
Development of supported intermetallic compounds: advancing the Frontiers of heterogeneous catalysis
Yuan-Jun
Song
ab,
Sijie
Guo
c,
Peng
Xia
ab,
Fei
Sun
ab,
Ze-Xian
Chen
ab,
Shi-Han
Yang
ab,
Xiao-Yang
Zhang
ab and
Tong
Zhang
 *abd
*abd
aSchool of Electronic Science and Engineering, Southeast University, Nanjing 210096, China. E-mail: tzhang@seu.edu.cn
bSuzhou Key Laboratory of Metal Nano-Optoelectronic Technology, Suzhou 215123, China
cDepartment of Chemical and Biomolecular Engineering, University of Tennessee, Knoxville, Tennessee 37996, USA
dKey Laboratory of Micro-Inertial Instrument and Advanced Navigation Technology, Ministry of Education, and School of Instrument Science and Engineering, Southeast University, Nanjing 210096, China
First published on 3rd October 2024
Abstract
Intermetallic compound (IMC) catalysts have garnered significant attention due to their unique surface and electronic properties, which can lead to enhanced catalytic performance compared to traditional monometallic catalysts. However, developing IMC materials as high-performance catalysts has been hindered by the inherent complexity of synthesizing nanoparticles with well-defined bulk and surface compositions. Achieving precise control over the composition of supported bimetallic IMC catalysts, especially those with high surface area and stability, has proven challenging. This review provides a comprehensive overview of the recent progress in developing supported IMC catalysts. We first examine the various synthetic approaches that have been explored to prepare supported IMC nanoparticles with phase-pure bulk structures and tailored surface compositions. Key factors influencing the formation kinetics and compositional control of these materials are discussed in detail. Then the strategies for manipulating the surface composition of supported IMCs are delved into. Applications of high-performance supported IMCs in important reactions such as selective hydrogenation, reforming, dehydrogenation, and deoxygenation are comprehensively reviewed, showcasing the unique advantages offered by these materials. Finally, the prevailing research challenges associated with supported IMCs are identified, including the need for a better understanding of the composition–property relationships and the development of scalable synthesis methods. The prospects for the practical implementation of these versatile catalysts in industrial processes are also highlighted, underscoring the importance of continued research in this field.
1. Introduction
Currently, one of the grand challenges for the heterogeneous catalysis community is the purposeful and selective manipulation of surface chemistry that destabilizes specific chemical moieties within an environment of many different chemical moieties.1–4 To achieve this goal, materials with sufficient complexity and tunability concerning their surface chemistry must be understood to the point that they can be rationally designed as catalysts. Thus, there is an urgent demand for fundamental connections between surface and catalytic chemistry over catalytic materials. However, many mainstream monometallic or TM–TM alloy catalysts often show ill-defined bulk and surface composition, so their surface chemistry cannot be rationally manipulated for specific catalytic reactions. Therefore, the synthesis of well-defined catalysts NCs with pure bulk phase and controllable surface has attracted tremendous attention from academic and industrial communities.Intermetallic compounds (IMCs) are a fascinating class of materials composed of two or more metallic elements combined in a specific, ordered atomic arrangement. Unlike the random, disordered structure in metal alloys, IMCs form an ordered crystal structure driven by a strong chemical attraction between the elements. This unique structure results in new electronic properties compared to the parent elements. It also gives rise to unique surface chemistry in IMCs, allowing for controlled surface reactivity towards C, H, and O across a wide range by adjusting bulk and surface composition. In contrast, the limited tunability of surface chemistry in alloys is due to the mutual solubility of the elements in a random manner. Therefore, IMCs have been suggested to be ideal catalysts in many reactions due to the possibility of control of catalytic chemistry.3,5–18 For example, many studies have illustrated the use of both precious group metals (PGM) and non-noble metal IMCs as catalysts in reactions that require either aggressive or gentle activation of saturated and unsaturated C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and N–O bonds, e.g., semi-hydrogenation, selective hydrogenation, heteroatom removal, reforming, electrocatalytic oxidation, partial hydrogenation, etc.2,3,13,15,17,19–29 Through these studies, it has been demonstrated that the inclusion of the p-block TM in the TM solid directly modifies and lessens surface reactivity towards CC bonds while building in enhanced reactivity towards bonds that contain oxygen, such as C–O, O–H, CO, N–O, etc.15,18,21,23,24,28,30 Other studies have also indicated that different bulk stoichiometries/phases of IMCs exhibit different catalytic performances. However, due to a lack of truly surface-sensitive surface composition analysis, this trend is still unclear.3,13,20–23
O, and N–O bonds, e.g., semi-hydrogenation, selective hydrogenation, heteroatom removal, reforming, electrocatalytic oxidation, partial hydrogenation, etc.2,3,13,15,17,19–29 Through these studies, it has been demonstrated that the inclusion of the p-block TM in the TM solid directly modifies and lessens surface reactivity towards CC bonds while building in enhanced reactivity towards bonds that contain oxygen, such as C–O, O–H, CO, N–O, etc.15,18,21,23,24,28,30 Other studies have also indicated that different bulk stoichiometries/phases of IMCs exhibit different catalytic performances. However, due to a lack of truly surface-sensitive surface composition analysis, this trend is still unclear.3,13,20–23
Unfortunately, the fundamental study and design of IMC materials as catalysts from an atomic and electronic level has proven to be challenging due to innate complexity and difficulties in synthesizing nanoparticles with well-defined bulk and surface compositions across a wide range of compositions, especially high surface area supported bimetallic compounds. In addition, since heterogeneous catalysis is based on surface science, consequently, significant experimental efforts have been dedicated to actualizing computational predictions in this field. Specifically, particular attention has been paid to the development of synthesis techniques for oxide-supported IMC nanoparticles of phase-pure bulk and controlled particle surface composition. In contrast, the preparation of other heterogeneous catalysts, such as supported metal or metal oxide catalysts, may involve simpler methods, but they may not provide the same level of adjustability in structure and composition as IMCs. These catalysts usually prioritize the control of morphologies and particle size. The extra synthetic steps needed for IMCs enable the optimization of their electronic and surface properties to target specific catalytic applications. The ultimate goal of such synthesis is the catalysts with controllable bulk and surface composition that will enable comprehensive investigation of their effects on surface and catalytic chemistry. The ultra-high vacuum (UHV) technique is regarded as the most exact tool to study the surface chemistry of these materials which can rationally set the arrangement of atoms.2,31–33 It must be noted, however, that conducting UHV surface science studies is expensive, and operating the UHV system can be both challenging and time-consuming. Over the past several decades, numerous synthesis routes have been developed for supported IMCs.8,34–37 However, despite the availability of these methods, there is still a lack of a comprehensive summary that includes the mechanism of formation and control of bulk and surface composition. A summary of this nature is essential for a deep and thorough investigation of the true surface science of materials within the entire community. Therefore, to gain a clear understanding of the physical phenomena that dictate their bulk and surface composition as a function of constituent element selection, oxide support choice, and preparation environment, this review will focus on the recent progress of the high surface area supported IMC catalysts. Firstly, the common synthesis techniques of supported IMC nanoparticles will be discussed, including the key factors that influence their formation kinetics and well-defined examples. Then the surface manipulation of these catalysts will be delved. Furthermore, this review presents the applications of selected high-performance supported IMCs in various catalytic reactions, such as selective hydrogenation, reforming, dehydrogenation, and deoxygenation. Lastly, we summarize the prevailing research challenges associated with supported IMCs in terms of their bulk and surface control, catalytic chemistry, and applications as well as prospects.
2. Synthesis method of supported IMCs
2.1. Impregnation method
The most commonly used method for the synthesis of supported IMC nanoparticles is the impregnation method due to simple procedure. As shown in Fig. 1, firstly constituent element precursors are deposited on the support at the same time or in subsequent steps. In route 1, one of the constituent elements is first deposited on the support and reduced to pure metal, and then the secondary element precursor is added and reduced. After deposition, precursors of constituent elements are reduced under an H2 environment, then diffuse across the surface of the support and finally form a compound with a specific crystal structure. In route 2, A and B precursors are co-deposited. In both routes, since the element precursors are physically mixed with support, theoretically any precursors can be deposited on any supports, which makes this method universal for the preparation of different materials. However, many studies indicate nanoparticle IMC synthesized via impregnation methods often results in multi-phase systems where particles may be of disparate phases or polyphase individual particles.38–40 This is because the kinetics of constituent element incorporation into the growing IMC nanoparticle is a function of precursor reduction kinetics, diffusion kinetics of reduced precursor metal across the support, and the thermodynamic driving force for IMC formation. Frequently, additional annealing pretreatment is needed for the reorganization of bulk and surface. Additionally, adsorbate–metal interactions at the surface of the IMC particle during synthesis and under reaction conditions drive element segregation only at elevated temperatures where the thermodynamics of IMC formation are surmounted.41 Each of these effect factors can individually drive the formation of multiphase systems with off-stoichiometric surface compositions that inhibit well-defined fundamental studies, which has been informed in many studies.38,40–43 Thus, to understand how to control the bulk and surface composition using this method, these effect factors are reviewed and analyzed in detail in this section.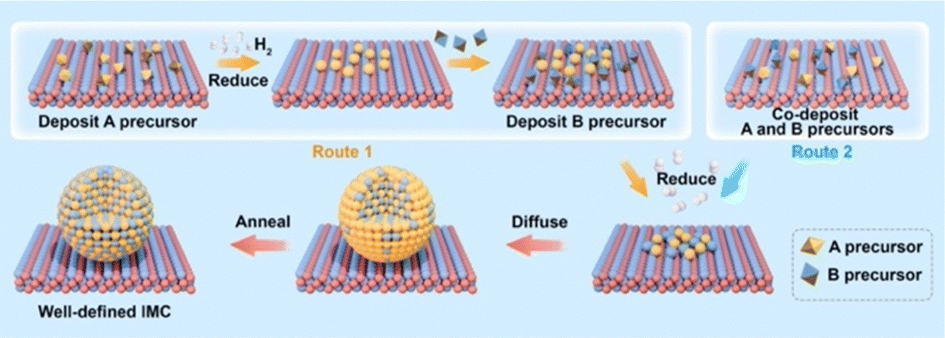 | ||
| Fig. 1 Schematic for the synthesis supported IMCs via impregnation method. | ||
2.1.1.1. Reduction kinetics. The reduction kinetics of constituent element precursors is complex, which is related to the reducibility of precursor, interaction between element and support, and reduction environment (such as H2 concentration and temperature). In the co-impregnation route, the simultaneous and complete reduction of both constituent elements is required to ensure the homogeneous distribution of atoms on the support surface, which will favor the formation of the pure phase. To reach this goal, one simple method is using high H2 concentration in the impregnation method or strong reduction agents in the chemical reduction method which can promote rapid reduction. As shown in Fig. 2, the study by Laursen groups shows that pure phase Ni + Ga/SiO2 IMC can be produced as the H2 concentration increases.8 The phenomenon of elevated H2 concentration promoted reduction kinetics was also observed in other studies.44,45 In addition, it is also noted the critical concentration of H2 for different stoichiometries phases depends on the choice of support and stability of phase. The effect of support on the reduction kinetics of precursors originates from the element–support interaction. It is found that utilization of inert support such as SiO2 and carbon can promote the formation of pure phase IMC, while utilization of more active support such as Al2O3 leads to multi-phase due to inhomogeneous reduction kinetics caused by the strong metal–support interaction and hydrophilic and acidic nature of support.5,8,46,47 This phenomenon was also observed in the synthesis studies of Pd3Pb over SiO2 and Al2O3 where phase pure Pd3Pb could be obtained over SiO2 but not over Al2O3 under the same reduction conditions.48,49
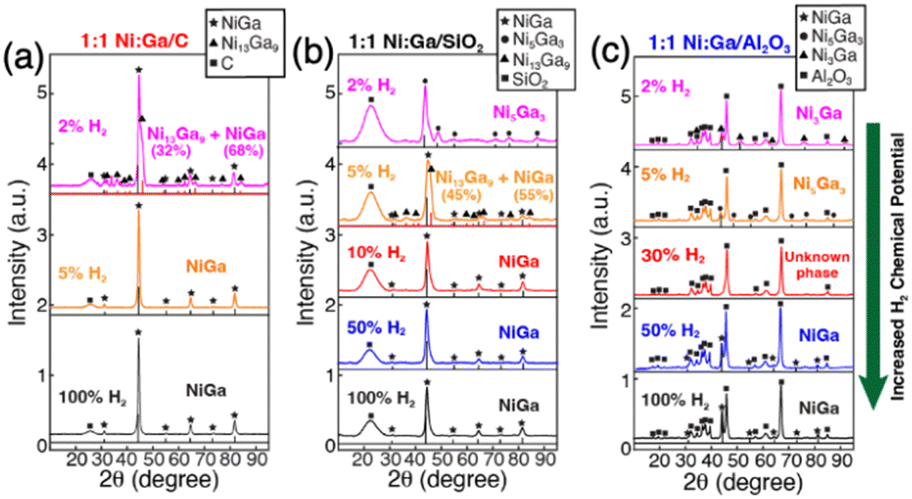 | ||
Fig. 2 Long-acquisition-time pXRD of (a) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ni:Ga/C; (b) 1:1 Ni:Ga/SiO2; and (c) 1:1 Ni:Ga/Al2O3 after reduction with different concentrations of H2 in Ar at 700 °C, which illustrated the effect of H2 chemical potential on bulk composition.8 Copyright 2022, Royal Society of Chemistry. :1 Ni:Ga/C; (b) 1:1 Ni:Ga/SiO2; and (c) 1:1 Ni:Ga/Al2O3 after reduction with different concentrations of H2 in Ar at 700 °C, which illustrated the effect of H2 chemical potential on bulk composition.8 Copyright 2022, Royal Society of Chemistry. | ||
On the other hand, in the sequent impregnation route, after the first element A precursor is deposited and reduced on the support, the later added secondary B element will grow following the mode of island growth, layer growth, or Stranski–Krastanov, depending on the strongness of A–B interaction vs. B-support interaction.50 If the A–B interaction is stronger than the B-support interaction, island growth will dominate. Otherwise, layer growth will occur. The Stranski–Krastanov growth is the combination of the previous two modes which often occurs when the lattices of solid and support mismatch. However, in either mode, it is easy to produce core–shell structures which are often unstable under harsh reaction conditions, and also lose the possible reaction sites dominated by the element in the core.51,52 Therefore, to fix this problem, usually, the first element precursor needs a rapid reduction to form tiny seeds so that their high surface energy can avoid aggregation of atoms. Similarly, the rapid reduction of secondary element precursor is also required to drive the coalescence with element A to form compounds with lower energy.
Another direct way to tune the reducibility of elements is the selection of different metal precursors. The common precursors are usually inorganic salts (such as nitrate, chloride, and acetates) and organic complexes (such as carboxylates, acetylacetonates, and phosphine), which show different reduction kinetics and significantly affect the bulk composition of IMCs. For example, Lagrow and co-workers showed the effect of precursor ligands on the synthesis of Pt + Ni and Pt + Cu compounds, which indicated the choice of precursors choice affected the nanoparticle sizes and bulk composition.53 Another study by the Saha group also showed that pure PtPb solid can be synthesized by using acetylacetonate salt while multi-phase was observed using chloroplatinic acid because of the strong metal–acetylacetonate interaction limiting the reduction.54
For the co-impregnation, similar reduction kinetics of constituent elements will promote the rapid and complete reduction leading to the formation of IMCs with pure bulk phase. Otherwise, compounds with multi-phase will be observed. For example, in the work by the Laursen group, the reducibility of different nickel and gallium salts and their effects on the formation of Ni + Ga IMCs were systematically investigated, which indicates NiGa with pure bulk phase can be synthesized when the nickel nitrate and gallium nitrate were used due to their similar reduction kinetics.8 A similar phenomenon was encountered in a study focused on Pd-based IMCs synthesis only pure phase PdBi/Al2O3 was produced but Pd3Sn and Pd3Pb showed multi-phase bulk compositions due to large different reduction kinetics of Sn and Pb salt with Pd salts48 (see Fig. 3). This figure also shows pure Co3Sn2/Al2O3, Ni3Sn/Al2O3, and Ni3Sn2/Al2O3 were obtained as a result of similar reduction potential between Co or Ni and Sn salts. In addition, the choice of solvent also affects the reduction of metal salts, especially for reactive metals such as early transition metals (TMs) and large p-elements which will react with water to form unreducible complexes or precipitation. In those cases, organic solvents will be utilized. For example, Zhang et al. found in the Fischer–Tropsch synthesis on Co/SiO2 using dehydrated ethanol as solvent can increase the conversion compared to using water and 95% ethanol caused by the formation of cobalt complex in water and strong interaction with support leading to difficult reduction.55 Moreover, to overcome the slow and incomplete issues caused by low reducibility or strong metal–support interaction, a high reduction temperature can be applied.
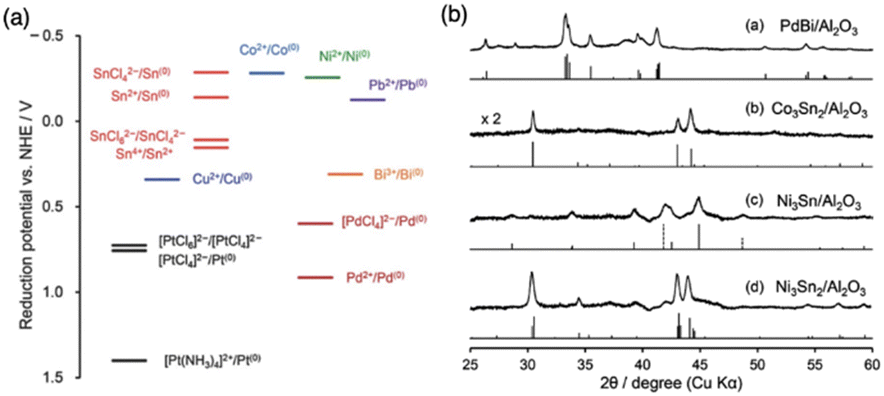 | ||
| Fig. 3 (a) Energy diagram of standard reduction potentials of the chemicals used in this study and related ones. (b) XRD patterns of (1) PdBi/Al2O3, (2) Co3Sn2/Al2O3, (3) Ni3Sn/Al2O3, and (4) Ni3Sn2/Al2O3 prepared by hydrogen reduction at 600 °C and their reference materials.48 Copyright 2013, Royal Society of Chemistry. | ||
2.1.1.2. Diffusion kinetics. After the reduction step, reduced atoms diffuse across the support surface and coalesce into a solid. The diffusion kinetics of reduced metal is also a function of the support and pretreatment environment. Off-stoichiometric IMC compounds will be produced if element diffusion is limited. To overcome the diffusion barrier, sufficient energy is needed which can be provided by either high temperature or high driving force. Under high temperatures, diffusion kinetics of reduced elements are surmounted leading to free movement of atoms on the surface and sitting at the lowest energy position. However, this tends to drive the surface segregation or sintering as well as the disordered phase.56–58 Thus, high-temperature pretreatment should be carefully employed. Interestingly, some studies suggest that the presence of atomic H can promote the formation of compounds with pure phase because the H spillover effect can reduce interaction between elements and support enhancing the diffusion.8,59–61 In addition, some studies indicate utilization of relatively inert support can decrease the diffusion barriers of elements leading to more readily formation of compounds, however, causing sintering and large nanoparticle size.8,62–64 For example, as shown in Fig. 4, the work by the Laursen group on the synthesis of Ni + Ga IMCs confirms the trend of diffusion barriers as a function of support activity. This trend is evident by the formation of pure phase NiGa IMCs at lower temperatures over more inert carbon support compared to the SiO2 support but with larger particle size (see Fig. 4a–d).8 Similarly, He et al. reported that the particle size of Pd over hydrotalcite support is much smaller than when using MgO and Al2O3 supports, which have more inert reactivity.62 Huang group also found the effect of support on the particle size in the synthesis of PtSn (1
:1) compounds, showing that PtSn over mesoporous silica wells (MSWs) has a smaller size with 3.9 nm than when using mesoporous silica support (SBA-15) with 6–8 nm (see Fig. 4e–g).65
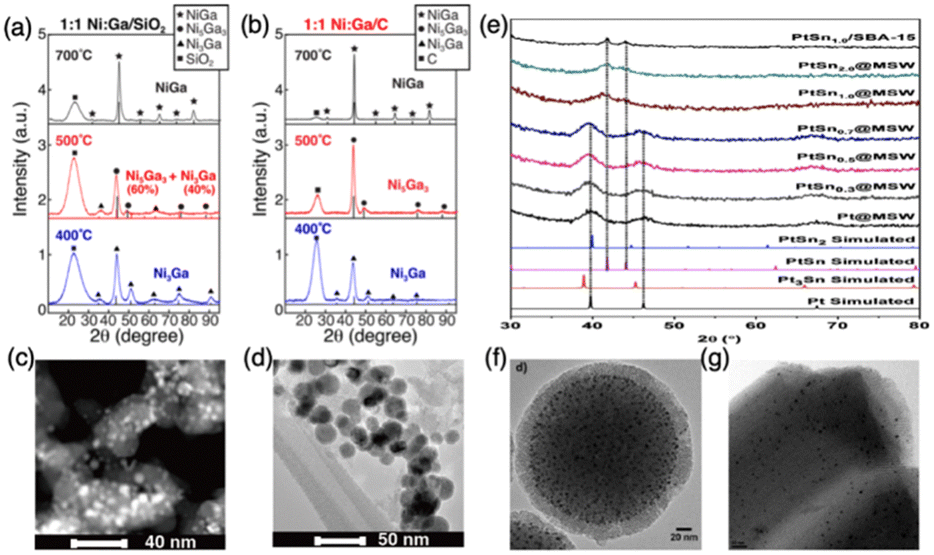 | ||
| Fig. 4 (a) XRD of 1:1 NiGa/SiO2 formed at different temperatures; (b) XRD of 1:1 NiGa/C formed at different temperatures; (c) TEM of 1:1 NiGa/SiO2 formed at 700 °C; (d) TEM of 1:1 NiGa/C formed at 700 °C; (e) PXRD patterns of the PtSnx@MSW, x = 0, 0.30, 0.50, 0.70, 1.0 and 2.0; (f) TEM of PtSn1.0/MSW; (g) TEM of PtSn1.0/SBA-15. (a)–(d) are from ref. 8, Copyright 2022, Royal Society of Chemistry, and (e)–(g) are from ref. 65, Copyright 2020, WILEY-VCH Verlag GmbH & Co.KGaA. | ||
2.1.1.3. Thermodynamics of formation. The complex thermodynamics of IMC formation can be signified by the phase relative stability and pretreatment temperature. According to the Gibbs free energy of a specific phase. In the binary element system, the stability of the phase is correlated to temperature, usually the most stable phase with the lowest enthalpy will be produced firstly at low temperature and different phases can transform at critical temperature. When the temperature is increased during reduction or annealing, the phase initially tends to change from a disordered state to an ordered state. For instance, a pure phase multi-principal element IMC was produced by rapid annealing at 1100 K for 5 mins which is sufficient to complete the transition from the disordered structure to ordered and pure phase.66 Once the phase transformation critical temperature is reached, a further increase in temperature will cause the phase to transform from ordered to disordered. This means that controlling the temperature is essential for controlling the bulk composition. However, the high temperature tends to drive the formation of disordered atomic arrangement due to the higher entropy than the ordered phase. Conversely, at low temperatures, the ordered phase is thermodynamically favored. Therefore, the phase transformation as a function of reduction or annealing temperature is consistent with the order of phase stability.
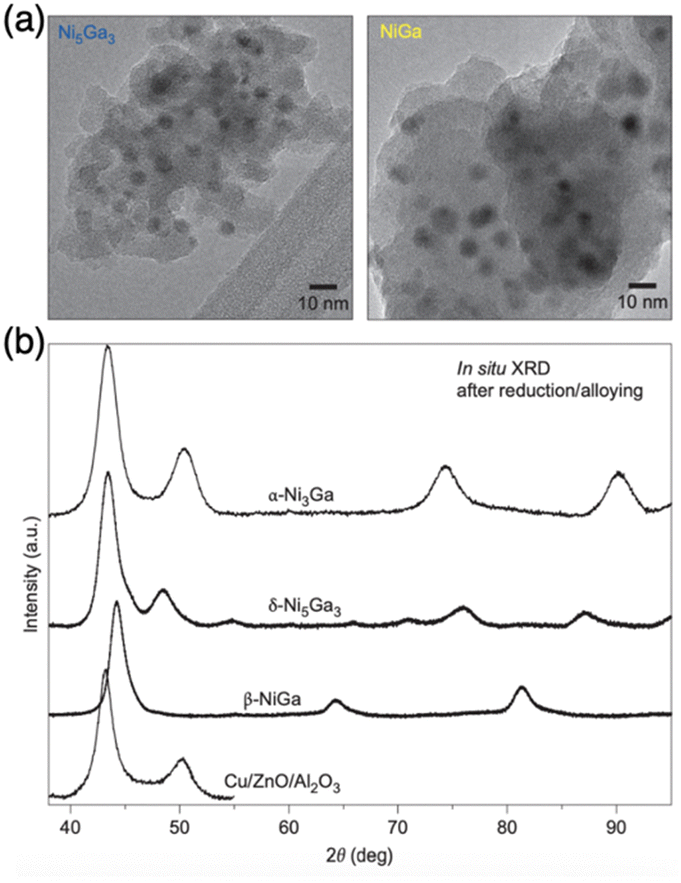 | ||
| Fig. 5 (a) TEM images of Ni5Ga3 and NiGa and (b) in situ XRD patterns of Ni3Ga, Ni5Ga3, and NiGa intermetallic compounds as well as Cu/ZnO/Al2O3.3 Copyright 2014, Springer Nature Limited. | ||
The ability to synthesize well-defined supported IMCs with fine-tuned composition and nearly ideal catalytic performance through the impregnation method has been demonstrated. Additionally, the development of novel IMCs further highlights the importance of understanding IMC growth processes for future design and utilization. While the impregnation method also has some drawbacks. One of the limitations is the potential difficulty in controlling the particle size and distribution of the IMCs, which can impact their catalytic properties. Furthermore, the process of achieving surface composition resembling the bulk material through adjustment of pretreatment conditions or additional annealing pretreatments can be time-consuming and may not always result in the desired composition. These drawbacks highlight the need for further research and development to optimize the impregnation method for IMC synthesis.
2.2. Strong metal–support interaction induced IMC formation
As discussed in Section 2.1, strong metal–support interaction (SMSI) may cause multiphase, but if the support is reducible such as TiO2, Ga2O3, and In2O3, this kind of unfavorable phenomenon can induce the formation of IMC at high reduction temperature or ultrahigh vacuum. Many studies have demonstrated the successful synthesis of IMCs based on SMSI.61,70,71 SMSI term was first coined by the Tauster group in the late 1970s and originated from the observation of dramatically reduced H2 and CO adsorption over TiO2 supported PGMs under high-temperature reduction conditions due to the formation of PGMs–Ti bonding.46,72,73 Subsequently, the studies on SMSI prevailed for a while but were suppressed because of limited characterization techniques for the investigation of dynamic processes on the metal–support interface. In recent years, advanced characterization technologies such as X-ray photoelectron spectroscopy (XPS) and in situ high-resolution transmission electron microscopy (HRTEM) have developed allowing a deep understanding of the SMSI mechanism, thus SMSI attracts boosting attention again in the heterogeneous catalyst community. More meaningfully, the discovery of SMSI brings about new avenues for the rational design of heterogeneous catalysts due to the significant effect on the surface and catalytic chemistry of catalysts. Despite the efforts of many researchers, it should also be noted that SMSI mechanisms are still not exactly clear because of the complex dynamics of the interface. Therefore, this review focuses on the formation of IMCs based on SMSI under high-temperature reduction conditions.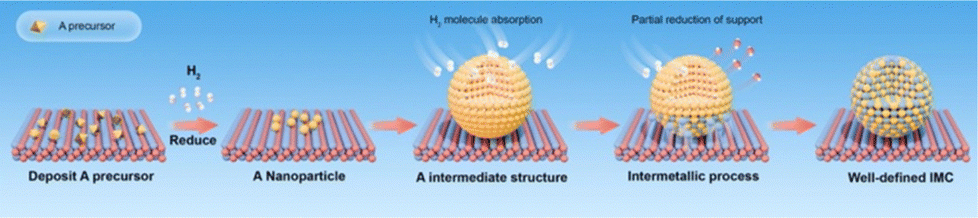 | ||
| Fig. 6 Schematic for the synthesis of supported IMCs via SMSI. | ||
To enable the control of the bulk composition of IMCs, the amount of metal B exacted from support needs to be adjusted by changing the reduction conditions. In the example of the formation of Pd + In IMCs by SMSI using In2O3 as support, as the reduction temperature increases, the In concentration increases.61 As shown in Fig. 7, pure phase PdIn and Pd2In3 were produced when reduced at 300 °C and 390 °C, respectively. But when the reduction temperature increases to 550 °C, a mixture of Pd3In7 phase and pure In metal was observed due to the complete reduction of In2O3. A similar phenomenon was encountered in the synthesis of Pd + Ga/Ga2O3 that a more Ga-rich Pd + Ga phase was formed as the reduction temperature increased.70,82
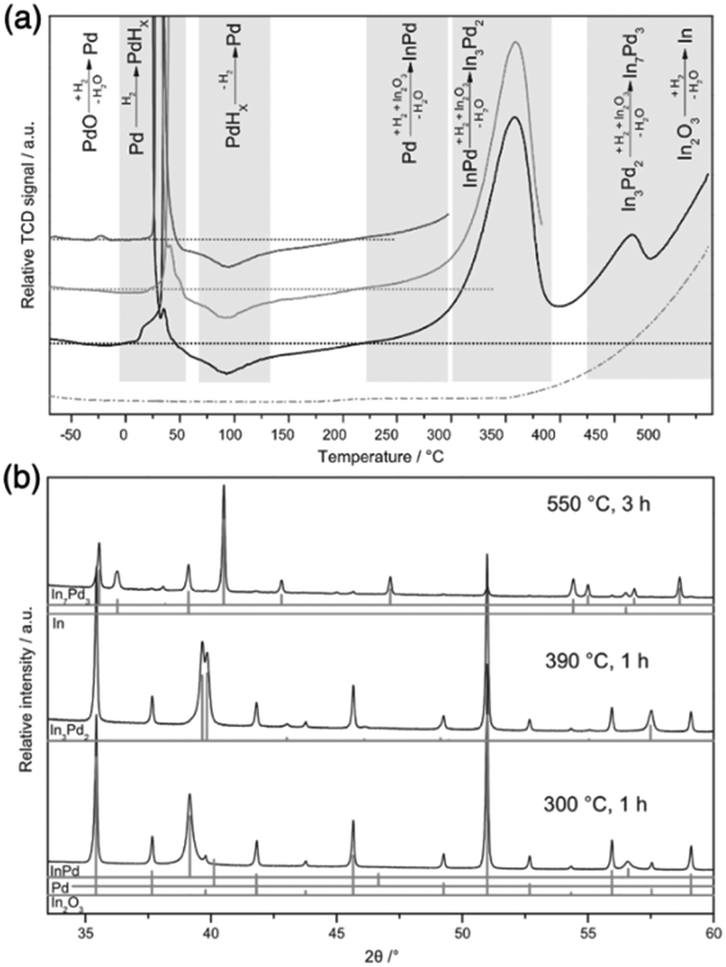 | ||
| Fig. 7 (a) TPR measurements of PdO/In2O3 (solid lines) and In2O3 (dashed line) with 5 °C min−1 in 10 vol% H2/Ar and (b) XRD patterns of PdO/In2O3 after the indicated reductive treatments.61 Copyright 2016, Elsevier Inc. | ||
| Catalyst | IMC phase | Pretreatment condition | Particle size | Reaction |
|---|---|---|---|---|
| Pd/In2O361 | PdIn | Reduction at 300 °C | 84 nm | Steam reforming of methanol |
| Pd2In3 | Reduction at 390 °C | 90 nm | ||
| Pd3In7 | Reduction at 550 °C | 104 nm | ||
| Pd/α-Ga2O370 | Pd2Ga | Reduction at 250 °C | 6 nm | Steam reforming of methanol, hydrogenation of acetylene, and CO2 hydrogenation |
| Pd5Ga3 | Reduction at 550 °C | 5 nm | ||
| Pd/β-Ga2O370 | Pd2Ga | Reduction at 310 °C | 8 nm | Methanol steam reforming, hydrogenation of acetylene, CO2 hydrogenation |
| Pd5Ga3 | Reduction at 565 °C | 7 nm | ||
| Pd and Pt supported on ZnO, Ga2O3, and In2O374 | PdZn, Pd5Ga3, PdGa5, PtZn, Pt5Ga3, and PtIn2 | Reduction at 500 °C | — | Steam reforming of methanol, dehydrogenation of methanol |
| Pt supported on SiO2, Al2O3, and CeO276 | Pt3Si | Reduction at 600 °C | 10–15 nm | — |
| Pt3Al | ||||
| Pt3Ce | ||||
| Ni/SiO277 | Ni3Si | Reduction at 850 k | <5 nm | — |
| Pd/SiO278 | Pd2Si | Reduction at 800 k | 4.4 nm | — |
| Pt/Al2O380 | Pt3Al | Epitaxial growth method | 5–10 nm | Hydrogen cyanide synthesis |
| Pd/Ga2O383,84 | Pd5Ga2 | Reduction at 250 °C | 8.3 nm | Steam reforming of methanol |
| Pd5Ga2 | Reduction at 300 °C | 8.3 nm | ||
| Pd5Ga2 | Reduction at 400 °C | 11.0 nm | ||
| Pd/ZnO86 | PdZn | Reduction at 400 °C | 1.5–6 nm | CO2 hydrogenation |
| Pd/ZnO87 | PdZn | Reduction at 400 °C | — | Steam reforming of methanol |
| Pd/ZnO88 | PdZn | Reduction at 400 °C | 10.4 nm | CO2 hydrogenation |
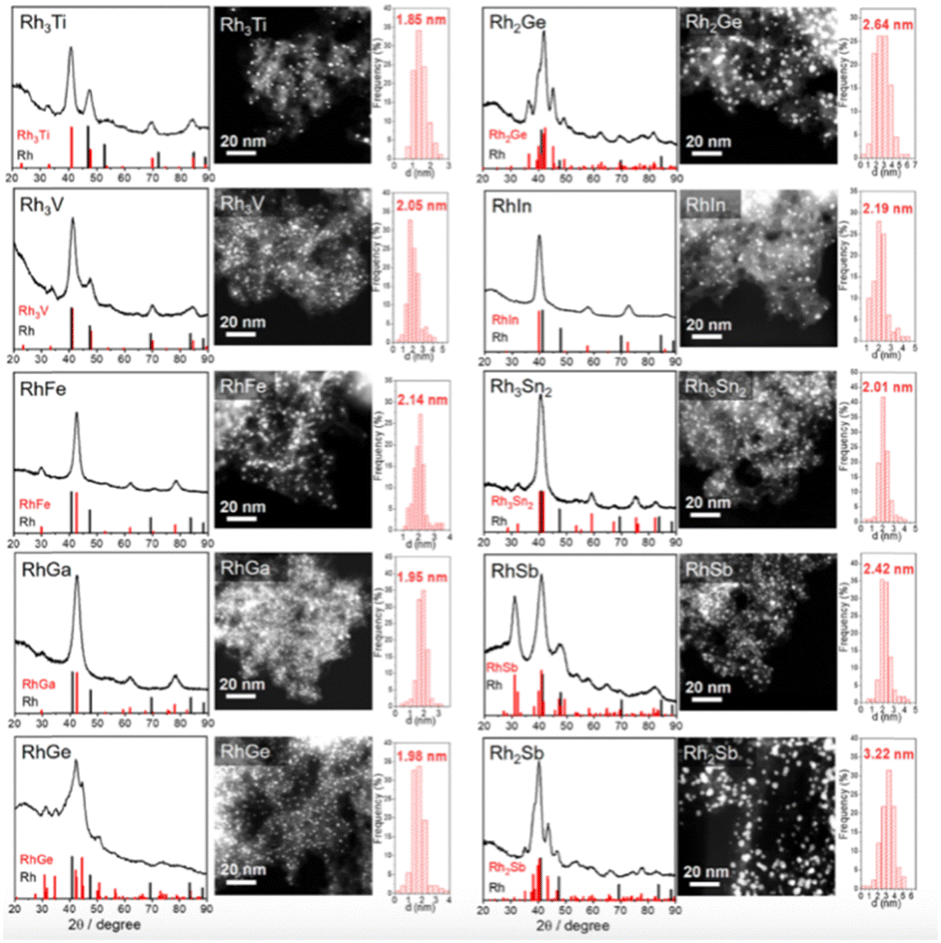 | ||
| Fig. 8 XRD patterns, HAADF-STEM images, and corresponding particle size distribution of Rh-based IMCs supported on S–C.91 The standard peaks of Rh and the corresponding Rh-based intermetallic are shown in the XRD patterns. Copyright 2022, American Chemical Society. | ||
Overall, the formation of IMCs through SMSI represents a versatile strategy for tailoring the properties of heterogeneous catalysts. By elucidating the mechanisms underlying SMSI-induced IMC formation, researchers can design catalysts with enhanced performance and stability for a wide range of chemical transformations. However, this approach shows significant drawbacks. Firstly, this method is only suitable for specific systems, mostly those composed of reducible oxide support and PGMs, which limits the compositional space choice for catalyst design. In addition, high-temperature reduction conditions which are mostly higher than 500 °C tend to cause nanoparticle sintering and surface segregation leading to less control of the surface composition.
2.3. Precipitation method
Compared to the impregnation method, the precipitation method is more advanced and can produce supported IMCs with higher loading and more well-dispersed nanoparticles. The precipitation method can be classified as deposition precipitation and co-precipitation according to the addition order of constituent element precursors and support. As shown in route 1 of Fig. 9, the deposition precipitation method involves adding one of the constituent element precursors first, which then reacts with an additional substance (such as NaOH, KOH, Na2CO3, and urea) to form a precipitation complex, usually in the form of metal hydroxide. This complex is then attracted to the support surface due to electrostatic interaction or hydroxyl bonding.93–95 The secondary constituent element precursor is added afterward and deposited by adjusting pH to form precipitation. Some studies have confirmed the effect of the addition order of metal salt on the bulk phase and catalytic performance.96–98 On the other hand, in route 2 of Fig. 9, the co-precipitation method involves the simultaneous addition of metal salts and the support material, requiring careful control of process conditions to prevent inhomogeneous nucleation and ensure proper dispersion dynamics. Similar to the impregnation method, after the precipitation and washing stage, the dried precipitate undergoes pretreatments such as reduction and annealing for the formation of IMCs as well as control of bulk and surface composition. Furthermore, due to the precipitation method being carried out using a significant volume of aqueous solution, sometimes chemical reduction agents like NaBH4 and LiHBEt3 are utilized to reduce the metal complex to metallic solids. For example, Magno et al. reported the synthesis of intermetallic Pt/Bi and Pt/Pb nanoparticles using NaBH4 as a reducing agent.99 Dey synthesized Ag–Co/C IMCs by reduction of AgNO3 and Co(NO3)2 by NaBH4.100 For the less reduceable metal salts, a stronger reducing agent is selected. Armbruster and co-workers synthesized Al2O3 supported PdGa and Pd2Ga using LiHBEt3 instead of NaBH4 to ensure complete reduction of GaCl3.101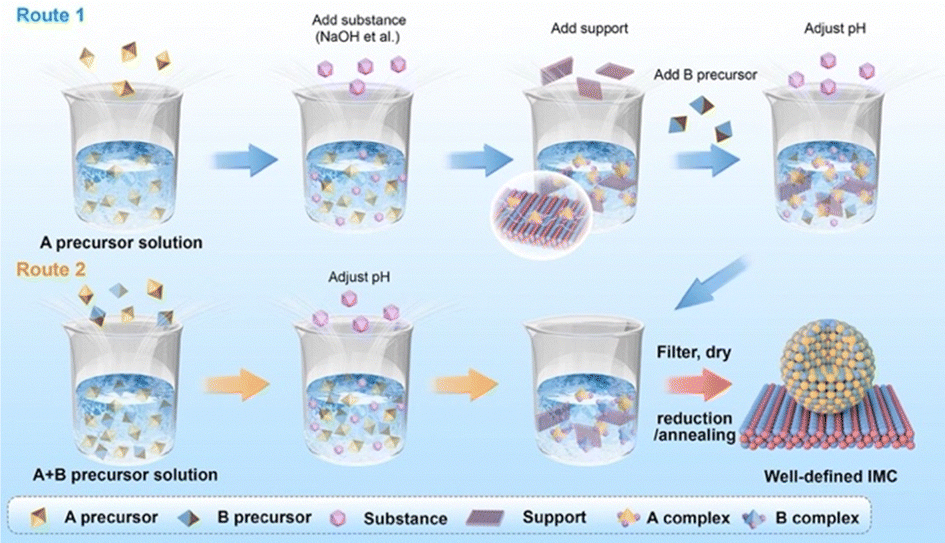 | ||
| Fig. 9 Schematic for the synthesis of supported IMCs via precipitation method. | ||
Second, the precipitation rate and deposition of precursors on the support need to be controlled to a point where the metal complex precipitation just forms and simultaneously attracts the support. Otherwise, over-precipitation or weak interaction with support can cause less loading and aggregation of nuclear which affects dispersion and particle size. Each metal salt has an individual hydrolysis curve as the function of pH and temperature, so nucleation will not occur if not reach the precipitation point. On the other hand, the point of zero charge (PZC) of support is defined as the pH value at which zero electric charge density of the support surface, which is an important parameter to feature the ability of the support surface for ion adsorption. The PZC of commonly used supports are listed in Table 2.103,104 When the pH value is lower than PZC, the support surface is positively charged and attracts anions and vice versa. Therefore, in the deposition synthesis method, to generate the adsorption site of the metal hydroxide complex on the support, the pH of the solution should be different from the PZC of the support.105 In the synthesis of Al2O3 supported Ni + Ga IMCs by Laursen group, the pH was firstly adjusted to 3.9 for precipitation of gallium nitrate, and then added to Al2O3 support with 8–10 PZC.5,8 After full deposition of gallium hydroxide by stirring, the nickel nitrate was added and pH was adjusted to 7. In Fig. 10a, XRD results indicate the formation of NiGa and Ni3Ga pure phases suggesting the successful synthesis. Pt + Fe/C IMCs (pure Pt3Fe and PtFe phases, see Fig. 10b–e) were prepared by dissolving H2PtCl6 and FeSO4 into ethylene glycol and adding NaOH to adjust pH at 11.106 Similarly, PtCo/C was synthesized by adding NaOH to the solution of as-prepared Pt/C and Co(NO3)2 solution to adjust pH at 10 and using ethylene glycol as a reducing agent.107
| Support | α-Al2O3 | γ-Al2O3 | SiO2 | TiO2 | Fe2O3 | CeO2 | ZnO | MgO | Activated carbon |
|---|---|---|---|---|---|---|---|---|---|
| PZC range | 8–10 | 8–10 | 2–4 | 4–7 | 5–8 | 6 | 8–10 | 10.8–12 | 2–4 |
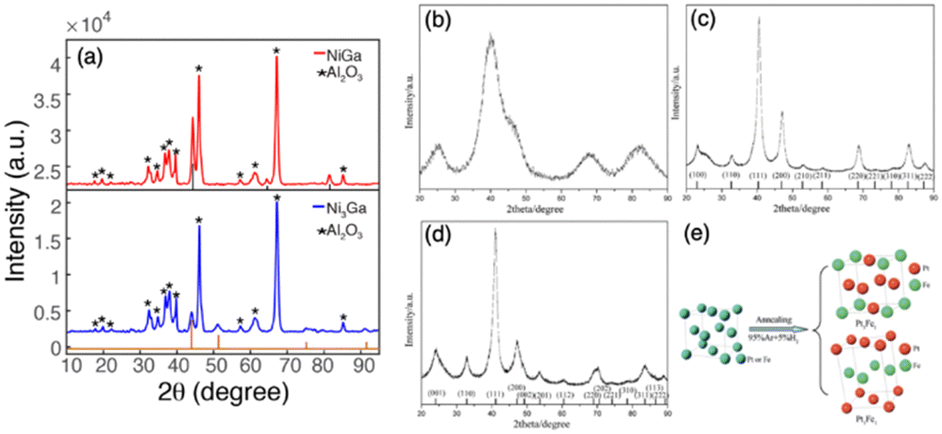 | ||
| Fig. 10 Powder XRD patterns of (a) NiGa/Al2O3 and Ni3Ga/Al2O3,5,8 Copyright 2018, American Chemical Society and Copyright 2022, Royal Society of Chemistry. (b) Typical as-prepared supported chemically disordered Pt–Fe nanoparticles; (c) ordered Pt3Fe1/C; vertical lines show the peak positions of ordered intermetallic Pt3Fe1 (JCPDS No. 89-2050); (d) ordered Pt1Fe1/C; vertical lines show the peak positions of ordered intermetallic Pt1Fe1 (JCPDS No. 65-1051); and (e) schematic showing phase transformation during annealing. (b)–(e) are from ref. 106, Copyright 2012, Royal Society of Chemistry. | ||
Furthermore, in the precipitation method, the reaction temperature can significantly affect the precipitation rate. Generally, higher temperatures can accelerate the precipitation rate and promote the interaction between metal complexes and supports. This is because increased diffusion allows for better mixing of the components and facilitates the adsorption of metal species onto the support surface. However, excessively high temperatures can lead to issues such as particle agglomeration or Ostwald ripening where larger particles grow at the expense of smaller ones.108 Therefore, proper control of temperature is essential to prevent these undesirable effects and ensure uniform dispersion of the supported metal on the catalyst support. Controlled growth of IMCs in the pretreatment steps, such as reduction and annealing, is also challenging. This has been discussed in the impregnation method section, so we will not repeat it here.
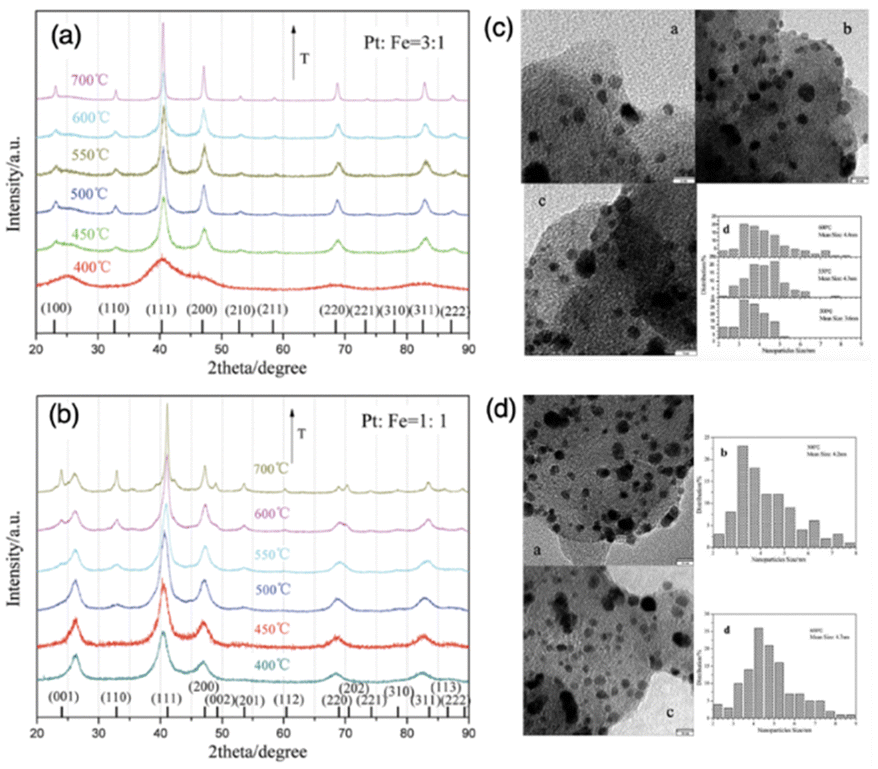 | ||
| Fig. 11 Powder XRD patterns of as-prepared supported (a) Pt3Fe1 and (b) Pt1Fe1 electrocatalysts annealed under a reducing atmosphere at a series of temperatures. TEM images and size distribution of (c) supported Pt3Fe1 and (d) Pt1Fe1 electrocatalysts annealed under a reducing atmosphere at different temperatures.106 Copyright 2012, Royal Society of Chemistry. | ||
2.4. Chemical vapor deposition
Beyond those above liquid-phase synthesis routes, supported IMCs can also be obtained via the chemical vapor deposition (CVD) method. As shown in Fig. 12, in a typical CVD process, the support material is exposed to A precursor vaporization in a closed ultra-high vacuum reactor first. Subsequently, the A atoms are reduced under a flow of H2, followed by the introduction of vapor of the B precursor onto the A/support system, allowing the two metals to directly react with each other. This reaction leads to the formation of a solid material with an atomically ordered crystal structure. Most importantly, since this method provides the ability to facilitate direct reactions between metal precursors on the support surface, it enables the rational control of composition by adjusting the concentration of precursor gases, while also generating well-dispersed and stable materials. These contribute to the creation of IMCs with tailored surface and catalytic properties.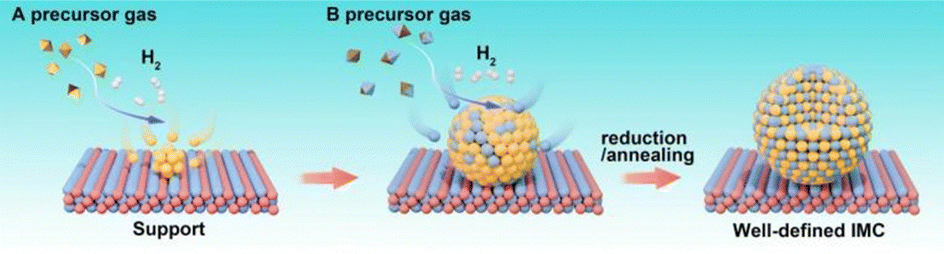 | ||
| Fig. 12 Schematic for the synthesis of supported IMCs via chemical vapor deposition method. | ||
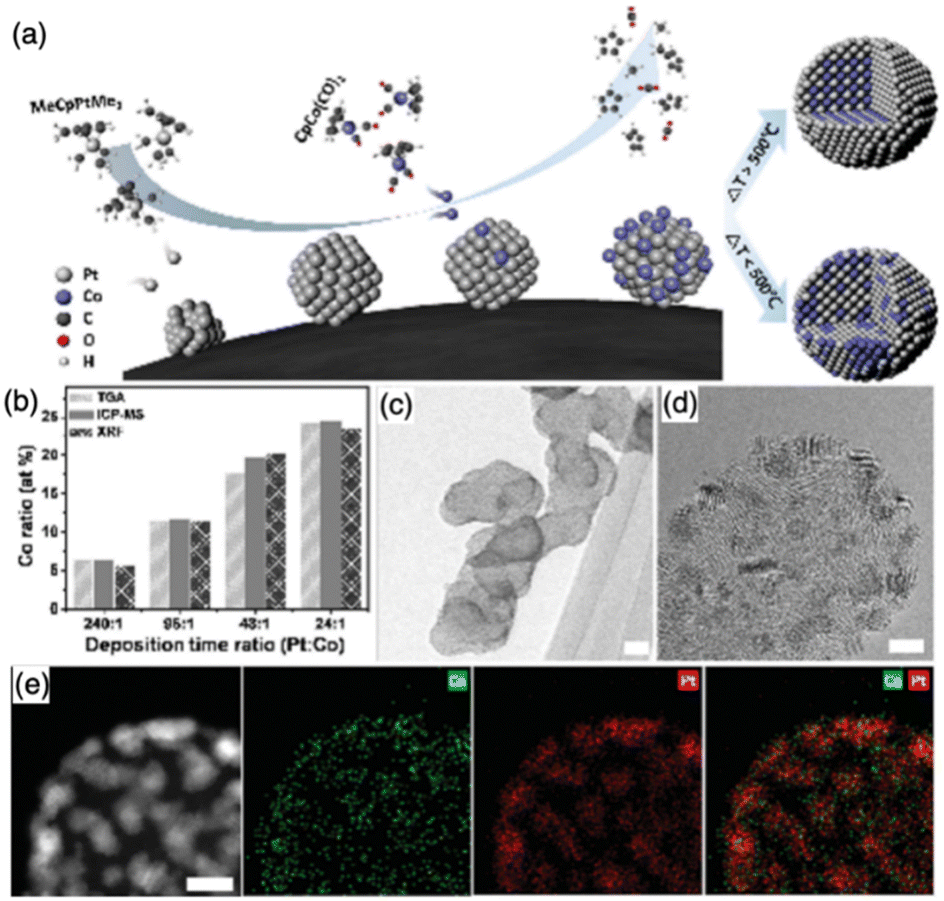 | ||
| Fig. 13 (a) Schematic for the synthesis of Pt–Co bimetallic nanoparticles on a carbon black support; (b) Pt versus Co composition control via deposition time control analyzed with TGA, ICP-MS, and XRF; (c) and (d) HR-TEM images of the as-deposited Pt and Co nanoparticles. Scale bars represent 20 and 3 nm, respectively; and (e) HAADF image and EDS maps of Co, Pt, and the composite of Pt and Co. Scale bar represents 3 nm.118 Copyright 2016, WILEY-VCH Verlag GmbH & Co. KGaA. | ||
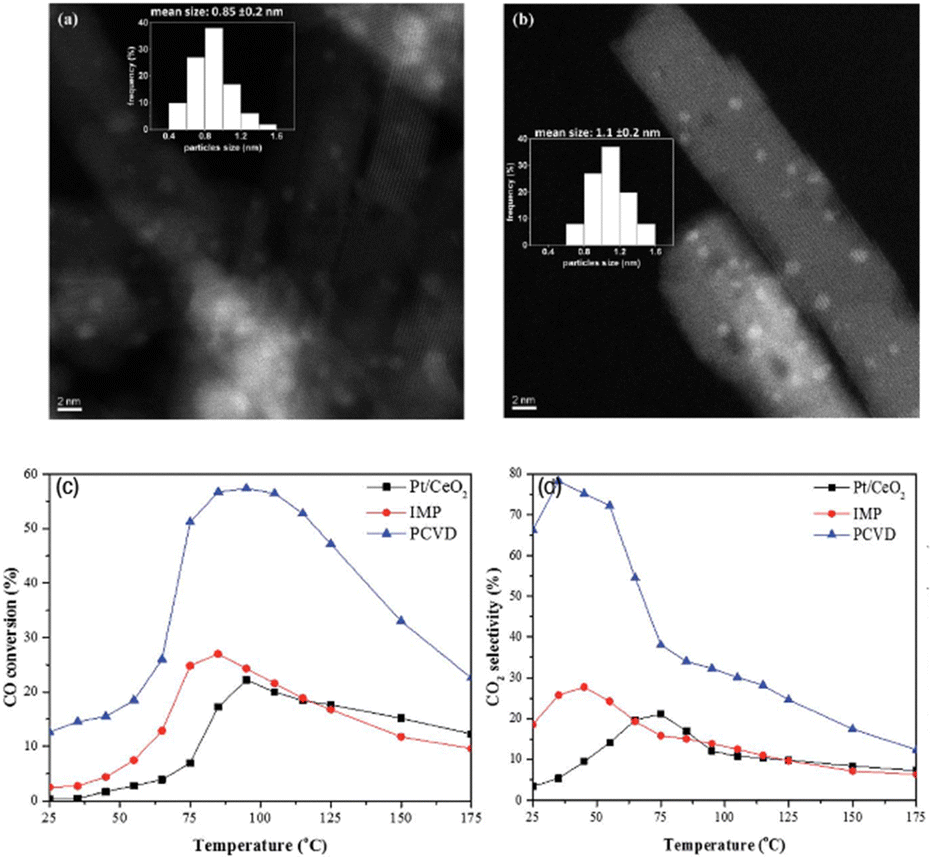 | ||
| Fig. 14 HAADF-STEM images and particle size distribution histogram (inset) of (a) Pt/CeO2 and (b) Pt3Co/CeO2, and comparison of the catalytic activity of Pt3Co/CeO2–PCVD, Pt3Co/CeO2–IMP, and Pt/CeO2 samples in CO PROX (c) CO conversion and (d) CO2 selectivity. The samples are annealed at 550 °C for 6 h under a hydrogen flow.117 Copyright 2017, Royal Society of Chemistry. | ||
2.5. Irradiation-based synthesis methods
New synthesis techniques, such as irradiation-based synthesis methods, have emerged for fabricating IMCs, which offer unique advantages over traditional techniques compared to conventional methods. Radiolysis has recently garnered significant attention for its simple operation and effective control over the composition of IMC nanoparticles. As shown in Fig. 15, during the radiosynthesis process, constituent element precursors in an aqueous solution are exposed to radiation to absorb energy. This radiation induces water radiolysis, leading to the production of hydrated electrons and radicals. These radicals then reduce the precursors, forming nanoparticles that are deposited on the support by electrostatic force. Afterward, the reduced metal atoms come together to form nuclei, which then grow into IMC nanoparticles.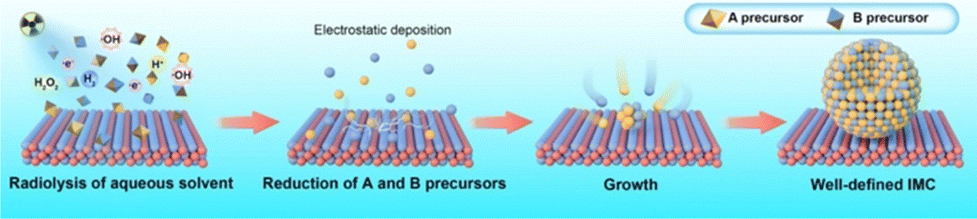 | ||
| Fig. 15 Schematic for the synthesis of supported IMCs via radiolysis method. | ||
:1 to 1:9. Interestingly, the surface can also be manipulated by changing the nominal precursor loading, which is difficult to achieve by using conventional methods such as impregnation method causing surface segregation. Similar to other methods, the bulk composition of IMCs obtained by radiation also is affected by the choice of support. Kugai et al. further studied the synthesis of Pt + Cu on three oxide supports (Fe2O3 and two CeO2 from different sources) by electron beam irradiation and confirmed this conclusion.128 Over Fe2O3, CuO was observed due to the stronger interaction between Cu and oxide support than CeO2 support, which caused a detrimental effect on the CO oxidation activity. To promote the formation of Pt–Cu/C IMC, they used ethylene glycol as the stabilizer to prevent Cu clusters from oxidation reaction.131 Other researchers also used stabilizers (such as PVP, PVA, propanol, and SDS) to solve the oxidation issues caused by the dissolved oxygen and containing O radicals generated during radiolysis which may hinder the production of IMCs with uniform sizes. The Al–Ni IMC was prepared through γ-irradiation of an aqueous precursor solution containing AlCl3 and NiCl2 in the presence of 2-propanol.132 Similarly, Au–Pt IMC was synthesized by exposing aqueous solutions of KAuCl4 and H2PtCl6 using a 13C6+ ion beam in the presence of PVA.133
 | ||
| Fig. 16 TEM images of (a) and (b) Pt9Cu/Fe2O3; (c) and (d) PtCu9/Fe2O3; and colloidal Al–Ni nanoparticles for precursor concentration of 5 × 10−5 mol mL−1 synthesis at a dose of (e) 60 kGy and (f) 100 kGy. (a)–(d) are from ref. 126. Copyright 2011, Elsevier Inc. and (e) and (f) are from ref. 132. Copyright 2012, Elsevier Inc. | ||
Some studies indicate the dose rate of irradiation plays a crucial role in determining the rates of metal ion reduction, nucleation, and formation during the synthesis process leading to the control of the composition of IMCs.134,135 High dose rates are associated with rapid reduction of metal precursors, leading to the formation of small nanoparticles with a narrow size distribution. Conversely, lower dose rates result in the formation of larger particles due to faster aggregations compared to the reduction process. For example, Al–Ni IMC nanoparticles synthesized by Abedini et al. showed a reduced particle size from 32.7 nm at 60 kGy to 4.4 nm at 100 kGy dose as shown in Fig. 16e and f.132
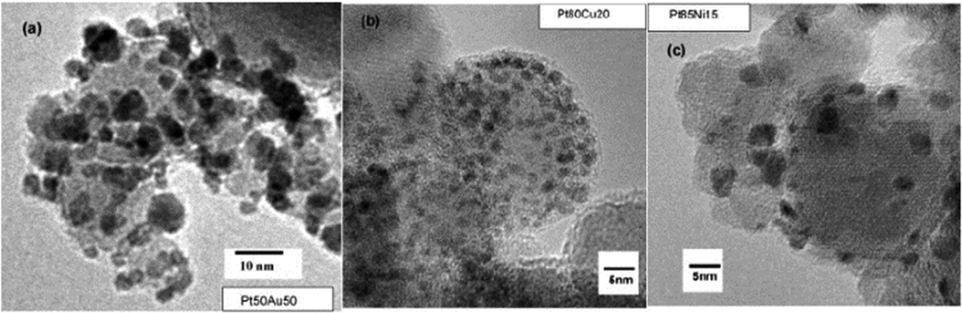 | ||
| Fig. 17 TEM micrographs of bimetallic (a) PtAu; (b) PtCu; and (c) PtNi grains supported on γ-Fe2O3 particles.136 Copyright 2010, Elsevier Inc. | ||
The growing number of IMCs discovered through radiolysis indicates the potential for exploring and designing new IMCs using this method. However, some elements may be sensitive to radiation, leading to the formation of undesired by-products or hindering the creation of the desired compounds. This can impact the stability and properties of the synthesized intermetallic compounds. Additionally, conducting radiolysis experiments requires expensive equipment and resources, making this method less cost-effective compared to other synthesis techniques. As a result, controlling the irradiation process on a large scale poses challenges, limiting industrial production. In conclusion, these limitations hinder the selection of materials suitable for radiolysis synthesis.
3. Manipulation of surface composition
The surface properties of supported IMCs play a critical role in determining their functional performance in applications such as catalysis, energy conversion and storage, and electronics. However, there has been limited research on manipulating and controlling the surface composition of these materials, especially supported IMCs. In addition, it is needed to improve how to characterize the surface composition of IMC. Some researchers are currently using techniques like XPS and EDS line scans, which only give us information from the top several nanometers of the material. It would be more appropriate to use techniques like HS-LEIS, which are becoming more common. However, there is still a requirement for more widespread use of these techniques, as they can greatly enhance our understanding of the relationship between the surface composition of IMCs and catalytic chemistry. One commonly used approach for the control of surface composition is high-temperature annealing, which can induce significant changes to the surface structure and composition. During annealing, the elevated temperatures provide the necessary energy for surface atoms to overcome diffusion barriers and rearrange into more thermodynamically stable configurations. This can drive the formation of new and ordered surfaces but also with the risk of the segregation or enrichment of specific elemental species.56–58 By carefully tuning the annealing temperature and time, researchers have demonstrated the ability to engineer the exposure of active sites, the ratio of different surface species, and the extent of surface ordering. These structural and compositional changes can significantly impact the catalytic activity, selectivity, and stability of the supported intermetallic compounds.For example, the Laursen group synthesized well-defined Ni + Ga/SiO2 and Co + Ga/SiO2 IMCs with pure phases using the impregnation method.8 By adjusting the reduction pretreatment conditions or incorporating additional annealing pretreatments, the surface compositions resembling the bulk material were achieved as characterized using high sensitivity low energy ion scattering (HS-LEIS) (see Fig. 18a and b). HS-LEIS of as-reduced and after-annealing samples of 3:1 and 1:1 NiGa on SiO2 shows the transformation from gallium-rich surfaces of particles to a composition that more closely resembles bulk after annealing at 700 °C for 12 hours. This fine-tuning of the surface chemistry towards carbon, hydrogen, and oxygen enabled us to achieve nearly ideal selectivity ratios of H2/CO and H2/CO2.7 Additionally, the particle size of the phase-pure IMCs could be adjusted through an annealing pretreatment, as illustrated in Fig. 18c. The as-reduced IMCs typically had a particle size of around 2–5 nm, and this could be increased to 4–8 nm after annealing for 12 hours at 700 °C under an atmosphere of either pure Ar or 2% hydrogen in Ar. The surprising stability of the IMCs against sintering is exemplified as well in these results. Another example is, the Kuhn group synthesized Ni3Zn/SiO2 using the impregnation method.139 They optimize the surface composition while retaining the bulk Ni3Zn structure by tuning the Zn precursor concentration with high-temperature annealing at 600 °C, which was confirmed by XRD and HS-LEIS characterizations (see Fig. 18d). The average particle size was around 4–6 nm (see Fig. 18e). They also reported different activities and H2/CO ratios were achieved by tailoring the surface composition Ni–Zn/SiO2. The study by Li et al. focused on the Pt3Fe/C and PtFe/C and also indicated the annealing effect on the surface composition at different temperatures (400–700 °C).106
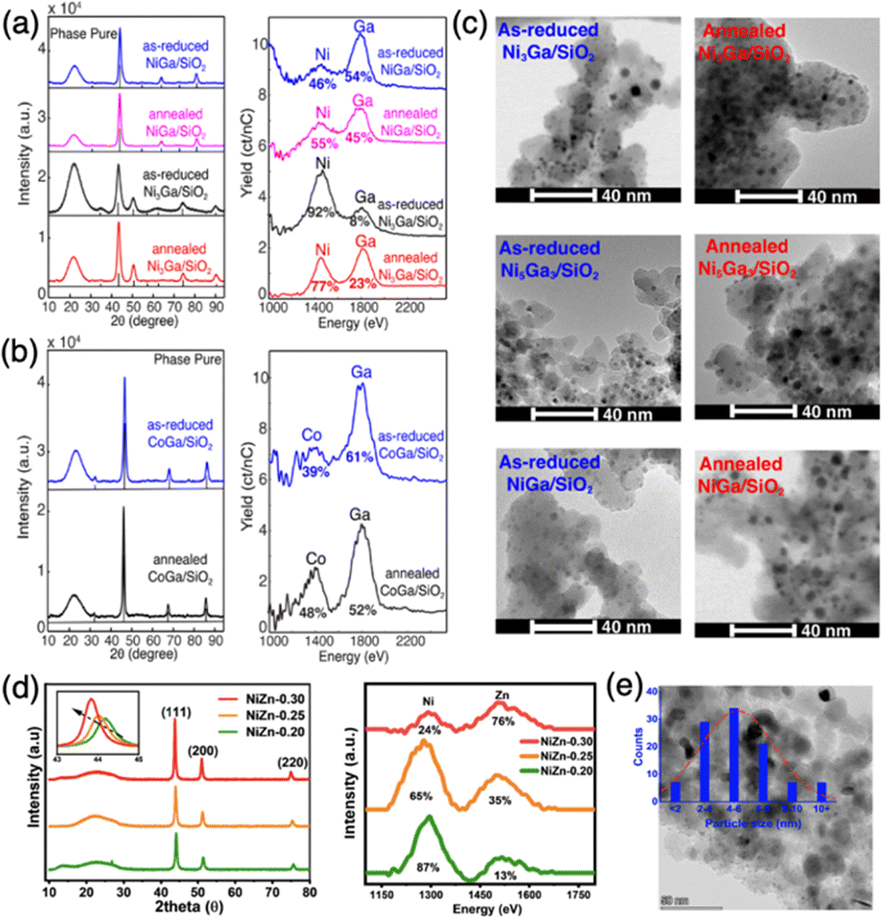 | ||
| Fig. 18 Demonstration of well-defined IMCs with phase pure bulk and controllable particle surface composition. XRD and HS-LEIS characterizations of (a) as-reduced and freeze-annealed NiGa/SiO2, and as-reduced and direct-annealed Ni3Ga/SiO2; (b) as-reduced and freeze-annealed CoGa/SiO2; and (d) Ni3Zn/SiO2; Electron micrographs of (c) as-reduced and annealed Ni + Ga/SiO2 and (e) Ni3Zn/SiO2. (a)–(c) are from ref. 8, Copyright 2022, Royal Society of Chemistry and (d) and (e) are from ref. 139, Copyright 2024, American Chemical Society. | ||
Besides thermal annealing, researchers have also explored chemical pretreatment methods such as selective leaching for surface manipulation. Selective leaching with acids and bases can remove certain surface components, thereby exposing the underlying intermetallic structures and their active sites. The choice of leaching agent, concentration, and leaching duration can be optimized to achieve the desired surface composition and morphology, leading to enhanced catalytic performance and improved stability under reaction conditions. For example, Kovnir et al. used ammonia solution with different pH to manipulate the surface of PdGa and Pd3Ga7 IMCs which can selectively etch the Ga. For both samples, XPS analysis indicates as the pH value increased the surface concentration of Ga decreased with the retained bulk phase, but the selectivity of ethylene reduced because of the generation of Pd ensembles.11 However, this technique is rarely employed in the cases of supported IMCs. In the future, researchers should put more effort into precise control over the surface properties of supported intermetallic compounds, enabling the optimization of their performance in a wide range of applications, from catalysis and energy storage to sensing and microelectronics.
In addition, it should be noted that IMCs will also encounter oxidation issues, leading to the formation of an oxidation layer on the surface under the atmosphere, similar to metal and alloy nanoparticles. However, this is often not a concern when testing their catalytic performance in situ, as the test can be done in a controlled environment that avoids contact with oxygen. The ordered structure and strong chemical bonding in IMCs result in improved stability and better oxidation resistance than pure metals.140,141 Moreover, intentionally forming an oxidation layer at room temperature with 1% O2 through calcination can protect the catalysts and allow for easy reduction before characterizations, if necessary (such as XPS and LEIS for surface composition measurement) and catalytic performance testing. Therefore, the heterogeneous catalysis community is generally not overly worried about the oxidation issue with IMCs, as in situ testing can bypass this problem. However, oxidation during a reaction containing oxygen should be a focus, as it may change the surface and bulk composition of the IMC catalyst, leading to deactivation.
4. High performance of well-defined IMCs in catalysis
Improving surface chemistry to selectively manipulate specific chemical components within a complex environment is a crucial goal in heterogeneous catalysis. Traditional catalysts often lack the necessary surface chemistry for many catalytic reactions, which has led to the exploration of IMCs as catalyst materials. Understanding the relationship between the composition and surface chemistry of IMC nanocatalysts and their catalytic performance is essential for designing effective catalysts. This focus on composition-sensitive catalytic performance in various reactions will provide valuable insights. In this section, we will explore how the bulk and surface of IMC nanocatalysts affect their catalytic performances such as selective hydrogenation, reforming, deoxygenation, and dehydrogenation. Furthermore, we will delve into the underlying reaction mechanism of IMCs and compare them with metals. This comparison can help us understand the fundamental reasons behind the impressive catalytic performance of IMCs, thereby facilitating the development of next-generation catalytic materials with improved activity, selectivity, and stability.4.1. Selective hydrogenation
Hydrogenation is crucial in many industrial processes, but controlling it can be difficult. Selective hydrogenation, as shown in Fig. 19a, involves reducing only a specific target functional group while leaving all other functional groups in the substrate unsaturated, and is particularly important. For example, in the synthesis of fine chemicals, various functional groups such as –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, –CO, –NO2, –CN, –COOR, and –CONH2 can be selectively reduced by clean and inexpensive hydrogen to form their corresponding alkenes, alcohols, and amines. Extensive research on PGM or pure TM has shown their inability to fully control hydrogenation, often leading to the production of saturated products such as saturated alcohols or alkanes due to their strong surface reactivity towards CC bonds and facile hydrogenation kinetics. For example, Pd/CNTs showed 75% selectivity towards saturated aldehyde production in the selective hydrogenation of cinnamaldehyde, with a TOF of 1.0 s−1.142 Similarly, Ru/CNTs exhibited 52% selectivity of saturated aldehyde in the cinnamaldehyde hydrogenation with a TOF of 0.17 s−1.143 Additionally, Ni/C showed 36% selectivity towards saturated alcohol formation in the cinnamaldehyde hydrogenation.144
C, –CO, –NO2, –CN, –COOR, and –CONH2 can be selectively reduced by clean and inexpensive hydrogen to form their corresponding alkenes, alcohols, and amines. Extensive research on PGM or pure TM has shown their inability to fully control hydrogenation, often leading to the production of saturated products such as saturated alcohols or alkanes due to their strong surface reactivity towards CC bonds and facile hydrogenation kinetics. For example, Pd/CNTs showed 75% selectivity towards saturated aldehyde production in the selective hydrogenation of cinnamaldehyde, with a TOF of 1.0 s−1.142 Similarly, Ru/CNTs exhibited 52% selectivity of saturated aldehyde in the cinnamaldehyde hydrogenation with a TOF of 0.17 s−1.143 Additionally, Ni/C showed 36% selectivity towards saturated alcohol formation in the cinnamaldehyde hydrogenation.144
 | ||
| Fig. 19 Schematic for different catalytic reactions: (a) selective hydrogenation; (b) reforming; (c) dehydrogenation; and (d) deoxygenation. | ||
In contrast, many studies have shown that IMCs with controlled surface chemistry, focused on C, H, and O through the selection of specific constituent elements, are capable of offering better control over hydrogenation processes. Specifically, IMCs exhibit enhanced hybridization, leading to attenuated surface reactivity towards C that facilitates limited hydrogenation of CC bonds. For example, a study from the Wei group showed the successful synthesis of pure phase Al2O3 supported Ni + Sn IMCs with different stoichiometries including Ni3Sn, Ni3Sn2, and Ni3Sn4.113 These catalysts were tested in the selective hydrogenation of furfural and showed catalytic activity follows Ni > Ni3Sn > Ni3Sn2 > Ni3Sn4 while the selectivity towards furfuryl alcohol follows Ni3Sn4 (99%) = Ni3Sn2 (99%) > Ni3Sn (75%) > Ni (2%) (see Fig. 20a–e). The FTIR characterizations of furfural adsorption over Ni and Ni + Sn IMCs in Fig. 20f suggested that Ni can activate both CC and CO bonds while only CO activations were observed over Ni + Sn IMCs leading to improved selectivity. The DFT calculations of furfural adsorption also confirmed the strong activation towards CC and CO over Ni causing overhydrogenation (see Fig. 20f).113 Similarly, Casella group prepared Pt + Sn supported on SiO2. The results showed that increasing the Sn concentration in the bulk of Pt + Sn IMC compound led to enhanced selectivity towards unsaturated alcohol. Notably, the highest TOF was recorded for PtSn0.2 (0.620 s−1) which is much higher than the performance of pure Pt catalyst (0.067 s−1).145 Similarly, studies by M. Consonni also showed pure phase PtZn/ZnO catalysts achieve 81% selectivity towards croton alcohol in selective hydrogenation of crotonaldehyde.146 Equally noteworthy, the PdIn/Al2O3 catalysts enhanced selectivity and catalytic activity in acetylene hydrogenation reactions.69 The selectivity towards ethylene was increased to as high as 80% using PdIn/Al2O3 with nearly 100% conversion and TOF of 0.8 s−1 while the selectivity and TOF over pure Pd/Al2O3 were 0% and 0.5 s−1, respectively. Additionally, a study by Chen et al. showed Ni10In/SiO2 and Ni6In/SiO2 catalysts displaying nearly 70% selectivity toward ethylene production in the selective hydrogenation of acetylene as well as the significantly improved stability than Ni/SiO2.40 The TG-DTA results suggested that the deactivation of Ni was caused by coking due to aggressive carbon affinity, which was overcome by incorporating Sn to reduce the surface reactivity towards C. Similarly, the introduction of Sn to NiSn/SiO2 even led to an impressive 96% selectivity towards ethylene production due to the completely new surface chemistry than Ni.123 In addition, Zhang et al. showed that the pure bulk phase Pt3Ti/TiO2 showed improved selectivity towards 1-phenylethanol (90%) compared with Pt/TiO2 (70%) in selective hydrogenation of acetophenone.147 Furthermore, studies from the Ding group illustrated that CuPd/SiO2 catalysts exhibited over 90% selectivity towards alkene production in selective hydrogenation of acetylene and butadiene at 100% conversion, compared with that Cu is inactive in hydrogenation reaction and Pt run full hydrogenation, illustrating new electronic structure formation.148
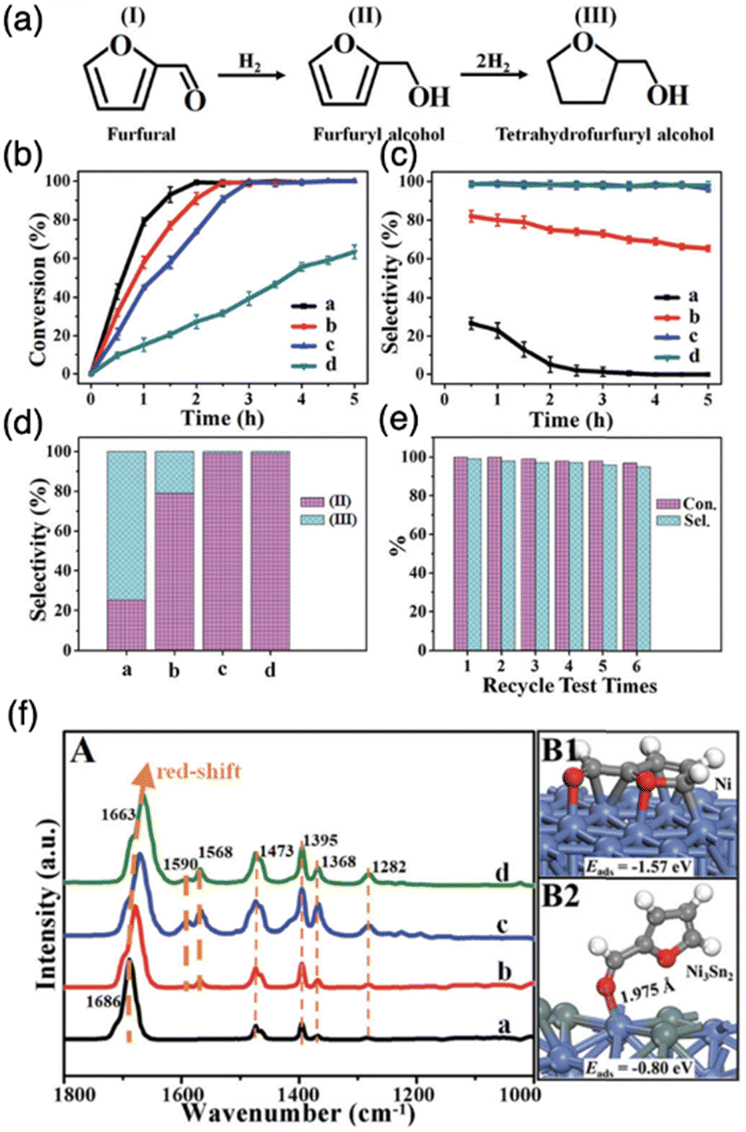 | ||
| Fig. 20 (a) Reaction pathways in the hydrogenation of furfural: (I) furfural, (II) furfuryl alcohol, and (III) tetrahydrofurfuryl alcohol; (b) catalytic conversion; (c) corresponding selectivity vs. reaction time for furfural hydrogenation (d) a comparison of selectivity toward each product at the same conversion (40%) over various catalysts: (a Ni, b Ni3Sn1, c Ni3Sn2, and d Ni3Sn4); (e) catalytic conversion and selectivity vs. recycling time over the Ni3Sn2 catalyst; and (f) in situ FT-IR spectra of (A) furfural adsorption over various samples: (a Ni, b Ni3Sn1, c Ni3Sn2, and d Ni3Sn4) recorded from 1800–1000 cm−1 after a furfural flow for 15 min at 100 °C and subsequent flushing with He for 15 min. The optimal adsorption structures of (B1) Ni and (B2) Ni3Sn2.113 Copyright 2019, Royal Society of Chemistry. | ||
In conclusion, the comprehensive research and diverse catalyst compositions discussed underscore the potential for improved control and selectivity in hydrogenation processes, paving the way for advancements in industrial applications and the synthesis of fine chemicals.
4.2. Reforming
The catalytic wet or dry reforming of hydrocarbons and oxygenated feedstocks is crucial for producing hydrogen and synthesis gas (H2/CO) for the chemical industry (see Fig. 19b). However, there is still a lack of comprehensive understanding of the reaction mechanisms involved, which hinders the development of advanced catalysts. The challenges in catalyst design stem from the reactivity of the catalyst surface during the initial activation of reactants and the subsequent surface reaction steps that affect product selectivity and the formation of coke or TM carbide. The reactivity of saturated hydrocarbons often requires metal catalysts with high carbon affinity to facilitate C–H bond activation. However, this can lead to the formation of coke or TM carbides if the carbon species are not efficiently coupled with the oxidizer supply or oxidation steps necessary for CO or CO2 production. The interaction with C and O on the catalyst surface is crucial for determining CO versus CO2 selectivity and linking efforts to enhance C–H activation and catalyst stability with overall product selectivity. For example, Pt/Al2O3 achieved 50% conversion of methane and CO2 at 550 °C, with a TOF of 1.7 s−1 in dry reforming of methane. However, the catalyst rapidly deactivated within 10 hours because of high surface reactivity towards C which is imbalanced with the oxygen supply and oxidation of carbon species.149 Similarly, Ni/Al2O3 achieved 60% methane conversion and 66% CO2 conversion at 700 °C showing rapid deactivation due to coke formation on the surface.150 In the study of methanol steam reforming, the Cu catalyst achieved 90% methanol conversion and 50% H2O conversion, with a CO/CO2 ratio greater than 9:1. However, Cu suffered from sintering issues due to the instability.151
To address these challenges and attain superior catalytic performance, IMCs with carefully managed the balance of surface reactivity towards C, H, and O have proven to be capable of controlling product distribution. Furthermore, by fine-tuning reaction kinetics, these IMCs display improved stability. For example, the study by the Laursen group on Ni + Ga/SiO2 IMCs (including NiGa, Ni3Ga, and Ni5Ga3) achieved nearly ideal selectivity towards either H2/CO2 and H2/CO, improved stability at comparable catalytic activity to established catalysts by manipulating TM-to-p-element ratio and surface composition.7 The selectivity and TOF rates of propane in propane steam reforming over Ni + Ga/SiO2 IMCs and pure metals were summarized in Table 3. It was observed that Ni + Ga IMCs exhibited higher selectivity for H2 with comparable TOF than pure metals, even when the reaction was carried out at lower temperatures. Then, the Laursen group carried out a comprehensive computational surface science study to understand the origin of this special catalytic performance. Calculation results indicate the activation of the hydrocarbon and oxidizer as well as hydrogenation kinetics and oxidation kinetics could be rationally controlled as a function of IMC bulk and surface composition. This control was the origin of the experimentally observed control over product selectivity (CH4vs. H2 and CO vs. CO2). In addition, the investigation of correlations between the d-band center and surface chemistry of Ni + Ga IMCs illustrated the strong electronic effects that lead to unique surface chemistry significantly different from pure metals. In a similar vein, a study by Haghofer et al. highlighted the significance of the bulk compositions of Pd + Ga in the catalytic performance of methanol steam reforming reactions (see Fig. 21).82 PdGa exhibited 75% CO2 selectivity and 14% conversion at 523 K, whereas Pd2Ga displayed notably different performance, demonstrating 90% selectivity to CO2 and 58% conversion. It is worth noting that both PdGa and Pd2Ga exhibited significantly improved catalytic performance compared to pure Pd. Similarly, the study from the Linic group revealed that NiSn/YSM catalysts with a pure phase exhibited improved stability compared to the pure Ni catalysts.152 In addition, PdIn/In2O3, synthesized by Neumann et al. for methanol steam reforming, not only demonstrated high CO2 selectivity of up to 98% but also excellent stability for as long as 100 hours.61 However, the activity of Pd + In/SiO2 IMCs decreased as the bulk concentration indium increased (2.7, 0.22, 0.12, and 0.04 (mmol MeOH)/(mmol Pd*h) for Pd, PdIn, Pd2In3, and Pd3In7, respectively). Similarly, the study by Fottinger showed that PdZn/ZnO achieves a high 90% selectivity towards CO2 production and 10% CO byproduct in methanol wet reforming with 36% conversion at 523 K.153 They guessed this kind of interesting catalytic performance of PdZn/ZnO is caused by many factors, such as particle size effect, electronic structure, and different contributions of reaction site to reaction pathways. In the studies of dry reforming of methane, Ni60Ga40/Al2O3 achieved 75% CH4 conversion and 85% CO2 conversion at 800C with close to 1:1 H2/CO ratio.154 In summary, the synthesis of various IMCs has yielded promising results in bolstering catalyst stability and enhancing product selectivity in diverse reforming reactions.
| Catalyst | Temp. (°C) | Selectivity (H2)(%) | TOF (s−1) (propane) | Ref. |
|---|---|---|---|---|
| As-reduced Ni3Ga/SiO2 | 400 | 70 | 1.08 | 7 |
| Annealed Ni3Ga/SiO2 | 400 | 72 | 0.43 | 7 |
| As-reduced Ni5Ga3/SiO2 | 400 | 65 | 0.25 | 7 |
| Annealed Ni5Ga3/SiO2 | 400 | 70 | 0.09 | 7 |
| As-reduced Ni5Ga3/SiO2 | 600 | 75 | 9.34 | 7 |
| Annealed Ni5Ga3/SiO2 | 600 | 70 | 3.37 | 7 |
| As-reduced NiGa/SiO2 | 600 | 74 | 11.58 | 7 |
| Annealed NiGa/SiO2 | 600 | 72 | 4.64 | 7 |
| Ni/Al2O3 | 550 | 56 (37% yield) | 0.65 | 155 |
| Rh/Al2O3 | 475 | 61 | 1.27 | 156 |
| Rh/CeO2 | 700 | 78 (97.5% yield) | 1.7 | 157 |
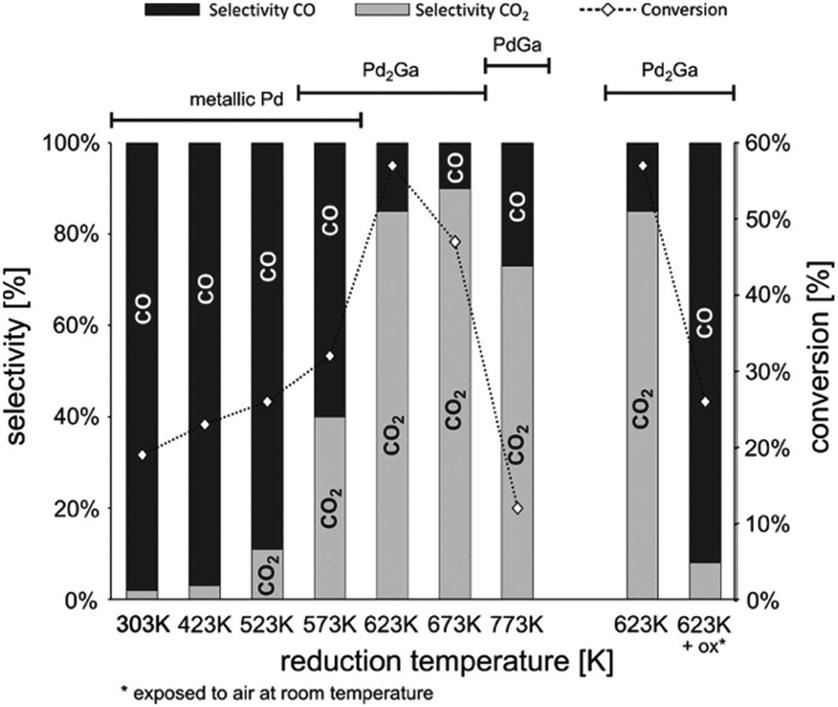 | ||
| Fig. 21 Initial selectivity (bars) and total reaction rate (diamonds) during methanol conversion over Pd/Ga2O3 as a function of the reduction temperature (left). The effect of air exposure on selectivity and total rate is shown on the right. Treaction = 523 K, mcatalyst = 20 mg, pCH3OH = pH2O = 30 mbar, total flow = 20 mL min−1.82 Copyright 2012, Elsevier Inc. | ||
4.3. Dehydrogenation
Light olefins, such as propylene and ethylene, are extremely important compounds in the chemical industry. They are used as raw materials for producing a wide variety of chemicals and crucial chemical intermediates. The demand for these fundamental building blocks has been steadily increasing in recent years, with all major markets experiencing positive growth in both demand and production. However, effectively adsorbing and activating saturated hydrocarbons and then desorbing olefins on the surfaces of heterogeneous catalysts remains a challenging task. Traditional heterogeneous catalysts, such as Pt and metal oxides, used in dehydrogenation reactions face significant challenges. The strong interaction between metal and alkenes can lead to over-dehydrogenation, and coking can also occur. Because of the thermal instability at high reaction temperatures, sintering of catalysts is often encountered. Additionally, these catalysts are prone to poisoning by contaminants and are expensive, especially noble metals like Pt, making them less suitable for long-term industrial applications. For instance, studies on propane dehydrogenation using Pt/Al2O3 show that as particle size increases, selectivity towards propylene decreases while conversion increases, with the highest TOF of 4.4 s−1 observed at 0.05% Pt loading. However, coke formation and sintering cause a gradual decline in catalytic activity over time.158 Similarly, CrOx/Al2O3 initially achieved 45% selectivity towards propylene, but deactivation began within 2 hours, leading to a rapid drop to 0% conversion within 10 hours.159In recent years, IMCs have garnered increased attention due to their capacity for controlled surface reactivity with C and H. This controlled surface reactivity helps to regulate dehydrogenation kinetics and inhibits coke formation (see Fig. 19c). The Laursen group reported that the Ni3Ga/Al2O3 catalyst has shown excellent performance in catalyzing the dehydrogenation of propane and ethane. It exhibited over 90% selectivity in producing propene and ethylene, respectively. When compared with established commercialized catalysts, the Ni3Ga/Al2O3 catalyst showed comparable TOF for propane and ethane.5,6 Additionally, the comparison presented in Fig. 22a illustrated the TOF propane and selectivity of propylene over most Pd-based IMC catalysts were higher than pure Pt.158 Furukawa et al. also investigated the catalytic chemistry of a series of Pt-based IMCs supported on SiO2 in the dehydrogenation of cyclohexane and n-butane (see Fig. 22b and c).119 The addition of a secondary element to Pt can disrupt the ensemble of Pt which drives hydrogenolysis and C–C breaking reaction pathway, leading to undesirable side reactions and catalyst deactivation. This modification can also alter the surface chemistry and electronic structure of the catalyst to aid in the desorption of the desired alkene product, helping to slow down the overall deactivation process. Therefore, Pt3Sn/SiO2 and PtGe/SiO2 showed higher selectivity of alkenes (benzene and butene) than pure Pt and other IMCs (Pt3Zn, Pt3Co, PtCo, Pt3Tl2, and PtSn), which is potential because of the reduced surface reactivity towards C. They also investigated the control of catalytic performance over Pd-based IMCs system by incorporating different secondary elements into Pd. It was found that Pd3Bi exhibited the highest selectivity for N-benzylidenebenzylamine in the dehydrogenation of dibenzylamine due to surface reactivity towards C, which is attributed to the ensemble effect of large Bi element.160 The Ryoo group found that PtZn/zeolite and PtGa/zeolite catalysts achieved over 95% selectivity and 25 hours of stability in propane dehydrogenation.161 Similarly, research by the Linic group indicated that PtSn/SiO2 catalysts achieved about 67% conversion with selectivity to propylene of more than 99%.162 Additionally, Wu et al. demonstrated that PdIn/zeolites exhibited increased selectivity towards ethylene in the dehydrogenation of ethane, with selectivity near 100% and TOF of 0.26 s−1.163 In comparison, a monometallic Pd catalyst showed a much lower dehydrogenation selectivity of 53% and a TOF of 0.03 s−1. Takanabe group utilized NiZn/Al2O3 in the selective dehydrogenation of methylcyclohexane to toluene, achieving nearly 100% selectivity for toluene production and even higher TOF than Ni/Al2O3.164 DFT analysis shows that the addition of Zn primarily occupies the highly reactive sites of Ni, like corner and edge sites, to decrease the surface reactivity towards C, which enhances the desorption of toluene and prevents C–C bond breaking. Lastly, the research by Yashima et al. showed that Ni–Sn/SiO2 IMCs prepared by the CVD method with different stoichiometry exhibited high benzene selectivity (>99%) and varying conversion rates for cyclohexane dehydrogenation.25
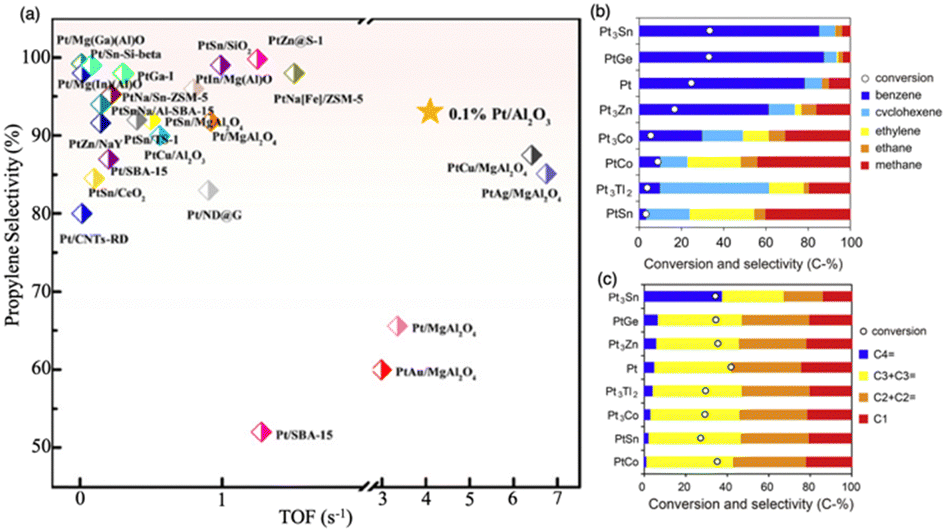 | ||
| Fig. 22 (a) Comparison of TOF of propane and selectivity over Pt-based catalyst,158 Copyright 2020, American Chemical Society. Conversion and product distribution of (b) cyclohexane dehydrogenation to benzene and (c) n-butane dehydrogenation to butenes over Pt/SiO2 and Pt–M/SiO2 catalysts.119 Copyright 2013, Elsevier Inc. | ||
4.4. Deoxygenation
Catalytic deoxygenation is a key process in the production of biofuels, which involves converting biomass-derived oxygenates into hydrocarbons suitable for use as transportation fuels. This process is crucial for upgrading bio-oils, which are typically rich in oxygenated compounds, into hydrocarbon fuels with properties similar to conventional petroleum fuels (see Fig. 19d). Upgrading heavily oxygenated feedstocks has become a significant focus, requiring the design of catalytic materials that exhibit a uniquely balanced surface chemistry towards carbon, oxygen, and hydrogen. The ideal catalysts should selectively or completely remove –OH groups while limiting C–C bond cleavage and preventing over-hydrogenation of valuable olefin products. Research has shown that monometallic PGM, PGM-derived PGM + TM alloys, and zeolites are unsuitable for these reactions because of their limited tunability, insufficient stability as well as high cost. In the use of reduced metal catalysts, large selections have been investigated in glycerol, glucose, and other polyol deoxygenation (Pt, Pd, Ru, Ir, Rh, Cu, and Ni).165–167 These studies highlight the inappropriate surface chemistry of pure reduced metals in this catalysis, yet isolated two metals with higher surface oxophilicity, Cu and Ru, that were most capable in driving oxygen removal. However, control of hydrogenation activity was poor and selectivity towards fully saturated HC products was still dominant.166 Similarly, in the deoxygenation of furfural, Cu/SiO2 at 300 °C resulted in 14% selectivity for 2-methylfuran production.168 Aqueous phase reduction of glucose over a selection of metals also showed that Ru exhibited oxophilicity appropriate to remove all OH groups, but also favored complete hydrogenation to saturated alkane products.167 Many other metals drove C–C cleavage and sorbitol production.167 Similar results were found for linear C5 and C6 polyols.165 On the other hand, zeolites and some pure metals with high surface oxophilicity require greatly elevated temperatures and pressure to drive oxygen removal resulting in catastrophic breakdown of the reactants and favored saturated HC production.169–172 These conditions can also contribute to oxidation and coke formation, ultimately deactivating the catalyst. For example, the deoxygenation of anisole for aromatic production over Ni on different oxide supports was employed at 3 bar and 300 °C but only achieved as high as 70% selectivity towards saturated alkanes.173 Similarly, a series of supported Pt catalysts also showed the production of unselective propylbenzene in the deoxygenation of 4-propylphenol at 573 K and 20 bars.174 It is clear that no pure reduced metals exhibit the ideally balanced surface reactivity towards C, O, and H to drive these reactions nor do they present the systematic tunability needed to tune product distributions.On the other hand, IMCs exhibit more favorable surface chemistry and enhanced compositional stability and seem promising in meeting this requirement. For example, the Chen group reported the enhanced deoxygenation activity of Ni + Ga/SiO2 and Ni + Zn/SiO2 compared to Ni/SiO2.175–177Fig. 23a illustrates that, at a similar anisole conversion (∼31%), the selectivity to benzene was increased from 75.2% over Ni/SiO2 to 83.0% over Ni6Ga/SiO2 and 92.6% on Ni3Ga/SiO2, respectively. The TOF values of Ni + Ga/SiO2 (0.95 s−1 for Ni6Ga/SiO2 and 15.3 s−1 for Ni3Ga/SiO2) were also much higher than Ni/SiO2 (0.3 s−1). As depicted in Fig. 23b, the higher surface reactivity towards O over Ni6Ga/SiO2 and Ni3Ga/SiO2, in comparison to Ni, results in their preference for complete cleavage of the C–O bond to produce benzene, while Ni generates partially deoxygenated products (phenol). Similarly, the TOF values of Ni + Zn/SiO2 were also higher than that of Ni/SiO2 (see Fig. 23c). This study revealed a volcano trend in Fig. 23d when examining the deoxygenation of methyl laurate over Ni + Zn/SiO2. At 330 °C, with the decrease of the Ni/Zn ratio, the selectivity to C11 and C12 hydrocarbons increased to the maximum (93.1%) on Ni8Zn1. However, it quickly decreased as the Ni/Zn ratio further decreased. This is because the high oxophilicity and reduced carbon affinity of Zn can facilitate the hydrodeoxygenation pathway over Ni + Zn. Nevertheless, an excessively high concentration of Zn leads to weak catalytic activity. On the other hand, the high surface reactivity towards C over Ni causes the C–C breaking leading to C6–C10 products. This phenomenon was also observed in heptanoic acid deoxygenation over Pd and PdSn. Pd preferred C–C cleavage, yet PdSn promoted singular OH removal to form heptanal.38 In addition to influencing the product distribution, the addition of Sn to Pd also enhanced the stability. This is evident by the change of conversion and TOF at the time on stream. The TOF of Pd/SiO2 was reduced by 71% from 0.0045 s−1 at 1.5 h to 0.0013 TOF s−1 at 10 hours. Notably, the overall activity over PdSn/SiO2 was slightly decreased after 10 hours. These results clearly illustrate the role of bulk composition in the surface chemistry of IMCs as a result of geometric and electronic effects. The research conducted by Khalit et al. showcased the utilization of NiZn/C in the deoxygenation of waste cooking oil, resulting in over 90% hydrocarbon production and the highest selectivity in C15 production.178 Furthermore, studies by He et al. demonstrated that the CoZn/ZnO catalyst was applied in the deoxygenation of 5-hydroxymethylfurfural, resulting in near 100% conversion with 90% selectivity towards 2,5-dimethylfuran (DMF) production.179 Furthermore, Ni3Sn2/Al2O3 with pure phase synthesized by Shu et al. exhibited yields of aiming alkanes reaching about 99% in the deoxygenation of methyl octanoate and methyl palmitate, respectively.112
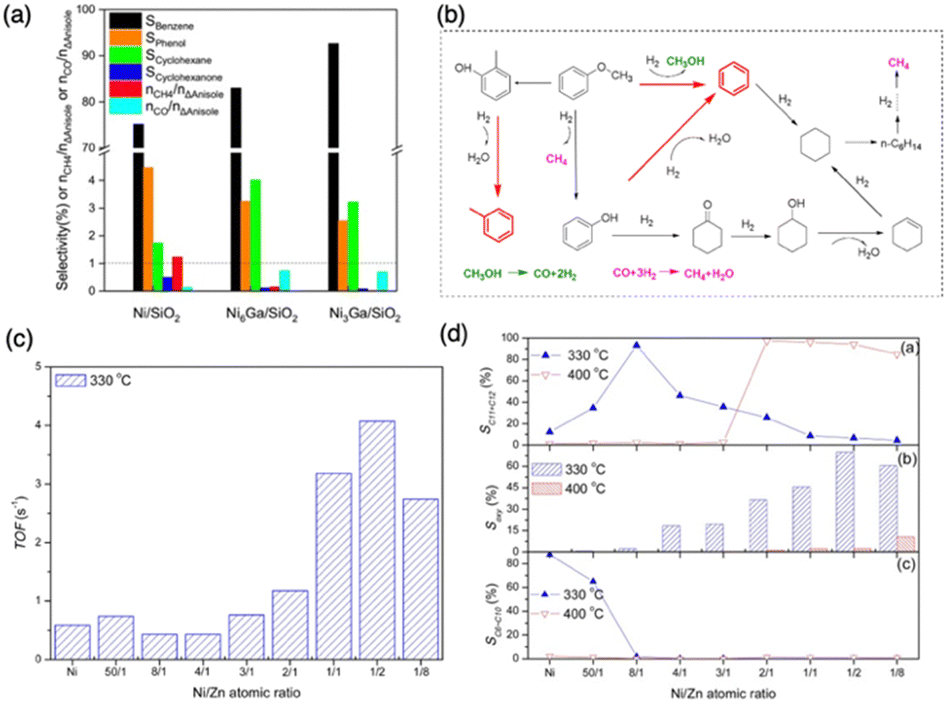 | ||
| Fig. 23 (a) Product distribution on Ni/SiO2, Ni6Ga/SiO2, and Ni3Ga/SiO2 at similar anisole conversion (31 ± 4%); (b) scheme of the reaction mechanism of deoxygenation of anisole.177 Copyright 2019, Elsevier. (c) TOF and (d) product selectivity of deoxygenation of methyl laurate on different NixZny catalysts.176 Copyright 2018, Elsevier. | ||
5. Concluding remarks and outlook
The development of supported IMC catalysts has emerged as a promising strategy to address the growing demand for high-performance materials in a wide range of energy and chemical conversion processes. The ability to precisely control the bulk and surface compositions of these compounds with ordered nanostructures has enabled significant improvements in catalytic activity, selectivity, and stability compared to conventional monometallic and alloy catalysts. This review examines the remarkable progress made in developing synthetic approaches capable of tailoring the structural and compositional features of supported IMC catalysts. Techniques such as impregnation, precipitation, CVD, and radiolysis synthesis have facilitated the fabrication of supported IMC catalysts with tunable compositions, leading to specific surface chemistries that can be leveraged in different reactions. Furthermore, the incorporation of these IMCs into support materials like carbon, metal oxides, and zeolites has further enhanced their catalytic performance by improving surface area, dispersion, and stability properties.Despite these advancements, several challenges remain to be addressed before the widespread adoption of supported IMC catalysts in industrial applications. Firstly, it is necessary to develop a universal method for the supported IMC catalysts that can be applied to all combinations of TM + TM and TM + p-elements. This would enable the systematic exploration and optimization of a wide range of IMC compositions, leading to the discovery of new high-performance catalysts. Secondly, controlling the surface composition of supported IMCs is critical for the surface reaction. However, few studies have focused on this aspect due to the difficulty in manipulating the surface composition through pretreatment and the challenges in accurately characterizing the true surface structure. Advanced in situ characterization techniques, combined with computational modeling, can provide valuable insights into the dynamic changes in catalyst structure and composition during operation, as well as the underlying reaction mechanism. Additionally, supported IMC catalysts often require complex and costly synthesis methods, which can make it challenging to scale up and make economically viable compared to more traditional catalyst formulations. Developing scalable synthesis methods for supported IMC catalysts is a major challenge, as it requires precise control of both the bulk and surface compositions when scaling up the synthesis. Furthermore, achieving high dispersion of small IMC nanoparticles on the support material at larger scales is difficult, yet critical for maximizing the active surface area for high catalytic activity. Factors to consider in addressing these issues include the selection of appropriate precursor materials and supports, the synthesis techniques and pretreatment conditions, and the evaluation of long-term stability and renewability. Careful consideration of these parameters is necessary to develop IMC-based catalysts that can effectively balance performance, scalability, and cost for practical industrial applications. Equally important is the need to improve the long-term stability of supported IMC catalysts under harsh reaction conditions, such as high temperatures, pressures, and corrosive environments. Overcoming these challenges will be essential for realizing the full translational potential of supported IMC catalysts in large-scale industrial applications.
Additionally, the inability to discover and develop yet compositionally more complex heterogeneous catalysts arises from a lack of understanding of connections between electronic structure and surface and catalytic chemistry, therefore, which will be a crucial aspect of future research. This insight can guide the rational design of next-generation supported IMC catalysts with optimal performance from the electronic level upwards. Continued research and development in this field are expected to accelerate the deployment of supported IMC catalysts with transformative improvements in the efficiency and sustainability of various energy and chemical technologies. For instance, the use of supported IMC catalysts in chemical synthesis processes could enable more efficient and selective transformations, reducing the energy and resource inputs required. In photocatalytic applications, the tailored electronic and optical properties of IMCs could enhance light harvesting and charge separation, leading to improved solar-to-fuel conversion efficiencies. Similarly, the incorporation of IMCs into fuel cell and electrolysis technologies could boost the performance and durability of these energy conversion and storage devices, ultimately contributing to a more sustainable energy landscape. Beyond surface and catalytic science, understanding how to control and optimize the properties of well-defined solids will favorably impact the materials, sensors, electronics, and optics community.
Author contributions
Conceptualization, T. Z.; writing – original draft preparation, Y. S., and S. G.; writing – review & editing, P. X., F. S., Z. C., S. Y., and X. Z.; supervision, T. Z.; project administration, T. Z.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by NSFC (Grant No. 22102023 and 62275047), the National Key R&D Program of China (Grant No. 2021YFB2800700), Jiangsu Province Excellent Post-Doctoral Program (Grant No. 2024ZB424), the Postdoctoral Fellowship Program of CPSF (Grant No. GZC20240251), and Natural Science Foundation of Jiangsu Province (Grant BK20220816).Notes and references
- J. Meng, H. Liu, J. Xu, Y. Lou, H. Sun, B. Jiang, Y. Liu, H. Qin, S. Dou and H. Yu, SusMat, 2024, e221 CrossRef CAS
.
- C. Breinlich, J. Haubrich, C. Becker, A. Valcarcel, F. Delbecq and K. Wandelt, J. Catal., 2007, 251, 123–130 CrossRef CAS
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS
- L. Zhang, L. Xue, B. Lin, Q. Zhao, S. Wan, Y. Wang, H. Jia and H. Xiong, ChemSusChem, 2022, 15, e20210294 Search PubMed
- Y. He, Y. Song, D. A. Cullen and S. Laursen, J. Am. Chem. Soc., 2018, 140, 14010–14014 CrossRef CAS
- Y. He, Y. Song and S. Laursen, ACS Catal., 2019, 9, 10464–10468 CrossRef CAS
- Y. Song, Y. He and S. Laursen, ACS Catal., 2020, 10, 8968–8980 CrossRef CAS
- Y. Song, Y. He and S. Laursen, Catal. Sci. Technol., 2022, 12, 3568–3581 RSC
- Y. Song and S. Laursen, J. Catal., 2019, 372, 151–162 CrossRef CAS
- C. S. Spanjers, J. T. Held, M. J. Jones, D. D. Stanley, R. S. Sim, M. J. Janik and R. M. Rioux, J. Catal., 2014, 316, 164–173 CrossRef CAS
- K. Kovnir, J. Osswald, M. Armbrüster, D. Teschner, G. Weinberg, U. Wild, A. Knop-Gericke, T. Ressler, Y. Grin and R. Schlögl, J. Catal., 2009, 264, 93–103 CrossRef CAS
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, J. Am. Chem. Soc., 2010, 132, 14745–14747 CrossRef
- A. Onda, T. Komatsu and T. Yashima, Phys. Chem. Chem. Phys., 2000, 2, 2999–3005 RSC
- S. Saadi, B. Hinnemann, S. Helveg, C. Appel, F. Abild-Pedersen and J. Norskov, Surf. Sci., 2009, 603, 762–770 CrossRef CAS
- R. Rodiansono, M. D. Astuti, S. Khairi and S. Shimadzu, Bull. Chem. React. Eng. Catal., 2016, 11, 1–9 CrossRef CAS
- L. Piccolo and L. Kibis, J. Catal., 2015, 332, 112–118 CrossRef CAS
- M. Krajci and J. Hafner, J. Catal., 2011, 278, 200–207 CrossRef CAS
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS
- H. Zhao and B. E. Koel, Surf. Sci., 2004, 572, 261–268 CrossRef CAS
- I. Sharafutdinov, C. F. Elkjær, H. W. Pereira De Carvalho, D. Gardini, G. L. Chiarello, C. D. Damsgaard, J. B. Wagner, J.-D. Grunwaldt, S. Dahl and I. Chorkendorff, J. Catal., 2014, 320, 77–88 CrossRef CAS
- J. Zhou, Y. Yang, C. Li, S. Zhang, Y. Chen, S. Shi and M. Wei, J. Mater. Chem. A, 2016, 4, 12825–12832 RSC
- J. Shabaker, D. Simonetti, R. Cortright and J. Dumesic, J. Catal., 2005, 231, 67–76 CrossRef CAS
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180–191 CrossRef CAS
- C. Rudolf, B. Dragoi, A. Ungureanu, A. Chirieac, S. Royer, A. Nastro and E. Dumitriu, Catal. Sci. Technol., 2014, 4, 179–189 RSC
- A. Onda, T. Komatsu and T. Yashima, J. Catal., 2004, 221, 378–385 CrossRef CAS
- Y. Ma, Y. Xu, M. Demura, D. H. Chun, G. Xie and T. Hirano, Catal. Lett., 2006, 112, 31–36 CrossRef CAS
- C. Li, Y. Chen, S. Zhang, J. Zhou, F. Wang, S. He, M. Wei, D. Evans and X. Duan, ChemCatChem, 2014, 6, 824–831 CrossRef CAS
- C. Li, Y. Chen, S. Zhang, S. Xu, J. Zhou, F. Wang, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2013, 25, 3888–3896 CrossRef CAS
- T. Komatsu, T. Kishi and T. Gorai, J. Catal., 2008, 259, 174–182 CrossRef CAS
- X. Chen, Y. Ma, L. Wang, Z. Yang, S. Jin, L. Zhang and C. Liang, ChemCatChem, 2015, 7, 978–983 CrossRef CAS
- G. Hamm, T. Schmidt, J. Breitbach, D. Franke, C. Becker and K. Wandelt, Surf. Sci., 2004, 562, 170–182 CrossRef CAS
- D. I. Jerdev, A. Olivas and B. E. Koel, J. Catal., 2002, 205, 278–288 CrossRef CAS
- J. Chen and B. Fruhberger, Surf. Sci., 1996, 367, L102–L110 CrossRef CAS
-
B. A. T. Mehrabadi, S. Eskandari, U. Khan, R. D. White and J. R. Regalbuto, Advances in Catalysis, Elsevier, 2017, vol. 61, pp. 1–35 Search PubMed
- M. J. Ndolomingo, N. Bingwa and R. Meijboom, J. Mater. Sci., 2020, 55, 6195–6241 CrossRef CAS
- P. Munnik, P. E. De Jongh and K. P. De Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS
- S. Penner and M. Armbrüster, ChemCatChem, 2015, 7, 374–392 CrossRef CAS
- N. Kaylor, J. Xie, Y.-S. Kim, H. N. Pham, A. K. Datye, Y.-K. Lee and R. J. Davis, J. Catal., 2016, 344, 202–212 CrossRef CAS
- G. Saravanan, H. Abe, Y. Xu, N. Sekido, H. Hirata, S. Matsumoto, H. Yoshikawa and Y. Yamabe-Mitarai, Langmuir, 2010, 26, 11446–11451 CrossRef CAS
- Y. Chen and J. Chen, Appl. Surf. Sci., 2016, 387, 16–27 CrossRef CAS
- T.-W. Song, C. Xu, Z.-T. Sheng, H.-K. Yan, L. Tong, J. Liu, W.-J. Zeng, L.-J. Zuo, P. Yin, M. Zuo, S.-Q. Chu, P. Chen and H.-W. Liang, Nat. Commun., 2022, 13, 6521 CrossRef CAS
- J. Llorca, J. Catal., 1995, 156, 139–146 CrossRef CAS
- J. Llorca, P. R. De La Piscina, J.-L. G. Fierro, J. Sales and N. Homs, J. Mol. Catal. A: Chem., 1997, 118, 101–111 CrossRef CAS
- F. Chen, W. Lv, G. Zhou, Z. Liu, M. Chu and X. Lv, Int. J. Hydrogen Energy, 2024, 55, 502–511 CrossRef CAS
- M.-H. Bai, H. Long, S.-B. Ren, D. Liu and C.-F. Zhao, ISIJ Int., 2018, 58, 1034–1041 CrossRef CAS
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS
- C.-J. Pan, M.-C. Tsai, W.-N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S.-Y. Cheng and B.-J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154–186 CrossRef CAS
- S. Furukawa, K. Ozawa and T. Komatsu, RSC Adv., 2013, 3, 23269 RSC
- T. Komatsu, Appl. Catal., A, 2003, 251, 315–326 CrossRef CAS
- J. Huang, L. Ji, X. Li, X. Wu, N. Qian, J. Li, Y. Yan, D. Yang and H. Zhang, CrystEngComm, 2022, 24, 3230–3238 RSC
- H. Yin, Z. Ma, M. Chi and S. Dai, Catal. Today, 2011, 160, 87–95 CrossRef CAS
- L. Wan, X. Zhang, J. Cheng, R. Chen, L. Wu, J. Shi and J. Luo, ACS Catal., 2022, 12, 2741–2748 CrossRef CAS
- A. P. LaGrow, K. R. Knudsen, N. M. AlYami, D. H. Anjum and O. M. Bakr, Chem. Mater., 2015, 27, 4134–4141 CrossRef CAS
- D. Saha, E. D. Bøjesen, A. H. Mamakhel, M. Bremholm and B. B. Iversen, ChemNanoMat, 2017, 3, 472–478 CrossRef CAS
- Y. Zhang, Y. Liu, G. Yang, S. Sun and N. Tsubaki, Appl. Catal., A, 2007, 321, 79–85 CrossRef CAS
- B. M. Vogelaar, P. Steiner, T. F. Van Der Zijden, A. D. Van Langeveld, S. Eijsbouts and J. A. Moulijn, Appl. Catal., A, 2007, 318, 28–36 CrossRef CAS
- N. Zanganeh, V. K. Guda, H. Toghiani and J. M. Keith, ACS Appl. Mater. Interfaces, 2018, 10, 4776–4785 CrossRef CAS
- C. Wang, G. Wang, D. Van Der Vliet, K.-C. Chang, N. M. Markovic and V. R. Stamenkovic, Phys. Chem. Chem. Phys., 2010, 12, 6933 RSC
- K. Mori, N. Hashimoto, N. Kamiuchi, H. Yoshida, H. Kobayashi and H. Yamashita, Nat. Commun., 2021, 12, 3884 CrossRef CAS PubMed
- M. Vaarkamp, F. S. Modica, J. T. Miller and D. C. Koningsberger, J. Catal., 1993, 144, 611–626 CrossRef CAS
- M. Neumann, D. Teschner, A. Knop-Gericke, W. Reschetilowski and M. Armbrüster, J. Catal., 2016, 340, 49–59 CrossRef CAS
- Y. He, J. Fan, J. Feng, C. Luo, P. Yang and D. Li, J. Catal., 2015, 331, 118–127 CrossRef CAS
- O. S. Alexeev and B. C. Gates, Ind. Eng. Chem. Res., 2003, 42, 1571–1587 CrossRef CAS
- X. Yang, D. Chen, S. Liao, H. Song, Y. Li, Z. Fu and Y. Su, J. Catal., 2012, 291, 36–43 CrossRef CAS
- R. V. Maligal-Ganesh, Y. Pei, C. Xiao, M. Chen, T. W. Goh, W. Sun, J. Wu and W. Huang, ChemCatChem, 2020, 12, 3022–3029 CrossRef CAS
- M. Cui, C. Yang, S. Hwang, M. Yang, S. Overa, Q. Dong, Y. Yao, A. H. Brozena, D. A. Cullen, M. Chi, T. F. Blum, D. Morris, Z. Finfrock, X. Wang, P. Zhang, V. G. Goncharov, X. Guo, J. Luo, Y. Mo, F. Jiao and L. Hu, Sci. Adv., 2022, 8, eabm4322 CrossRef CAS
- S. Furukawa, K. Takahashi and T. Komatsu, Chem. Sci., 2016, 7, 4476–4484 RSC
- X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286–293 CrossRef CAS PubMed
- Y. Cao, Z. Sui, Y. Zhu, X. Zhou and D. Chen, ACS Catal., 2017, 7, 7835–7846 CrossRef CAS
- L. Li, B. Zhang, E. Kunkes, K. Föttinger, M. Armbrüster, D. S. Su, W. Wei, R. Schlögl and M. Behrens, ChemCatChem, 2012, 4, 1764–1775 CrossRef CAS
- X. Du, Y. Huang, X. Pan, B. Han, Y. Su, Q. Jiang, M. Li, H. Tang, G. Li and B. Qiao, Nat. Commun., 2020, 11, 5811 CrossRef CAS
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS
- S. Tauster, J. Catal., 1978, 55, 29–35 CrossRef CAS
- N. Iwasa, T. Mayanagi, N. Ogawa, K. Sakata and N. Takezawa, Catal. Lett., 1998, 54, 119–123 CrossRef CAS
- R. Lamber and N. I. Jaeger, J. Appl. Phys., 1991, 70, 457–461 CrossRef CAS
- S. Penner, D. Wang, D. S. Su, G. Rupprechter, R. Podloucky, R. Schlögl and K. Hayek, Surf. Sci., 2003, 532–535, 276–280 CrossRef CAS
- R. Lamber, N. Jaeger and G. Schulz-Ekloff, Surf. Sci., 1990, 227, 268–272 CrossRef CAS
- R. Lamber, N. Jaeger and G. Schulz-Ekloff, J. Catal., 1990, 123, 285–297 CrossRef CAS
- T. Ren-Yuam, W. Rong-An and L. Li-Wu, Appl. Catal., 1984, 10, 163–172 CrossRef
- T. Baczynska, L. Kepinski, M. Seweryniak, J. Wrzyszcz and M. Zawadzki, Catal. Lett., 1997, 44, 217–219 CrossRef CAS
- J. Wang, A.-H. Lu, M. Li, W. Zhang, Y.-S. Chen, D.-X. Tian and W.-C. Li, ACS Nano, 2013, 7, 4902–4910 CrossRef CAS
- A. Haghofer, K. Föttinger, F. Girgsdies, D. Teschner, A. Knop-Gericke, R. Schlögl and G. Rupprechter, J. Catal., 2012, 286, 13–21 CrossRef CAS
- S. Penner, H. Lorenz, W. Jochum, M. Stöger-Pollach, D. Wang, C. Rameshan and B. Klötzer, Appl. Catal., A, 2009, 358, 193–202 CrossRef CAS
- H. Lorenz, S. Penner, W. Jochum, C. Rameshan and B. Klötzer, Appl. Catal., A, 2009, 358, 203–210 CrossRef CAS
- S. E. Collins, J. J. Delgado, C. Mira, J. J. Calvino, S. Bernal, D. L. Chiavassa, M. A. Baltanás and A. L. Bonivardi, J. Catal., 2012, 292, 90–98 CrossRef CAS
- M. Bowker, N. Lawes, I. Gow, J. Hayward, J. R. Esquius, N. Richards, L. R. Smith, T. J. A. Slater, T. E. Davies, N. F. Dummer, L. Kabalan, A. Logsdail, R. C. Catlow, S. Taylor and G. J. Hutchings, ACS Catal., 2022, 12, 5371–5379 CrossRef CAS
- N. Iwasa, S. Masuda, N. Ogawa and N. Takezawa, Appl. Catal., A, 1995, 125, 145–157 CrossRef CAS
- H. Bahruji, M. Bowker, G. Hutchings, N. Dimitratos, P. Wells, E. Gibson, W. Jones, C. Brookes, D. Morgan and G. Lalev, J. Catal., 2016, 343, 133–146 CrossRef CAS
- E. Castillejos-López, G. Agostini, M. Di Michel, A. Iglesias-Juez and B. Bachiller-Baeza, ACS Catal., 2017, 7, 796–811 CrossRef
- P. Yin, Q. Yan and H. Liang, Angew. Chem., 2023, 135, e202302819 CrossRef
- P. Yin, M.-X. Chen, M. Zuo, L.-L. Zhang and H.-W. Liang, ACS Mater. Lett., 2022, 4, 1350–1357 CrossRef CAS
- C.-L. Yang, L.-N. Wang, P. Yin, J. Liu, M.-X. Chen, Q.-Q. Yan, Z.-S. Wang, S.-L. Xu, S.-Q. Chu, C. Cui, H. Ju, J. Zhu, Y. Lin, J. Shui and H.-W. Liang, Science, 2021, 374, 459–464 CrossRef CAS
- X. Jia, Q. Chu, P. Jiang, Z. Han, T. Li and M. Wang, Catal. Lett., 2024, 154, 4955–4969 CrossRef CAS
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2020, 120, 3890–3938 CrossRef CAS
- S. Singh, R. Kumar, H. D. Setiabudi, S. Nanda and D.-V. N. Vo, Appl. Catal., A, 2018, 559, 57–74 CrossRef CAS
- L. S. Carvalho, C. L. Pieck, M. C. Rangel, N. S. Figoli, C. R. Vera and J. M. Parera, Appl. Catal., A, 2004, 269, 105–116 CrossRef CAS
- F. E. López-Suárez, A. Bueno-López, K. I. B. Eguiluz and G. R. Salazar-Banda, J. Power Sources, 2014, 268, 225–232 CrossRef
- F. E. López-Suárez, M. Perez-Cadenas, A. Bueno-López, C. T. Carvalho-Filho, K. I. B. Eguiluz and G. R. Salazar-Banda, J. Appl. Electrochem., 2015, 45, 1057–1068 CrossRef
- L. M. Magno, W. Sigle, P. A. Van Aken, D. G. Angelescu and C. Stubenrauch, Chem. Mater., 2010, 22, 6263–6271 CrossRef CAS
- S. Dey, S. Pramanik, P. Chakraborty, D. K. Rana and S. Basu, J. Appl. Electrochem., 2022, 52, 247–258 CrossRef CAS
- M. Armbrüster, G. Wowsnick, M. Friedrich, M. Heggen and R. Cardoso-Gil, J. Am. Chem. Soc., 2011, 133, 9112–9118 CrossRef
- B. M. Leonard, Q. Zhou, D. Wu and F. J. DiSalvo, Chem. Mater., 2011, 23, 1136–1146 CrossRef CAS
- M. A. Álvarez-Merino, M. A. Fontecha-Cámara, M. V. López-Ramón and C. Moreno-Castilla, Carbon, 2008, 46, 778–787 CrossRef
- M. Kosmulski, Adv. Colloid Interface Sci., 2016, 238, 1–61 CrossRef CAS PubMed
- Y. Yuan, Z. Yang, W. Lai, L. Gao, M. Li, J. Zhang and H. Huang, Chem. - Eur. J., 2021, 27, 16564–16580 CrossRef CAS PubMed
- X. Li, L. An, X. Wang, F. Li, R. Zou and D. Xia, J. Mater. Chem., 2012, 22, 6047 RSC
- J. Lu, Z. Liu, X. Zeng, L. Zhang, W. Fang, W. Gao, Y. Yin, X. Zhu, T. Yang, X. Du, C. Wang, H. Chen and L. Zhao, Energy Fuels, 2024, 38, 2387–2395 CrossRef CAS
- P. Sahu and B. L. V. Prasad, Langmuir, 2014, 30, 10143–10150 CrossRef CAS PubMed
- A. Ota, E. L. Kunkes, I. Kasatkin, E. Groppo, D. Ferri, B. Poceiro, R. M. Navarro Yerga and M. Behrens, J. Catal., 2012, 293, 27–38 CrossRef CAS
- A. Ota, M. Armbrüster, M. Behrens, D. Rosenthal, M. Friedrich, I. Kasatkin, F. Girgsdies, W. Zhang, R. Wagner and R. Schlögl, J. Phys. Chem. C, 2011, 115, 1368–1374 CrossRef CAS
- J. Xin, H. Cui, Z. Cheng and Z. Zhou, Appl. Catal., A, 2018, 554, 95–104 CrossRef CAS
- S. Shu, Z. Wang, X. Zhang, H. Shi, S. Feng and J. Chen, Mol. Catal., 2023, 540, 113056 CrossRef CAS
- Y. Yang, L. Chen, Y. Chen, W. Liu, H. Feng, B. Wang, X. Zhang and M. Wei, Green Chem., 2019, 21, 5352–5362 RSC
- X. Chen, M. Li, J. Guan, X. Wang, C. T. Williams and C. Liang, Ind. Eng. Chem. Res., 2012, 51, 3604–3611 CrossRef CAS
- S. Furukawa and T. Komatsu, ACS Catal., 2016, 7, 735–765 CrossRef
- S. Saedy, D. Palagin, O. Safonova, J. A. Van Bokhoven, A. A. Khodadadi and Y. Mortazavi, J. Mater. Chem. A, 2017, 5, 24396–24406 RSC
- S. Saedy, D. Palagin, O. Safonova, J. A. Van Bokhoven, A. A. Khodadadi and Y. Mortazavi, J. Mater. Chem. A, 2017, 5, 24396–24406 RSC
- D. S. Choi, A. W. Robertson, J. H. Warner, S. O. Kim and H. Kim, Adv. Mater., 2016, 28, 7115–7122 CrossRef CAS
- S. Furukawa, A. Tamura, K. Ozawa and T. Komatsu, Appl. Catal., A, 2014, 469, 300–305 CrossRef CAS
- T. Komatsu and A. Tamura, J. Catal., 2008, 258, 306–314 CrossRef CAS
- T. Komatsu and Y. Fukui, Appl. Catal., A, 2005, 279, 173–180 CrossRef CAS
- T. Komatsu, M. Mesuda and T. Yashima, Appl. Catal., A, 2000, 194–195, 333–339 CrossRef CAS
- A. Onda, T. Komatsu and T. Yashima, J. Catal., 2001, 201, 13–21 CrossRef CAS
- R. P. Doherty, J.-M. Krafft, C. Méthivier, S. Casale, H. Remita, C. Louis and C. Thomas, J. Catal., 2012, 287, 102–113 CrossRef CAS
- S. Chettibi, R. Wojcieszak, E. H. Boudjennad, J. Belloni, M. M. Bettahar and N. Keghouche, Catal. Today, 2006, 113, 157–165 CrossRef CAS
- J. Kugai, R. Kitagawa, S. Seino, T. Nakagawa, Y. Ohkubo, H. Nitani, H. Daimon and T. A. Yamamoto, Appl. Catal., A, 2011, 406, 43–50 CrossRef CAS
- J. Belloni, M. Mostafavi, H. Remita, J.-L. Marignier and A. M.-O. Delcourt, New J. Chem., 1998, 22, 1239–1255 RSC
- J. Kugai, T. Moriya, S. Seino, T. Nakagawa, Y. Ohkubo, H. Nitani, Y. Mizukoshi and T. A. Yamamoto, Appl. Catal., B, 2012, 126, 306–314 CrossRef CAS
- Z. Guo, Y. Chen, L. Li, X. Wang, G. L. Haller and Y. Yang, J. Catal., 2010, 276, 314–326 CrossRef CAS
- J. Grand, S. R. Ferreira, V. De Waele, S. Mintova and T. M. Nenoff, J. Phys. Chem. C, 2018, 122, 12573–12588 CrossRef CAS
- J. Kugai, E. Dodo, S. Seino, T. Nakagawa, T. Okazaki and T. A. Yamamoto, J. Nanopart. Res., 2016, 18, 62 CrossRef
- A. Abedini, F. Larki, E. Saion, A. Zakaria and M. Zobir Hussein, Radiat. Phys. Chem., 2012, 81, 1653–1658 CrossRef CAS
- H. Remita, I. Lampre, M. Mostafavi, E. Balanzat and S. Bouffard, Radiat. Phys. Chem., 2005, 72, 575–586 CrossRef CAS
- K. Čubová and V. Čuba, Radiat. Phys. Chem., 2020, 169, 108774 CrossRef
- I. Călinescu, D. Martin, D. Ighigeanu, A. Gavrila, A. Trifan, M. Patrascu, C. Munteanu, A. Diacon, E. Manaila and G. Craciun, Open Chem., 2014, 12, 774–781 CrossRef
- T. A. Yamamoto, T. Nakagawa, S. Seino and H. Nitani, Appl. Catal., A, 2010, 387, 195–202 CrossRef CAS
- K.-D. Seo, S.-D. Oh, S.-H. Choi, S.-H. Kim, H. G. Park and Y. P. Zhang, Colloids Surf., A, 2008, 313–314, 393–397 CrossRef
- D.-S. Yang, K.-S. Sim, H.-D. Kwen and S.-H. Choi, J. Ind. Eng. Chem., 2012, 18, 538–545 CrossRef CAS
- O. Johnson, Y. He, I. St. Pierre-Charles, J. Richter, B. Joseph and J. N. Kuhn, ACS Catal., 2024, 14, 7746–7755 CrossRef CAS
- J. T. L. Gamler, H. M. Ashberry, S. E. Skrabalak and K. M. Koczkur, Adv. Mater., 2018, 30, 1801563 CrossRef PubMed
- E. Antolini, Appl. Catal., B, 2017, 217, 201–213 CrossRef CAS
- W. S. Lamme, J. Zečević and K. P. de Jong, ChemCatChem, 2018, 10, 1552–1555 CrossRef CAS
- Y. Wang, Z. Rong, Y. Wang, P. Zhang, Y. Wang and J. Qu, J. Catal., 2015, 329, 95–106 CrossRef CAS
- L. Ceatră, O. C. Pârvulescu, I. Rodríguez Ramos and T. Dobre, Ind. Eng. Chem. Res., 2016, 55, 1491–1502 CrossRef
- A. B. Merlo, B. F. Machado, V. Vetere, J. L. Faria and M. L. Casella, Appl. Catal., A, 2010, 383, 43–49 CrossRef CAS
- M. Consonni, D. Jokic, D. Yu Murzin and R. Touroude, J. Catal., 1999, 188, 165–175 CrossRef CAS
- X. Zhang, W. Shi, Y. Li, W. Zhao, S. Han and W. Shen, ACS Catal., 2023, 13, 4030–4041 CrossRef CAS
- P. Zhai, D. A. Cullen and K. Ding, Chem. Eng. J., 2024, 480, 148238 CrossRef CAS
- F. Pompeo, N. N. Nichio, M. M. V. M. Souza, D. V. Cesar, O. A. Ferretti and M. Schmal, Appl. Catal., A, 2007, 316, 175–183 CrossRef CAS
- D. San-José-Alonso, J. Juan-Juan, M. J. Illán-Gómez and M. C. Román-Martínez, Appl. Catal., A, 2009, 371, 54–59 CrossRef
- M. Khzouz, E. I. Gkanas, S. Du and J. Wood, Fuel, 2018, 232, 672–683 CrossRef CAS
- E. Nikolla, J. Schwank and S. Linic, J. Catal., 2007, 250, 85–93 CrossRef CAS
- K. Föttinger, Catal. Today, 2013, 208, 106–112 CrossRef
- X. Liu, F. Jiang, K. Liu, G. Zhao, J. Liu and H. Xu, Chem. Eng. J., 2024, 487, 150351 CrossRef CAS
- S. Natesakhawat, O. Oktar and U. S. Ozkan, J. Mol. Catal. A: Chem., 2005, 241, 133–146 CrossRef CAS
- Y. Li, X. Wang, C. Xie and C. Song, Appl. Catal., A, 2009, 357, 213–222 CrossRef CAS
- J. Thormann, L. Maier, P. Pfeifer, U. Kunz, O. Deutschmann and K. Schubert, Int. J. Hydrogen Energy, 2009, 34, 5108–5120 CrossRef CAS
- W. Zhang, H. Wang, J. Jiang, Z. Sui, Y. Zhu, D. Chen and X. Zhou, ACS Catal., 2020, 10, 12932–12942 CrossRef CAS
- L. Zhang, K. Chen, H. Chen, X. Han, C. Liu, L. Qiao, W. Wu and B. Yang, Chem. Eng. J., 2024, 483, 149366 CrossRef CAS
- T. Komatsu and S. Furukawa, Mater. Trans., 2015, 56, 460–467 CrossRef CAS
- S. W. Han, H. Park, J. Han, J.-C. Kim, J. Lee, C. Jo and R. Ryoo, ACS Catal., 2021, 11, 9233–9241 CrossRef CAS
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed
- Z. Wu, E. C. Wegener, H.-T. Tseng, J. R. Gallagher, J. W. Harris, R. E. Diaz, Y. Ren, F. H. Ribeiro and J. T. Miller, Catal. Sci. Technol., 2016, 6, 6965–6976 RSC
- A. H. Al-ShaikhAli, A. Jedidi, D. H. Anjum, L. Cavallo and K. Takanabe, ACS Catal., 2017, 7, 1592–1600 CrossRef CAS
- T. Soták, T. Schmidt and M. Hronec, Appl. Catal., A, 2013, 459, 26–33 CrossRef
- Z. Wu, H. Yan, S. Ge, J. Gao, T. Dou, Y. Li, A. C. K. Yip and M. Zhang, Catal. Commun., 2017, 92, 80–85 CrossRef CAS
- C. Liu, C. Zhang, K. Liu, Y. Wang, G. Fan, S. Sun, J. Xu, Y. Zhu and Y. Li, Biomass Bioenergy, 2015, 72, 189–199 CrossRef CAS
- S. Sitthisa, T. Sooknoi, Y. Ma, P. B. Balbuena and D. E. Resasco, J. Catal., 2011, 277, 1–13 CrossRef CAS
- A. N. Kay Lup, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, J. Ind. Eng. Chem., 2017, 56, 1–34 CrossRef CAS
- Z. Y. Zakaria, N. A. S. Amin and J. Linnekoski, Biomass Bioenergy, 2013, 55, 370–385 CrossRef CAS
- A. Corma, G. Huber, L. Sauvanaud and P. Oconnor, J. Catal., 2008, 257, 163–171 CrossRef CAS
- Z. Y. Zakaria, J. Linnekoski and N. A. S. Amin, Chem. Eng. J., 2012, 207–208, 803–813 CrossRef CAS
- Y. Yang, C. Ochoa-Hernández, V. A. De La Peña O’Shea, P. Pizarro, J. M. Coronado and D. P. Serrano, Appl. Catal., B, 2014, 145, 91–100 CrossRef CAS
- S. Ohta, O. Komai and H. Hanakawa, Mod. Rheumatol., 2014, 24, 144–149 CrossRef CAS PubMed
- X. Niu, L. Wang and J. Chen, Catal. Commun., 2020, 140, 106001 CrossRef CAS
- Z. Pan, R. Wang and J. Chen, Appl. Catal., B, 2018, 224, 88–100 CrossRef CAS
- Y. Zheng, N. Zhao and J. Chen, Appl. Catal., B, 2019, 250, 280–291 CrossRef CAS
- W. N. Adira Wan Khalit, T. S. Marliza, N. Asikin-Mijan, M. S. Gamal, M. I. Saiman, M. L. Ibrahim and Y. H. Taufiq-Yap, RSC Adv., 2020, 10, 37218–37232 RSC
- Z.-H. He, C.-S. Jiang, Z.-Y. Wang, K. Wang, Y.-C. Sun, M.-Q. Yao, Z.-H. Li and Z.-T. Liu, Sustainable Energy Fuels, 2020, 4, 4558–4569 RSC
| This journal is © The Royal Society of Chemistry 2025 |