. These newly appeared peaks are attributed to the structural changes derived from the protonation of polymers corresponding to CNH+ stretching vibrations. The differences in peak shifts for protonated p(TT-TII-PD) and protonated p(TT-TII-2,6ND) suggest that p(TT-TII-2,6ND) forms a stronger protonated imine bond, leading to a delayed degradation rate.
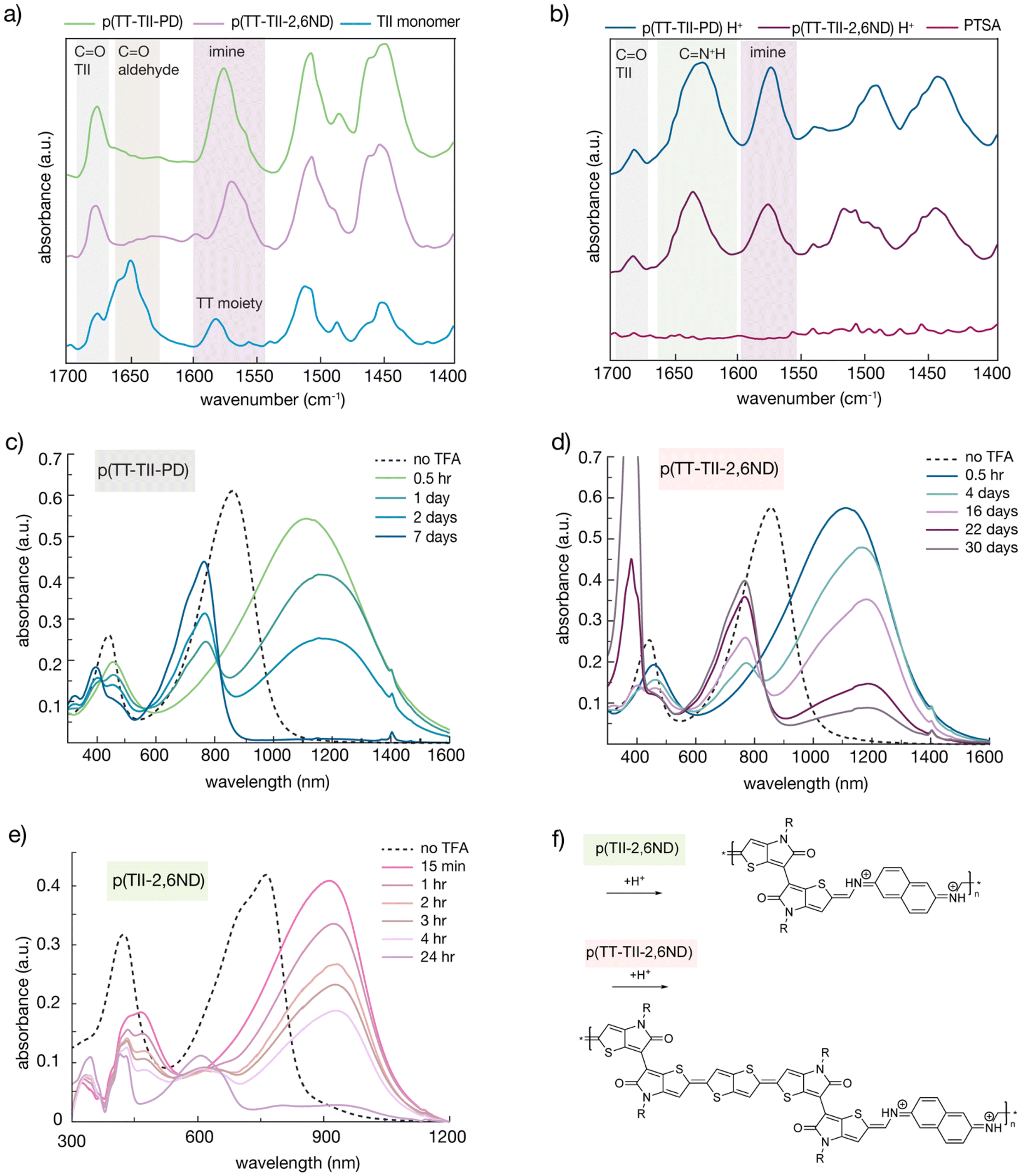 | ||
| Fig. 3 Evidence of quinoidal structure improving chemical stability. Infrared spectroscopy (IR) on (a) pristine and protonated p(TT-TII-PD) and (b) pristine and protonated p(TT-TII-2,6ND). Time-dependent UV-vis-NIR absorption profiles for (c) p(TT-TII-PD), (d) p(TT-TII-2,6ND) and (e) p(TII-2,6ND). (f) Results of DFT calculations showing coplanarity of pristine and protonated model compound based on p(TT-TII-2,6ND). | ||
To quantitatively investigate the degradation of p(TT-TII-PD) and p(TT-TII-2,6ND) at room temperature, time-dependent UV-Vis-NIR absorption spectroscopy was used (Fig. 3c, d and S11†). In the experimental setup, 250 μL of a 1 M trifluoroacetic acid (TFA) solution (aq) was added to each polymer solution (0.5 mg of polymers in 50 mL of chlorobenzene). Notably, after the TFA addition, both p(TT-TII-PD) and p(TT-TII-2,6ND) immediately displayed substantially red-shifted absorption profiles by approximately 250 nm (Fig. 3c and d). Specifically, p(TT-TII-2,6ND) exhibited a more red-shifted absorption compared to p(TT-TII-PD) due to the extended π-conjugation of the naphthalene unit in p(TT-TII-2,6ND) compared to the benzene unit in p(TT-TII-PD). We hypothesize the red-shifted absorption occurs as a result of imine bond protonation, causing a shift from the aromatic state to quinoidal state. This hypothesis is further supported by other protonated poly(azomethine)s in the literature, which undergo a bond length alternation from an aromatic to quinoidal structure upon acidification.40–42 Furthermore, the TII unit has an inherent tendency to form the quinoidal structure,26 as it facilitates bond length alternation, ultimately producing delocalized π-conjugation along the polymer chain.
From the UV-vis-NIR absorption profiles of the protonated polymers, we estimated the energy gaps to be 0.8–0.9 eV, highlighting an overall reduction in band gap energy from the aromatic to quinoidal form for both polymers. Over time, the red-shifted peaks gradually diminish, transitioning to the absorption spectrum of the monomer. We observed the complete degradation of p(TT-TII-PD) into the monomer within a week, whereas p(TT-TII-2,6ND) exhibited much slower degradation rates, with the main polymer absorption peak remaining prominent for several weeks. A reduction in electrostatic repulsion between positively charged imine species of the larger π-system of the naphthalene unit relative to the benzene unit may explain this large difference in degradation rates, whereby the naphthalene unit stabilizes the protonated form of p(TT-TII-2,6ND). Of other thing to note is the significant contribution from the TT unit on the rate of degradation. For example, our previously reported polymer, p(TII-2,6ND), degraded fully in 24 hours (Fig. 3e). In contrast, p(TT-TII-2,6ND), which contains the TT unit in the redesigned monomer, degrades after 30 days (Fig. 3d). This stark difference in degradation rate can be explained by the TT moiety having a strong tendency to stabilize the quinoidal form of the polymer through the positively charged CNH+ bonds by widely spreading π-conjugation throughout the polymer backbone. Another difference to note is the Mn between p(TT-TII-2,6ND) (7.6 kDa) and p(TII-2,6ND) (19.2 kDa). In general, polymers bearing small Mn degrade quicker. However, our results indicate that chemical modification of the polymer backbone has a greater effect on degradation rates compared to Mn since the Mn value of p(TT-TII-2,6ND) is smaller than that of p(TII-2,6ND). As such, our study indicates that quinoid stabilization has a greater impact on poly(azomethine) degradation rates in the presence of acid compared to molecular weight.
To estimate the structural change around the imine bond connection between TII unit and aromatic ring, four model compounds representing the pristine and protonated polymers (Fig. S12a†) were used to perform geometry optimizations using density functional theory (DFT) calculations, where all computations were performed at the theoretical level of B3LYP-D3/6-31G** level.43 Branched long alkyl chains were replaced with methyl groups and we assumed the trans-orientation for two imine bonds flanked by a central aromatic ring, with all sulfurs of the TII moiety oriented in a zigzag conformation for a more feasible model to compute. When comparing pristine and protonated molecules (TII-PD with TII-PD-H and TII-2,6ND with TII-2,6ND-H), bond length alternations resulting from protonation were observed between the thiophene part of the TII unit and imine bond. Specifically, differences were observed between the 1st–5th and 11th–15th bonds for TII-PD and TII-PD-H and 1st–5th and 14th–18th bonds for TII-2,6ND and TII-2,6-ND-H, in which the double bond is elongated, and the single bond is shortened after protonation at the nitrogen atom of the imine moiety (Fig. S12b†). Moreover, the bond length alternation on the TII unit via protonation could potentially explain the red-shifted absorption peaks in the UV-vis-NIR absorption spectra during polymer degradation. Regarding the central aromatic units (benzene vs. naphthalene) there were no noticeable bond length alternations; however, imine bonds (5th and 11th for TII-PD-H and 5th and 14th for TII-2,6ND-H) were largely elongated owing to the protonation. Considering the coplanarity of the model compounds (Fig. 3f and S12c†), the torsion angles between central benzene or naphthalene units and flanked TII unit were both 30.8° for TII-PD and 37.2° and 34.1° for TII-2,6ND, respectively. By contrast, the protonated polymers, TII-PD-H and TII-2,6ND-H, displayed reduced torsion angles of both 20.0° for TII-PD-H and 18.0° and 18.1° for TII-2,6ND-H. This remarkably planarized conformation suggests enhanced intermolecular charge carrier transport through quinoidal resonance due to acid mediated protonation. Overall, the observations from DFT calculations between TII and the imine bond in both polymer structures provides clear evidence related to quinoidal bond length alternation affecting degradation rates.
