
Conduction band photonic trapping via band gap reversal of brookite quantum dots using controlled graphitization for tuning a multi-exciton photoswitchable high-performance semiconductor†
Sanjiv
Sonkaria
 *a,
Tae Woo
Lee
b,
Aniket
Kumar
c,
Soo-Kyung
Hwang
d,
Piotr G.
Jablonski
ef and
Varsha
Khare
*a
*a,
Tae Woo
Lee
b,
Aniket
Kumar
c,
Soo-Kyung
Hwang
d,
Piotr G.
Jablonski
ef and
Varsha
Khare
*a
aSoft Foundry Institute, College of Engineering, Seoul National University, Seoul, Republic of Korea. E-mail: ssonkaria64@snu.ac.kr; khare@snu.ac.kr
bDepartment of Materials Science and Engineering, Seoul National University, Seoul, Republic of Korea
cInnovoja Sustainable Solutions, White Rock, BC, Canada
dLaboratory of Adhesion & Bio-Composites, Major in Environmental Materials Science, Seoul National University, Republic of Korea
eLaboratory of Behavioral Ecology, School of Biological Sciences, Seoul National University, Seoul, Republic of Korea
fMuseum and Institute of Zoology, Warsaw, Poland
First published on 9th November 2024
Abstract
Brookite exists as the metastable phase of titania and often mediates the transformation of anatase to rutile. The photocatalytic competence of brookite relative to polymorphs anatase and rutile has generally been considered structurally and energetically unfavourable for reasons that remain largely unknown and unchallenged. However, the process of phase transformation and performance related cooperativity among all three polymorphs has recently unlocked alternative directions for exploring brookite photovoltaics. Here, we demonstrate the programmable re-configuration of anatase to quantum confined reduced graphene (rGO)–brookite and show it is entirely modulated by surface-driven effects. Key components to this mechanism suggest that the self-assembly of rGO–brookite quantum dots is defect driven through pathways that favour a direct-to-indirect band gap reversal resulting from the graphitization of brookite. The accompaniment of new bandgap characteristics under quantum confinement introduce new hybridized energy states at the graphitic carbon–brookite juncture by modulation of the intrinsic sp2 character to extrinsic sp3 clusters intermediate to graphene quantum dots (GQDs) and graphene oxide quantum dots (GOQDs). Evidenced by the intercalation of photochromic/fluorescent carbazole and anthracene moieties within the rGO framework by self-assembly, we show that the acquired fluorescence and luminescence properties of rGO–brookite are multi-emissive and reversibly quenchable under light excitation and from solvent polarity differences. Further, tuning the excitonic response of rGO–brookite by modulation of the photoluminescence (PL) signal intensity signifies coordinated interaction between localised carbazole and benz(a)anthracene moities which can undergo further structural refinement to adapt more optimally to both internal and external energy waves. Distinguishable by a large red-shift in the photoluminescent emission peak at λ479 nm in the NIR region, we infer that a photoelectron sink driven by the quantum confinement of a narrow band gap of 0.78 eV formed from the orbital overlap of unoccupied interfacial sites promotes strong e−h+ coupling in the hybridized defect structure imposing a high charge separation by hindering e−h+ recombination. Modulation of interlayer spacing between rGO sheets and the synergy of complexation between intercalated carbazole/benz(a)nthracene can be adapted to achieve rapid photodegradation characteristics for DSSC applications.
Introduction
The photoelectrochemical potential of dye-sensitized solar cells (DSSCs)1 is rooted in material capability and requires that the conduction band of semiconductor nanocrystals2 be photochemically aligned at complex material interfaces to engage in optimal charge transfer and recycling events. Rising interest in the field of DSSCs since its inception in 19913 has been dominated by aspects of material tunability driven by cost reduction performance. The ability of photovoltaic devices to generate high photocurrents has largely been weighted against the applicability of different semiconductor types in improving photon adsorption which is consequential to enhancing electronic excitation. Dye adsorption at the electrode surface however, could be a limiting factor to photocurrent enhancement if charge diffusion characteristics at the semiconductor/dye interface are hindered by physical and chemical considerations. This argument can also be extended to the electrolyte interface. Despite such concerns, material fabrication is a critical feature in determining the degree of tunability aided by novel surface modifications arising from material hybridizations and processing routes. This opens possibilities to explore semiconductor material phases that have been largely ignored as potential alternatives to DSSCs.Among the several polymorph types known to exist as variants of titanium oxide,4 TiO2-anatase and rutile are generally accepted as the photocatalyst of choice over brookite for the development of DSSC based applications. The relatively sluggish performance of the crystalline form of brookite is further widened when compared to the binary performance of rutile and anatase which is rate enhanced through a mechanistic cooperative effect. For example, lattice matching of anatase-TiO2/H-rutile-TiO2 through optimal band alignment in the heterophase yields high quantum efficiencies measurable by H2 evolution under UV light irradiation.5 Identifiably, brookite suffers relatively poor stability characteristics readily undergoing phase transformation to rutile and demonstrating poorer charge transfer properties while negatively impacting electron–hole recombination kinetics due to reduced charge separation. Recently, these conclusions have been challenged by reviewing the photocatalytic properties of mixed phase investigations of anatase–brookite TiO2 which were reported to exceed the CO2 photoreduction by single phase anatase or brookite.6 Enhanced photocatalytic activity has also been observed for the mixture of anatase–brookite outperforming anatase nanoparticles alone. In the same study, the phase transformation of anatase–brookite to rutile showed a dependency on brookite particles, different to the direct transformation of anatase and brookite to rutile in the absence of the mixture.7 Such observations may be evidential of atomistic changes at the anatase–brookite interfacial boundary enhancing key photovoltaic properties of the hybrid TiO2 nanocrystal. Surface exposure of helium to all three polymorphs provides a model to investigate surface induced modifications to compare the effect of defects with defect free polymorph nanocrystals. The nature of such defects in oxides strongly relate to oxygen vacancy and interstitial8 Ti3+ sites which are positioned between the valence and conduction bands generating trapped electrons and thus altering localised charge carrier characteristics. Optoelectronic differences are associated with shifts across high and low energy bands spanning ultraviolet, visible and infrared regions with strong prospects for bandgap engineering.9 Multiphase heterojunctions in conjunction with electron/electron hole pairs can considerably alter charge carrier and stability characteristics of brookite.10 Brookite however, is not considered a viable photocatalytic material unaided by other polymorphic forms.11
Under conditions of low pH, the brookite phase dominates the transformation of anatase to rutile12 favoured by acidic conditions. A study by Lin et al.13 however proposes that brookite crystals are not intrinsically inert and that the selective exposure of faceted crystal faces can generate highly active photoactive brookite nanocrystals under low basicity. For the current work, we rationalised that multi-reactant mediums comprising of specific cationic (basic) and anionic (acidic) chemical entities may provide important opportunities to stabilize intermediate crystalline phases of brookite undergoing phase transformation to more stable TiO2 polymorphs. The recruitment of building blocks from the surroundings must align well to catalytically exposed surface energies of TiO2 to overcome energy barriers to drive polymer assembled growth. One-pot synthetic processes may inherently differ from multi-step processes in steering surface dynamics to desired outcomes. A rarity in exploring the ‘non-classical’ route to the stabilisation of the brookite metastable phase is allowing the thermal integration of multicomponent assemblies synthesised in isolation of each other using a common reaction medium.
This approach took the investigation on a route to explore the synergy between non-competing paths to surface polymer assembly in a highly acidic cationic–anionic ionic liquid (CA-IL) reaction medium induced catalytically by two independent reactions at the interface of a (1) calcinated bulk anatase TiO2 precursor and the (2) graphene oxide phase. Under identical reaction conditions, polymer nucleation at the surface of (1) and (2) from the interfacial interaction with CA-IL is facet dependent and polymer selectivity is contingent on energy barriers at surface exposed catalytic sites. We were interested in examining if the process of introducing secondary polymer nucleation sites seeded by the union of (1) and (2) was thermally driven by altering the trajectory of polymer faceted growth through pathway convergence. Secondly, whether the cationic–anionic acidic reaction medium was effective in the stabilisation of metastable phase of TiO2 (intermediate to anatase and rutile). Thirdly, whether controlled faceted growth of multi-assembled structures via self assembly provide accessibility to synergise oriented assembly between all components thereby enhancing charge transfer properties.
From the perspective of bioeletrocatalytic hybrid interfaces found in nature, dynamic changes in response to light are often central to generating highly charge separated states to drive charge across steep gradients. Such multi-interface bio-transformations use light-to-chemical energy conversions reversibly to operate photoisomerization-type switching events14 or show capability to adapt surface exposed structures through charge migration by using external energy sources. This requires hybridization of diverse material interfaces to enhance the electronic and optical properties to elicit new functionalities. Charge migration and exchange between TiO2 photoactive centers could be enhanced through the energy alignment of electron–proton donor–acceptor pairs close to the semiconductor interface. In nature, light sensitive chromomorphic groups and fluorescent sensitive structures act as photosensors with the ability to undergo chemical transformation through the acceptance or release of charge. The realisation to acquiring more favourable photonic structures controlled through self-assembly could be better guided by chemical fuels originating from precursor building blocks selected from complex heat-driven reaction environments and more notably, using charge separated ionic liquids driving the assembly of charge sensitive structures. Conceptually, the non-equilibrium nature of such supramolecular geometries15 offers a degree of controllable programmability and energy dependence directed from external sources with the possibility of introducing new functionalities such as light sensitive photoswitchability.
We show that the acid–base character of the CA-IL reaction medium was strongly influential in arresting the brookite phase of TiO2 strongly hindering its transformation to rutile. A strong synergy is observed in polymer assembly at the surface of brookite and rGO suggesting that the forces driving polymer self-assembly are highly influenced both by the chemical environment and faceted growth. Our inference is supported by the growth of anthracene during the phase transition from anatase to brookite and secondly, the assembly of both anthracene and carbazole by reduced graphene oxide (rGO) as independent processes in CA-IL. The remarkable overlap in catalytic synergy towards controlled polymerizations showed further selectivity by complexation of brookite and rGO in suppressing the faceted growth of carbazole as the dominant structure in favour of benzanthracene. This process enabled precision polymer synthesis for improving the charge transfer properties of brookite. We infer that the intercalation of a low bandgap rGO–brookite organic semiconductor complex (>1 eV) quantum confined within a layered architecture of rGO sheets by chemically fuelled polyaniline bridges is morphologically important, for an efficiently designed charge transfer complex. Further, the work provides some meaningful clue towards the existence of non-equilibrium assemblies that are dynamic in nature. We propose that the size of the energy barrier for charge transfer at the rGO-carbazole and rGO-benzanthracene interface is made feasible by carriers that diminish the barrier height. The photoluminescent assisted conjugation of monomers carbazolyl and anthracene to form carbazolyl-phenyl-anthracene demonstrates surface adaptable behaviour but the complexation occurs by a mechanism that remains elusive.
Materials and methods (incomplete)
Materials
All materials and reagents were purchased from Daejung Chemicals, Sigma-Aldrich Alfa Aesar and Merck unless otherwise stated. 1-ethyl 1-methylpyrrolidinium Chloride (BMIMCl) (C7H16NCl) was purchased from lolitec (Iolitec, Ionic Liquids Technologies GmbH).Synthesis of calcinated TiO2 anatase and brookite TiO2 QDs, reduced graphene oxide (rGO)-CA-IL and rGO–(brookite) TiO2
In step A, an anatase polymorph of titanium(IV) was used as a precursor for the preparation of brookite quantum sized particles. In a sol gel process, 20 mL of isopropanol was added to the titanium isopropoxide precursor (1.0 g) in the presence of glycerol (1.0 g). The isopropanol–precursor blend was added dropwise to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) water–isopropanol mixture and the mixture was added to 1-1-butyl 1-methylpyrrolidinium after melting at 120 °C (m.p ∼117 °C) for 24 h. The pH of the resulting mixture was adjusted by hydrochloric acid. The preparation was continuously stirred for 1 h until a yellow transparent gel was obtained and subsequently dried at 105 °C over a period of several hours. After heating, a black crystal was formed. Finally, the crystals were calcinated in air at 500 °C. In step B, 0.004 g of graphene oxide (GO) was added to 2.0 g of 1-butyl 1-methylpyrrolidinium and glycerol (1.9 g) and heated to 120 °C for 24 h. In the final step, the controlled graphitization of TiO2 was achieved by combining the formulations from step A and step B under stirring for 24 h at R.T.
:1 (v/v) water–isopropanol mixture and the mixture was added to 1-1-butyl 1-methylpyrrolidinium after melting at 120 °C (m.p ∼117 °C) for 24 h. The pH of the resulting mixture was adjusted by hydrochloric acid. The preparation was continuously stirred for 1 h until a yellow transparent gel was obtained and subsequently dried at 105 °C over a period of several hours. After heating, a black crystal was formed. Finally, the crystals were calcinated in air at 500 °C. In step B, 0.004 g of graphene oxide (GO) was added to 2.0 g of 1-butyl 1-methylpyrrolidinium and glycerol (1.9 g) and heated to 120 °C for 24 h. In the final step, the controlled graphitization of TiO2 was achieved by combining the formulations from step A and step B under stirring for 24 h at R.T.
Characterisation
Wide-angle X-ray scattering (WAXS) and X-ray diffraction (XRD) were performed using a D8 advanced XRD system from Bruker at the National Instrumentation Center for Environmental Management (NICEM) of Seoul National University (SNU) (CuKα radiation for 2θ ranging from −100° to +168° (WAXS) and 10° to 85° (XRD). High resolution transmission electron microscopy (HRTEM) images were recorded by an analytical TEM JEM-2100F from JEOL Ltd. with a resolution of 0.10 nm for lattice imaging and 0.23 nm for point imaging. Elemental composition of nanostructures were measured by Energy Dispersive X-ray Spectroscopy (EDS) attached to Field-Emission Scanning Electron Microscope (FESEM) Supera 55VP from CarlZeiss. X-ray photoelectron spectroscopy (XPS) (Kratos AXIS-Hsi) and UV-Visible (Agilent Cary 60 UV-Vis Spectrophotometer) measurements were conducted at the Research Institute of Advanced Materials (RIAM) of Seoul National University. Fluorescent images were captured using a institutionally housed Olympus FV1200 confocal microscope.PL measurements and transient decay kinetics (FlouTime 300, PicoQuant) were performed using excitation wavelength of 260 nm and Raman spectroscopy (LabRAM HR Evolution, Horiba) scattering measurements were performed at RIAM. Elemental composition was measured by Fourier Transform Infrared Spectroscopy (FT-IR) was performed using Vertex-80V/Hyperion2000 from Bruker instruments. Photocatalytic activity measurements were performed under UV–vis conditions. Samples were dissolved in 0.5 mM N719 dye solution and incubated for 2 h in the dark to facilitate dye absorption.
Results and discussion
In a multicomponent synthetic process separated by a 3-stage procedure, the method initially involved the pre-treatment of TiO2 anatase and graphene oxide (GO) at 120 °C in separate reactions using the same ionic liquid composition comprising charge separated cationic and anionic ions (1-ethyl 1-methylpyrrolidinium chloride). The approach ensured that both the anatase TiO2 polymorph and rGO were sufficiently monophasic prior to mixing at the same temperature under reducing conditions. Fig. 1 summarizes the synthetic steps designated as A, B and C.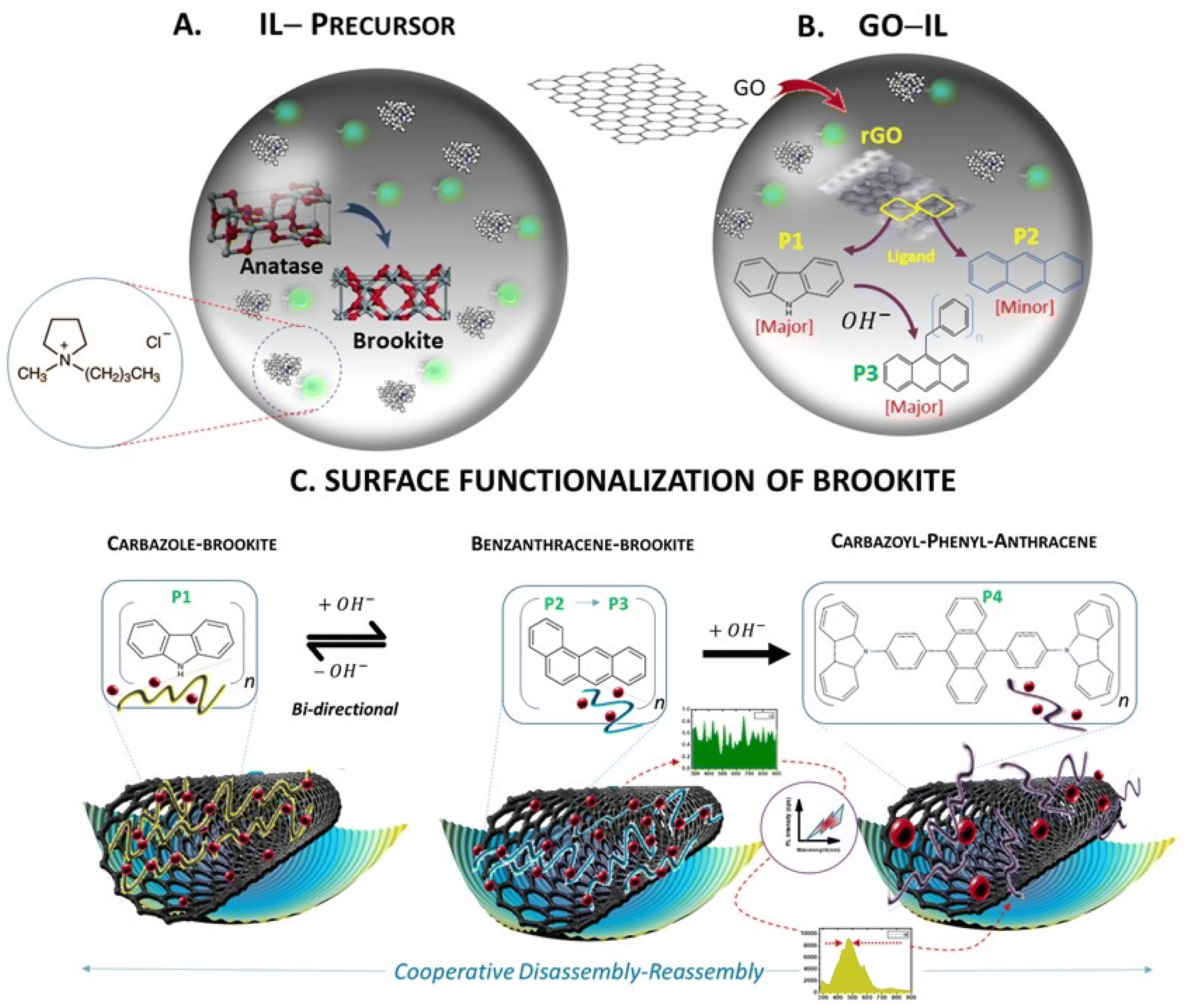 | ||
| Fig. 1 A schematic summary of the synthetic steps for the assembly of GO functionalised brookite quantum sized particles. The calcinated anatase phase of the resulting structure almost entirely retained the tetragonal monocrystalline configuration of anatase (99%) with an intensely low biphasic crystalline signal characteristic of a suppressed brookite phase (1%). (A) Exposure of calcinated TiO2 to 1-ethyl 1-methylpyrrolidinium (C7H16NCl) comprising a highly acidic charge separated cationic–anionic reaction medium (CA-IL) at X °C initiated the transformation of the anatase phase to brookite crystals. (B) The charge induced modification of GO at the CA-IL-GO interface drives the catalytic cyclisation of ring aromaticity at the carbocatalytic surface of GO leading to ‘far-equilibrium’ surface co-polymer structures (C) hierarchical thermally induced merger of brookite and surface functionalised rGO. Solvent and photo-assisted modulation of surface polymer structure-functionality enabling mobility and transfer of charge as an efficient photocatalyst. | ||
Self-assembled stabilisation of a metastable phase of brookite disguised within the anatase polymorph configuration
Titanium isopropoxide Ti(O-i-Pr)4 was used as a starting precursor at a calcination temperature of 250 °C. Following calcination of the reactive isopropoxide moieties under mild reduction, X-ray diffraction analysis showed TiO2 nanoparticles were almost entirely monophasic consistent with a tetragonal anatase configuration (99%) but was accompanied by a predominately suppressed brookite orthorhombic as minority phase (1%) with lattice parameters a = 5.4558 Å, b = 9.1819 Å, c = 5.1429 Å (JCPDS- 00-016-0617, space group Pcab) (Fig. 2a–d). The disintegration of the peak intensities in the deconvoluted form shown in the XRD profile substantiates the complete absence of the rutile phase but strongly supports the growth of a nanocrystalline anatase phase along the [101]. Although the growth parameters entirely favours anatase, the thermodynamic phase stability between both polymorphs is sufficient to reveal the overlapping diffraction peaks corresponding to interatomic d-spacing between polymorphs anatase and brookite. The process of calcination underlines the notion that phase transformation is intrinsically driven by temperature favouring the domination of anatase or rutile in the mixed phase.16 While rapid growth distinguishes the spontaneous growth of lower to higher energy surfaces, it is noteworthy that brookite shows preferential growth along the lattice plane [111] accounting for 33% of the total surface area for predicted morphologies.17 Phase transformation among the TiO2 polymorphs has been reported to be size dependent in earlier investigations and this phenomenon may be explained by differences in thermodynamic stabilities. The particle size calculated (using scherrer formula) from the major crystallographic peak corresponding to the anatase phase resides in the range of 10–20 nm. This size range however unexpectedly identifies well with stability range of brookite18 of the lower energy facet [111] implying that external factors might readily influence and favour polymorph growth of suppressed crystalline states over dominant phases. This may be particularly applicable for driving the reverse transformation of anatase to brookite which is energetically favoured by a factor of 14-fold compared to the configuration change from brookite to rutile. In this context, the synergy between high and low energy facets7 along the [111] direction of growth is now being recognised as an important route to stabilise brookite19 but continues to be a largely unexplored synthetic doorway to establish more easily accessible routes to acquire monophasic brookite photocatalysts. Nevertheless, reforming the synergistic relationship between active polymorphic phases by tuning the surface energy landscape is proving to be effective in liberating brookite mediated catalysis.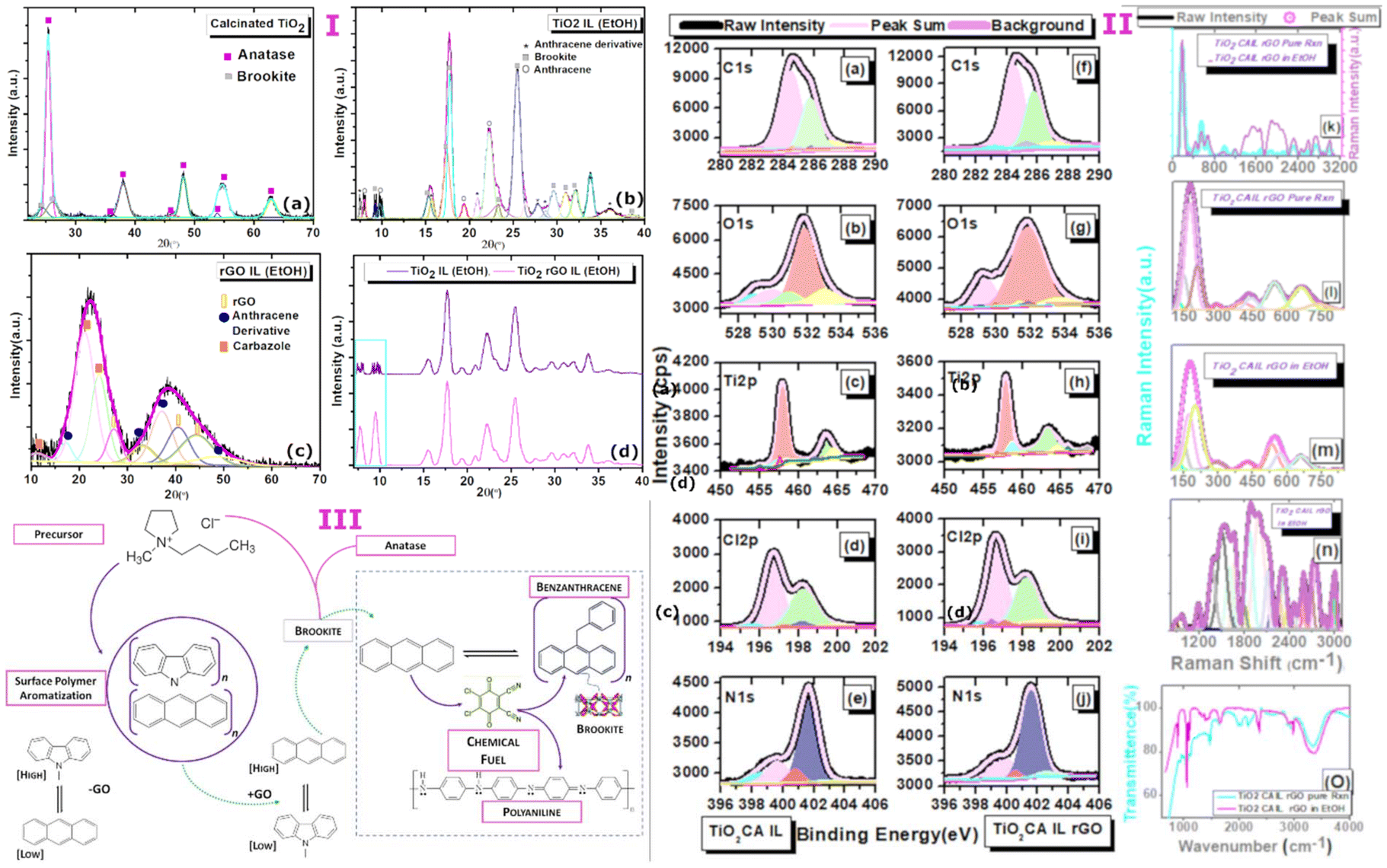 | ||
| Fig. 2 (I) X-ray diffraction profile of (a) calcinated TiO2 anatase and diffraction patterns of (b) CA-IL treated reduced graphene oxide in solvent, (c) CA-IL treated calcinated TiO2 (brookite) in the presence and absence of solvent and (d) synthetic merger of brookite and surface functionalised rGO embedded in the solvent environment. [(II) (a–j)] Binding energies from X-ray photoelectron spectroscopy (XPS) elemental profiles comparing TiO2 CA-IL (–rGO) and TiO2 CA-IL rGO (+rGO) (brookite). Raman spectroscopy of TiO2 CA-IL rGO in (k–n) in the absence and presence of ethanol in the wavelength range of 150–3000 cm−1 and fourier-transform infrared spectroscopy (FTIR) spectroscopy of (O) of TiO2 CA-IL in in the absence and presence of ethanol. (III) A Schematic showing precursor chemistry driving surface polymer aromatization of cyclic hydrocarbons modulated by rGO and the chemically fuel driven assembly of benzanthracene from anthracene at the brookite surface. | ||
The exposure of calcinated TiO2 from step A to a strongly anionic–cationic ionic liquid reaction medium resulted unexpectedly in a near-complete monophasic phase transformation from anatase to brookite with the stabilisation and subsequent arrest of the orthorhombic configuration with particle size of the order 3–12 nm (Fig. 2a). Abandonment of the anatase configuration marked by the complete and unusual shift to the brookite structure deviates from the commonly reported anatase–brookite bi-crystalline structure.6 The dramatic phase change is likely controlled by reconfiguration of landscape energies of exposed crystal facets at the anatase–brookite interface which is strongly indicative of tuneable forces. The reverse phase is possible by overcoming surface energy barriers at relatively lower temperatures than would be expected for conversion from anatase.
The crystal phase dependency evidenced here by a change in the chemical environment under moderate heat (120 °C) eliciting single-crystal reversibility occurs with synergistic precision. The XRD pattern was visibly coherent with the brookite phase with the loss of anatase crystallinity observed in the absence of the CA-IL. The orthogonal to orthorhombic phase switching however was accompanied by polymers identified as high intensity anthracene and anthracene derivatives of lower intensity diffraction peaks (Fig. 2 (I), 2a–d). The most intense peak in the transformation of brookite crystals is visible at 2θ of 16.5°, 25.5° with respect to planes [110] and [111]. Further, the cross-over stability from anatase to brookite is marked visibly by the multi-faceted alignment of particle growth along planes [010], [100], [121], [200], [130], [025] and [102] and may signify a closely balanced coordination of energies over a small size distribution. The fact that brookite is abundantly stable across broad areas of the crystalline surface likely indicates that the structural evolution of the brookite phase is largely occurring between the transforming particle and the CA-IL environment.
The full width half maximum (FWHM) along [110] is significantly diminished in comparison to [111] and the narrowing of FWHM is indicative of sharper lattice planes and reduced stress while peak broadening signifies reduction in the crystalline size and increase in atomic stresses. This observation is consistent with the decrease in particle size from 10 to 2.8 nm and growth confinement is a direct consequence of surface functionalization of brookite aligned along the [111] growth trajectory. We rationalized that the acid–base character of the charge separated cationic–anionic reaction medium was sufficiently well suited to selectively trap the brookite polymorph phase under chemically homogeneous conditions defying conventional routes favouring a tetragonal configuration.
Here, peak broadening also relates to the introduction of lattice imperfections due to interfacial stresses at defect sites imposing growth restrictions around nucleation of brookite particles predictably through the formation of electron-rich structures as evidenced here. The chemical polymerization of anthracene and its derivatives as aryl ringed structures has strong implications as TiO2-conjugated conducting surface polymers in semiconducting blends and will be discussed in more detail with reference to Step C. It is noteworthy however, that the multifaceted brookite nanostructure as a supporting catalyst can effectively promote the hydrogenation of surface adsorbed heteroaromatic rings comprising C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds such as pyrrolidinium (5 membered N-heterocycle) to generate fused or bicyclic benzene rings in the form of anthracene and their polycyclic variants under moderate reaction conditions. However, XRD analysis does not provide any insight of how the assembly of the ‘far from equilibrium’ carbocyclic rings and the role of the Cl− ions present in the reaction medium affect the stabilisation of brookite crystals and their growth via defect driven Ti–N or Ti–Cl associations. The lack of profile depth among these elements might be indicative of largely amorphous regions which are more intimately related to unresolved and undetectable minor phases indistinguishable from the bulk. The energy X-ray spectroscopic (EDS) compositional map of rGO–brookite shown in Fig. S1(a) (ESI†) reveals the elemental pattern from distribution of carbon, chlorine, titanium and oxygen at the brookite/rGO interface (Fig. S1(b)–(f), ESI†).
N bonds such as pyrrolidinium (5 membered N-heterocycle) to generate fused or bicyclic benzene rings in the form of anthracene and their polycyclic variants under moderate reaction conditions. However, XRD analysis does not provide any insight of how the assembly of the ‘far from equilibrium’ carbocyclic rings and the role of the Cl− ions present in the reaction medium affect the stabilisation of brookite crystals and their growth via defect driven Ti–N or Ti–Cl associations. The lack of profile depth among these elements might be indicative of largely amorphous regions which are more intimately related to unresolved and undetectable minor phases indistinguishable from the bulk. The energy X-ray spectroscopic (EDS) compositional map of rGO–brookite shown in Fig. S1(a) (ESI†) reveals the elemental pattern from distribution of carbon, chlorine, titanium and oxygen at the brookite/rGO interface (Fig. S1(b)–(f), ESI†).
Graphene oxide as a multi-faceted tuneable carbocatalyst for the co-polymerization of charge donor–acceptors
Interestingly in step B, the effect of graphitization of the pyrrolidinium–chloride rich IL (CA-IL) reaction medium as the second precursor to step C is characterised by spontaneous polymer self-assembly driven chemically by the reduction of GO to rGO in a heat induced reaction. N-heteroaromatization at the GO–CA-IL interface is strongly apparent through the self-assembly of both anthracene and carbazole at the surface of rGO. It is probable that exposure of rGO to the highly acidic Cl− content is highly favourable to altering the surface potential of rGO allowing the migration of charge between electron-deficient pyrrolidinium-N as the acidified precursor binding to acceptor sites on the rGO surface. The binding association between opposing charges on the carbon support and their engagement in proton transfer is a prerequisite to polymerisation. It is envisaged that the surface functionalisation of the rGO surfaces via (1) aromatic cyclisation of the N-linked short alkyl chain moieties extending the pyrrole ring to carbazole and (2) fusion of the 6-memebred rings to form anthracene both occur independently as evidenced by the preferred orientation of polymer growth from the XRD profile. The XRD profile (Fig. 2 (I) a–d) shows the effective use of reduced graphene oxide in catalytically influencing the restructuring of pyrrolidinium building blocks at the rGO boundary and thus enabling the co-catalysis of energetically related but divergent polycyclic polymers. The broad peaks aligned with rGO shown in the deconvoluted XRD pattern are indicative of high energy growth defects resulting from applied stresses from the complex interfacial composition of the reaction medium. The hybridization of extrinsic carbon and nitrogen bonds and their direct incorporation into the graphene framework results in geometrical changes around the growth polymers along the carbon network of sheets. Carbazole self assembles as the dominant polymer along [013] and [014] with clear evidence of anthracene as the secondary polymer with peak intensity along [310] direction and minor growth along [014], [025] and [231]. The FWHM of anthracene and carbazole (Fig. 2 (I) a–d) is comparably larger compared to the FWHM of brookite alone in the reaction medium which broadly signifies a shift towards amorphicity or at best, a state of poor crystallinity and narrowing of particle nucleation size. The broad nature of rGO peak reflections at 2θ of 27 °C, 40.5 °C and 44.5 °C indexed to planes [002], [100] and [101] show an overlapping diffraction pattern substantiating cooperative growth of the polymer at the rGO surface. De-convolution of the WXAS profile (Fig. 2 (I) a–d) indicates that carbazole is the dominant phase in the copolymer system and a quantitative assessment of the relative peak areas shows the large phase (58%) to be assigned to carbazole with orthorhombic symmetry (S.G.-Pnam, a = 7.779 Å, b = 5.722 Å, c = 19.15 Å)20 accompanied by a smaller phase (24%) specific to monoclinic anthracene and its derivatives (a = 8.561 Å, b = 6.036 Å, c = 13.506 Å, β = 125°, a = 7.910 Å, b = 6.430 Å, c = 23.960 Å, β = 99°) while 18% is indefinable with rGO.Step C involves the synthetic fusion of step A (brookite stabilisation in CA-IL) and GO treated CA-IL from step B. Further, evidence from X-ray diffraction data (Fig. 2 (I) a–d) revealed that the basic architecture of the pyrrolidinium cation as the starting building block permits ring opening and undergoes heat-induced aromatization via the short alkyl chains coupled to reactive ring nitrogen. The geometrical change to the pyrrolidinium structure occurs by ring addition strengthening the heterocyclic chemistry to carbazole. Cyclisation around the 5-membered ring parent is strongly supported by a core Ti–N bond character (Ti 2p; 457.93 eV) around the nitrogen atom of the pyrrole ring of carbazole in the absence of rGO. Further, the magnitude of the Ti 2p BE correlates well with the BE of N 1s (401.6 eV; 61.89%) with respective peak intensities of Ti 2p and N 1s around 52 and 58.95% (Fig. 2 (II) c, h and e, j). Orbital overlap between Ti- and N- also correlates to the formation of oxygen vacancies shown for Ti–graphene interfaces21,22 and driven by a substantial increase in Ti3+/Ti4+ ratio resulting from the bulk reduction of the isopropoxide precursor to nano and quantum states mediated by anatase. The influence of the thermally driven nitrogen-rich cationic–anionic reaction medium was noticeably surface directed with the percentage atomic Ti3+ concentrations quantifiably around 59% (BE Ti 2p; 457.8 eV, 58.95%) (Fig. 2 (II) c, h). Binding energies of the C 1s spectrum further support a pyrrole-like framework (BE C 1s; 284.46 eV, 57.04%) (Fig. 2 (II) a and f). The substantial C 1s peak area (∼57%) is in strong agreement with N 1s peak area (∼59%) assigned to Ti–N bonding sites confirming the expectation that the BEs of C 1s and N 1s BEs originate from the pyrrolic ring. Pyrrole ring adsorption at oxygen deficient Ti3+ sites evidenced by Ti–N binding energies predicts the loss of the propyl and methyl moieties from the pyloric nitrogen that allows its chemical transformation to an active pyrrole derivative via an unprotected nitrogen ring. The exposure of the electron-rich heteroaromatic framework to a high concentration of electro-deficient Ti3+ vacancy sites is consistent with the nucleophilic substitution of N-linked hydrocarbon moieties. This configuration favours the central pyrrole ring of carbazole directed through the interaction of the nitrogen lone pair and TiO2 heterochemistry (C4H4N–Ti).
Despite the high surface exposure of Ti3+ defects embedded within the brookite structure, the effect of combining brookite from step A with rGO-carbazole-anthracene from step B, the selective suppression of carbazole was observed in the XRD profile in the mixing of A and B in the presence of rGO but not in its absence The spectral change was accompanied by polymer switching from anthracene to benzanthracene. The higher order Bragg reflections in the XRD profiles highlight the differences between TiO2 CA-IL and TiO2–CA-IL–rGO indicating the direct interaction between brookite and rGO as a key step in the synergistic selection of an anthracene polymer derivative while minimising carbazole to basal levels. Fluorinated ionic liquids have been used to suppress the growth of crystalline facets with the ability to direct the control of anatase-to-rutile transitions with high selectivity23 by dominating the growth suppression of [112] facet. The relationship between semi-conductor electrocatalytic surfaces and their interaction with primary and secondary modulators which (1) initially inhibit or divert the growth of crystallographic facets by interacting with active sites (site 1) and further (2) a secondary modulator that is also sensitive to catalyst surface structure (site 2) but operates as a waste chemical fuel to drive the assembly of ‘out-of-equilibrium’ structures. We propose that chloride-rich aqueous structures acting as chemical fuels use energy from their surroundings to self-assemble and influence facets via oxidation/reduction events and thus influencing both surface polymer growth and stoichiometric relationships among them.
Chemically fuelled synergy orchestrated at the rGO–brookite interface for aligning surface-to-sub-surface charge transfer
A detailed examination of XPS data provides insight into the complexity of substrate-selective catalytic processes and the structural and functional relationships between spectroscopically detectable chemical structures that result from those events from chemical building blocks. A major clue to the origin of co-assembled heterocyclic and non-heterocyclic structures comes from the identity of a photo-excitable dichloro-dicyano benzoquinone (DDQ) derivative (scheme, Fig. 2 (III)) which is coupled to N 1s BEs of 399.3 and 399.5 eV (Fig. 2 (II) e and j) irrespective of the presence of GO. The quinone functional group is also supported by Raman peak at 1660 cm−1 (Fig. 2 (II) k–n) and a slightly shifted peak at 2978 cm−1 corresponding to 2,3-dichloro-5,6-dicyano-1,4-benzoquinone revealing the cyano (1994–2164 cm−1) and chloro (550–794 cm−1) pendant groups of benzoquinone in TiO2 decorated rGO. The importance of DDQ to its role in steering the facet-controlled growth between copolymers carbazole and anthracene is strongly evident in the brookite/GO copolymer mix. (Further discussions are provided as supplementary text for this section). The wide-angle X-ray scattering (WAXS) profile shown in (Fig. 2 (I) a–d) depicts the nature of the interplay between polymer suppression and growth modulation by addition of polymer bound rGO from step B. Here, in the presence of ethanol as the polar solvent, the diffracted intensity of carbazole is sharply reduced to 15% (4.8-fold decrease) while the anthracene peak modified to benzanthracene increases to 29% (1.2-fold decrease) in the presence of rGO and brookite (51%). We assert that access to controllable polymer conversion at the rGO–brookite interface is mediated by the high reduction potential of quinone. This is afforded by interchangeable oxidation states allowing the transfer of charge for dehydrogenative and oxidative couplings and aromatization or cyclisation of C–H, C–C, C–O and C–N bonds at electron-rich sites. The suppression in the diffraction peak of carbazole occurs through its modification to the donor–acceptor 9-vinyl(-9H-carbazole)-tetrachloro, 1,2 benzoquinone [(–CH(CH12H8N)CH2)]n (OC6ClxO) (scheme, Fig. 2 (III)) is evidenced by the slightly higher N 1s BE of 401.6 and 401.5 eV relative to unmodified carbazole with BEs 400.6 eV (−rGO) and 400.5 eV (+rGO) (Fig. 2 (II) e and j) accompanied by a variable degree of ring chlorination (x = 2 or 4) by atleast 2-fold as observable from weak Raman signal. Hence, a facet dependent selectivity favouring benzanthracene over carbazole is strongly observed at the rGO–brookite interface. DDQ assisted vinylation of carbazole is also noticeable, introducing the potential for further reactivity and possible aromatization of the monomer. In the presence of strong oxidants, the transition from sp3 to sp2 hybridization of GO occurs by the loss of oxygen at C–O sites24 and this finding is corroborated by the spectral decrease of C–O bond vibrations in TiO2 decorated rGO shown by the Raman bands in the range 889–1017 cm−1 peaking at 975 cm−1 (Fig. 2 (II) k–n) by atleast 2-fold as observable from weak Raman signal. The formation of new electron-rich C–H bonds [CH2 and CHCH] more strongly identifiable from the Raman peaks (550–794 cm−1) (Fig. 2 (II) k–n) and subsequent release of CO (BE; C 1s 282.69 eV) (Fig. 2 (II) a and f) and an increased water peak around 1652 cm−1 in the presence of GO in the FT-IR peak (Fig. 2 (II), O) which may to some extent be complexed as acidified water nH2O·Cl (BE; Cl 2p 198.1 eV) (Fig. 2 (II) I and j). In view of the evidence demonstrating that polymer assembly is surface directed, the localised conversion of polymers occurring at polymer/rGO junctions in the presence of self-assembled DDQ oxidants is more inclined to be driven by C–C bond rotation and H migration via hole defects25 permitting surface rearrangement. The catalytic transformation of anthracene to benz(a)ntharcene agrees well the cyclohydrogenation of the GO surface with elimination of two hydrogens [–2H] which may assist in the ring aromatization of anthracene to benz(a)ntharcene (scheme, Fig. 2 (III)) via C–C ring coupling in a polar solvent environment. An intermediary clue to the possible fate of 9-vinyl(-9H-carbazole)-tetrachloro, 1,2 benzoquinone (Fig. 2 (III)) is the complete loss of the N 1s BE around 401.6 eV (Fig. 2 (II) e and j) at the rGO/brookite interface favouring the formation of polyaniline identified from the N 1s BE of 402.4 eV.
The conversion path involves the electrocyclic intermediary 1,4,4-trimethyl-2,3-diazabicyclo(3.2.2)non-2-ene N,N′-dioxide (scheme, Fig. 2 (III)) which shares the same BE with structure DDQ (N 1s BE; 401.6 eV) and surface characteristics of a conjugated protonated forms of amine (–NH2+) and imine (NH2+) complexed with unprotonated amine (–NH) and imine (NH–) groups.26 The biopolaron character of the ringed structure comprising of cationic nitrogen pairs [N+N+] in close proximity redistributed in the conjugated ring structure of the polyaniline chain (scheme, Fig. 2(III)) and are located across the centre of the polymer. The probable distortion caused by the close proximity of the pair (–NH2+) within localised regions of the lattice likely contributes to its destabilisation and susceptibility to the structural rearrangement in the presence of GO. The relatively small peak area associated with the intermediary structures is indicative of its transitory state to polyaniline. Polyaniline is stabilised by π-conjugation within the rGO layers forming the backbone of the lattice through the stacking of layers. The Fourier transform infrared (FTIR) band vibrations of the complexation of polyaniline at brookite/rGO interface in solvent (Fig. 2 (II) O) is characterised by the quinoid ring architecture (1644 cm−1) with associated ring vibrations across C–N/C–H bands (1314 and 1088 cm−1).27 Further, C–C, (1466 cm−1)28 CC, (1631 cm−1) and CN+ (1466 cm−1) band stretches of the quinoid moiety of polyaniline is also evidential to the complex structure. In addition, stretching vibration between the benzoid C–CN (1298 cm−1) quinoid CN+ (1237 cm−1) and N+H rings signify the inclusion of a polaronic and bi-polaronic structure to the overall assembly.
The ratio (ID/IG) of the Raman active peaks of rGO at 1372 cm−1 (D) and 1506 cm−1 (G) is a good indicator and measure of structural defects. A 2-fold decrease in [(ID/IG)−ethanol]/[(ID/IG)+ethanol] ratio of brookite decorated rGO (162.7/260.9) demonstrates that the solvent environment substantially increases complex stability by improving the alignment between polyaniline and rGO and the overall graphitic nature of the polymer. As discussed earlier, the substantiated loss of the rGO peaks 2θ = 27, 40.5 and 44.5 °C visible in the XRD profile of rGO (no longer observed at the brookite/rGO interface) is a direct consequence of the shift to benz(a)ntharcene which signifies the surface association of anthracene with rGO. The uniformity and alignment of the peaks of neat brookite with brookite/rGO functionalised benz(a)ntharcene convincingly argues in support of a reconfigured rGO polymer framework with the ability to strongly intercalate uncoordinated brookite particles organised by linear or circular layers. It further is anticipated that the effect of solvent could also increase the hole scavenging rate of TiO2 as observed in other systems.29
Defect driven confinement of brookite
An unexpected and unique observation in the precursor chemistry leading to the fabrication and arrest of brookite relates to size selectivity of particles. The low-dimensional confinement of particles in the range of 2–2.8 nm shown by the particle size distribution shown in Fig. 3n was achievable for brookite which often represents a considerable synthetic challenge in the fabrication of narrower band-gap semiconductors that are intrinsically broad—a problem well recognised for TiO2 polymorphs. To better understand the surface dynamics of the growth leading to the brookite morphology, 3D surface analysis of the regional space surrounding the trapped crystalline brookite phase of the HRTEM image uncovers the transient nature of the pre-crystallization nucleation phase. | ||
| Fig. 3 Evolution of brookite quantum particles at the GO–polymer interface. (a) HRTEM lattice pattern of the tubular like growth of rGO polymer complexes. Polymer evolution and growth at the polymer–rGO interface corresponds to carbazole/Anthracene lattice directions as shown by the atomic arrangement of the FTT patterns in insets (b–e) with a possible (f) amorphous liquid origin. (g) The growth restriction of brookite, rGO and benzanthracene to quantum scales of (h–j) depicts one-dimensional complexed morphologies. (p) Low resolution and (o) high resolution rGO decorated polymer complexes directed by (k–m) multifaceted crystallographic faces. (n) A histogram analysis of the particle size distribution of brookite–rGO particles showing the highest frequency resides in the range of quantum confinement. | ||
The initial reduction of GO in CA-IL results in heterostructures (Fig. 3h) defined by the overall surface energy at the intersection of GO and carbazole/anthracene complex. The reduction of GO to rGO under continuous heat is observed to accelerate the ‘rolling’ of rGO sheets forming a tubular-like structures of dimensions 100–300 nm (Fig. 3j, inset). We propose that the nucleation and growth of carbazole/anthracene particles is synchronous to the helical-tube-like folding of rGO—a process which facilitates the strong polymer-driven encapsulation of TiO2 brookite particles within the enclosed architecture. A restrictive growth regime imposes spherical to 1-dimensional expansion in concert with polymer growth inhibition under quantum confinement. The growth constraint of anthracene to scales <3.0 nm (Fig. 3g) below the quantum size which may have an amorphous origin (Fig. 3f).
These events maybe better related to the surface progression of polymer growth at the rGO–brookite interface. Fig. 3a shows lattice image of the tubular-like growth of rGO and the diffraction patterns immediate to the surface of rGO provides a spatial frequency content of the surroundings using Fast Fourier Transform (FFT) generated from different sections of HRTEM image indicated withing circles and rectangle. The atomic arrangement here further supports the presence of benzanthracene and the growth dynamics with anthracene in the local regions of the FFT (Fig. 3, inset b and c). Presence of carbazole and rGO along with benzanthracene is evidenced by the region dissected form surface of the overall HRTEM image (Fig. 3d and e). The features resemble amorphous growth patterns leading to the tuneable confinement of brookite crystals TiO2 originating from a readily formed amorphous network (Fig. 3f and Fig. S2, ESI†) indicative of multiple nucleations. Anthracene evolves in the amorphous region while benzanthracene originates from non-amorphous regions as represented by the reciprocal space patterns of the diffraction spots for respective heterointerfaces. A detailed examination of the low-dimensional bright field image of brookite decorated rGO shown from 100 nm (Fig. 3p) to higher resolution images down to 5 nm (Fig. 3o) shows that the rGO–brookite interface is shaped by several crystallographic planes (Fig. 3, inset k–m) through a process that involve multifaceted cooperativity which is likely solvent dependent.
This finding might signal a role for Cl− in the phase stability of brookite along [110] and [111]. The functional directionality of the polymer switch may be rationalised by a number of advantages that (1) enhances the surface area of brookite substantially in allowing the benzene moiety of anthracene to be easily integrated within the extensive GO network, (2) increased surface–surface interactions facilitated by π–π stacking of rGO layers in a well patterned framework and (3) permitting polymer embedded photon responsive TiO2 centers to capture energy driving the (4) spontaneous transfer of charge. This implies a synergistic relationship between the spontaneous assembly and disassembly of carbazole and benzanthracene respectively and a change in the solvent environment plays an important role in regulating the chemical transformation. From a biological perspective, organic building blocks undergo disassembly–assembly processes of co-assembled structures30 and are accompanied by both structural and morphological changes relating to non-covalent interactions in supramolecular structures.31 The notion that the short hydrocarbons liberated as intermediates can act as precursors for extending the Ti–pyrrole ring framework at the organometallic interface by forming non-equilibrium structures by self-assembly is conceptually important to the design new photonic structures. Such pathways are typically aligned to interfacial surface energies driving disassembly and assembly of non-equilibrium structures.
Examination of the surface features by 3D surface analysis provides a topographic visual of information that remains inaccessible by direct HRTEM imaging to distinguish regional differences at the crystalline and non-crystalline interface. In the transmission electron microscopy imaging of the trapped crystalline brookite phase, the transient nature of pre-crystallization nucleation phase of amorphous growth patterns is observed in the 3D surface analysis of the transmission microscopy image shown (Fig. 3f). The 2D to 3D projection map provides an image of the surface topography by height separation of more elevated non-crystalline regions of an amorphised depth of 130–140 nm (violet region) and crystalline orientations of depth 40–95 nm (green to light blue). The thickness difference points to the distribution of entanglements of the amorphous region as large dense areas of surface polymers comprising the carbazole/benzanthracene network controlled by their thickness.32 These observational differences are consistent which changes during the dynamic interplay between the mobile amorphous, semicrystalline and crystalline transition phases. Slowed crystal growth due to a high degree of entanglement of evolving polymer chain segments in restricted spaces and is potentially reinforced by ring aromatization and Ti–N ligand chemistry increasing the possibility polymer caged ring compartmentalisation.33 Previously we have seen that the availability of subsurface Ti3+ catalytic states and their bond association with nitrogen species can intrinsically alter particle growth characteristics by driving the formation ring size geometries. The particle size distribution of brookite (Fig. 3n) is sizeably weighted towards confinement demonstrating the need to understand and apply key ‘growth restrictive’ regimes in fabrication methods. The peculiar arrest of the brookite polymorph phase is symbolic of changes in the landscape chemistry typified by slowed crystallization kinetics and growth confinement substantially delaying or preventing polymorph conversion. This suggests that barriers to nucleation favouring pathways to rutile formation are strongly dependent on (1) control of amorphous-to-crystalline states and their growth trajectories (pre-crystallization pathway selectivity) and (2) arrest of precursor states (3) the effect of secondary nucleation of contrasting surfaces such as polymers regulating confinement and nucleation growth through the binding of self-assembled building elements at oxygen vacancy defect sites. Polymer assembly is intimately linked to metal catalytic sites that use chemical building blocks derived from cationic–anionic reaction environments. Identifiably, the order of dimensionality is a consequence of these combined events. Quantum confinement of TiO2 may be indicative of covalent bond distortion lying at the Ti–N bond interface of pyrrole and brookite reminiscent of stress-induced polymer at the Ti–N bond interface of pyrrole and brookite and is reminiscent of stress-induced polymer forming non-equilibrium structures by self-assembly is conceptually important to the design of new photonic structures. Such pathways are typically aligned to interfacial surface energies driving disassembly and assembly of non-equilibrium structures.
Examination of the surface features by 3D surface analysis provides a topographic visual of information that remains inaccessible by direct HRTEM imaging to distinguish regional differences at the crystalline and non-crystalline interface. In the transmission electron microscopy imaging of the trapped crystalline brookite phase, the transient nature of pre-crystallization nucleation phase of amorphous growth patterns is observed in the 3D surface analysis of the electron transmission microscopy image shown (Fig. 3f). The 2D to 3D projection map provides an image of the surface topography by height separation of more elevated non-crystalline regions of an amorphised depth of 130–140 nm (violet region) and crystalline orientations of depth 40–95 nm (green to light blue). The thickness difference points to the distribution of entanglements of the amorphous region as large dense areas of surface polymers comprising the carbazole/benzanthracene network controlled by their thickness.32 These observational differences are consistent which changes during the dynamic interplay between the mobile amorphous, semicrystalline and crystalline transition phases. Slowed crystal growth due to a high degree of entanglement of evolving polymer chain segments in restricted spaces is potentially reinforced by ring aromatization and Ti–N ligand chemistry increasing the possibility of polymer caged ring compartmentalisation.33 Previously we have seen that the availability of subsurface Ti3+ catalytic states and their bond association with nitrogen species can intrinsically alter particle growth characteristics by driving the formation ring size geometries. The particle size distribution of brookite (Fig. 3n) is sizeably weighted towards confinement demonstrating the need to understand and apply key ‘growth restrictive’ regimes in fabrication methods. The peculiar arrest of the brookite polymorph phase is symbolic of changes in the landscape chemistry typified by slowed crystallization kinetics and growth confinement substantially delaying or preventing polymorph conversion. This suggests that barriers to nucleation favouring pathways to rutile formation are strongly dependent on (1) control of amorphous-to-crystalline states and their growth trajectories (pre-crystallization pathway selectivity) and (2) arrest of precursor states (3) the effect of secondary nucleation of contrasting surfaces such as polymers regulating confinement and nucleation growth through the binding of self-assembled building elements at oxygen vacancy defect sites. Polymer assembly is intimately linked to metal catalytic sites that use chemical building blocks derived from cationic–anionic reaction environments. Identifiably, the order of dimensionality is a consequence of these combined events. Quantum confinement of TiO2 may be indicative of covalent bond distortion lying at the Ti–N bond interface of pyrrole and brookite reminiscent of stress-induced polymerisation at the Ti–N bond interfaces of pyrrole and brookite and is reminiscent of polymer directed confinement33 compounded by growth inhibition via N doping. Surface and sub-surface states located close to bulk regions indicated unperturbed accessibility to modification by exposure to the cationic–anionic ionic liquid (CA-IL) environment in the rGO-IL brookite preparation after thermal treatment. Such binding energies associated with the formation of Ti3+ oxygen vacancies (Ov) are strongly indicative of defect traps (Ti2O3) forming a mixed Ti2O3/TiO2 lattice. The synthetic approach allows for a rationalized procedure for the surface stabilisation and functionalisation of TiO2 brookite. In the absence of rGO, synergistic changes in the chemical state of the brookite surface may play a significant role in phase selection of the polymorph relative to both anatase and rutile. The dominant binding energy peak originating from the Ti 2p spectra is assigned to 457.8 eV (Ti 2p; 457%) and signifies a high concentration of Ti2O3 defect states. The high occurrence of oxygen deficiency states shows that the cationic configuration of Ti2O3 defects is strongly synergistic with binding energies between the nitrogen and carbon species at the Ti3+/O interface.
Multifaceted growth of brookite in CA-IL assists quantum size confinement and band gap reduction and band gap reversal
Fig. 4(a) shows a comparison of the UV-vis profile of rGO functionalised (TiO2-CA-IL + rGO) and non-functionalised (TiO2-CA-IL) brookite. The graphitization of brookite is corroborated by the spectral red-shift from 228.4 to 237.3 nm correlating to an intensity increase from 0.5 to 0.8. To estimate the absorption characteristics of functionalised and non-functionalised forms of brookite, we compared the spectra to the solar spectral behaviour based on specific Air Mass (AM) coefficients at AM0 (solar flux without atmospheric interference) and AM1.5 (solar flux@air mass of 1.5 at 48 °C relative to AM0) as standards to define solar energy fields at the Earth's surface. With respect to functionalised rGO brookite, the absorption path of TiO2-CA-IL + rGO intersects AM1.5 at 0.68 and plateaus in the visible-infrared range (450–850 nm) corresponding to an absorption magnitude of 52.3%. This compares to an intensity crossing of 0.94 (6% absorption) for non-functionalised brookite relative to AM0 from 850 nm. For visual clarity, Fig. 4(b) shows a 3D representation of the relative absorption of functionalised and non-functionalised forms of brookite relative to AM0 and AM1.5. This result correlates well to a significant change in the lattice parameters of rGO functionalised brookite indicated by several higher angle diffraction peaks at (2θ = 27.1°, 50.5° and 44.6°) to a single low angle (2θ < 10°) (Fig. 2 (I) a–d) dominated by its surface functionalisation with benzanthracene at the TiO2 interface along the d002 crystallographic face. The d002 growth trajectory of graphene oxide is reminiscent of the low angle growth pattern and increased interlayer spacing along the d002 plane.34 Linearization of the UV absorption curves conforming to the Tauc equation αhv = B(hv − Eg)n where α is the absorption coefficient, h is the Planck constant, v is the frequency of light and B is a constant was used to elucidate the distribution of energy of states. Treatment of the data effectively distinguishes between Tauc exponents (n) 0.5 and 2 which adhere to a non-linear and linear relationship with respect to the value of n. A plot of photon energy (eV) versus (αhv)2 (eV cm−1) Fig. 4(c and d) for TiO2 CA-IL in the presence of rGO shows linearity when n = 2 and is strongly indicative of an indirect semiconductor which is consistent with the characteristic of brookite. Extrapolation of the slope of [(αhv)2/hv] provides the indirect optical band energy Eg at the intercept yielding a value of 0.78 eV. This lies below the extrapolated band gap of 1.16 eV for TiO2 CA-IL in the absence of rGO. Here, we borrow the notion developed by Zhang et al.35 in their analysis to explain the probable differences in photo-generated electrons between direct and indirect routes to energy transfer at the level of photons and phonons. The inability of indirect semi-conductors to participate directly in e−h+ recombination processes unlike direct band gap semiconductors usually signify differences in the charge carrier properties coupled to their respective propagation of phonon (mechanical) or photon (electromagnetic) energy states. In case of the former, an increase in the lifetime decay of e−h+ recombination events is a consequence of diffusion and prolonged reaction times with the effect of enforcing charge separations over longer time periods and thus enhancing photocatalytic competence. The band edge of rGO–brookite that resides in the UV region diminishingly extends into the visible range.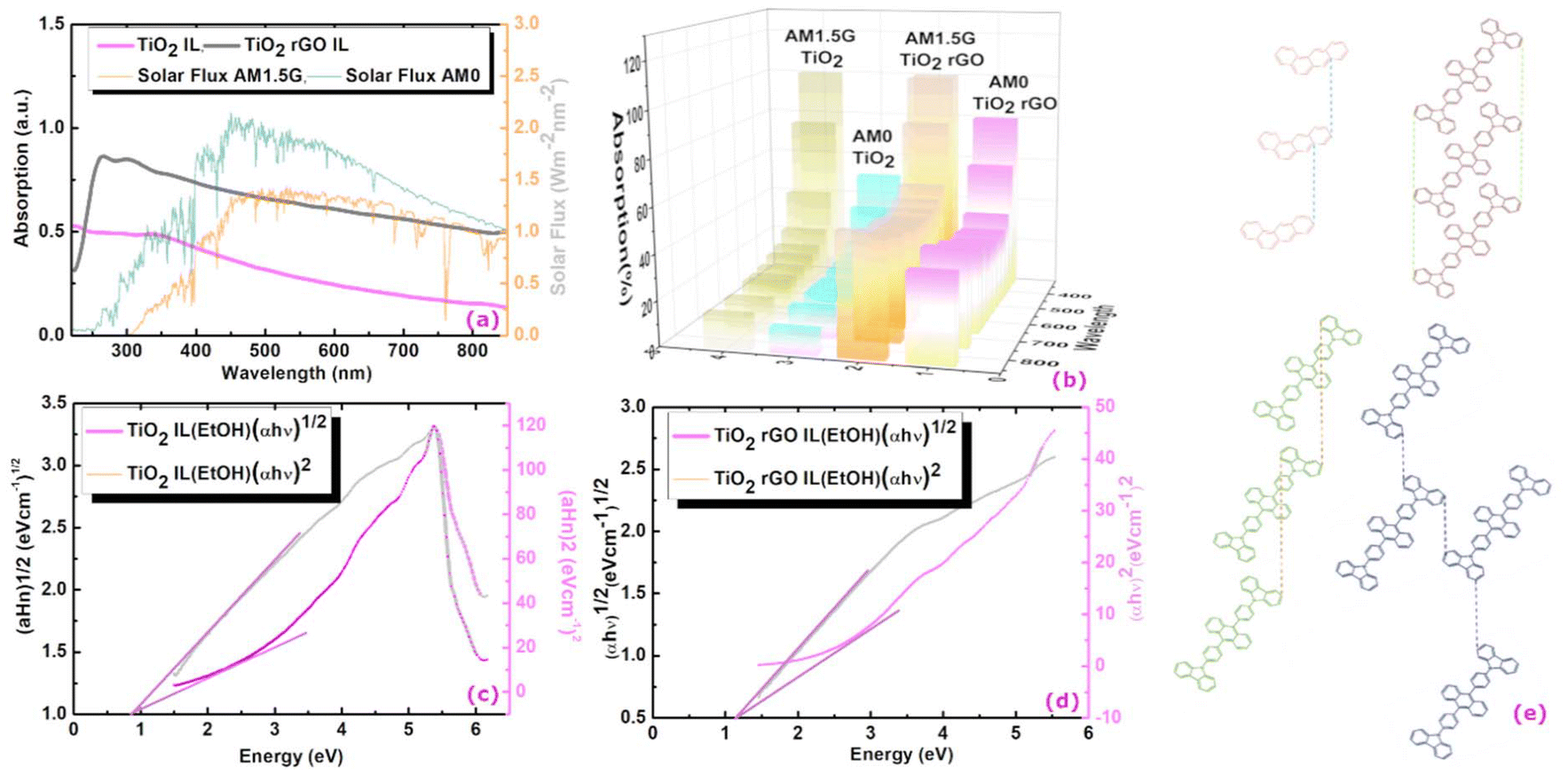 | ||
| Fig. 4 (a) UV-vis absorption spectra of rGO–brookite. Brookite exhibits stronger ultraviolet absorption properties in the presence of rGO to brookite in the absence of rGO. A performance comparison of the absorptive properties of brookite to the solar flux generated AM0 (without atmospheric interference) and AM1.5 (solar flux@air mass of 1.5 at 48 °C relative to AM0) corresponded to 6 and 52.3% respectively represented as a (b) 3D comparative plot of a. (c and d) Adherence of non-functionalised and functionalised brookite to the Tauc equation [αhv = B(hv − Eg)n] distinguishable by linearity (red line) of the extrapolated slope (band gap energy, Eg) versus non-linearity (black line) demonstrating that complexation of TiO2 (brookite) with rGO results in switching from direct to indirect optical transition semiconductor behaviour. The morphological transition from anatase to brookite correlates to a change in the Tauc exponent (n) from n = 0.5 (direct band gap) to n = 2 (indirect band gap) yielding a quantifiable band gap of 0.78 eV and the result agrees well with quantum sized brookite particles confined within rGO layers. (e) Possible pairing symmetry for π-conjugated electronic configuration of anthracene and carbozyl phenyl anthracene predicted from Hückel tight-binding calculations.38 Orbital interactions involving the same or different connection sites of repeated conjugated assemblies produce distinct stacking patterns among overlapping carbons of valence and conduction bands strongly influencing band gap structures. | ||
An important consideration influencing charge transfer properties for ringed structures is the degree of aromaticity in which ring structures must be cyclic, conjugated, and flat in nature and the number of π electrons must conform to and satisfy [4n + 2]π Hückel's rule. This describes a delocalised system of p-linked orbitals positioned in parallel allowing their interaction and this arrangement emphasizes the importance of a planar structure while [4n] π electronic structures are anti-aromatic. Here, we speculate that the moderate ID/IG ratio (193.8/282) evident from Raman spectroscopy analysis (Fig. 2 (II) k–n) might infer that (1) rGO defect sites result from the loss of atomic carbon from the framework and (2) may also reflect a monocyclic ringed hydrocarbon conforming to the [4n] π electronic structure composed of an odd number of conjugated π electrons represented by a singlet system of valence bonds or an odd system of polycyclic hydrocarbon atoms (n ≥ 7). This gives rise to non-bonding orbitals, resonance energies and a considerable number of localised hole defect heterogeneities across the rGO framework.36 Reversibility in aromaticity of the ππ* excited state of [4n + 2]π electron configuration to antiaromatic behaviour is particularly documented for benzene for its role in photo-assisted transformation to new structures coupled to its ability for H atom abstraction. The self-assembly property benz(a)anthracene to carbozyl phenyl anthracene evidenced here is representative of the fusion of the polymerisation of aromatic carbazole and benz(a)nthracene. In the context of the Baird rule, the merger is a result of the excited-state antiaromaticity of [4n + 2]π electrons of benz(a)nthracene with aromatic carbazole units of the larger polycyclic conjugated hydrocarbon structure. This is consequential to a rearrangement in electron delocalisation across an extended structure of conjugated units which further adapts to a decreased band gap.37 The expansion of benzanthracene end capped with carbazole moieties cater for increased stability conducive to a lower energy structure with increased electron mobility across shared p-orbitals. This offers a distinct advantage of tuning ππ* excitable states and their adaptability to aromatic and antiaromatic states of matter leading to their engagement of useful electronic configurations defined by their interactions.
Furthermore, the structural arrangement among monomer units in polymeric organic semiconductors and the underlying connectivity to band gap type might be predictable from pairing symmetry around bonding orbitals. A theoretical study based on extend Hückel tight-binding calculations, the stacking geometry of conjugated structures is a determining factor in band gap selection. The findings stipulate the alignment of carbon–carbon atoms in a parallel type assembly conforming to a direct overlapping of the plane or a skewed arrangement (Fig. 4e) in which the ring structures are shifted along the plane resulting in a misaligned carbon–carbon bonding arrangement. The probable transition from a skewed C–C atom anthracene structure to a non-skewed C–C aligned structure shown for carbozyl phenyl anthracene or a zig-zag orientation (Fig. 4e) may provide some structural significance from a direct to an indirect band gap inversion.38 The surface conjugation and stacking arrangement over GO layers and the interaction with surface conjugated structures can dramatically affect the band gap properties at the GO–brookite interface. The possible interaction of the lattice oxygen of TiO2 with GO detectable by XPS (O 1s, 529.2 eV) (Fig. 2 (II) b and g) together with the depth of C–C interactions (C 1s, 284.5 eV) (Fig. 2 (II) d and i) might be indicative of the involvement of photoelectron trapping of oxygen at the GO edge favouring charge transfer at the expense of electron–hole recombination. Doping of the TiO2 precursor clearly facilitates the homogenous restructuring of GO from a 0-dimensional material to a 2D structure with interchangeable bonding configurations passivated by N-chemistry under quantum confinement. Band confinement at the TiO2–GO interface strongly correlates with the departure of atomic vacancies localised around Ti–N and Ti 2p3/2 (Ti3+) bonding sites corresponding to BEs of 458.6, 465.88 and 455.74 eV (Fig. 2 (II) c and h) in the presence of GO. The finding may have considerable overlap with the decrease in the O/C ratio for low bandgap materials which coincide with the increased aromaticity of GO nanosheets and the narrowing of HOMO–LUMO transitions often associated with the clustering of GO sheets.39 The bulk reduction of bivalent (Ti4+) to trivalent (Ti3+) centers signifying the formation of oxygen vacancies following calcination of the TiO2 anatase precursor in the presence of bulk phase dopants correlates well to binding energies of surface state defects. It would therefore not be out of context to suggest that the ring distortions within the rGO framework are the result of disorder-induced arrangements dictated by the patterns of association at these sites. A reduced bandgap has been correlated to changes in the carbon–oxygen orbital hybridization occupied between the bandgaps of rGO and brookite. Substantial reductions in the BE at Ti–N defect sites correlates well to increases in Ti–O–C associations sites (O 1s, 531.7 eV) (Fig. 2 (II) b and g) between Ti with graphitic carbon via Ti–O− activation following functionalisation with rGO. This may be consequential to changing the O/C ratio affecting the chemical state of carbon and electron localisation and possibly reducing e−/h+ recombination. Ito et al.40 proposes stronger bridging between π-bonds restricting electronic states at graphitic carbon resulting in a direct to indirect bandgap transition. We extend this idea to the passivation of N-occupied defect sites resulting in the replacement of Ti–N (77%) to a Ti–O–C configuration (62%) increasing the rGO–brookite lattice oxygen intercalation from 15.2 to 21.98%. This is consistent with the increase in rGO from a zero-band gap material to a finite state while reducing the bandgap of TiO2.
An important consequence arising from the intercalation of rGO and brookite is the separation between the valence and conduction bands of rGO leading to an opening in the bandgap characteristics from 0 to 0.78 eV at the rGO–brookite intersection. The introduction of chemical modifications within the confined band structure while transitioning from GO to rGO through disruption of ordered sp2 to disordered sp3 defects drives the progression of new discrete energy states in the conduction band and emergent luminescent properties resulting from hybridized states impacting e−h+ recombination. Such events are evidenced by significant changes in luminescent properties universally coupled to dimensional confinement, size, shape and chemical composition altering the C/O landscape. To judge the range of energies associated with rGO–brookite particles, we imaged the photo-responsivity of the hybrid material by increasing the amount of rGO–brookite to a fixed intensity of UV light over a short exposure time. Instantaneous enhancement in the luminescent behaviour of the material with increasing concentration (Fig. 5(a–d)) indicated that the measured photoresponse was intrinsic to its structure. We conclude that strong luminescence under UV irradiation supports a quantum confined multicomponent assembly consistent with restricted growth patterns coupled to a multifaceted assembly as observed earlier. Further, to determine the origin of strong luminescence, initial exposure of rGO–brookite particles to different excitation wavelengths (λex) demonstrated multi-emissive behaviour generating blue, green and red fluorescence shown in (Fig. 5(e–g)) indicative of differing electronic transitory states and multiplicity of excitable species of rGO–brookite. Although the rGO–brookite nanoparticles show stronger green emission characteristics, multiple excitable shifts from cyan, yellow, orange and pyrol were also visible across longer emitting wavelengths indicative of broad luminescence in a single solvent. The multi-emissive nature of rGO–brookite provides some insight into the quantum dot size variation revealing a population of variable energy states for red, green and blue regions (Fig. 5(e–g)). The observation implies a degree of photo-switchability among the distinctive emitting species permissible through a small energy barrier of separation for energetically closely related assemblies through self-assembly. We assert that these events are important to understanding how existing surface structures such as rGO-derived carbazole and their energy configurations relate to newly evolving surface functionalities and barriers to their state of interchangeability under extrinsic excitation. It is notable that the C 1s spectral profile of rGO–brookite shows considerable resemblance to the C 1s spectrum of graphene quantum dots (GQDs).41 However, the reduced oxidised form of graphene shows a more strongly pronounced shoulder indicating less dominance from the sp2 spectral carbon peak overlap and more alliance with graphene oxide quantum dots (GOQDs) reflecting a greater diversity of oxygen functional groups, particularly C–O residing around 286 eV. This observation points to the existence of a rGO crystalline carbon structure intermediate to GQDs and GOQDs. The origin of the prominent green fluorescent emission (Fig. 5e) however, is indicative of oxygen functional groups ascribed to GOQDs while blue fluorescence is largely emitted by GQDs (Fig. 5g). This observation might be consistent with the UV spectral pattern in line with the intrinsic behavioural properties of rGO–brookite.
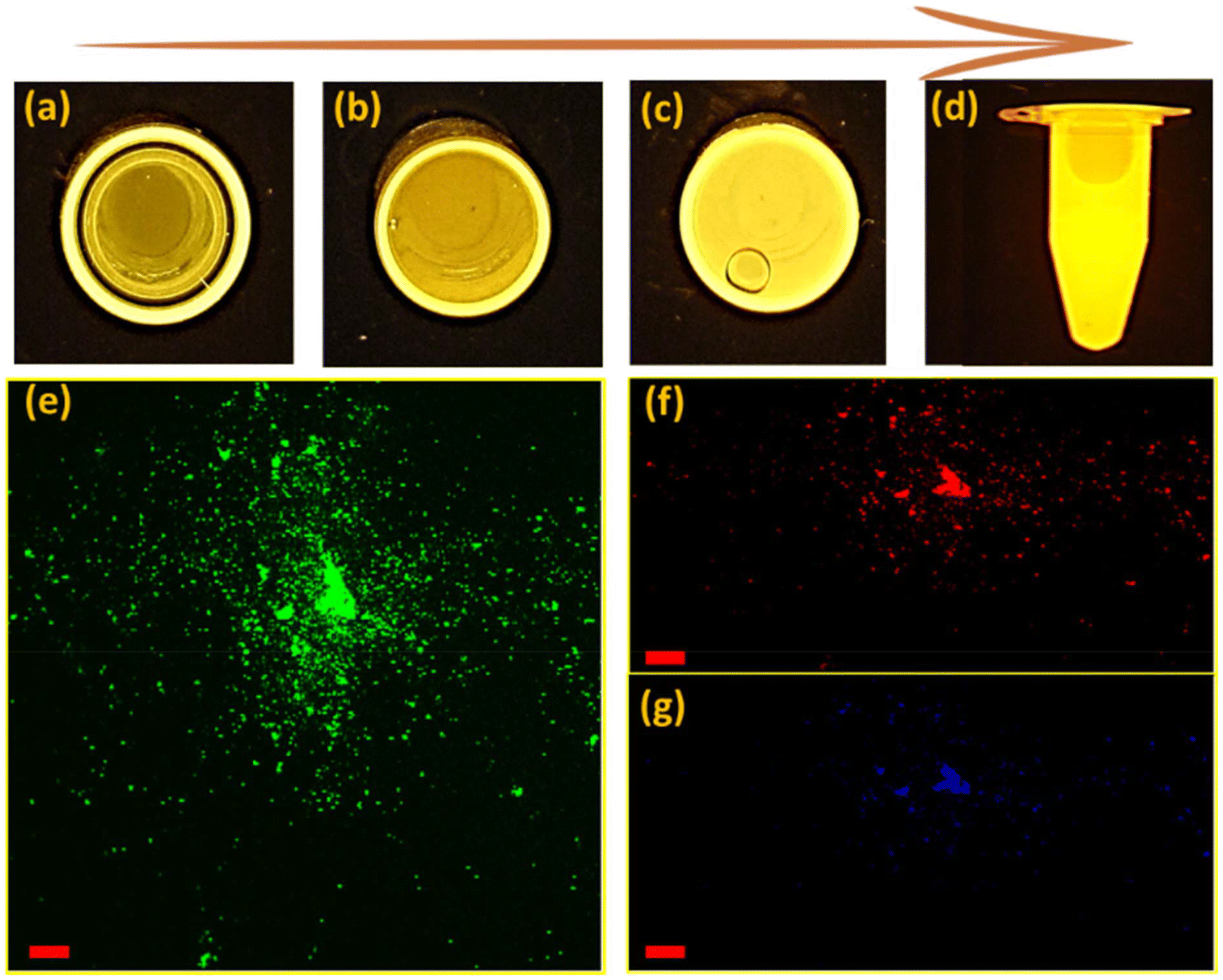 | ||
| Fig. 5 Luminescent and fluorescent images of rGO–TiO2 (brookite). Increase in the luminescence of quantum GO–brookite particles is correlated to the enhancement in intensity of particle concentration from (a) to (d) resulting from the quantum confinement of GO and TiO2 particles. The multi-emissive nature of the particles signifying photoswitchable energy states at the rGO-brookite interface. | ||
Photo-switchable multi-emissive properties of rGO–brookite
The emissive properties are further tuneable from the donor–acceptor units of surface bound carbazole and anthracene residing above or within intercalated sites of rGO exhibiting both photochromic and fluorescent signals. The absorption peaks of the UV-vis spectra of rGO–brookite (Fig. 6a) shows there is considerable complexation between rGO and brookite which differ significantly from the pristine forms reported to occur around 23142 and 210 nm respectively. The complexation is evidenced by a strong redshift to 265 nm from the non-complexed form (231 nm) with a characteristic high-to-low energy tail synonymous with rGO extending across the visible and infra-red regions of the spectrum. A partially masked 335 nm absorption band at the peak tail of rGO shows resemblance to carbazole and the underlying structural association with the rGO surface42 through self-assembly. Further, the rGO-coupled anthracene peak at 355 nm (ref. 43) resides close to the carbazole band indicative of the close energy configurations between both structures. Substantial increase in the ID/IG ratio of rGO–brookite from 0 in the absence of rGO to 0.7 in the presence of rGO (Fig. 2 (II) k–n) reflects the degree of graphitization evidenced by the increase in the sp3/sp2 ratio indicating an enhanced defect state correlated to the C 1s BE's at 284.4 (56.7%) and 285.7 (33.7%) (Fig. 2 (II) d and i) signifying the association with functional C–O/C–N and –CO respectively. The broad extrinsic emission properties of the hybrid material following graphitization of brookite likely originate from structural and geometrical changes introduced at the rGO–brookite interface with the possibility of linker sites and conjugation distances between rGO sheets. This hold the potential to be modulated by the interaction of carbazole and moieties regulating excitation thresholds via charge transfer processes. We also explored indications for the modulation of the bandgap energy at the rGO–brookite interface in view of the peak shifts in the in the 2D band in the Raman spectra of rGO. Since band gap the represents the optimum energy difference in electronic transitions between the valence and conduction bands of semiconductors, dramatic changes to the band structure of brookite and rGO suggests that is highly responsive and adaptable to the functional nature of the polymer surface of rGO at interfaces. Hetero-restructuring at strained interfaces is a prerequisite to lattice matching, energy harmonization and band alignment through structural intercalation and band gap tuning. Such properties are inherent to the modifiable surface of rGO, high surface area and multiplicity in layering of graphene. A distinguishable feature of rGO is the bandgap modulation via the disruption of sp2 bonds accessed from the rGO edge bonds by oxidation of functional groups and anchoring of pendant group chemistries. This remarkable character is marked by a non-luminescent to a luminescent switchable state and a multi-emissive state inherently achievable via a flexible band gap. The ability of rGO to distort the structural configuration of brookite through intercalation as a composite via bond hybridization in ex situ is evidenced by the appearance of a strong C–O bond ascribed to C 1s peak at 288.5 eV between Ti and carbon atomic framework of rGO (Fig. 2 (II) d, i and c, h). Peak shifts around 100 cm−1 in the 2D band in the Raman spectra of rGO from 2500 cm−1 is observed at 2552, 2596 and 2698 cm−1 and is indicative of structural modifications to the oxidised graphene carbonaceous framework while the 2D band at 2700 cm−1 is symptomatic of sp3 defects. The Ramen scattering of excited electrons arise from the defect-phonon interactions and their properties characteristically shape the 2D scattering patterns44 and stacking of rGO layers.
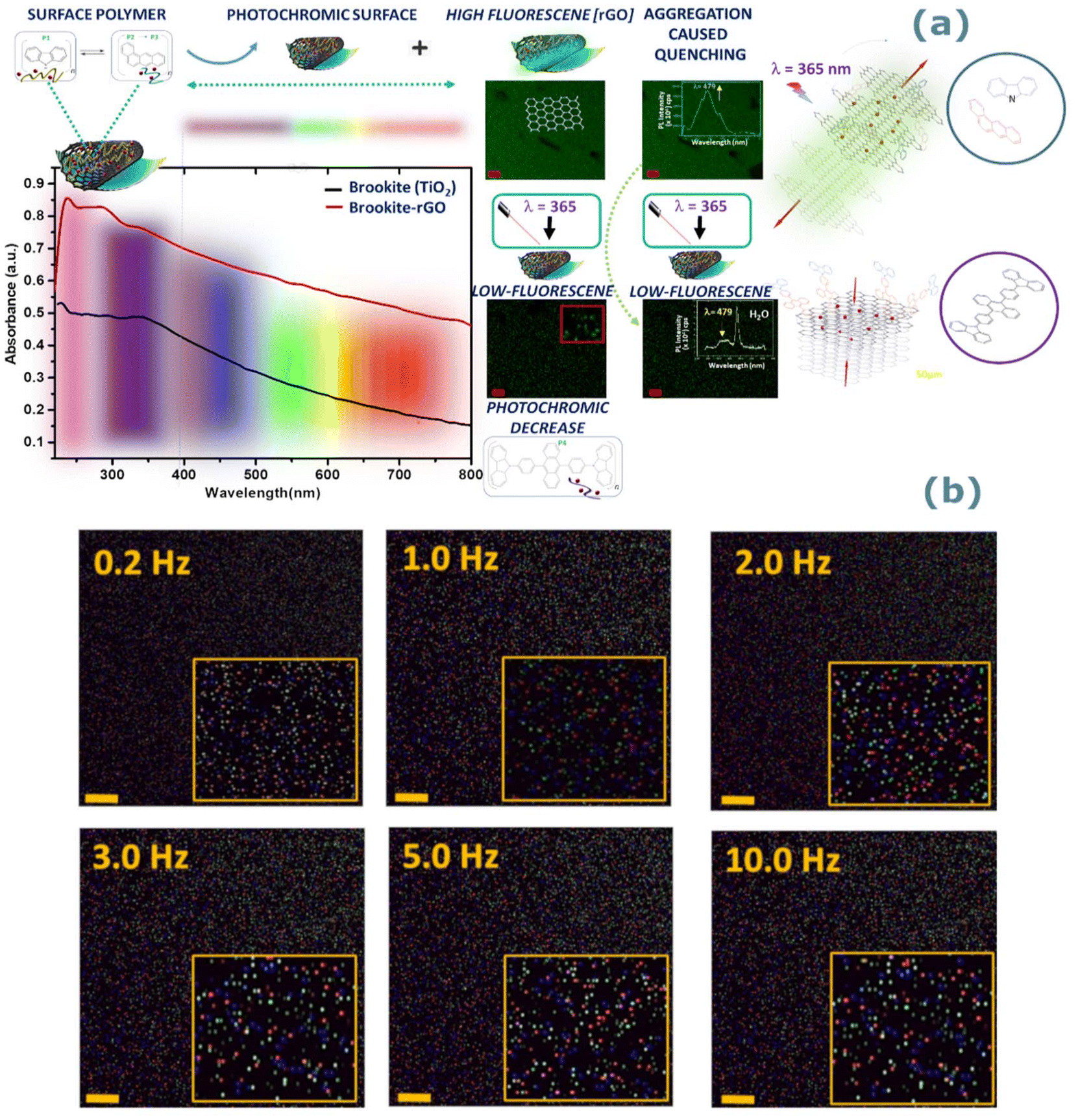 | ||
| Fig. 6 Photochromic and luminescent properties of rGO–brookite. (a) Comparative UV-vis absorption spectra of brookite and low bandgap rGO–brookite nanoparticles. The coupling of carbazole and anthracene moieties to the GO surface at 335 and 355 nm respectively extend across the UV-vis region of the spectrum. Further densely populated fluorescent rGO–brookite particles in ethanol (and inset) show the strongly emissive multi-peak PL spectrum (λex = 265 nm) with a peak maximum at λ479 nm (9.252 × 103 cps) Loss in fluorescence intensity after UV-irradiation at 365 nm reveals dynamics between excitable and non-excitable states linked to extrinsic and intrinsic properties and a state of photo-switchability. Inset shows a solvent triggered quenching effect from a change in solvent environment from ethanol to water resulting in a 1000-fold drop in PL signal and complete loss of the peak maximum (λ479 nm). Non-quenched and quenched states of rGO–brookite is inherent to interlayer-compression and decompression to an extrinsically driven sp2-to-sp3 arrangement away from the more stable sp2 configuration. (b) Fluorescent imaging of rGO–brookite demonstrating size dependency of particles modulated by frequency changes under a fixed voltage of 0.5 V measured between 0.2–10 Hz. | ||
The inherent photochromic and fluorescent nature of rGO-bound carbazole and anthracene moieties and the proximity of immobilised states can potentially show photoswitching between excited and quenched states mediated by energy transfer processes or charge transfer aided by the rGO matrix. This possibility arises for reversible interchangeability or hybridization between the inter-switching of excited and non-excited states. Fluorescent and photochromic tuning is therefore achievable by employing both photochromic and fluorescent property enhancements by utilising spacer units which exploit conjugated chemical features large enough to permit the transfer of charge without interference from overlapping photochromic and fluorescent peak emissions. Hybridised structures arising from the union of carbazole and anthracene or their derivatives may possibly satisfy the requirements for luminescence through the decoupling of emitted peaks.
The possibility of controlling material luminescence is highly desirable for bandgap tuneable materials. We tested the intrinsic photo-switching characteristics of self-assembled rGO–brookite by introducing (1) a solvent change replacing ethanol with water and following changes in the photoluminescence (PL) signal and (2) by monitoring wavelength emissive properties intrinsic to its quenched or no-quenched state under UV irradiation at λ365 nm and (3) exploring how differences in size related observations could be inferred to such interpretations. We observed a considerable loss in fluorescence signal after excitation at λ365 nm (Fig. 6a). The residual fluorescence signal however is indicative of multi-emissive components with intrinsic emissions modulated by the spacing properties of rGO intercalated by photochromic moieties via self-assembly. A more insightful interpretation emerges from a change of the solvent from a high-polar to a low-polar environment. The complete abolishment of the PL signal in water compared to ethanol (Fig. 6a) advocates that the weak polarity of water substantially suppresses the number of emitting fluorophores causing ground-state silencing under excitation. Two distinguishable features in the PL profile is the immediate loss of the maximum PL peak at λmax at 479 nm exceeding 1000-fold drop in the PL count (counts per s) (Fig. 6a). Nano domains associated with the peak position at 479 nm provide elemental clues about the morphological pattern intermediate to the intrinsic π–π* sp2 configuration and the extrinsic character comprising n–π* sp3 clusters consistent with chemical functionalities which show sharp deviation from the stable sp2 character. The unambiguous assignment of NIR green emissions bands occurring at 490 and 520 nm to edge defect states of GQDs to COOH/C–OH, CO/C–O chemical functionalities45 reveal a more comprehensive understanding of GQDs surface trends under a range of λex. The progressive GQD PL peak migration from 266 nm redshifted to positions at 422 and 448 nm generate respective barriers to the excitation of mixed intrinsic/extrinsic states solely to extrinsic states supporting the idea of intrinsic to extrinsic state transitioning. Here, rGO–brookite is evidently distinguishable from GQDs by virtue of a comparatively larger red shift PL emission from 416 nm (in case of pristine GQ)46 to 479 nm (Fig. 6a) towards the NIR region with a decreased dependency on the sp2 configuration. The sharp digression to a quenched fluorescent state under irradiation at λ365 nm and abolishment of the PL signal under reduced polarity (Fig. 6a) is strongly indicative of the involvement of n–π* (hexagonal carbon units denoted by n) and oxygen defect absorption regions through the introduction of new energy bands. Although the dominance of the λ479 nm peak is clearly visible, the multi-peak origin characterised by a considerably broad full width at half maximum (FWHM) (Fig. 6a) does not exclude the possibility of a subdomain population suggesting differences in their PL origin photoinduced to follow a sp2-to-sp3 configuration. Shared characteristics with GQDs and GOQDs hold the presumption that the sp2-to-sp3 arrangement is likely intermediate to both. PL peaks undergoing a redshift transition from 436 to 487 at λ325 nm show size dependent behaviour affecting particle diameter.41 With this observation, we analogize cooperative events in graphite that undergo interlayer compression from sliding to intra-layer buckling mediated key sp2-to-sp3 configurations.46 We speculate that the collapse of the PL peak at 479 nm is symptomatic of the loss of fluorescence from the interruption subdomains resulting from prolonged exposure at λ365 as depicted by Fig. 6 showing the transition of rGO–brookite to a compressed form upon UV exposure. We used fluorescence microscopy imaging to explore the phenomenon of size driven excitability to correlate events to the accepted view supporting size dependent PL behaviour largely influenced by quantum confinement effects. We expect that the emissive properties of rGO–brookite is likely to be intrinsically coupled to the fluorescent and photochromic properties of carbazole/anthracene moieties and the pairing arrangements described in Fig. 4e. The affect of intercalation may also influence the overall luminescent behaviour of assemblies. In Fig. 6b, we selected a range of frequency-voltage signals to explore size modulation effects on rGO–brookite particles by visualizing the multi-emissive properties (enlarged view shown in the inset). Particle size showed diminished size properties at 1.0 Hz with the voltage set at 0.5 V. With increased scaling of the frequency, particle size increased from 2.0–10 Hz indicating that size change in response to frequency-voltage stress is tuneable with a uniform size distribution. Laser guided transformation is inherently modulated by changes in particle size driven by electronic forces leading to deformation, charge carrier multiplication and selective phonon excitation which necessarily implicates sp2-to-sp3 governing events and the creation of e−h+ pairs. Reduced bandgap at the site of carbon–TiO2 orbital overlap not only operate as non-covalent interlayer connectors during lone-pair donation into empty π* orbitals at oxygen defect sites but are potentially key targets for phonon interactions and recycling of photo excited electrons within unoccupied high energy conduction bands. Emissive trapping effects could significantly select for events conducive to red-shift behaviour. The luminescent and fluorescent properties of rGO–brookite particles and the induced chemical functionalization of rGO and the property change at the rGO interface potentially hinders the non-radiative nature of pristine rGO sheets exhibiting much weaker emission properties arising from higher e–h pairing recombination events.
Multi-exciton responsive material property of quantum confined brookite for photo-efficient dye degradation
We investigated if the reduced band gap intrinsic to the rGO–brookite structure could lead to an emission enhancement of the photo-active polymer conjugated surface by tuning excitonic response of the annealed surface under PL. Following excitation at 265 nm, the PL spectrum shows that the absorption edge of π-conjugated brookite polymer semiconductor blend extends across the visible range (380–740 nm) corresponding to the photonic energy range of 2.75–1.98 eV. The direct-to-indirect bandgap transition marks a very weak wavelength dependence of the PL peaks at lower intensities (I1–I6.5) in the presence of rGO (Fig. 7). This might signal a possible transition from the monomer to a multi-layered configurational change from the merger of rGO and TiO2 originating from an almost flat energy band. The PL modulation profiles demonstrate changes in the spectral profile shape and shifts in the peak wavelength and intensity peaks after exposing rGO–brookite (step 3) to increasing photon excitations (Fig. 7 1I-8I). With increasing energy, the rGO–brookite structure is reconfigured as a NIR red-shifted band peaking in the photonic band range of 2.75–3.26 with a distinct peak around 479.5 nm (Fig. 7; 8I) in ethanol. The intensification and considerable narrowing of the PL emission peak of the progressive plots of 1I – I8 (Fig. 7) may be reflective of the decreased heterogeneity of quantum confined rGO–TiO2 clusters driving a reduction in the size separation among of rGO quantum dot population. Non-uniformity in the quantum dot population coupled with weak confinement reveals a strong tendency to drive a band gap shift at the rGO–brookite interface that potentially optimizes a charge e−h+ separated state through quantum dot size selection.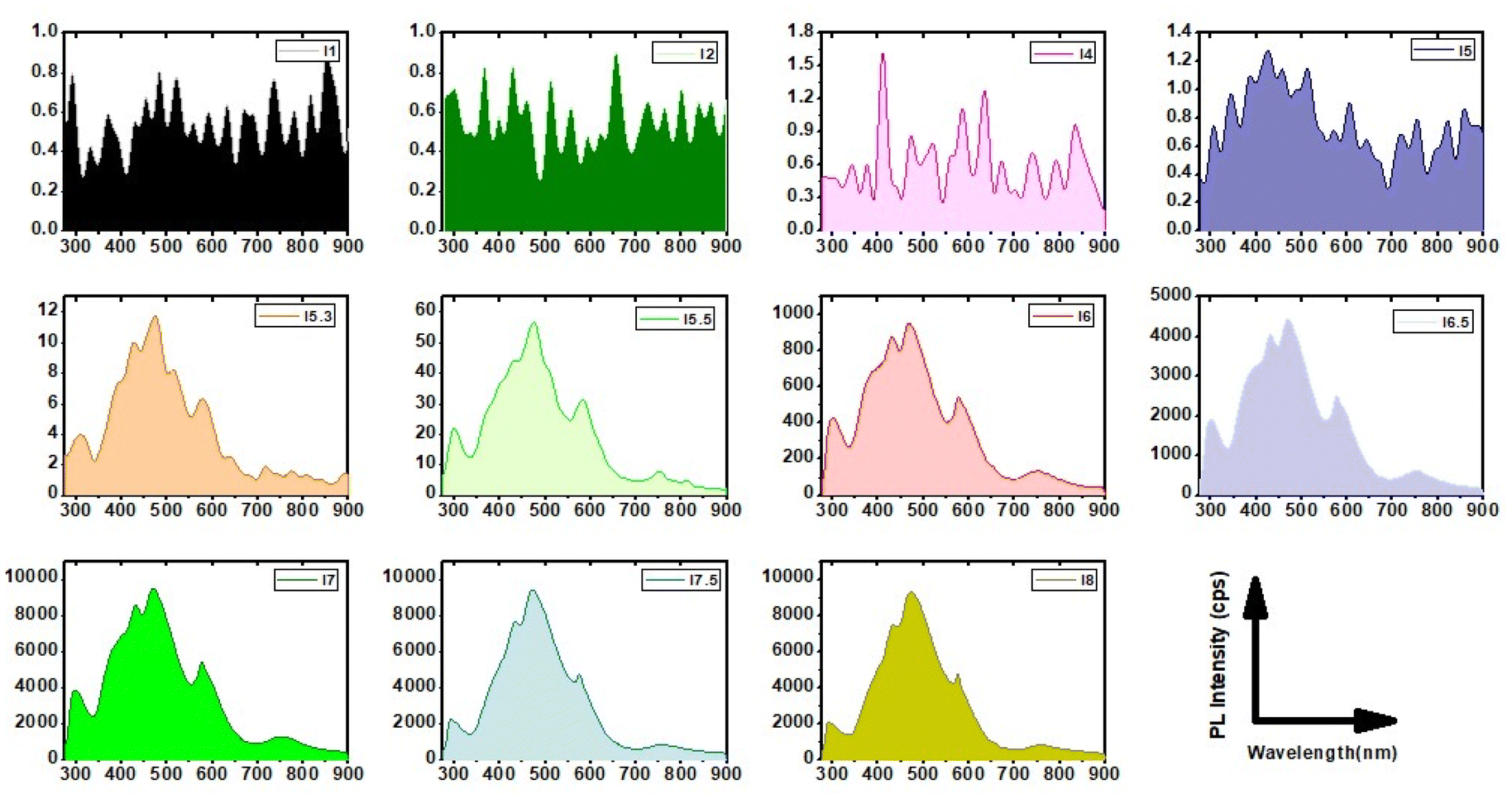 | ||
| Fig. 7 Tuning the excitonic response of rGO–brookite. Marked changes in the shape and PL intensity of rGO–brookite reflects the evolution of the exciton population as a function of wavelength emission under excitation at 265 nm with increasing laser excitation power from I1 to I8. | ||
The steady-state absorption of rGO–brookite reveals a phenomenological occurrence which potentially defines the structural adaptation between switchable polymers carbazole and benzanthracene. We propose that the external photon excitation energy shapes the evolution of an excited state structure which is representative of an overlapping vibronic wavefunction corresponding to a dominant new emission peak of 479.5 nm from time-resolved spectroscopic analysis of decay curves in the nanosecond (ns) range and time-correlated single photon counting (TCSPC) at room temperature. The excited state dynamics of the newly localised peak of the main emitting species marks a stability that is indicative of the PL emission of a carbazolyl-phenyl-anthracene crystal reportedly assigned at 445 nm in CH2CH2 and 550 nm as a film47 which bears some resemblance to the electrodeposition of carbazole onto rGO visible at 450 nm.42 Evidence in support of an excimer necessitates the formation of a photoinduced half-dimer between anthracene and carbazole which does not exist as a ground state structure as shown by the XRD profile (Fig. 2 (I) a–d) and the excited state is transitory and short lived at least across the nanoscale time period. The multi-fluorescence properties of rGO–brookite can be assigned to the decay kinetics of three exponentials with decay constants τ1, τ2 and τ3 (Fig. S3 and Table S1, ESI†). The time decay kinetics of a multi-exponential fit for each peak in the PL profile in the range of 280–900 nm with photon intensity 5.3 reveals the highest peak intensity (peak 3) of 9.41 × 103 cps. The time-correlated single photon (TCSPC) fast decay time (τ1) of 0.40 ns (Fig. 7; I5) correlates well with a lifetime of 0.45 ns (Chen et al.)47 indicating fluorescence emission of an excitable species and two slower phase components of 2.64 (τ2) and 8.91 (τ3) ns at lower intensity. However, a longer time constant τ4 at 44.93 ns is also detectable. The time constant τ1 (ns) for rapid decay is a relatively short-lived species and contributes to 4.23% of the overall emission which is substantially lower compared to the slower decaying components τ2 and τ3 (ns) representing 50.84 and 44.93% of the total PL respectively. Resolution of time constants correlate well to three emissions bands and relative contributions.
However, π-stacking through molecular coupling between weakly conjugated structures increases the excimer lifetime and correlates well with increasing exposure of excited photons measured on the time scale of seconds. This observation fits well with the self-photodimerisation of anthracene and in this case, with heteroarenes such as carbazole and is consistent with the broad emission properties in the range of 400–600 nm which is typical of anthracene derivatives.48 A key feature in controlling excimer properties in the excited state different to its structural configuration in the ground state is governing the π–π distance of host–guest interactions with increased stacking density at the brookite surface. Hence, the increased photon signals become the major route to drive new polymer associations marking the divergence of pathways away from a chemical to a photochemical dependent reaction path.
The photocatalytic efficiency of the self-assembled surface functionalised rGO–brookite polymer was demonstrated by the interaction of the photocatalyst with the chromophoric dye, N719.
The rapid release of the degradation products was monitored spectroscopically over a timescale of minutes. This is indicative of a multi-component structure that is well assembled to operate as an electron-donor–acceptor complex at the TiO2–polymer interface enabled synergistically via rGO. Epoxy and hydroxyl groups residing within and at the graphitic surface are identifiably key enablers in the charge transfer process by permitting non-carbon chemisorbed functional groups to interact with O-rich and H-rich energy intermediates. Hence, hydroxyl groups positioned at the centre of epoxy groups for example result in unpaired electrons. This is conducive to nitric and nitrous (NO)x groups and NO3 binding configurations detectable with BEs around 396.8 eV (N 1s) (Fig. 2 (II) e–j) which support the finding that nitrogen oxides show stronger chemisorption with rGO than GO49 and can strongly influence degradation performance.50 The richly acidic environment due to the presence of Cl− can induce oxygen binding sites for the nucleation and vertical growth of polyaniline wrapped between inter-spaced rGO sheets.51 A 2-fold increase in the ID/IG defect ratio (Fig. 2 (II) 2k–n) correlates well with a ‘bottom-to-top’ sheet-to-sheet like architecture providing anchoring sites for rGO π–π stacking of layers. Such defects can potentially serve as local sites for the nucleation of alternate benzoid/quinoid ring conjugation of polyaniline stabilised via sheet-assisted vertical alignment.52 The wired-like architecture is consistent with the sheet-like stacking and is an example of how self-assembled structures can cater for long-order interfacial contact and connectivity to effectively assist in photoelectron transport from the surface to the interlayers of rGO. The photocatalytic decrease of the N719 peak (333 nm) from 1.237 × 10−4 to 5.399 × 10−5 by brookite/rGO polyaniline composite under light irradiation represents a 56% loss in the dye stability in 30 min and is comparably superior in efficiency to the performance of brookite in the absence of rGO (36% peak loss in 90 min) (Fig. 8). The efficiency difference is indicative of a high absorption coefficient of dye molecules but also highlights a structural–functional synergy between rGO, polyaniline and brookite that is reflective of the conversion efficiency.
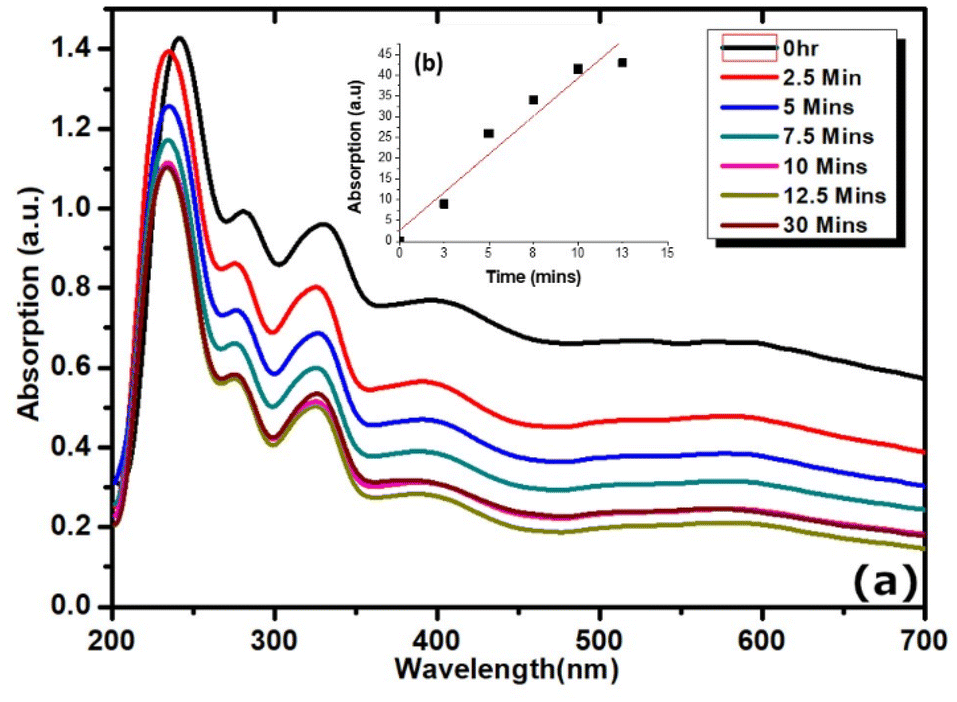 | ||
| Fig. 8 (a) Time dependent catalytic N719 dye degradation by photoactivated rGO–brookite. Dye degradation was monitored by loss of the N719-dye desorbed peak. The rate of dye degradation is given by the linearity of the slope shown in insert (b) and is plotted as a function of the decrease in the dye absorbance peak at 333 nm. The structural loss of the dye from the dye–rGO–brookite interaction occurs at an efficiency rate of 3.7 min−1 with an estimated loss of 68% of the dye at the brookite interface. | ||
The energy alignment between reduced graphitic carbon orbitals and Ti atoms of brookite becomes critical in facilitating Ti–O–C bonds and is strongly matched with interfacial bonding. In the context of an direct-to-indirect band gap reversal, enhancement in the crossover from a semimetallic to semiconductor state is achieved through band positioning of the lowest orbital of carbon located below the conduction band of TiO2.53 Closely matched band structures here would also imply a longer life time of photogenerated carriers increasing carrier decay kinetics54 observable in the photoactive behaviour of rGO–brookite (τ4 = 44.93 ns) which is attributed to stronger electronic coupling in the hybridized band structure at the carbon–Ti2+/Ti3+ overlap, particularly at oxygen vacancy interfacial sites. In this scenario, lowest unoccupied orbitals in the conduction band can operate as a photoelectron sink potentially impeding e−h+ recombination53 tuned through localized graphitic regions in rGO framework. Such a working hypothesis is further evidenced by a large PL redshift at λ479 (NIR) and a small interfacial band gap. The higher dye degradation rate may primarily be seeded in the charge separation of e−h+ properties as a prominent feature of the excited state. PL measurements suggest that the dynamics between self-assembled polymers could be critical to the charge carrier mobility at the surface and the intercalation of polyaniline between rGO sheets could significantly favour the transfer of charge to the conduction band of brookite. Efficient dye degradation reported here is interfaced well with antiaromatic holes positioned in rGO layered sheets and its alignment with photo-induced carbazoyl phenyl anthracene. The potential for an extended shell-like structure formed from a carbazoyl phenyl anthracene network shaped into surface shell-like morphologies resembling tubular structures is shown by the TEM image in Fig. 3. An increase in the PL count-rate by the order of 103 corroborates a substantially high e−h+ separation (Fig. 8) and provides the potential to drive e−h+ through defects assisted by polyanline bridges structured through rGO layers to reach the valence of bands of rGO-embedded brookite via the dye oxidation–reduction cycle. We hypothesize that proton abstraction and subsequent transfer may have the effect of increasing di- and tri-vacancy sites causing proton migration by an alternate route facilitated by the photo-oxidised state to overcome energy barriers (Fig. S4, ESI†). This depth-range in the d-spacing of GO (0.77–0.81 nm)55 and the inclusion of polyaniline has the potential to considerably increase the interlayer between successive carbon sheets. The spacing observed along the d002 plane agrees with the 2D–3D projection map of an observable depth of 130–140 nm (Fig. 3f).
Concluding comments
We show that the high acid–low basic chemical environment selectively drives the reorganization of the Ti-bonds to the polymorph phase of brookite. Defect driven complexation of brookite with rGO leads to the synergistic resurgence of rGO-intercalated brookite as a strongly luminescent and multi-emissive semiconductor under the quantum confinement of a small interfacial bandgap. Stronger electronic coupling between graphitic carbon of the rGO framework and the conduction band of brookite is accompanied by a band gap reversal from a direct to an indirect configuration facilitating enhanced dye degradation performance under UV illumination. Our synthetic approach proposes a chemically fuelled multi-component photoswitchable TiO2 carbocatalyst with intrinsic emissive properties originating from a high electron–hole excitonic separation while substantially lowering electron–hole recombination. In the broader context of this finding, the photocatalytic modulation of high performing photo-responsive materials such as titanium nitride under conditions of quantum confinement facilitated by graphene oxide as precursor materials, could be effective routes to novel photocatalytic materials in the future.Author contributions
Conceptualisation and methodology: S.S, V.K, writing original draft: S.S and V.K., data curation: A.K, S.S, V.K and S.K.H, data interpretation and data analysis: A.K, S.S and V.K writing—manuscript editing and reviewing: A.K, S.S, T.W.L, S.K.H, V.K and P.G.J, resources: S.S, V.K, T.W.L and P.G.J, supervision and funding aquistion: S.S, V.K. All authors read and approved the final manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
This work acknowledges support from the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (No. 2022R1A2C1006090, 2017R1A2B4008801), and NRF Basic Research Program in Science and Engineering by the Ministry of Education (No. 2017R1D1A1B03036226). P. G. J. and V. K. also thankfully acknowledge the support from the National Convergence Research of Scientific Challenges through the National Research Foundation of Korea (NRF), funded by Ministry of Science and ICT (No. 2021M3F7A1017476). S. S. and V. K. gratefully acknowledge the Soft Foundry Institute for support.References
- M. Kokkonen,
et al., Advanced research trends in dye-sensitized solar cells, J. Mater. Chem. A, 2021, 9, 10527–10545 RSC
.
- M. Kim, M. Choi, S. Choi and S. Jeong, Semiconductor Nanocrystals: Unveiling the Chemistry behind Different Facets, Acc. Chem. Res., 2023, 56, 1756–1765 CrossRef CAS PubMed
-
S. N. Karthick, et al., Dye-Sensitized Solar Cells: History, Components, Configuration, and Working Principle, in Interfacial Engineering in Functional Materials for Dye–Sensitized Solar Cells, 2019, pp. 1–16. DOI:10.1002/9781119557401.ch1
- Z. Hiroi, Inorganic Structural Chemistry of Titanium Dioxide Polymorphs, Inorg. Chem., 2022, 61, 8393–8401 CrossRef CAS PubMed
- X. Ruan,
et al., Favorable Energy Band Alignment of TiO2 Anatase/Rutile Heterophase Homojunctions Yields Photocatalytic Hydrogen Evolution with Quantum Efficiency Exceeding 45.6%, Adv. Energy Mater., 2022, 12, 2200298 CrossRef CAS
- H. Zhao, L. Liu, J. M. Andino and Y. Li, Bicrystalline TiO2 with controllable anatase-brookite phase content for enhanced CO2 photoreduction to fuels, J. Mater. Chem. A, 2013, 1, 8209–8216 RSC
- T. A. Kandiel, L. Robben, A. Alkaim and D. Bahnemann, Brookite versus anatase TiO2 photocatalysts: Phase transformations and photocatalytic activities, Photochem. Photobiol. Sci., 2013, 12, 602–609 CrossRef CAS
- L. Liu, H. Zhao, J. M. Andino and Y. Li, Photocatalytic CO2 Reduction with H2O on TiO2 Nanocrystals: Comparison of Anatase, Rutile, and Brookite Polymorphs and Exploration of Surface Chemistry, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS
- A. Chaves,
et al., Bandgap engineering of two-dimensional semiconductor materials, npj 2D Mater. Appl., 2020, 4, 29 CrossRef CAS
- P. Szołdra,
et al., Effect of brookite on the photocatalytic properties of mixed-phase TiO2 obtained at a higher temperature, Mater. Sci. Eng., B, 2023, 287, 1–11 Search PubMed
- A. Di Paola, M. Bellardita and L. Palmisano, Brookite, the least known TiO2 photocatalyst, Catalysts, 2013, 3, 36–73 Search PubMed
- Y. Hu, H. L. Tsai and C. L. Huang, Effect of brookite phase on the anatase-rutile transition in titania nanoparticles, J. Eur. Ceram. Soc., 2003, 23, 691–696 Search PubMed
- H. Lin,
et al., Synthesis of High-Quality Brookite TiO2 Single-Crystalline Nanosheets with Specific Facets Exposed: Tuning Catalysts from Inert to Highly Reactive, J. Am. Chem. Soc., 2012, 134, 8328–8331 CrossRef CAS PubMed
- F. Wang, X. Liu and I. Willner, Integration of photoswitchable proteins, photosynthetic reaction centers and semiconductor/biomolecule hybrids with electrode supports for optobioelectronic applications, Adv. Mater., 2013, 25, 349–377 Search PubMed
- I. Aprahamian and S. M. Goldup, Non-equilibrium Steady States in Catalysis, Molecular Motors, and Supramolecular Materials: Why Networks and Language Matter, J. Am. Chem. Soc., 2023, 145, 14169–14183 CrossRef CAS PubMed
- M. G. Kim,
et al., Effects of Calcination Temperature on the Phase Composition, Photocatalytic Degradation, and Virucidal Activities of TiO2 Nanoparticles, ACS Omega, 2021, 6, 10668–10678 CrossRef CAS PubMed
- F. Lai, M. Zhao and S. Tong, Surface energy calculation of brookite TiO2 using Inverse Wulff Construction, J. Phys.: Conf. Ser., 2023, 2553, 1–5 CrossRef
- H. Zhang and J. F. Banfield, Understanding polymorphic phase transformation behavior during growth of nanocrystalline aggregates: Insights from TiO2, J. Phys. Chem. B, 2000, 104, 3481–3487 CrossRef CAS
- A. Mamakhel, J. Yu, F. Søndergaard-Pedersen, P. Hald and B. B. Iversen, Facile synthesis of brookite TiO2 nanoparticles, Chem. Commun., 2020, 56, 15084–15087 RSC
- S. N. Trzaska and F. Olbrich, Crystal structure of carbazole - 1,4,7,10-tetraoxacyclododecane, C 12H9N · C8H16O4, Z. Kristallogr. – New Cryst. Struct., 2008, 223, 457–458 CAS
- J. Lee,
et al., Preparation and electrochemical performance of titanium nitride-graphene nanocomposite with high Ti contents and tailored morphology, Curr. Appl. Phys., 2019, 19, 961–967 CrossRef
- P. Han,
et al., Graphene nanosheet–titanium nitride nanocomposite for high performance electrochemical capacitors without extra conductive agent addition, J. Mater. Chem., 2012, 22, 24918–24923 RSC
- E. C. Kohlrausch,
et al., Selective suppression of {112} anatase facets by fluorination for enhanced TiO2 particle size and phase stability at elevated temperatures, Nanoscale Adv., 2021, 3, 6223–6230 RSC
- K. Morioku, N. Morimoto, Y. Takeuchi and Y. Nishina, Concurrent Formation of Carbon-Carbon Bonds and Functionalized Graphene by Oxidative Carbon-Hydrogen Coupling Reaction, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed
- C. Ma,
et al., Controllable conversion of quasi-freestanding polymer chains to graphene nanoribbons, Nat. Commun., 2017, 8, 1–7 CrossRef
- A. Roy, S. Mukhopadhyay, P. S. Devi and S. Sundaram, Polyaniline Layred Rutile TiO2 Nanorods as Alternative Photoanode in Dye Sensitized Solar Cells, ACS Omega, 2019, 4, 1130–1138 CrossRef CAS
- A. Roy, S. Mukhopadhyay, P. S. Devi and S. Sundaram, Polyaniline-Layered Rutile TiO2 Nanorods as Alternative Photoanode in Dye-Sensitized Solar Cells, ACS Omega, 2019, 4, 1130–1138 CrossRef CAS PubMed
- C. Ferrag, M. Noroozifar, A. R. Modarresi-Alam and K. Kerman, Graphene oxide hydrogel electrolyte for improving the performance of electropolymerized polyaniline solar cells, J. Power Sources, 2022, 542, 231796 CAS
- A. A. Barragan,
et al., Photochemistry of Plasmonic Titanium Nitride Nanocrystals, J. Phys. Chem. C, 2019, 123, 21796–21804 CAS
- W. Ji,
et al., Expanding the Structural Diversity and Functional Scope of Diphenylalanine-Based Peptide Architectures by Hierarchical Coassembly, J. Am. Chem. Soc., 2021, 143, 17633–17645 CAS
- J. Xia,
et al., Disassembly and reassembly of diphenylalanine crystals through evaporation of solvent, J. Colloid Interface Sci., 2021, 599, 661–666 CAS
- R. Kurz,
et al., Interplay between Crystallization and Entanglements in the Amorphous Phase of the Crystal-Fixed Polymer Poly(ε-caprolactone), Macromolecules, 2018, 51, 5831–5841 CAS
- D. Choi,
et al., Quantum scale biomimicry of low dimensional growth: An unusual complex amorphous precursor route to TiO2 band confinement by shape adaptive biopolymer-like flexibility for energy applications, Sci. Rep., 2019, 9, 1–15 CrossRef
- A. H. Wazir and I. W. Kundi, Synthesis of Graphene Nano Sheets by the Rapid Reduction of Electrochemically Exfoliated Graphene Oxide Induced by Microwaves, J. Chem. Soc. Pak., 2016, 38, 11–16 CAS
- J. Zhang, P. Zhou, J. Liu and J. Yu, New understanding of the difference of photocatalytic activity among anatase, rutile and brookite TiO2, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC
- J. R. Dias and J. Aihara, Antiaromatic holes in graphene and related graphite defects, Mol. Phys., 2009, 107, 71–80 CrossRef CAS
- K. L. Woon, A. Ariffin, K. W. Ho and S. A. Chen, Effect of conjugation and aromaticity of 3,6 di-substituted carbazoles on triplet energy and the implication of triplet energy in multiple-cyclic aromatic compounds, RSC Adv., 2018, 8, 9850–9857 RSC
- D. K. Seo and R. Hoffmann, Direct and indirect band gap types in one-dimensional conjugated or stacked organic materials, Theor. Chem. Acc., 1999, 102, 23–32 Search PubMed
- Q. Mei,
et al., Graphene Oxide: From Tunable Structures to Diverse Luminescence Behaviors, Adv. Sci., 2019, 6, 1–16 Search PubMed
- J. Ito, J. Nakamura and A. Natori, Semiconducting nature of the oxygen-adsorbed graphene sheet, J. Appl. Phys., 2008, 103, 113712-1–113712-5 CrossRef
- S. H. Song,
et al., Highly Efficient Light-Emitting Diode of Graphene Quantum Dots Fabricated from Graphite Intercalation Compounds, Adv. Opt. Mater., 2014, 2, 1016–1023 CrossRef CAS
- J. D. Mangadlao,
et al., Grafted carbazole-assisted electrodeposition of graphene oxide, ACS Appl. Mater. Interfaces, 2015, 7, 10266–10274 CrossRef CAS PubMed
- I. Filpponen, H. Sadeghifar and D. S. Argyropoulos, Photoresponsive cellulose nanocrystals, Nanomater. Nanotechnol., 2011, 1, 34–43 CAS
- K. Das, L. Gabrielli and L. J. Prins, Chemically Fueled Self-Assembly in Biology and Chemistry, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed
- G. Rajender and P. K. Giri, Formation mechanism of graphene quantum dots and their edge state conversion probed by photoluminescence
and Raman spectroscopy, J. Mater. Chem. C, 2016, 4, 10852–10865 RSC
- H. Yoon,
et al., Intrinsic Photoluminescence Emission from Subdomained Graphene Quantum Dots, Adv. Mater., 2016, 28, 5255–5261 CrossRef CAS PubMed
- Y. H. Chen,
et al., Insight into the mechanism and outcoupling enhancement of excimer-Associated white light generation, Chem. Sci., 2016, 7, 3556–3563 RSC
- A. Das,
et al., Dynamics of Anthracene Excimer Formation within a Water-Soluble Nanocavity at Room Temperature, J. Am. Chem. Soc., 2021, 143, 2025–2036 Search PubMed
- Y. Ma,
et al., Strong electrostatic adsorption approach to the synthesis of sub-three nanometer intermetallic platinum–cobalt oxygen reduction catalysts, Nano Energy, 2021, 79, 105465 Search PubMed
- S. S. Sambaza, A. Maity and K. Pillay, Polyaniline-Coated TiO2 Nanorods for Photocatalytic Degradation of Bisphenol A in Water, ACS Omega, 2020, 5, 29642–29656 Search PubMed
- Y. Liu, R. Deng, Z. Wang and H. Liu, Carboxyl-functionalized graphene oxide–polyaniline composite as a promising supercapacitor material, J. Mater. Chem., 2012, 22, 13619–13624 RSC
- Y. Zhang, J. Liu, Y. Zhang, J. Liu and Y. Duan, Facile synthesis of hierarchical nanocomposites of aligned polyaniline nanorods on reduced graphene oxide nanosheets for microwave absorbing materials, RSC Adv., 2017, 7, 54031–54038 RSC
- P. N. O. Gillespie and N. Martsinovich, Origin of Charge Trapping in TiO2/Reduced Graphene Oxide Photocatalytic Composites: Insights from Theory, ACS Appl. Mater. Interfaces, 2019, 11, 31909–31922 CrossRef CAS
- Y. Zhang, R. Dai and S. Hu, Study of the role of oxygen vacancies as active sites in reduced graphene oxide-modified TiO2, Phys. Chem. Chem. Phys., 2017, 19, 7307–7315 RSC
- T. N. A. B. T. A. Mutalib,
et al., Properties of polyaniline/graphene oxide (PANI/GO) composites: effect of GO loading, Polym. Bull., 2021, 78, 4835–4847 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03616f |
| This journal is © The Royal Society of Chemistry 2025 |