
Theoretical insights and design of MXene for aqueous batteries and supercapacitors: status, challenges, and perspectives
Jun
Zhao
 a,
Ninggui
Ma
a,
Tairan
Wang
a,
Yuhang
Wang
a,
Bochun
Liang
a,
Yaqin
Zhang
a,
Shuang
Luo
a,
Yu
Xiong
a,
Qianqian
Wang
a and
Jun
Fan
*ab
a,
Ninggui
Ma
a,
Tairan
Wang
a,
Yuhang
Wang
a,
Bochun
Liang
a,
Yaqin
Zhang
a,
Shuang
Luo
a,
Yu
Xiong
a,
Qianqian
Wang
a and
Jun
Fan
*ab
aDepartment of Materials Science and Engineering, City University of Hong Kong, Hong Kong, China. E-mail: junfan@cityu.edu.hk
bDepartment of Mechanical Engineering, City University of Hong Kong, Hong Kong, China
First published on 29th October 2024
Abstract
Aqueous batteries and supercapacitors are promising electrochemical energy storage systems (EESSs) due to their low cost, environmental friendliness, and high safety. However, aqueous EESS development faces challenges like narrow electrochemical windows, irreversible dendrite growth, corrosion, and low energy density. Recently, two-dimensional (2D) transition metal carbide and nitride (MXene) have attracted more attention due to their excellent physicochemical properties and potential applications in aqueous EESSs. Understanding the atomic-level working mechanism of MXene in energy storage through theoretical calculations is necessary to advance aqueous EESS development. This review comprehensively summarizes the theoretical insights into MXene in aqueous batteries and supercapacitors. First, the basic properties of MXene, including structural composition, experimental and theoretical synthesis, and advantages in EESSs are introduced. Then, the energy storage mechanism of MXene in aqueous batteries and supercapacitors is summarized from a theoretical calculation perspective. Additionally, the theoretical insights into the side reactions and stability issues of MXene in aqueous EESSs are emphasized. Finally, the prospects of designing MXene for aqueous EESSs through computational methods are given.
 Jun Zhao | Jun Zhao is currently a doctoral candidate in the Department of Materials Science and Engineering, City University of Hong Kong, supervised by Prof. Jun Fan. He received her M.S. degree from the School of Chemical Engineering, Tianjin University. His current research interests are first-principles calculation of MXene materials in the energy storage field. |
 Jun Fan | Dr. Jun Fan received her bachelor's and doctoral degrees from Tsinghua University and Princeton University, respectively. She is a tenured associate professor and doctoral supervisor at City University of Hong Kong. Her research interests mainly include (1) two-dimensional material nanopore sequencing of proteins/DNA/RNA, the mechanism of the influence of nanomaterials on cell membranes, and the regulation of protein conformational conversion; (2) research on two-dimensional materials in the field of energy storage and catalysis; (3) the application of machine learning in force field development and material design. |
1. Introduction
Industrial development and population growth have led to energy shortages and environmental issues, posing significant obstacles to human progress.1,2 Therefore, developing efficient, reliable, safe, and environmentally friendly energy storage systems is crucial to address these problems.3–5 Renewable energy sources, such as solar and wind, are intermittent, necessitating effective storage solutions for stable and continuous energy supply.3,6 Electrochemical energy storage systems (EESSs), including batteries and supercapacitors, play a key role in capturing, storing, and delivering energy.4,7–9 Among these, aqueous EESSs, especially aqueous batteries and supercapacitors, have garnered significant attention due to their inherent safety, environmental protection, and cost-effectiveness.7,8,10,11Compared to traditional non-aqueous EESSs, aqueous EESSs have multiple advantages.12–14 First, using water as an electrolyte solvent reduces flammability and toxicity, enhancing safety. Second, aqueous EESSs generally have higher ionic conductivity, leading to better device rate performance. Third, the materials and processes for aqueous electrolytes are cheaper, making these systems more economically feasible. However, aqueous EESSs face challenges, such as a narrow electrochemical window, electrode material corrosion, irreversible metal dendrite growth, and low energy density.15–17
Recently, MXene materials have shown promise in addressing the issues of aqueous EESSs.10,11,13 Since their first synthesis, MXenes have attracted great interest due to their unique properties, such as high electrical conductivity, excellent mechanical strength, hydrophilicity, and tunability. They have been used in energy storage, biology, sensors, catalysis, optics, electromagnetic shielding, etc.18–24 These unique properties enable MXene to serve as the intermediates of aqueous EESSs, exhibiting excellent performance.11,13 Abundant surface sites of MXene provide storage or redox sites for charge carriers, enhancing the energy density of EESSs.25,26 Besides, their layered structure facilitates rapid ion insertion and de-insertion, contributing to excellent rate performance and inhibiting dendrite formation.27,28 The inherent high conductivity of MXene ensures efficient electron transport, essential for high power density and fast charging.29 In short, these properties together make MXenes highly effective electrode materials in aqueous batteries and supercapacitors.
Further development of MXene materials for aqueous EESSs requires understanding their properties and energy storage mechanisms at the atomic scale. Compared to experimental methods, theoretical calculations and modeling are indispensable tools with the advantages of low cost, high efficiency, and precision.30,31 Computational methods such as density functional theory (DFT) and molecular dynamics (MD), multi-physics fields, etc. provide valuable insights into the electronic structure and synthesis, electric field regulation, surface interactions, and charge storage mechanisms of MXene.25,30,32–35 However, comprehensive reviews of theoretical calculations for understanding and designing MXene materials for aqueous EESSs are rare. Therefore, we review the current status, challenges, and perspectives in this field to promote the development of more efficient aqueous EESSs.
2. Structural composition, synthesis, and properties of MXene
2.1. Structural composition of MXene
MXene is a class of 2D materials composed of transition metal (TM) carbides and nitrides, attracting widespread attention for their unique layered structure and tunable chemical composition.36–38 As shown in Fig. 1(a), MXene typically has a multi-layer structure, obtained by selectively etching A elements (e.g., Al, Ga, Sn) in the MAX phases, which can then be further peeled into a single-layer structure.38 MXene shares a similar hexagonal close-packed crystal structure and P63/mmc symmetry with the MAX phases.24 In Fig. 1(b), the general chemical formula of MXene is Mn+1XnTx, where M represents the early TM, which can be a single metal or an ordered/disordered mixture of metals; X is carbon and/or nitrogen, and n ranges from 1 to 4 depending on the number of TM layers; Tx indicates the surface functional groups determined by the preparation method.39 This structural and compositional diversity endows MXenes with varied physical, chemical, electronic, and optical properties, giving them significant potential in energy storage applications.11,13,24,40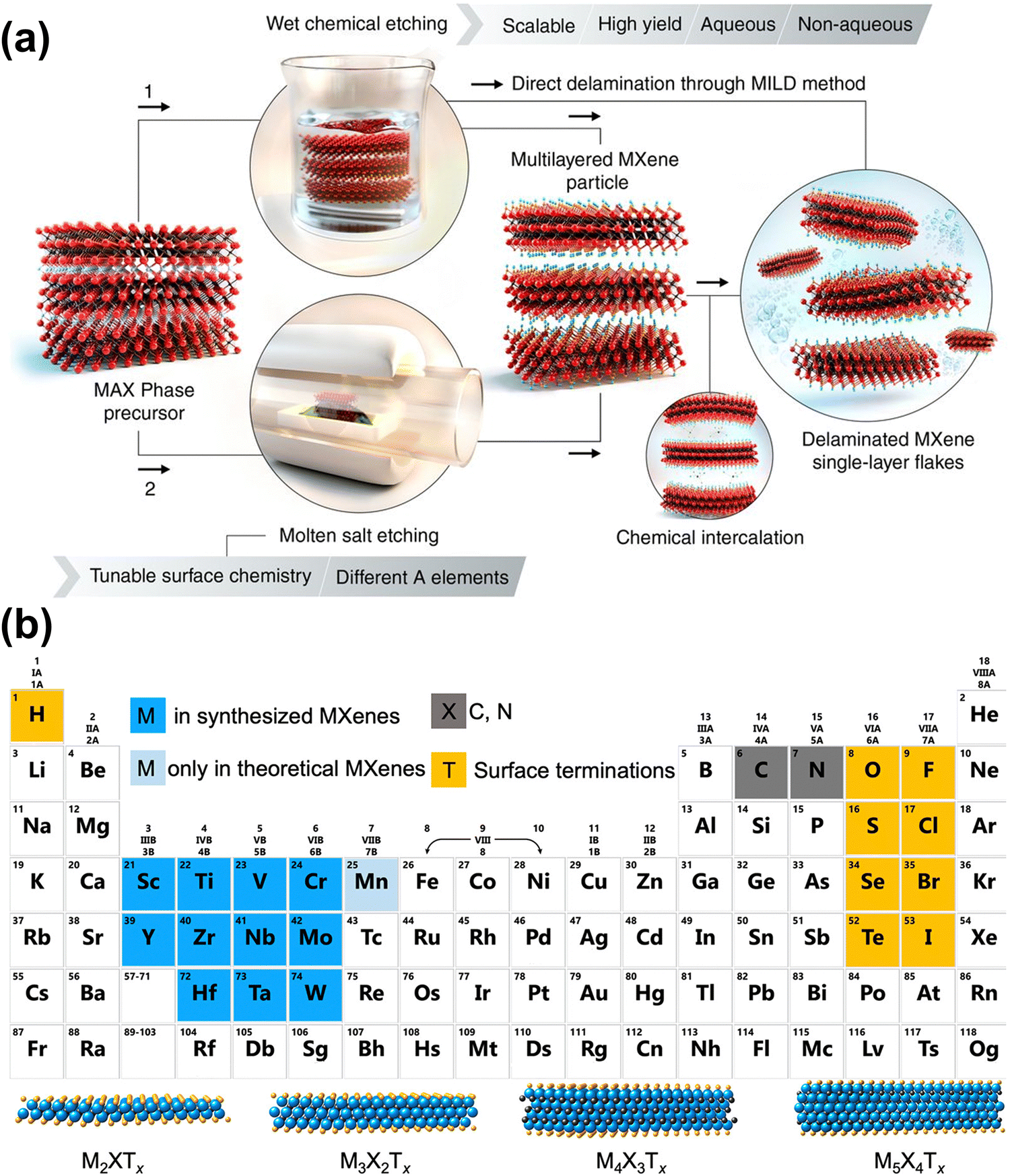 | ||
| Fig. 1 (a) Schematic illustration of two approaches to produce MXene by removal of A layers from MAX phases and related layered compounds.24 Reproduced with permission ref. 24. Copyright 2021, AAAS. (b) Periodic table showing compositions of MXene. Elements used to build MXene are color-coded. The schematics of four typical structures of MXene are presented at the bottom.39 Reproduced with permission ref. 39. Copyright 2021, American Chemical Society. | ||
The compositional diversity of MXene underpins their unique properties. MXene can be composed of various TMs, such as III B (Sc, Y), IV B (Ti, Zr, and Hf), V B (V, Nb, and Ta), VI B (Cr, Mo, and W), and VII B (Mn, verified only by calculation) elements.24,39,41,42 These metals can be present as single-layer, double-layer, or multi-layer in the MXene structure.38 Besides a single metal composition, various TM elements can be introduced at the metal site, including bimetallic and high-entropy MXene.24,30,43,44 The metals in bimetallic MXenes can form ordered and solid solution MXene.45,46 Ordered bimetallic MXene are divided into in-plane ordered (i-MXene) and out-of-plane ordered (o-MXene) structures. Selective etching of TM elements in i-MXene can produce non-stoichiometric in-plane ordered vacancy-type MXene.47 High entropy alloy MXenes contain more than 4 TM elements occupying the M site.30 So far, only a few high-entropy MXenes have been successfully synthesized, but their unique mechanical and electrochemical properties show great potential in batteries and supercapacitors.44,47,48
The X element in MXene is generally C, N, or a mixture of both. Currently, experimentally synthesized MXenes are mainly carbides, with only a few nitrides.49 Although a few nitrides have been synthesized, they exhibit some special properties. For example, compared to Ti2CTx, Ti2NTx exhibits better structural stability as well as electrocatalytic N2 reduction reaction (NRR) activity.50 Additionally, the choice of X element affects the electronic structure and conductivity of MXenes, with nitrides generally exhibiting higher band gaps than carbides.47 For carbonitrides, the occupation of C and N atoms at the X site is generally random, independent of their stoichiometric ratio. Carbonitrides have been proven useful in EESSs.51
The surface termination group of MXene is very important, which greatly affects the interface properties and thus its performance in electrocatalysis and EESSs.13,52,53 MXene also has a wide range of surface functional groups, including VA, VIA, and VIIA group elements.24,38 The type and distribution of these surface functional groups can be adjusted through different synthesis methods. The surface groups can be mixed functional groups such as OH, O, and F atoms, or mixed F, O, and Cl atoms; they can also be single functional groups, such as Ti2CO2, Ti2CCl2, etc.38,41 Furthermore, in-plane or out-of-plane ordering at surface sites has not been observed experimentally. However, theoretical calculations show that out-of-plane ordered MXenes based on surface functional group atoms are stable (often called Janus MXene) and have special electronic, magnetic, and optical properties.54
2.2. Synthesis of MXene
Screening and prediction of MAX phases. As mentioned previously, MXene is synthesized from the precursor MAX phase. Therefore, the composition, structural characteristics, and stability of MAX directly affect the properties of MXene.38 To date, monometallic, bimetallic ordered, bimetallic solid solution and high entropy alloy MAX precursors (shown in Fig. 2(a)) have been synthesized experimentally.44,65–68 However, as the types of sites (M, A, X sites) and disorder increase, the chemical space of MAX phases expands significantly. Computational screening and prediction can discover new thermodynamically stable MAX phase structures with various compositions, guiding the synthesis of MXene with special properties.
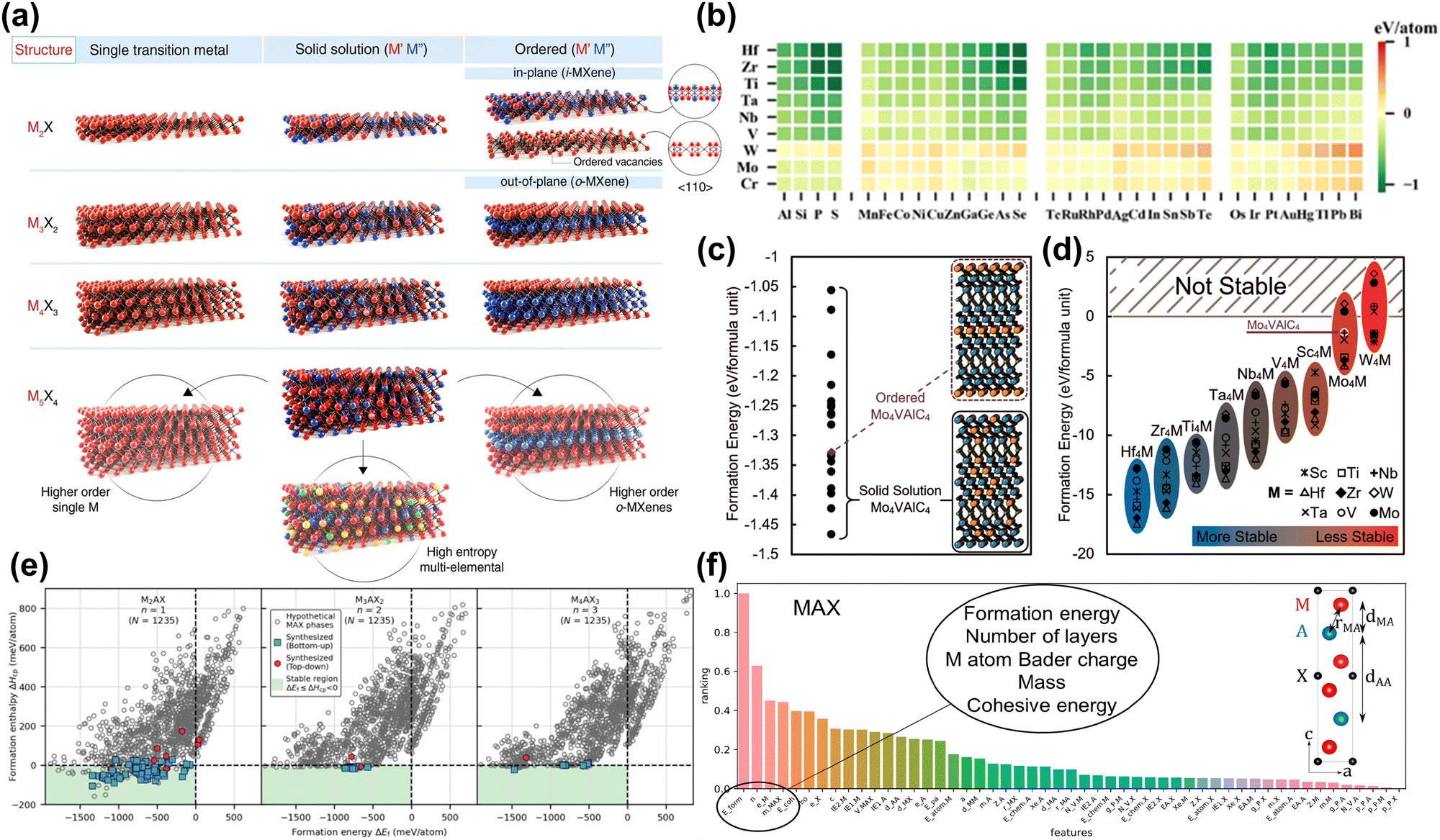 | ||
| Fig. 2 (a) Schematic illustration of the MXene structures.24 Reproduced with permission ref. 24. Copyright 2021, AAAS. (b) ΔEf for 288 M2AC phases displayed as a heatmap for 9 metals and 32 different elements at the A site.69 Reproduced with permission ref. 69. Copyright 2023, Elsevier. (c) Formation energies for solid-solution Mo4VAlC4 configurations (black dots) compared to ordered Mo4VAlC4 (purple diamond). (d) DFT calculated formation energies of ordered M′4M′′AlC4 MAX phases. Most are predicted to be stable except for some Mo and W compositions.70 Reproduced with permission ref. 70. Copyright 2019, American Chemical Society. (e) Calculated formation enthalpy ΔHcp as a function of formation energy ΔEf for ternary Mn+1AXn phases with n = 1, 2, 3. Hypothetical MAX phases are given by grey circles and synthesized MAX phases by blue squares (bottom-up synthesis) and red hexagons (top-down synthesis). The green area marks the region of thermodynamic stability fulfilling ΔHcp < 0 (and ΔEf < 0).71 Reproduced with permission, ref. 71. Copyright 2024, Elsevier. (f) Feature importance ranking for MAX. Layer distances and bond lengths are labeled in the unit cell diagrams.72 Reproduced with permission, ref. 72. Copyright 2017, American Chemical Society. | ||
To predict the stability of MAX phases, researchers often use formation energy (ΔEf) and formation enthalpy (ΔHcp) as evaluation indicators.69,73–76 Their calculation equations are as follows:
| ΔEf(Mn+1AXn) = E(Mn+1AXn) − (n + 1)μM − μA − nμX | (1) |
| ΔHcp(MAX) = E(MAX) − E(set of most competing phases) | (2) |
Calculating ΔEf involves comparing the total energy of the MAX phase with the chemical potential of its constituent elements (generally regarded as the ground state crystal structure energy of the elements). As shown in Fig. 2(b), Wu et al. evaluated the stability of 288 MAX phases based on formation energy through high-throughput computational screening, finding that 222 of these phases have negative formation energy.69 In addition, the formation energy is related to the number of valence electrons of the M-site metal, with S2C and Ti2C MAX showing the best stability. However, experimental results show that only 50 MAX phases have been synthesized, indicating that this is not a suitable stability criterion. Because only the energy change of MAX decomposing into elemental phases is considered, this method overestimates stability.30 Similarly, various research groups use formation energy to assess the stability of MAX phases.75,77 Notably, this method is reasonable when evaluating the stability of MAX phases with the same elements. Fig. 2(c) and (d) show stable ordered/disordered multi-metal (M site) MAX phases can be quickly discovered by this method.70
To ensure accurate predictions, the energy difference between the MAX phase and all possible competing phases in the M-A-X system must be evaluated. One possible solution is to evaluate the ΔHcp. This method can accurately evaluate the stability of existing MAX phases and guide the synthesis of new ones. In Fig. 2(e), Dahlqvist et al. evaluated the stability of 3705 MAX phases by calculating the ΔHcp.71 The green area is a theoretically stable area based on the criteria of ΔEf ≤ ΔHcp < 0. This shows that using ΔHcp to judge the possibility of MAX phase synthesis is more consistent with the experimental results compared to ΔEf. In addition, for MAX phases of mixed elements, such as bimetallic ordered/disorder phases and high entropy phases, configurational entropy caused by the element arrangement needs to be considered. Dahlqvist et al. also used a special quasi-random structure method to simulate the distribution of chemically disordered bimetallic sites at M sites.32 When configurational entropy was not considered (at 0 K), the solid solution MAX phase was unstable. However, after introducing configurational entropy (at 1173 K), a stable disordered MAX phase with the lowest energy was found. Through predicting stable MAX phases, bimetallic MAX phases and high entropy phases have been synthesized successfully in experimental work.48,78–80
Machine learning (ML) is a branch of artificial intelligence that focuses on building models that analyze and predict data. In materials science, ML is used to accelerate the discovery of new materials and predict material properties, thereby changing traditional research methods. For example, experimental testing and computational modeling are often time- and resource-intensive and therefore inefficient for exploring the vast space of possible material configurations. ML leverages large datasets generated by experiments or high-throughput computations to more quickly identify new materials with desirable properties. Researchers could use ML methods to accelerate the discovery of stable MAX phases. Frey et al. adopted a positive and unlabeled ML framework to predict the MAX phase.72 Positive and unlabeled machine learning is used to deal with training models using only positive and unlabeled data. This method is beneficial in scenarios where negative examples are scarce or unavailable. By leveraging the available positive instances and treating unlabeled data with sophisticated techniques, this method effectively identifies patterns and makes predictions.81 Characteristics related to thermodynamic stability, bond strength, and charge distribution were found to be most important in model predictions. In particular, as shown in Fig. 2(f), formation energy, number of layers, Bader charge of the M atoms, mass of MAX, and cohesive energy were key features for predicting the likelihood of MAX phase synthesis. Through prediction, 111 MAX phases were found to be synthesized. However, using ΔEf as an important descriptor introduces bias in the composition predictions.30
Computationally driven synthesis of MXene. Once a thermodynamically stable MAX phase is identified, the next step is the synthesis of MXenes. This process involves selective stripping of the A element from the MAX phase to obtain a 2D MXene structure.38 Computational studies are crucial for understanding and optimizing the processes of converting MAX phases into MXene. This includes modeling the chemical exfoliation process, assessing the thermodynamic stability of MXene, and predicting the feasibility of synthesizing new MXenes.
Similar to calculating the stability of the MAX phase, the ΔEf of MXene can be calculated by taking into account the chemical potentials of its constituent elements. Hu et al. used this calculation method to obtain a negative ΔEf for Sc2C, suggesting it is stable and can be synthesized.82 However, synthesizing this MXene experimentally remains unachievable. As another example, Wyatt et al. predicted 20 bimetallic o-MXenes that could be synthesized through this calculation method, but most MXenes still cannot be synthesized.83 As stated above, this calculation method is flawed and needs to consider competing phases. Afterward, Ashton et al. calculated the formation energies of 54 MXenes considering competing phases, as shown in Fig. 3(a). However, all formation energies are positive (unstable), attributed to the metastable properties of 2D materials. Moreover, there is a lack of correlation between the calculated formation energy trends and experimental results.84 In fact, MXene synthesis involves stripping the MAX phase, a process ignored by these two calculation methods.
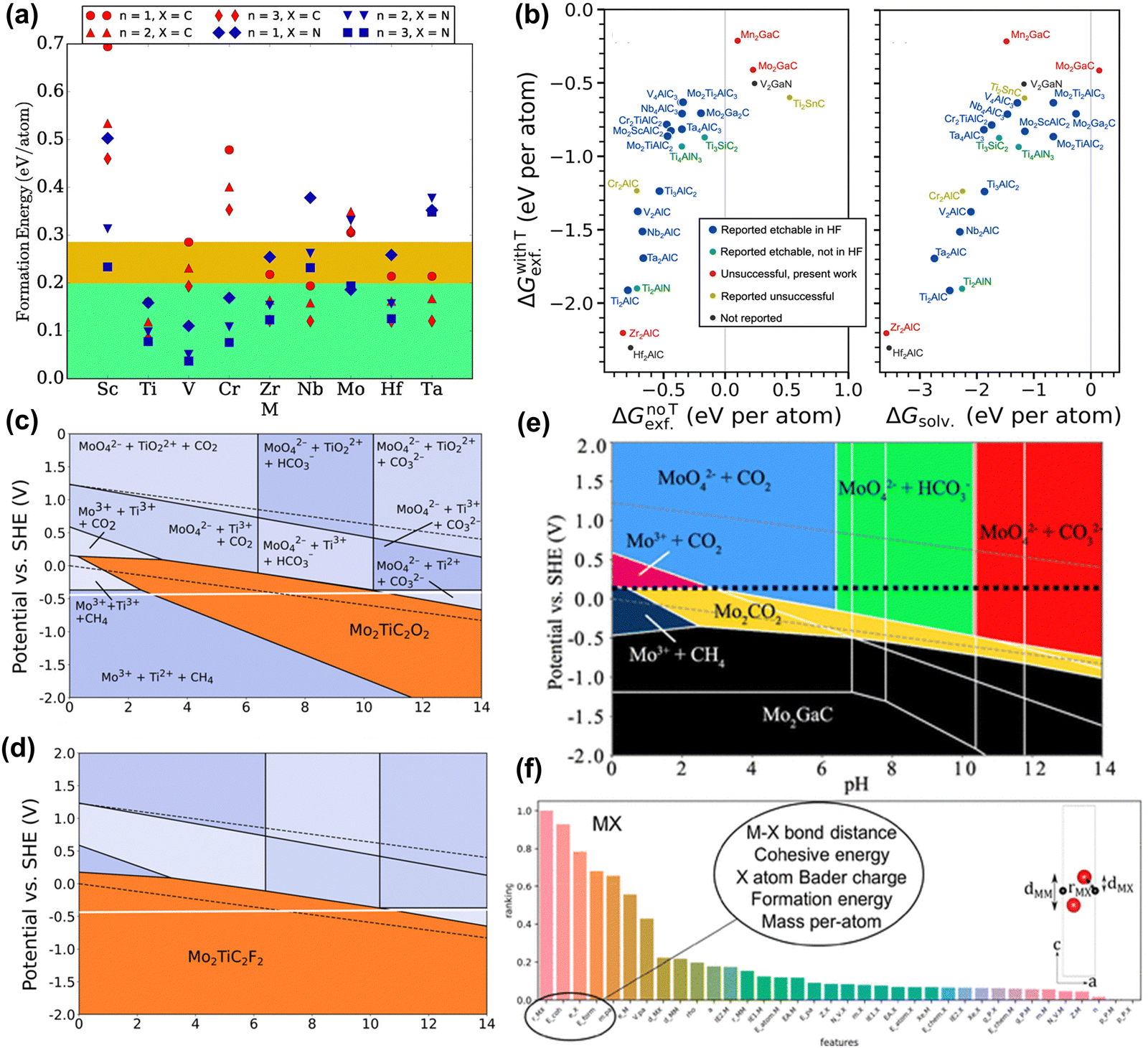 | ||
| Fig. 3 (a) Formation energies for Mn+1XnO2, MXene relative to the lowest energy mixture of competing bulk phases. The green region highlights the general 0.2 eV per atom threshold observed for 2D material stability, and the yellow region highlights the 0.285 eV per atom formation energy of the V2CO2 MXene, the highest of those that have been synthesized.84 Reproduced with permission, ref. 84. Copyright 2016, American Chemical Society. (b) Exfoliation free energies for the chemical exfoliation of MAX phases (left) when including termination groups (ΔGwithTexf) on the resulting MXene compared to the exfoliation free energy without terminations (ΔGnoTexf) and (left) the solvation free energies (ΔGsolv).85 Reproduced with permission, ref. 85. Copyright 2023, Springer Nature. Etching Pourbaix diagram for Mo2TiAlC2, showing regions where (c) Mo2TiC2O2, and (d) Mo2TiC2F2 are stable (orange segments). The dashed black lines indicate the stability range of water. The solid white line shows PZC.86 Reproduced with permission, ref. 86. Copyright 2020, American Chemical Society. (e) Etching diagrams of 2D Mo2CO2 for Mo2GaC. The PZC values are shown by the dashed black lines.87 Reproduced with permission, ref. 87. Copyright 2019, American Chemical Society. (f) Feature importance ranking for MXene models. Layer distances and bond lengths are labeled in the unit cell diagrams.72 Reproduced with permission, ref. 72. Copyright 2019, American Chemical Society. | ||
Therefore, some studies consider the MAX to MXene synthesis process as being achieved by mechanical exfoliation.30 The calculation formula of this process is as follows:
| ΔGexf = ΔEexf = [E(Mn+1Xn) + E(A) − E(Mn+1AXn)]/N | (3) |
This process does not take into account real reactions, such as the formation of functional groups. Therefore, predicting MXene synthesis using this calculation method is difficult. For example, Kahazaei et al. calculated 82 MAX phases and predicted 13 MAX phases that are easily exfoliated, but they could not experimentally synthesize the corresponding MXene phases.88
To obtain more accurate predictions, a correct description of the experimental synthesis process is necessary, fully considering the chemical potential of the etched elements A and Tx. Jonas et al. considered how to construct chemical potential in HF, and also considered the stripping energy with or without functional groups.85 The results in Fig. 3(b) showed that regardless of functional groups, most MXene phases can be synthesized through exfoliation, differing from the experimental results. This discrepancy arises because MAX may always dissolve to form new species instead of MXene when etched by HF (as shown in Fig. 3(c) and (d)). As another example, Seong et al. considered AlF3, SiF4, and H2 molecules during the HF etching process of the high-entropy MAX phase (A = Si, Al).48 The results show that the chemical exfoliation of Al is thermodynamically favorable, while the opposite is true for Si. Despite the successful synthesis of new MXene phases, the etching process still does not consider competing phases (i.e., dissolved products).
The Pourbaix diagram is a calculation method that fully considers competing phases and explores the electrochemical stability of materials. Ashton et al. considered the stability of MXene and MAX phases under the influence of electrode potential (U) and pH (Fig. 3(e)).87 The Pourbaix diagram reveals the potential and pH conditions under which Mo2CO2 is stable and can be synthesized. Tsounis et al. considered the overall stability of MXene materials. By calculating the Pourbaix diagrams of Mo2CO2 and V2CO2, MXene has the best thermodynamic stability in the region of high pH and negative potential.89 In another work, Caffery et al. used Pourbaix diagrams to predict the possibility of electrochemical etching to improve o-MXene production.86 They found that high yields of (Mo, Ti)2CTx were possible because they were stable over a wide range of pH and U. Meanwhile, the stability window of F-functionalized MXene is higher than that of an O-functionalized one. Although electrochemical etching is not a universal method for synthesizing MXene, these works can give valuable information on the etching reaction process.
ML is also used in the synthetic discovery of MXene. Instead of constructing the correct synthesis reaction process, ML needs to provide easily accessible descriptors. Frey et al. used ML methods to obtain 20 MAX/MXene combinations that were predicted to be synthesized (Fig. 3(f)).72 The most important features include M–X bond length, cohesive energy and formation energy, atomic mass, and Bader charge of M and X atoms. However, this result is considered controversial due to the mechanical peeling energy involved. More precise descriptors such as vacancy formation, chemical stripping energy, etc. need to be developed in the future.30
(1) Adjustability. The general chemical formula of MXene is Mn+1XnTx, where any variable can be altered, providing a wide chemical composition space. This is the primary distinction from other 2D materials.92 Therefore, MXene can serve as the cathode, anode, separator, or electrolyte additive in EESSs.10,11,13,93
(2) High specific surface area. MXenes have extremely high specific surface areas, providing more active sites per unit of mass or volume. For EESSs, this enhances the contact area between the electrode material and the electrolyte, increasing the rate and efficiency of the chemical reaction, and boosting the theoretical capacity of energy storage.94
(3) Excellent mechanical flexibility. The structure of MXene materials remains intact even under strong bending and stretching, crucial for flexible energy storage devices.95 When MXene is used as an intercalation electrode, volume changes during ion insertion and extraction are mitigated and their lifetime is enhanced.96,97 In addition, theoretical calculations show that tensile strain imparts additional physical and chemical properties to MXene, such as improving electrocatalytic water splitting reactions.98
(4) Layered structure and fast ion diffusion. The layered structure of MXene provides a convenient diffusion channel for ions, crucial for the rapid insertion and de-intercalation of charge carriers. This improves the rate performance and cycle stability of the battery, extending the battery life. For example, DFT calculations show that MXene can provide a lower ion diffusion barrier (i.e., rate capability) for aqueous Zn-ion batteries.27
(5) Excellent electronic conductivity. Compared to other 2D materials, MXene has superior electronic conductivity.99 This property is crucial for energy storage systems, ensuring rapid electron transfer, reducing energy loss, and increasing the charge and discharge rate.100
(6) Excellent hydrophilicity. The surface of MXene can be occupied by –OH and –O functional groups, giving it excellent hydrophilicity and better wettability between the MXene electrode and the aqueous electrolyte. Additionally, the hydrophilic MXene surface can induce uniform metal nucleation, inhibiting dendrite formation.13
(7) Abundant redox sites. TM sites in MXene can provide oxidative activity. Besides, MXene provides additional pseudo-capacitance in acidic electrolyte supercapacitors due to abundant O surface sites.11
In short, due to these excellent characteristics, MXene materials can be used to design high-capacity, long-life, and high-rate aqueous EESSs, providing valuable strategies for other fields.
3. Aqueous metal-ion batteries
Aqueous metal ion battery systems are divided into monovalent metal ion systems (e.g. Li+, Na+, K+) and multivalent metal ion (e.g. Mg2+, Zn2+, Al3+) systems according to the valence state of the charge carrier.16,17 Among them, although monovalent metal ions have mature battery technology, their relatively low theoretical capacity and higher reactivity with water limit their application in aqueous batteries.12,13 Multivalent metal ions are gaining attention due to their cost advantages from high abundance and capacity benefits from multi-electron transfer processes, making them suitable for large-scale energy storage devices.15,16 However, as shown in the Pourbaix diagram of Fig. 4(a)–(c), the narrow electrochemical window of the aqueous electrolyte (∼1.23 V) limits the use of metals (e.g. Al) as anodes.101 Only metallic Zn is an ideal candidate for aqueous batteries due to its high theoretical capacity (5851 mA h cm−3), low potential (−0.76 V vs. SHE), high abundance, environmental friendliness, and nontoxicity.13,15,102 However, aqueous Zn-ion batteries (AZIBs) still face issues such as side reactions (e.g. hydrogen evolution reaction (HER), corrosion), uncontrollable dendrites, host material structural degradation and slow diffusion kinetics.15,103–107 Recently, MXene was found to alleviate the problems of AZIBs.108–110 Therefore, we explore the role of MXene in AZIBs from a theoretical calculation perspective.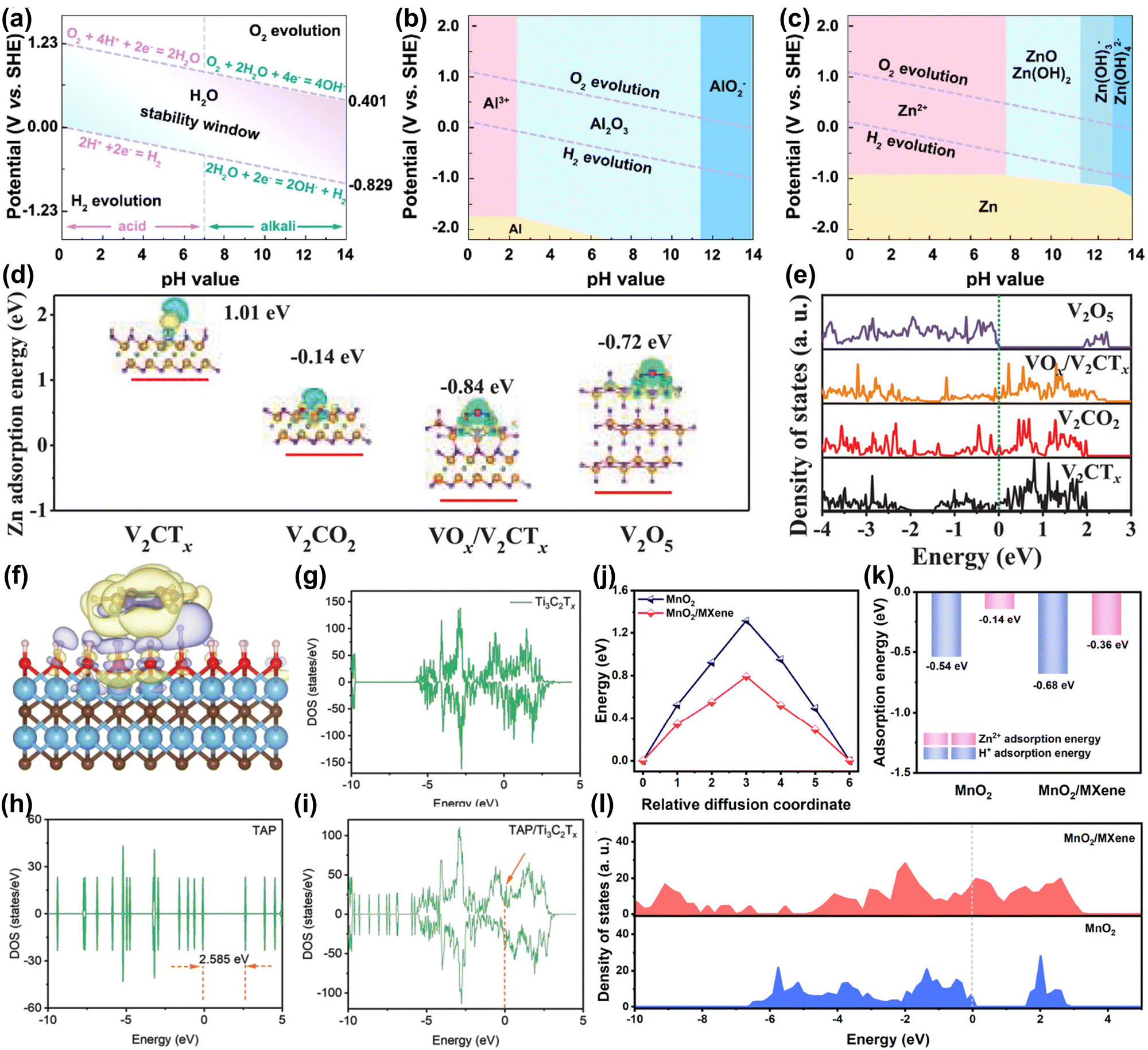 | ||
| Fig. 4 Pourbaix diagram of (a) aqueous electrolyte, metallic (b) Al and (c) Zn in aqueous electrolyte at 25 °C.13 Reproduced with permission, ref. 13. Copyright 2022, Elsevier. Zn2+ adsorption energy, charge density difference (d), and DOS (e) of V2CTx, V2CO2, VOx/V2CTx, and V2O5. (V: orange; C: greyish-green; H: red; F: grey; O: purple; Zn: pink).111 Reproduced with permission, ref. 111. Copyright 2021, John Wiley and Sons. Charge density difference (f) of TAP/Ti3C2Tx. DOS of Ti3C2Tx (g), TAP (h), and TAP/Ti3C2Tx (i).93 Reproduced with permission, ref. 93. Copyright 2022, John Wiley and Sons. Diffusion energy barriers (j) and absorption energies (k) of H+ and Zn2+ on the surface of MnO2 and MnO2/MXene. (l) DOS for MnO2 and MnO2/MXene.112 Reproduced with permission, ref. 112. Copyright 2023, American Chemical Society. | ||
3.1. MXene in cathodes
Commonly used AZIB cathode materials include organic materials, metal oxides, Prussian blue analogs, etc.13 However, these materials mainly face problems such as structural phase transition and degradation, poor conductivity, and poor rate performance.12,113 The main reasons are: (1) highly active materials cause side reactions like water decomposition and metal dissolution.14,113 (2) The insertion and extraction of high charge density Zn2+ cause structural bending and collapse. Additionally, the slow diffusion of Zn2+ and subsequent ion accumulation lead to irreversible phase transition.14,113,114 (3) Materials like organic compounds inherently have low electronic conductivity.115 Therefore, developing new materials is an effective way to alleviate these problems.Liu et al. proposed using highly conductive V2CTx materials and generating high-valent VOx by an in situ electrochemistry method to address the low conductivity and the ion diffusion rate issues of V-based cathode materials.111 In Fig. 4(d), theoretical calculations showed that original V2CTx has difficulty adsorbing Zn2+ (adsorption energy +1.01 eV). When a VOx/V2CTx heterostructure forms, the adsorption energy for Zn2+ is −0.84 eV, which accelerates the redox kinetics of stored Zn ions. Moreover, in Fig. 4(e), the high electronic state occupancy near the Fermi level indicates that the electrode has high electronic conductivity. The material achieved a specific capacity of 358 mA h g−1 at a high current density of 30 A g−1. Wang et al. designed imine-based tris(aza)pentacene (TAP)/MXene composites to alleviate the problems of poor conductivity and structural degradation of organic electrodes.93 In Fig. 4(f), theoretical calculations reveal that the strong electron transfer interaction between Ti3C2Tx and TAP helps maintain the structure of TAP. The density of state (DOS) results in Fig. 4(g–i) show that introducing Ti3C2Tx enhances the conductivity of the electrode. Ultimately, the electrode achieves a long cycle life (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times) and a high-capacity retention rate (81.6%). Besides, Wang et al. used MXene to synergistically improve the ion/electron transfer kinetics and structural stability of MnO2 (Fig. 4(j and k)).112 Calculation results showed that MXene/MnO2 has a strong adsorption capacity and low diffusion energy barrier for H+/Zn2+. Besides, the heterostructure increases the electronic state density of electrons at the Fermi level and promotes the transfer of electrons. Wang et al. constructed a MOF/MXene heterostructure material to achieve durable and fast Zn-ion batteries. Although porous 2D MOF is an ideal storage and transport material for Zn ions, its inherent low conductivity leads to low capacity and power density. Theoretical calculations show that MXene can improve the conductivity of MOFs. Therefore, MOF/MXene exhibited a superior electrochemical performance (260.1 mA h g−1 at 0.1 A g−1).116
000 times) and a high-capacity retention rate (81.6%). Besides, Wang et al. used MXene to synergistically improve the ion/electron transfer kinetics and structural stability of MnO2 (Fig. 4(j and k)).112 Calculation results showed that MXene/MnO2 has a strong adsorption capacity and low diffusion energy barrier for H+/Zn2+. Besides, the heterostructure increases the electronic state density of electrons at the Fermi level and promotes the transfer of electrons. Wang et al. constructed a MOF/MXene heterostructure material to achieve durable and fast Zn-ion batteries. Although porous 2D MOF is an ideal storage and transport material for Zn ions, its inherent low conductivity leads to low capacity and power density. Theoretical calculations show that MXene can improve the conductivity of MOFs. Therefore, MOF/MXene exhibited a superior electrochemical performance (260.1 mA h g−1 at 0.1 A g−1).116
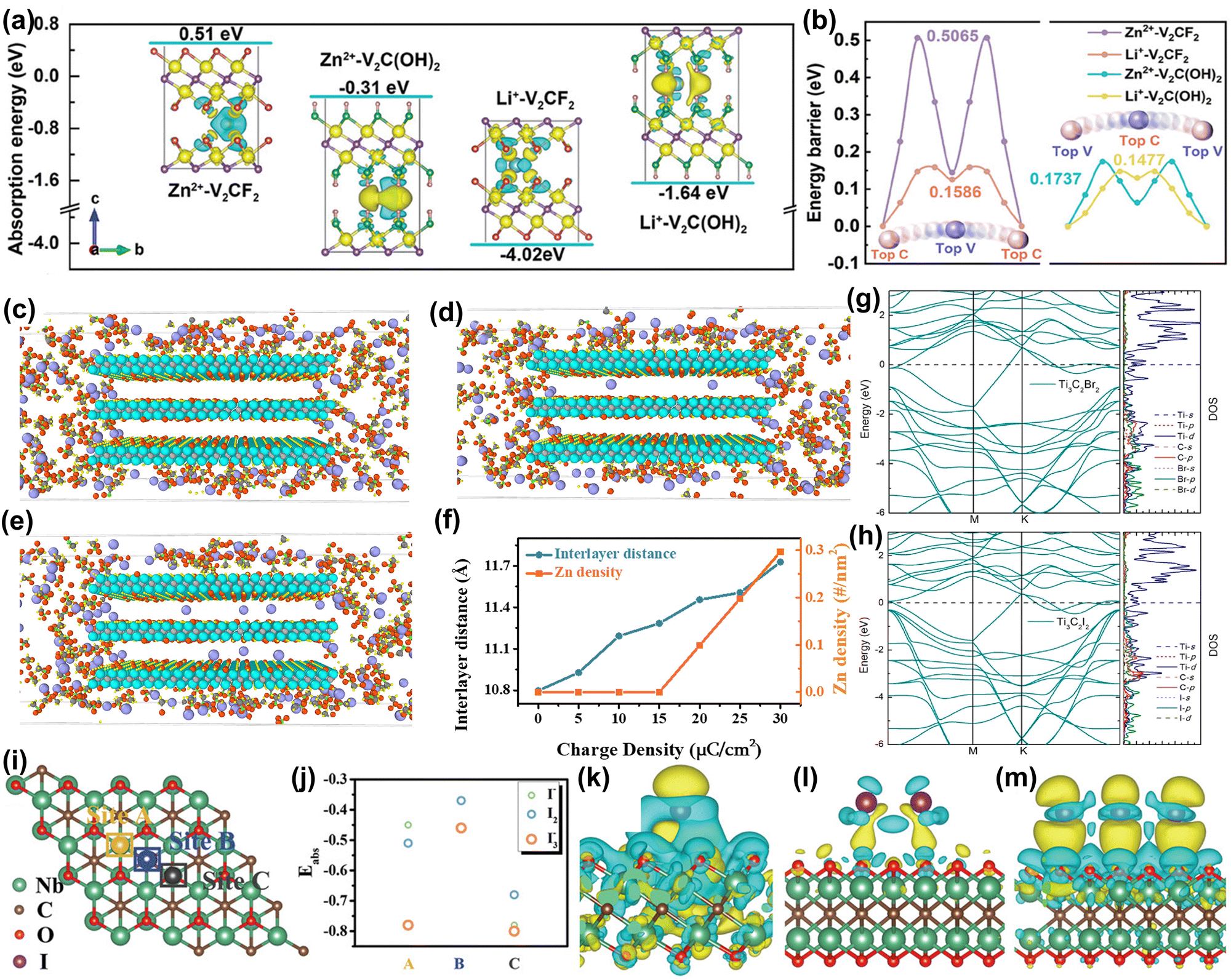 | ||
| Fig. 5 (a) The absorption energies of Li+/Zn2+ with V2CF2 and V2C(OH)2 models. Insets provide the corresponding differential charge density diagrams, where the yellow and blue domains represent electron accumulation and depletion, respectively. (b) The migration energy barriers of Li+/Zn2+ in V2CF2 and V2C(OH)2 lattices.117 Reproduced with permission ref. 117. Copyright 2024, John Wiley and Sons. Configurations of the Nb2CTx cathode were assigned with a charge density of (c) 15 μC cm−2, (d) 20 μC cm−2, and (e) 30 μC cm−2, where the purple, grey, cyan, red, yellow, and green spheres represent Zn, C, Nb, O, F, and S atoms, respectively. (f) Dependence of interlayer distance and inserted Zn density on the charge density of the Nb2CTx cathode. Zn2+ can only enter the interlayer when the charge density increases to 20 μC cm−2.118 Reproduced with permission, ref. 118. Copyright 2021, Cell Press. Electronic structure of (g) Ti3C2Br2 and (h) Ti3C2I2.119 Reproduced with permission, ref. 119. Copyright 2021, American Chemical Society. (i) Crystal model of the Nb2CTx host with the proposed adsorption sites. (j) Calculated adsorption energy of iodine species (I−, I2, and I3−) on Nb2CTx flakes at different sites. Final optimized charge-density-difference patterns of I− (k), I2 (l), and I3− (m) on the Nb site. The iso-surface value is set to be 0.001 e Å−3.120 Reproduced with permission ref. 120. Copyright 2021, John Wiley and Sons. | ||
In addition, MXene electrodes usually exhibit capacitive behavior, leading to a rapid drop in output voltage during discharge, which limits their energy density. Li et al. activated MXene battery behavior by 2.4 V high-voltage scanning, and the Nb2CTx cathode gradually showed stable Zn2+ storage behavior.118 The battery behavior of MXene results in a 92% higher capacity compared to capacitive behavior. To understand how high-voltage scanning triggers the voltage platform, MD simulation was used. By applying classical mechanics, MD is a computational simulation method used to study the physical movements of atoms and molecules, and provides insights into the structure, dynamics, and thermodynamics of complex molecular systems over time. Simulation results in Fig. 5(c–f) showed that Zn2+ ions can only enter the cathode interlayer when the applied charge density increases to 20 μC cm−2. At lower charge density, the ions cannot overcome the resistance to achieve insertion/extraction and are only adsorbed on the surface or edge area, showing typical pseudocapacitive behavior. In short, high voltage triggers the initial insertion/extraction of Zn2+ ions, achieving the high energy density of the MXene positive electrode.
Li et al. proposed a new method to directly synthesize Ti3C2 MXene with single, binary, and ternary halogen end groups (such as –Cl, –Br, –I, –BrI, and –ClBrI). Among these, Ti3C2Br2 and Ti3C2I2 showed clear discharge platforms and high specific capacities.119 The calculated band structure in Fig. 5(g) and (h) shows strong hybridization of Ti d, C p, and Br/I p orbitals in the energy range of −2 to −6 eV, indicating that the outermost Ti atoms form strong coordination bonds with carbon and halogen atoms. In addition, Li et al. used a simple electrodeposition strategy to insert and confine iodine species between MXene layers, effectively suppressing the shuttle effect.120 Calculation results in Fig. 5(i) and (j) showed that the adsorption of I species on the MXene surface is spontaneous due to high adsorption stability. Fig. 5(k–m) shows that differential charge density results indicate a strong electronic interaction between I species and the MXene surface, beneficial for achieving fast redox kinetics. Finally, the long cycle life and high-rate capability of Zn–I2 aqueous batteries were achieved. Similarly, they found that this method can be applied to the Zn–Br2 aqueous battery system, achieving high energy density and low-temperature cycle stability.122 Calculation results showed that this is due to the strong electronic interaction between the MXene surface and the Br species, which benefits the fixation of Br2 and the rapid transfer of electrons.
3.2. MXene in anodes
Metallic Zn is an ideal anode for AZIBs due to its high abundance, safety, suitable potential, and high capacity. However, there are still some challenges for Zn metal anodes: (1) uneven nucleation of metal Zn can lead to dendrite formation and promote side reactions.103,123,124 (2) Acidic solutions can exacerbate metal corrosion, HER, and passivation effects.12 Among them, the uncontrollable dendrite problem of Zn anode is the main obstacle for AZIBs. At present, various strategies, such as regulating the interfacial electric field, Zn deposition process, Zn coordination environment, etc., are used to solve this problem.125–127 Recently, researchers have applied MXene materials to anodes to enhance Zn metal anode performance, mitigate Zn dendrite growth and side reactions, and elucidate the mechanism of MXene action through computational simulation.26–29,35,109,128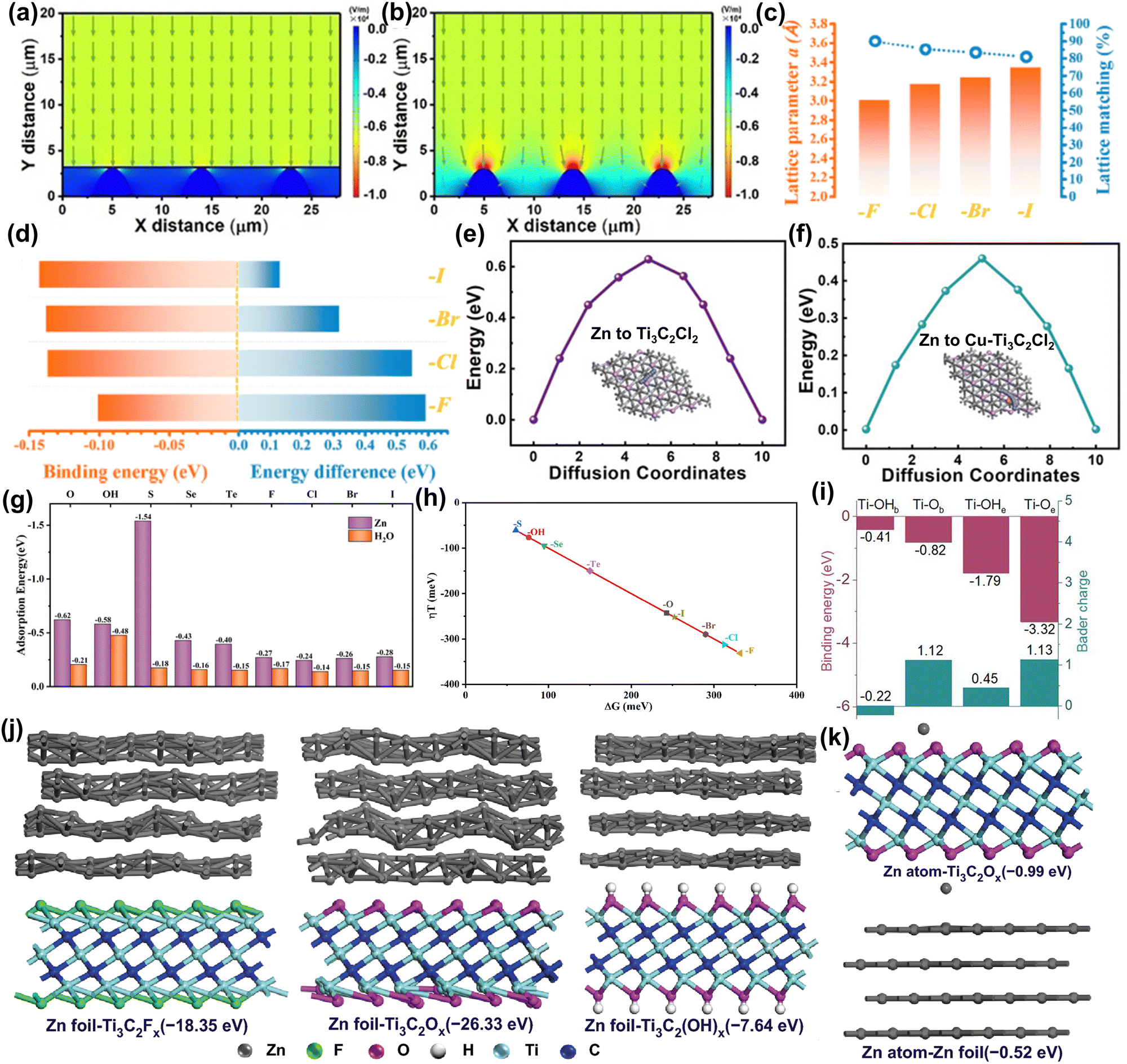 | ||
| Fig. 6 Models of the electric field distributions for (a) MXene-coated Zn foil and (b) pure Zn.109 Reproduced with permission, ref. 109. Copyright 2021, John Wiley and Sons. (c) Lattice parameters vs. calculated lattice matching ratios of halogen-terminated Ti3C2(F/Cl/Br/I)2. (d) Calculated binding energy between two Zn atoms on different MXene surfaces vs. the energy difference for the later Zn atom spread on an MXene surface or stacked on the previous Zn atom.26 Reproduced with permission, ref. 26. Copyright 2021, American Chemical Society. Diffusion energy of Zn on (e) Ti3C2Cl2 and (f) Cu–Ti3C2Cl2 for the original, transition, and final states.27 Reproduced with permission, ref. 27. Copyright 2023, John Wiley and Sons. (g) The adsorption energy of Zn ions and water on MXene surfaces with different functional groups. (h) The volcano plot of the calculated overpotential (η = ±ΔG/e0) vs. ΔG on Ti3C2Tx.28 Reproduced with permission, ref. 28. Copyright 2024, John Wiley and Sons. (i) Binding energies and charge transfer number between the Zn atom and functional groups (Ti–OHb, Ti–OHe, Ti–Ob, and Ti–Oe) on Ti3C2.129 Reproduced with permission, ref. 129. Copyright 2023, American Chemical Society. Calculation models corresponding binding energies of Zn foil (j) adsorbed on Ti3C2Tx and Zn atoms (k) absorbed on Zn foil, Ti3C2Ox.130 Reproduced with permission, ref. 130. Copyright 2021, Springer Nature. | ||
MXene has shown an important role in protecting Zn metal anodes, but the protection mechanism of different functional groups is still unclear. Therefore, Luo et al. used DFT calculations and MD simulations to study the protection mechanism of MXene with nine different surface functional groups (−O, –OH, –S, –Se, –Te, –F, –Cl, –Br, and –I).28 The study found that all tested MXene materials showed good electrical conductivity, structural stability, and stronger adsorption capacity for Zn ions relative to water molecules (Fig. 6g). In particular, MXene containing sulfur (–S) and oxygen (–O) functional groups showed high adsorption energy for Zn2+, promoting uniform Zn ion deposition and reduced water contact with the anode, thus enhancing the stability of the anode. In terms of HER side-reaction, as shown in Fig. 6(h), halogen-terminated MXene showed a strong inhibitory effect with a higher absolute value of ΔG. In short, by rationally designing the surface functional groups of MXene, a dendrite-free and side-reaction-free Zn anode can be achieved.
3.3. MXene for separators and electrolytes additives
In addition to being used in cathode and anode electrodes of AZIBs, MXene can also be used as separators and electrolyte additives to improve the performance of AZIBs. To address the inevitable zinc dendrite formation of AZIBs, Bu et al. proposed using a polypropylene separator wrapped with mesoporous Ti3C2 MXene with active edge sites to eliminate dendrites.129 The edge sites in Ti3C2 can convert the formed dead zinc (Zn0) dendrites back into electroactive zinc ions (Zn2+) through spontaneous redox reactions. DFT calculation results in Fig. 6(i) show that the binding energy between the edge Ti–O sites and Zn atoms is the highest, and the Bader charge results show that Zn transfers more electrons to MXene. This means that they can extract electrons from Zn0 dendrites more effectively, promoting the dendrite digestion process. This method enables AZIB to exhibit an ultra-long cycle life of 2200 hours. Sun et al. explored the role of Ti3C2Tx as an electrolyte additive in AZIBs.130 They found that MXene additives provide abundant Zn-affinity groups (such as –OH, –F, and –O) and good conductivity. DFT calculations in Fig. 6(j) and (k) show that all functionalized MXene have a strong binding ability with Zn, among which O-terminated MXene can provide seed sites for uniform nucleation of Zn ions. The weak binding energy between Zn ions and substrates induces only a small number of nucleation sites. In short, MXene electrolyte additives can control the Zn ion nucleation and growth to inhibit the formation of zinc dendrites.4. Aqueous supercapacitors
In addition to batteries, supercapacitors are also a key component of electrochemical energy storage systems.132,133 Due to the fast charge response, supercapacitors show higher power density, although the energy density is low.132 For aqueous supercapacitors, although they have advantages in price, safety, and environmental protection, the inherent narrow electrochemical window further limits the improvement of energy density.133–135 To achieve wider applications, they still need to make breakthroughs in energy density.132 In recent years, MXene has greatly improved the performance of aqueous supercapacitors due to its unique advantages (as described in Section 2.2.3).134,136,137 First, most MXenes are highly conductive metallic materials characterized by a high density of states at the Fermi level.24Second, the layered two-dimensional structure of MXenes facilitates rapid ion transport and provides redox reaction sites, resulting in high rates and long cycle life.11 Additionally, the transition metal sites and surface functional groups of MXenes are adjustable, allowing for both double-layer capacitance and rapidly reversible redox reactions at or near the surface (i.e., pseudo-capacitor).10 Finally, the hydrophilic surface of MXenes facilitates efficient charge storage and enables rapid charge and discharge cycles.13Table 1 summarizes the capacitance data for various materials in aqueous electrolytes. The results indicate that MXenes exhibit ultra-high capacitance and rate performance. Given the promising potential of MXenes in aqueous supercapacitors, a thorough understanding of their energy storage mechanisms is essential for advancing supercapacitor technology. Therefore, this discussion begins with the charge storage mechanism of supercapacitors, exploring the capacitance mechanism of MXenes in detail through theoretical calculations.
| Materials | Electrolyte | Capacitance (scan rate) | Stability (retention/cycles/scan rate) | Energy density (W h kg−1) | Power density (W h kg−1) | Ref. |
|---|---|---|---|---|---|---|
| Co9S8 | 3 M KOH | 1775 at 4 A g−1 | 91.4% after 2000 cycles at 16 A g−1 | 49.9 at 4 A g−1 | 900 at 4 A g−1 | 138 |
| MoS2 | 0.5 M Li2SO4 | 350 F g−1 at 5 mV s−1 | 88.0% after 10000 cycles at 5 A g−1 |
50 at 1 A g−1 | 1000 at 1 A g−1 | 139 |
| PEDOT | 1 M KCl | 117 F g−1 at 100 mV s−1 | 86.0% after 1000 cycles at 0.2 A g−1 | 11.4 at 0.2 A g−1 | 100 at 0.2 A g−1 | 140 |
| PPy/Graphene | 1 M H2SO4 | 626 F g−1 at 0.22 A g−1 | 75.4% after 5000 cycles at 4 A g−1 | 21.7 | 110 | 141 |
| Co-MOF | 3 M KOH | 534 F g−1 at 1 A g−1 | 99% after 5000 cycles at 5 mV s−1 | 9.62 at 4 mA cm−2 | 1454 at 4 mA cm−2 | 142 |
| ZnCo2O4–MnO2 | 3 M KOH | 2057 F g−1 at 1 A g−1 | 99% after 5000 cycles at 15 A g−1 | — | 143 | |
| Ti3C2Tx | 3 M H2SO4 | 210 F g−1 at 10 V s−1 | — | 144 | ||
| Ti3C2Tx | 1 M H2SO4 | 429 F g−1 at 1 A g−1 | 89% after 5000 cycles at 10 A g−1 | 29.2 at 1 A g−1 | 320 at 1 A g−1 | 145 |
4.1. Supercapacitor charge storage mechanism
Battery systems can store higher energy density based on Faraday reactions but are limited by the diffusion capacity of charge carriers, resulting in low charge and discharge rates.146 In contrast, supercapacitors achieve fast charge and discharge rates due to a fast charge response but have low energy density.147 Supercapacitors can be classified into electrical double-layer capacitors (EDLCs) and pseudo-capacitors according to the charge storage mechanism of the electrodes.148,149The charge storage of EDLCs is not a Faraday process. Charge is stored and released only by physical adsorption and desorption of electrolyte ions on the surface of electrode materials (as shown in Fig. 7(a)). In this energy storage conversion process, the formation of the electrical double-layer (EDL) takes a very short time, and no electron exchange occurs on the EDL, so high power density and fast charging and discharging can be achieved. This surface charge storage process will cause only slight volume changes in the electrode material during long-term operation, thereby achieving excellent cycle stability.132
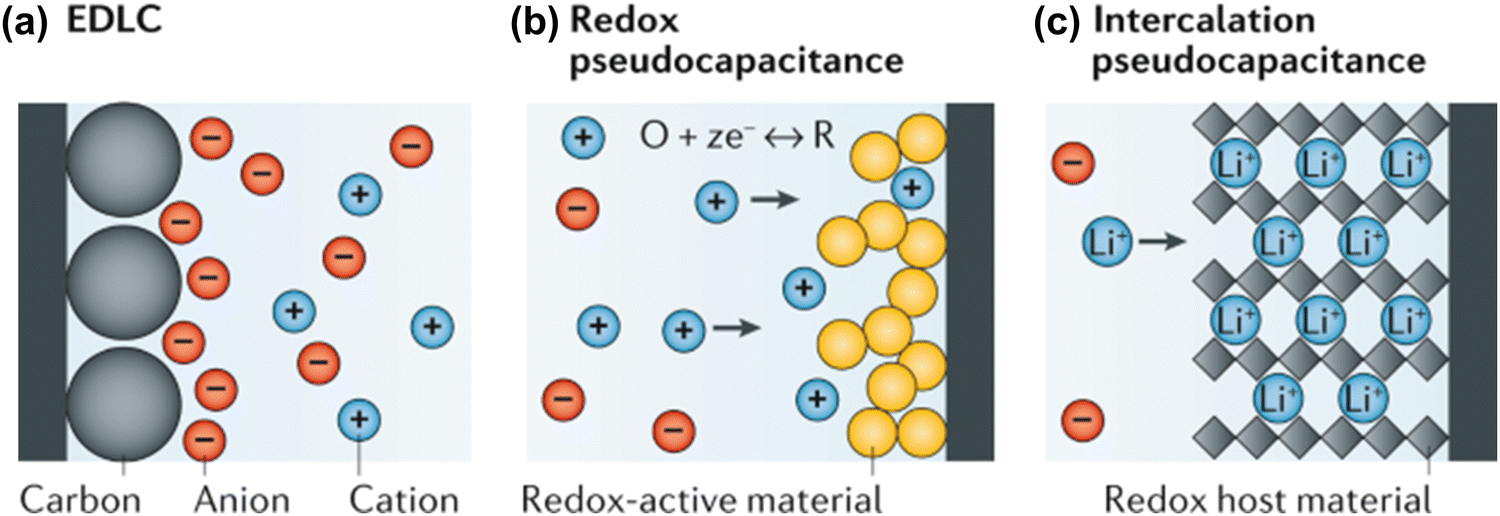 | ||
| Fig. 7 Electrochemical charge-storage mechanisms: (a) ELDC, (b) Redox pseudo-capacitance and (c) intercalation pseudo-capacitance.149 Reproduced with permission, ref. 149. Copyright 2020, Springer Nature. | ||
Pseudo-capacitors achieve capacitive charge storage through the Faraday process. Because the charge transfer process occurs on or near the surface and is not controlled by semi-infinite diffusion, its charge and discharge rate are comparable to that of EDLCs. Pseudo-capacitors are mainly divided into two types according to their working mechanism: redox pseudo-capacitors and intercalation pseudo-capacitors.149 Redox capacitors are when ions are adsorbed on the interface of electrode materials and undergo a charge transfer process with nearby electrode materials (i.e., the redox reaction process shown in Fig. 7(b)), thereby achieving energy storage.
Intercalation pseudo-capacitors are capacitors that store energy by inserting electrolyte ions into the interlayer or pores of electrode materials, which involves a charge transfer process (as shown in Fig. 7(c)). The electrode reaction of the battery is also an ion intercalation process similar to that of the intercalation pseudo-capacitor. Unlike the electrode reaction of the battery, the intercalation pseudo-capacitor does not involve a phase change reaction during the Faraday redox reaction and is not diffusion-controlled due to the rapid ion transport process.53
4.2. Computational understanding of MXene for aqueous supercapacitors
Neutral and alkaline electrolytes. The hydrated metal cations confined in 2D MXene nanosheets are believed to improve capacitance by forming an EDL. First, Sugahara et al. found that water molecules confined in the MXene slits can enhance the EDLC, with the larger-size Li+-hydration layer providing the most capacitance (Fig. 8(a)).150 Besides, they used the three-dimensional reference interaction site model (3D-RISM) to evaluate the hydration structure of various alkali ions in the MXene microslit. 3D-RISM is a computational approach used to study molecular solvation and interactions at the atomic level. It allows for detailed analysis of solvation structures, free energies, and molecular interactions.151 The results show that the highest water density is found in the Li+-intercalated MXene in Fig. 8(b and c). Also, in Fig. 8(d and e), they observed that the direction of the electric field of Li intercalation was reversed inside the intercalation hydration layer, meaning a negative dielectric constant of the hydration layer, leading to enhanced capacitance. This phenomenon is caused by the resonance effect between the dipole polarization of water molecules and the external electric field, excessively shielding the external electric field. Ando et al. revealed the charge storage mechanism of MXene electrodes intercalated with hydrated metal cations through DFT calculations.94 From an electronic perspective, fully hydrated metal cation intercalation only produces EDL capacitance behavior, attributed to the hydrated shell shielding the orbital coupling between cations and MXene (Fig. 8(f), left). In contrast, partially or completely de-solvated cations are coupled with MXene electronic orbits, resulting in pseudo-capacitance (as shown in the right figure of Fig. 8(g)). Besides, Simon et al. pointed out that due to the small number of intercalated metal cations, the resulting pseudo-capacitance can be ignored compared to EDLC.133
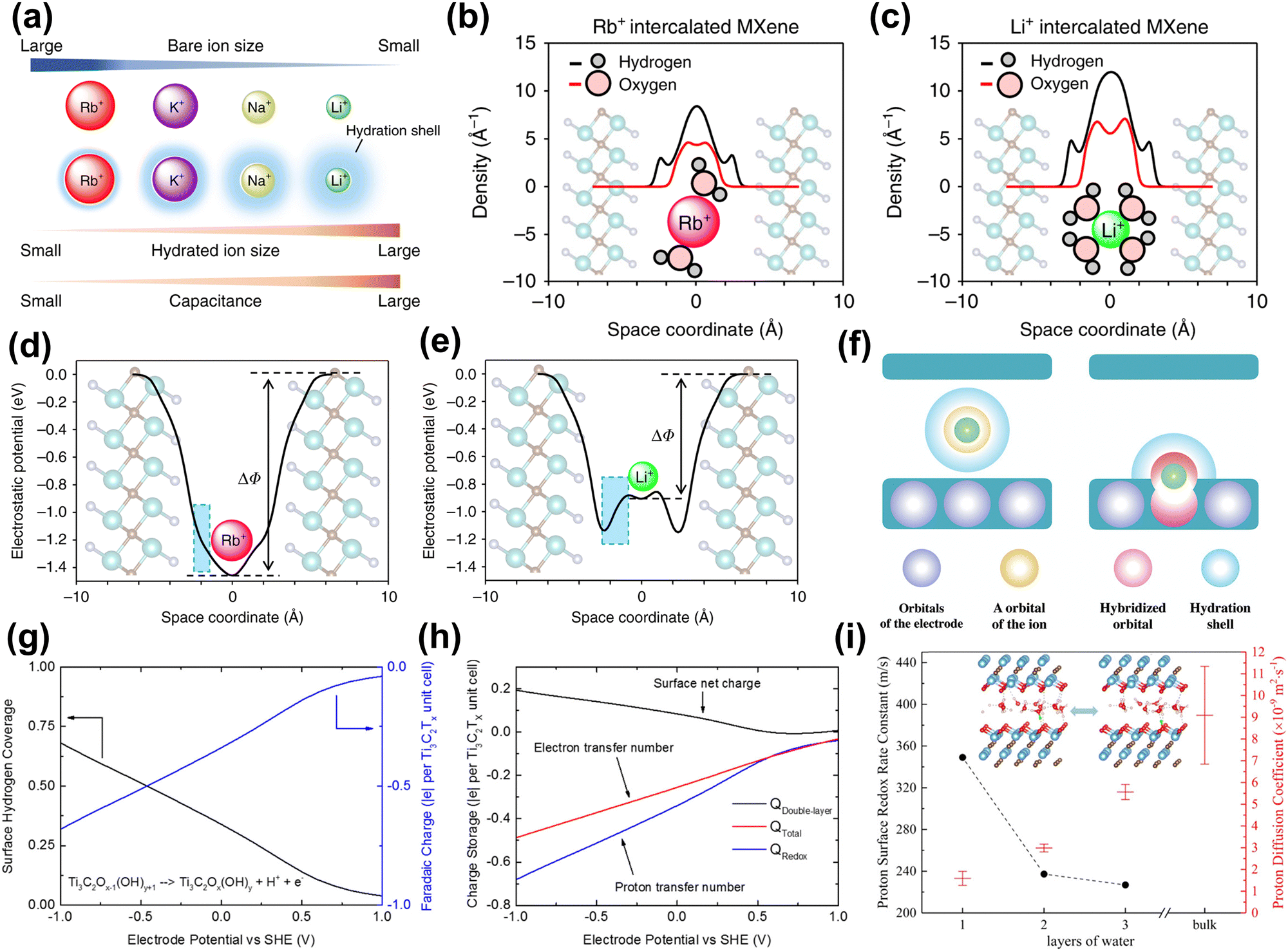 | ||
| Fig. 8 (a) Orders of bare-ion size, hydrated ion size, and observed capacitance. (b) and (c) 3D-RISM calculation results for the hydrated ions confined in the MXene microslit. Hydrogen and oxygen atomic density profiles along the c axis (perpendicular to the MXene layers) in Rb+ intercalated and Li+ intercalated Ti2CTx·nH2O. The optimized n values are 0.8, and 1.35 for the Rb+ intercalated and Li+ intercalated MXene, respectively. (d) and (e) Theoretical calculation for the electrostatic potential profile of Rb+- and Li+-intercalated Ti2CTx·nH2O.150 Reproduced with permission, ref. 150. Copyright 2019, Springer Nature. (f) Schematic pictures of the capacitive and pseudocapacitive conditions formed inside the MXene electrodes. Green sphere represents a cation. Purple, yellow, red, and blue spheres indicate orbitals of an electrode, orbital of the ion, hybridized orbital of the electrodes and cations, and the hydration shell.94 Reproduced with permission, ref. 94. Copyright 2020, John Wiley and Sons. (g) Average H coverage of Ti3C2Tx in 1 M H2SO4 at different electrode potentials. (h) Faradaic charge (blue, to balance proton transfer), EDL charge (black, due to surface net charge), and total charge (red, net electron transfer number) stored at different electrode potentials.34 Reproduced with permission, ref. 34. Copyright 2018, American Chemical Society. (i) The average number of water molecules hydrogen-bonded to the hydronium ion for different layers of water with protons confined in Ti3C2O2 layers. The inset is a schematic diagram of the structure of the intercalated water-MXene model.25 Reproduced with permission, ref. 25. Copyright 2019, American Chemical Society. | ||
Acidic electrolytes. Compared with alkaline (or neutral) electrolytes, MXene electrodes under acidic electrolytes exhibit higher capacitance due to pseudo-capacitance. This is because the proton undergoes a rapid redox reaction with the MXene surface. Jiang's group revealed the surface redox process of Ti3C2Tx (T = O, OH) MXene in 1 M H2SO4 electrolyte at constant-electrode potential through DFT calculations and implicit solvation models.34 The calculation results in Fig. 8(h) showed that based on the experimental electrochemical window (0.5 V to −1.0 V vs. SHE), the net surface charge of 0.2|e| per unit MXene was observed, which means that the redox reaction dominates the capacitance. After that, they further investigated the redox process of Ti3C2O2 with protons under the explicit solvent model (acidic electrolyte).25 As shown in Fig. 8(i), the study found that in interlayer-restricted water, protons are rapidly and reversibly exchanged between the MXene surface and interfacial water molecules. In addition, the surface redox rate is related to the number of intercalated water layers, and single-layer water has the highest redox rate.
Ji et al. calculated the work function consistent with a standard hydrogen electrode (SHE) and DOS to obtain the intrinsic capacitance (i.e., quantum capacitance) of Ti2CT2.152 The calculation results show that the predicted pseudo-capacitance of O-functionalized MXene is 56.69 F g−1 in a neutral solution. In addition, they calculated the integrated capacitance of Ti2CF2 and Ti2CO2 in Na ion capacitors (Fig. 9(a)), reaching 291 F g−1 and 252.2 F g−1, respectively. Combined with the low diffusion barrier of Na ions, MXene is considered to be a good intercalation pseudo-capacitor material. In another work, they used a similar method to explore the effects of terminals (F, O, and OH) and coordinated atoms on capacitance. The results showed that O-functionalized MXene mainly exhibits pseudocapacitive behavior in aqueous electrolytes due to a large number of unoccupied partial DOS (PDOS) of Ti atoms above the Fermi level within the electrolyte window (Fig. 9(b)). In contrast, bare-, F-, and OH-functionalized MXene exhibit EDL characteristics. In addition, the calculation method of quantum capacitance has been used to explore other MXene materials.153–156
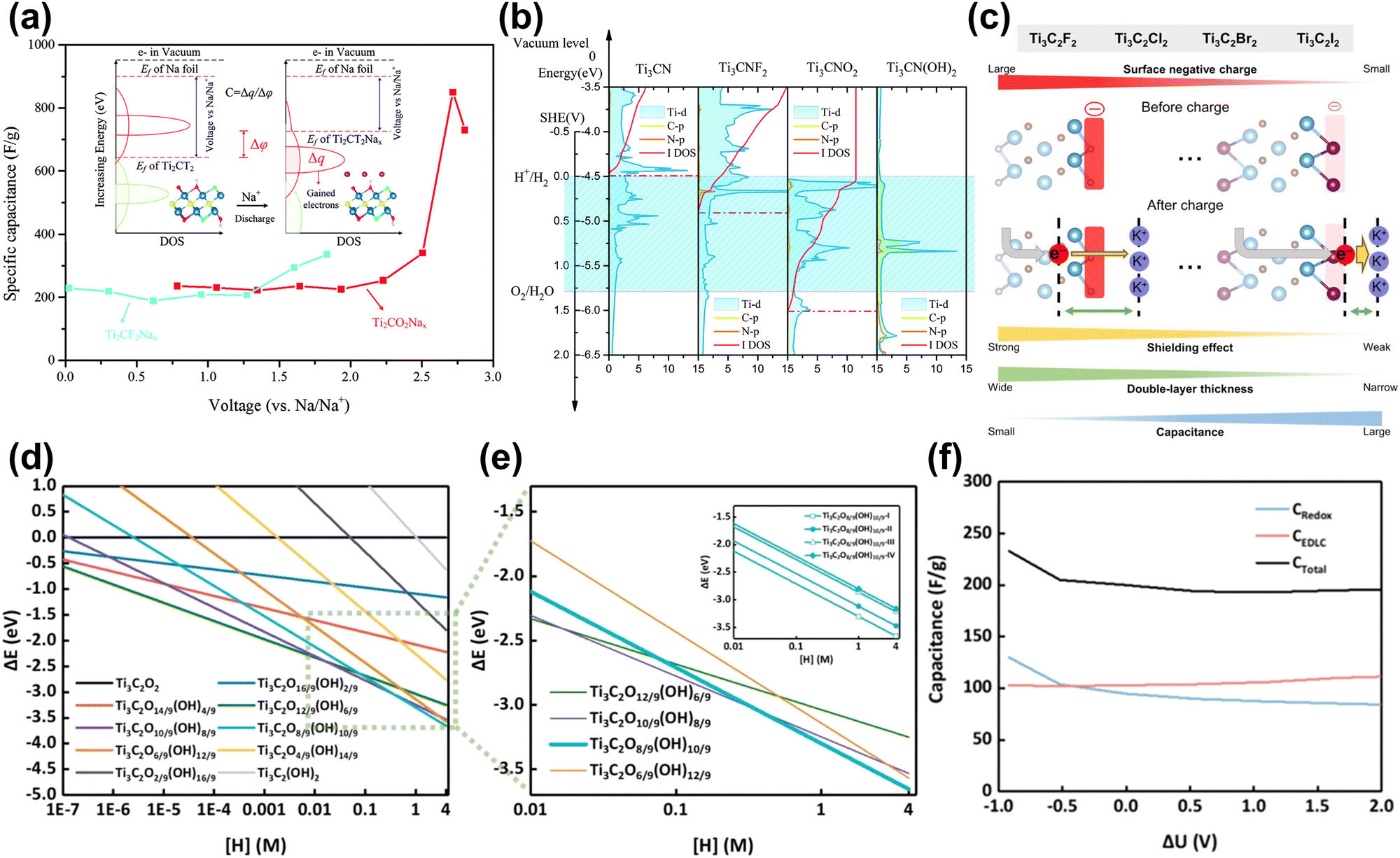 | ||
| Fig. 9 (a) Specific capacitance of Ti2CO2 and Ti2CF2 nanosheets, and the calculated method is illustrated as the inset.152 Reproduced with permission, ref. 152. Copyright 2016, Royal Society of Chemistry. (b) Relative PDOS and integral DOS of the d-orbitals of Ti atoms referenced to SHE. The yellow and red lines show the PDOS of C atoms and N atoms, respectively. The red dot lines are the Fermi level positions. The blue region represents the electrolyte window. The Fermi level of Ti3CN(OH)2 cannot be marked due to outside of the y-axis range, which lies approximately 1.82 eV relative to the vacuum level.157 Reproduced with permission, ref. 157. Copyright 2016, Royal Society of Chemistry. (c) Schematics showing the electronic impact of the MXene electrode surface on EDL capacitances depending on the surface functional groups.158 Reproduced with permission, ref. 158. Copyright 2022, American Chemical Society. (d) Surface Pourbaix diagram of MXene surfaces with various –OH groups as a function of the concentration of hydrogen ions. (e) Dashed-rectangle box-highlighted Pourbaix diagram of MXene surfaces under strongly acidic conditions. The formation energy of each surface is indicated by thin lines, while the most stable surfaces are represented by bold lines. The Pourbaix diagram of the most stable surface with different -OH group distributions Ti3C2O8/9(OH)10/9-(I, II, III or IV) is the inset. (f) Predicted capacitance performance.159 Reproduced with permission, ref. 159. Copyright 2024, American Chemical Society. | ||
The real charging and discharging process is more complicated and requires additional constraints. Cheng et al. introduced implicit solvent models and constant electrode potentials into the theoretical calculations of supercapacitors.34 They examined the point of zero charge (PZC) of MXene electrodes at different hydrogen coverage ratios. The results showed that in the redox-dominated region (<0.5 V vs. SHE in Fig. 8(h)), the integrated capacitance of Ti3C2Tx was ∼230 F g−1 close to the experimental value. In subsequent work, they used this method and combined it with high-throughput computational screening to find a series of MXene electrodes with high capacitance, among which Ti2N exhibited the highest specific capacitance of over 450 F g−1.160 Shimada et al. calculated the EDLC of halogen (F, Cl, Br, I) functionalized MXene by the ESM-RISM method (a hybrid DFT-solvation method).158,161 The results showed that the capacitance per unit surface area increases with the increase of the halogen atomic number (F < Cl < Br < I). This is because the functional groups with low electronegativity help introduce electrode electrons into the EDL region, thereby increasing the capacitance (Fig. 9(c)). Zheng et al. then used Pourbaix diagrams in Fig. 9(d and e) to study the distribution of functional groups on the surface of MXene in acidic electrolytes and calculated the EDLC and pseudo-capacitance based on the real surface. The results in Fig. 9(f) showed that the theoretical capacitance of MXene was in strong acid ranges from 200 F g−1 to 233 F g−1. In addition, Wang et al. considered the charge storage and release process during capacitor operation.162 They predicted that the capacitance of the original Ti3C2 could be as high as 2131 F g−1.
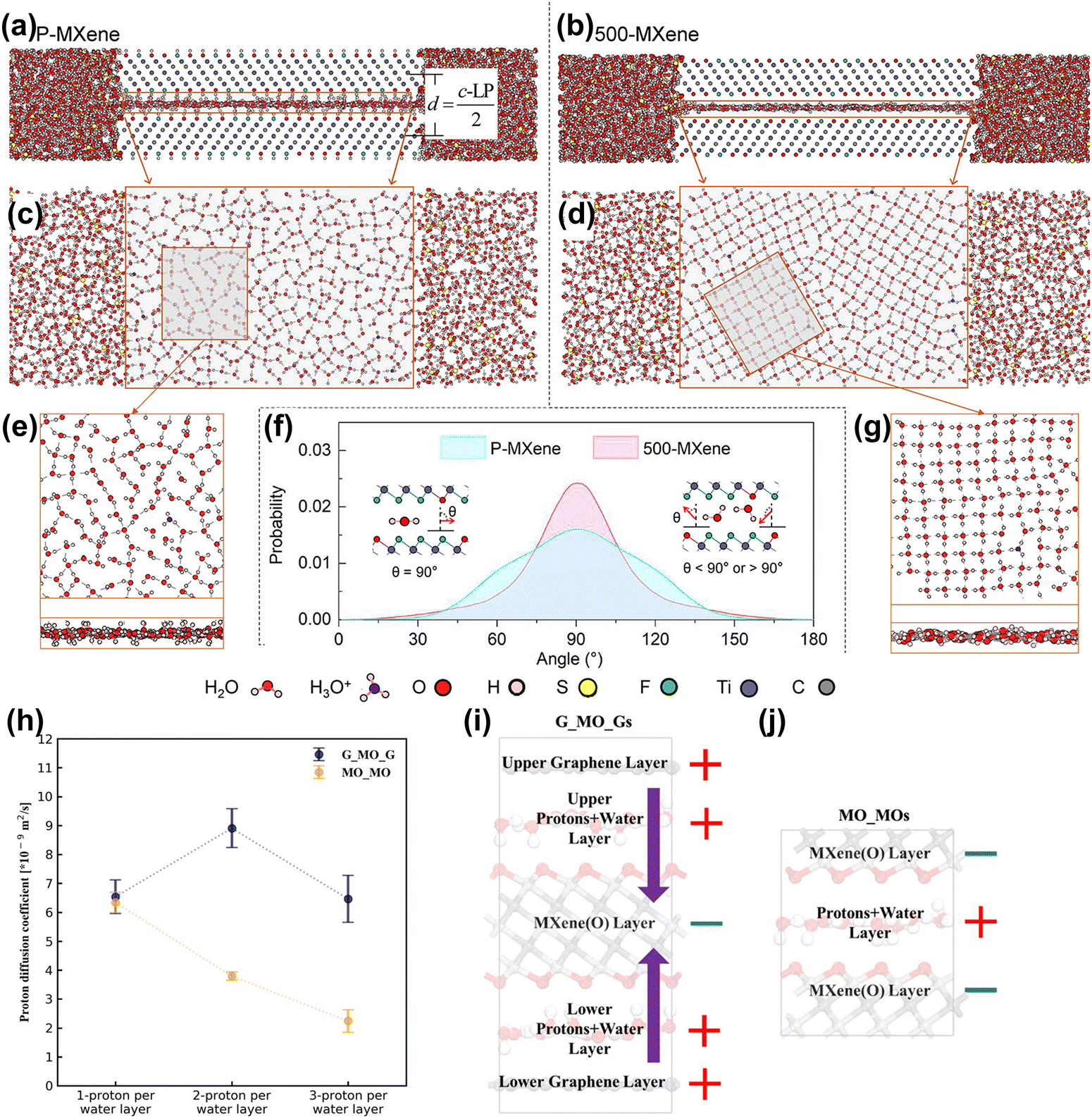 | ||
| Fig. 10 Side view of (a) P-MXene and (b) 500-MXene layers with surrounding electrolytes. Top view of the water molecules between the (c) P-MXene and (d) 500-MXene layers. MXene atoms are not shown here. (e and g) Close-up top view and side view of water molecule distribution between the P-MXene and 500-MXene layers. (f) Comparison of probability profiles of dipole orientation of water molecules inside P-MXene and 500-MXene layers. θ gives the angle between the water molecular dipole moment and the electrode surface normal.163 Reproduced with permission, ref. 163. Copyright 2020, American Chemical Society. (h) Proton diffusivity at the Ti3C2O2-graphene (G_MO_G) and Ti3C2O2–Ti3C2O2 (MO_MO) interfaces vs. different amounts of intercalated protons. A schematic of the charge distribution (± signs) and interfacial electric field (purple arrows): (i) water/hydronium confined in the Ti3C2O2-graphene (G_MO_G) interfaces and (j) water/hydronium confined between Ti3C2O2–Ti3C2O2 (MO_MO) layers.164 Reproduced with permission, ref. 164. Copyright 2021, American Institute of Physics. | ||
5. Challenges
Researchers have demonstrated that using MXene as electrodes can significantly improve the performance of aqueous energy storage systems.11,13,136 However, some issues remain to be addressed. On the one hand, the highly active surface of MXene further promotes the water-splitting side reaction, resulting in a shortened effective electrochemical window. On the other hand, the structure of MXene is unstable and dynamic under conditions of solution and electric field. In response to these challenges, we reviewed computational studies on MXene in water splitting reactions and stability, which will help to rationally design MXene electrodes for aqueous EESSs.5.1. Water-splitting reaction of MXene
A major challenge in aqueous energy storage systems is the water-splitting reaction.16,134,135 As a side reaction, it consumes energy and produces gas during charging and discharging, leading to electrolyte decomposition and corrosion of electrode materials and reducing the electrochemical window and stability.11 Moreover, the good performance of MXene materials in the HER further limits the performance of energy storage devices.144,166 Therefore, a deep understanding of the behavior of MXene in the HER is crucial for the design and optimization of aqueous batteries and supercapacitors.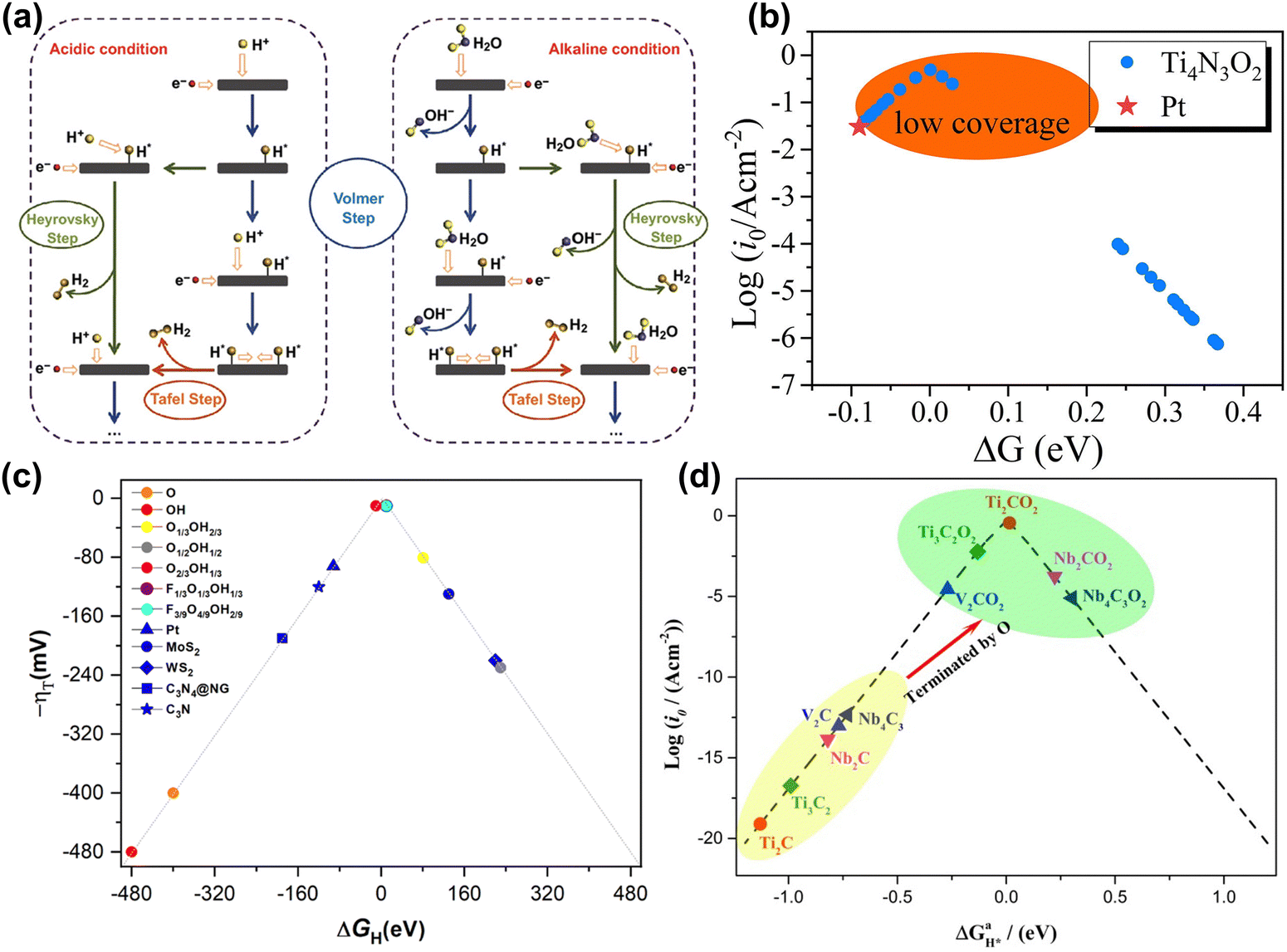 | ||
| Fig. 11 (a) Schematic pathways for the hydrogen evolution reaction under acidic and alkaline conditions.167 Reproduced with permission, ref. 167. Copyright 2018, Springer Nature. (b) Volcano curve of the exchange current density i0 of Ti4N3O2 as a function of ΔG. The value of Pt is inserted by a red star for comparison.172 Reproduced with permission, ref. 172. Copyright 2020, Elsevier. (c) Volcano plot of -η vs. ΔGH on the different terminated Ti3C2 surface models, including fully O- and OH-terminated cases, as well as binary situations represented by the O1/3OH2/3, O1/2OH1/2, and O2/3OH1/3 models, and ternary ones as in the F1/3O1/3OH1/3 and F3/9O4/9OH2/9 models. In addition, reference values for Pt, MoS2, WS2, C3N4@NG, and C3N are included for comparison.173 Reproduced with permission, ref. 173. Copyright 2023, Royal Society of Chemistry. (d) Volcano curve of the exchange current (i0) as a function of the average Gibbs free energy of hydrogen adsorption (ΔGH*).174 Reproduced with permission ref. 174. Copyright 2017, American Chemical Society. | ||
Generally, researchers evaluate the HER performance by calculating the Gibbs free energy (ΔG) of MXene hydrogen adsorption intermediates, as it is considered the rate-controlling step of the reaction based on the Sabatier principle and Brønsted–Evans–Polanyi relationship.175–177 The closer ΔG is to 0, the higher the HER catalytic activity.172 Some research showed that O-terminated MXene tends to exhibit higher HER performance. In Fig. 11(b), Yang et al. found that O-functionalized Ti4N3O2 MXene showed higher HER activity than Pt catalysts at low H coverage.172 Besides, Abraham et al. compared the HER activity of F, Cl, O, S functionalized MXene and found that the ΔG value of MXene with an O functional group was mainly distributed around 0.178 Moreover, the surface compositions of MXene under real conditions are often a mixture of multiple elements, such as O, F, and OH, which play an important role in the HER process. Meng et al. used DFT calculations to investigate the surface composition of MXene at specific pH and U. In Fig. 11(c), they found that the surface functional groups are usually mixed, and the higher the oxygen concentration in the mixed functional groups, the better the HER activity.173
Adjusting the M site, X site, and layer thickness of MXene will also significantly affect HER activity. In Fig. 11(d), Gao et al. found that when the proton coverage was 1, the HER activity of Nb2CO2, Nb4C3O2, and V2CO2 was lower than that of Ti3C2O2 and Ti2CO2.174 Bai et al. calculated the HER performance of a series of O-functionalized carbides and nitrides by changing the metal elements. The results indicated that Nb2NO2 and Ti2NO2 are promising HER electrocatalysts.179
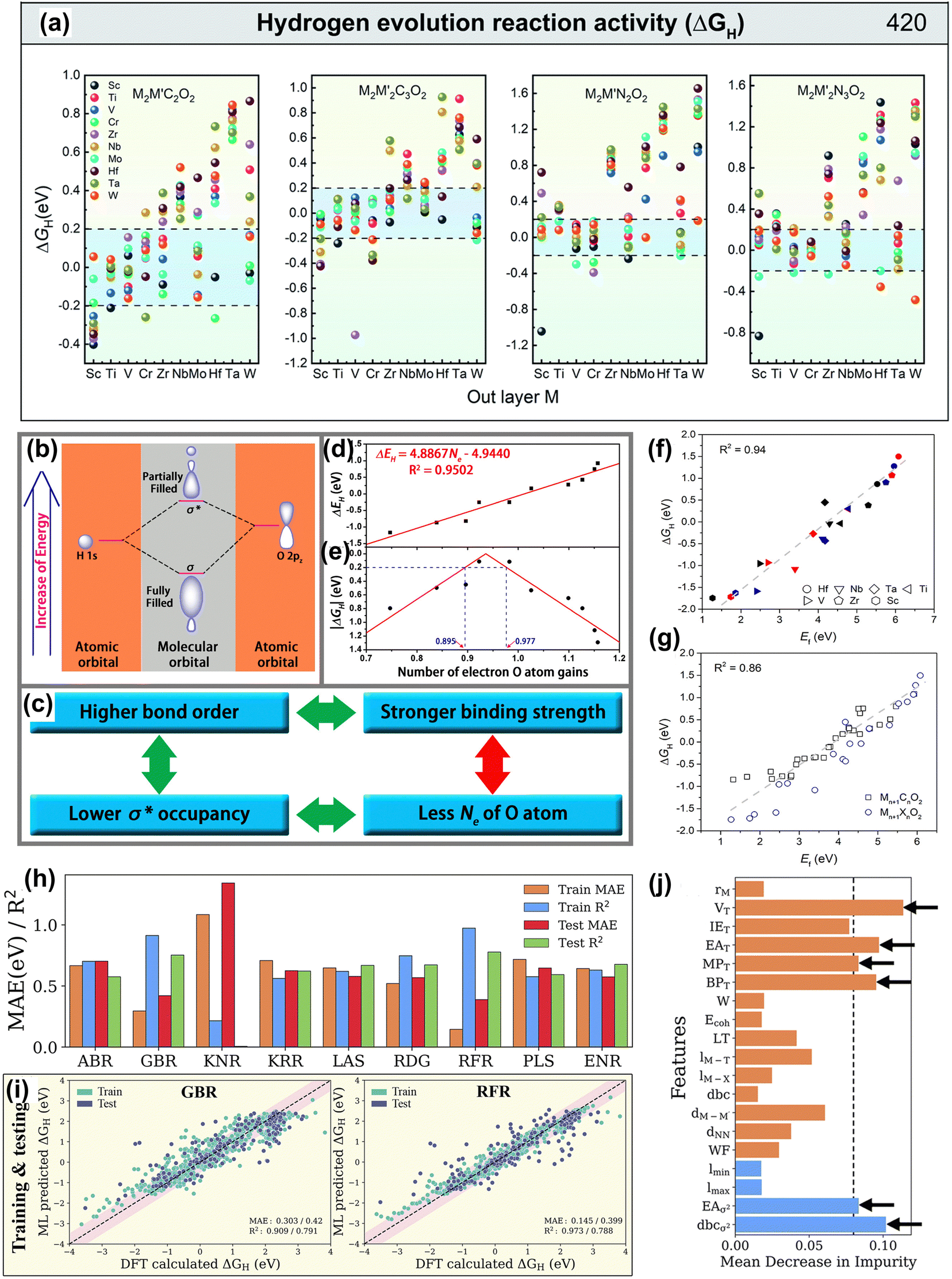 | ||
| Fig. 12 (a) ΔGH distribution of hydrogen adsorbed by M2M′X2 and M2M′2X3O2, and the black dotted line range represents |ΔGH| < 0.2 eV of MXene on equilibrium H coverage.187 Reproduced with permission, ref. 187. Copyright 2020, Royal Society of Chemistry. (b) Schematic of the formation of the H–O bond on the surface of O-terminated MXene, where the linear combination of a H 1s orbital and an O 2pz orbital forms a fully filled, low-energy bonding orbital (σ) and a partially filled, high-energy antibonding orbital (σ*). (c) The derivation of the correlation between the Ne of the O atom and the H-O binding strength. The calculated (d) ΔEH and (e) |ΔGH| as a function of the number of electron O atom gains (Ne), where the R2 is 0.95, indicating the high linear correlation between the Ne and ΔEH.188 Reproduced with permission, ref. 188. Copyright 2016, American Chemical Society. (f) Linear relationship between the oxygen vacancy formation energy (Ef) and free energy of hydrogen adsorption (ΔGH) of Mn+1NnO2. The black, red, and blue symbols represent M2XO2, M3X2O2 and M4X3O2, respectively. (g) The linear relationship between Ef and ΔGH of both Mn+1CnO2 and Mn+1NnO2. The gray dashed line represents linear fitting of the data.189 Reproduced with permission, ref. 189. Copyright 2018, American Chemical Society. (h) Mean absolute error (MAE) and coefficient of determination (R2 score) of the ABR, ENR, GBR, KNR, KRR, LAS, PLS, RFR and RDG algorithms using primary (atomistic, structural and electronic indicators) and statistical function-processed features. (i) Parity plots of the best-performing RFR and GBR models using the DFT dataset of hydrogen adsorption Gibbs free energies (ΔGH). The pink-shaded regions indicate a deviation of up to 0.5 eV. (j) Feature importance from the mean decrease in impurity for the GBR model with RFE-HO-LOO, evaluated via 20-fold cross-validation.178 Reproduced with permission, ref. 178. Copyright 2023, Royal Society of Chemistry. | ||
In the design of MXene materials, descriptors help understand the relationship between material properties and structure.190,191 This strategy accelerates materials discovery and provides a deep understanding of the intrinsic properties, guiding optimization and application. For example, Ling et al. screened highly active HER materials based on O-functionalized MXene by establishing a simple descriptor.188 They found a linear relationship in Fig. 12(b–e) between the number of electrons acquired by surface oxygen atoms (Ne) and the free energy of hydrogen adsorption (ΔGH). Moreover, in Fig. 12(f), Jiang et al. found a linear relationship between the oxygen vacancy ΔEf and ΔGH.189 Moreover, ΔEf is also linearly related to the binding strength of Li atoms on MXene, offering a new perspective in the MXene battery materials search.
Furthermore, new materials meeting design requirements can be quickly discovered through ML algorithms and new descriptors. Wang et al. combined five feature descriptors and the Adaboost algorithm to identify the HER activity trend of 2D ordered binary alloy MXene materials.187 The AdaBoost algorithm is a powerful ensemble learning technique that combines multiple weak classifiers to form a strong classifier, enhancing the model's performance.192 Zheng et al. used the random forest algorithm by constructing multiple decision trees to efficiently predict the ΔGH of MXene materials through simple element characteristics and screened out active catalysts with a ΔGH of close to 0.193 Besides, as shown in Fig. 12(g–j), Abraham established a HER performance data set of 4500 MXenes.178 The gradient boosting regressor algorithm with specific parameters showed the best predictive performance for ΔGH. This algorithm combines the predictions of multiple weak learners (usually decision trees) to improve prediction accuracy by minimizing the loss function via gradient descent.194 Among descriptors, the number of valence electrons of terminal atoms is the most important feature descriptor. In short, HTC and ML methods play an indispensable role in the screening and design of MXene electrode materials.
5.2. Stability of MXene in aqueous solution
The stability of MXene in aqueous solutions is crucial as it directly affects performance and lifespan in energy storage devices. However, some studies found MXene has poor stability in aqueous solutions, being corroded by water molecules and oxidatively degraded into TM oxides.31,195 This structural degradation leads to a loss of electrochemical performance, reducing device lifespan. To overcome this problem, understanding the atomic-level mechanisms of the oxidation process is necessary. In recent years, researchers have used theoretical calculations to reveal atomic details of MXene degradation. These insights help design MXene electrodes with enhanced stability in aqueous solutions.Early experimental studies found that MXene is easily oxidized and degraded in humid air or aqueous solution environments, hindering its application.195 Huang et al. found through spectroscopic characterization that water is the key to MXene oxidation.196 Water “attacks” the metal in MXene, forming oxidized Ti species and leading to degradation. Inspired by this, Wu et al. used first-principles MD to explore how MXene degrades in water.197 The results in Fig. 13(a) show that water molecules attack MXene through adsorption and pull out Ti atoms. The water molecules are then deprotonated to form Ti–OH, reconstructing the surface. At the same time, the formed Ti–OH substances continue to react with water molecules, causing irreversible degradation.
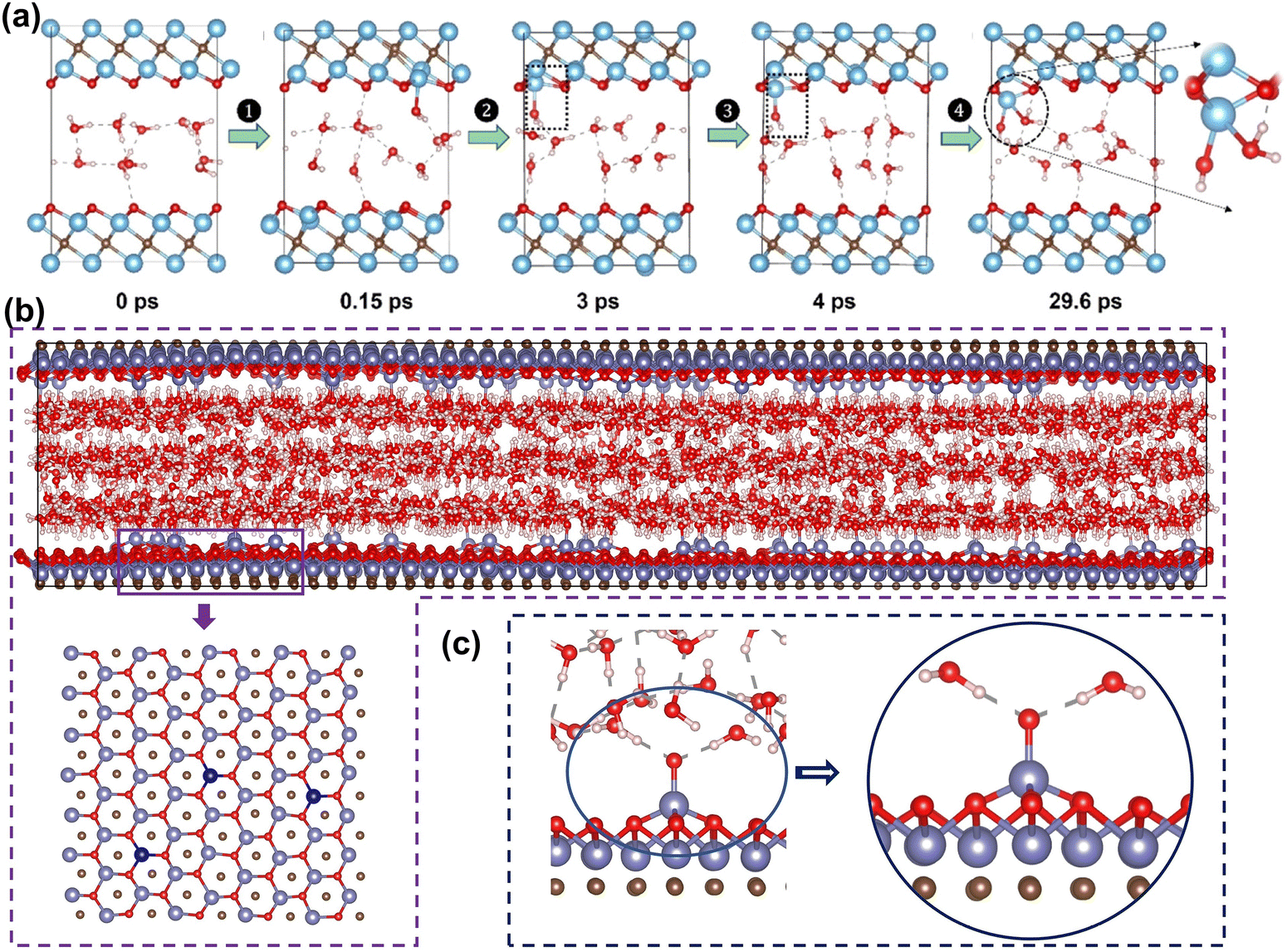 | ||
| Fig. 13 (a) AIMD simulations of two layers of water confined in Ti3C2O2 at 300 K: the figures here are the representative snapshots.197 Reproduced with permission ref. 197. Copyright 2022, American Chemical Society. (b) The positions of V atoms in vanadium oxides (dark blue atoms). (c) The vanadium oxide with two water molecules.198 Reproduced with permission, ref. 198. Copyright 2023, John Wiley and Sons. | ||
Hou et al. further studied the oxidation process of MXene in large aqueous solutions on the nanosecond time scale using a neural network potential.198 Deep neural network potentials provide accurate potential energy surfaces by learning high-fidelity quantum mechanical calculations and capturing complex atomic interactions. These models can achieve quantum mechanics and MD scale accuracy, observing molecular behavior over long time-scales and large atomic scales.199 As shown in Fig. 13(b and c), the V–O species formed by MXene and water are evenly distributed. The outermost O atom of the V–O species can form hydrogen bonds with two water molecules, preventing the nearby vanadium atoms from being attacked by water molecules. Besides, H generated by water decomposition also prevents MXene oxidation. These protection mechanisms cause the degree of MXene oxidation to decay exponentially over time, consistent with experimental observations. In another study, Song et al. studied the effects of vacancies and F functional groups on the stability of Ti3C2O2 MXene.200 They found that F functional groups can slow down water erosion. In addition, Nesterova et al. used enhanced sampling MD to discover that water attacks on metals in MXene depend on the coordination environment of the metal site and the chemical composition of the MXene surface. Edge and defect sites are believed to enhance water adsorption and subsequent MXene degradation. Zhao et al. calculated that the oxidation process preferentially starts from the edge of the nanosheet.201 Marquize et al. found that MXene with vacancies and edge sites exhibits strong water chemical adsorption.202
The atomic-level mechanism of MXene degradation in aqueous solutions was revealed through theoretical calculations. Improving the stability of MXene has become increasingly important. Researchers have proposed promising strategies from a computational perspective to alleviate MXene degradation. Some reports suggest that MXene hydrolysis products such as protons and oxides can prevent water from continuing to attack the MXene surface and thus slow down the degradation rate.197,198 Zhao et al. confirmed that the acidic system is more beneficial to the stability of MXene.201 Surface modification of MXene has proven to be another effective method. Nesterova et al. proposed that MXene stability can be improved by reducing the MXene work function and avoiding loosely anchored oxygen-coordinated Ti sites.203 Song et al. found that increasing the coverage of F functional groups on the MXene surface can prevent water oxidation.200
5.3. Electrochemical stability of MXene
In aqueous batteries and supercapacitors, the electrochemical stability of MXene electrode materials is significantly affected by performance at different pH values and potential U.89 The distribution and composition of MXene surface functional groups, and the overall structure, change with potential and pH conditions, directly affecting the electrochemical performance of the electrode. However, it is very difficult to experimentally obtain the surface composition under real conditions. Therefore, quickly exploring MXene stability under different conditions through theoretical calculations is crucial for the design and optimization of aqueous batteries and supercapacitors.Ibragimova et al. studied the distribution and composition of surface functional groups of Ti2C and Ti3C2 MXene, finding that a mixture of O, OH, and F will be formed on the surface.204 The composition of MXene depends on pH, temperature, and work function. Lopez et al. and Meng et al. also concluded that mixed surface functional groups are beneficial.173,205 Ibragimova further studied other MXene and found that the distribution of mixed functional groups has little to do with the type of metal, carbon, or nitrogen species, and the number of atomic layers of MXene (Fig. 14(a)).206 Wei et al. suggested that the stability of double-TM ordered MXenes is closely related to the type of TM atoms in the outermost layer.207 Gao et al. demonstrated that the surfaces of Ti2C, V2C, and Ti3C2 are composed of O and OH in the standard state, while Nb2C is completely functionalized by O atoms.174 In another study, Fredrickson examined the effects of water intercalation, various functional groups, and applied potential on the stability of Ti2C and Mo2C MXenes.208 They found that MXenes are always functionalized with a layer of O functional groups when the external potential is zero, while bare MXenes are unstable at any external potential (Fig. 14(b and c)). Bo et al. found that pH controls surface termination.159 Acidic pH produces an initial MXene surface with a specific functional group distribution beneficial to capacitive performance.
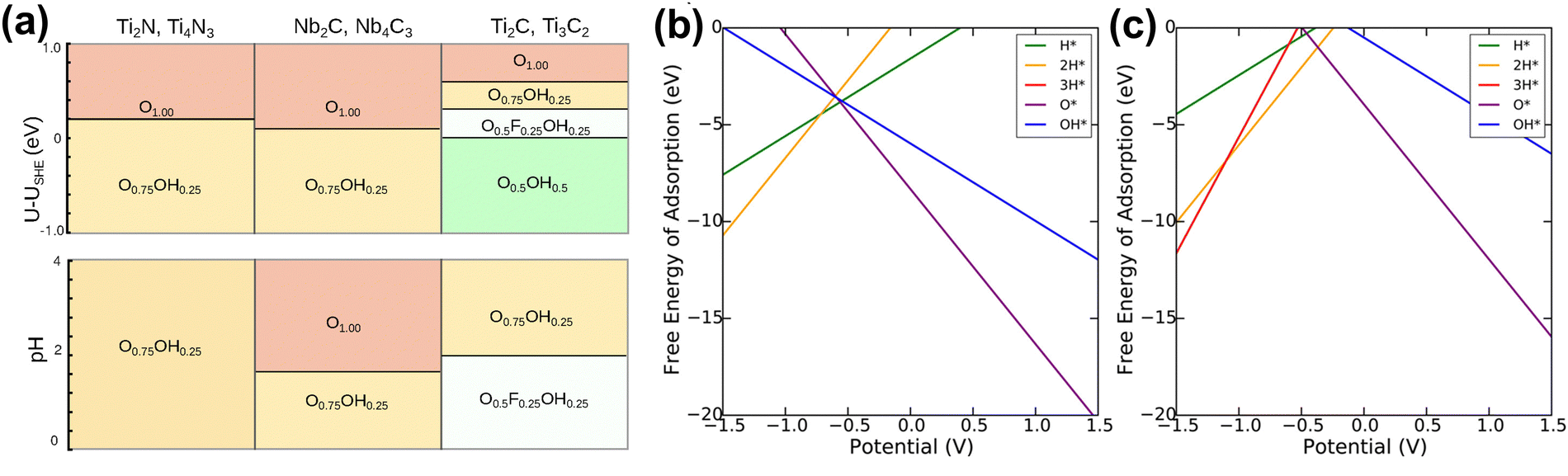 | ||
| Fig. 14 (a) Summary of stable compositions for all systems as a function of the open-circuit potential (at pH 0; top panel) and pH (at U–USHE = 0V; bottom panel).206 Reproduced with permission, ref. 206. Copyright 2021, American Chemical Society. Calculated Pourbaix diagrams for (b) Ti2C and (c) Mo2C MXene. Only the lowest free energies at a given potential are labeled by the functionalization.208 Reproduced with permission, ref. 208. Copyright 2016, American Chemical Society. | ||
6. Conclusion and perspectives
With the development of high-performance computing and ML, computational simulations have become crucial for designing and discovering new materials. This is exemplified by the computational insights into MXene materials used in aqueous batteries and supercapacitors. First, theoretical calculations and high-throughput screening of thermodynamically stable MAX precursors guided the experimental synthesis of new MXene phases. Second, multi-scale theoretical simulations elucidated MXene's working mechanisms in aqueous energy storage systems, including Zn deposition inhibition, activation of electrochemical reaction sites, and pseudocapacitive mechanisms. Finally, new MXene materials can be rationally designed to address issues in aqueous energy storage systems, such as the HER. Although computational simulations have achieved significant success in these fields, a substantial gap remains between simulations and actual reactions, necessitating more advanced and comprehensive calculations and modeling. To address this challenge, we propose focusing on the following aspects.(1) Enhance the complexity of the simulation system. As computing power increases, the dimensional explosion issue caused by system complexity will be mitigated, allowing researchers to consider more realistic systems. For MXene synthesis, current computational systems overly simplify reaction conditions, making it challenging to extend to complex experimental processes. Therefore, additional factors such as etchant type, solution pH, reaction temperature, and kinetics must be considered to establish a comprehensive synthesis theory. For energy storage, simulations must also account for the complexity of MXene surface chemistry and aqueous solutions, including functional group mixing, electrolyte pH, and intercalation thickness.
(2) Employ advanced computational methods. For electrochemical energy storage and catalysis processes, reactions occur under bias conditions, yet most DFT calculations are constant electron simulations. Constant potential simulations, which better mimic electrochemical reactions, allow for clearer insights into the double-layer effect at interfaces, charge transfer processes, and solvent structures.
(3) ML deep potential. ML deep potential provides an accurate and efficient potential energy surface by learning particle motion behavior in AIMD, promising to accelerate MXene research in aqueous energy storage systems. ML potential achieves quantum mechanics and MD scale accuracy, observing molecular behavior over long time-scales and large atomic scales. This understanding aids in comprehending the long-term evolution of MXene electrodes in aqueous devices. Thus, developing robust ML potentials to predict MXene behavior under various electrochemical conditions is crucial. Combining AIMD with advanced sampling methods like metadynamics enhances the exploration of unknown potential energy surfaces, achieving precise descriptions.
(4) Selecting the best descriptors and ML models. Choosing the best descriptors and ML models is vital for accurately predicting MXene's electrochemical properties. Essential descriptors include surface composition, electronic structure, and chemical composition. Dimensional reduction techniques, such as recursive feature elimination and principal component analysis, identify the most relevant descriptors, enhancing model accuracy. Incorporating domain knowledge into descriptors ensures physical relevance to MXene's properties. For ML models, complexity and interpretability must be considered; ensemble methods like random forests or gradient boosting offer strong performance and insights. Future research should develop unique descriptors and interpretable ML models, optimizing hyperparameters to achieve the best prediction performance and promote rational MXene design in aqueous energy storage systems.
(5) High-throughput computational screening and database establishment. High-throughput computational screening systematically explores numerous candidate materials, making it a powerful strategy for discovering and optimizing materials. For MXene, this method can identify compositions, surface terminations, and structural modifications with optimal electrochemical performance. Building high-throughput computational data into accessible MXene databases will significantly advance application research. To enhance usability, researchers should establish standardized data formats and incorporate real-time updates of ongoing research. Integrating these databases with ML tools can predict new MXene compositions for aqueous batteries and supercapacitors.
(6) Multiscale simulation. Multiscale simulation methods are crucial for bridging the gap between atomic-scale phenomena and macroscopic device performance. This review examines how MXene materials impact the electrochemical behavior of aqueous energy storage systems using DFT calculations, classical MD simulations, and COMSOL simulations. Future research should develop integrated multiscale models that combine quantum mechanical calculations with continuum-scale simulations. This integration can provide a comprehensive understanding of performance-limiting factors and degradation mechanisms in MXene-based devices, ultimately leading to the design of MXene materials with enhanced performance and durability.
List of abbreviations
| 2D | Two-dimensional |
| 3D-RISM | Three-dimensional reference interaction site model |
| AIMD | Ab initio molecular dynamics |
| AZIB | Aqueous Zn-ion batteries |
| DFT | Density functional theory |
| DOS | Density of state |
| EDLCs | Electrical double-layer capacitors |
| EDL | Electrical double-layer |
| EESS | Electrical energy storage systems |
| ΔEf | Formation energy |
| ΔG | Gibbs free energy |
| ΔGH | Free energy of hydrogen adsorption |
| HER | Hydrogen evolution reaction |
| HTC | High-throughput calculation |
| ΔHcp | Formation enthalpy |
| i-MXene | In-plane ordered MXene |
| MOF | Metal–organic frameworks |
| PEDOT | Poly(3,4-Ethylenedioxythipohene) |
| PPy | Polypyrrole |
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Grants Council of Hong Kong (CityU 11305919 and 11308620), CityU project (7005635), and NSFC/RGC Joint Research Scheme N_CityU104/19. Hong Kong Research Grant Council Collaborative Research Fund: C1002-21G and C1017-22G. This research made use of the computing resources of the X-GPU cluster supported by the Hong Kong Research Grant Council Collaborative Research Fund: C6021-19 EF.References
- A. M. Omer, Renewable Sustainable Energy Rev., 2008, 12, 2265–2300 CrossRef CAS.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- J. Mitali, S. Dhinakaran and A. Mohamad, Energy Storage Sav., 2022, 1, 166–216 CrossRef CAS.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CAS.
- M. M. Rana, M. Uddin, M. R. Sarkar, S. T. Meraj, G. Shafiullah, S. Muyeen, M. A. Islam and T. Jamal, J. Energy Storage, 2023, 68, 107811 CrossRef.
- Y. Liang, C. Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J. Q. Huang, D. Yu, Y. Liu, M. M. Titirici and Y. L. Chueh, InfoMat, 2019, 1, 6–32 CrossRef CAS.
- G. Liang, F. Mo, X. Ji and C. Zhi, Nat. Rev. Mater., 2021, 6, 109–123 CrossRef CAS.
- Z. Pan, X. Liu, J. Yang, X. Li, Z. Liu, X. J. Loh and J. Wang, Adv. Energy Mater., 2021, 11, 2100608 CrossRef CAS.
- Z. Yin, J. Zhao, D. Luo, Y. Y. Chin, C. T. Chen, H. Chen, W. Yin, Y. Tang, T. Yang and J. Ren, Adv. Sci., 2024, 2307397 CrossRef CAS.
- M. Okubo, A. Sugahara, S. Kajiyama and A. Yamada, Acc. Chem. Res., 2018, 51, 591–599 CrossRef CAS PubMed.
- J. Xu, J. You, L. Wang, Z. Wang and H. Zhang, Sustainable Mater. Technol., 2022, 33, e00490 CrossRef CAS.
- N. Zhang, X. Chen, M. Yu, Z. Niu, F. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC.
- Y. Tian, Y. An, J. Feng and Y. Qian, Mater. Today, 2022, 52, 225–249 CrossRef CAS.
- F. Wan, X. Zhou, Y. Lu, Z. Niu and J. Chen, ACS Energy Lett., 2020, 5, 3569–3590 CrossRef CAS.
- M. Li, Z. Li, X. Wang, J. Meng, X. Liu, B. Wu, C. Han and L. Mai, Energy Environ. Sci., 2021, 14, 3796–3839 RSC.
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598–616 CrossRef CAS.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- X. Li, Z. Huang, C. E. Shuck, G. Liang, Y. Gogotsi and C. Zhi, Nat. Rev. Chem., 2022, 6, 389–404 CrossRef PubMed.
- B. Lu, Z. Zhu, B. Ma, W. Wang, R. Zhu and J. Zhang, Small, 2021, 17, 2100946 CrossRef CAS.
- Y. Pei, X. Zhang, Z. Hui, J. Zhou, X. Huang, G. Sun and W. Huang, ACS Nano, 2021, 15, 3996–4017 CrossRef CAS PubMed.
- A. Morales-Garcia, F. Calle-Vallejo and F. Illas, ACS Catal., 2020, 10, 13487–13503 CrossRef CAS.
- B. Fu, J. Sun, C. Wang, C. Shang, L. Xu, J. Li and H. Zhang, Small, 2021, 17, 2006054 CrossRef CAS.
- A. Iqbal, P. Sambyal and C. M. Koo, Adv. Funct. Mater., 2020, 30, 2000883 CrossRef CAS.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS.
- Y. Sun, C. Zhan, P. R. Kent, M. Naguib, Y. Gogotsi and D.-E. Jiang, ACS Appl. Mater. Interfaces, 2019, 12, 763–770 CrossRef.
- X. Li, M. Li, K. Luo, Y. Hou, P. Li, Q. Yang, Z. Huang, G. Liang, Z. Chen and S. Du, ACS Nano, 2021, 16, 813–822 CrossRef.
- Y. Li, Q. Zhu, M. Xu, B. Zang, Y. Wang and B. Xu, Adv. Funct. Mater., 2023, 33, 2213416 CrossRef CAS.
- H. Luo, J. Jiang, M. Li, K. Sun and Y. Zheng, J. Colloid Interface Sci., 2024, 654, 289–299 CrossRef CAS.
- D. Zhao, Z. Li, D. Xu and Z. Yang, Adv. Funct. Mater., 2024, 2316182 CrossRef CAS.
- J. Zhou, M. Dahlqvist, J. Björk and J. Rosen, Chem. Rev., 2023, 123, 13291–13322 CrossRef CAS PubMed.
- T. Wu and D.-E. Jiang, MRS Bull., 2023, 48, 253–260 Search PubMed.
- M. Dahlqvist and J. Rosen, Nanoscale, 2020, 12, 785–794 RSC.
- S. Luo, N. Ma, J. Zhao, Y. Wang, Y. Zhang, Y. Xiong and J. Fan, J. Mater. Sci. Technol., 2024, 199, 145–155 CrossRef.
- C. Zhan, M. Naguib, M. Lukatskaya, P. R. Kent, Y. Gogotsi and D.-E. Jiang, J. Phys. Chem. Lett., 2018, 9, 1223–1228 CrossRef CAS PubMed.
- J. Yu, C. Chen, F. Shi, R. Li, F. Chen, J. Tang, K. C. Chan and Z.-L. Xu, Energy Storage Mater., 2023, 63, 102966 CrossRef.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. Man Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137–1140 CrossRef CAS PubMed.
- V. Kamysbayev, A. S. Filatov, H. Hu, X. Rui, F. Lagunas, D. Wang, R. F. Klie and D. V. Talapin, Science, 2020, 369, 979–983 CrossRef CAS PubMed.
- K. R. G. Lim, M. Shekhirev, B. C. Wyatt, B. Anasori, Y. Gogotsi and Z. W. Seh, Nat. Synth., 2022, 1, 601–614 CrossRef CAS.
- Y. Gogotsi and Q. Huang, ACS Nano, 2021, 15, 5775–5780 CrossRef CAS.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- X. Zhan, C. Si, J. Zhou and Z. Sun, Nanoscale Horiz., 2020, 5, 235–258 RSC.
- D. Gandla, Z. Zhuang, V. V. Jadhav and D. Q. Tan, Energy Storage Mater., 2023, 102977 CrossRef.
- I. Hussain, M. Ahmad, O. J. Kewate, A. Hanan, F. Bibi, M. S. Javed, I. Shaheen and K. Zhang, ChemSusChem, 2024, e202400283 CrossRef CAS PubMed.
- Z. Du, C. Wu, Y. Chen, Z. Cao, R. Hu, Y. Zhang, J. Gu, Y. Cui, H. Chen and Y. Shi, Adv. Mater., 2021, 33, 2101473 CrossRef CAS PubMed.
- R. Meshkian, Q. Tao, M. Dahlqvist, J. Lu, L. Hultman and J. Rosen, Acta Mater., 2017, 125, 476–480 CrossRef CAS.
- M. Dahlqvist, J. Lu, R. Meshkian, Q. Tao, L. Hultman and J. Rosen, Sci. Adv., 2017, 3, e1700642 CrossRef.
- M. Sokol, V. Natu, S. Kota and M. W. Barsoum, Trends Chem., 2019, 1, 210–223 CrossRef CAS.
- H. W. Seong, M. S. Lee and H. J. Ryu, J. Mater. Chem. A, 2023, 11, 5681–5695 RSC.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- S.-H. Seok, Y. Sim, J.-H. Han, Y. H. Jin, Y. Chae, J. Park and S.-Y. Kwon, Cell Rep. Phys. Sci., 2023, 4, 101582 CrossRef CAS.
- C. Fan, P. Zhang, R. Wang, Y. Xu, X. Sun, J. Zhang, J. Cheng and C. Xu, Curr. Nanosci., 2021, 17, 2–13 CrossRef CAS.
- M. Zubair, M. M. U. Hassan, M. T. Mehran, M. M. Baig, S. Hussain and F. Shahzad, Int. J. Hydrogen Energy, 2022, 47, 2794–2818 CrossRef CAS.
- P. Bhojane, J. Energy Storage, 2022, 45, 103654 CrossRef.
- E. M. D. Siriwardane and J. Hu, J. Phys. Chem. C, 2021, 125, 12469–12477 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, MXenes, Jenny Stanford Publishing, 2011, pp. 15–29 Search PubMed.
- T. Li, L. Yao, Q. Liu, J. Gu, R. Luo, J. Li, X. Yan, W. Wang, P. Liu and B. Chen, Angew. Chem., Int. Ed., 2018, 57, 6115–6119 CrossRef CAS.
- A. Feng, Y. Yu, F. Jiang, Y. Wang, L. Mi, Y. Yu and L. Song, Ceram. Int., 2017, 43, 6322–6328 CrossRef CAS.
- W. Sun, S. Shah, Y. Chen, Z. Tan, H. Gao, T. Habib, M. Radovic and M. Green, J. Mater. Chem. A, 2017, 5, 21663–21668 RSC.
- X. Xie, Y. Xue, L. Li, S. Chen, Y. Nie, W. Ding and Z. Wei, Nanoscale, 2014, 6, 11035–11040 RSC.
- M. W. Barsoum and T. El-Raghy, J. Am. Ceram. Soc., 1996, 79, 1953–1956 CrossRef CAS.
- W. Wang, I. Ruiz, S. Guo, Z. Favors, H. H. Bay, M. Ozkan and C. S. Ozkan, Nano Energy, 2014, 3, 113–118 CrossRef CAS.
- M. Naguib, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2021, 33, 2103393 CrossRef CAS.
- O. Salim, K. Mahmoud, K. Pant and R. Joshi, Mater. Today Chem., 2019, 14, 100191 CrossRef CAS.
- Y. Wei, P. Zhang, R. A. Soomro, Q. Zhu and B. Xu, Adv. Mater., 2021, 33, 2103148 CrossRef CAS.
- P. P. Michałowski, M. Anayee, T. S. Mathis, S. Kozdra, A. Wójcik, K. Hantanasirisakul, I. Jóźwik, A. Piątkowska, M. Możdżonek and A. Malinowska, Nat. Nanotechnol., 2022, 17, 1192–1197 CrossRef PubMed.
- M. Han, K. Maleski, C. E. Shuck, Y. Yang, J. T. Glazar, A. C. Foucher, K. Hantanasirisakul, A. Sarycheva, N. C. Frey and S. J. May, J. Am. Chem. Soc., 2020, 142, 19110–19118 CrossRef CAS PubMed.
- Z. Liu, E. Wu, J. Wang, Y. Qian, H. Xiang, X. Li, Q. Jin, G. Sun, X. Chen and J. Wang, Acta Mater., 2014, 73, 186–193 CrossRef CAS.
- N. Kubitza, A. Reitz, A.-M. Zieschang, H. Pazniak, B. Albert, C. Kalha, C. Schlueter, A. Regoutz, U. Wiedwald and C. S. Birkel, Inorg. Chem., 2022, 61, 10634–10641 CrossRef CAS.
- E. Wu, Y. Zhang, M. Li, Y. Li, K. Liang, S. Du and Q. Huang, Materialia, 2023, 27, 101676 CrossRef CAS.
- G. Deysher, C. E. Shuck, K. Hantanasirisakul, N. C. Frey, A. C. Foucher, K. Maleski, A. Sarycheva, V. B. Shenoy, E. A. Stach and B. Anasori, ACS Nano, 2019, 14, 204–217 CrossRef PubMed.
- M. Dahlqvist, M. W. Barsoum and J. Rosen, Mater. Today, 2024, 72, 1–24 CrossRef.
- N. C. Frey, J. Wang, G. I. N. Vega Bellido, B. Anasori, Y. Gogotsi and V. B. Shenoy, ACS Nano, 2019, 13, 3031–3041 CrossRef CAS PubMed.
- M. Dahlqvist, B. Alling, I. A. Abrikosov and J. Rosén, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024111 CrossRef.
- M. Dahlqvist, B. Alling and J. Rosén, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 220102 CrossRef.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- M. Rougab and A. Gueddouh, J. Phys. Chem. Solids, 2023, 176, 111251 CrossRef CAS.
- S. Aryal, R. Sakidja, M. W. Barsoum and W. Y. Ching, Phys. Status Solidi B, 2014, 251, 1480–1497 CrossRef CAS.
- P. Eklund, M. Dahlqvist, O. Tengstrand, L. Hultman, J. Lu, N. Nedfors, U. Jansson and J. Rosén, Phys. Rev. Lett., 2012, 109, 035502 CrossRef.
- R. Syamsai, J. R. Rodriguez, V. G. Pol, Q. Van Le, K. M. Batoo, S. F. Adil, S. Pandiaraj, M. Muthumareeswaran, E. H. Raslan and A. N. Grace, Sci. Rep., 2021, 11, 688 CrossRef CAS PubMed.
- A. Mockuté, M. Dahlqvist, J. Emmerlich, L. Hultman, J. M. Schneider, P. O. Persson and J. Rosén, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 094113 CrossRef.
- G. Niu, M. C. Du Plessis, T. Sakai, Y. Ma and M. Sugiyama, Advances in neural information processing systems, 2016, 29, 1–9 Search PubMed.
- Q. Hu, H. Wang, Q. Wu, X. Ye, A. Zhou, D. Sun, L. Wang, B. Liu and J. He, Int. J. Hydrogen Energy, 2014, 39, 10606–10612 CrossRef CAS.
- B. C. Wyatt, A. Thakur, K. Nykiel, Z. D. Hood, S. P. Adhikari, K. K. Pulley, W. J. Highland, A. Strachan and B. Anasori, Nano Lett., 2023, 23, 931–938 CrossRef CAS PubMed.
- M. Ashton, K. Mathew, R. G. Hennig and S. B. Sinnott, J. Phys. Chem. C, 2016, 120, 3550–3556 CrossRef CAS.
- J. Björk, J. Halim, J. Zhou and J. Rosen, npj 2D Mater. Appl., 2023, 7, 5 CrossRef.
- N. M. Caffrey, J. Phys. Chem. C, 2020, 124, 18797–18804 CrossRef CAS.
- M. Ashton, N. Trometer, K. Mathew, J. Suntivich, C. Freysoldt, S. B. Sinnott and R. G. Hennig, J. Phys. Chem. C, 2019, 123, 3180–3187 CrossRef CAS.
- M. Khazaei, A. Ranjbar, K. Esfarjani, D. Bogdanovski, R. Dronskowski and S. Yunoki, Phys. Chem. Chem. Phys., 2018, 20, 8579–8592 RSC.
- C. Tsounis, P. V. Kumar, H. Masood, R. P. Kulkarni, G. S. Gautam, C. R. Müller, R. Amal and D. A. Kuznetsov, Angew. Chem., Int. Ed., 2023, 62, e202210828 CrossRef CAS.
- H. Zhu, Z. Liang, S. Xue, X. Ren, X. Liang, W. Xiong, L. Gao and A. Liu, Ceram. Int., 2022, 48, 27217–27239 CrossRef CAS.
- Q. Zhu, J. Li, P. Simon and B. Xu, Energy Storage Mater., 2021, 35, 630–660 CrossRef.
- K. S. Novoselov, A. Mishchenko, A. Carvalho and A. Castro Neto, Science, 2016, 353, aac9439 CrossRef CAS PubMed.
- X. Wang, Y. Liu, Z. Wei, J. Hong, H. Liang, M. Song, Y. Zhou and X. Huang, Adv. Mater., 2022, 34, 2206812 CrossRef CAS PubMed.
- Y. Ando, M. Okubo, A. Yamada and M. Otani, Adv. Funct. Mater., 2020, 30, 2000820 CrossRef CAS.
- Z. Ling, C. E. Ren, M.-Q. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676–16681 CrossRef CAS.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall’Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS.
- J. Zhao, N. Ma, T. Wang, N. Li, Y. Wang and J. Fan, J. Mater. Chem. A, 2022, 10, 21611–21621 RSC.
- N. Ma, N. Li, Y. Zhang, T. Wang, J. Zhao and J. Fan, Appl. Surf. Sci., 2022, 590, 153149 CrossRef CAS.
- J. Zhang, N. Kong, S. Uzun, A. Levitt, S. Seyedin, P. A. Lynch, S. Qin, M. Han, W. Yang and J. Liu, Adv. Mater., 2020, 32, 2001093 CrossRef CAS.
- Y. T. Liu, P. Zhang, N. Sun, B. Anasori, Q. Z. Zhu, H. Liu, Y. Gogotsi and B. Xu, Adv. Mater., 2018, 30, 1707334 CrossRef.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS.
- J. Chen, W. Zhao, J. Jiang, X. Zhao, S. Zheng, Z. Pan and X. Yang, Energy Storage Mater., 2023, 59, 102767 CrossRef.
- K. Guan, L. Tao, R. Yang, H. Zhang, N. Wang, H. Wan, J. Cui, J. Zhang, H. Wang and H. Wang, Adv. Energy Mater., 2022, 12, 2103557 CrossRef CAS.
- Y. Li, C. B. Musgrave III, M. Y. Yang, M. M. Kim, K. Zhang, M. Tamtaji, Y. Cai, T. W. Tang, J. Wang and B. Yuan, Adv. Energy Mater., 2024, 14, 2303047 CrossRef CAS.
- X. Li, M. Li, Q. Yang, G. Liang, Z. Huang, L. Ma, D. Wang, F. Mo, B. Dong and Q. Huang, Adv. Energy Mater., 2020, 10, 2001791 CrossRef CAS.
- N. Zhang, S. Huang, Z. Yuan, J. Zhu, Z. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2021, 60, 2861–2865 CrossRef CAS PubMed.
- X. Li, Q. Li, Y. Hou, Q. Yang, Z. Chen, Z. Huang, G. Liang, Y. Zhao, L. Ma and M. Li, ACS Nano, 2021, 15, 14631–14642 CrossRef CAS.
- Y. Liu, Y. Jiang, Z. Hu, J. Peng, W. Lai, D. Wu, S. Zuo, J. Zhang, B. Chen and Z. Dai, Adv. Funct. Mater., 2021, 31, 2008033 CrossRef CAS.
- Y. Wang, L. Liu, Y. Wang, J. Qu, Y. Chen and J. Song, ACS Nano, 2023, 17, 21761–21770 CrossRef PubMed.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- H. Zhang, J. Wang, Q. Liu, W. He, Z. Lai, X. Zhang, M. Yu, Y. Tong and X. Lu, Energy Storage Mater., 2019, 21, 154–161 CrossRef.
- H. Zhang, Y. Fang, F. Yang, X. Liu and X. Lu, Energy Environ. Sci., 2020, 13, 2515–2523 RSC.
- Y. Wang, J. Song and W. Y. Wong, Angew. Chem., Int. Ed., 2023, 135, e202218343 CrossRef.
- C. Chen, T. Wang, X. Zhao, A. Wu, S. Li, N. Zhang, X. Qu, L. Jiao and Y. Liu, Adv. Funct. Mater., 2024, 34, 2308508 CrossRef CAS.
- X. Li, X. Ma, Y. Hou, Z. Zhang, Y. Lu, Z. Huang, G. Liang, M. Li, Q. Yang and J. Ma, Joule, 2021, 5, 2993–3005 CrossRef CAS.
- M. Li, X. Li, G. Qin, K. Luo, J. Lu, Y. Li, G. Liang, Z. Huang, J. Zhou and L. Hultman, ACS Nano, 2021, 15, 1077–1085 CrossRef CAS PubMed.
- X. Li, N. Li, Z. Huang, Z. Chen, G. Liang, Q. Yang, M. Li, Y. Zhao, L. Ma and B. Dong, Adv. Mater., 2021, 33, 2006897 CrossRef CAS PubMed.
- H. Tian, T. Gao, X. Li, X. Wang, C. Luo, X. Fan, C. Yang, L. Suo, Z. Ma and W. Han, Nat. Commun., 2017, 8, 14083 CrossRef CAS.
- X. Li, N. Li, Z. Huang, Z. Chen, Y. Zhao, G. Liang, Q. Yang, M. Li, Q. Huang and B. Dong, ACS Nano, 2021, 15, 1718–1726 CrossRef CAS PubMed.
- G. Liang, Z. Tang, B. Han, J. Zhu, A. Chen, Q. Li, Z. Chen, Z. Huang, X. Li and Q. Yang, Adv. Mater., 2023, 35, 2210051 CrossRef CAS.
- C. Li, X. Xie, S. Liang and J. Zhou, Energy Environ. Sci., 2020, 3, 146–159 CAS.
- Z. Cai, Y. Ou, B. Zhang, J. Wang, L. Fu, M. Wan, G. Li, W. Wang, L. Wang and J. Jiang, J. Am. Chem. Soc., 2021, 143, 3143–3152 CrossRef CAS PubMed.
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1903675 CrossRef.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu and M. J. Hourwitz, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
- X. Zhu, X. Li, M. L. K. Essandoh, J. Tan, Z. Cao, X. Zhang, P. Dong, P. M. Ajayan, M. Ye and J. Shen, Energy Storage Mater., 2022, 50, 243–251 CrossRef.
- F. Bu, Z. Sun, W. Zhou, Y. Zhang, Y. Chen, B. Ma, X. Liu, P. Liang, C. Zhong and R. Zhao, J. Am. Chem. Soc., 2023, 145, 24284–24293 CrossRef CAS.
- C. Sun, C. Wu, X. Gu, C. Wang and Q. Wang, Nano-Micro Lett., 2021, 13, 1–13 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- T. Guo, D. Zhou, L. Pang, S. Sun, T. Zhou and J. Su, Small, 2022, 18, 2106360 CrossRef CAS PubMed.
- M. Yu, Y. Lu, H. Zheng and X. Lu, Chem. – Eur. J., 2018, 24, 3639–3649 CrossRef CAS.
- W. Zheng, L. Yang, P. Zhang, V. Natu, Z. Sun, J. Rosen and M. W. Barsoum, Energy Storage Mater., 2023, 103037 CrossRef.
- Q. Shan, X. Mu, M. Alhabeb, C. E. Shuck, D. Pang, X. Zhao, X.-F. Chu, Y. Wei, F. Du and G. Chen, Electrochem. Commun., 2018, 96, 103–107 CrossRef CAS.
- J. Pu, Z. Wang, K. Wu, N. Yu and E. Sheng, Phys. Chem. Chem. Phys., 2014, 16, 785–791 RSC.
- X. Geng, Y. Zhang, Y. Han, J. Li, L. Yang, M. Benamara, L. Chen and H. Zhu, Nano Lett., 2017, 17, 1825–1832 CrossRef CAS PubMed.
- R. Ravit, J. Abdullah, I. Ahmad and Y. Sulaiman, Carbohydr. Polym., 2019, 203, 128–138 CrossRef CAS PubMed.
- M. Hou, M. Xu, Y. Hu and B. Li, Electrochim. Acta, 2019, 313, 245–254 CrossRef CAS.
- S. R. Gurav, G. R. Chodankar, S. A. Sawant, U. V. Shembade, A. V. Moholkar and R. G. Sonkawade, J. Energy Storage, 2023, 73, 109254 CrossRef.
- V. S. Kumbhar and D.-H. Kim, Electrochim. Acta, 2018, 271, 284–296 CrossRef CAS.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum and P. Simon, Nat. Energy, 2017, 2, 1–6 Search PubMed.
- Z. Zhang, Z. Yao, X. Zhang and Z. Jiang, Electrochim. Acta, 2020, 359, 136960 CrossRef CAS.
- G. E. Blomgren, J. Electrochem. Soc., 2016, 164, A5019 CrossRef.
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gomez-Romero, Chem. Soc. Rev., 2015, 44, 1777–1790 RSC.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2020, 5, 5–19 CrossRef.
- A. Sugahara, Y. Ando, S. Kajiyama, K. Yazawa, K. Gotoh, M. Otani, M. Okubo and A. Yamada, Nat. Commun., 2019, 10, 850 CrossRef PubMed.
- A. Kovalenko and F. Hirata, Chem. Phys. Lett., 1998, 290, 237–244 CrossRef CAS.
- X. Ji, K. Xu, C. Chen, B. Zhang, Y. Ruan, J. Liu, L. Miao and J. Jiang, Phys. Chem. Chem. Phys., 2016, 18, 4460–4467 RSC.
- R.-Z. Zhang, X.-H. Cui, S.-S. Li, X.-H. Li and H.-L. Cui, J. Mol. Liq., 2022, 345, 118263 CrossRef CAS.
- Y. Xin and Y.-X. Yu, Mater. Des., 2017, 130, 512–520 CrossRef CAS.
- H.-T. Yan, X.-H. Li, M.-Z. Liu, X.-H. Cui, S.-S. Li and H.-L. Cui, Vacuum, 2022, 201, 111094 CrossRef CAS.
- S.-H. Yin, X.-H. Li, R.-Z. Zhang and H.-L. Cui, FlatChem, 2024, 100632 CrossRef CAS.
- W. Zhang, C. Cheng, P. Fang, B. Tang, J. Zhang, G. Huang, X. Cong, B. Zhang, X. Ji and L. Miao, Phys. Chem. Chem. Phys., 2016, 18, 4376–4384 RSC.
- T. Shimada, N. Takenaka, Y. Ando, M. Otani, M. Okubo and A. Yamada, Chem. Mater., 2022, 34, 2069–2075 CrossRef CAS.
- Z. Bo, Y. Chen, Q. Yu, J. Yan, K. Cen and Z. Liu, J. Phys. Chem. C, 2024, 128, 2352–2361 CrossRef CAS.
- C. Zhan, W. Sun, P. R. Kent, M. Naguib, Y. Gogotsi and D.-E. Jiang, J. Phys. Chem. C, 2018, 123, 315–321 CrossRef.
- S. Nishihara and M. Otani, Phys. Rev. B, 2017, 96, 115429 CrossRef.
- L. Wang, J. Wang, Z. Zhang, L. Wang, W. Wang, J. Liu, Z. Hong, K. Cho and W. Wang, J. Mater. Chem. A, 2019, 7, 16231–16238 RSC.
- H. Shao, K. Xu, Y.-C. Wu, A. Iadecola, L. Liu, H. Ma, L. Qu, E. Raymundo-Piñero, J. Zhu and Z. Lin, ACS Energy Lett., 2020, 5, 2873–2880 CrossRef CAS.
- L. Xu and D.-e Jiang, J. Chem. Phys., 2021, 155, 234707 CrossRef CAS.
- J. Wen, Q. Fu, W. Wu, H. Gao, X. Zhang and B. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 7087–7095 CrossRef.
- Y. Sui and X. Ji, Chem. Rev., 2021, 121, 6654–6695 CrossRef CAS.
- J. Wei, M. Zhou, A. Long, Y. Xue, H. Liao, C. Wei and Z. J. Xu, Nano-Micro Lett., 2018, 10, 1–15 CrossRef.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS.
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2017, 1, 173–180 CrossRef.
- Y. Jiang, T. Sun, X. Xie, W. Jiang, J. Li, B. Tian and C. Su, ChemSusChem, 2019, 12, 1368–1373 CrossRef CAS.
- H. Yang, Y. Ma, X. Lv, B. Huang and Y. Dai, J. Catal., 2020, 387, 12–16 CrossRef CAS.
- L. Meng, L.-K. Yan, F. Viñes and F. Illas, J. Mater. Chem. A, 2023, 11, 6886–6900 RSC.
- G. Gao, A. P. O’Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin and M. Mavrikakis, J. Catal., 2002, 209, 275–278 CrossRef.
- H. Ooka, J. Huang and K. S. Exner, Front. Energy Res., 2021, 9, 654460 CrossRef.
- B. M. Abraham, P. Sinha, P. Halder and J. K. Singh, J. Mater. Chem. A, 2023, 11, 8091–8100 RSC.
- X. Bai, C. Ling, L. Shi, Y. Ouyang, Q. Li and J. Wang, Sci. Bull., 2018, 63, 1397–1403 CrossRef CAS PubMed.
- R. Anand, A. S. Nissimagoudar, M. Umer, M. Ha, M. Zafari, S. Umer, G. Lee and K. S. Kim, Adv. Energy Mater., 2021, 11, 2102388 CrossRef CAS.
- D. Kan, D. Wang, X. Zhang, R. Lian, J. Xu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 3097–3108 RSC.
- B. Wei, Z. Fu, D. Legut, T. C. Germann, S. Du, H. Zhang, J. S. Francisco and R. Zhang, Adv. Mater., 2021, 33, 2102595 CrossRef CAS.
- J. Zheng, X. Sun, C. Qiu, Y. Yan, Z. Yao, S. Deng, X. Zhong, G. Zhuang, Z. Wei and J. Wang, J. Phys. Chem. C, 2020, 124, 13695–13705 CrossRef CAS.
- D. Jin, L. R. Johnson, A. S. Raman, X. Ming, Y. Gao, F. Du, Y. Wei, G. Chen, A. Vojvodic and Y. Gogotsi, J. Phys. Chem. C, 2020, 124, 10584–10592 CrossRef CAS.
- M. Pandey and K. S. Thygesen, J. Phys. Chem. C, 2017, 121, 13593–13598 CrossRef CAS.
- Z. Zeng, X. Chen, K. Weng, Y. Wu, P. Zhang, J. Jiang and N. Li, npj Comput. Mater., 2021, 7, 80 CrossRef CAS.
- X. Wang, C. Wang, S. Ci, Y. Ma, T. Liu, L. Gao, P. Qian, C. Ji and Y. Su, J. Mater. Chem. A, 2020, 8, 23488–23497 RSC.
- C. Ling, L. Shi, Y. Ouyang and J. Wang, Chem. Mater., 2016, 28, 9026–9032 CrossRef CAS.
- W. Jiang, X. Zou, H. Du, L. Gan, C. Xu, F. Kang, W. Duan and J. Li, Chem. Mater., 2018, 30, 2687–2693 CrossRef CAS.
- B. Wang and F. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202111026 CrossRef CAS.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- Y. Freund and R. E. Schapire, J. Comput. Syst. Sci., 1997, 55, 119–139 CrossRef.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- J. H. Friedman, Ann. Stat., 2001, 1189–1232 Search PubMed.
- R. A. Soomro, P. Zhang, B. Fan, Y. Wei and B. Xu, Nano-Micro Lett., 2023, 15, 108 CrossRef CAS.
- S. Huang and V. N. Mochalin, ACS Nano, 2020, 14, 10251–10257 CrossRef CAS.
- T. Wu, P. R. Kent, Y. Gogotsi and D.-E. Jiang, Chem. Mater., 2022, 34, 4975–4982 CrossRef CAS.
- P. Hou, Y. Tian, Y. Xie, F. Du, G. Chen, A. Vojvodic, J. Wu and X. Meng, Angew. Chem., Int. Ed., 2023, 135, e202304205 CrossRef.
- L. Zhang, J. Han, H. Wang, R. Car and W. E, Phys. Rev. Lett., 2018, 120, 143001 CrossRef CAS.
- H. Song and D.-e Jiang, Nanoscale, 2023, 15, 16010–16015 RSC.
- X. Zhao, A. Vashisth, J. W. Blivin, Z. Tan, D. E. Holta, V. Kotasthane, S. A. Shah, T. Habib, S. Liu and J. L. Lutkenhaus, Adv. Mater. Interfaces, 2020, 7, 2000845 CrossRef CAS.
- E. Marquis, F. Benini, B. Anasori, A. Rosenkranz and M. C. Righi, Nano Convergence, 2023, 10, 16 CrossRef CAS PubMed.
- V. Nesterova, V. Korostelev and K. Klyukin, J. Phys. Chem. Lett., 2024, 15, 3698–3704 CrossRef CAS PubMed.
- R. Ibragimova, M. J. Puska and H.-P. Komsa, ACS Nano, 2019, 13, 9171–9181 CrossRef CAS.
- M. López, K. S. Exner, F. Viñes and F. Illas, Adv. Theory Simul., 2023, 6, 2200217 CrossRef.
- R. Ibragimova, P. Erhart, P. Rinke and H.-P. Komsa, J. Phys. Chem. Lett., 2021, 12, 2377–2384 CrossRef CAS.
- B. Wei, Z. Fu, D. Legut, T. C. Germann, Q. Zhang, S. Du, H. Zhang, J. S. Francisco and R. Zhang, J. Phys. Chem. C, 2021, 125, 4477–4488 CrossRef CAS.
- K. D. Fredrickson, B. Anasori, Z. W. Seh, Y. Gogotsi and A. Vojvodic, J. Phys. Chem. C, 2016, 120, 28432–28440 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |