
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer-confined synthesis of gram-scale high-entropy perovskite fluoride nanocubes for improved electrocatalytic reduction of nitrate to ammonia†
Guohao
Xue
a,
Tianlu
Wang
a,
Hele
Guo
*ab,
Nan
Zhang
 a,
Claire J.
Carmalt
c,
Johan
Hofkens
b,
Feili
Lai
*bd and
Tianxi
Liu
*a
a,
Claire J.
Carmalt
c,
Johan
Hofkens
b,
Feili
Lai
*bd and
Tianxi
Liu
*a
aThe Key Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, P. R. China. E-mail: txliu@jiangnan.edu.cn
bDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, Leuven 3001, Belgium. E-mail: hele.guo@kuleuven.be; feili.lai@kuleuven.be
cDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
dState Key Laboratory of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: feililai@sjtu.edu.cn
First published on 25th October 2024
Abstract
High-entropy perovskite fluoride (HEPF) has gradually attracted attention in the field of electrocatalysis due to its unique properties. Although traditional co-precipitation methods can efficiently produce HEPF, the resulting catalysts often lack regular morphology and tend to aggregate extensively. Here, nanocubic K(CuMgCoZnNi)F3 HEPF (HEPF-2) was successfully prepared on a gram-scale by a polyvinylpyrrolidone (PVP)-confined nucleation strategy. Benefiting from its large electrochemically active surface area and well-exposed active sites, the HEPF-2 demonstrates dramatically enhanced electrocatalytic activity in electrocatalytic nitrate reduction to ammonia, leading to an improved ammonia yield rate (7.031 mg h−1 mgcat.−1), a high faradaic efficiency (92.8%), and excellent long-term stability, outperforming the irregular HEPF nanoparticles (HEPF-0) prepared without the assistance of PVP. Our work presents an efficient and facile method to synthesize perovskite fluorides with a well-defined structure, showing great promise in the field of high-performance electrocatalysis.
New conceptsHigh entropy perovskite fluorides (HEPFs) offer significant potential in electrochemical reactions due to their customizable elemental composition, diverse active sites, and structural flexibility. However, current preparation methods, such as ball milling and hydrothermal methods, are time-consuming and energy-intensive. Ball milling usually produces nanomaterials with irregular shapes and large sizes, while hydrothermal methods are challenging for large-scale production. Our research introduces a polyvinylpyrrolidone confined nucleation strategy to prepare gram-scale nanocubic K(CuMgCoZnNi)F3 HEPF (HEPF-2). In solution, the abundant and evenly distributed pyrrolidone groups on the PVP polymer chains serve as nucleation sites, promoting the uniform nucleation and growth of HEPF. This approach significantly enhances the electrochemically active surface area, reduces charge transfer resistance, and optimizes the surface potential of HEPF, thereby greatly improving the yield rate and faradaic efficiency for electrocatalytic nitrate reduction to ammonia. This strategy not only enhances the catalytic activity of HEPF but also provides a scalable and efficient method for the synthesis of high-entropy nanomaterials. |
Introduction
Ammonia (NH3) plays a pivotal role in contemporary society, particularly in the fields of agriculture, medicine, chemicals, and energy storage applications.1–4 Nevertheless, the extensive production of NH3 through the Haber–Bosch process since the 20th century has presented notable environmental concerns. This method requires high temperature (400–500 °C) and pressure (10–30 MPa), as well as a significant amount of hydrogen obtained from natural gas, resulting in considerable fossil fuel consumption and carbon dioxide emissions.5–8 To achieve carbon neutrality goals for energy preservation and environmental protection, it is essential to prioritize the exploration of environmentally friendly and sustainable methods for NH3 production.9–11 In recent years, the electrocatalytic nitrogen reduction reaction (eNRR) has been extensively studied as a method for NH3 synthesis. However, the slow dissociation process of the exceptionally stable N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonds (binding energy of 941 kJ mol−1) has led to a relatively low NH3 yield rate and faradaic efficiency (FE) in this reduction process.12–15 This limitation hinders its ability to meet industrial requirements and may potentially result in unreliable experimental quantification due to trace impurities.16,17 Conversely, due to the considerable solubility of nitrate (NO3−) and the relatively weak N
N triple bonds (binding energy of 941 kJ mol−1) has led to a relatively low NH3 yield rate and faradaic efficiency (FE) in this reduction process.12–15 This limitation hinders its ability to meet industrial requirements and may potentially result in unreliable experimental quantification due to trace impurities.16,17 Conversely, due to the considerable solubility of nitrate (NO3−) and the relatively weak N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond (with a binding energy of 204 kJ mol−1), the electrocatalytic nitrate reduction reaction (eNITRR) has arisen as an alternative to the eNRR process, offering substantial improvements in both NH3 yield rate and FE.18,19 Additionally, NO3− is also a prevalent pollutant in various types of wastewater.20,21 Consequently, eNITRR not only offers a prospective path for sustainable NH3 production but also aids in the denitrification of wastewater, thereby assisting in the restoration of the disrupted nitrogen cycle.
O double bond (with a binding energy of 204 kJ mol−1), the electrocatalytic nitrate reduction reaction (eNITRR) has arisen as an alternative to the eNRR process, offering substantial improvements in both NH3 yield rate and FE.18,19 Additionally, NO3− is also a prevalent pollutant in various types of wastewater.20,21 Consequently, eNITRR not only offers a prospective path for sustainable NH3 production but also aids in the denitrification of wastewater, thereby assisting in the restoration of the disrupted nitrogen cycle.
High-entropy materials have demonstrated significant potential in electrocatalysis, particularly in electrochemical reactions, owing to their numerous intrinsic active sites, intricate electronic structures, and random element distribution.22–26 Within the realm of high-entropy materials, high-entropy perovskites (HEPs), specifically high-entropy perovskite oxides, have recently attracted interest in electrocatalysis due to their customizable elemental composition, varied active sites, and structural resilience.27–29 However, conventional preparation techniques for high-entropy perovskite oxides typically involve high-temperature sintering (usually up to 900 °C), which is energy-intensive and not fully reproducible. Substituting oxygen with fluorine can enhance the stability of perovskites since the highly electronegative fluorine (F) atoms (with an electronegativity of 3.98) bonded to metal in fluoride perovskites can create highly polarized metal–F bonds.22,30 This characteristic results in milder preparation conditions for fluoride perovskites, effectively inhibiting the self-reduction of catalysts and enhancing stability.31 Besides, ABF3-type perovskite fluorides with a robust open framework structure and intersecting tetragonal chains are beneficial for the rapid migration of ions.32 Moreover, fluoride perovskites exhibit wider band gaps and higher conductivities than oxides, which can promote the electron transfer of eNITRR, thereby improving the yield rate and FE of ammonia.22 However, the current methods for preparing high-entropy perovskite fluoride (HEPF) are mostly ball milling or hydrothermal methods, which are limited by poor product uniformity, difficulty in controlling particle size, complex post-treatment processes, safety concerns, and challenges in scaling up production.33 Therefore, developing a facile and efficient new method for large-scale preparation of nano-sized HEPF is of great significance.
Herein, a nanocubic K(CuMgCoZnNi)F3 HEPF (HEPF-2) was successfully fabricated on a gram-scale by a polyvinylpyrrolidone (PVP)-confined nucleation strategy. In solution, the abundant and uniformly distributed pyrrolidone groups on the PVP molecular chain were used as nucleation sites to facilitate uniform nucleation and growth of HEPF. Due to its larger electrochemical active surface area and well-exposed active sites, HEPF-2 exhibits markedly improved electrocatalytic activity for eNITRR, resulting in an improved ammonia yield rate (7.031 mg h−1 mgcat.−1), a high FE (92.8%), and excellent long-term stability. These metrics are significantly better than those of irregular HEPF nanoparticles (HEPF-0) synthesized without PVP assistance. This research work explores an efficient method to synthesize perovskite fluorides with a well-defined structure on a gram-scale, which shows great application prospects in the field of electrocatalytic nitrate reduction.
Results and discussion
As shown in Fig. 1, the traditional co-precipitation method for preparing HEPF undergoes a random nucleation and growth process, resulting in the formation of irregular HEPF nanoparticles. However, the PVP-confined nucleation strategy can promote the uniform nucleation and growth of HEPF, resulting in the formation of nanocubic K(CuMgCoZnNi)F3 HEPF. HEPF-0, HEPF-1, HEPF-2, and HEPF-3 were prepared, corresponding to the PVP mass fractions of 0%, 5%, 10%, and 15%, respectively. Powder X-ray diffraction (PXRD) patterns of HEPF samples exhibit eight diffraction peaks at 2θ ≈ 22.2°, 31.5°, 38.8°, 45.0°, 50.6°, 55.9°, 65.4°, and 74.4° corresponding to the (100), (110), (111), (200), (210), (211), (220), and (310) crystal planes of HEPF, consistent with prototype orthorhombic-type perovskites of KZnF3, indicating that the Cu2+, Mg2+, Co2+, and Ni2+ ions are all integrated into KZnF3 to form a new high-entropy K(CuMgCoZnNi)F3 solid solution (Fig. 2a). However, an impurity peak that appeared in the XRD pattern of HEPF-3 at 33.3° indicates the failed synthesis of pure HEPF (inset of Fig. 2a). Fig. 2b shows the ideal cubic structure of K(CuMgCoZnNi)F3. The B site cations (Cu2+, Mg2+, Co2+, Zn2+, and Ni2+) have 6 coordination numbers and occupy the center of the tightly packed octahedron, leading to the formation of a three-dimensional skeleton structure by sharing corner points. The large sphere in the center is the A-site cation (K+), which is in the cavity formed by 8 octahedra. The transmission electron microscopy (TEM) image reveals the disordered structure of HEPF-0 with severe agglomeration (Fig. S1, ESI†). In contrast, HEPF-1 predominantly displays a nanocubic structure with nanoparticle size at around 100 nm, while the agglomeration is still evident (Fig. S2, ESI†). Meanwhile, HEPF-2 exhibits good dispersion and a well-defined nanocubic structure with nanoparticle size under 100 nm (Fig. 2c and d). The lattice fringes of the HEPF-2 nanoparticles were measured to be 0.394 nm, corresponding to the d-spacing of the (100) plane of the cubic crystal structure (Fig. 2e). Additionally, the high-angle annular dark-field scanning TEM (HAADF-STEM) image of the HEPF-2 and corresponding energy-dispersive X-ray spectroscopy (EDS) elemental mappings demonstrate the uniform distributions of K, Cu, Mg, Co, Zn, Ni, and F in HEPF nanocubes, as depicted in Fig. 2f, further proving the formation of single-phase K(CuMgCoZnNi)F3 solid solution.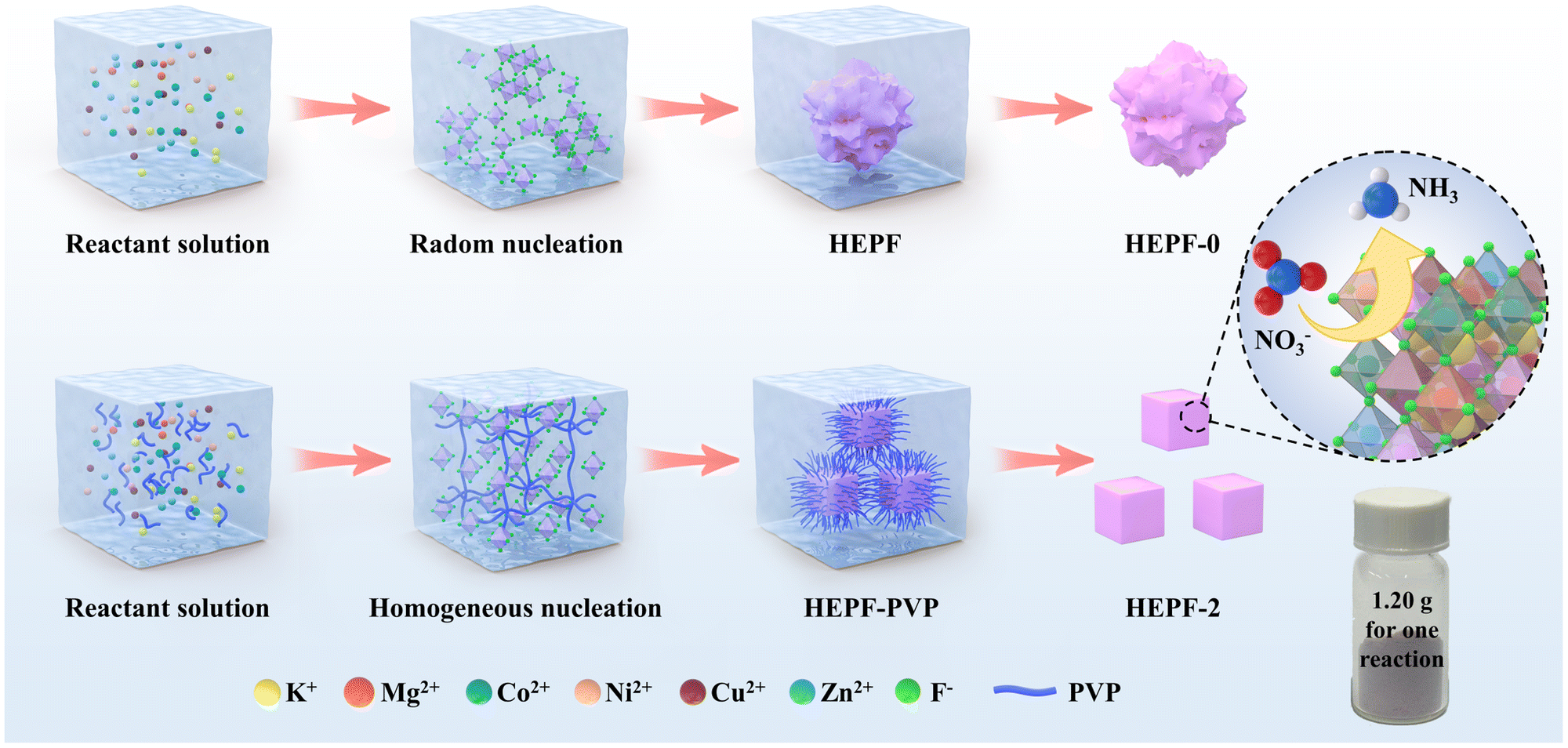 | ||
| Fig. 1 Schematic diagram of the traditional co-precipitation method and the PVP-confined nucleation strategy for preparing HEPF nanocubes. | ||
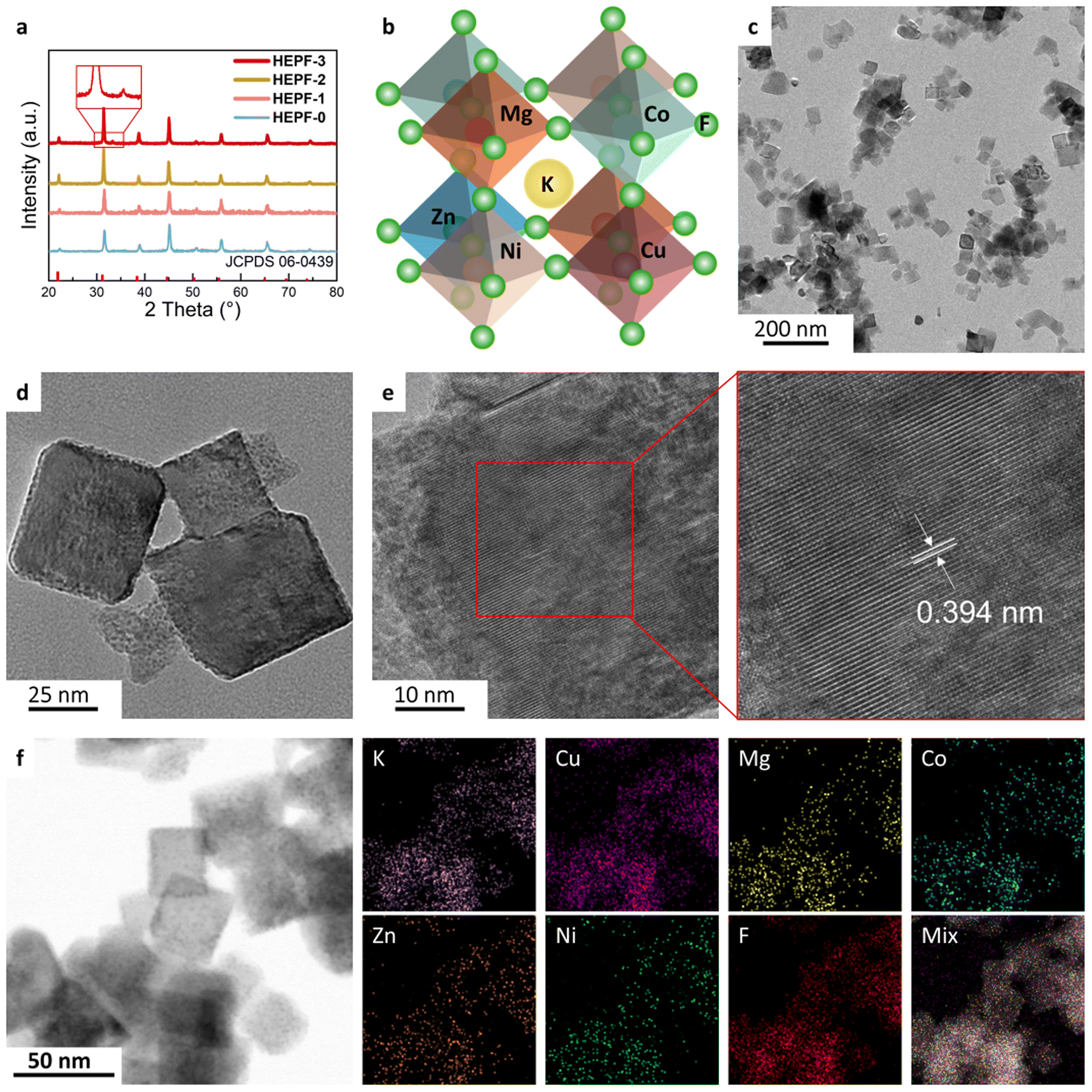 | ||
| Fig. 2 (a) XRD patterns of HEPF samples with different PVP contents. (b) Schematic presentation of the HEPF structure. (c) and (d) TEM and (e) HRTEM images of HEPF-2. (f) HAADF-STEM image and corresponding EDS elemental mappings of HEPF-2. | ||
X-ray photoelectron spectroscopy (XPS) was performed to elucidate the chemical compositions and states of HEPF-2 and HEPF-0. As shown in Fig. S3 and S4 (ESI†), the elements of K, Cu, Mg, Co, Zn, Ni, and F are clearly presented in the XPS spectra of HEPF-2 and HEPF-0. Fig. 3a shows the high-resolution F 1s spectra of HEPF-2 and HEPF-0, where the peaks at 684.80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV are ascribed to the M–F bonds.34 The high-resolution Cu 2p spectra of HEPF-0 and HEPF-2 are displayed in Fig. 3b. Both of them can be deconvoluted into six peaks, which are related to the Cu2+ 2p3/2 (936.90 eV), Cu2+ 2p1/2 (956.60 eV), Cu+ 2p3/2 (933.15 eV), Cu+ p1/2 (953.10 eV), and two satellite peaks.35 Notably, the Cu+/Cu2+ ratio in HEPF-2 is calculated to be 1.97, which is significantly higher than that in HEPF-0 (1.19). This indicates that the HEPF-2 contains more Cu+, which is beneficial for the activation of NO3−.36,37 The high-resolution Mg 1s spectra of HEPF-2 and HEPF-0 (Fig. 3c) show that the Mg element exists in the form of Mg2+ (1304.65 eV).38 The high-resolution Co 2p spectra of both HEPF-2 and HEPF-0 (Fig. 3d) can be divided into four distinct peaks belonging to Co2+ 2p3/2 (782.77 eV), Co2+ 2p1/2 (798.95 eV) species, and two satellite peaks.39,40 Moreover, the high-resolution Zn 2p spectra of HEPF-2 and HEPF-0 in Fig. 3e show the Zn 2p1/2 spin orbit at 1045.54eV and the Zn 2p3/2 orbit at 1022.32 eV.41–43Fig. 3f shows the high-resolution Ni 2p spectra of HEPF-2 and HEPF-0, both of which can be divided into four distinct peaks belonging to Ni2+ 2p3/2 (857.65 eV), Ni2+ 2p1/2 (876.09 eV) species, and two satellite peaks (863.60 and 882.50 eV).39,40,44 The high-resolution K 2p spectrum of HEPF-2 (Fig. S5, ESI†) can be deconvoluted into two peaks, which are related to the K 2p3/2 (292.79 eV) and K 2p1/2 (295.54 eV).45 In addition, the high-resolution N 1s XPS spectrum of HEPF-2 shows no signal (Fig. S6, ESI†), indicating that PVP can be completely removed by the washing process.
eV are ascribed to the M–F bonds.34 The high-resolution Cu 2p spectra of HEPF-0 and HEPF-2 are displayed in Fig. 3b. Both of them can be deconvoluted into six peaks, which are related to the Cu2+ 2p3/2 (936.90 eV), Cu2+ 2p1/2 (956.60 eV), Cu+ 2p3/2 (933.15 eV), Cu+ p1/2 (953.10 eV), and two satellite peaks.35 Notably, the Cu+/Cu2+ ratio in HEPF-2 is calculated to be 1.97, which is significantly higher than that in HEPF-0 (1.19). This indicates that the HEPF-2 contains more Cu+, which is beneficial for the activation of NO3−.36,37 The high-resolution Mg 1s spectra of HEPF-2 and HEPF-0 (Fig. 3c) show that the Mg element exists in the form of Mg2+ (1304.65 eV).38 The high-resolution Co 2p spectra of both HEPF-2 and HEPF-0 (Fig. 3d) can be divided into four distinct peaks belonging to Co2+ 2p3/2 (782.77 eV), Co2+ 2p1/2 (798.95 eV) species, and two satellite peaks.39,40 Moreover, the high-resolution Zn 2p spectra of HEPF-2 and HEPF-0 in Fig. 3e show the Zn 2p1/2 spin orbit at 1045.54eV and the Zn 2p3/2 orbit at 1022.32 eV.41–43Fig. 3f shows the high-resolution Ni 2p spectra of HEPF-2 and HEPF-0, both of which can be divided into four distinct peaks belonging to Ni2+ 2p3/2 (857.65 eV), Ni2+ 2p1/2 (876.09 eV) species, and two satellite peaks (863.60 and 882.50 eV).39,40,44 The high-resolution K 2p spectrum of HEPF-2 (Fig. S5, ESI†) can be deconvoluted into two peaks, which are related to the K 2p3/2 (292.79 eV) and K 2p1/2 (295.54 eV).45 In addition, the high-resolution N 1s XPS spectrum of HEPF-2 shows no signal (Fig. S6, ESI†), indicating that PVP can be completely removed by the washing process.
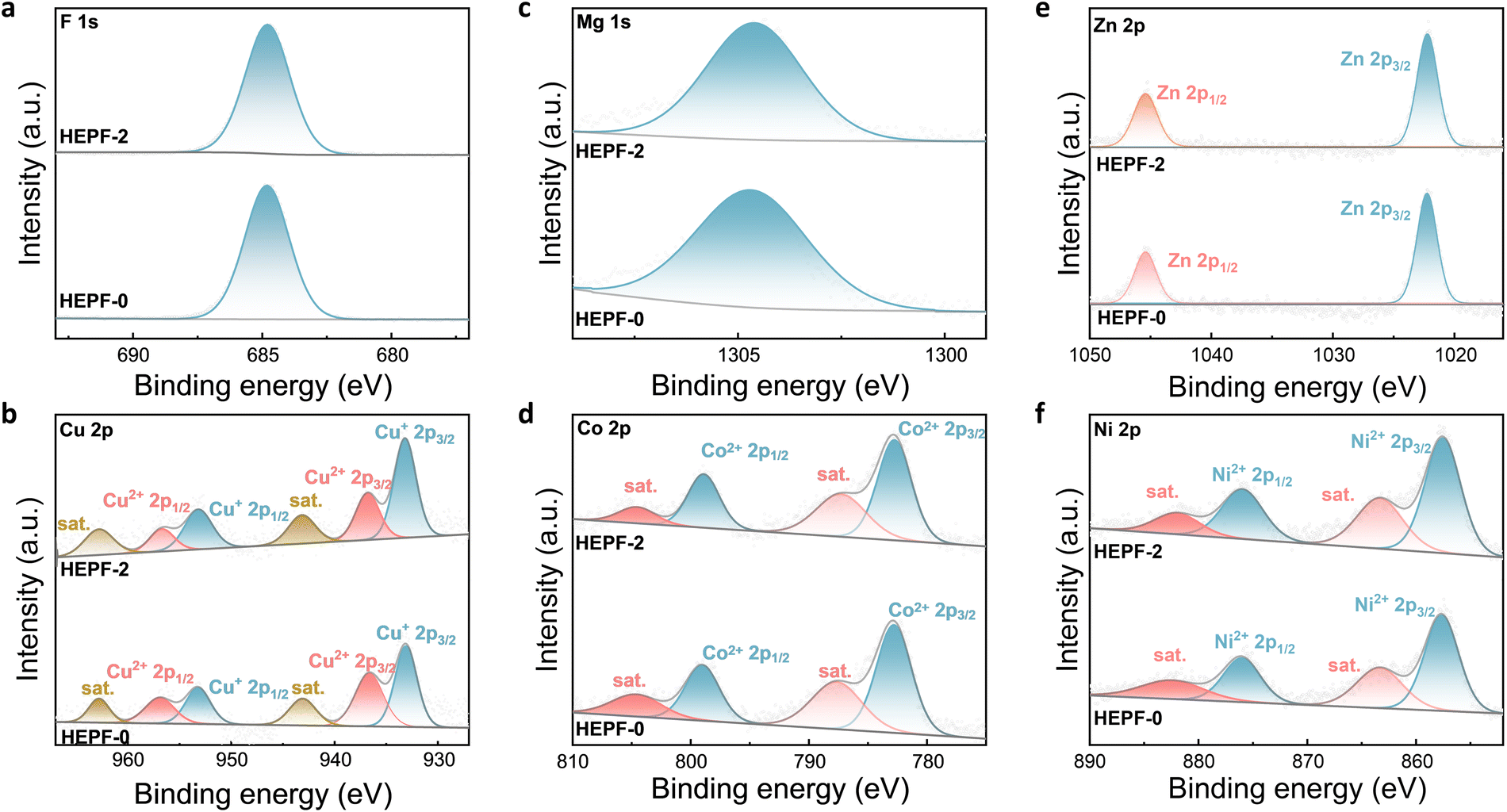 | ||
| Fig. 3 High-resolution (a) F 1s, (b) Cu 2p, (c) Mg 1s, (d) Co 2p, (e) Zn 2p, and (f) Ni 2p XPS spectra of HEPF-2 and HEPF-0 catalysts. | ||
To investigate the eNITRR activity of the HEPF catalysts, a series of detailed electrochemical tests were performed in 0.5 M K2SO4 electrolyte with 0.1 M KNO3 using a three-electrode system. The linear sweep voltammetry (LSV) curves shown in Fig. 4a and Fig. S7 (ESI†) indicate that the current densities of the HEPF-2 and HEPF-0 catalysts in the electrolyte with 0.1 M KNO3 are significantly higher than that in the electrolyte without KNO3, suggesting the electroreduction of KNO3. The eNITRR activity of the HEPF catalysts was further evaluated by monitoring the NH3 yield after chronoamperometry at various potentials from −0.5 to −0.9 V versus the reversible hydrogen electrode (vs. RHE) for 1 h (Fig. S8, ESI†). The NH3 yields of the HEPF catalysts were determined by measuring the ultraviolet-visible (UV-Vis) absorption curves at different potentials based on the calibration curves between absorbance and NH3 concentration (Fig. S9, ESI†). The HEPF-2 catalyst demonstrates a notable NH3 yield rate of 7.031 mg h−1 mgcat.−1 and a maximum FE value of 92.8% at −0.6 V vs. RHE, substantially surpassing the performance of HEPF-1 (5.682 mg h−1 mgcat.−1 and 83.0%) and HEPF-0 catalyst (5.160 mg h−1 mgcat.−1 and 75.2%), as illustrated in Fig. 4b and c. At reduction potentials above −0.6 V, insufficient driving force causes intermediate desorption, hindering their conversion to NH3 and lowering the FE value. Below −0.6 V, the intensified HER process can also decrease the FE value. As a result, the FE value of the HEPF-2 catalyst exhibits a volcanic trend, peaking at −0.6 V. To confirm the accuracy of the measurement, we also quantified the NH3 production using the nuclear magnetic resonance (NMR) method (Fig. S10, ESI†). The results are similar to those obtained by the UV-Vis method (Fig. S11, ESI†). Fig. 4d shows that the NH3 yield rate and FE value of each cycle fluctuate slightly but remain stable, suggesting excellent electrocatalytic stability of HEPF-2 for potential practical applications. The long-term stability of the HEPF-2 was also evaluated through chronoamperometry testing (Fig. 4e). The current density of HEPF-2 shows remarkable stability even after 15 h testing. The XRD, TEM, and XPS analyses provide further evidence supporting the morphological and structural stabilities of HEPF-2 during eNITRR (Fig. S12–S14, ESI†). The XRD pattern demonstrates the stable crystal structure, the TEM image reveals no alterations in nanocubic morphology, and the XPS spectra indicate no valence changes in the HEPF-2 catalyst before and after the catalytic reaction. These results confirm that HEPF-2 remains stable without undergoing structural reconstruction under eNITRR conditions. To confirm the nitrogen source involved in NH3 synthesis, isotope labelling experiments were carried out using electrolytes with 0.1 M K15NO3 or K14NO3, respectively. The identification of 15NH4+ and 14NH4+ products was confirmed through 1H NMR spectra, distinguished by the chemical shifts of the doublet and triplet couplings, respectively. Fig. 4f presents the 1H NMR spectrum of 15NH4+ obtained using an electrolyte containing 0.1 M K15NO3, revealing two distinct peaks of 15NH4+ at 6.93 and 7.11 ppm. In contrast, the 1H NMR spectrum of the electrolyte with 0.1 M K14NO3 after the electrocatalytic reaction exhibits a triple characteristic peak of 14NH4+ at 6.90, 7.03, and 7.16 ppm. This isotope labelling experiment conclusively confirms that the nitrogen source in the synthesized NH3 originates from the nitrate instead of any other contaminants. The remarkable eNITRR activity achieved by HEPF-2 is comparable to that of state-of-the-art high-entropy materials documented in the literature (Table S1, ESI†), highlighting that HEPF is a very promising class of electrocatalyst for eNITRR.
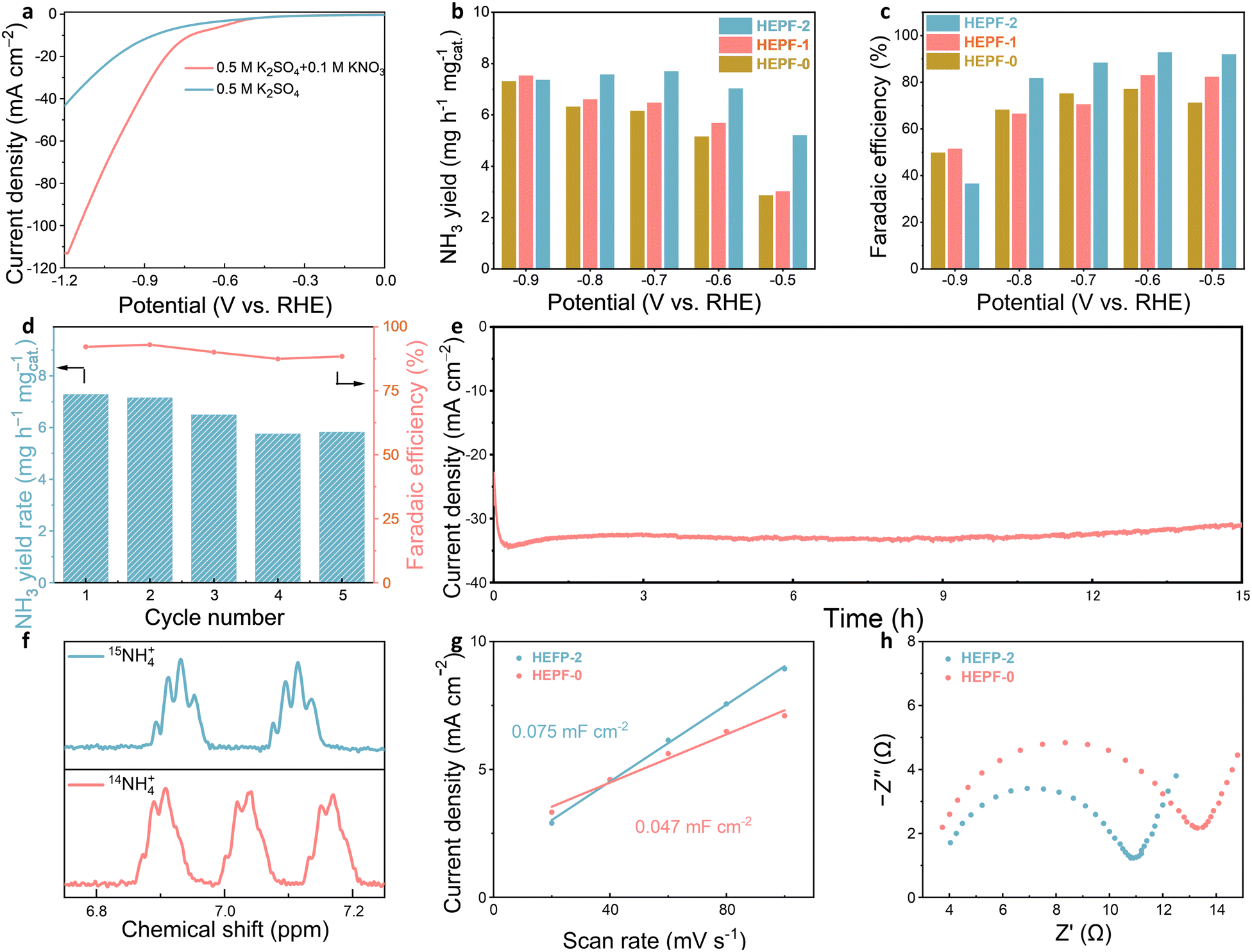 | ||
| Fig. 4 (a) LSV curves for HEPF-2 in 0.5 M K2SO4 electrolytes with and without KNO3, (b) NH3 yield rates and (c) FE values of HEPF-0, HEPF-1 and HEPF-2 under different potentials. (d) NH3 yield rates and FE values of HEPF-2 at −0.6 V vs. RHE for 5 cycles. (e) Long-term stability test of the HEPF-2 catalyst at −0.6 V vs. RHE in electrolyte with 0.1 M KNO3. (f) 1H NMR spectra of NH4+ prepared with K15NO3 and K14NO3 as nitrogen sources. (g) The Cdl values of the HEPF-2 and HEPF-0 catalysts. (h) Nyquist plots of HEPF-2 and HEPF-0 catalysts. | ||
The overall activity of the electrocatalyst is influenced by the number of active sites and the intrinsic activity of each site. To characterize the electrochemically active surface areas (ECSAs) of the HEPF-2 and HEPF-0 catalysts, capacitive currents were measured at various scan rates (ranging from 20 to 100 mV s−1) in non-Faraday potential regions (Fig. S15 and S16, ESI†). The linear fitting results unveil that HEPF-2 (0.075 mF cm−2) shows a much higher double-layer capacitance (Cdl) value than HEPF-0 (0.047 mF cm−2). Further electrochemical impedance spectroscopy (EIS) analysis for HEPF-2 and HEPF-0 was conducted to explore the differences in charge transfer resistance. As shown in Fig. 4h, the values of charge transfer resistance for HEPF-2 and HEPF-0 are 9 and 12 Ω, respectively. Furthermore, a zeta potential analysis was performed. As shown in Fig. S17 (ESI†), HEPF-2 has a more positive potential (23.55 mV) than that of HEPF-0 (22.74 mV), which indicates that HEPF-2 exhibits a stronger adsorption capacity for negatively charged NO3− and promotes the desorption of positively charged NH4+. The outstanding eNITRR activity of HEPF-2 is largely attributed to its well-defined nanocubic structure, large ECSA, low charge transfer resistance, positive surface potential, and the synergistic effect of its multi-element composition. Additionally, based on the review of the existing literature on eNITRR applications,46 Fig. S18 (ESI†) presents a schematic of the possible eNITRR pathways on HEPF. These pathways involve three key stages: (1) adsorption of nitrate (NO3− → *NO3); (2) surface reactions of intermediates, including multi-step deoxygenation and hydrogenation; and (3) desorption of ammonia (*NH3 → NH3).
Conclusion
In summary, we successfully prepared HEPF with a nanocubic structure on a gram-scale by a facile PVP-confined nucleation process. Due to its larger electrochemical active surface area and well-exposed active sites, HEPF-2 exhibits significantly enhanced electrocatalytic activity in eNITRR, resulting in an improved ammonia productivity (7.031 mg h−1 mgcat.−1), a high faradaic efficiency (92.8%), and an excellent long-term stability, outperforming HEPF-0 prepared without the use of PVP. This work paves the way for designing nano-HEPFs tailored for advanced electrocatalytic reactions, thereby expanding the scope for their application in various fields of technology and sustainable development.Experimental section
Preparation of HEPF catalysts
The HEPF samples were synthesized by a PVP-confined nucleation method. Specifically, 30 mmol of KF was dissolved in 50 mL of aqueous solution containing X% (X = 0, 5, 10, 15) of PVP, marked as solution A. Subsequently, 10 mmol metal salts (2 mmol CuAc2, 2 mmol MgAc2·4H2O, 2 mmol CoAc2·4H2O, 2 mmol ZnAc2·2H2O, and 2 mmol NiAc2·4H2O) were dissolved in 20 mL of X% PVP aqueous solution, marked as solution B. Solutions A and B were heated to boiling, and then solution B was slowly added dropwise to solution A, leading to the generation of a large amount of pink precipitate immediately. The resulting pink product was further aged for 4 h to obtain a uniform particle size. The precipitate was then collected by high-speed centrifugation at 10000 rpm and washed several times with deionized water and anhydrous ethanol to eliminate residual PVP and impurities. Finally, HEPF-0, HEPF-1, HEPF-2, and HEPF-3 were obtained after vacuum drying at 60 °C for 20 h, corresponding to the PVP mass fractions of 0%, 5%, 10%, and 15%, respectively.
Author contributions
Guohao Xue: methodology, validation, formal analysis, investigation, data curation, writing – original draft. Tianlu Wang: investigation, data curation. Hele Guo: conceptualization, methodology, data curation, validation, writing – original draft. Nan Zhang: formal analysis. Claire J. Carmalt: resources, writing – review & editing. Johan Hofkens: resources, writing – review & editing. Feili Lai: formal analysis, resources, conceptualization, project administration, supervision, writing – review & editing. Tianxi Liu: writing – review & editing, resources, supervision, project administration, funding acquisition.Data availability
The data supporting this article have been included in the figures and tables, as well as a part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 12411530102, 52303151, 52161135302, 52211530489), the Royal Society (IEC\NSFC\233810), and the Research Foundation of Flanders (FWO, No. G0F2322N, VS06523N, 1298323N).References
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, Chem. Eng. J., 2021, 403, 126269 CrossRef CAS.
- W. Chen, X. Yang, T. Huang, Y. Li, X. Ren, S. Ye, Q. Zhang and J. Liu, Compos. Commun., 2023, 43, 101715 CrossRef.
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187 CrossRef CAS PubMed.
- M. Xu, F. Xu, K. Zhu, X. Xu, P. Deng, W. Wu, W. Ye, Z. Sun and P. Gao, Compos. Commun., 2022, 29, 101037 CrossRef.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- X. Wu, A. Ma, D. Liu, X. Li, Y. Zhou, A. T. Kuvarega, B. B. Mamba, H. Li and J. Gui, Sci. China Mater., 2023, 66, 4367–4376 CrossRef CAS.
- A. Paliwal, C. D. Bandas, E. S. Thornburg, R. T. Haasch and A. A. Gewirth, ACS Catal., 2023, 13, 6754–6762 CrossRef CAS.
- N. R. Singstock and C. B. Musgrave, J. Am. Chem. Soc., 2022, 144, 12800–12806 CrossRef CAS PubMed.
- W. Zong, H. Gao, Y. Ouyang, K. Chu, H. Guo, L. Zhang, W. Zhang, R. Chen, Y. Dai, F. Guo, J. Zhu, Z. Zhang, C. Ye, Y.-E. Miao, J. Hofkens, F. Lai and T. Liu, Angew. Chem. Int. Ed., 2023, 62, e202218122 CrossRef CAS PubMed.
- K. Chu, F. Liu, J. Zhu, H. Fu, H. Zhu, Y. Zhu, Y. Zhang, F. Lai and T. Liu, Adv. Energy Mater., 2021, 11, 2003799 CrossRef CAS.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- T. Zhang, W. Zong, Y. Ouyang, Y. Wu, Y.-E. Miao and T. Liu, Adv. Fiber Mater., 2021, 3, 229–238 CrossRef CAS.
- Y. Zhong, H. Xiong, J. Low, R. Long and Y. Xiong, eScience, 2023, 3, 100086 CrossRef.
- N. Zhou, Z. Wang, N. Zhang, D. Bao, H. Zhong and X. Zhang, ACS Catal., 2023, 13, 7529–7537 CrossRef CAS.
- M. De Ras, L. Hollevoet, J. A. Martens, T. Liu, B. M. Nicolai, M. L. A. T. M. Hertog, J. Hofkens and M. B. J. Roeffaers, Green Chem., 2024, 26, 1302–1305 RSC.
- Y. Wang, L. Zhang, Y. Niu, D. Fang, J. Wang, Q. Su and C. Wang, Green Chem., 2021, 23, 7594–7608 RSC.
- K. Hadjiivanov and H. Knözinger, Phys. Chem. Chem. Phys., 2000, 2, 2803–2806 RSC.
- K. Chu, W. Zong, G. Xue, H. Guo, J. Qin, H. Zhu, N. Zhang, Z. Tian, H. Dong, Y.-E. Miao, M. Roeffaers, J. Hofkens, F. Lai and T. Liu, J. Am. Chem. Soc., 2023, 145, 21387–21396 CrossRef CAS.
- Y. Qie, J. Gao, S. Li, M. Cui, X. Mao, X. Wang, B. Zhang, S. Chi, Y. Jia, Q.-H. Yang, C. Yang and Z. Weng, Sci. China Mater., 2024, 67, 2941–2948 CrossRef CAS.
- T. Wang, H. Chen, Z. Yang, J. Liang and S. Dai, J. Am. Chem. Soc., 2020, 142, 4550–4554 CrossRef CAS PubMed.
- H. Guo, Z. Guo, K. Chu, W. Zong, H. Zhu, L. Zhang, C. Liu, T. Liu, J. Hofkens and F. Lai, Adv. Funct. Mater., 2023, 33, 2308229 CrossRef CAS.
- H. Chen, K. Jie, C. J. Jafta, Z. Yang, S. Yao, M. Liu, Z. Zhang, J. Liu, M. Chi, J. Fu and S. Dai, Appl. Catal., B, 2020, 276, 119155 CrossRef CAS.
- H. Qiao, X. Wang, Q. Dong, H. Zheng, G. Chen, M. Hong, C.-P. Yang, M. Wu, K. He and L. Hu, Nano Energy, 2021, 86, 106029 CrossRef CAS.
- S. Huang, H. Wu, Y. Chen, Z. Zhao, X. Liu, Y. Deng and H. Zhu, Compos. Commun., 2023, 38, 101510 CrossRef.
- K. Chu, J. Qin, H. Zhu, M. De Ras, C. Wang, L. Xiong, L. Zhang, N. Zhang, J. A. Martens, J. Hofkens, F. Lai and T. Liu, Sci. China Mater., 2022, 65, 2711–2720 CrossRef CAS.
- T. Wang, J. Fan, C.-L. Do-Thanh, X. Suo, Z. Yang, H. Chen, Y. Yuan, H. Lyu, S. Yang and S. Dai, Angew. Chem., Int. Ed., 2021, 60, 9953–9958 CrossRef CAS.
- M. Chen, N. Kitiphatpiboon, C. Feng, A. Abudula, Y. Ma and G. Guan, eScience, 2023, 3, 100111 CrossRef.
- Y. Li and R. Ding, Nano Energy, 2024, 124, 109430 CrossRef CAS.
- J. Zhang, Y. Ye, Z. Wang, Y. Xu, L. Gui, B. He and L. Zhao, Adv. Sci., 2022, 9, 2201916 CrossRef CAS.
- P. Goel, S. Sundriyal, V. Shrivastav, S. Mishra, D. P. Dubal, K.-H. Kim and A. Deep, Nano Energy, 2021, 80, 105552 CrossRef CAS.
- H. Wu, Q. Lu, Y. Li, M. Zhao, J. Wang, Y. Li, J. Zhang, X. Zheng, X. Han, N. Zhao, J. Li, Y. Liu, Y. Deng and W. Hu, J. Am. Chem. Soc., 2023, 145, 1924–1935 CrossRef CAS PubMed.
- C.-Z. He, Y.-X. Zhang, J. Wang and L. Fu, Rare Met., 2022, 41, 3456–3465 CrossRef CAS.
- D. Kim, Y. J. Lee and K. H. Ahn, Compos. Commun., 2022, 30, 101093 CrossRef.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- Y. Xue, Q. Yu, Q. Ma, Y. Chen, C. Zhang, W. Teng, J. Fan and W. Zhang, Environ. Sci. Technol., 2022, 56, 14797–14807 CrossRef CAS.
- S. Mei, B. Xiang, S. Guo, J. Deng, J. Fu, X. Zhang, Y. Zheng, B. Gao, K. Huo and P. K. Chu, Adv. Funct. Mater., 2023, 34, 2301217 CrossRef.
- L. Qin, Y. Liu, S. Zhu, D. Wu, G. Wang, J. Zhang, Y. Wang, L. Hou and C. Yuan, J. Mater. Chem. A, 2021, 9, 20405–20416 RSC.
- S. Guddehalli Chandrappa, P. Moni, D. Chen, G. Karkera, K. R. Prakasha, R. A. Caruso and A. S. Prakash, ACS Appl. Energy Mater., 2021, 4, 13425–13430 CrossRef CAS.
- S. Dong, A. Niu, K. Wang, P. Hu, H. Guo, S. Sun, Y. Luo, Q. Liu, X. Sun and T. Li, Appl. Catal., B, 2023, 333, 122772 CrossRef CAS.
- Y. Wang, Y. Kang, Y. Miao, M. Jia, S. Long, L. Diao, L. Zhang, D. Li and G. Wu, Compos. Commun., 2024, 48, 101890 CrossRef.
- Y. Wang, N. Li, H. Liu, J. Shi, Y. Li, X. Wu, Z. Wang, C. Huang, K. Chen, D. Zhang, T. Wu, P. Li, C. Liu and L. Mi, Adv. Fiber Mater., 2023, 5, 2002–2015 CrossRef CAS.
- S. C. Sekhar, B. Ramulu, S. J. Arbaz, M. Nagaraju and J. S. Yu, Adv. Fiber Mater., 2024, 5, 1229–1240 CrossRef.
- R. Sawyer, H. W. Nesbitt and R. A. Secco, J. Non-Cryst. Solids, 2012, 358, 290–302 CrossRef CAS.
- T. Hu, C. Wang, M. Wang, C. M. Li and C. Guo, ACS Catal., 2021, 11, 14417–14427 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00341a |
| This journal is © The Royal Society of Chemistry 2025 |