Charge carrier transport properties
To assess the charge carrier transport properties of the polymer, FET devices were fabricated with a bottom-gate, top-contact configuration with Au source and drain electrodes. The active layer was spin-coated onto n-doped Si substrates treated with octadecyltrimethoxysilane (OTMS) (300 nm SiO2 layer).44 The spin-coated films were then heat-annealed at 250 °C for 15 minutes in a glovebox. Subsequently, Au source/drain electrodes were deposited onto the active layer under vacuum, with a channel length of 50 μm and a channel width of 1000 μm. All measurements were conducted under vacuum, and the mobility values were estimated from the saturation regimes, as summarized in Table 3. Thin films of p(TT-TII-PD) and p(TT-TII-2,6ND) exhibited typical p-channel behaviors, as anticipated from HOMO and LUMO levels estimated from the redox and the optical properties (Fig. 4a, c and S13a, b†). The average hole mobilities for all polymers were on the order of 10−3 cm2 V−1 s−1, comparable to our previously reported polymers.29 We note that the low on–off current ratio around 102–103 was due to unintentional doping by oxygen during solution preparation under ambient conditions.26,45 Compared to our previously reported polymers,29 we observed one order of magnitude lower for the on–off current ratio. This is attributed to the insertion of the TT unit between TII units, which favors an aromatic to quinoidal transformation, resulting in a smaller energy gap.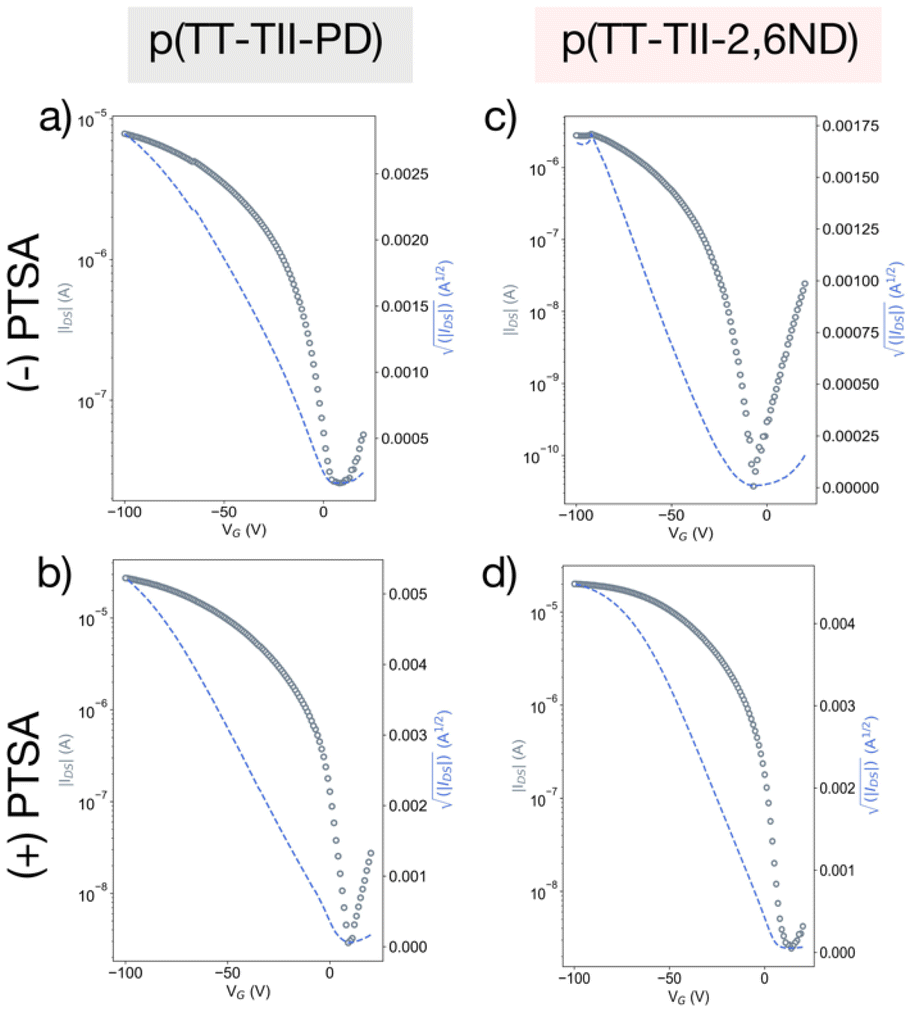 | ||
| Fig. 4 Typical transfer curves of pristine ((−)PTSA) and protonated ((+)PTSA) thin films: (a) pristine p(TT-TII-PD), (b) protonated p(TT-TII-PD), (c) pristine p(TT-TII-2,6ND), and (d) protonated p(TT-TII-2,6ND). | ||
To examine the impact of protonation on charge carrier properties, FET devices of fully protonated polymers were measured by the same method as the pristine ones (Table 3). To prepare the protonated thin films, polymers were dissolved in a saturated PTSA/chloroform solution (14.7 mmol L−1, ca. 15 equiv. to the repeating unit of polymers) and spin coated onto n-doped Si substrates. For p-type semiconducting polymers, acid doping is often used to improve device performance because the addition of radical cations on the polymer backbone leads to increased carrier concentration.46–48 In the case of p(TT-TII-PD) and p(TT-TII-2,6ND) without gate bias modulation, acid mediated doping increases the average IDS values from 90.0 to 243.1 nA for p(TT-TII-PD) and from 11.5 to 193.2 nA for p(TT-TII-2,6ND), respectively. Thereby, both protonated thin films displayed the improved FET performance with hole mobilities as high as 10−2 cm2 V−1 s−1, a whole order of magnitude greater compared to pristine polymers (Fig. 4b and d).
Thin film microstructure
To gain further insight on charge carrier transport associated with the interchain packing in thin films annealed at 250 °C, measurements were conducted using grazing incidence wide-angle X-ray scattering (GIWAXS). Fig. 5 and S14† show GIWAXS patterns and one-dimensional (1-D) profiles, with extracted parameters summarized in Tables S1 and S2.† The GIWAXS two-dimensional (2-D) patterns (Fig. 5a and c) reveal that pristine polymer thin films adopted similar microstructures. In both out-of-plane and in-plane directions, all thin films exhibited a mixed bimodal orientation with strong (100) lamellar scattering up to (200) order diffraction, showing both edge-on and face-on mixed orientations. We note that (100) peaks of all polymers were overlapped with X-ray leakage from the beam stopper. The respective out-of-plane and in-plane lamellar distances were 26.2 Å and 26.4 Å for p(TT-TII-PD) and 28.2 Å and 27.2 Å for p(TT-TII-2,6ND), respectively. Additionally, both polymers showed an (010) diffraction peak in out-of-plane direction corresponding to π-stacking peaks, and the presence of out-of-plane (010) peak of both polymers indicates face-on dominant bimodal polymer chain alignment. To examine the impact of protonation on the interchain packing and orientation of the polymers, GIWAXS measurements of fully protonated polymers were taken and the results summarized in Table S2.† Considering the results of the GIWAXS 2D patterns and 1D profiles, all protonated thin films exhibited edge-on and face-on mixed bimodal orientation (Fig. 5b, d and S14†). We observed no change in polymer orientation and unaltered thin film microstructures upon the addition of PTSA to p(TT-TII-PD) and p(TT-TII-2,6ND). The respective out-of-plane and in-plane lamellar distances were 25.4 Å and 26.7 Å for p(TT-TII-PD), and 23.7 Å and 26.2 Å for p(TT-TII-2,6ND), in which these approximately shorter values imply compact lamellar packing compared to pristine thin films. Interestingly, the π-stacking distances estimated from the out-of-plane (010) diffraction peak of protonated p(TT-TII-PD) and p(TT-TII-2,6ND) are approximately 3.5 Å and slightly shorter than that of pristine polymers, which is originating from the planarized backbone of protonated polymer chains as evidenced by theoretical calculations. When comparing the relative intensity of out-of-plane and in-plane in thin films of p(TT-TII-PD) and p(TT-TII-2,6ND), we note a large intensity in the out-of-plane direction; therefore, both thin films prefer an edge-on dominant ordering which is preferential for charge carrier transport because the channel direction is lateral in FET. This shift towards more edge-on packing in PTSA-doped thin films suggests that the aromatic to quinoidal transformation induces more compact lamellar packing as shown in similar aromatic to quinoidal transformations of TT-based polymers.33 Additionally, it is surprising that p(TT-TII-PD) and p(TT-TII-2,6ND) exhibited smaller lamellar distances compared to pristine thin films, as this implies PTSA molecules would be implanted into amorphous region. These results suggest that PTSA doping has a positive influence on the charge carrier transport for both p(TT-TII-PD) and p(TT-TII-2,6ND).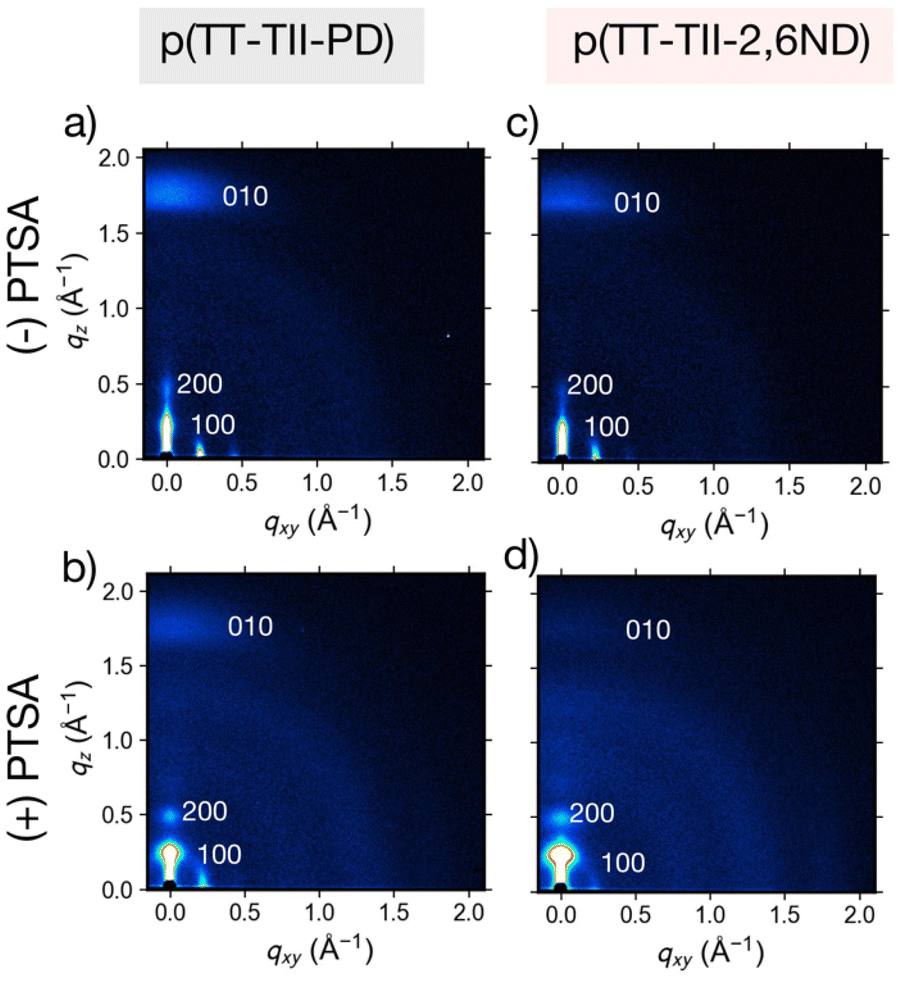 | ||
| Fig. 5 GIWAXS patterns of pristine ((−)PTSA) and protonated ((+)PTSA) thin films: (a) p(TT-TII-PD), (b) protonated p(TT-TII-PD), (c) p(TT-TII-2,6-ND), and (d) protonated p(TT-TII-2,6ND). | ||
To study the origin of different FET performances between pristine and protonated polymers, we examined the thin film morphology by atomic force microscopy (AFM). Compared to our previous report regarding p(TII-PD) and p(TII-2,6ND),29 we observed quite different morphology in AFM height images (Fig. 6). Thin films of p(TII-PD) and p(TII-2,6ND) showed continuous nanofibril or bundle-like networks with void-like structure, whereas thin films of pristine p(TT-TII-PD) and p(TT-TII-2,6ND) exhibited densely packed granular morphology. This implies that the TT unit insertion has a large impact on thin film morphology. AFM results provide evidence of early microstructure degradation of the polymer thin films. Pristine p(TT-TII-PD) features a densely pack granular morphology; however, the addition of PTSA results in the loss of defined granules and an overall reduction in surface height. A similar observation is also noted with p(TT-TII-2,6ND), where the surface height also decreases, defined granules are lost, and small holes appear; these observations are consistent with other reported semiconducting polymer systems when subjected to degradation conditions.49,50 The differences in surface morphology between doped and undoped polymer films suggest the polymers are at an intermediate stage between the quinoidal transformation (λmax ∼1100 nm) and polymer degradation (λmax ∼780 nm) (Fig. 3c and d). In view of the FET performance, the protonated polymers exhibited smaller domain size and CCL values (Table S2†) than pristine polymers, which typically worsen charge transport. However, the AFM images of the protonated thin films show nanofibrils (Fig. 6b and d), which may serve as interconnect contacts between the grain boundaries. In contrast, the AFM images of the pristine thin films show a granular morphology. In addition, protonated polymers showed slightly compact π-stacking distances derived from planarized polymer geometry with doped charge carrier which increases charge carrier concentration. Overall, we note that the protonation of p(TT-TII-PD) and p(TT-TII-2,6ND) leads to higher FET performance compared to pristine p(TT-TII-PD) and p(TT-TII-2,6ND), as supported by thin film morphology observations obtained from AFM and GIWAXS.
 | ||
| Fig. 6 AFM height images of pristine ((−)PTSA) and protonated ((+)PTSA) thin films: (a) p(TT-TII-PD), (b) protonated p(TT-TII-PD), (c) p(TT-TII-2,6-ND), and (d) protonated p(TT-TII-2,6ND). | ||
Conclusions
In this study, two imine-based semiconducting polymers, p(TT-TII-PD) and p(TT-TII-2,6ND), were synthesized and characterized to evaluate the structure–property relationship of aromatic to quinoidal transformations on degradation kinetics. Both polymers were synthesized by an imine polycondensation reaction using TT-(TII-CHO)2 with two diamine monomers (PD and 2,6ND). The two polymers showed similar optical and redox properties, and insertion of the TT unit along the polymer backbone was found to elevate HOMO and lower LUMO levels. Our degradation studies revealed red-shifted UV-vis-NIR absorption profiles upon acidification of both polymers associated with the shift from an aromatic to quinoid structure. The TT insertion between TII units allowed for the quinoid form to dominate the polymer structures upon acid doping, and consequently stabilized positively charged protonated species along the polymer backbone, resulting in significantly slower degradation rates compared to our previously reported polymers. The pristine thin films of p(TT-TII-PD) and p(TT-TII-2,6ND) exhibited p-type properties with mobilities on the order of 10−3 cm2 V−1 s−1. Protonating the thin films using PTSA resulted in mobility values one order of magnitude higher compared to pristine films due to planarized molecular geometries and improved thin film microstructures. Overall, the contribution of aromatic to quinoidal transformation on the polymer backbone has a large impact on acid-mediated degradation kinetics and charge carrier mobility. We hope that the exploration of aromatic to quinoidal transformations in this study will shape the way structure–property relationships are utilized during the design of degradable organic semiconducting polymers, opening avenues for precise control over degradation kinetics and semiconductor lifetimes.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by JSPS KAKENHI grant numbers JP 22K05047 (for M. A.) the Natural Sciences and Engineering Research Council (NSERC) of Canada (H. T., RGPIN2021-03554; A. U., CGS-D scholarship) and the University of Toronto's Acceleration Consortium from the Canada First Research Excellence Fund (H. T., CDG13-2023). The synchrotron radiation experiments were performed at the BL40B2 beamline in SPring 8 with the approval of JASRI (proposal no. 2023B1123). The authors thank Dr Masato Koizumi (Open Facility Center, Institute of Science Tokyo) in the Material Analysis Division for ESI-HRMS spectra, and Dr Hiroyasu Masunaga and Dr Noboru Ohta (Japan Synchrotron Radiation Research Institute; JASRI) for assistance in the GIWAXS experiment. Lastly, the authors thank Ms Yurika Kashino (Institute of Science Tokyo) for useful discussion about DFT calculations.References
- W. B. Han, J. H. Lee, J. W. Shin and S. W. Hwang, Advanced Materials and Systems for Biodegradable, Transient Electronics, Adv. Mater., 2020, 32(51), 1–17 Search PubMed.
- E. W. C. Chan, X. Sun and J. Travas-Sejdic, Recent Progress and Future Prospects in Transient Polymer Electronics, Macromolecules, 2023, 56(11), 3755–3773 CrossRef.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Transient Electronics: Materials and Devices, Chem. Mater., 2016, 28(11), 3527–3539 CrossRef.
- A. Uva, S. Michailovich, N. S. Y. Hsu and H. Tran, Degradable π-Conjugated Polymers, J. Am. Chem. Soc., 2024, 146(18), 12271–12287 CrossRef PubMed.
- G. Garbay, L. Giraud, S. M. Gali, G. Hadziioannou, E. Grau, S. Grelier, E. Cloutet, H. Cramail and C. Brochon, Divanillin-Based Polyazomethines: Toward Biobased and Metal-Free π-Conjugated Polymers, ACS Omega, 2020, 5(10), 5176–5181 CrossRef PubMed.
- T. R. Martin, L. Rynearson, M. Kuller, J. Quinn, C. Wang, B. Lucht and N. R. Neale, Conjugated Imine Polymer Synthesized via Step-Growth Metathesis for Highly Stable Silicon Nanoparticle Anodes in Lithium-Ion Batteries, Adv. Energy Mater., 2023, 13(13), 2203921 CrossRef.
- A. Uva, A. Lin and H. Tran, Biobased, Degradable, and Conjugated Poly(Azomethine)s, J. Am. Chem. Soc., 2023, 145(6), 3606–3614 CrossRef.
- H. Tran, S. Nikzad, J. A. Chiong, N. J. Schuster, A. E. Peña-Alcántara, V. R. Feig, Y.-Q. Q. Zheng and Z. Bao, Modular Synthesis of Fully Degradable Imine-Based Semiconducting p-Type and n-Type Polymers, Chem. Mater., 2021, 33 (18), 7465–7474 CrossRef.
- N. S. Y. Hsu, A. Lin, A. Uva, S. H. Huang and H. Tran, Direct Arylation Polymerization of Degradable Imine-Based Conjugated Polymers, Macromolecules, 2023, 56(21), 8947–8955 CrossRef.
- B. R. Varju and D. S. Seferos, Direct Heteroarylation Polymerization of a π-Conjugated Polymer with Degradable 1,2,4-Oxadiazole Linkers, Polym. Chem., 2022, 13(46), 6386–6392 RSC.
- H. Huang, W. Xie, Q. Wan, L. Mao, D. Hu, H. Sun, X. Zhang and Y. Wei, A Self-Degradable Conjugated Polymer for Photodynamic Therapy with Reliable Postoperative Safety, Adv. Sci., 2022, 9(4), 2104101 CrossRef PubMed.
- T. Repenko, A. Rix, S. Ludwanowski, D. Go, F. Kiessling, W. Lederle and A. J. C. Kuehne, Bio-Degradable Highly Fluorescent Conjugated Polymer Nanoparticles for Bio-Medical Imaging Applications, Nat. Commun., 2017, 8(1), 1–8 Search PubMed.
- F. Jansen, P. A. Schuster, M. Lamla, C. Trautwein and A. J. C. Kuehne, Biodegradable Polyimidazole Particles as Contrast Agents Produced by Direct Arylation Polymerization, Biomacromolecules, 2021, 22(12), 5065–5073 CrossRef.
- J. A. Chiong, Y. Zheng, S. Zhang, G. Ma, Y. Wu, G. Ngaruka, Y. Lin, X. Gu and Z. Bao, Impact of Molecular Design on Degradation Lifetimes of Degradable Imine-Based Semiconducting Polymers, J. Am. Chem. Soc., 2022, 144(8), 3717–3726 CrossRef PubMed.
- H. Park, Y. Kim, D. Kim, S. Lee, F. S. Kim and B. J. Kim, Disintegrable N-Type Electroactive Terpolymers for High-Performance, Transient Organic Electronics, Adv. Funct. Mater., 2022, 32(2), 2106977 CrossRef.
- T. Lei, M. Guan, J. Liu, H. C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T. C. Huang, L. Shao, K. T. Cheng, J. B. H. Tok and Z. Bao, Biocompatible and Totally Disintegrable Semiconducting Polymer for Ultrathin and Ultralightweight Transient Electronics, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(20), 5107–5112 CrossRef.
- G. S. Liou, H. Y. Lin, Y. L. Hsieh and Y. L. Yang, Synthesis and Characterization of Wholly Aromatic Poly(Azomethine)s Containing Donor–Acceptor Triphenylamine Moieties, J. Polym. Sci., Part A: Polym. Chem., 2007, 45 (21), 4921–4932 CrossRef.
- J. A. Chiong, L. Michalek, A. E. Peña-Alcántara, X. Ji, N. J. Schuster and Z. Bao, Degradable Semiconducting Polymers without Long-Range Order for on-Demand Degradation of Transient Electronics, J. Mater. Chem. C, 2023, 11(43), 15205–15214 RSC.
- K. A. Bartlett, A. Charland-Martin, J. Lawton, A. L. Tomlinson and G. S. Collier, Azomethine-Containing Pyrrolo[3,2-b]Pyrrole Copolymers for Simple and Degradable Conjugated Polymers, Macromol. Rapid Commun., 2023, 45, 2300220 CrossRef.
- A. Charland-Martin and G. S. Collier, Understanding Degradation Dynamics of Azomethine-Containing Conjugated Polymers, Macromolecules, 2024, 57(13), 6146–6155 CrossRef.
- N. Kleinhenz, L. Yang, H. Zhou, S. C. Price and W. You, Low-Band-Gap Polymers That Utilize Quinoid Resonance Structure Stabilization by Thienothiophene: Fine-Tuning of HOMO Level, Macromolecules, 2011, 44(4), 872–877 CrossRef.
- J. Huang and G. Yu, Recent Progress in Quinoidal Semiconducting Polymers: Structural Evolution and Insight, Mater. Chem. Front., 2021, 5(1), 76–96 RSC.
- H. Hwang, D. Khim, J. M. Yun, E. Jung, S. Y. Jang, Y. H. Jang, Y. Y. Noh and D. Y. Kim, Quinoidal Molecules as a New Class of Ambipolar Semiconductor Originating from Amphoteric Redox Behavior, Adv. Funct. Mater., 2015, 25(7), 1146–1156 CrossRef.
- Y. Kim, H. Hwang, N.-K. Kim, K. Hwang, J.-J. Park, G.-I. Shin, D.-Y. Kim, Y. Kim, H. Hwang, K. Hwang, J.-J. Park, G.-I. D. Shin and N.-K. Kim, π-Conjugated Polymers Incorporating a Novel Planar Quinoid Building Block with Extended Delocalization and High Charge Carrier Mobility, Adv. Mater., 2018, 30(22), 1706557 CrossRef.
- A. Velusamy, C. H. Yu, S. N. Afraj, C. C. Lin, W. Y. Lo, C. J. Yeh, Y. W. Wu, H. C. Hsieh, J. Chen, G. H. Lee, S. H. Tung, C. L. Liu, M. C. Chen and A. Facchetti, Thienoisoindigo (TII)-Based Quinoidal Small Molecules for High-Performance n-Type Organic Field Effect Transistors, Adv. Sci., 2021, 8(1), 2002930 CrossRef.
- T. Hasegawa, M. Ashizawa, J. Hiyoshi, S. Kawauchi, J. Mei, Z. Bao and H. Matsumoto, An Ultra-Narrow Bandgap Derived from Thienoisoindigo Polymers: Structural Influence on Reducing the Bandgap and Self-Organization, Polym. Chem., 2016, 7(5), 1181–1190 RSC.
- D. Yoo, T. Hasegawa, M. Ashizawa, T. Kawamoto, H. Masunaga, T. Hikima, H. Matsumoto and T. Mori, N-Unsubstituted, Thienoisoindigos: Preparation, Molecular Packing and Ambipolar Organic Field-Effect Transistors, J. Mater. Chem. C, 2017, 5(10), 2509–2512 RSC.
- L. Shen, X. Gao, Z. Chang, C. Zhang, Y. Li, J. Lu, Q. Meng and Q. Wu, Sufficient Driving Force for Quinoidal Isoindigo-Based Diradicaloids with Tunable Diradical Characters, Phys. Chem. Chem. Phys., 2023, 26(3), 2529–2538 RSC.
- N. Nozaki, A. Uva, H. Matsumoto, H. Tran and M. Ashizawa, Thienoisoindigo-Based Recyclable Conjugated Polymers for Organic Electronics, RSC Appl. Polym., 2024, 1(2), 163–171 RSC.
- I. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planells, H. Bronstein, J. W. Rumer, D. I. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J. C. Flores, H. Sirringhaus and I. McCulloch, Thieno[3,2-b]Thiophene Flanked Isoindigo Polymers for High Performance Ambipolar OFET Applications, Adv. Funct. Mater., 2014, 24(45), 7109–7115 CrossRef.
- Y. Li, C. Y. Chang, Y. Chen, Y. Song, C. Z. Li, H. L. Yip, A. K. Y. Jen and C. Li, The Effect of Thieno[3,2-b]Thiophene on the Absorption, Charge Mobility and Photovoltaic Performance of Diketopyrrolopyrrole-Based Low Bandgap Conjugated Polymers, J. Mater. Chem. C, 2013, 1(45), 7526–7533 RSC.
- Y. Li, S. P. Singh, P. Sonar, Y. Li, S. P. Singh, P. Sonar and Y. Li, A High Mobility P-Type DPP-Thieno[3,2-b]Thiophene Copolymer for Organic Thin-Film Transistors, Adv. Mater., 2010, 22(43), 4862–4866 CrossRef PubMed.
- J. Huang, S. Lu, P. A. Chen, K. Wang, Y. Hu, Y. Liang, M. Wang and E. Reichmanis, Rational Design of a Narrow-Bandgap Conjugated Polymer Using the Quinoidal Thieno[3,2- b]Thiophene-Based Building Block for Organic Field-Effect Transistor Applications, Macromolecules, 2019, 52(12), 4749–4756 CrossRef.
- J. Mei and Z. Bao, Side Chain Engineering in Solution-Processable Conjugated Polymers, Chem. Mater., 2014, 26(1), 604–615 CrossRef.
- L. Giraud, S. Grelier, E. Grau, L. Garel, G. Hadziioannou, B. Kauffmann, É. Cloutet, H. Cramail and C. Brochon, Synthesis and Characterization of Vanillin-Based π-Conjugated Polyazomethines and Their Oligomer Model Compounds, Molecules, 2022, 27(13), 4138 CrossRef.
- H. Zheng, H. Ye, X. Yu and L. You, Interplay between N→π* Interactions and Dynamic Covalent Bonds: Quantification and Modulation by Solvent Effects, J. Am. Chem. Soc., 2019, 141 (22), 8825–8833 CrossRef PubMed.
- H. B. Michaelson, The work function of the elements and its periodicity, J. Appl. Phys, 1997, 48(11), 4729–4733 CrossRef.
- H. Jin, K. K. Kim, S. Park, J. H. Rhee, H. Ahn, D. J. Kim, K. K. Kim, J. H. Noh, T. S. Kim, E. Y. Shin and H. J. Son, Chemically Recyclable Conjugated Polymer and One-Shot Preparation of Thermally Stable and Efficient Bulk-Heterojunction from Recycled Monomer, Adv. Funct. Mater., 2023, 2304930 CrossRef CAS.
- R. Xia, C. Li, X. Yuan, Q. Wu, B. Jiang and Z. Xie, Facile Preparation of a Thienoisoindigo-Based Nanoscale Covalent Organic Framework with Robust Photothermal Activity for Cancer Therapy, ACS Appl. Mater. Interfaces, 2022, 14(17), 19129–19138 CrossRef CAS PubMed.
- J. Gasiorowski, E. D. Głowacki, B. Hajduk, M. Siwy, M. Chwastek-Ogierman, J. Weszka, H. Neugebauer and N. S. Sariciftci, Doping-Induced Immobile Charge Carriers in Polyazomethine: A Spectroscopic Study, J. Phys. Chem. C, 2013, 117(6), 2584–2589 CrossRef.
- P. Nitschke, B. Jarząbek, A. E. Bejan and M. D. Damaceanu, Effect of Protonation on Optical and Electrochemical Properties of Thiophene-Phenylene-Based Schiff Bases with Alkoxy Side Groups, J. Phys. Chem. B, 2021, 125(30), 8588–8600 CrossRef PubMed.
- B. Hu, X. Zhu, X. Chen, L. Pan, S. Peng, Y. Wu, J. Shang, G. Liu, Q. Yan and R. W. Li, A Multilevel Memory Based on Proton-Doped Polyazomethine with an Excellent Uniformity in Resistive Switching, J. Am. Chem. Soc., 2012, 134(42), 17408–17411 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Ito, A. A. Virkar, S. Mannsfeld, H. O. Joon, M. Toney, J. Locklin and Z. Bao, Crystalline Ultrasmooth Self-Assembled Monolayers of Alkylsilanes for Organic Field-Effect Transistors, J. Am. Chem. Soc., 2009, 131(26), 9396–9404 CrossRef.
- K. Takimiya, I. Osaka and M. Nakano, π-Building Blocks for Organic Electronics: Revaluation of “Inductive” and “Resonance” Effects of π-Electron Deficient Units, Chem. Mater., 2014, 26(1), 587–593 CrossRef.
- H. Yano, K. Kudo, K. Marumo and H. Okuzaki, Fully Soluble Self-Doped Poly(3,4-Ethylenedioxythiophene) with an Electrical Conductivity Greater than 1000 S Cm -1, Sci. Adv., 2019, 5(4), 9492–9504 CrossRef.
- W. Zhao, J. Ding, Y. Zou, C. A. Di and D. Zhu, Chemical Doping of Organic Semiconductors for Thermoelectric Applications, Chem. Soc. Rev., 2020, 49(20), 7210–7228 RSC.
- A. I. Hofmann, R. Kroon, L. Yu and C. Müller, Highly Stable Doping of a Polar Polythiophene through Co-Processing with Sulfonic Acids and Bistriflimide, J. Mater. Chem. C, 2018, 6(26), 6905–6910 RSC.
- A. Tournebize, P. O. Bussière, P. Wong-Wah-Chung, S. Thérias, A. Rivaton, J. L. Gardette, S. Beaupré and M. Leclerc, Impact of UV-Visible Light on the Morphological and Photochemical Behavior of a Low-Bandgap Poly(2,7-Carbazole) Derivative for Use in High-Performance Solar Cells, Adv. Energy Mater., 2013, 3(4), 478–487 CrossRef.
- I. F. Domínguez, P. D. Topham, P. O. Bussière, D. Bégué and A. Rivaton, Unravelling the Photodegradation Mechanisms of a Low Bandgap Polymer by Combining Experimental and Modeling Approaches, J. Phys. Chem. C, 2015, 119(4), 2166–2176 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedure, NMR, TGA, DSC, UV-vis-NIR, CV, FET mobility, GIWAXS, and AFM data. See DOI: https://doi.org/10.1039/d4lp00310a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |