
Tailoring catalysis at the atomic level: trends and breakthroughs in single atom catalysts for organic transformation reactions
Devendra
Sharma†
 ,
Devanshu
Sajwan†
,
Shubhankar
Mishra
,
Ashrumochan
Gouda
,
Prerna
Mittal
,
Priyanka
Choudhary
,
Bhagyashree Priyadarshini
Mishra
,
Sahil
Kumar
and
Venkata
Krishnan
*
,
Devanshu
Sajwan†
,
Shubhankar
Mishra
,
Ashrumochan
Gouda
,
Prerna
Mittal
,
Priyanka
Choudhary
,
Bhagyashree Priyadarshini
Mishra
,
Sahil
Kumar
and
Venkata
Krishnan
*
School of Chemical Sciences and Advanced Materials Research Center, Indian Institute of Technology Mandi, Kamand, Mandi 175075, Himachal Pradesh, India. E-mail: vkn@iitmandi.ac.in
First published on 5th December 2024
Abstract
The utilization of precise materials in heterogeneous catalysis will provide various new possibilities for developing superior catalysts to tackle worldwide energy and environmental issues. In recent years, single atom catalysts (SACs) with excellent atom utilization and isolated active sites have progressed dramatically as a thriving sector of catalysis research. Additionally, SACs bridge the gap between homogeneous and heterogeneous catalysts and overcome the limitations of both categories. Current research on SACs is highly oriented towards the organic synthesis of high-significance molecules with promising potential for large-scale applicability and industrialization. In this context, this review aims to comprehensively analyze the state-of-the-art research in the synthesis of SACs and analyze their structural, electronic, and geometric properties. Moreover, the unprecedented catalytic performance of the SACs towards various organic transformation reactions is succinctly summarized with recent reports. Further, a detailed summary of the current state of the research field of SACs in organic transformation is discussed. Finally, a critical analysis of the existing challenges in this emerging field of SACs and the possible countermeasures are provided. We believe that SACs have the potential to profoundly alter the chemical industry, pushing the boundaries of catalysis in new and undiscovered territory.
 Devendra Sharma | Devendra Sharma received his BSc degree in Chemistry from Delhi University, and MSc degree from National Institute of Technology, Warangal. Later, he joined the Indian Institute of Technology Mandi, India, as a PhD scholar in the research group of Prof. Venkata Krishnan in 2021. He was awarded the prestigious Prime Minister Research Fellowship from the Ministry of Education, Government of India. His research work is on the design and development of heterogeneous catalysts for organic transformation reactions and plastic upcycling. |
 Devanshu Sajwan | Devanshu Sajwan received his BSc degree in Chemistry (Hons.) from Doon University, Uttarakhand, in 2022 and has completed his MSc degree in Chemistry in 2024 from the Indian Institute of Technology Mandi, India, wherein he worked on his master's thesis project under the guidance of Prof. Venkata Krishnan. His research interest lies in the design and development of heterogeneous catalysts for photocatalytic nitrogen fixation and plastic upcycling. |
 Shubhankar Mishra | Shubhankar Mishra received his BSc degree in Chemistry from Midnapore College in 2022 and MSc degree from National Institute of Technology, Warangal, in 2024. Subsequently, he joined the Indian Institute of Technology Mandi, India, as a project assistant in the research group of Prof. Venkata Krishnan. His research interest lies in the design and development of efficient metal-based heterogeneous catalysts for organic transformation reactions. |
 Ashrumochan Gouda | Ashrumochan Gouda received his BSc degree in Chemistry from Rayagada Autonomous College and MSc degree from Visvesvaraya National Institute of Technology, Nagpur. Later, he joined the Indian Institute of Technology Mandi, India, as a project scientist in the research group of Prof. Venkata Krishnan in 2023. His research interest lies in the design and development of heterogeneous catalysts for hydrogen evolution and plastic upcycling reactions. |
 Prerna Mittal | Prerna Mittal received her BSc degree in Chemistry (Hons.) from Delhi University and has recently completed her MSc degree in Chemistry in 2024 from the Indian Institute of Technology Mandi, India, wherein she worked on her master's thesis project under the guidance of Prof. Venkata Krishnan. Her research interest lies in the design and development of metal phosphide-based heterogeneous catalysts for organic transformation reactions. |
 Venkata Krishnan | Venkata Krishnan received his PhD in Chemistry from the University of Stuttgart, Germany, in 2006. Subsequently, he worked as a postdoctoral researcher at the University of Pennsylvania, USA, and then as a research associate at the National Institute for Materials Science, Japan. He then joined the Indian Institute of Technology Mandi, India, in 2012 and is currently working as a Full Professor of Chemistry. His research interests are in the field of heterogeneous catalysis for energy and environmental applications. |
1. Introduction
Catalysis, known as the engine of industrial chemistry, is now contributing to the production of over 90% of all chemicals, materials, and green fuels. Hence, the need for novel catalytic technologies keeps rising to develop a sustainable future for modern society.1,2 In general, catalysis can be classified as homogeneous catalysis, bio-catalysis and heterogeneous catalysis. Despite their high selectivity and excellent mass transfer, homogeneous catalysis and bio-catalysis suffer from thermal stability and product–catalyst separation.3–6 In this regard, heterogeneous catalysis is highly significant as the catalysts not only provide a variety of active sites for the catalytic reaction but also ensure high-yielding conversions into selective products and easy product–catalyst separation.7 Moreover, the multiphasic and multicomponent heterogeneous catalysts provide a distinct environment to the reaction and rely on surface-mediated bond activation between the reactants and the catalyst.8 Due to these excellent advantages, heterogeneous catalysis accounts for almost 80% of the industrial catalytic processes and has been employed in various research fields like water splitting, nitrogen fixation, CO2 reduction, biomass conversion and organic transformation reactions via photocatalytic, electrocatalytic, photo-electrocatalytic and thermo-catalytic techniques.9–20 Hence, it has been evident that pioneering studies in heterogeneous catalytic systems play a vital role in the development of chemical industries.Metal and alloy nanoparticles (NPs) are the kind of functional metal-based catalysts that have shown strong potential in various catalytic chemical transformation and energy conversion applications.3 Since the heterogeneous catalysis relies on the exposed metal active centers, the metal NP inaccessible by the reactant molecules remains un-utilized. Furthermore, the agglomeration of metal NPs causes broad size distribution and unusual morphologies and hence may contain multiple active sites with varying performance. This heterogeneity affects product selectivity by limiting the utilization of metal active sites. Therefore, the size of the metal NP is a dominant factor affecting the catalytic reactivity and product specificity. In this regard, a reduction in the size of metal NPs is thought to be an effective way to increase catalytic performance (Fig. 1). This is because of the tremendous increase in exposed surface atoms and modulation in the atomic structure at the surface, electronic structure and extent of surface defects. However, when particle size decreases, metals’ surface free energy rises noticeably, encouraging the aggregation of tiny clusters. So, to avoid aggregation of metal species, the metal species can be supported on suitable support materials that interact with the metal species to produce stable, finely dispersed metal clusters with high catalytic activity.21 Therefore, by controlling the particle size distribution, support materials, and different crystal structures, the efficiency, yield and selectivity of a reaction can be effectively controlled. As an example, bulk Au is highly inert for organic transformation reactions but on reducing the size to the NP level, it exhibits promising activity owing to the surface plasmon resonance and alteration in the electronic structure.22,23 In this context, as the next-generation catalyst, single atom catalysts (SACs) have a recent breakthrough in interdisciplinary research.
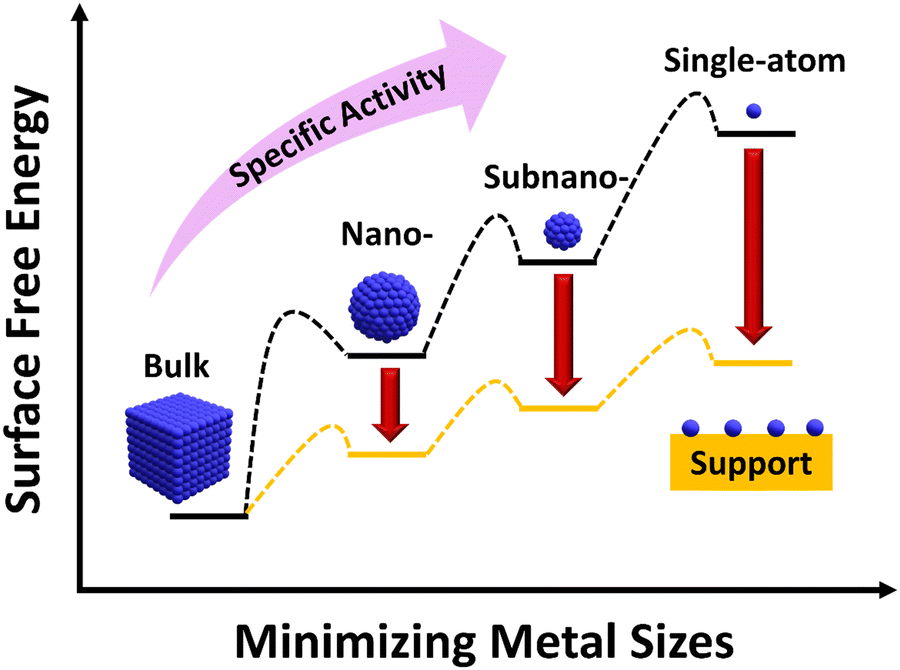 | ||
| Fig. 1 Correlation between the metal size and surface free energy of the metal. Adapted with permission.21 Copyright 2013, American Chemical Society. | ||
SACs are proven to be an elegant class of materials endowed with outstanding catalytic properties compared to nano and sub-nano metallic counterparts owing to excellent properties like 100% atom utilization and highly exposed surface sites.24–26 Also, the enhanced activity is attributed to the special unsaturated co-ordination environment that resembles a molecular complex. This unique unsaturated coordination around the metal atoms provides the scope for tailoring the acidic/basic sites, electron density, and metal–support interactions, which play a vital role in enhancing the catalytic activity.27 Therefore, the SACs are equipped with the ability to shrink the gap between homogeneous and heterogeneous catalysis.21,28
The isolated metal centers possess strong covalent interaction with the support materials, hence they exhibit high stability even under harsh reaction conditions. Due to the tunable charge over the metal, SACs exhibit maximum catalytic performance. Moreover, the combined effect of the unique single atom distribution and the absence of metal–metal bonds is ascribed to the high selectivity of SACs.29 In addition, SACs can also be considered as nanoenzymes as they have the ability to provide information on the function of enzymes in biocatalysis.30 Another intriguing fact is that there can be various types of metallic active sites under the domain of SACs which involve single metal sites, bimetallic sites and single metal double sites which also affect the product distribution.27 Inspired by these intriguing properties, tunability and characteristics of SACs, this review delves into the current state of research on the synthesis of SACs based on metals (noble metals and non-noble metals) and non-metals. Also, a discussion on the sophisticated microscopy and X-ray absorption spectroscopy which are used to characterize and confirm the single atoms has been included. As SACs find excellent utility in organic transformation reactions, an extensive discussion on various reactions has been made followed by a detailed summary of the review and current scenario of the research field. At last, several key challenges in the research field have been identified and discussed along with the possible solutions which can help the readers to analyze the scope of this field which can lead to pioneering works and even industrialization of the technology.
1.1. Brief history of SACs
The concept of designing isolated metal species-based catalysts can be traced back to 1995, when Maschmeyer et al.31 grafted the titanium (Ti) metallocene complex onto the inner wall of mesoporous silica MCM-41 and demonstrated the catalyst to be highly selective towards epoxidation of cyclohexene. In 1999, Iwasawa and co-workers obtained a Pt/MgO catalyst in which Pt clusters were coordinated onto the MgO surface by Pt–O bonds and showed better catalytic activity for propane combustion than the isolated Pt NPs.32 Later on, Heiz et al.33 in 2001 fabricated nano-assembled Pdn (1 ≤ n ≤ 30) clusters over MgO(100) by the soft-landing method. The cluster size of Pd was precisely controlled and it was found that the number of Pd atoms in the cluster highly affects the selectivity of acetylene polymerization. Further in 2003, Stephanopoulos et al.34 discovered that atomically dispersed nonmetallic Au and Pt sites on CeO2 participated in the water–gas shift (WGS) reaction, whereas the respective NPs were unable to carry out the same reaction. In 2005, Xu et al.35 synthesized a Au/ZrO2 catalyst that was demonstrated to be highly selective towards 1,3-butadiene hydrogenation, and various spectroscopic and chemisorption methods were used to analyze the bonding. However, various advanced techniques were introduced afterward to deduce the monodispersed metal ions deposited over the supports. Aberration-corrected scanning transmission electron microscopy (AC-STEM), X-ray absorption spectroscopy (XAS) and extended X-ray absorption fine structure (EXAFS) were developed as the main characterization methods for determining the SACs. In this regard, Lee et al.36 in 2007 fabricated Pd-decorated Al2O3 single site catalysts and characterized them with combined AC-STEM and XAS to show the existence of Pd single sites. The well-dispersed single Pd atoms were confirmed from the AC-STEM images and the formation of the Pd–O bond rather than the Pd–Pd bond was confirmed by EXAFS. The Pd/Al2O3 single site catalysts were employed for alcohol oxidation and exhibited better catalytic performance than the corresponding nanocrystals. Further, in 2011, Zhang and co-workers for the first time introduced the term SACs by synthesizing robust Pt/FeOx SACs using a co-precipitation strategy and employed the catalyst for selective oxidation of CO.37 This study performed combined AC-STEM, XAS and Fourier transform infrared (FTIR) spectroscopy to explicitly validate the Pt atomic dispersion over FeOx. Additionally, to reveal the Pt single atom stabilization over FeOx, the authors conducted density functional theory (DFT) calculations and explained the exceptional CO oxidation activity of Pt/FeOx SACs from DFT calculations. Therefore, this report proved to be a pioneering study in the field of single atom catalysis and since then, there has been an upswing in research on single atom catalysis for a variety of applications by the global scientific community. Further, a discussion of these characterization methods and their utilization has been made in the synthesis section.1.2. Scope of the review
Within a decade, SACs have gained tremendous attention in heterogeneous catalysis, leading to numerous fascinating discoveries. To date, several excellent review articles have been reported in the literature on single atom catalysis, but most of them are usually categorized on the basis of different synthesis methods like pyrolysis route, wet deposition, gas phase techniques, electrochemical methods, etc. for various applications.4,21,24,38–41 Wang et al.24 reported a perspective about the bonding in SACs and how it affects the catalytic performance in various industrially relevant reactions. Further, Lang and co-workers reported the synthesis methods and application of metal oxide based SACs for reactions like water gas shift (WGS), oxidation, reduction, etc.38 Moreover, Kaiser et al.4 provided a compositional encyclopedia on SACs ranging from elemental diversity, milestones of the field, and recent advances in technology. In contrast to the published literature in this field, this review focuses on the element-based synthesis of SACs which have therefore been categorized as metal and non-metal based SACs (MSACs and NOMSACs) wherein the metal-based SACs are further split into noble metal and non-noble metal based SACs (NMSACs and NNMSACs). As the research on non-metal-based SACs is very limited and rarely discussed in the existing review articles, their elaborate discussion in this review article broadens the scope of the review. Furthermore, various advanced characterization techniques used to examine the successful synthesis of the SACs are explained in the synthesis section. Afterward, the excellent catalytic potential of the different SACs towards thermally driven organic transformation reactions, such as hydrogenation reactions, hydroformylation reactions, coupling reactions, bond cleavage reactions, and oxidation reactions, is solely discussed in detail. Lastly, this review provides a comprehensive summary and primary loopholes in the existing research on SACs especially focusing on the synthesis and large-scale implementation of this research field along with potential solutions that can surely assist the researchers working in this field to fill the voids in the existing state of research.It is a well-known fact that the development of chemical industries primarily relies on advancements in catalysis, which is responsible for almost 90% of global consumer product manufacturing. As SACs are promising heterogeneous catalysts in various fields, the significance of this detailed review can be understood by the fact that heterogeneous catalysis contributes 80% to the catalytic process, followed by homogeneous catalysis (15%) and bio-catalysis (5%), as shown in Fig. 2(a).4 Further, this review presents a wide scope and timely discussion on single atom-based catalysts for their utility in different catalysis fields as this research domain has shown excellent growth between 2014 and 2024 as depicted in Fig. 2(b). Further, a significant proportion of the research work on SACs has used different organic transformation processes like hydrogenation, oxidation, hydroformylation, water gas shift, etc., as keywords, which further depict the timeliness of the review article (Fig. 2(c)). Therefore, this review article provides deep insight into the state-of-the-art current research on SACs and will be helpful for both industrial and academic researchers working in the field of organic transformation reactions using SACs to develop various pioneering reaction protocols in the near future.
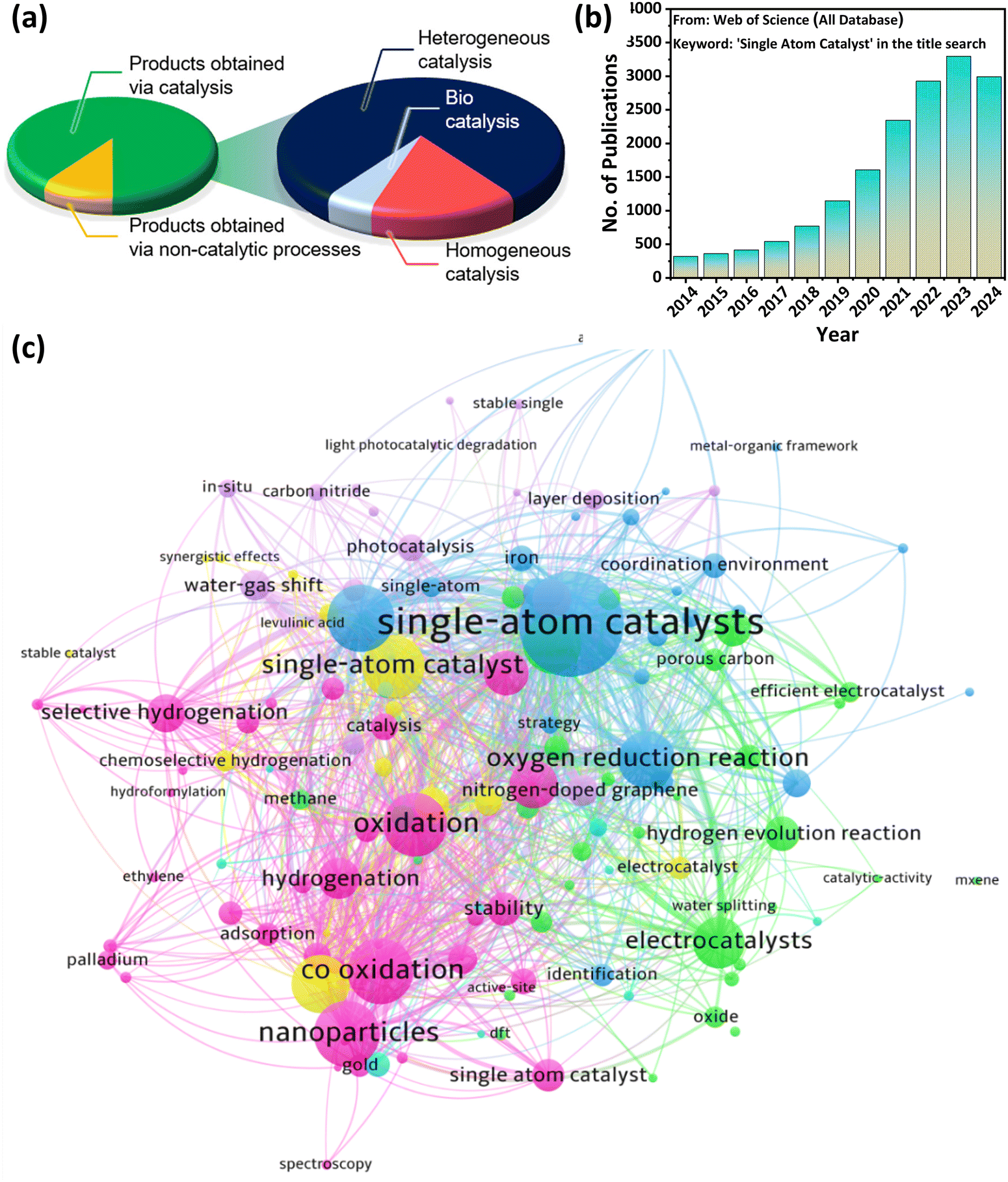 | ||
| Fig. 2 (a) Pie chart showing the importance of catalysis and distribution of catalyst type employed to obtain products. Reproduced with permission.4 Copyright 2020, American Chemical Society. (b) Number of publications per year on SACs from 2014 to 2024 (data obtained on Oct. 26, 2024, from Clarivate Analytics) and (c) network data visualization of recent publications on single atom catalysis. The most frequently used keywords in works published from 2014 to 2024 using the search terms “single atom catalyst” were visualized by VOSviewer. | ||
2. Material synthesis
One of the most significant areas of research is now being focused on investigating and advancing synthetic techniques for SACs. The isolated single atoms readily transfer and aggregate into particles due to their high surface energy. Consequently, it is challenging to fabricate SACs and sustain metal species’ atomic dispersion under practical synthesis and reaction conditions. Constant efforts are being made to overcome this challenge of obtaining highly dispersed single atoms. In this section, a detailed elaboration of current advances in the synthesis of SACs based on noble metals, non-noble metals and non-metals has been made.2.1. Metal based SAC synthesis (MSACs)
Zhang et al.37 synthesized Pt atoms supported on FeOx using a facile coprecipitation method using a Pt/Fe ratio of 1/1430 and 0.17 wt% of Pt loading. The synthesis was performed with a highly tuned coprecipitation temperature and pH to ensure the anchoring and isolation of Pt atoms onto the defects present on the surface of FeOx. It was reported that a very low wt% of Pt loading was used to ensure the isolation of Pt atoms and prevent them from aggregating. A similar catalyst with 2.5 wt% of Pt loading was also synthesized to examine the properties of both catalysts.37 It was observed from the High-Angle Annular Dark Field (HAADF) images that in the case of 0.17 wt% Pt1–FeOx, individual Pt atoms were seen to be uniformly distributed on the support (Fig. 3(a)). The sub ångström-resolution HAADF image shown in Fig. 3(b) further shows that the Pt atoms occupy the exact positions of Fe atoms and the density of Pt single atoms was calculated to be 0.07 Pt nm−2 which closely resembled the actual loading. Contrastingly, Pt single atoms, Pt rafts (2D), and Pt clusters (3D) were observed in the HDAAF images of the 2.5 wt% Pt–FeOx catalyst, which suggested the importance of controlling the loading to maintain single atom dispersion.37 Shan et al.44 reported an excellent study of integrating interactive Pt single atoms into the lattice of various metal oxides (Co3O4, Mn5O8, NiO, Fe2O3), and structural diversification was critically elucidated (Fig. 3(c)). The catalyst synthesis was carried out using metal–organic frameworks (MOFs) of different metals, which included Co-ZIF-67, Mn-ZIF-8, Ni-UMOFNs, and MIL-100 (Fe). These MOFs were subjected to an ion exchange process using K2PtCl6 as a Pt source under stirring conditions for 3 h, followed by washing and drying. Further, the obtained precipitate was pyrolyzed at 300 °C in air for 4 h to obtain all the Pt-integrated metal oxide catalysts.44 The synchrotron-based Pt L3-edge EXAFS spectra were used to describe the local coordination structure of the Pt sites. In the case of Pt–Co3O4, two peaks at 1.6 and 2.7 Å were observed, which can be attributed to the Pt–O coordination and the co-existence of Pt–Co and Pt–Pt interactions, respectively. The results for PtSA–Co3O4 showed the lack of second shell Pt–Pt co-ordination, which proved the isolation of the Pt single atoms. Further, HDAAF-STEM was used to observe the incorporation of Pt into the lattice of various metal oxides. The HDAAF-STEM analysis proved the topology of Pt–Co3O4, Pt–Mn5O8, Pt–NiO and Pt–Fe2O3 to be cristobalite (crs), face-centered cubic-hexagonal-primitive cubic (fcu-hex-pcu), face centered cubic (fcu) and body centered cubic (bcu-x), respectively, as shown in Fig. 3(d–g).44
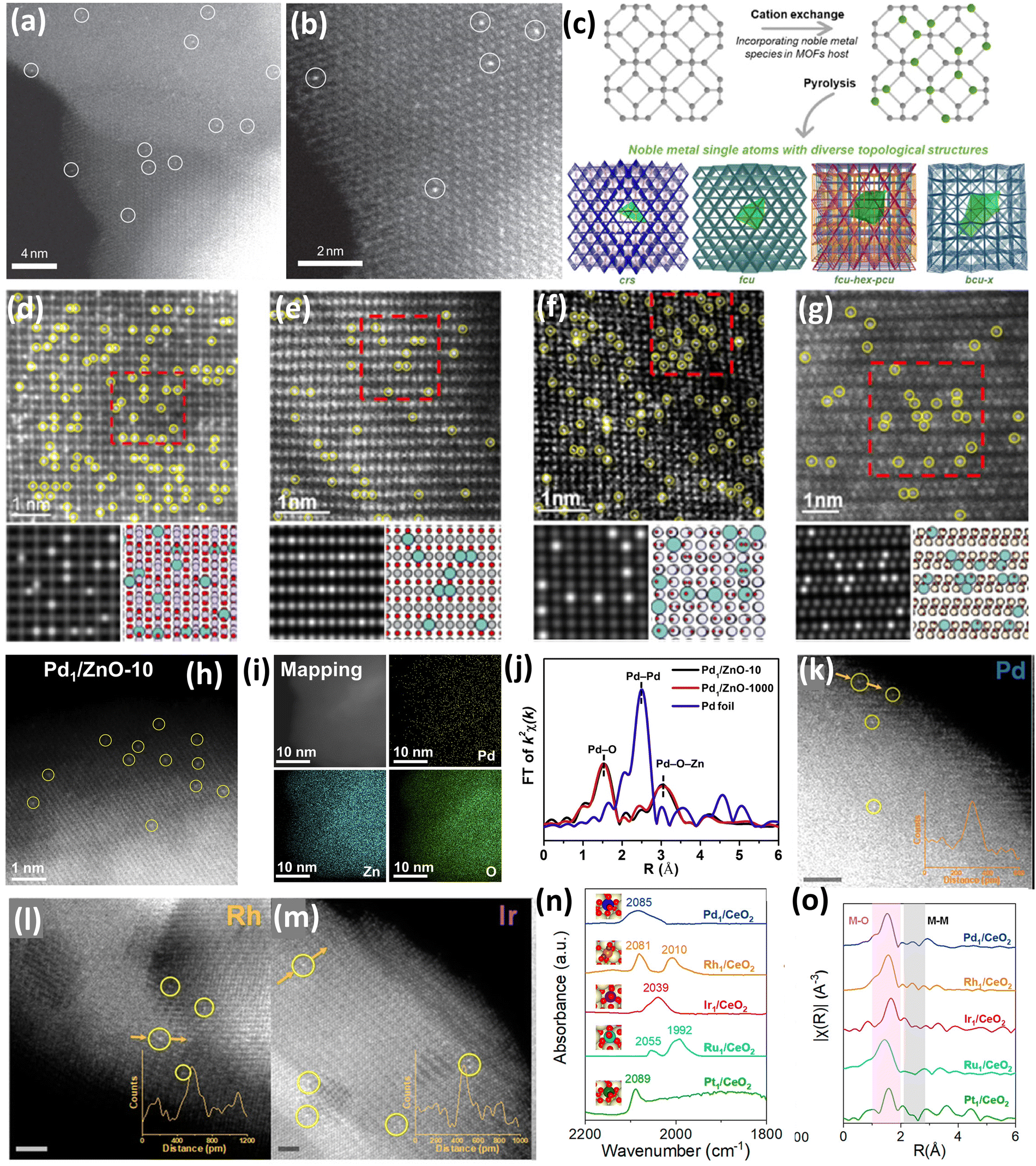 | ||
| Fig. 3 (a) HDAAF image of Pt1–FeOx and (b) subångström-resolution HAADF image of Pt1–FeOx. Reproduced with permission.37 Copyright 2011, Nature Publishers. (c) Schematic representation of integrating Pt into the lattice of metal oxides and HDAAF-STEM images of (d) Pt–Co3O4, (e) Pt–Mn5O8, (f) Pt–NiO, and (g) Pt–Fe2O3. Reproduced with permission.44 Copyright 2022, American Chemical Society. (h) HDAAF-STEM image of Pd1/ZnO-10, (i) elemental mapping of Pd1/ZnO-10, and (j) EXAFS spectra of Pd foil, Pd1/ZnO-10, and Pd1/ZnO-1000. Reproduced with permission.48 Copyright 2020, Cell Press Publishers. HDAAF-STEM images of (k) Pd–CeO2, (l) Rh–CeO2, and (m) Ir–CeO2, and (n) DRIFTS spectra and (o) EXAFS of noble metal supported CeO2 catalysts. Reproduced with permission.49 Copyright 2022, American Chemical Society. | ||
Further, He et al. reported the large-scale synthesis of Pd supported on a zinc oxide (ZnO) support using a facile mechanochemical method involving ball milling followed by a calcination process of the commercially available zinc acetylacetonate (Zn(acac)2) and palladium acetylacetonate (Pd(acac)2).48 Initially, Pd(acac)2 and Zn(acac)2 were mixed homogeneously using ball milling with a Pd(acac)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn(acac)2 ratio of 1:400 for 10 h at 400 rpm. It is important to remember that using a common ligand, such as acetylacetonate (acac), guarantees high compatibility between Pd(acac)2 and Zn(acac)2, which promotes Pd(acac)2 diffusion into the bulk Zn(acac)2 during the ensuing milling operation. In the subsequent step, the mixture was calcinated at 400 °C for 2 h, transforming the metal acetylacetonate structure to corresponding metal oxides to yield Pd supported ZnO (Pd1–ZnO). Further, the Pd loading % calculated from inductively coupled plasma optical emission spectroscopy (ICP-OES) was 0.25 wt%, closely resembling the theoretically calculated value (0.29 wt%).48 The HDAAF-STEM images (Fig. 3(h)) revealed the presence of individual Pd atoms over the ZnO surface whereas the high homogeneity of Pd atoms over the ZnO surface was confirmed by the energy dispersive X-ray (EDX) analysis as shown in Fig. 3(i). Furthermore, the dispersion state of Pd species was analyzed by EXAFS (Fig. 3(j)), and two peaks at 1.6 and 3.0 Å were observed, which corresponded to the Pd–O and Pd–O–Zn contributions, respectively. The absence of Pd–Pd contribution was confirmed by the absence of any peak at 2.5 Å, which suggested the dominant presence of Pd atoms dispersed homogeneously over the ZnO surface with very low aggregation. Further, this strategy exhibited an excellent scaling-up effect and the catalyst can be synthesized up to 1000 g without any impact on the properties, which is highly beneficial as the primary issue with SACs is the bulk synthesis.48
:Zn(acac)2 ratio of 1:400 for 10 h at 400 rpm. It is important to remember that using a common ligand, such as acetylacetonate (acac), guarantees high compatibility between Pd(acac)2 and Zn(acac)2, which promotes Pd(acac)2 diffusion into the bulk Zn(acac)2 during the ensuing milling operation. In the subsequent step, the mixture was calcinated at 400 °C for 2 h, transforming the metal acetylacetonate structure to corresponding metal oxides to yield Pd supported ZnO (Pd1–ZnO). Further, the Pd loading % calculated from inductively coupled plasma optical emission spectroscopy (ICP-OES) was 0.25 wt%, closely resembling the theoretically calculated value (0.29 wt%).48 The HDAAF-STEM images (Fig. 3(h)) revealed the presence of individual Pd atoms over the ZnO surface whereas the high homogeneity of Pd atoms over the ZnO surface was confirmed by the energy dispersive X-ray (EDX) analysis as shown in Fig. 3(i). Furthermore, the dispersion state of Pd species was analyzed by EXAFS (Fig. 3(j)), and two peaks at 1.6 and 3.0 Å were observed, which corresponded to the Pd–O and Pd–O–Zn contributions, respectively. The absence of Pd–Pd contribution was confirmed by the absence of any peak at 2.5 Å, which suggested the dominant presence of Pd atoms dispersed homogeneously over the ZnO surface with very low aggregation. Further, this strategy exhibited an excellent scaling-up effect and the catalyst can be synthesized up to 1000 g without any impact on the properties, which is highly beneficial as the primary issue with SACs is the bulk synthesis.48
An excellent strategy was reported by Pu et al.49 for the synthesis of various noble metals’ (Pd, Ir, Rh, Ru, and Pt) single atoms supported on nanoceria (CeO2) for the combustion of methane by synergistically employing the single atom active site and the oxygen vacancies (OV) available due to the Ce3+/Ce4+ redox cycle. All the catalysts were synthesized by a facile hydrothermal method followed by calcination with very slight variations.49 The HDAAF-STEM analysis confirmed that the predominantly exposed facet of CeO2 is (111), which was used in density functional theory (DFT) studies (Fig. 3(k–m)). Moreover, in the in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of CO adsorption over dehydrated catalysts, only the linear adsorption peaks for CO on atomic noble metals were observed, as depicted in Fig. 3(n). The EXAFS studies (Fig. 3(o)) revealed the presence of M–O contributions in all the catalysts, with the absence of M–M contributions suggesting the presence of single atoms of noble metals over CeO2.49
Wu et al.50 successfully synthesized a Pt single atom catalyst supported on an organometal halide perovskite, FAPbBr3−xIx (FA = CH(NH2)2) due to its excellent photocatalytic properties and it was applied for photocatalytic hydrogen evolution via hydrohalic acid splitting. The catalyst was synthesized by a self-adsorption and photo-reduction process (Fig. 4(a)). Initially, FAPbBr3 was formed by a facile coprecipitation method followed by ion exchange with iodide ions in HBr/HI mixed haloid acid under light irradiation to speed up the reaction to form the FAPbBr3−xIx perovskite. Following these steps, the Pt immobilization was carried out by adding H2PtCl6·6H2O in a HBr/HI solution saturated with the perovskite solute and suspended with FAPbBr3−xIx particles under rapid stirring.50 After light irradiation, Pt gets reduced and Pt atoms get uniformly distributed by substituting the formamidinium groups (FA). The HDAAF-STEM studies confirmed the presence of Pt atoms dispersed over the perovskite support (Fig. 4(b)), which tend to aggregate when the Pt loading is increased (Fig. 4(c)). Further, the Pt L3-edge X-ray absorption near-edge structure (XANES) spectrum shown in Fig. 4(d) revealed that the white line intensity of 1.8-Pt/FAPbBr3−xIx which indicates the oxidation state (OS) of Pt is in between the intensities of Pt foil and H2PtCl6·6H2O which confirms that the Pt carries some positive charge. Also, EXAFS studies (Fig. 4(e)) showed no contribution of Pt–Pt, which indicates the non-existence of Pt particles or clusters. The Pt atoms are present at the surface FA positions by coordinating with Br and I, which was found by DFT studies as depicted in Fig. 4(f).50
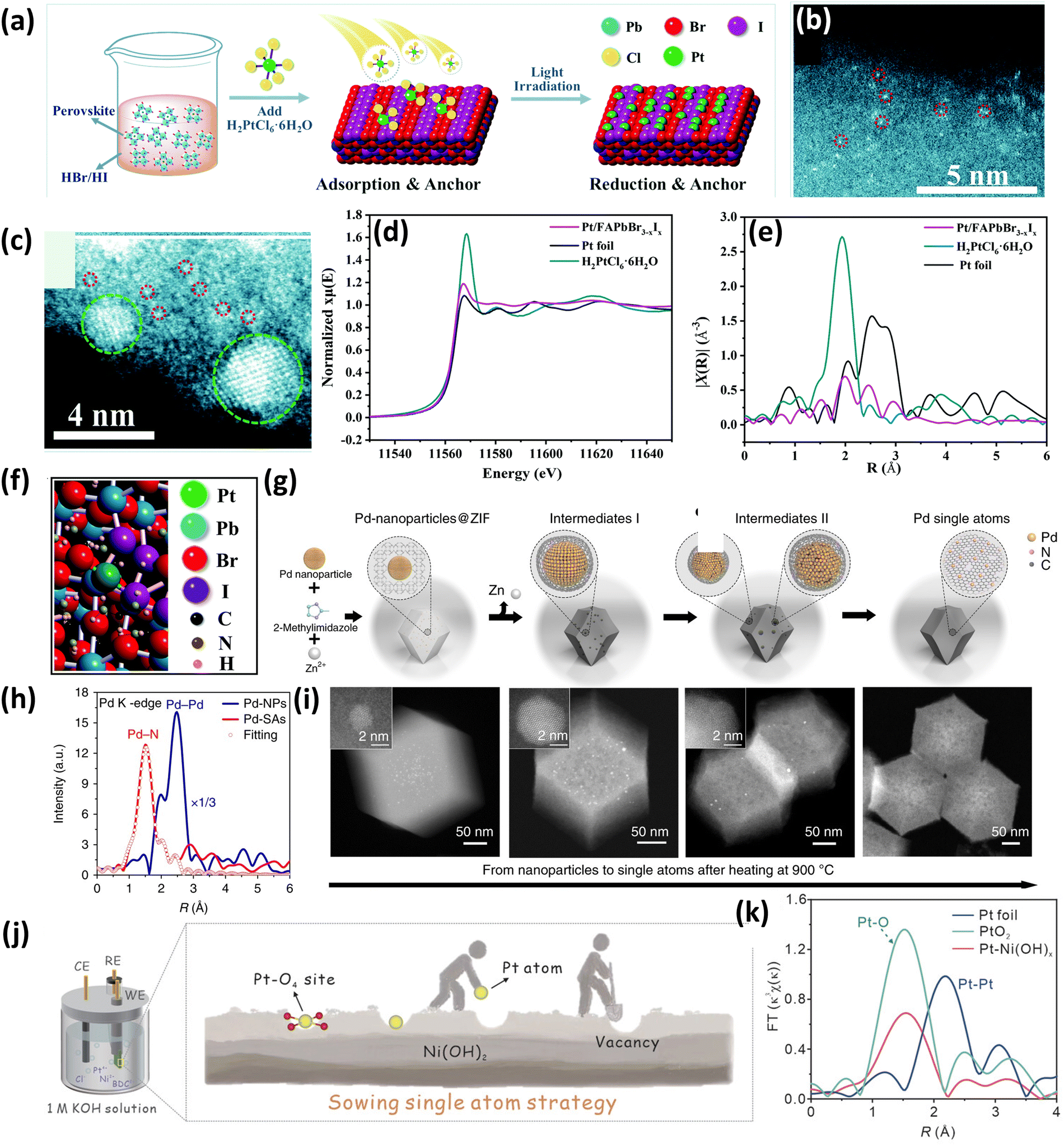 | ||
| Fig. 4 (a) Synthesis scheme of the Pt/FAPbBr3−xIx SAC, HDAAF-STEM image of (b) 0.7 wt% Pt/FAPbBr3−xIx and (c) 2.1 wt% Pt/FAPbBr3−xIx, (d) normalized XANES spectra at the Pt L3-edge of Pt foil, H2PtCl6 and 1.8-Pt/FAPbBr3−xIx, (e) the k3-weighted FT spectra from EXAFS, and (f) simulated structure of Pt/FAPbBr3−xIx. Reproduced with permission.50 Copyright 2022, Royal Society of Chemistry. (g) Synthesis scheme of Pd–CN, (h) FT of k3-weighted Pd K-edge EXAFS for Pd-NPs and Pd-SAs, and (i) HDAAF-STEM images of the transformation of NPs to SAs. Reproduced with permission.51 Copyright 2018, Nature Publishers. (j) Synthesis scheme of Pt–Ni(OH)x and (k) FT-EXAFS spectra of Pt foil, PtO2, and Pt–Ni(OH)x. Reproduced with permission.52 Copyright 2023, Wiley VCH. | ||
It is well known that the SACs and ultrafine clusters tend to aggregate at elevated temperatures but an unexpected phenomenon was reported by Wei et al.51 which involved the transformation of noble metal NPs into single atoms above 900 °C under an inert atmosphere (Fig. 4(g)). The authors utilized nitrogen-doped carbon (CN), which was derived from the zeolite imidazolate framework (ZIF-8), as a support material to study the transformation of NPs to single atoms. Initially, the ZIF-8 crystals were grown around Pd NPs by mixing zinc nitrate, Pd NPs, and 2 methyl imidazole to form the Pd@ZIF-8 composite, which was then heated at 900 °C in an inert atmosphere for 3 h. The aberration-corrected scanning electron microscopy (AC-SEM) confirmed the existence of Pd atoms dispersed uniformly over the CN matrix. Also, the EXAFS studies revealed the absence of Pd–Pd contributions, which confirmed the presence of Pd single atoms on the surface of the support, as shown in Fig. 4(h). Furthermore, from HDAAF-STEM images it was observed that the particles aggregate during the first 30 min, and after 1.5 h, the crystalline Pd converts into an amorphous phase. All the remaining NPs were digested after 3 h, and the HDAAF-STEM confirmed the absence of Pd NPs or clusters (Fig. 4(i)). In this reported phenomenon, sintering does take place, but the larger particles intensively collide with each other as well as the substrate to eventually become smaller and smaller into single atoms.51
In 2023, a versatile sowing strategy for hyper-low Pt loading (0.17 wt%) was reported by Li and coworkers using vacancies as sites for Pt loading for hydrogen evolution reaction (Fig. 4(j)).52 The catalyst synthesis was performed by an electrochemical reduction method in two steps: (1) derivatization of the Ni-BDC MOF (BDC, benzene dicarboxylic acid) into Ni(OH)x followed by, (2) introduction of Pt atoms. The Ni-BDC MOF nanosheets were grown vertically on the acid-treated nickel foam followed by etching in basic medium, and under the driving force of an electric field, the BDC organic linker was slowly removed from the nanosheets. Eventually, Ni(OH)x is formed once the nanosheets containing the metal cations get in contact with the hydroxyl anions in the alkaline electrolyte.52 In the BDC removal process, which is a very violent reaction, it was proposed that leaching of a certain amount of Ni2+ cations is observed which resulted in Ni2+ vacancies. Further, these Ni2+ vacancies act as potential holes or sites for the Pt single atoms when the platinum source is introduced to the electrolytic system. On recording the photoluminescence spectra of Ni2+ vacancies, it was observed that Ni(OH)x exhibited higher intensity as compared to Pt–Ni(OH)x which suggested that Pt atoms occupy the vacant sites in Pt–Ni(OH)x. Further, the EXAFS and DFT studies confirmed that the Pt atom is coordinatively unsaturated with four oxygen (Pt–O4) and the absence of Pt–Pt contributions in the EXAFS spectra confirmed the successful incorporation of Pt atoms in the Ni2+ vacant sites (Fig. 4(k)).52 Another study focusing on the design of SACs for hydrogen evolution was reported by Zhu et al.53 which included the synthesis of Pt single atoms supported on the edge of 2D layered Ni(OH)2 denoted as Pt–Ni(OH)2-E using the in situ electrodeposition technique. It was observed that Pt–Ni(OH)2-E has a higher intrinsic catalytic activity and a better electron affinity than Pt SACs anchored on the basal plane of Ni(OH)2 (Pt–Ni(OH)2-BP). This promotes strong adsorption and quick dissociation toward water molecules, resulting in excellent enhancement in hydrogen evolution studies at relatively low overpotentials.53
Further, Li et al.54 developed a movable printing method for the synthesis of precious metal SACs (Pd, Pt, Rh, Ir, and Ru) (PMSACs) supported on nitrogen-doped carbon nitride (CN) for hydrogen evolution reaction and hydrogen oxidation reaction (Fig. 5(a)). Initially, the noble metal chloride (0.03 mmol) was added to HCl solution (4.80 wt%) containing melamine (9 g) followed by vigorous stirring and sonication. Afterwards, the solvent was evaporated, and the precipitate was heated at 550 °C for 2 h to obtain the first anchored noble metals on C3N4 (C3N4-PM). These obtained materials (1.5 g) were added to a solution of tris buffer (50 mL) followed by ultrasound treatment. Afterward, dopamine hydrochloride was added to ensure the formation of polydopamine-coated C3N4-PM. Further, the carbonization process was carried out via pyrolysis at 750 °C for 2 h to form nitrogen-rich CN which acts as a support for the second anchoring of the PM single atoms (SA-PM/CNs).54 Therefore, utilizing this facile synthesis technique, highly dispersed single atoms were obtained on the nitrogen doped CN support. The nitrogen-rich environment enhances the anchoring of single atoms. The HDAAF-STEM images of SA-PM/CNs shown in Fig. 5(b–f) revealed the presence of bright spots which confirms the presence of single atoms on the CN support. The FT-EXAFS of SA-Pt/CN revealed a peak at 1.56 Å attributed to the Pt–N contribution which confirms the interaction between Pt and N from the N-rich CN support with four nitrogen atoms surrounding one Pt atom (Fig. 5(g)). Also, the Ru K-edge EXAFS revealed a peak for the Ru–N contribution and absence of Ru–Ru contributions as shown in Fig. 5(h). Similar observations were made for other catalysts also and it was observed that the Ru and Pt-based catalysts were highly active in hydrogen reactions as compared to their NP counterparts.54
 | ||
| Fig. 5 (a) Schematic synthesis scheme of PMSACs supported on nitrogen-doped CN, (b)–(f) HDAAF-STEM images of SA-Pd/CN, SA-Pt/CN, SA-Rh/CN, SA-Ir/CN, and SA-Ru/CN, respectively, (g) k3 weighted FT-EXAFS spectra of SA-Pt/CN and Pt foil, and (h) k3 weighted FT-EXAFS spectra of SA-Ru/CN, Ru foil, and RuO2. Reproduced with permission.54 Copyright 2023, Wiley VCH. (i) EDX image of Pt in VR-UCN@Pt and (j) partly enlarged XANES N K-edge for V-UCN. Reproduced with permission.55 Copyright 2023, American Chemical Society. (k) TEM image of Pt/WO3−x-R and (l) R-space spectra from FT-EXAFS. Reproduced with permission.56 Copyright 2023, Elsevier Publishers. | ||
Zhai et al.55 employed electronic regulation in Pt SACs using local coordination state adjustments with nitrogen vacancy-rich C3N4 as a support for increased photocatalytic hydrogen evolution performance. Initially, three different types of C3N4 (VR-UCN, VM-UCN, and VL-UCN) with varying nitrogen vacancies were prepared via thermal condensation followed by exfoliation using 3-amino-1,2,4-triazole as a vacancy regulator. The three N vacancy C3N4, namely. VR-UCN, VM-UCN, and VL-UCN, exhibited 12.4, 7.9, and 2.0% missing N atoms, respectively. Further, Pt loading on the supports was carried out by a liquid phase reaction followed by annealing under an Ar atmosphere. It was observed from the HDAAF-STEM and EDX mapping (Fig. 5(i)) images that the Pt atoms are uniformly dispersed on the UCN support, but the sole role of N vacancies in trapping the Pt atoms was proved false by the ICP analysis as the VR-UCN, VM-UCN, and VL-UCN catalysts exhibited 2.32, 2.29, and 2.34 wt% Pt loading.55 As VR-UCN contains an excess amount of N vacancies, the Pt atoms get anchored only on the N vacant sites, whereas in the case of VM-UCN and VL-UCN, a second nitrogen adsorption site is provided by N2C apart from the nitrogen vacant sites due to the large surface area of vacancy rich UCN. This was confirmed by the XANES spectra (Fig. 5(j)), which revealed that the N2C peak intensity increased progressively in the case of VM-UCN and VL-UCN as compared to VR-UCN signifying that the N vacancy concentration is slashed, and N2C sites are the partially reconstructed region. Therefore, the authors synthesized the V-UCN@Pt SAC with two different types of configurations, Pt–C2N and Pt–N2, which exhibited excellent catalytic activity towards photocatalytic hydrogen evolution as Pt–C2N. This was due to the short carrier delivery channel for Pt–C2N and decreased H* desorption energy, which makes it easy for H+ to couple with the electrons.55
Further, Li et al.56 designed and developed Au single atoms trapped in OV-rich WO3−x for the demethoxylation of guaiacols derived from lignin. The Pt–WO3−x catalyst was synthesized by an in situ redox method. Initially, WO3−x was prepared by an easy hydrothermal method using tungstic acid hydroxylamine hydrochloride, hexadecyltrimethylammonium bromide and thiourea as precursors. The as-synthesized WO3−x was anchored with Au using HAuCl4 and the solution was conditioned at 60 °C for 5 h followed by reduction with hydrogen to increase the OV (Pt/WO3−x-R).56 The Pt/WO3−x-R catalyst showed nanorod like morphology as revealed by the HRTEM analysis as shown in Fig. 5(k) whereas the presence of Au atoms dispersed uniformly was revealed by HDAAF-STEM images. Further, the EXAFS studies of the catalyst revealed no contribution due to the Au–Cl or Au–Au interactions, which confirmed the effective reduction of Au3+ and the presence of atomic Au on the WO3−x support (Fig. 5(l)). The XANES studies proved that the intensity of Au/WO3−x-R was found to be between that of Au3+ and Au foil. This suggests that the Au atoms carried partial negative charges and that the Au–O coordination in Au/WO3−x-R differed from that in Au2O3.56
Over the past few years, a significant focus has also been given to the advancements in the synthesis of porous organic polymer (POP) supported SACs for various reactions.57–62 This is primarily due to the excellent scope of design and tunability over porosity, composition, and morphology in POP supports which helps in the controlled fabrication of SACs at the molecular level. Catalytically active metal centers found in POPs provide easy control over the atomic arrangement. A path toward precise control of the local coordination environment and electrical characteristics of single atom active centers is made possible by the porous material's ability to fine-tune the chelating and steric capabilities of the ligand.63,64 In this context, an interesting synthesis approach was reported by He et al.65 for the synthesis SACs through the precursor dilution strategy for various hydrogenation reactions. Specifically, metal-chelated tetraphenyl porphyrin (TPP) was used as a metal source and was copolymerized with excess free TPP as a diluent followed by pyrolysis. The dilution prevents the metal atoms from aggregating during the subsequent high-temperature pyrolysis by increasing the mean distance between them scattered across the as-prepared polymer matrix to obtain different M–N–C SACs. In the Pt1–N–C catalyst, the TEM, STEM, and AC-HDAAF-STEM microscopic analysis revealed high dispersion of Pt atoms and no Pt NPs. Further, EXAFS studies showed the presence of Pt–N and Pt–N–C contributions attributed to the peaks at 1.7 and 2.5Å, respectively. The Pt coordination number with N was 3.4 which confirmed that one Pt atom is coordinated to three or four N atoms. This precursor dilution strategy was also proved to be highly versatile with various other metals providing high atomic dispersion.65 Further, Shen et al.66 reported the efficient synthesis of porous organometallic polymers (POMPs) as single site catalysts with varying porosity and Ir metal content via a direct knitting strategy for the N-formylation of amines. In the typical synthesis process, the bis-N-heterocyclic carbene (NHC)–Ir complex was used as a precursor to synthesize various POMPs with formaldehyde dimethyl acetal (FDA) and benzene in the presence of FeCl3. The HDAAF-STEM with EDX analysis revealed that Ir was present as single sites and not as NPs which proved to be beneficial in the N-formylation of amines with CO2 and H2. Therefore, this report provided a promising avenue to regulate the porosity and adsorption capacity of POMP based single site catalysts in the N-formylation reaction.66
Wang and co-workers reported a PPH3 built-in POP with anchored Pd single atoms for the telomerization of 1,3 butadiene to synthesize 1-methoxy-2,7-octadiene (1-MOD).67 Instead of monodentate phosphine and NHC ligands, the PPH3 ligand was chosen owing to its structural simplicity and easy availability. The PPH3 built-in POP served as a solid ligand to bond with the single metallic sites to design a specifically defined ligated metal environment which helps in enhancing the ligand controlled organic transformation. In a specific synthesis experiment, the POP was mixed with Pd(OAc)2 in THF solvent and this mixture was stirred at room temperature under a N2 atmosphere for 24 h. The morphological studies confirmed the uniform dispersion of Pd atoms and absence of metal atom aggregation with a Pd loading of 0.79 wt% in Pd1@POP. The XANES Pd K-edge position of Pd1@POP was found close to Pd(OAc)2, confirming the +2 valence state of Pd which was also consistent with XPS observations. Also, the FT-EXAFS spectra of Pd1@POP revealed the contribution of Pd–O at 1.60Å and no Pt![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Pt contributions were observed which further showed uniform Pd atom distribution.67 Therefore, these recent reports on the synthesis of POP supported SACs provide an excellent catalyst development approach with high scope of tunability for specific applications which will surely be investigated to a further extent in the future research on SACs.
Pt contributions were observed which further showed uniform Pd atom distribution.67 Therefore, these recent reports on the synthesis of POP supported SACs provide an excellent catalyst development approach with high scope of tunability for specific applications which will surely be investigated to a further extent in the future research on SACs.
In conclusion, the discussed latest literature presents various facile synthetic methods from lab scale to bulk production for the effective and efficient synthesis of noble metal-based SACs with unprecedented catalytic activity. The cost of noble metals is relatively high and does not seem to be reducing in the near future, but the catalytic activity of these noble metal-based SACs is unmatched. Therefore, the development of cost-effective and highly promising synthetic strategies with low energy consumption should be focused on, lowering the overall cost of the process. These efforts can surely lead to the large scale industrialization of NMSACs which will be highly advantageous for the development of the chemical industry.
Recently, Zhang et al.69 reported the synthesis of Fe SACs (Fe–N4) with different types of pores (micropores, mesopores, and hierarchical pores), as shown in Fig. 6(a), and utilized the synthesized catalysts for oxygen reduction reactions (ORR). It was observed that the metal–nitrogen–carbon catalysts having hierarchically distributed pores offer efficient geometry for ORR. Nevertheless, the catalytic performance dictated by distinct and interdependent sites resulting from structural heterogeneity is especially difficult to comprehend. Therefore, for the synthesis of the Fe SAs-HP catalyst, coal tar pitch (CTP), which contains polycyclic aromatic hydrocarbons (PAH), was chosen because it is a high-quality carbon source used widely in energy applications. Due to the strong π–π interactions between the heme chloride macrocycle and PAH, heme chloride was carefully chosen as the Fe source. This allowed the Fe atoms to be distributed evenly into carbon networks and prevented the agglomeration of unwanted metallic iron NPs.69 Afterwards, the carbon substrate was allowed to react with ZnO, sacrificial templates and NH3 atmosphere at high temperature to form an adequate amount of mesopores and micropores to immobilize the Fe atoms effectively. In the experimental synthesis of Fe SAs-HP, an encapsulation–pyrolysis–evaporation synthesis path was followed. Initially, solution A was prepared by mixing CTP, melamine, and ZnO in dimethyl formamide (DMF) and solution B was prepared by mixing heme chloride in DMF solvent. Furthermore, solution B was dropwise added to solution A followed by stirring for 24 h and heating at 920 °C for 2 h in an NH3 atmosphere. The aberration-corrected HDAAF-STEM analysis showed the separated dispersion character of the Fe atoms which was indicated by their ability to penetrate the carbon matrix without aggregating (Fig. 6(b)). It was evident from the enlarged AC-STEM images (Fig. 6(c)) of the mesopores that Fe pairs resulting from structural heterogeneity were present.69
 | ||
| Fig. 6 (a) Synthesis scheme of Fe SACs with a different type of pores, (b) HDAAF-STEM image of Fe SAs-HP, (c) zoomed AC-STEM image of Fe Sas-HP. Reproduced with permission.69 Copyright 2024, Nature Publishers. (d) Synthesis scheme of Fe-N4SP SAC, (e) HDAAF-STEM of Fe–N4SP/NPS-HC. Reproduced with permission.70 Copyright 2024, Royal Society of Chemistry. (f) Ni@TiO2 SAC for enhanced H2 storage in MgH2, (g) Ni K-edge XANES spectra, and (h) Ni K-edge EXAFS of Ni0.034@TiO2 SAC. Reproduced with permission.71 Copyright 2024, American Chemical Society. | ||
Liu et al.70 reported the synthesis of the Fe–N4SP SAC via the multi-shell synergistic effect to boost the ORR of rechargeable zinc–air battery cathodes. The suggested structure could disrupt the symmetric structure of Fe–N4 sites by reorganizing electrons and enhancing spin polarization, which was effectively demonstrated by theoretical computations and experimental validations. In the typical synthesis method shown in Fig. 6(d), ZIF-8/FePc@PZS was initially developed by mixing iron phthalocyanine (FePc) into the ZIF-8 MOF and then coating its exterior with a homogeneous layer of the phosphonitrile polymer (PZS). This technique resulted in the encapsulation of FePc into the ZIF-8 cavity without altering the crystal structure. Further, Fe–N4SP/NPS-HC was later synthesized by acid etching and one-step pyrolysis of ZIF-8/FePc@PZS, where sufficient P and S elements in PZS were synchronously doped into the carbon shell, and doped N acted as the effective anchoring sites for the Fe atom.70 To alter the electrical structure of Fe–N4, the S element was also directly and axially linked to the Fe atom. The HRTEM and HDAAF-STEM images revealed the presence of uniformly distributed Fe single atoms throughout the hollow carbon shell with 1.33% of Fe present, as shown in Fig. 6(e). The XANES analysis revealed that the average valence of Fe atoms was +2 to +3 as the Fe K-edge absorption of Fe–N4SP/NPS-HC was situated between those of FePc and Fe2O3. In the k3-weighted FT-EXAFS, the contributions of Fe–N and Fe–S were observed with coordination numbers of 4 and 1.1 whereas Fe–Fe contributions were absent. The original Fe–N4 sites’ symmetry interactions were broken as a result of the core Fe being shifted slightly out of the Fe–N4 plane by the distinct axial S-coordination and neighboring P-coordination.70
In 2024, Huang and coworkers71 synthesized Ni single atoms loaded on TiO2 as a support with extremely high catalytic performance and thermal stability for catalyzing hydrogen storage in MgH2 (15wt%-Ni0.034@TiO2) as depicted in Fig. 6(f). The combined effect of single atom Ni, multiple OV, and multivalent Tix+ is responsible for the superior catalytic effect. Ni plays a crucial role in this process, speeding up the transfer of electrons between Mg2+ and H− while weakening the Mg–H bonds. The Nix@TiO2 catalysts were synthesized in different compositions by using a hot molten salt method. Initially, 20 mg of NiCl2·6H2O, 1.1 g of KCl, 0.9 g of NaCl and 1 g of TiO2 were mixed and grinded thoroughly followed by sintering at 500 °C for 2 h at a ramp rate of 10 °C min−1. The resultant powder was washed with deionized water thrice to ensure the removal of any residual impurities and at last, the powder was dried to obtain the Nix@TiO2 catalysts.71 The XRD patterns revealed three prominent peaks for the TiO2 anatase phase and no diffraction peak for Ni was observed. The edge length of 20–30 nm and a d-spacing of 0.3611 nm were calculated from the HRTEM analysis of the Ni0.034@TiO2 catalyst. The HDAAF-STEM images revealed the appearance of Ni in the brighter regions as compared to the case of Ti due to the larger molecular weight of Ni. Also, the Ni K-edge XANES spectra of Ni0.034@TiO2 were different from that of Ni foil and NiO, which suggested the diverse configurations of Ni, and specifically, an increase in the oxidation state of Ni was observed due to the shifting of Ni0.034@TiO2 XANES towards higher energy than in the case of Ni foil (Fig. 6(g)). Finally, the EXAFS studies (Fig. 6(h)) showed the presence of Ni–Ni contributions in Ni foil which were absent in Ni0.034@TiO2 confirming the presence of Ni single atoms over the TiO2 support.71
Similar to Fe SACs, Cu SACs have also been promisingly studied for electrocatalytic ORR and found to be excellent alternatives for the traditional Pt catalysts.72–74 In this context, Yang et al.72 developed the Cu–N–C SAC via a two-step synthetic process involving pyrolysis and etching followed by metal anchoring and thermal activation, as shown in Fig. 7(a). In the first step, the authors used 2,4-diaminopyridine (DAP) as a C and N source with silica as a template for the synthesis of the N-doped carbon support. For the synthesis of the N-doped carbon support, ammonium peroxydisulfate was used for the polymerization of DAP (PDAP) and further pyrolysis of PDAP was done at 800 °C for 2 h in an NH3/He atmosphere and etching with HF to remove silica. In the next step, successful anchoring of Cu2+ was carried out, followed by NH3 treatment at 140 °C. Interestingly, the Cu content calculated by X-ray photoelectron spectroscopy (XPS) (4.2 wt%) was found to be double that calculated by ICP (2 wt%) which was attributed to the presence of Cu atoms predominantly on the surface.72 The electron energy loss spectra (EELS) from a single atomic site with brighter contrast in HAADF imaging showed C, N, and Cu signals (Fig. 7(b)). Additionally, it was revealed from the elemental mapping (Fig. 7(c)) that the N atoms surround the Cu atoms, showing the most possible coordination of Cu with N or C, in accordance with the proposed M–Nx coordination structure for these types of catalysts. Further, the theoretical modeling of all the possible configurations concluded that the Cu–N4 configuration resembled the experimental XAS results. Therefore, the catalyst exhibited Cu–N4 configuration which transforms into Cu–N3 and further to OH–Cu–N2 under ORR reaction conditions as confirmed by combining the XAS and theoretical results.72
 | ||
| Fig. 7 (a) Synthesis scheme of the Cu–N–C SAC, (b) EEL spectra from single Cu sites in the HDAAF image, and (c) EELS map for N + Cu. Reproduced with permission.72 Copyright 2021, American Chemical Society. (d) Plot of Cu loading vs. Cu density and (e) Cu K-edge XANES spectra for various Cu SACs, Cu2O, and Cu foil. Reproduced with permission.75 Copyright 2023, American Chemical Society. (f) Synthesis strategy of CoML and CoMM SACs. Reproduced with permission.76 Copyright 2023, American Chemical Society. | ||
Further, Jin et al.75 reported the density-dependent activity of Cu SACs in the organic transformation of benzene to phenol. The study involved the synthesis of various Cu SACs with different Cu atom densities (0.1 to 2.4 atoms nm−2) and it was found that the hydroxylation reaction was directly proportional to the Cu atom density (Fig. 7(d)). In the case of high atom density, the interactions between Cu atoms changed the electronic structure of Cu, which resulted in stronger adsorption of the ˙OH radical. In the catalyst synthesis, the Cu1–N3O1 configuration of the Cu SAC was synthesized using a facile polycondensation followed by a pyrolysis method. Initially, cyanuric acid, phytic acid, melamine, L-alanine, and Cu(NO3)2 were mixed in water and a polymerization process was carried out to synthesize nanosheets.75 In the second step, pyrolysis was done where phytic acid, L-alanine, melamine, and cyanuric acid served as oxygen, carbon, and nitrogen sources. The TEM images of all the catalysts showed a laminar structure, and a uniform distribution of Cu was seen even at the highest loading (21.3 wt%) without any NPs or clusters, which was further confirmed by AC-HDAAF-STEM. The absorption threshold positions of the Cu SAC in the XANES spectra shown in Fig. 7(e) lie in between Cu2O and copper phthalocyanine which confirmed the valence of Cu to be in between that of Cu1+ and Cu2+. Also, the FT-EXAFS showed the contribution of Cu–N/O evident by a peak at ∼1.5 Å whereas no peak was observed for Cu–Cu, which suggested negligible interaction between Cu atoms and uniformly distributed Cu atoms over the N, O-doped carbon support.75
An excellent study to enhance the high-density loading of Co single atoms was reported by Kumar et al.76 by using a macromolecule-assisted SAC synthetic route for oxygen evolution reactions (Fig. 7(f)). The authors successfully achieved high loading (10.6 wt%) of Co single atoms over a graphitic network rich in pyridinic nitrogen. The condensation of melem species (CoMM) and cobalt phthalocyanine (CoPc) synthesized the nitrogen-rich carbon support. It is to be noted that the amalgamation of the CoPc and melem (C6N7) core in CoMM causes a rise in the N content, which is predominated by the pyridinic nitrogen. The structure of CoMM revealed that the C:N ratio was 1:1 with periodic arrangement of carbon and nitrogen.76 The substrates prepared from melamine and melem were named CoCML and CoCMM, respectively, and the corresponding Co SACs were named CoML and CoMM, respectively. Initially, CoCML and CoCMM exhibited nanorods with Co NPs at the tip. Remarkably, isolated Co–N4 single atom sites incorporated in the C–N framework were obtained when CoPc was utilized as a preconfined source of Co. Even at very high loadings, HRTEM, AC-STEM and EELS analysis revealed no aggregation of Co atoms signifying the presence of Co atoms. The Raman studies confirmed the absence of Co oxides evident by the vibrational peaks for metallic Co with separated D and G bands.76 Further, Li and coworkers synthesized a variety of Co SACs by tuning the local nitrogen environment (Co–Nx(C), x = 2, 3, and 4) to study their potential as a mimic for oxidase.77 The study concludes that the oxidase resembling activity of Co-based SACs can be successfully optimized by tuning the N environment. In the typical catalyst synthesis, the Co/Zn ZIF MOF was prepared using 2-methylimidazole as an organic linker. Afterward, the obtained Co/Zn ZIF was heated at 800 °C (5 °C min−1) and 900 °C (5 °C min−1) in an Ar atmosphere for 3 h to synthesize Co–N4 and Co–N3, respectively.77 For the synthesis of Co–N2, the obtained Co/Zn ZIF was heated at 800 °C for 1 h, 900 °C for 1 h and 1000 °C for 1 h respectively at a 5 °C min−1 ramp rate under an Ar atmosphere. The Co–N3 catalysts exhibited the best oxygen adsorption structure and strong formation of reactive oxygen species (ROS) among the investigated single atom Co catalysts, providing the preferred oxidase-like catalytic activity.77
Further, Zhang et al.78 reported a facile synthesis route for developing Ti single atoms supported on reduced graphene oxide (rGO) for enhancement in the energy level alignment in perovskite solar cells (Fig. 8(a)). The synthesized SAC Ti1–rGO contains Ti single atoms anchored uniformly by the O atoms present on the rGO support in Ti1O4–OH configuration. This configuration exhibited the capability to alter the electronic properties of rGO mediated by charge transfer between Ti single atoms and rGO. The catalyst was synthesized by a facile chemisorption process where titanium diisopropoxide bis(acetyl acetonate) was used as a Ti source and mixed with rGO followed by chemisorption to yield the Ti–rGO SAC.78 The XPS analysis revealed the presence of residual oxygen in the form of carboxyl, hydroxyl, and epoxide. These species provide efficient active sites for the homogeneous anchoring of Ti single atoms. The Ti1–rGO SAC exhibited ultrathin and wrinkled sheet-like morphology similar to rGO thereby retaining the flexibility of rGO. Additionally, the uniform dispersity of Ti single atoms as bright spots was confirmed by the HDAAF-STEM analysis. The coordination of Ti with O of rGO was confirmed by the EELS analysis as the O EELS map was gathered around Ti atoms (Fig. 8(b)). Further, Ti K-edge XANES spectra confirm that the OS of Ti in Ti1–rGO is approximately +4 as the absorption edge energy of Ti1–rGO is falling between that of TiC and TiO2 as shown in Fig. 8(c).78 Therefore, this study provides an excellent protocol to enhance the properties of rGO by using Ti single atoms.
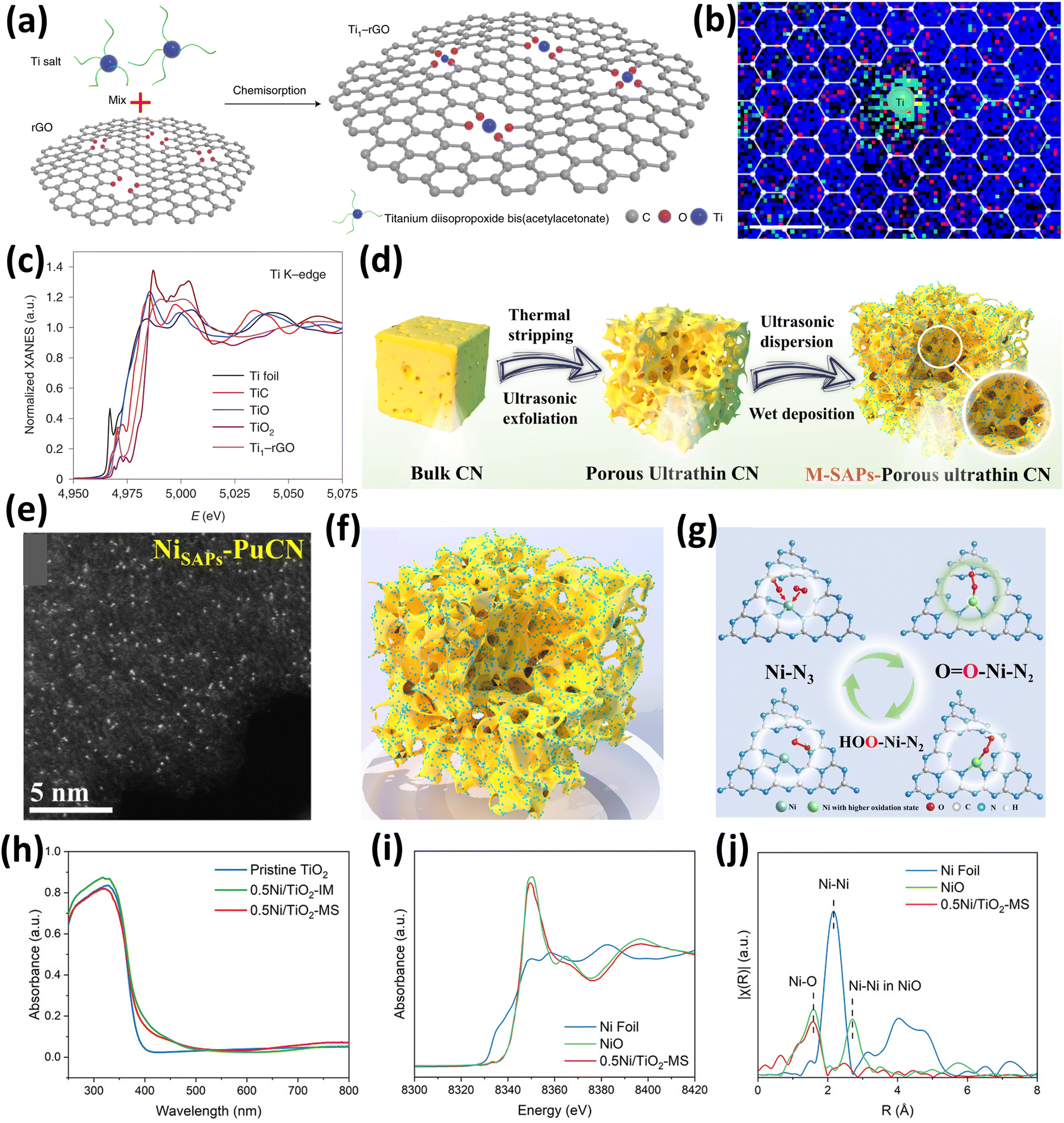 | ||
| Fig. 8 (a) Synthesis scheme of Ti1–rGO, (b) EELS map for C, O, and Ti of Ti1–rGO, and (c) Ti K-edge XANES spectra. Reproduced with permission.78 Copyright 2021, Nature Publishers. (d) Schematic synthesis scheme for M-PUCN, (e) AC-HDAAF-STEM image of NiSAPs-PUCN, (f) structure of NiSAPs-PUCN, and (g) change in the configuration of Ni during the photocatalytic H2O2 production reaction. Reproduced with permission.79 Copyright 2023, Nature Publishers. (h) UV-DRS absorbance spectra of TiO2, 0.5Ni/TiO2-MS, and 0.5Ni/TiO2-IM, and (i) Ni K-edge XANES spectra and (j) Ni K-edge EXAFS spectra of 0.5Ni/TiO2-MS. Reproduced with permission.80 Copyright 2023, Wiley VCH. | ||
Furthermore, the facile development and synthesis of Ni single atoms anchored on graphitic C3N4(g-C3N4) was reported by Zhang et al.79 for its application in photocatalytic H2O2 production using water as a medium. In the typical catalyst synthesis shown in Fig. 8(d), the initial steps were to adjust the microtopography of the C3N4 bulk (BCN) substrate by utilizing thermal stripping and ultrasonic exfoliation, to obtain porous ultrathin C3N4 (PUCN). The synthesized PUCN exhibited numerous active sites for the effective and efficient loading of Ni single atoms, which was carried out using a wet precipitation method along with continuous ultrasonic treatment. The ultrasonication conditions ensure the homogeneous dispersion of single atoms and also further exfoliate PUCN to create other sites for anchoring the single atoms.79 The same synthesis strategy was followed for various metals to synthesize M-PuCN (M = Fe, Co, Ni, Cu, Zn, Sr, W, Pt). It was observed from the morphological analysis that, as compared to PUCN, Ni-PUCN exhibited a more porous structure and thinner undulating folds. The AC-HDAAF-STEM images and EDS mapping also confirmed the presence of well-dispersed Ni single atoms (bright spots) on the PUCN support, as shown in Fig. 8(e and f). Crucially, it was directly observed by in situ synchrotron XAS and Raman spectroscopy that after O2 adsorption, the initial Ni–N3 sites dynamically change into high valent O1–Ni–N2 sites and then proceed to form a crucial *OOH intermediate before ultimately forming HOO–Ni–N2 (Fig. 8(g)). The overall studies further show that the evolution of the active site structure improves H2O2 generation activity and selectivity by lowering the energy barrier of *OOH formation and suppressing the OO bond cleavage.79
Moreover, SACs have also been extensively used in the catalytic transformation of biomass-derived compounds into value-added chemicals.81–83 One such biomass-derived compound is glycerol which is obtained in high amounts as a byproduct of biodiesel production.84 In this context, Xiong and coworkers reported the conversion of glycerol to glycolaldehyde by C–C cleavage using a light-driven Ni single atom supported TiO2 catalyst which works under mild conditions utilizing air as an oxidant.80 The 0.5Ni/TiO2 catalyst was synthesized by two methods, the molten salt method (0.5Ni/TiO2-MS) and the impregnation method (0.5Ni/TiO2-IM), but higher activity towards glycerol conversion was shown by the catalyst prepared by the molten salt method. Therefore, in the molten salt method, the Ni single atoms are anchored over TiO2 in metal salts at high temperatures, followed by thorough washing with water to remove any leftover impurities.80 The XRD analysis revealed the absence of any diffraction peaks for metallic Ni or nickel oxide, and the UV-visible diffuse reflectance spectroscopy (UV-DRS) studies showed the band gap of the catalyst to be 3 eV with no changes in the onset edge in the molten salt or impregnation method (Fig. 8(h)). The non-existence of Ni in the form of clusters or NPs was confirmed by EDX and HDAAF-STEM analysis which showed the presence of Ni atoms uniformly dispersed over the TiO2 support. The absorption edge of 0.5Ni/TiO2-MS in the XANES spectra was found close to NiO which confirmed the oxidation state of the Ni species to be +2 as shown in Fig. 8(i). Further, the Ni K-edge FT-EXAFS (Fig. 8(j)) showed the contribution of Ni–O interactions with a coordination number of 5.2 which elucidated that the NiO5 species are selectively formed. No observation of the Ni–Ni or Ni–O–Ni interaction was observed which confirmed the presence of Ni as single atoms over TiO2.80
Further, Li et al.85 reported the synthesis of Zn single atoms supported on a bipyridine functionalized POP via a copolymerization synthesis strategy for the N-formylation reaction of amines with CO2 and PhSiH3. In typical synthesis experiments, a free radical copolymerization reaction was performed between divinyl-bipyridine and divinyl-benzene monomers which was followed by a complexation step with Zn(OAc)2. The free radical copolymerization was chosen because of various advantages like easy operations, well defined active centres, and metal-free polymer synthesis.86,87 Also, the SACs prepared from this method frequently have a large specific surface area, a hierarchically porous structure, and an incredibly flexible framework, giving the polymeric catalysts “quasi-homogeneous” catalytic properties. The Zn content in all the synthesized catalyst was found out by ICP analysis and ranged from 2.04 to 6.09 wt%. Furthermore, AC-HDAAF-STEM analysis revealed isolated bright spots representing the existence of Zn as atoms and not aggregated NPs suggesting the formation of SACs.85 Additionally, Wang and co-workers reported the synthesis of a non-heme Mn single site catalyst supported on the skeleton of POPs and having M–N4 configuration for the aliphatic C–H oxidation and CC epoxidation reactions.88 The series of catalysts were prepared via radical polymerization under solvothermal conditions by reacting divinyl benzene and Mn complexes possessing a vinyl functionalized N4 ligand in varying proportions (Mn-1@POP-40 and Mn-1@POP-20). Using non-local DFT, it was revealed that the synthesized catalysts exhibit hierarchical porosity which enhances the reactant and product transport in the catalytic system. The morphological studies (HDAAF-STEM and EDX) showed the presence of individual Mn–N4 complexes over the porous POP support suggesting the effective formation of the single site catalyst. In comparison to homogeneous analogues, the Mn single site catalyst was stabilized for catalytic reactions by its inherent single site and confined reaction mode.88
Dong et al.59 reported an excellent strategy to incorporate Ni single atoms in different microenvironments into one POP support material for photocatalytic CO2 reduction. The authors synthesized a novel POP with simultaneous integration of the salphen complex and metallothalocyanine to form one extended framework denoted as NiPc–MPOP. The two immobilized entities in this framework consisted of single atoms having different –N4 and –N2O2 configurations, respectively. The existence of single atoms in two different configurations provided multiple active sites for the reaction and a scope of clear comparative study between various SACs with different configurations which will help to find out the intrinsic catalytic centre. The catalysts were synthesized by the polycondensation reaction of nickel(II) 2,3,9,10,16,17,23,24-octakis (amino) phthalocyanine ((NH2)8NiPc) with 2,6-diformylphenol (DFP) in a mixture of dimethylacetamide (DMAc) and mesitylene with aniline as a regulating agent, followed by treating the obtained samples with Ni(OAc)2·4H2O.59 In the Fourier transform infrared (FTIR) spectroscopy results, a neighboring peak was observed at 1557 cm−1 after the incorporation of Ni atoms which was attributed to the binding of CN groups to the metal centres. The O 1s XPS spectra showed two peaks for C–O and M–O bonding which was ascribed to the successful incorporation of extra metallic sites in the salphen pocket. In the FT-EXAFS spectra of NiPc–NiPOP, no peaks corresponding to the Ni–Ni interaction was observed which eliminated the possibility of the co-existence of two metal atoms existing in the salphen unit.59 Lastly, these reports on the synthesis of POP framework-based SACs provide an excellent understanding of the role of the framework type support in tuning the physicochemical properties of the catalyst which plays a crucial role in the enhancement of the efficiency and performance of the catalytic system.
In conclusion, this section provides an elaborate discussion of the various synthetic routes for the facile development of NNMSACs which are highly active in various applications due to their excellent physicochemical properties. There has been promising attention paid to designing effective protocols for enhancing the NNMSACs’ research field as they serve as highly economical and abundant alternatives to cost-ineffective NMSACs. They exhibit several advantages like high tunability for reaction selectivity, high metal–support interactions, and high atomic utilization.80 These properties help to retain the catalytic activity of the NNMSACs with highly dispersed single atoms serving as active sites, which adds to the advantage of this research field.
2.2. Non-metal based SAC synthesis (NOMSACs)
As per the status of the current research on SACs, an enormous amount of attention has been given to a wide range of metal active centers like noble metals and non-noble metals. In contrast, there has been very limited focus on the identification of non-metallic active centers which is primarily because of the hurdles in finding the non-metallic active sites and the appropriate reaction pathways.89 These catalysts offer highly economical and earth-abundant pathways with facile synthesis procedures and efficient regulation of the substrates.90 In this section, we will discuss the very few available reports on the synthesis of NOMSACs for various applications to understand the current state of this research field and what the future of this technology holds.In this regard, the first investigation on NOMSACs was reported by Fu et al.90 which involved the effective synthesis of phosphorus single atoms supported on intriguing single crystal Mo2C nanosheets with (001) plane exposure supported by carbon sheets (SAP-Mo2C-CS) for application in hydrogen evolution. The synthesized catalyst exhibited outstanding hydrogen evolution performance, and the P bonds with the surrounding Mo atoms create a localized electronic state due to the bidirectional regulation of P single atoms. In the catalyst synthesis, the first step was to synthesize single crystal Mo2C hexagonal nanosheets which was carried out by a facile calcination process.90 Afterwards, an easy pressure gas-assisted process was used in a PH3 atmosphere (0.18 MPa) at low operating temperatures for the synthesis of SAP-Mo2C. It was observed that the proposed synthesis method did not alter the morphology of Mo2C-CS but led to the anchoring of P single atoms on Mo2C-CS owing to the decomposition of PH3 on Mo2C-CS. The HDAAF-STEM studies confirmed the maintained crystallinity (d = 0.261 nm), whereas the AC-HDAAF-STEM analysis revealed the presence of P single atoms on the surface of Mo2C-CS in SAP-Mo2C-CS. It was revealed that the P atoms occupy the space group between three Mo atoms in the (001) crystal plane in order to form a stable structure, as this space group is more flexible. Further, the molar ratio of P atoms in Mo2C-CS was found to be 3.6% via TEM-EDS analysis.90 Also, the EELS spectrum of Mo was found to be not similar in Mo2C-CS and SAP-Mo2C-CS which suggested the presence of Mo in +2 oxidation state (OS) in Mo2C-CS and in +3 OS in SAP-Mo2C-CS due to some electron transfer to P atoms which are supported on the Mo2C surface. A similar observation was also made in the XANES spectra, where the Mo K-edge spectra shifted towards higher energy in SAP-Mo2C-CS as compared to Mo2C-CS. In the XANES spectra of P, three peaks were observed at 2146.8, 2148.7, and 2149.9 eV, which were attributed to P–Mo, P–C, and P–O. The intensity of the P–Mo peak in SAP-Mo2C-CS was high which confirmed that the P atoms are primarily coordinated to Mo atoms and have a very weak interaction with the carbon atoms.90 Therefore, this study provides an excellent investigation to identify the non-metal single atoms as catalytically active sites, which opens new opportunities in the field of SACs.
Further, Yang et al.89 reported the synthesis of P single atoms supported on the edges of graphene (P-SAC-NG) for the electroreduction of CO2 to CO. The SAC was efficiently designed by synthesizing a porous layered hierarchical precursor by self-assembling glyphosate and melamine (Fig. 9(a)). Subsequently, this precursor was subjected to pyrolysis, which yielded the P-SAC-NG SAC. Prior to the pyrolysis step, the self-assembly of melamine and glyphosate is highly required to obtain P-SAC-NG with high P content and porosity. Further, the SEM and TEM analysis confirmed the 3D interconnected architecture that resulted from the 2D porous graphene nanosheets as shown in Fig. 9(b and c). The HRTEM analysis showed that the P atoms can be seen at the edges of graphene as bright spots, whereas the uniform dispersion of P atoms was confirmed by AC-HDAAF-STEM images (Fig. 9(d)). The XPS and ICP-MS analysis revealed the absence of any metal species, which confirmed the successful anchoring of P heteroatoms. Furthermore, 31P solid state NMR showed peaks at chemical shift −5.3 ppm and −16.1 ppm for P-SAC-NG which resembled P atoms in PPh3 and OP(OH)Ph2, respectively.89 These results concluded that the P atoms are present in two oxidation states ascribed to C3–P(O) and C2–PO2. In the P K-edge XANES spectra of P-SAC-NG, the white line position appearing at 2153.6 eV was ascribed to 1s → 3p transition where the 3p orbital is empty for P in the +5 OS. The spectra were fitted to two absorption peaks related to the P–C (σ*) and P–O (π*) coordination, and this concluded that P atoms are present in two types of configurations in P-SAC-NG, 2C–PO(OH) or 3C–PO. At last, by combining the theoretical and experimental results it was concluded that the configuration of P atoms changes from 2C–PO(OH) to 2C–PO through proton coupled electron transfer in the CO2 to CO conversion (Fig. 9(e)). This transformation helps to decrease the activation energy of CO2 present at the P single atom sites to form the 2C–P(CO2)δ−O intermediate.89
 | ||
| Fig. 9 (a) Schematic synthesis diagram for the formation of P-SAC-NG, (b) SEM, (c) dark field TEM, (d) HDAAF-STEM image of P-SAC-NG, and (e) configuration change of P single atoms during degassing and exposure to CO2. Reproduced with permission.89 Copyright 2023, Royal Society of Chemistry. | ||
As mentioned earlier, experimental research in this field is lacking and is at its very nascent stages but contrastingly there have been few reports focusing on computational studies on NOMSACs. Ling et al. for the first time proposed the first principles calculations on B atoms supported on g-C3N4 (B/g-C3N4) for photocatalytic nitrogen fixation to ammonia (Fig. 10(a)).91 Boron was chosen because in sp2 hybridization, N2 can bind with two boron sites in an end-on manner, whereas in sp3 hybridization, this binding happens in a side-on manner due to the feasibility of the orbital symmetry. The electron exchange when the N2 molecule is attached to a boron atom supported on a suitable support material is shown in Fig. 10(b). In the structure of g-C3N4, the 2-fold coordinated nitrogen atoms serve as a site for B anchoring. In order to stabilize the B atom onto g-C3N4, two N–B bonds are created during this procedure, leaving one occupied and one unoccupied sp3 orbital. As a result, there will be significant contact between the gas phase N2 molecules and the attached B atoms.91 As anticipated, there is a very strong interaction among N2 and B/g-C3N4, with side-on adsorption having an adsorption energy of −1.04 eV and end-on adsorption having an adsorption energy of −1.28 eV (Fig. 10(c)). A sizable charge transfer between the attached B atom and N2 is visible for both adsorption patterns. It was discovered that gas phase N2 on B/g-C3N4 can be effectively reduced into NH3 by an enzyme process with an all-time low onset potential of 0.20 V. Notably, it is further shown that the extraordinarily high stability of the as-designed catalyst holds significant promise for synthesis.91
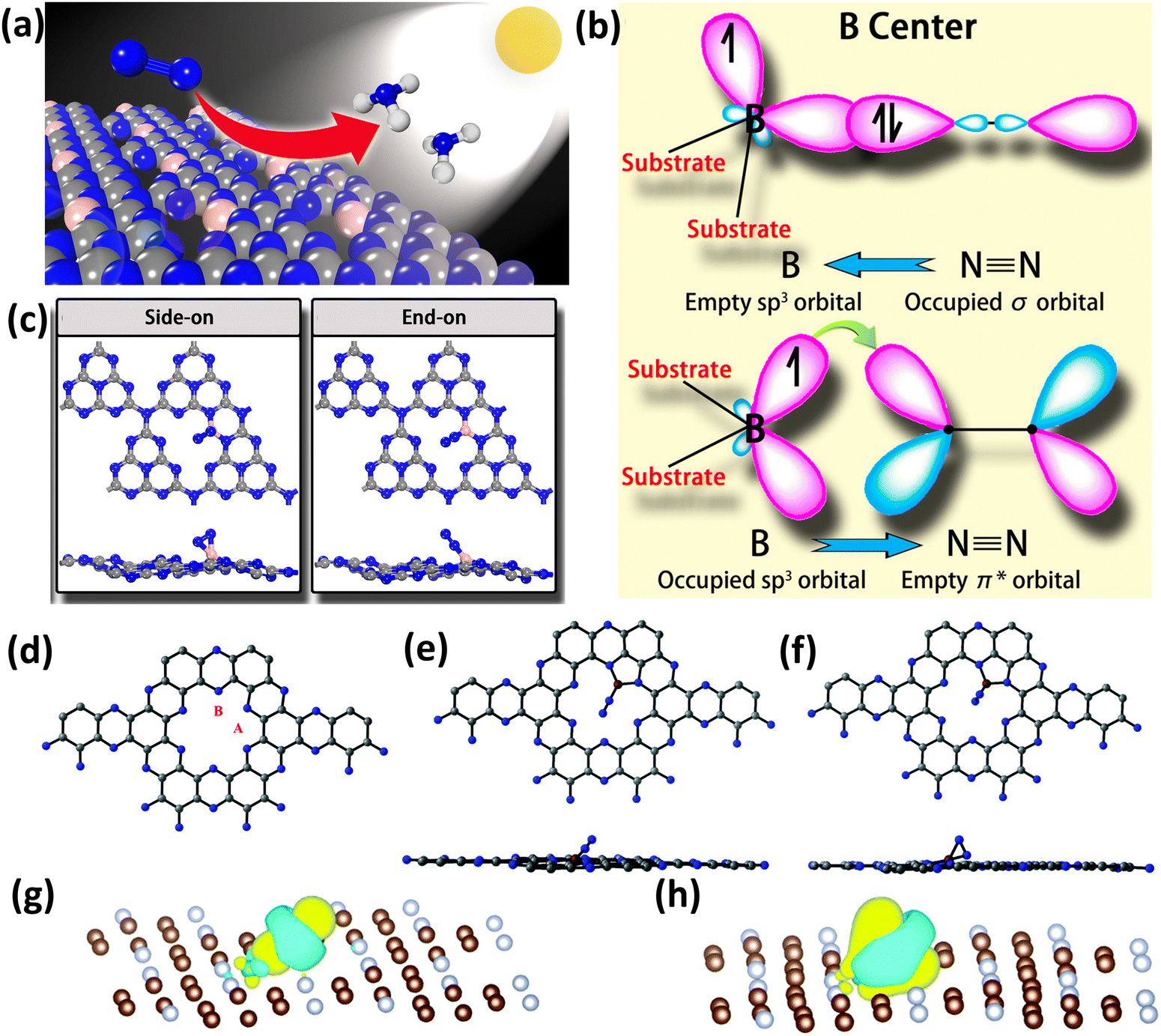 | ||
| Fig. 10 (a) B/g-C3N4 catalyst for photocatalytic nitrogen fixation, (b) binding of N2 on the boron atom, which is anchored on a suitable substrate, and (c) side-on and end-on binding modes of N2 on boron single atoms. Reproduced with permission.91 Copyright 2018, American Chemical Society. (d) Structure of C2N along with the possible binding site for the boron atom, (e) end-on binding of N2, (f) side-on binding of N2, (g) charge density difference in end-on binding, and (h) charge density difference in side-on binding. Reproduced with permission.92 Copyright 2019, Royal Society of Chemistry. | ||
Similarly, Bhattacharyya et al.92 reported first principles calculations for B single atoms anchored on the C2N monolayer (B/C2N) as a support for photocatalytic nitrogen fixation. The structure of C2N consisted of two-fold coordinated N atoms and three-fold coordinated C atoms (Fig. 10 (d)). The computational analysis showed that the binding energy of the boron atom at site A is −5.77 eV which is more than that at site B (−0.68 eV). In addition to high binding energy, the AIMD simulations confirmed the high thermal stability of the catalyst up to 800 K. It was found from the N2 adsorption calculations that the N2 molecule can feasibly interact with the boron atom in an end-on and side-on fashion with binding energies of −1.44 and −0.58 eV, respectively, as shown in Fig. 10(e and f).92 In both binding fashions, the N2 bond length increases due to the back bonding from metal d orbitals to the antibonding orbital of N2. It is to be noted that the charge density was entirely concentrated at the binding sites which tells about the promising bonding interaction in both types of bindings (Fig. 10(g and h)). In the end-on fashion, the reduction occurs via a distal or alternate mechanism, whereas in the side-on mode, reduction occurs through the enzymatic pathway. The theoretical analysis proved that the onset potential for the enzymatic pathway is 0.18 eV, which was comparatively much lower than that of distal (0.99 eV) and alternate pathways (0.71 eV). Therefore, these theoretical studies proposed B/C2N as a potential single atom photocatalyst with feasible formation for effective nitrogen fixation.92
3. Catalytic activity of SACs towards organic transformation reactions
Over the past few years, SACs have shown exceptional catalytic potential toward various catalytic applications, including hydrogen evolution,93–96 CO2 conversion,97–100 energy storage101–107 and organic transformation reactions.65,108–111 This part will provide a detailed discussion of the current scenario and developments in the research field of SACs for a wide range of organic transformation processes. SAC catalyzed reactions such as hydrogenation, hydroformylation, coupling, bond cleavage reactions, etc., have been elaborately discussed. These catalytic organic reactions hold industrial significance in the manufacturing of a diverse range of fine chemicals and value-added products which directly impacts the development of the chemical industry.65,112–1153.1. Hydrogenation reactions
Hydrogenation of organic functional groups is a crucial process in organic chemistry, which is extensively used in various industries, including fine chemicals and pharmaceuticals. While direct hydrogenation using molecular hydrogen (H2) is effective, it presents challenges such as the need for high hydrogen pressure, specialized reactors and strict safety measures. An alternative approach, i.e., transfer hydrogenation, offers several advantages, including enhanced safety, ease of handling, and broader applicability in various organic transformations.11 The use of SACs, made up of individual metal atoms spread out on a support material with different coordination environments, has attracted much interest due to their excellent catalytic properties and potential for use in hydrogenation reactions. SACs provide numerous benefits over traditional catalysts in hydrogenation reactions. The atomic-level dispersion of metal atoms results in a high surface area, increasing catalytic activity. This heightened activity enables greener and more sustainable processes by reducing reaction temperatures and energy consumption. Examples of hydrogenation processes using SACs include the chemoselective hydrogenation of alkynes, nitriles, nitroarenes, and carbonyl groups. These instances demonstrate the remarkable efficiency and selectivity of SACs in various chemical transformations.Herein, Liu et al.122 investigated the selective hydrogenation of 1-hexyne to 1-hexene using PdAu single atom alloy (SAA) catalysts. Additional investigation was carried out using surface science techniques and DFT to better understand the contribution of Pd atoms and the processes that lead to improved hydrogenation selectivity. The findings suggest that the variations in reactivity are due to the differences in energy barriers for over-hydrogenation of the terminal carbon atom. The kinetics of selectively hydrogenating 1-hexyne to 1-hexene was investigated in a batch reactor focusing on the initial reaction rate. The hydrogenation rate per surface Pd atom for Pd0.004Au-SAA is approximately six times lower than that for Pd NPs at 25 °C. Moreover, the Pd0.004Au-SAA catalyst demonstrates 85% selectivity for 1-hexene with complete conversion of 1-hexyne, while the selectivity of Pd NPs is approximately 50% at high reactant conversion. Additionally, DFT calculations were used to assess the hydrogenation barriers for 1-hexene on Pd(111) and PdAu(111) surfaces. The analysis was focused on the barrier for hydrogenation of the terminal carbon of 1-hexene to produce 2-hexyl, as this pathway exhibited a lower barrier compared to the formation of 1-hexyl on both surfaces. The energy barriers for hydrogenation were discovered to be almost the same on both surfaces, at 0.65 eV on Pd(111) and 0.61 eV on PdAu(111). Also, the adsorption energy of 1-hexene was similar on the two surfaces, with values of 1.30 eV on Pd(111) and 1.13 eV on PdAu(111), as depicted in Fig. 11(a). The profound comprehension of the selective hydrogenation process on PdAu single atom alloys (SAAs) obtained from this study offers valuable perspectives for the design of future catalysts.
 | ||
| Fig. 11 (a) Adsorption energy on Pd(111) and PdAu(111). Reproduced with permission.122 Copyright 2019, American Chemical Society. (b) Model catalyst diagrams for different loading scenarios. Reproduced with permission.118 Copyright 2022, American Chemical Society. (c) and (d) H2 spillover over physical mixtures. Reproduced with permission.123 Copyright 2019, American Chemical Society. | ||
Furthermore, Song et al.118 developed a highly effective ultralow Pd catalyst with the help of nickel-modified alumina. This catalyst leverages the benefits of Pd for hydrogen activation while utilizing Ni to modulate the adsorption of alkynes and alkenes on the catalyst surface. The addition of Ni substantially impacted the structure of the flaky alumina support and the distribution of Pd particles. Ni was evenly dispersed throughout the support in a highly scattered state. XPS revealed the existence of Ni, resulting in electron transfer from the Pd NP to the altered support. This transfer likely assisted in the attachment of electron-rich phenylacetylene to the electron-deficient palladium NP, ultimately improving performance at reduced temperature and pressure. Under specific conditions of low temperature and normal air pressure, phenylacetylene's semi-hydrogenation resulted in 98% conversion rate and 94% selectivity. The catalyst's performance remained consistent after 10 experimental cycles, while maintaining its original structure. Additionally, Fig. 11(b) displays model catalyst diagrams depicting various loading scenarios, such as monometallic Pd/γ-Al2O3, Ni@γ-Al2O3 and Pd/Ni@γ-Al2O3 catalysts. When the catalyst included only Pd as the active metal, phenylacetylene exhibited a high conversion rate but experienced notable over-hydrogenation. As the Pd loading decreased, the selectivity for styrene increased while the conversion rate remained constant. This indicates that lower Pd loading reduces the number of hydrogenation active sites, thereby improving styrene selectivity. When only Ni was actively involved, the reaction was not very active, resulting in a conversion of less than 30% of phenylacetylene. However, the selectivity for intermediate products was exceptionally high, exceeding 99%. This research sheds light on the development of supported ultra-low noble-metal based catalyst for semi-hydrogenation reaction. It proposes a technique to improve the hydrogenation selectivity of these catalysts by using supports that have been modified with a secondary metal.
In another study, Ma et al.123 reported that incorporating phosphorus (P) into a CeO2 support significantly reduces the electron density of Pt1 atoms. This was supported by data from DRIFTS on CO chemisorption, XAFS, and XPS. The Pt1 atom's electronic interactions with the phosphorus-modified neighboring oxygen atoms led to a significantly increased activity for the hydrogenation of cyclohexene, styrene, nitrobenzene, and phenylacetylene under mild conditions. The observed activity enhancement was up to 10 times more significant compared to that of Pt1 on CeO2. In the hydrogenation of compounds such as cyclohexene, styrene, nitrobenzene, and phenylacetylene, the Pt1/PO4–CeO2-500 catalyst demonstrated a significant increase in activity, up to ten times greater than that of Pt1/CeO2. The FT-IR analysis revealed that styrene has a stronger chemisorption on Pt1/PO4–CeO2-500 compared to Pt1/CeO2-500. Additionally, H2-TPR studies indicated that phosphorus doping in CeO2 enhanced hydrogen spillover capabilities, which contributed to the substantial improvement in hydrogenation activity. Fig. 11(c and d) reveals the H2 spillover over physical mixtures of Fe2O3 + Pt1/CeO2-500 and Fe2O3 + Pt1/PO4–CeO2-500.
Furthermore, the utility of metalloenzyme like M–N–C SACs is well-known in the field of organic transformation reactions for the production of fine chemicals but there is a lack of organic transformation studies using these SACs in aqueous media. In this regard, Liu et al.124 synthesized bioinspired hydrophobic Pd1–S–C SACs through a straightforward hydrothermal method. Inspired by the hydrophobic pockets of metalloenzymes, the authors used metal–sulfur coordination chemistry wherein the hydrophobic Pd1–S–C catalyst was synthesized by atomically dispersing Pd atoms over a S-doped C support. The significance of hydrophobicity was revealed by the molecular dynamics simulation which confirmed the boosted reaction kinetics enhancing the alkyne substrate around the catalyst in aqueous media. Additionally, it was shown that the electron-rich PdS4 single sites functioned better than the electron-deficient PdN4 single sites in terms of activating H2 molecules and desorbing C–C intermediates. Therefore, the Pd1–S–C catalyst successfully catalyzed the semi-hydrogenation of terminal alkynes in water, displaying high selectivity, wide applicability to different substrates and remarkable stability. The hydrophobic Pd1–S–C catalyst showed a remarkable selectivity of 94.4% for styrene with a phenylacetylene conversion of 97.2% in just 90 min. On the other hand, the Pd–S–C catalyst with NP dispersion exhibited a lower selectivity for alkenes (51.2%) at a comparable reaction conversion of 98.5%. Additionally, the Pd1–S–C catalyst stability was tested, and it was found that the recycled catalyst maintained its activity and selectivity for five consecutive runs without any significant degradation. Therefore, these studies provide an extensive investigation on designing highly efficient SACs with excellent hydrogenation properties for the hydrogenation of alkynes to form alkenes of high industrial importance.
In another work, He et al.65 presented an efficient method for synthesizing SACs using a precursor-dilution strategy. The process entails combining metalloporphyrin (MTPP) containing target metals with diluents such as tetraphenylporphyrin (TPP). This is followed by pyrolysis to create N-doped porous carbon-supported SACs (M1/N–C). Furthermore, the synthesis also achieved the creation of various catalysts with bi-metallic sites, different surface atom densities, and metal aggregation states. Pt1/N–C demonstrated remarkable chemoselectivity in the hydrogenation of 1-nitro-4-ethynylbenzene and 1-ethynyl-4-vinylbenzene. It selectively converted alkyne groups to alkenyl groups while preserving the –NO2 and –CCH2 groups. This resulted in 99% selectivity for 1-nitro-4-vinylbenzene, 99% selectivity for 1,4-divinylbenzene at approximately 20% conversion, 98% selectivity for 1-nitro-4-vinylbenzene, and 97% selectivity for 1,4-divinylbenzene at nearly 100% conversion. On the other hand, when using Pt–NPs/N–C as a catalyst, multiple products are produced because of the simultaneous hydrogenation of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH with –NO2 and –CCH with –CC. In contrast, the Pt1/N–C catalyst allows for the selective hydrogenation of –CCH over –NO2 and –CC. This selectivity is mainly attributed to the optimal combination of the relatively low catalytic activity of Pt single atoms on carbon supports and the high reactivity of terminal alkynes. Furthermore, constant efforts in these studies to synthesize high-selectivity catalysts will favor the high-purity chemical grade alkenes with low concentrations of highly unsaturated compounds (5–10 ppm).
CH with –NO2 and –CCH with –CC. In contrast, the Pt1/N–C catalyst allows for the selective hydrogenation of –CCH over –NO2 and –CC. This selectivity is mainly attributed to the optimal combination of the relatively low catalytic activity of Pt single atoms on carbon supports and the high reactivity of terminal alkynes. Furthermore, constant efforts in these studies to synthesize high-selectivity catalysts will favor the high-purity chemical grade alkenes with low concentrations of highly unsaturated compounds (5–10 ppm).
O bonds holds great industrial significance for the synthesis of a variety of platform chemicals. In the processes involving various functional groups like CC and CO, reaction protocols focusing on high selectivity becomes a major factor as it minimizes the side reactions, separation steps and substrate consumption.125–127 The development of highly selective catalysts especially for organic reactions is very challenging, especially, it is difficult to promote selective CO hydrogenation in the presence of CC bonds in an organic compound which often leads to low product yield. In this regard, fine-tuning of SACs focusing on high selectivity can serve as a potential strategy for selective carbonyl group hydrogenation.128–131
In this context, Chen et al.132 demonstrated that Zn–N–C SACs containing Zn–N3 groups can effectively catalyze the transformation of cinnamaldehyde (CAL) into cinnamyl alcohol, achieving a high conversion and selectivity of 95.5% and 95.4%, respectively, under optimized conditions. Through the use of DFT calculations, in situ FT-IR spectroscopy and isotopic labeling, it was revealed that the reactants, when co-adsorbed at the Zn sites, undergo a catalytic transformation through a Meerwein–Ponndorf–Verley mechanism. Further, DFT calculations indicate that the high catalytic activity of the Zn–N3 moieties is due to the optimum adsorption energy and favored reaction energy of the rate determining step at the Zn sites. Two possible pathways for the CTH reaction catalyzed by Zn–N–C SACs exist. The preferred pathway over the Zn–N3 site was confirmed by calculating the reaction free energy for each proposed pathway. For path A, thermodynamic reaction free energy was determined to be 0.946 eV, whereas path B exhibited a higher reaction energy barrier of 1.377 eV. Therefore, the catalytic transfer hydrogenation (CTH) reaction is more favorable for path A because of its lower reaction energy barrier. Nonetheless, when the Zn–N4 site was employed as the catalyst for the CTH reaction, the associated reaction energy barrier for path A enhanced to 1.395 eV, surpassing that for the Zn–N3 site. As a result, both the adsorption free energies of the reactants and the reaction energy barriers over the Zn–N3 and Zn–N4 sites demonstrated that Zn–N–C SACs containing the Zn–N3 site exhibit higher catalytic activity for the CTH reaction of CAL compared to those containing the Zn–N4 site as shown in Fig. 12(a and b).
 | ||
| Fig. 12 (a) and (b) Adsorption-free energies and the reaction energy barriers over the Zn–N3 and Zn–N4 sites. Reproduced with permission.132 Copyright 2024, American Chemical Society. (c) Proposed reaction mechanism of the Pd1+NPs/TiO2 catalyst towards hydrogenation of ketones and aldehydes. Reproduced with permission.133 Copyright 2020, Nature Publishers. | ||
In addition, Geng et al.133 explored the synergistic effect between the Pd single atom (Pd1) and nanoparticles (PdNPs) for the hydrogenation of ketones and aldehydes at room temperature. The Pd single atom/nanoparticles were supported on a mesoporous TiO2 support and denoted as Pd(1 + NPs)/TiO2. In the as-synthesized catalyst, it was proposed that Pd1 sites are responsible for the activation of the carbonyl group of ketones and aldehydes whereas PdNPs sites facilitate the dissociation of H2. The dissociated H atom on the surface of PdNPs migrated to the carbonyl group attached on the Pd1 sites of the Pd1+NPs/TiO2 catalyst to complete the hydrogenation reaction as depicted in Fig. 12(c). It was also observed that Pd1+NPs/TiO2 shows higher catalytic activity as compared to simple Pd1/TiO2, PdNPs/TiO2 and Pd/C catalysts. The control studies confirmed the role of synergism between the Pd single atom and nanoparticles in the hydrogenation reaction. Therefore, these recent reports provide a comprehensive understanding for the design and development of SACs with excellent activity for the hydrogenation of carbonyl compounds to yield valuable alcohols.
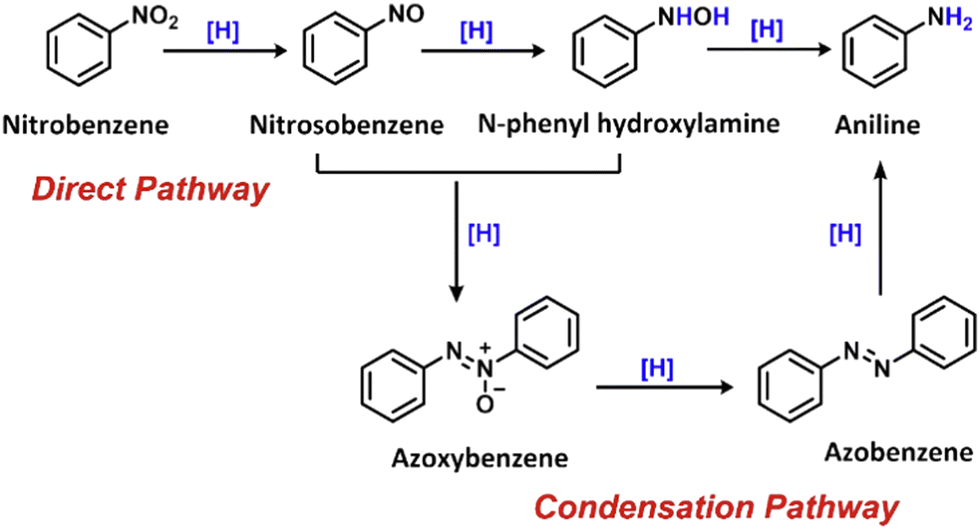 | ||
| Fig. 13 Reaction pathways of the conversion of nitrobenzene to aniline via direct and condensation pathways. Reproduced with permission.138 Copyright 2024, Elsevier Publishers. | ||
In another study, Zhang et al.142 developed atomically dispersed Pd1Ba1/Al2O3 double atom catalysts (DACs) using the ball-milling method. The use of individual Pd atoms (Pd1) with dispersed Ba atoms as electronic promoters (Pd1Ba1/Al2O3 DACs) results in a significant increase in activity, at least fourfold as compared to Pd1/Al2O3 SACs, for hydrogenating various substrates. Studies involving XPS, CO-DRIFTS and differential charge density studies suggested that the presence of Ba atoms enables electron transfer from Ba to Pd atoms, leading to the formation of electron-rich Pdδ+ species. Kinetic studies and H2–D2 exchange experiments also reveal that the presence of Ba reduces the activation energy and promotes H2 dissociation, resulting in exceptional catalytic activity. The time-dependent conversion of nitrobenzene using four different catalysts is shown in Fig. 14(a). Pd1/Al2O3 achieved a modest conversion rate of 13.1% after a 60-minute reaction period. In contrast, the conversion rate of nitrobenzene using Pd1Ba1/Al2O3 increased significantly from 1.5% to over 99.9% as the reaction time extended from 5 to 30 min. Pd1Ba1/Al2O3 demonstrated an impressive activity of 835 h−1, which is 5.8 times greater than that of Pd1/Al2O3, which recorded an activity of 145 h−1. Remarkably, the reaction maintained a selectivity of over 99% for aniline, with no byproducts detected. Additionally, the influence of temperature on the catalytic activity was evaluated. For Pd1Ba1/Al2O3, the activities at 30, 35, 40 and 45 °C were 411, 835, 1103 and 2142 h−1, respectively. In comparison, Pd1/Al2O3 showed activities of 39, 145, 193, and 508 h−1 at the same temperatures. Furthermore, the control studies with Ba1/Al2O3 and Al2O3 were also employed for the hydrogenation reaction and were found to be inactive as depicted in Fig. 14(b). These results demonstrate that introducing Ba single atoms significantly enhances the catalytic performance in nitrobenzene hydrogenation.
 | ||
| Fig. 14 (a) Time-dependent conversion of nitrobenzene and (b) control reactions with four different catalysts. Reproduced with permission.142 Copyright 2022, American Chemical Society. (c) Reaction mechanism of transfer hydrogenation of nitrobenzene with HCOOH. Reproduced with permission.143 Copyright 2022, American Chemical Society. (d) Comparison of hydrogenation activities from nitrobenzene to aniline for various catalysts (y-axis (logarithmic) shows the TOF numbers). Reproduced with permission.108 Copyright 2022, American Chemical Society. | ||
In another study using the porous organic framework, Su and colleagues143 were able to produce SACs by converting zeolitic imidazolate frameworks into a porous carbon-supported N, P dual-coordinated Mn catalyst through phosphatization followed by pyrolysis at 900 °C. The resulting Mn1–N/P–C catalyst, which had an atomic MnN2P structure, showed enhanced activity compared to a similar catalyst with an Mn–Nx structure in the transfer hydrogenation of nitroarenes. The catalytic performance of manganese (Mn)-based catalysts for the catalytic transfer hydrogenation (CTH) of nitrobenzene using formic acid as the model reaction was investigated. Initially, conducting the CTH without a catalyst resulted in no product formation. The nitrogen-doped carbon (NC) catalyst did not show any activity under similar conditions, suggesting that the N-doped carbon support alone cannot reduce nitrobenzene. On the other hand, the NPC catalyst, which incorporates a phosphorus source, yielded 12%, indicating that P doping improves its activity. The Mn1–N/P–C catalyst demonstrated outstanding performance, achieving a 99% yield. In contrast, the Mn1–N–C and Mn–C catalysts were nearly inactive under the same conditions. Additionally, catalysts with different Mn contents (Mn2–N/P–C and Mn3–N/P–C) showed lower activities with yields of 83% and 85%, respectively, highlighting the crucial role of P doping in enhancing catalytic performance. Mn1–N/P–C also showed high catalytic activity with various functional groups, yielding excellent results with –CH3, –F and –Cl groups. Other groups like OCH3 and COCH3 achieved 81% and 99% yields, albeit requiring an elongated reaction time, and the substrates with halogens also produced good yields. Additionally, several benzoheterocycle substrates including 7-nitro-1,2,3,4-tetrahydroquinoline, 7-nitrobenzimidazole, 5-nitroquinoline, 1-nitronaphthalene and 7-nitroindole yielded their corresponding products with good efficiency. The proposed mechanism is illustrated in Fig. 14(c). Firstly, the design of the N, P dual coordinated Mn structure involves a C support with a high density of N basic sites, which facilitates proton transfer and captures H+ from HCOOH, forming NH+. Additionally, Mn atoms due to their low d-electron density and empty orbitals can coordinate with HCOO− to form a Mn–formate intermediate. This intermediate subsequently decomposes to produce CO2 and Mn–H−in situ. Nitrobenzene attaches close to the catalyst surface, where it interacts with Mn–H− and NH+ to create aniline using a CTH method. It is important to note that phosphorus, with its strong electron-donating ability and larger atomic size, boosts electron spread and the positive partial charge on Mn atoms, improving how well the substance attaches. Ultimately, aniline effortlessly reacts with HCOOH to generate N-phenylformamide without the need for a catalyst.
Similarly, another hydrogenation of nitroarenes was reported by Li and co-workers by using protic solvents and external bases.108 Extensive research has demonstrated that the breaking of H2 at the metal center and the nearby coordination sphere is significantly improved by the process of deprotonation or H-shuttling, which involves bases or protic solvents. Furthermore, the resulting cobalt hydride species at the atomic level have been proven to be highly reactive in reducing nitro groups, displaying exceptional effectiveness and wide-ranging selectivity in the hydrogenation of different nitroarenes. Eventually, a catalytic process has been suggested to clarify how SACs achieve their activity, guiding the shaping of SACs by enhancing the surroundings of active sites. The Co/C-PAQ SACs (Co single atoms supported on co-coordinated poly(amino-quinone)) were tested using the hydrogenation of nitrobenzene to aniline as a representative reaction. Although precious metal catalysts such as Pd, Pt, and Ru have demonstrated high catalytic activity in this reaction, their restricted accessibility and high expense greatly hinder their widespread use. Therefore, there is a strong interest in finding alternatives using earth-abundant metals, although these typically require harsh reaction conditions (temperatures above 383 K, hydrogen pressures between 20 and 50 bar) and extended reaction times due to their lower activity. In this study, reactions were performed under milder conditions of 353 K and 10 bar of H2. Various cobalt catalysts were tested for their catalytic performance. The reactions resulted in very low product yields in the absence of a catalyst or with the metal-free sample C-PAQ. However, the Co/C-PAQ catalyst managed to convert nitrobenzene to aniline completely within 2 h using a very low catalyst loading (0.2 mol% Co). As a result, Co/C-PAQ exhibited an impressive TOF of 398 h−1, surpassing most reported values, as illustrated in Fig. 14(d).
Shi et al.144 synthesized a range of size-tunable Cu/Al2O3 catalysts, from single Cu atoms to 9.3 nm, using atomic layer deposition (ALD), drawing inspiration from the impressive selectivity and coking resistance of Pd SACs. These catalysts were extensively analyzed using aberration-corrected HAADF-STEM, XAS, in situ XPS, and in situ TGA. During the semi-hydrogenation of acetylene in an excess of ethylene, it was observed that the Cu1 SAC demonstrated higher ethylene selectivity, but lower activity compared to other samples. Notably, the Cu1 SAC exhibited exceptional stability, contrasting sharply with the rapid deactivation of Cu particle samples. In situ TGA revealed that the Cu1 SAC significantly reduced coke formation by approximately 89%. These findings highlight the substantial impact of reducing metal particle size to single atoms on catalytic performance, suggesting a promising approach for designing selective and stable catalysts.
In another study, Han et al.145 presented the isolation of contiguous platinum (Pt) atoms and the formation of Pt–Zn intermetallic NPs as a strategy to enhance the selectivity of Pt catalysts. The researchers created a catalyst called PtZn/HNCNT by isolating contiguous Pt atoms into single atoms and forming Pt–Zn intermetallic NPs supported on hollow nitrogen-doped carbon nanotubes. They confirmed the structure using advanced microscopy and spectrometry techniques. The PtZn/HNCNT catalyst exhibited remarkable performance, achieving a conversion rate of over 99% and a selectivity of 99% in the hydrogenation of 4-nitrophenylacetylene to 4-aminophenylacetylene. This outperformed comparison samples with Pt isolated single atomic sites (Pt/HNCNT) and Pt NPs (Pt/CN). Additionally, DFT calculations reveal that the positive Zn atoms aid in the adsorption of the nitro group and the Pt–Zn intermetallic nanoparticles kinetically enhance the hydrogenation of the nitro group. Furthermore, Tian et al.146 successfully synthesized a mesoporous graphitic carbon nitride (mpg-C3N4) supported dual-atom Pt2 catalyst. The catalyst showed exceptional performance in selectively converting nitrobenzene to aniline with a conversion rate of over 99%, surpassing the performance of other supported single Pt atoms and ultra-small Pt NPs. First-principles calculations suggested that the unique activity of the Pt2 entities is attributed to the easy dissociation of H2 facilitated by the diatomic nature of Pt and the effortless desorption of the product.
Synergistic effects in bimetallic heterogeneous catalysis have been widely studied, but their mechanisms at the atomic level remain unclear. In the study by Fu et al.,147 Ir2Mo2(CO)10(η5-C5H5)2 was activated on TiO2 in an Ar atmosphere resulting in the formation of DSAC Ir1Mo1/TiO2. This arrangement contains individual atoms of Ir (Ir1) and Mo (Mo1) attached to TiO2, which is distinct from the previously documented bimetallic structures formed through H2 treatment. The dual single atom configuration was confirmed using aberration-corrected STEM, XAFS spectroscopy, and DRIFTS. The Ir1Mo1/TiO2 catalyst exhibited significantly enhanced catalytic activity and chemoselectivity in the hydrogenation of 4-nitrostyrene (4-NS) to 4-vinylaniline (4-VA) compared to the SACs Ir1/TiO2 and Mo1/TiO2 prepared from single-metal precursors using the same process. When Mo1/TiO2 was used as the catalyst, no products were detected. However, Ir1Mo1/TiO2 exhibited an outstanding chemoselectivity of 96.3% for 4-VA with a complete conversion of 4-NS within one hour. In contrast, Ir1/TiO2 under identical reaction conditions demonstrated poor chemoselectivity, showing only 37.8% chemoselectivity for 4-VA at an 87.1% conversion rate of 4-NS. In addition, two different products were produced: 4-ethyl nitrobenzene resulted from the specific reduction of the CC group and 4-ethylaniline was obtained from the complete reduction of both the nitro and CC groups, with yields of 29.3 and 32.8% (Fig. 15(a)). DFT studies revealed that the catalytic hydrogenation of 4-nitrosobenzene on well-dispersed Ir1 and Mo1 atoms supported on TiO2 is a thermodynamically favorable process with a calculated reaction energy of −3.15 eV. The critical step in the reaction pathway is the deoxygenation of 4-NS, which requires overcoming a rate-determining energy barrier of 0.80 eV, significantly lower than that on Pd(111). This pathway effectively utilizes the unique roles of Mo1 and Ir1, with Ir1 facilitating the adsorption and dissociation of H2, and Mo1 aiding in the adsorption of 4-NS and the formation of key intermediates, leading to the formation of water and overall reaction completion (Fig. 15(b)). So, the synergistic effect between Ir1 and Mo1 sites contributes to the enhanced catalytic activity. Therefore, these excellent studies signify that the SACs possess a wide scope of development and large-scale utilization in the selective hydrogenation reactions of nitroarenes to valuable aniline-based chemicals.
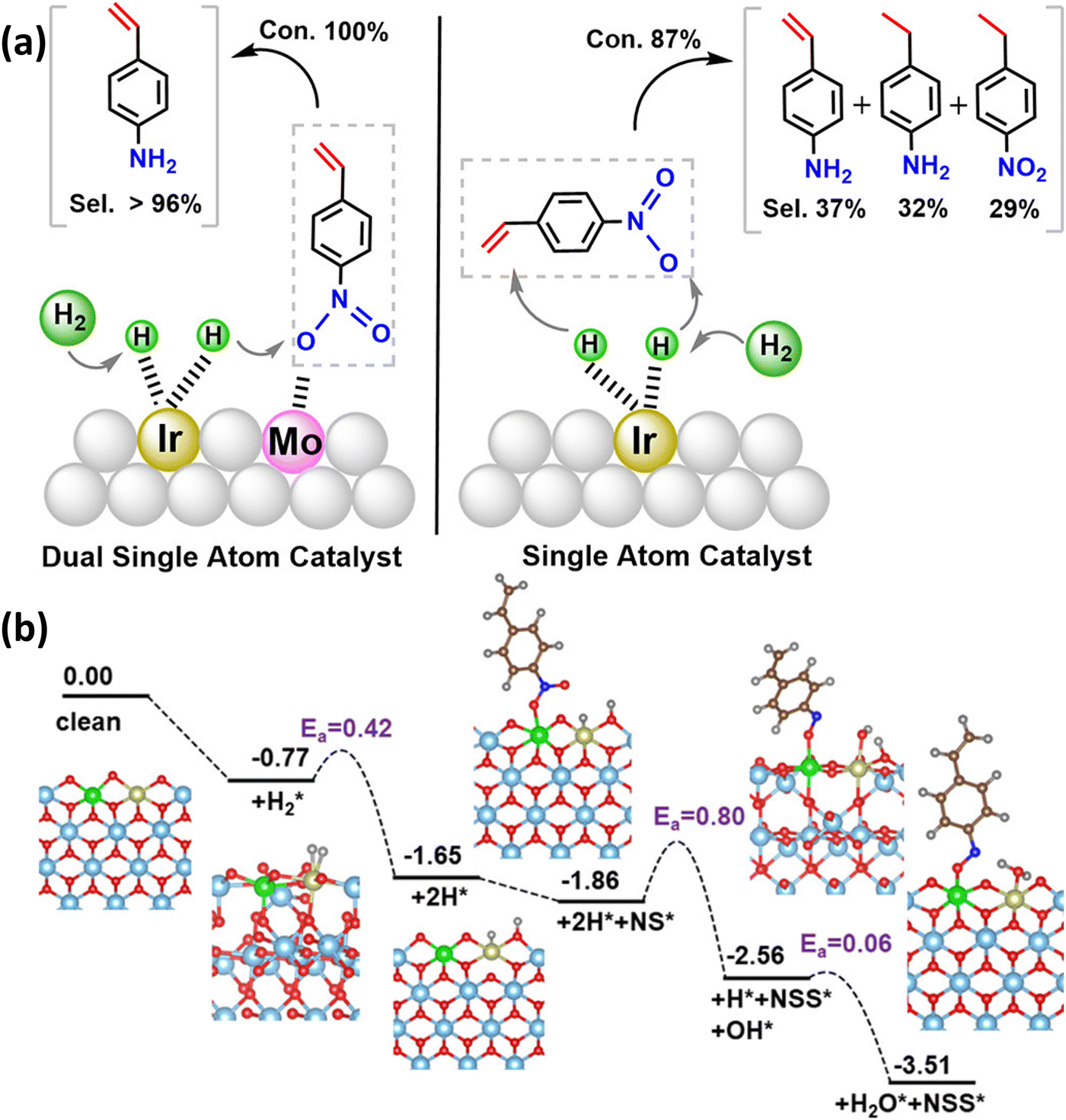 | ||
| Fig. 15 (a) Catalytic activity of Ir1Mo1/TiO2 and Ir1/TiO2 towards hydrogenation of nitroarenes and (b) energy profile for the conversion of 4-nitrostyrene to 4-nitrosostyrene on the surface of Ir1Mo1/TiO2 (Ti, Mo, Ir, O, H, N, and C atoms are represented by blue, green, gold, red, gray, deep blue, and brown balls, respectively, 4-nitrostyrene and 4-nitrosostyrene are represented by NS and NSS). Reproduced with permission.147 Copyright 2021, American Chemical Society. | ||
3.2. Hydroformylation reaction
Hydroformylation is basically a C–C coupling reaction used for the synthesis of aldehydes from olefins and syngas (a mixture of CO and H2). It is considered as one of the industrially important reactions for the synthesis of high value-added products, that is, aldehydes, from syngas in one step. Though the main product of hydroformylation reactions is aldehydes, in some cases, value-added ketones can also be formed.148–151 There are some crucial applications of aldehydes, like the synthesis of alcohols, amines, and carboxylic acids, and they also act as an important feedstock for dialkyl phthalates as plasticizers. The primary focus in the hydroformylation process is controlling regioselectivity, as the desired product, whether linear or branched aldehydes, depends on its intended application. Traditionally, homogeneous Rh catalysts with different phosphorous ligands were extensively used in hydroformylation reactions due to their high catalytic activity and selectivity. However, the homogeneous nature of Rh species limits their large-scale practical usage due to the difficulty in separation and recycling of the catalyst. Heterogeneous Rh based catalysts were also explored for this process because of their high stability and ease in catalyst recycling, but due to their low activity and selectivity, their application was limited as well. Due to these reasons, SACs have gained a lot of attention as a new field of heterogeneous catalysts. They combine the usage of homogeneous catalysts as isolated metal atoms and heterogeneous catalysts as grafted on support material to make a recyclable catalyst. It was also observed that the leaching problem of metal species was also minimized in the SACs. Additionally, phosphine ligands built-in copolymer-supported Rh catalysts were also explored for olefin hydroformylation which show very high regioselectivity and activity.149,152–157Recently, in 2021, Qin et al.152 reported hydroformylation of ethylene to produce 3-pentanone using Ru SACs supported on activated carbon having 83.3% selectivity and 5.9 mol molRu−1 h−1 yield of the product. To check the catalytic activity, both Ru SACs and Ru NPs were employed for the hydroformylation of ethylene at 150 °C using syngas and it was observed that instead of the conventional product of ethylene hydroformylation, i.e., propanal and propanol, 3-pentanone was formed with high selectivity when using Ru SACs whereas in the case of Ru NPs ethane selectivity was increased up to 52.1%. In 2022, Javier et al.156 also synthesized a Ru single-site catalyst supported on a N-doped carbon matrix for 1-hexene hydroformylation having high activity and regioselectivity. The reaction was performed at 150 °C using syngas and almost 92% of linear aldehyde was obtained as a major product. The turnover frequency (TOF) was calculated based on size and it was found that high TOF is due to single atoms present in the catalyst (Fig. 16(a)). In a synthesized catalyst, the nitrogen atom plays a dual role: formation of active sites and stabilization of active sites under different reaction conditions. In 2023, Zeng et al.153 also tried to synthesize a rhodium-based catalyst for the application of alkene hydroformylation where the phosphorous heteroatoms were impregnated into a porous carbon support (Rh/P-mC) (Fig. 16(b)). The model reaction was done with styrene by using syngas and toluene as a solvent at 120 °C for 24 h. It was found that linear aldehyde was formed as a major product, but as soon as the steric factor increases at CC, branched aldehyde was formed majorly.
 | ||
| Fig. 16 (a) Schematic representation of the hydroformylation of 1-hexene using the Ru@NC catalyst. Reproduced with permission.156 Copyright 2022, American Chemical Society. (b) Schematic illustration of hydroformylation of alkene using Rh/P-mC. Reproduced with permission.153 Copyright 2023, Elsevier Publishers. (c) Representation of the potential energy pathway for the formation of OV on the Rh/CeO2 surface (color legend: blue, Rh; white, H; red, O; creamy white, Ce) and (d) Gibbs free-energy pathway for hydroformylation reaction on Rh/CeO2 and Rh/CeO2-1Vo(B) catalysts. Reproduced with permission.154 Copyright 2023, American Chemical Society. | ||
Furthermore, Jurado et al.155 reported Rh SACs supported on graphitic carbon nitride (g-C3N4 or ECN). ECN allows the introduction of SACs in its s-triazine rings. The as-synthesized catalyst (Rh-ECN) was employed for the hydroformylation of styrene using syngas, and linear/branched aldehyde was formed in a 1.5 ratio. Moreover, Zheng et al.154 reported Rh SACs supported on CeO2 for the hydroformylation reaction of propylene. It was observed that when Rh/CeO2 was calcined at high temperature it showed a high number of OV which results in high activity. Further, DFT studies were also conducted and it was observed that the formation of OV occurred with the increasing calcination temperature. At first, the hydroxyl group was introduced on the surface of CeO2 and the Rh atoms were present on the bridge site and/or hollow site surrounded by 4 –OH groups. Then on increasing the temperature, initially surface –OH groups were released as H2O and other Rh sites were formed. On further increasing the temperature up to 600 °C, it led to the formation of Rh/CeO2, whereas when calcination was done up to 800 °C, drastic changes in oxygen vacancies were observed and led to the formation of Rh/CeO2-1Vo (Fig. 16(c)). The mechanism of propylene hydroformylation includes the following steps: CO desorption on HRh(CO)2, activation of propylene and hydrogenation, CO binding, CO insertion, hydrogenolysis, and reductive elimination of butanal. It includes the formation of 4 transition states. The Gibbs free energy profile diagram for the formation of butanal and propane from propylene using the catalysts Rh/CeO2 and Rh/CeO2-1Vo depicts that butanal formation has a low energy barrier for the rate-determining step compared to propane formation, meaning the reaction is highly chemoselective for butanal formation (Fig. 16(d)).
Further, Li and coworkers reported the hydroformylation of propene to yield linear butyraldehyde in a continuous fixed bed reactor over Rh-single atoms supported on the POP synthesized from the copolymerization reaction of vinyl biphephos and tris(4-vinphenyl) phosphane (3vPPh3) under solvothermal conditions, named Rh/CPOL-bp&P.158 These copolymer self-supported heterogeneous SACs showed excellent performance in the hydroformylation reaction of propene to yield butyraldehyde. Among all the catalysts, Rh/CPOL-1bp&10P performed the best with excellent regioselectivity (linear:branch ratio = 24), high TOF greater than 1200 h−1, and very high stability greater than 1000 h at 70 °C and 0.5 MPa. Further, the catalytic performance of the catalyst increased by increasing the ratio of vinyl biphephos:3vPPh3 till 0.13 which was due to the enhanced concentration of biphephos units with the promising stereoelectronic and steric environment in the skeleton. Additionally, the Rh loading of 0.05 wt% yielded the best results (TOF = 3290 h−1 and linear:branched = 65) but on further increasing the loading amount, a decrease in the overall activity was observed which suggests the importance of optimum concentration of catalytic centers.158 It was also observed that the reaction temperature has a positive impact on the TOF but negative impact on the linear:branched ratio up to 110 °C while increasing the temperature beyond this has a deteriorating impact on both the parameters. This highly effective solvent-free system was also observed to fit in the requirements of green industrial processing since it is easy to use and emits virtually no waste, in contrast to liquid–liquid biphasic (LLB) and ionic liquid (IL) systems.158 Therefore, this study provides a promising investigation on the design of effective SACs supported on porous framework supports for an industrially important reactions.
An interesting investigation was reported by Feng et al.159 for ethylene hydroformylation over a single site Rh anchored on a POP support made from the tri-4-vinyltriphenylphosphine (PPh3) monomer via solvothermal polymerization. The molecular structure of the Rh1-POP was confirmed to be HRh(CO)(PPh3-frame)3, which is comparable to the homogeneous HRh(CO)(PPh3)3 catalyst. In comparison to the analogous homogeneous catalyst, it is proposed that the abundant PPh3 in POPs provided the core metal ions with a rich electron environment and enhanced heterogeneous activity. Interestingly, the effect of sulfur poisoning and regeneration on the performance of the system was investigated as it is a key challenge in metal-based catalysts.159 In the catalytic experiments, the initial assessment of the catalytic system was done with a mixture of gas feed (CO/H2/C2H4 at 120 °C and 1.0 MPa) and it was observed that an excellent TOF of 4317 molCO molRh−1 h−1 was achieved at the time on stream (TOS) of 16 hours but the reaction system was highly poisoned after the H2S co-feed was used with 1000 ppm H2S, thereby decreasing the TOF to 229 h−1 within 3 h. It was reasoned that the decrease in the TOF after sulfur poisoning is due to the fact that severe electronic charge decrease was observed in the isolated Rh atoms and the phosphine ligand. Even though the H2S co-feed severely affected the reaction performance, the catalytic system could be self-recovered after withdrawing H2S which was not in the case of Rh NPs. In the hydrogenolysis of (C2H5CO)Rh(CO)(PPh3-frame)2, the rate-determining step gave H2S a chance to compete with H2 coordination.159 Since (SH)Rh(CO)(PPh3-frame)2 had the lowest Gibbs energy and C2H4 was unable to coordinate due to the high Gibbs free energy barrier, H2S coordination revolutionized Rh1/POPs and caused sulfur poisoning of Rh1/POPs in the H2S co-feed. Therefore, this report is promising for the advancement of sulfur-related chemistry and helps comprehend the structure–activity relationship of ethylene hydroformylation on a single-Rh-site from the perspectives of electronics and geometry.
3.3. Coupling reactions
The industrial importance of coupling reactions has been very well known from a long time, and regular efforts are made to develop efficient and economic reaction protocols for coupling reactions under mild conditions. In this regard, SACs have emerged as promising catalysts exhibiting excellent activity in various coupling reactions such as Sonogashira coupling, Heck coupling, Suzuki coupling, C–C coupling reaction of aryl halides, and so on. Sonogashira coupling is a C–C bond formation reaction that includes the bond formation between Csp2 and Csp and helps to introduce alkynyl moieties and shows application in the pharmaceutical industry.160 Suzuki cross coupling reaction is also a C–C bond formation reaction which was extensively studied by using SACs. Generally, in Suzuki coupling reactions, aryl bromides and iodides are used as substrates because of their high reactivity, and aryl chlorides though comparatively cheaper and readily available, find limited applications due to their chemical inertness, which is a major issue. Various research works have already been done to solve the problem of activation of aryl chlorides by using metal–organic frameworks, porous nanorods of CeO2 and other nanoporous catalysts. However, SACs show a higher TOF value than the catalysts that have already been reported.161C–C bond formation reaction was also studied using SACs. In 2020, Ding et al.162 reported a palladium SAC supported on bimetallic oxides (Pd–ZnO–ZrO2) for Suzuki C–C coupling reaction. The model reaction was performed between bromobenzene and p-tolylboronic acid using K2CO3 as a base and interestingly the reaction was performed under phosphine-free and open-air conditions at room temperature. Additionally, the solvents also affect the reaction conditions significantly. When the model reaction was performed using various organic solvents, it was found that the product was formed only in the case of water as a solvent. Vice et al.163 also synthesized a similar palladium SAC with some surface modification for the same Suzuki C–C coupling reaction between bromobenzene and p-tolylboronic acid. The modification was done using a catechol-type monolayer on the palladium surface (Pd/CeO2). This monolayer shows a dual advantage for the Suzuki C–C coupling reaction. It reduces the activation energy barrier and increases the catalytic activity rate 4 times as it shows the π–π interaction with substrates. Furthermore, Tao et al.164 also reported palladium-based SACs anchored in 3D-ordered macroporous ceria (Pd-SAs/3DOM-CeO2) for similar applications. The Pd single atoms initiate surface OV and get stabilized in CeO2 by overcoming the issue of leaching and aggregation. The synthesized catalyst was used for application in Suzuki coupling between phenylboronic acid and various aryl halides using K2CO3 as a base and a 1:1 mixture of EtOH and H2O as a solvent under ambient reaction conditions in an air atmosphere.
In 2023, Poier et al.160 synthesized a palladium SAC (Pd1@NC) for application in Sonogashira coupling reaction between iodobenzene and phenylacetylene by using CuI as a co-catalyst and PPh3 as a ligand. CuI plays an important role as a co-catalyst. It not only helps in alkyne activation but also promotes the transmetallation step. Meanwhile, the phosphine ligand enhances catalytic activity and affects reaction kinetics. Fig. 17(a) depicts the large batch of synthesized catalysts and their schematic structure at the particle and nanometer scale. The C–C coupling reaction between iodobenzene and phenylacetylene was studied using various different solvents and bases. The model reaction was done using NEt3 as the base and MeCN as the solvent at 80 °C for 24 h in an inert atmosphere. The same reaction conditions were employed for the cross-coupling reaction between iodoaniline and 2-methylbut-3yn-2-ol to produce the erlotinib intermediate (Fig. 17(b)). Recently, in 2024, Chen et al.161 reported Pd SACs supported on N-doped hollow carbon nanosheet assemblies (Pd/HCNAs) for Suzuki cross-coupling of aryl chlorides. This methodology solves the issue of chemical inertness of aryl chlorides under mild reaction conditions. The model reaction was done with chlorobenzene and phenylboronic acid by using Na2CO3 as a base and water and ethanol as solvent and catalyzed by the as-synthesized catalyst at 50 °C for 4 h. Recyclability studies were done with 3 catalysts: Pd1/HCNAs (having 1.0 wt% Pd-loading), Pd/HCNAs (having 3.0 wt% Pd-loading) and Pd/C. It was found that even after 5 cycles, Pd1/HCNAs showed good catalytic activity and conversion was still above 92%, whereas Pd/HCNAs and Pd/C showed a significant decrease in the yield, which were 16% and 82% respectively (Fig. 17(c)). Therefore, with a large scope of development, the existing studies involving the utilization of SACs in organic coupling reactions have shown excellent potential which will surely advance the synthesis of high value chemicals at an industrial level.
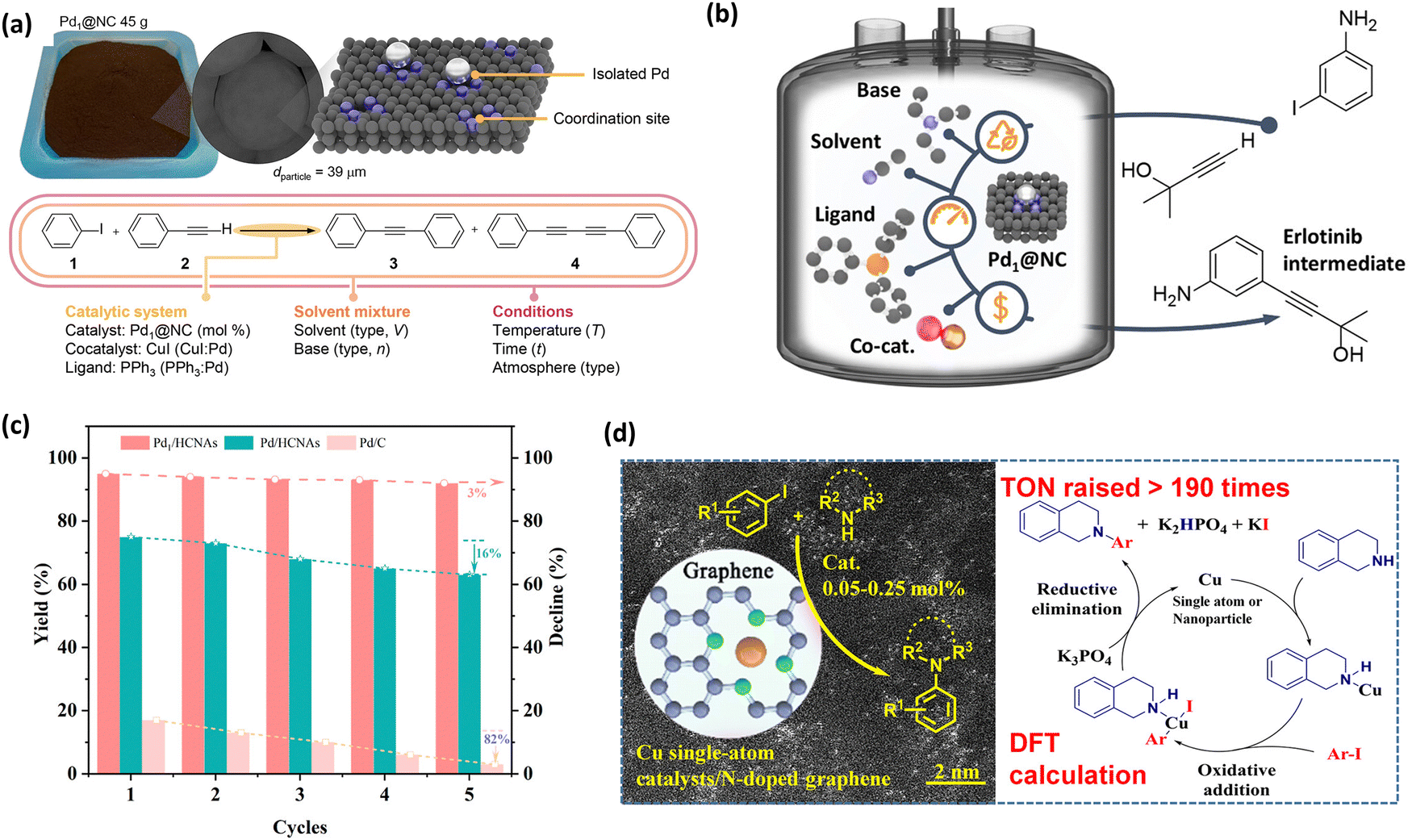 | ||
| Fig. 17 (a) Schematic representation of Sonogashira coupling of aryl iodides and acetylenes over Pd1@NC SACs and the formation of the Erlotinib intermediate and (b) representation of a large batch of the Pd1@NC catalyst and its structure at the particle and nanometer scale (color legend: blue, black, and gray represent N, C and Pd, respectively) and schematic depiction of the Sonogashira coupling for the synthesis of diphenylacetylene and the undesired Glaser homocoupling product. Reproduced with permission.160 Copyright 2023, American Chemical Society. (c) Recyclability studies using Pd1/HCNAs, Pd/HCNAs, and Pd/C catalysts. Reproduced with permission.161 Copyright 2024, American Chemical Society. (d) Schematic illustration of C–N bond formation reaction catalyzed by Cu SACs/NG along with the catalytic mechanism. Reproduced with permission.165 Copyright 2022, American Chemical Society. | ||
Conventionally, C–N bond formation reactions were reported only by using traditional homogeneous catalysts like copper powder, CuBr and CuI due to their low cost and less toxicity but a major drawback associated with them is difficulty in catalyst separation and in recyclability. However, as we have already discussed, these drawbacks can be overcome effectively by using SACs.165 In this context, Zhang et al.165 reported Cu SACs supported on N-doped graphene (Cu SACs/NG) for C–N bond formation reactions between iodobenzene and 1,2,3,4-tetrahydroisoquinoline (THIQ) using K3PO4 as a base and EtOH as a solvent for 36 h at 80 °C. Cu–N4 moieties in Cu SACs/NG resist leaching providing good recyclability and give more than 91% yield and a turnover number of 3800, performing far greater than the already reported catalysts. Based on DFT calculations, the reaction pathway was proposed which includes various steps: in the initial state (IS), THIQ reacts on the surface of Cu SACs/NG to form the first intermediate (IM1), which undergoes oxidative addition with aryl iodide to form the third intermediate state (IM3), which on further reductive elimination leads to the formation of the final state (FS) which is an aromatic aminated product (Fig. 17(d)).
Further, the direct synthesis of biphenyls via the oxidative homocoupling of benzene with O2 as the oxidant is very challenging, especially using heterogeneous catalysis due to the inert sp2 C–H bonds of benzene. In this context, Liu et al. reported the facile synthesis of Pd single atoms supported on a POP support (PdII@PDMS) with sulphonic acid and carboxyl acid groups for the homocoupling of benzene. In the catalytic reaction performed at 8 atm O2 pressure and 120 °C, the PdII@PDMS catalyst showed the best activity with excellent selectivity and yield of 98.3 and 26.1%, respectively. Owing to a very small amount of catalyst used in the reaction (Pd, 0.036 mol% based on benzene), a turnover number of 363 was obtained which was higher than that obtained for the homogeneous Pd(OAc)2 catalyst. The catalytic system exhibited excellent recyclability up to three reaction cycles (20.7% yield in the third cycle) and it was observed from the hot filtration test that no product formation takes place when the catalyst is removed, which confirms that the reaction happens at the surface of the catalyst and there is no role of leached Pd entities in the reaction. In the reaction mechanism, initially the benzene reactant migrates to the catalyst surface and accesses the Pd(O3SCF3)+ active sites which are formed by the interaction of Pd(OAc)2 and CF3SO3H. Furthermore, the σ-arylpalladium intermediate is generated by the electrophilic substitution followed by reaction with another benzene to form biphenyl Pd(II) species. At last, reductive elimination and desorption steps lead to the yield of the biphenyl product and the free catalyst surface. Hence, this report delves into the field of the synthesis of high performance SACs with controlled electronic and geometric features for a very challenging benzene aerobic homocoupling reaction.
3.4. Bond cleavage reactions
The most challenging task in organic reactions includes the selective cleavage of the C–C single bond, which is also a significant issue in biomass (specifically lignin) valorization. Lignin degradation into value-added products such as phenolic aldehydes, ketones and acids is still challenging due to its polymeric structure connected through C–C and C–O bonds. Selective cleavage of C–C linkage is more problematic compared to that of C–O linkage due to its high dissociation energy.111,166,167 Various research works have already been done in the field of oxidative depolymerization of lignin using various catalyst under thermal conditions. C–O bond cleavage is the most studied reaction as plenty of these linkages are present compared to C–C bond linkage and also due to its low dissociation energy.168,169 However, these methodologies suffer from various drawbacks due to the requirement of cost-effective non-noble catalysts. SACs, as discussed earlier due to the uniform active sites, high atomic efficiency, and the advantages of both heterogeneous and homogeneous catalysts offers to solve the above issues. Moreover, non-noble SACs can also be employed for selective C–O/C–C/CC bond cleavage thermally under ambient conditions.170 Furthermore, oxidative cleavage reaction of C–N bonds in N,N-dialkylamines or N-alkylamines into primary amines is another critical reaction in organic chemistry as amines act as precursors of high value-added materials such as drugs and agrochemicals. However, this methodology also faces some difficulties in the presence of 3 widely known competitive reactions: N-oxidation to produce N-oxides, carbonylation of α-C to amides and oxidation of C–C single bonds to enamines. The most influential step in C–N bond cleavage reactions includes the release of α-H in N-alkylamines which is thermodynamically unfavorable in thermal-catalytic methodology. So, SACs are one of the potential catalysts for the oxidative bond cleavage of C–N linkage.171
In 2019, Liu et al.168 reported a cobalt based SAC (Co–N–C) for the oxidative C–O bond (β-O-4) cleavage reaction used in lignin valorization. The model reaction was done using 2-(2-methoxyphenoxy)-1-phenylethanol (MPP-ol; which is a dimeric compound of lignin consisting of Cα–OH, Cα–Cβ and Cβ–O bonds), NaOH as a base and MeOH as a solvent at 150 °C for 4 h under air conditions to produce methyl benzoate and guaiacol as the major products along with a little amount of benzoic acid. However, adding NaOH enhances the esterification rate of benzoic acid with methanol and converts it into methyl benzoate. As depicted in Fig. 18(a) the as-synthesized catalyst (Co–N–C SAC) shows good recyclability with high catalytic activity and yield of the product up to 4 cycles and even after that the conversion only reduces from 95% to 82%. Garcia et al.169 also reported a similar kind of cobalt catalyst, Co–N–C, for the transfer hydrogenolysis of guaiacyl glycerol-β-guaiacyl ether (common linkage in lignin) using HCOOH as a hydrogen source using NEt3 which favors C–O bond cleavage reaction and a 1:1 mixture of EtOH and H2O as a solvent at 150 °C for 2 h. Guaiacol and isoeugenol (2-methoxy-4[(E)-1-prophenyl]phenol) were formed as the major products. It was found that among different ratios of synthesized catalyst (CoIINx), having a low cobalt content of 0.60 wt% shows the highest catalytic activity and selectivity towards β-O-4 (C–O) linkage.
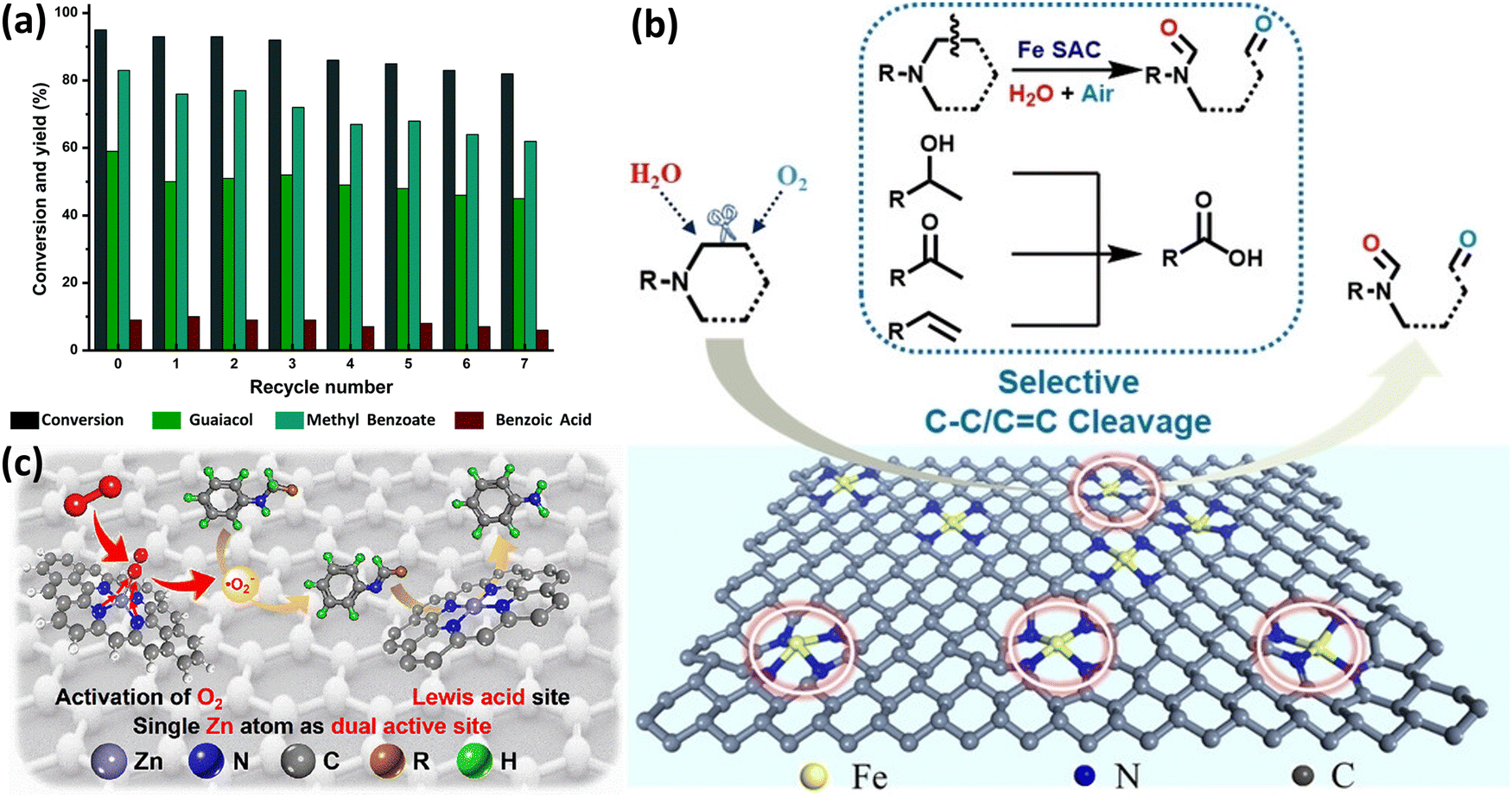 | ||
| Fig. 18 (a) Results of the recycling test over the Co–N–C catalyst. Reaction conditions: 20 mg catalyst, 0.2 mmol NaOH, 0.2 mmol MPP-ol, 5 mL MeOH, 1 MPa air, T = 150 °C, t = 4 h. Reproduced with permission.168 Copyright 2019, Royal Society of Chemistry. (b) Representation of the oxidative bond cleavage reaction of C–C/CC using the Fe–N–C SAC. Reproduced with permission.172 Copyright 2023, Wiley VCH. (c) Illustration of the oxidative C–N bond cleavage reaction in N-alkylamines and N,N-dialkylamines over ZnN4-SAC. Reproduced with permission.171 Copyright 2023, American Chemical Society. | ||
Recently, Qi et al.172 synthesized a non-noble Fe–N–C SAC consisting of highly dispersed iron centers for oxidative bond cleavage of C–C/CC in amines, secondary alcohols, ketones, and olefins using O2 and H2O thermally under milder conditions (Fig. 18(b)). The key to the successful reaction was the iron center that not only activates O2 but also H2O to produce 1O2 singlet and hydroxide species simultaneously. These species increase the product yield from <1% to >90% as they enhance the selectivity towards the C–C bond cleavage reaction, leading to high chemo-selectivity. In addition, Qin et al.173 synthesized an iron-based SAC containing the iron(III) nitride moiety (FeN3-SAC) which has the capability to show similar activity to enzymes and it was applied in the C–C bond cleavage reaction. This catalyst was proved to be effective being involved in both C–C bond cleavage and cyanation. The model reaction was performed using different types of reactants such as 1,2-diols, secondary alcohols and ketones to produce nitriles in high yield using O2 as an oxidant and NH3 as a nitrogen source. Interestingly, this methodology was found to be effective for the preparation of nitriles as it is cost-effective because only H2O is produced as a by-product and it did not involve any toxic solvents and reagents.
In 2023, Qin et al.171 reported a biomass-derived Zn SAC (ZnN4-SAC) for oxidative C–N bond cleavage reaction in N-alkylamines and N,N-dialkylamines in the presence 3 competitive reactions as already discussed. The model reaction was performed by using N-ethylaniline as a substrate along with 1,4-dioxane, NH3:H2O and O2 at 150 °C for 4 h to produce aniline as a major product. The synthesized catalyst successfully activates molecular O2 to produce reactive oxygen species (˙O2−). From DFT calculation also it was found that Zn atoms act as Lewis acidic sites and reduce the activation energy for the addition of the water molecule in the CN bond of the imine intermediate followed by cleavage of the C–N bond to generate aniline (Fig. 18(c)). In conclusion, the cleavage of C–C bonds in large organic compounds is highly beneficial as various small value-added chemicals can be obtained for multiple applications, and therefore, the growing use of SACs in this field is very much needed due to the high activity and selectivity which will aid the transition towards a sustainable chemical industry.
3.5. Oxidation reaction
In organic transformation reactions, the oxidation reaction is one of the important chemical processes that produce a variety of oxygenated molecules using different oxidizing agents such as molecular oxygen, dichromate, permanganate, and peroxide. To date, a variety of SACs have been explored for the oxidation of alcohols, hydrocarbons, silanes, etc.174,175 Selective oxidation of alcohols is one of the important industrial processes and is widely used in organic synthesis for the production of aldehydes. Some of the crucial applications of aldehydes have already been discussed previously like their ability to act as an intermediate and building blocks for several high value-added chemicals. However, these reactions have gained a lot of attention from researchers due to the efficient activation of molecular oxygen under mild reaction conditions. In general, the synthesis of new compounds having a singlet state from the reaction between organic molecules (mostly in singlet state) and molecular oxygen O2 (triplet ground state) is forbidden based on Winger's spin selection rules. Moreover, several methods have been developed to disperse single metal active sites on different supports like graphene, metals, metal oxides, and porous materials as the performance and electronic structure of SACs depend on the bonding between single metal atoms and the identity of the support.175–179 Selective oxidation of silanes using SACs was also studied extensively. It is regarded as one of the most important reactions as silanols have been used as building blocks in polymer chemistry. Conventionally, this reaction possesses several challenges like the formation of unwanted by-products like siloxanes and the usage of very strong oxidants. However, the current strategy for the selective oxidation of silanes using H2O as an oxidant has several advantages like low-cost and H2 as the only by-product.180 Oxidation of hydrocarbons to produce high value-added chemicals mainly ketones or alcohols as the main product has also been studied by using SACs. Acetophenone and its derivatives were mainly studied for their utilization in pharmaceuticals, agrochemicals, perfumes, etc. Conventionally, acetophenone was synthesized by Friedel-Craft acylation reaction and from the oxidation of styrene by KMnO4 but these were very challenging tasks as they led to the formation of by-products that were highly corrosive and toxic.181Recently, Li et al.177 reported a single atom cobalt catalyst atomically dispersed on nitrogen-doped graphene (Co-NG) for selective oxidation of benzyl alcohol to synthesize benzaldehyde having 94.8% conversion and 97.5% yield and the catalyst showed a TOF of over 500 h−1. The model reaction was done using DMF as a solvent, Co-NG as a catalyst, and O2 as an oxidant with a flow rate of 20 mL min−1 at 130 °C for 5 h. The same reaction was also tried by using air as an oxidant, but the catalytic efficiency was lower compared to that when O2 was used as the oxidant. Zhang et al.178 reported a ceria supported Au SAC (Au1/CeO2) for the synthesis of benzaldehyde from benzyl alcohol under solvent free conditions. The synthesized catalyst was employed in the selective oxidation of benzyl alcohol using O2 as an oxidant under solvent-free conditions at 150 °C for 8 h having 15.9% conversion and 98% selectivity with a TOF of approx. 3000 h−1. The proposed reaction pathway (Fig. 19(a)) for alcohol oxidation using Au1/CeO2 includes the following steps: oxidation of alcohol to aldehyde, formation of H2O as a by-product and formation of oxygen vacancies, adsorption of O2 at the vacancy site, again oxidation of alcohol to aldehyde, regeneration of the catalyst along with the formation of H2O as a by-product. Otake et al.179 synthesized single metal atom vanadium species incorporated in two highly crystalline MOFs: Hf-MOF-808-V [Hf6(μ3-O)4(μ3-OH)4(OH)6(H2O)6(BTC)2] and Zr-NU-1000-V [Zr6(μ3-O)4(μ3-OH)4(OH)4(H2O)4(TBAPy)2]∞ (H3BTC = benzene-1,3,5-tricarboxylic acid and H4TBAPy = 1,3,6,8-((p-benzoic-acid)pyrene). The model reaction was done by using 4-methoxybenzyl alcohol to produce 4-methoxybenzaldehyde using O2 as an oxidant, toluene as a solvent and triethylamine as a base at 105 °C for 8 h. The yield was calculated through GC yield by using mesitylene as an internal standard. The yield obtained with the two catalysts (Hf-MOF-808-V and Zr-NU-1000-V) was different (95% and 75% respectively) despite having the same selectivity (99%). Shang et al.176 reported atomic and nano-dispersed Pd catalysts on an Al2O3 support (Pd1/Al2O3 and Pd/Al2O3 respectively) for selective oxidation of cinnamyl alcohol by using O2 as an oxidant and toluene as a solvent at 80 °C for 8 h. The single atom Pd1/Al2O3 catalyst (92% conversion and 91% selectivity) showed high catalytic activity as compared to the nano Pd/Al2O3 catalyst (29% conversion and 89% selectivity). The proposed reaction pathway shown in Fig. 19(b) includes the following steps: (1) partial dehydrogenation reaction between Pd atoms and adsorbed allylic alcohols through charge transfer; (2) simultaneous generation of singlet O2 species; and (3) desired product formation by the reaction between singlet oxygen species and partially dehydrogenated intermediates.
 | ||
| Fig. 19 (a) Proposed mechanism for the oxidation of primary alcohols based on the Au1/CeO2 catalyst. Reproduced with permission.178 Copyright 2018, Wiley VCH. (b) The proposed reaction pathway for the oxidation of silane using H2O as an oxidant and the Au1/mpg-C3N4 catalyst. Reproduced with permission.180 Copyright 2018, Wiley VCH. (c) The proposed reaction mechanism for the oxidation of cinnamyl alcohol using Pd1/Al2O3. Reproduced with permission.176 Copyright 2020, Elsevier Publishers. | ||
SACs have also been explored for the oxidation of silanes and hydrocarbons as discussed earlier.180,181 In 2018, Chen et al.180 synthesized single site Au anchored on polymeric mesoporous graphitic carbon nitrides (Au/mpg-C3N4) for oxidation of silane using water as an oxidant. The model reaction was performed with diphenylmethyl silane using water as an oxidant and acetone as a solvent at 25 °C for 30 min. To check the catalytic activity of the synthesized catalyst, three parallel reactions were performed using three different Au catalysts, which include HAuCl4, Au nanoparticles and Au/mpg-C3N4, and a maximum yield of almost 99% was achieved by using single-site Au catalysts and the formation of siloxanes as a by-product was not observed. The proposed reaction pathway depicted in Fig. 19(c) includes three main steps: (1) formation of Si–AuIII–H by oxidation insertion of the single site Au1 catalyst in the Si–H bond; (2) formation of silanol and H2 gas; and (3) regeneration of the single atom Au1 catalyst. Further, Xiong et al.181 reported cobalt single atom catalysts (Co SACs) having 23.58% metal loading supported on carbon nitride for oxidation of hydrocarbons. The synthesized catalyst was employed in the oxidation of ethyl benzene for the production of acetophenone using air as an oxidant at 120 °C for 24 h. The catalyst showed high catalytic efficiency with 46% conversion, 97% selectivity and a TOF of 19.6 h−1. In conclusion, the oxidation of alcohols using SACs represents a significant advancement in organic transformation reactions. SACs, with their unique structure and high atom utilization, shows exceptional catalytic activity and selectivity for alcohol oxidation reactions. By harnessing the unique electronic properties of isolated metal atoms anchored on suitable supports, SACs enable efficient conversion of alcohols to aldehydes, ketones, or acids under mild conditions, often with minimal by-products. This approach not only aligns with green chemistry principles but also paves the way for designing more economical and sustainable catalytic processes in the future.
4. Summary, key challenges and their countermeasures
In summary, various metal and non-metal single atom based heterogeneous catalysts were utilized for the organic transformation reactions. The distinct structural and electronic properties of SACs were helpful for the selective conversion of reactants in organic reactions. Notably, in this review, we have comprehensively summarized the brief history of SACs, the synthesis of metal and non-metal SACs, and the advanced characterization techniques used to examine the successful synthesis of SACs. Additionally, the use of SACs towards organic transformation, such as hydrogenation reaction, coupling reaction, oxidation reaction, bond cleavage, etc., and the reaction mechanism are also discussed in detail. Despite much development in the field of single atom catalysis, there is still room for improvement and new opportunities, which are discussed as follows and represented in Fig. 20: | ||
| Fig. 20 Key future challenges to be focused upon to aim for industrialization in the field of single atom catalysis. | ||
(1) The current studies in the field of SACs have marked the potential of SACs in various applications ranging from thermal catalysis to photocatalysis. Considering the potential and impact of SACs on science, the primary challenge lies in their synthesis. It is difficult to stabilize single atoms due to their very high surface energy, which they tend to reduce by forming clusters or NPs that may or may not be as catalytically active as SACs. In this regard, future research should focus on developing highly accurate and reliable synthetic methods like trapping by defects, anchoring at coordination sites, mitigating thermal motion of molecules, etc., to obtain highly dispersed single atoms even at high loadings to use the SACs to their full potential. This will also help when this technology reaches the maturity to be considered for industrial deployment where the synthesis of bulk catalysts is required.
(2) It should be noted that during the synthesis of SACs, the selection of the mononuclear complexes should be critically made as per the properties of the support. Also, this will play a crucial role in the enhancement of metal–support interactions between the single atoms and the support, which truly enhances the catalytic activity due to effective and efficient charge transfer.
(3) The research on SACs has extensively focused on the synthesis of noble metal-based SACs due to their unmatched activity followed by the outstanding efforts to synthesize non-noble metal SACs as a potential replacement of costly noble metals. Apart from metallic SACs, there has been negligible focus on the identification of non-metal single atomic sites that are catalytically active due to the difficulty in elucidating the catalytically active sites. Therefore, there is a lot of scope in the field of non-metal-based SACs for a variety of applications which is highly expected soon.
(4) Many organic transformation reactions proceed under harsh conditions; therefore, SACs can be unstable under these conditions, which can cause challenges like metal leaching. Thus, the characterization (in situ and operando spectroscopy) of these catalysts under realistic operating conditions and post-catalysis changes in the metal loading and other properties is much needed for further detailed investigation. Eventually, progress in the spatially precise characterization of SACs would pave the way for the clear identification of the active sites. Also, the rigorous assessment of the stability of SACs under various reaction conditions compared to the NP and cluster-based systems is highly necessary to guide the catalyst design.
(5) It is very well known that large organic compounds with multiple functionalities like CC, CO, NO2, etc. are used in the industries as substrates for various platform chemicals, and non-selective hydrogenation of these functional groups leads to various additional steps in the chemical process which adds up to the cost and efforts. Therefore, it is highly desired that future studies should focus more on designing highly selective SACs for hydrogenation reactions to prevent over-hydrogenation of the substrate and avoid additional steps like product separation. These efforts will bring down the cost, feedstock amount, efforts, and energy requirement of the overall process.
(6) The efforts related to the large-scale synthesis of SACs are limited, with only a few reports available. This is because of the time-consuming synthetic procedures and the requirement of costly starting materials and sophisticated characterization instruments. Therefore, additional investigation is still necessary to maximize the synthesis process's scalability and guarantee that the final SACs perform consistently and steadily over batches. Furthermore, efforts are needed to develop SACs with a high metal content as low metal loadings continue to limit their application in various organic transformation processes.
(7) It is very well known that various factors, including coordinating atoms, support material, coordination number, arrangement, and metal species, influence the performance of SACs. Despite being the most widely utilized method of creating SACs today, experimental trial-and-error synthesis necessitates significant laboratory work and time for material investigation, particularly when done without supervision or prescreening. In this regard, computational screening and machine learning can be highly beneficial. Also, the creation of sophisticated high-throughput processes and big data mining tools will enable quick and effective screening of SACs and improve our knowledge of the links between their structure and activity. Furthermore, one of the next research hotspots in this area will be the use of machine learning to direct and optimize the synthesis and structural optimization of SACs, which will encourage the rapid development of the research area of SACs.
In a nutshell, this review focuses on the recent advancements in the synthesis of SACs and their utilization in organic transformation reactions. In addition, various key challenges and their possible countermeasures have been highlighted. So, this review is highly relevant to academic and industrial researchers working in the field of single atom-based heterogeneous catalysis and organic transformation processes.
Author contributions
Conceptualization and supervision: D. S. and V. K. Investigation, resources, and visualization: D. S. and V. K. Writing original draft: all authors. Review and editing: D. S., D. S., and V. K.Data availability
Most of the data that support the findings of this study are available in the article.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are thankful to IIT Mandi for the facilities provided. D. S. and S. K. acknowledge the Prime Minister's Research Fellowship (PMRF) from the Ministry of Education (MoE), Government of India, for the fellowship. B. P. M. acknowledges the Science and Engineering Research Board, Department of Science and Technology (DST), Government of India, for the National Postdoctoral Fellowship (NPDF).References
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef.
- I. Fechete, Y. Wang and J. C. Védrine, Catal. Today, 2012, 189, 2–27 CrossRef CAS.
- J. M. Thomas, Angew. Chem., Int. Ed. Engl., 1988, 27, 1673–1691 CrossRef.
- C. M. Friend and B. Xu, Acc. Chem. Res., 2017, 50, 517–521 CrossRef CAS PubMed.
- B. P. Mishra and K. Parida, J. Mater. Chem. A, 2021, 9, 10039–10080 RSC.
- D. Sharma, P. Choudhary, S. Kumar and V. Krishnan, Small, 2023, 19, 2207053 CrossRef CAS.
- D. Sharma, P. Choudhary, P. Mittal, S. Kumar, A. Gouda and V. Krishnan, ACS Catal., 2024, 14, 4211–4248 CrossRef CAS.
- A. Kumar, P. Choudhary, A. Kumar, P. H. Camargo and V. Krishnan, Small, 2022, 18, 2101638 CrossRef CAS.
- M. Bellardita, V. Loddo, F. Parrino and L. Palmisano, ChemPhotoChem, 2021, 5, 767–791 CrossRef CAS.
- J. Zhang, Y. Ji, P. Wang, Q. Shao, Y. Li and X. Huang, Adv. Funct. Mater., 2020, 30, 1906579 CrossRef CAS.
- J.-H. Yang, M. Peng, D.-D. Zhai, D. Xiao, Z.-J. Shi, S. Yao and D. Ma, ACS Catal., 2022, 12, 2898–2906 CrossRef CAS.
- A. M. Grigoras, F. Valentini, L. Latterini and L. Vaccaro, Green Energy Environ., 2024 DOI:10.1016/j.gee.2024.07.003.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- D. Sajwan, A. Semwal, J. Rawat, H. Sharma and C. Dwivedi, Mater. Today: Proc., 2023, 73, 180–188 CAS.
- A. Semwal, D. Sajwan, J. Rawat, L. Gambhir, H. Sharma and C. Dwivedi, Environ. Sci. Pollut. Res., 2023, 30, 45827–45839 CrossRef CAS.
- J. Rawat, D. Sajwan, S. V. Garimella, H. Sharma and C. Dwivedi, NanoTrends, 2023, 2, 100008 Search PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- H. Wei, S. K. Loeb, N. J. Halas and J.-H. Kim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15473–15481 CrossRef CAS.
- S. Loeb, C. Li and J.-H. Kim, Environ. Sci. Technol., 2018, 52, 205–213 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- B. B. Sarma, F. Maurer, D. E. Doronkin and J.-D. Grunwaldt, Chem. Rev., 2023, 123, 379–444 CrossRef CAS.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- J.-Y. Chen, Y. Xiao, F.-S. Guo, K.-M. Li, Y.-B. Huang and Q. Lu, ACS Catal., 2024, 14, 5198–5226 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- Y. Li, Y. Zhou, C. Shang, M. Yousaf, Z. Guo and S. Guo, Acc. Mater. Res., 2022, 3, 1160–1172 CrossRef CAS.
- L. Jiao, H. Yan, Y. Wu, W. Gu, C. Zhu, D. Du and Y. Lin, Angew. Chem., 2020, 132, 2585–2596 CrossRef.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159–162 CrossRef CAS.
- K. Asakura, H. Nagahiro, N. Ichikuni and Y. Iwasawa, Appl. Catal., A, 1999, 188, 313–324 CrossRef CAS.
- S. Abbet, A. Sanchez, U. Heiz and W.-D. Schneider, J. Catal., 2001, 198, 122–127 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- X. Zhang, H. Shi and B. Q. Xu, Angew. Chem., 2005, 117, 7294–7297 CrossRef.
- S. F. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- H. Yan, C. Su, J. He and W. Chen, J. Mater. Chem. A, 2018, 6, 8793–8814 RSC.
- W.-H. Li, J. Yang, D. Wang and Y. Li, Chem, 2022, 8, 119–140 CAS.
- V. B. Saptal, V. Ruta, M. A. Bajada and G. Vilé, Angew. Chem., Int. Ed., 2023, 62, e202219306 CrossRef CAS PubMed.
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 RSC.
- L. Zhang, L. Xue, B. Lin, Q. Zhao, S. Wan, Y. Wang, H. Jia and H. Xiong, ChemSusChem, 2022, 15, e202102494 CrossRef CAS PubMed.
- J. Shan, C. Ye, C. Zhu, J. Dong, W. Xu, L. Chen, Y. Jiao, Y. Jiang, L. Song and Y. Zhang, J. Am. Chem. Soc., 2022, 144, 23214–23222 CrossRef CAS.
- P. Zhou, Y. Chao, F. Lv, J. Lai, K. Wang and S. Guo, Sci. Bull., 2020, 65, 720–725 CrossRef CAS PubMed.
- Z. Chen, J. Liu, M. J. Koh and K. P. Loh, Adv. Mater., 2022, 34, 2103882 CrossRef CAS.
- D. Sajwan, A. Sharma, M. Sharma and V. Krishnan, ACS Catal., 2024, 14, 4865–4926 CrossRef CAS.
- X. He, Y. Deng, Y. Zhang, Q. He, D. Xiao, M. Peng, Y. Zhao, H. Zhang, R. Luo and T. Gan, Cell Rep. Phys. Sci., 2020, 1, 100004 CrossRef.
- T. Pu, J. Ding, X. Tang, K. Yang, K. Wang, B. Huang, S. Dai, Y. He, Y. Shi and P. Xie, ACS Appl. Mater. Interfaces, 2022, 14, 43141–43150 CrossRef CAS.
- Y. Wu, Q. Wu, Q. Zhang, Z. Lou, K. Liu, Y. Ma, Z. Wang, Z. Zheng, H. Cheng and Y. Liu, Energy Environ. Sci., 2022, 15, 1271–1281 RSC.
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang and L. Zheng, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS.
- Q. Li, Q. Zhang, W. Xu, R. Zhao, M. Jiang, Y. Gao, W. Zhong, K. Chen, Y. Chen and X. Li, Adv. Energy Mater., 2023, 13, 2203955 CrossRef CAS.
- C. Jin, L. Huo, J. Tang, S. Li, K. Jiang, Q. He, H. Dong, Y. Gong and Z. Hu, Small, 2024, 20, 2309509 CrossRef CAS PubMed.
- R. Li, D. Wu, P. Rao, P. Deng, J. Li, J. Luo, W. Huang, Q. Chen, Z. Kang and Y. Shen, Carbon Energy, 2023, 5, e294 CrossRef CAS.
- H. Zhai, P. Tan, M. Jiang, M. Zhang, R. Ren, R. Sa and J. Pan, ACS Catal., 2023, 13, 8063–8072 CrossRef CAS.
- S. Li, K. Wu, D. Wang, H. Yang, Z. Shen and W. Wang, Appl. Surf. Sci., 2023, 638, 158128 CrossRef CAS.
- H. Li, F. Pan, C. Qin, T. Wang and K. J. Chen, Adv. Energy Mater., 2023, 13, 2301378 CrossRef CAS.
- Z. Ren, Y. Liu, Y. Lyu, X. Song, C. Zheng, S. Feng, Z. Jiang and Y. Ding, J. Catal., 2019, 369, 249–256 CrossRef CAS.
- X. Y. Dong, Y. N. Si, Q. Y. Wang, S. Wang and S. Q. Zang, Adv. Mater., 2021, 33, 2101568 CrossRef CAS.
- J. Li, W. Zeng, L. Wang, G. Shi and D. Wang, Chem. Eng. J., 2023, 474, 145642 Search PubMed.
- Y. Liu, Y. Zhou, J. Li, Q. Wang, Q. Qin, W. Zhang, H. Asakura, N. Yan and J. Wang, Appl. Catal., B, 2017, 209, 679–688 Search PubMed.
- M. B. Buendia, S. Kegnæs and S. Kramer, Adv. Synth. Catal., 2020, 362, 5506–5512 CrossRef CAS.
- C. Qian, W. Zhou, J. Qiao, D. Wang, X. Li, W. L. Teo, X. Shi, H. Wu, J. Di and H. Wang, J. Am. Chem. Soc., 2020, 142, 18138–18149 CrossRef CAS.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- X. He, Q. He, Y. Deng, M. Peng, H. Chen, Y. Zhang, S. Yao, M. Zhang, D. Xiao and D. Ma, Nat. Commun., 2019, 10, 3663 CrossRef.
- Y. Shen, Q. Zheng, Z. N. Chen, D. Wen, J. H. Clark, X. Xu and T. Tu, Angew. Chem., Int. Ed., 2021, 60, 4125–4132 CrossRef CAS.
- Z. Wang, S. Hasegawa, K. Motokura, S. Kuang and Y. Yang, Ind. Eng. Chem. Res., 2023, 62, 3151–3156 CrossRef CAS.
- L. Li, J. Xu, Q. Zhu, X. Meng, H. Xu and M. Han, Dalton Trans., 2024, 53, 1915–1934 RSC.
- P. Zhang, H.-C. Chen, H. Zhu, K. Chen, T. Li, Y. Zhao, J. Li, R. Hu, S. Huang and W. Zhu, Nat. Commun., 2024, 15, 2062 CrossRef CAS PubMed.
- J. Liu, W. Chen, S. Yuan, T. Liu and Q. Wang, Energy Environ. Sci., 2024, 17, 249–259 RSC.
- J. Zhang, W. Wang, X. Chen, J. Jin, X. Yan and J. Huang, J. Am. Chem. Soc., 2024, 146, 10432–10442 CrossRef CAS PubMed.
- J. Yang, W. Liu, M. Xu, X. Liu, H. Qi, L. Zhang, X. Yang, S. Niu, D. Zhou and Y. Liu, J. Am. Chem. Soc., 2021, 143, 14530–14539 CrossRef CAS PubMed.
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- S. Ma, Z. Han, K. Leng, X. Liu, Y. Wang, Y. Qu and J. Bai, Small, 2020, 16, 2001384 CrossRef CAS.
- H. Jin, P. Cui, C. Cao, X. Yu, R. Zhao, D. Ma and W. Song, ACS Catal., 2023, 13, 1316–1325 CrossRef CAS.
- P. Kumar, K. Kannimuthu, A. S. Zeraati, S. Roy, X. Wang, X. Wang, S. Samanta, K. A. Miller, M. Molina and D. Trivedi, J. Am. Chem. Soc., 2023, 145, 8052–8063 CrossRef CAS PubMed.
- Z. Li, F. Liu, C. Chen, Y. Jiang, P. Ni, N. Song, Y. Hu, S. Xi, M. Liang and Y. Lu, Nano Lett., 2023, 23, 1505–1513 CrossRef CAS PubMed.
- C. Zhang, S. Liang, W. Liu, F. T. Eickemeyer, X. Cai, K. Zhou, J. Bian, H. Zhu, C. Zhu and N. Wang, Nat. Energy, 2021, 6, 1154–1163 CrossRef CAS.
- X. Zhang, H. Su, P. Cui, Y. Cao, Z. Teng, Q. Zhang, Y. Wang, Y. Feng, R. Feng and J. Hou, Nat. Commun., 2023, 14, 7115 CrossRef CAS PubMed.
- L. Xiong, H. Qi, S. Zhang, L. Zhang, X. Liu, A. Wang and J. Tang, Adv. Mater., 2023, 35, 2209646 CrossRef CAS PubMed.
- S. De, A. S. Burange and R. Luque, Green Chem., 2022, 24, 2267–2286 RSC.
- Y. Lu, Z. Zhang, H. Wang and Y. Wang, Appl. Catal., B, 2021, 292, 120162 CrossRef CAS.
- S. Tian, W. Gong, W. Chen, N. Lin, Y. Zhu, Q. Feng, Q. Xu, Q. Fu, C. Chen and J. Luo, ACS Catal., 2019, 9, 5223–5230 CrossRef CAS.
- S. Bagheri, N. M. Julkapli and W. A. Yehye, Renewable Sustainable Energy Rev., 2015, 41, 113–127 CrossRef CAS.
- Y. Li, Y. Chen, Y.-L. Wan, R.-S. Wang, H. Wang and Y.-Z. Lei, J. CO2 Util., 2022, 65, 102214 CrossRef CAS.
- S. Kramer, N. R. Bennedsen and S. Kegnæs, ACS Catal., 2018, 8, 6961–6982 CrossRef CAS.
- Q. Sun and F.-S. Xiao, Acc. Mater. Res., 2022, 3, 772–781 CrossRef CAS.
- B. Wang, J. Lin, Q. Sun, C. Xia and W. Sun, ACS Catal., 2021, 11, 10964–10973 CrossRef CAS.
- H. B. Yang, C.-Q. Xu, S. Baskaran, Y.-R. Lu, C. Gu, W. Liu, J. Ding, J. Zhang, Q. Wang and W. Chen, EES Catal., 2023, 1, 774–783 RSC.
- W. Fu, Y. Wang, W. Tian, H. Zhang, J. Li, S. Wang and Y. Wang, Angew. Chem., 2020, 132, 23999–24007 CrossRef.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS.
- K. Bhattacharyya and A. Datta, Phys. Chem. Chem. Phys., 2019, 21, 12346–12352 RSC.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- J. Wei, K. Xiao, Y. Chen, X.-P. Guo, B. Huang and Z.-Q. Liu, Energy Environ. Sci., 2022, 15, 4592–4600 RSC.
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang and C. He, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- A. Pei, R. Xie, Y. Zhang, Y. Feng, W. Wang, S. Zhang, Z. Huang, L. Zhu, G. Chai and Z. Yang, Energy Environ. Sci., 2023, 16, 1035–1048 RSC.
- X. Su, X.-F. Yang, Y. Huang, B. Liu and T. Zhang, Acc. Chem. Res., 2018, 52, 656–664 CrossRef.
- L. Gong, D. Zhang, C. Y. Lin, Y. Zhu, Y. Shen, J. Zhang, X. Han, L. Zhang and Z. Xia, Adv. Energy Mater., 2019, 9, 1902625 CrossRef CAS.
- S. Wang, L. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2023, 16, 2759–2803 RSC.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS.
- W. Li, Z. Guo, J. Yang, Y. Li, X. Sun, H. He, S. Li and J. Zhang, Electrochem. Energy Rev., 2022, 5, 9 CrossRef CAS.
- L. Zhang, Y. Wang, Z. Niu and J. Chen, Small Methods, 2019, 3, 1800443 CrossRef.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef PubMed.
- P. Wang, D. Zhao and L. Yin, Energy Environ. Sci., 2021, 14, 1794–1834 RSC.
- X. Zheng, P. Li, S. Dou, W. Sun, H. Pan, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 2809–2858 RSC.
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC.
- B. Xiao, Z. Sun, H. Zhang, Y. Wu, J. Li, J. Cui, J. Han, M. Li, H. Zheng and J. Chen, Energy Environ. Sci., 2023, 16, 2153–2166 RSC.
- M. Li, C. Zhang, Y. Tang, Q. Chen, W. Li, Z. Han, S. Chen, C. Lv, Y. Yan and Y. Zhang, ACS Catal., 2022, 12, 11960–11973 CrossRef CAS.
- Y. Xue, Q. Yu, Q. Ma, Y. Chen, C. Zhang, W. Teng, J. Fan and W.-X. Zhang, Environ. Sci. Technol., 2022, 56, 14797–14807 CrossRef CAS.
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 RSC.
- W. Liu, Y. Chen, H. Qi, L. Zhang, W. Yan, X. Liu, X. Yang, S. Miao, W. Wang and C. Liu, Angew. Chem., 2018, 130, 7189–7193 CrossRef.
- K. Kumari, P. Choudhary, D. Sharma and V. Krishnan, Ind. Eng. Chem. Res., 2022, 62, 158–168 CrossRef.
- P. Choudhary, K. Kumari, D. Sharma, S. Kumar and V. Krishnan, ACS Appl. Mater. Interfaces, 2023, 15, 9412–9420 CrossRef CAS.
- P. Choudhary, S. S. Chauhan, D. Sharma, S. Kumar and V. Krishnan, Chem. Eng. J., 2023, 475, 146055 CrossRef CAS.
- S. Kumar, P. Choudhary, D. Sharma, D. Sajwan, V. Kumar and V. Krishnan, ChemSusChem, 2024, e202400737 CrossRef.
- Z. Sun, S. Wang and W. Chen, J. Mater. Chem. A, 2021, 9, 5296–5319 RSC.
- A. V. Rassolov, I. S. Mashkovsky, G. N. Baeva, G. O. Bragina, N. S. Smirnova, P. V. Markov, A. V. Bukhtiyarov, J. Wärnå, A. Y. Stakheev and D. Y. Murzin, Nanomaterials, 2021, 11, 3286 CrossRef CAS PubMed.
- X. Song, F. Shao, Z. Zhao, X. Li, Z. Wei and J. Wang, ACS Catal., 2022, 12, 14846–14855 CrossRef CAS.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Havecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86–89 CrossRef CAS PubMed.
- R. Gao, J. Xu, J. Wang, J. Lim, C. Peng, L. Pan, X. Zhang, H. Yang and J.-J. Zou, J. Am. Chem. Soc., 2021, 144, 573–581 CrossRef.
- A. J. McCue, C. J. McRitchie, A. M. Shepherd and J. A. Anderson, J. Catal., 2014, 319, 127–135 CrossRef CAS.
- J. Liu, M. B. Uhlman, M. M. Montemore, A. Trimpalis, G. Giannakakis, J. Shan, S. Cao, R. T. Hannagan, E. C. H. Sykes and M. Flytzani-Stephanopoulos, ACS Catal., 2019, 9, 8757–8765 CrossRef CAS.
- Y. Ma, B. Chi, W. Liu, L. Cao, Y. Lin, X. Zhang, X. Ye, S. Wei and J. Lu, ACS Catal., 2019, 9, 8404–8412 CrossRef CAS.
- W. Liu, J. Liu, X. Liu, H. Zheng and J. Liu, ACS Catal., 2022, 13, 530–539 CrossRef.
- Y. Cao, B. Chen, J. Guerrero-Sánchez, I. Lee, X. Zhou, N. Takeuchi and F. Zaera, ACS Catal., 2019, 9, 9150–9157 CrossRef CAS.
- Y. Fan, C. Zhuang, S. Li, Y. Wang, X. Zou, X. Liu, W. Huang and G. Zhu, J. Mater. Chem. A, 2021, 9, 1110–1118 RSC.
- G. Guo, Y. Zhang, H. Zhang, X. Zhang, X. He and H. Ji, Ind. Eng. Chem. Res., 2023, 62, 19585–19591 CrossRef CAS.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2019, 120, 683–733 CrossRef PubMed.
- M. Deng, D. Wang and Y. Li, Appl. Catal., A, 2023, 119423 CrossRef CAS.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., 2016, 128, 8459–8463 CrossRef.
- S. Shao, Y. Yang, K. Sun, S. Yang, A. Li, F. Yang, X. Luo, S. Hao and Y. Ke, ACS Catal., 2021, 11, 12146–12158 CrossRef CAS.
- J. Chen, Y. Xia, Y. Ling, X. Liu, S. Li, X. Yin, L. Zhang, M. Liang, Y.-M. Yan and Q. Zheng, Nano Lett., 2024, 24, 5197–5205 CrossRef CAS PubMed.
- L. Kuai, Z. Chen, S. Liu, E. Kan, N. Yu, Y. Ren, C. Fang, X. Li, Y. Li and B. Geng, Nat. Commun., 2020, 11, 48 CrossRef.
- P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445–2465 RSC.
- H. Feng, W. Liu, L. Wang, E. Xu, D. Pang, Z. Ren, S. Wang, S. Zhao, Y. Deng and T. Liu, Adv. Sci., 2024, 2304908 CrossRef CAS PubMed.
- F. Cao, W. Ni, Q. Zhao, L. Wang, S. Xue, Y. Li, D. Kong, M. Wu and L. Zhi, Appl. Catal., B, 2024, 346, 123762 CrossRef CAS.
- Z. Li, X. Lu, C. Guo, S. Ji, H. Liu, C. Guo, X. Lu, C. Wang, W. Yan and B. Liu, Nat. Commun., 2024, 15, 3195 CrossRef CAS.
- D. Sharma, P. Choudhary, S. Kumar and V. Krishnan, J. Colloid Interface Sci., 2024, 657, 449–462 CrossRef CAS.
- W. Liu, L. Zhang, W. Yan, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, Chem. Sci., 2016, 7, 5758–5764 RSC.
- H. Yan, X. Zhao, N. Guo, Z. Lyu, Y. Du, S. Xi, R. Guo, C. Chen, Z. Chen and W. Liu, Nat. Commun., 2018, 9, 3197 CrossRef PubMed.
- C. Cao and W. Song, ACS Mater. Lett., 2020, 2, 1653–1661 CrossRef CAS.
- Y. Zhang, Y. Cheng, X. Wang, Q. Sun, X. He and H. Ji, ACS Catal., 2022, 12, 15091–15096 CrossRef CAS.
- T. Su and C. Cai, ACS Appl. Mater. Interfaces, 2022, 14, 55568–55576 CrossRef CAS.
- X. Shi, Y. Lin, L. Huang, Z. Sun, Y. Yang, X. Zhou, E. Vovk, X. Liu, X. Huang and M. Sun, ACS Catal., 2020, 10, 3495–3504 CrossRef CAS.
- A. Han, J. Zhang, W. Sun, W. Chen, S. Zhang, Y. Han, Q. Feng, L. Zheng, L. Gu and C. Chen, Nat. Commun., 2019, 10, 3787 CrossRef PubMed.
- S. Tian, B. Wang, W. Gong, Z. He, Q. Xu, W. Chen, Q. Zhang, Y. Zhu, J. Yang and Q. Fu, Nat. Commun., 2021, 12, 3181 CrossRef CAS.
- J. Fu, J. Dong, R. Si, K. Sun, J. Zhang, M. Li, N. Yu, B. Zhang, M. G. Humphrey and Q. Fu, ACS Catal., 2021, 11, 1952–1961 CrossRef CAS.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li and M. Wang, Nat. Commun., 2016, 7, 14036 CrossRef CAS.
- J. Amsler, B. B. Sarma, G. Agostini, G. Prieto, P. N. Plessow and F. Studt, J. Am. Chem. Soc., 2020, 142, 5087–5096 CrossRef CAS.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. T. Cui, Y. Tan, B. Qiao, L. Li and A. Wang, Angew. Chem., Int. Ed., 2016, 55, 16054–16058 CrossRef CAS.
- H. Gong, X. Zhao, Y. Qin, W. Xu, X. Wei, Q. Peng, Y. Ma, S. Dai, P. An and Z. Hou, J. Catal., 2022, 408, 245–260 CrossRef CAS.
- T. Qin, Y. Dang, T. Lin, B. Mei, B. Wu, X. Li, S. Li, Z. Jiang, Z. Tang and L. Zhong, Green Chem., 2021, 23, 9038–9047 RSC.
- G. Zeng, K. Liang, Y. Huang, C. Liu, H. Chen, D. Wang, J. Ma and Z. Dong, Mol. Catal., 2023, 550, 113548 CrossRef CAS.
- Y. Zheng, Q. Wang, Q. Yang, S. Wang, M. J. Hülsey, S. Ding, S. Furukawa, M. Li, N. Yan and X. Ma, ACS Catal., 2023, 13, 7243–7255 CrossRef CAS.
- L. Jurado, J. Esvan, L. A. Luque-Álvarez, L. F. Bobadilla, J. A. Odriozola, S. Posada-Pérez, A. Poater, A. Comas-Vives and M. R. Axet, Catal. Sci. Technol., 2023, 13, 1425–1436 RSC.
- F. J. Escobar-Bedia, M. Lopez-Haro, J. J. Calvino, V. Martin-Diaconescu, L. Simonelli, V. Perez-Dieste, M. J. Sabater, P. Concepción and A. Corma, ACS Catal., 2022, 12, 4182–4193 CrossRef CAS.
- P. Gao, G. Liang, T. Ru, X. Liu, H. Qi, A. Wang and F.-E. Chen, Nat. Commun., 2021, 12, 4698 CrossRef CAS PubMed.
- C. Li, L. Yan, L. Lu, K. Xiong, W. Wang, M. Jiang, J. Liu, X. Song, Z. Zhan and Z. Jiang, Green Chem., 2016, 18, 2995–3005 RSC.
- S. Feng, M. Jiang, X. Song, P. Qiao, L. Yan, Y. Cai, B. Li, C. Li, L. Ning and S. Liu, Angew. Chem., Int. Ed., 2023, 62, e202304282 CrossRef CAS.
- D. Poier, D. F. Akl, E. Lucas, A. R. Machado, G. Giannakakis, S. Mitchell, G. Guillén-Gosálbez, R. Marti and J. Pérez-Ramírez, ACS Sustainable Chem. Eng., 2023, 11, 16935–16945 CrossRef CAS.
- Y. Chen, H. Zhu, X. Ding, H. Wang, W. Qiu, J. Song and S. Pang, ACS Appl. Nano Mater., 2024, 7, 8063–8073 CrossRef CAS.
- G. Ding, L. Hao, H. Xu, L. Wang, J. Chen, T. Li, X. Tu and Q. Zhang, Commun. Chem., 2020, 3, 43 CrossRef CAS PubMed.
- A. Vice, N. Langer, B. Reinhart and O. Kedem, Inorg. Chem., 2023, 62, 21479–21486 CrossRef CAS PubMed.
- X. Tao, R. Long, D. Wu, Y. Hu, G. Qiu, Z. Qi, B. Li, R. Jiang and Y. Xiong, Small, 2020, 16, 2001782 CrossRef CAS PubMed.
- Y. Zhang, S. Ye, M. Gao, Y. Li, X. Huang, J. Song, H. Cai, Q. Zhang and J. Zhang, ACS Nano, 2022, 16, 1142–1149 CrossRef CAS.
- T. Cui, L. Ma, S. Wang, C. Ye, X. Liang, Z. Zhang, G. Meng, L. Zheng, H.-S. Hu and J. Zhang, J. Am. Chem. Soc., 2021, 143, 9429–9439 CrossRef CAS.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS.
- S. Liu, L. Bai, A. P. van Muyden, Z. Huang, X. Cui, Z. Fei, X. Li, X. Hu and P. J. Dyson, Green Chem., 2019, 21, 1974–1981 RSC.
- D. Bautista-García, D. Macias-José, P. Aguillón-Rodríguez, O. Pérez-Reyes and C. Ortiz-Cervantes, New J. Chem., 2023, 47, 6164–6170 RSC.
- K. Li, Q. Ge, Y. Liu, L. Wang, K. Gong, J. Liu, L. Xie, W. Wang, X. Ruan and L. Zhang, Energy Environ. Sci., 2023, 16, 1135–1145 RSC.
- J. Qin, B. Han, X. Lu, J. Nie, C. Xian and Z. Zhang, JACS Au, 2023, 3, 801–812 CrossRef CAS PubMed.
- H. Qi, S. Mao, J. Rabeah, R. Qu, N. Yang, Z. Chen, F. Bourriquen, J. Yang, J. Li and K. Junge, Angew. Chem., Int. Ed., 2023, 62, e202311913 CrossRef CAS PubMed.
- J. Qin, B. Han, X. Liu, W. Dai, Y. Wang, H. Luo, X. Lu, J. Nie, C. Xian and Z. Zhang, Sci. Adv., 2022, 8, eadd1267 CrossRef CAS PubMed.
- Q. Zhang and J. Guan, Nano Res., 2022, 15, 38–70 CrossRef CAS.
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang and J. Lim, Nat. Commun., 2021, 12, 5235 CrossRef CAS.
- Q. Shang, N. Tang, H. Qi, S. Chen, G. Xu, C. Wu, X. Pan, X. Wang and Y. Cong, Chin. J. Catal., 2020, 41, 1812–1817 CrossRef CAS.
- M. Li, S. Wu, X. Yang, J. Hu, L. Peng, L. Bai, Q. Huo and J. Guan, Appl. Catal., A, 2017, 543, 61–66 CrossRef CAS.
- T. Li, F. Liu, Y. Tang, L. Li, S. Miao, Y. Su, J. Zhang, J. Huang, H. Sun and M. Haruta, Angew. Chem., Int. Ed., 2018, 57, 7795–7799 CrossRef CAS PubMed.
- K.-i Otake, Y. Cui, C. T. Buru, Z. Li, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 8652–8656 CrossRef CAS PubMed.
- Z. Chen, Q. Zhang, W. Chen, J. Dong, H. Yao, X. Zhang, X. Tong, D. Wang, Q. Peng and C. Chen, Adv. Mater., 2018, 30, 1704720 CrossRef PubMed.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
Footnote |
| † D. S. and D. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |