
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational pore engineering reveals the relative contribution of enzymatic sites and self-assembly towards rapid ferroxidase activity and mineralization: impact of electrostatic guiding and cage-confinement in bacterioferritin†
Akankshika
Parida‡
 ,
Gargee
Bhattacharyya‡
,
Swagatika
Mallik‡
and
Rabindra K.
Behera
*
,
Gargee
Bhattacharyya‡
,
Swagatika
Mallik‡
and
Rabindra K.
Behera
*
Department of Chemistry, National Institute of Technology, Rourkela – 769008, Odisha, India. E-mail: beherarabi@nitrkl.ac.in; Fax: +91-661-2462651; Tel: +91-661-2462980
First published on 20th January 2025
Abstract
The self-assembled ferritin protein nanocage plays a pivotal role during oxidative stress, iron metabolism, and host–pathogen interaction by executing rapid iron uptake, oxidation and its safe-storage. Self-assembly creates a nanocompartment and various pores/channels for the uptake of charged substrates (Fe2+) and develops a concentration gradient across the protein shell. This phenomenon fuels rapid ferroxidase activity by an upsurge in the substrate concentration at the catalytic sites. However, it is difficult to segregate the relative contributions of the catalytic sites and self-assembly towards rapid ferroxidase/mineralization activity owing to the inherent self-assembly propensity of ferritins. In the current work, 3-fold pore electrostatics of bacterioferritin from Mycobacterium tuberculosis were rationally altered by site-directed mutagenesis to generate self-assembled (E121A and E121Q) and assembly-defective (E121K and E121F) variants. In comparison to the autoxidation of Fe2+ in buffer, the assembly-defective variants exhibited significantly faster ferroxidase/mineralization activity and O2 consumption kinetics due to their functional catalytic sites, but failed to level-up with the self-assembled variants even at 100-fold higher Fe2+ concentration. Only the self-assembled variants exhibited cooperativity in iron oxidation, maintained biomineral solubility, and protected DNA against the Fenton reaction. This report highlights the concerted effect of self-assembly and ferroxidase sites that propels the rapid Fe2+ uptake, its oxidation and biomineralization in bacterioferritin. The findings also establish the importance of electrostatic guiding and nanoconfinement offered by ferritin self-assembly towards its enzymatic activity and antioxidative properties. Moreover, this work identifies the key electrostatic interactions (“hot-spots”) at the subunit contact points that control the cage/pore formation, impart cage stability and influence ferritin's natural functions. Manipulation of hot-spot residues can be further extended towards the encapsulation of cargo, for various bio-medical applications, by strategically inducing its disassembly and subsequent reassembly through adjustments in ionic strength. This would bypass the need for extreme/harsh reaction conditions and minimize the loss of cargo/protein.
Introduction
Confinement and compartmentalization are linked to the origin of life and evolution of life-forms; the transition from the ‘primordial soup’ to the cellular life and prokaryotes to eukaryotes.1,2 These phenomena are critical to life functions; starting from ATP synthesis to heart pumping and brain functioning, due to the establishment of concentration gradients of protons/ions across cellular/intracellular membranes.3,4 Cell compartments ensure the segregation of the chemical environment, allowing specific reactions to take place that would otherwise be unfeasible in a diluted aqueous medium.5–7 Therefore, confinement and compartmentalization not only accelerate chemical/bio-chemical reactions, but also separate the bio-catalysts and substrates in distinct compartments (both spatial and temporal) unless and until required.3 The formation of selective barriers/boundaries separating chemical environments is, in most cases, a consequence of self-assembly, such as the self-assembled lipid membranes.1,8Ferritin proteins, the cellular iron repositories, are another prime example of such self-assembled systems (Fig. 1).9–12 They efficiently sequester and concentrate free Fe2+ and catalyze its conversion to a ferric-oxy-hydroxide mineral, thereby protecting cells from oxidative damage.10,13 Ferritins safeguard the iron mineral within their hollow nanocompartment, shielding it from the cytosol while allowing regulated iron release in response to cellular needs.14 Thus, these evolutionarily ubiquitous protein nanocages essentially serve a dual function: as a detoxification machinery and an iron reserve in biological systems.9,15 The 24 canonical subunits of ferritin establish a protein shell (∼2 nm) having an outer diameter of ∼12 nm, surrounding an ∼8 nm internal protein cavity, connected with the external environment by various symmetric pores/channels; six 4-fold pores, eight 3-fold pores, and twelve 2-fold pores.16–18 In addition to 4-3-2 symmetry, bacterial ferritins also possess 24 asymmetrical B-pores (Fig. 1A).19–21 The di-iron catalytic site (ferroxidase centers, Fox) is located at the center of a subunit which comprises four α-helices (A, B, C, and D) forming a bundle and a short E-helix (Fig. 1B).22,23 However, hetero-polymeric ferritins co-assembled from both catalytic and non-catalytic subunits are also found in mammals24,25 and certain bacteria such as Pseudomonas aeruginosa.26 Ferritin is mostly found inside the cell (in the cytosol and mitochondria/nucleus) and its expression is regulated translationally by iron levels.9,27 However, a tiny fraction of ferritin is also present in the serum that correlates with the body iron stores and is often used as a biomarker to assess iron status, particularly under iron deficiency and normal conditions (in the absence of inflammation/infection).28,29
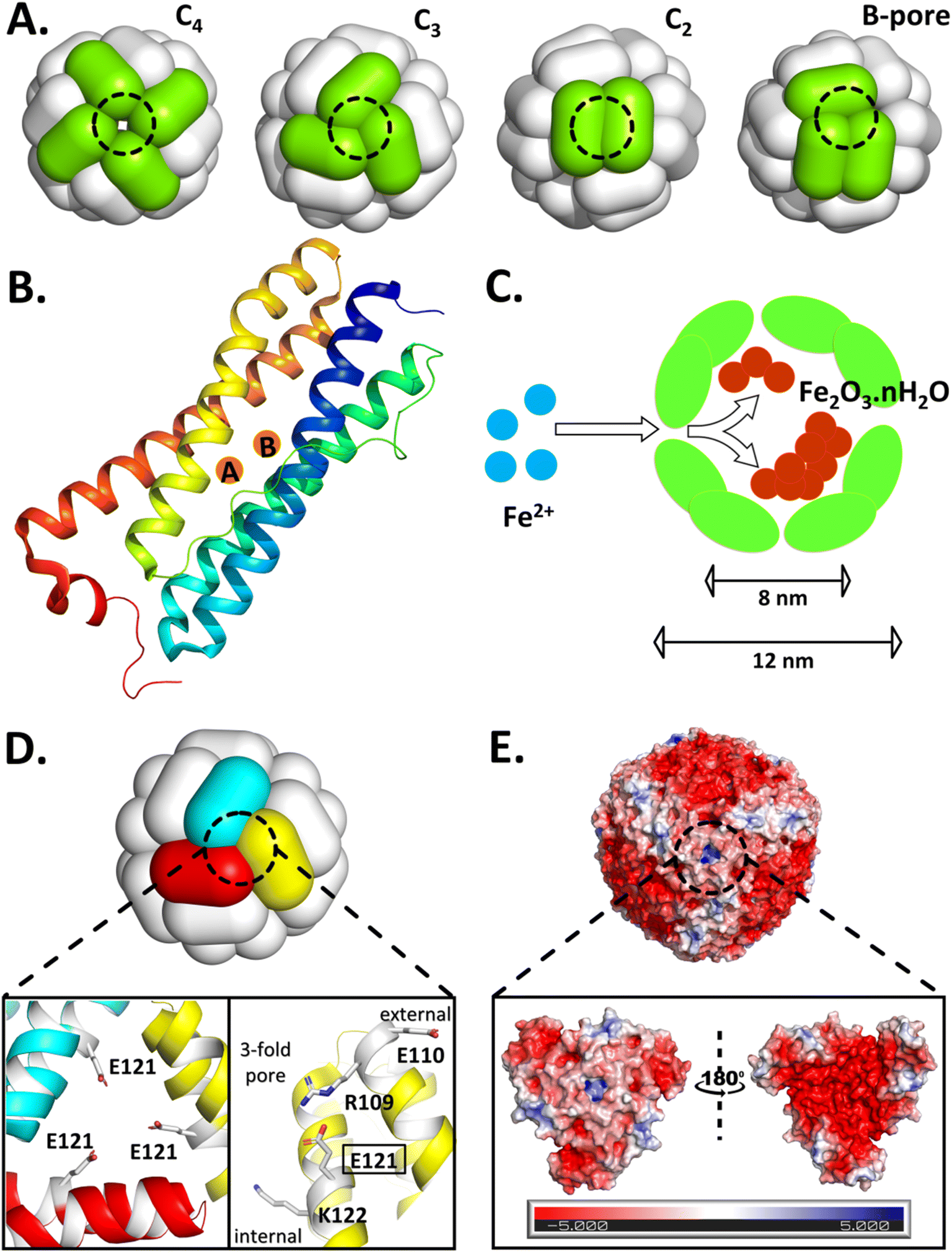 | ||
| Fig. 1 Self-assembled ferritin with its symmetric/asymmetric pores, subunits bearing di-iron oxidoreductase sites, caged-iron mineral and the structure/electrostatics of the targeted 3-fold pore of Mtb BfrA. (A) Schematic representation of the 24-meric nanocage architecture with symmetric (C4, C3, and C2) and asymmetric (B-) pores. (B) A single subunit of ferritin with di-iron ferroxidase sites. (C) Sequestration of free Fe2+ and the synthesis of iron-mineral within the ferritin nanocompartment. (D) 3-fold pore of Mtb BfrA, where the enlarged section highlights the targeted E121 residue, located in the middle, orienting towards the center of the 3-fold pore (left) and the alternate array of positive and negative residues present on a single subunit (right), possibly critical to self-assembly and cage stability. (E) Surface electrostatics of the Mtb BfrA nanocage viewed along the 3-fold pore. The enlarged section illustrates a close-up view of the electrostatics at/around the 3-fold pore, as viewed from the exterior (left) and interior (right) of the protein nanocage. The internal surface possesses a greater negative charge density. The Mtb BfrA structures are generated using PyMOL and the Poisson–Boltzmann electrostatic potential (expressed in units of ±5kBT/e, where kB is the Boltzmann constant, T is the temperature, and e is the electronic charge) is calculated using the APBS tool (PDB ID: 3UOI). | ||
When the ferritin-monomers self-assemble to form the protein nanocage, they create well-defined pores/channels that generate electrostatics, setting the stage for rapid uptake of charged substrates (Fe2+) to the Fox centers followed by oxidation/mineralization (Fig. 1C) via electrostatic guiding/focusing.19,22,30–34 This phenomenon has also been observed in few biological systems such as superoxide dismutase (SOD)35,36 and acetylcholinesterase (AcChoEase),37,38 where electrostatics steer the charged substrates, O2˙− and AcCh+, towards their respective active sites to achieve catalytic perfection, close to diffusion-controlled limits (kcat/kM: SOD ∼109 M−1 s−1 and AcChoEase ∼108 M−1 s−1).36,39 The importance of electrostatics is also reported in O2˙− bio-sensing by cytochrome c (a basic heme-protein: pI ∼10.0) where the placement of positively charged amino acids dictates the heme reduction rate by O2˙− either by reinforcing or misguiding the anion approach.40 Similar electrostatically enhanced oriented diffusion of charged substrates reinforces the buildup of an effective Fe2+ concentration inside the ferritin cavity, resulting in a concentration gradient across the protein shell. As a consequence, the enzymatic sites of ferritin possibly gain access to around 100–1000 times higher Fe2+ concentration in comparison to the autoxidation of Fe2+ in buffer (where ferritin is absent). The ferroxidase centers of ferritin leverage this effective substrate concentration to efficiently facilitate rapid iron oxidation, mineralization, and detoxification.13,41 However, the inherent self-assembly tendency of ferritins42 makes it difficult to distinguish the relative contribution of enzymatic sites and self-assembly in enhancing the overall ferroxidase activity and iron mineralization.
Iron is indispensable for most organisms owing to its involvement in various electron transfer reactions in key metabolic activities like nitrogen fixation, ATP synthesis during respiration (oxidative phosphorylation) and photosynthesis (photophosphorylation) and many essential cellular processes such as gene regulation, oxygen storage/transport, DNA synthesis, etc.3,43–45 However, despite its necessity (in μM to mM levels), Fe2+ can be highly toxic due to its ability to catalyze the formation of reactive oxygen species (ROS) like OH˙ radicals via Fenton reaction (∼104 M−1 s−1 at pH 7.0),46,47 which can cause cellular damage due to its high oxidizing ability (Em,7 ∼ +2.3 V) and hence its high reactivity (k ∼109−10 M−1 s−1; τ < 1 μs).5 Moreover, the poor solubility of Fe3+ (10−18 M at pH 7.0) in an aerobic environment makes it less bioavailable despite its abundance.3 Therefore, controlled iron acquisition/utilization is not only critical for maintaining iron homeostasis but also for avoiding its toxicity.9,11,48
Pathogenic bacteria, like any other organism, also need iron to survive and proliferate.49–52 Therefore, bacteria/pathogens have developed their own mechanism to acquire and store iron. Mycobacteriumtuberculosis (Mtb), the causative agent of tuberculosis, is one such deadly pathogen that has been a threat to humanity for centuries.53,54 During Mtb infection, the human body creates iron-limiting conditions, subjecting Mtb to a low-iron environment during its growth in human macrophages and lungs.55,56 For its survival and pathogenicity, Mtb expresses two iron storage proteins, namely, a heme-bound bacterioferritin (BfrA)53,57 and a non-heme binding ferritin (BfrB) with differential roles.54,58 In Mtb BfrA, the asymmetrical B-pores (lined with Asp and Glu) are crucial for iron translocation; the symmetrical 4-fold pores are involved in overall cage stability;19 however, the role of symmetrical 3-fold pores is not explored. Interestingly, the 3-fold pores of Mtb BfrA are lined with alternate layers of negative- and positively charged amino acid residues (Glu110, Arg109, Glu121, and Lys122), exhibiting positive electrostatics when viewed from outside due to Arg109 (Fig. 1D and E). These 3-fold pores are proposed to act as conduits for anions necessary for charge neutrality and mineral stability.19,20 Moreover, these alternate layers of opposite charges possibly provide essential electrostatic interactions (“hot-spots”) at the subunit contact points, necessary for cage assembly and its stability. However, this is in contrast to the 3-fold pores of most eukaryotic ferritins, which exhibit negative electrostatics due to the presence of an array of acidic amino acids like Asp and Glu, primarily involved in Fe2+ uptake.10,11,22
In addition to the intrinsic self-assembly property,42 most ferritin cages are exceptionally stable59 and thus, isolation of single/oligomeric assembly units in their folded form is difficult, despite substitution of multiple amino acids.19 Although, ferritins have been exploited for various applications by tuning their assembly/disassembly, they require extreme conditions like pH/denaturants/pressure.10,42,60 But these harsh methods involve loss of protein and generate assembly units in unfolded forms, which are unsuitable for studying the impact of self-assembly on bacterioferritin functionality: iron sequestration, ferroxidase activity, biomineralization and anti-oxidative properties. In the current work, the E121 residue at the center of the 3-fold pore interface was rationally substituted by site-directed mutagenesis to generate a series of mutations: E121A, E121Q, E121K, and E121F. The electrostatic/steric alterations resulted in both self-assembled (E121A and E121Q) and assembly-defective (E121K and E121F) variants, regardless of proper/efficient overexpression and folding. To understand the role of self-assembly, comparisons were made between these two sets of variants based on rapid ferroxidase activity and mineralization reaction, cooperativity, iron incorporation capacity, anti-oxidative properties, and thermal/conformational stability. Moreover, to assess the role of catalytic centers, autoxidation of the substrate in buffer was carried out alongside its catalytic oxidation by the ferritin variants. The study not only comprehends the role of electrostatic guiding in the rapid Fe2+ uptake and delivery to the Fox centers but also demonstrates the role of the nanocompartment in self-assembled cages that increases the effective iron concentration, facilitating the Fe2+ oxidation/mineralization process. This work can also be extended towards understanding the impact of nanocompartmentalization/confinement and electrostatic guiding on the reactivity of various supramolecular assemblies and enzymes along with ion transport through transmembrane proteins. In addition, this study may help in controlling Mtb proliferation by disrupting its Fe2+ sequestration/storage mechanism via manipulation of its iron uptake routes.
Methods
Materials
The Mycobacterium tuberculosis bacterioferritin A gene (Rv1876) cloned into pET21c was donated by Dr Anil. K. Tyagi and Dr Garima Khare from the University of Delhi South Campus. Isopropyl-1-thio-β-D-galactopyranoside (IPTG), FeSO4·7H2O, ferrozine, and 3-(N-morpholino) propane sulfonic acid (MOPS) were purchased from Sigma. Bovine serum albumin (BSA) was sourced from Thermo Scientific. Ethidium bromide (EtBr) was purchased from SRL and gadolinium acetate tetrahydrate (uranyl acetate alternative) was purchased from TED PELLA. Stock solutions of FeSO4 were freshly prepared in 1 mM HCl prior to respective experiments.Synthesis of BfrA variants by site-directed mutagenesis
Mtb BfrA mutants were generated by site-directed mutagenesis (using a QuikChange Kit from Stratagene-Agilent) and the mutations were confirmed by DNA sequencing. Oligonucleotide primers (Table S1†) used for site-directed mutagenesis were procured from Integrated DNA Technology (IDT).Preparation of recombinant bacterioferritins
Recombinant WT Mtb BfrA and its variants were overexpressed in E. coli [BL21(λDE3)] using a standard protocol (0.5 mM IPTG, 37 °C and 200 rpm) and purified using Q-Sepharose anion exchange chromatography, in accordance with earlier reports.57 The purity and assembly state of the collected fractions were assessed by 12.5% (w/v) denaturing and 6% (w/v) non-denaturing polyacrylamide gel electrophoresis (PAGE) techniques. Purified protein fractions were concentrated by ultrafiltration with a 10 kDa cutoff membrane (Millipore). The concentrated proteins were quantified using Bradford assay (Bio-Rad).E121K and E121F variants were also synthesized under slow over-expression conditions (Table S2†) with a lower IPTG concentration, lower incubation temperatures and using modified Luria-Bertani medium with betaine/sorbitol, following earlier reports.61
Pyridine hemochromagen assay
The heme content of WT BfrA and its variants was determined spectrophotometrically using previously reported methods.62 Briefly, a mixture of pyridine, NaOH, and potassium ferricyanide was mixed with the heme-proteins followed by the addition of sodium dithionite. The heme content was calculated from the UV-vis absorption spectra using the differential extinction coefficient (Δε = 23.98 mM−1 cm−1) between the reduced and oxidized states of the heme-protein.63Size exclusion chromatography (SEC) for assessing the assembly-state of the BfrA variants
The assembly-state of WT Mtb BfrA and its variants was analyzed by size exclusion chromatography. The as-purified protein samples were eluted through a Sephacryl S-300 HR (GE Healthcare) column pre-equilibrated with 20 mM Tris buffer (pH 8.0) containing 100 mM NaCl.Iron incorporation ability of recombinant bacterioferritins
The iron incorporation ability of the synthesized WT Mtb BfrA and its variants was assessed as per earlier reports.15 Briefly, freshly prepared FeSO4 solution was added to WT and its variants in 100 mM MOPS buffer (pH 7.0). Similarly, for assessing the iron loading capacity, aliquots of FeSO4 solution were sequentially added to ∼2 μM bacterioferritin samples at 2 hour intervals to achieve 500–4500 Fe/cage and the samples were subsequently incubated at room temperature for 2 hours and kept overnight at 4 °C.15,64Mineralized Mtb BfrA variants with differential Fe/cage were electrophoresed in 6 or 7% (w/v) non-denaturing polyacrylamide gels and the formation of iron mineral within the recombinant protein cages was visualized via Prussian blue precipitation, as reported earlier.15 Briefly, the gel was treated with a freshly prepared mixture of 2% K4Fe(CN)6 and 2% 11.6 M HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to allow the formation of the Prussian blue precipitate.15 The same gel was then thoroughly washed with distilled water and stained with Coomassie to visualize the corresponding protein bands.
:1, v/v) to allow the formation of the Prussian blue precipitate.15 The same gel was then thoroughly washed with distilled water and stained with Coomassie to visualize the corresponding protein bands.
Circular dichroism analysis
Far-UV (195–250 nm) and near-UV (250–350 nm) circular dichroism (CD) spectra at room temperature were obtained using a Jasco J-1500 CD spectrometer in 2 and 5 mm quartz cuvettes, respectively. 6 μM subunit and 6 μM cage or cage equivalent of the protein samples in 10 mM Tris buffer (pH 7.0) were used for secondary and tertiary structure analyses, respectively.Transmission electron microscopy (TEM)
The WT BfrA and its variants (1 mg ml−1) were mineralized, as described above, to achieve ∼480 Fe/cage. The TEM sample grids were prepared and stained with a 2% gadolinium acetate tetrahydrate (an alternative to uranyl acetate) solution, as described earlier.42 The grids were dried and then imaged using a FEI Tecnai G2 TF30-ST transmission electron microscope, equipped with a LaB6 electron gun operating at 300 keV.Dynamic light scattering (DLS) analysis
DLS analyses were carried out as per earlier reports.59 Briefly, a zetasizer (Malvern Zetasizer 90, Malvern, Netherland) was used to measure the hydrodynamic diameters of WT BfrA and its variants. All the protein samples (0.1 mg ml−1) were prepared in 100 mM MOPS (pH 7.0) and processed through 0.22 μm syringe filters prior to DLS measurements.Mineralized samples were prepared by sequential addition of 48 μM FeSO4 solution to ∼0.5 mg ml−1 (∼1 μM cage or cage equivalent) protein solution, with intermittent incubation periods of 15 min to achieve 100 and 480 Fe/cage before proceeding with DLS analysis.
Stopped-flow rapid kinetic measurements of Fe2+ oxidation and mineralization
Rapid kinetic measurements of Fe2+ oxidation (ferroxidase activity) in self-assembled and assembly-defective Mtb BfrA variants were monitored by measuring the change in absorbance: A350nm, for all types of [Fe3+–O]x species; A650nm, for di-ferric peroxo like species (DFP), as reported earlier.19,53 For a single-turnover (48 Fe/cage), equal volumes of 4.16 μM protein cage or cage equivalent in 100 mM MOPS buffer (pH 7.0) were mixed with freshly prepared 200 μM FeSO4 solution in a rapid mixing stopped-flow system (Hi-Tech SF61MX). The initial rates for single turnover were determined from the linear fitting of the initial data points (up to 0.03 s) from the respective kinetic traces.The rate constants for the formation of [Fe3+–O]x species were determined by fitting the kinetic traces at 350 nm using the double exponential equation19 (eqn (1)).
| At = A1 × ek1t + A2 × ek2t + A∞ | (1) |
Similarly, the progress curves for the formation of [Fe3+–O]x species were monitored for multiple-turnovers using two different concentrations of protein (1.04 and 2.08 μM) in 100 mM MOPS (pH 7.0) and 100 mM MES (pH 6.0) by mixing desired amounts of Fe2+ solutions.
The rate constants for the formation (k1) and decay (k2) of DFP-like transient species were obtained from the kinetic traces at 650 nm for self-assembled and assembly-defective Mtb BfrA variants using the modified Bateman equation (eqn (2)).
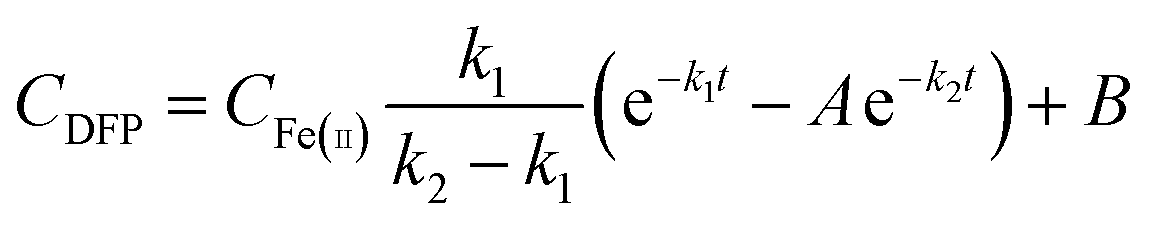 | (2) |
Titration measurements were performed in a stopped-flow spectrophotometer to monitor the formation of [Fe3+–O]x species at 350 nm by mixing 2 μM protein cage with increasing amounts of freshly prepared Fe2+ solutions to achieve ∼6–96 Fe/cage. The initial rates were determined by the linear fitting of the data points (within 0.03 s). To extract the kinetic parameters, the initial rate vs. [Fe2+] plot was subjected to non-linear fitting to the Hill equation, expressed as Vi = Vmax[Fe2+]n/(Km + [Fe2+]n) using Origin 8.5 Pro, where Vi = initial rate; Vmax = maximum reaction rate; n = Hill coefficient, which indicates the degree of cooperativity; Km = constant, analogous to the Michaelis constant (KM).
Quantification/detection of in situ generated H2O2 during the ferroxidase/mineralization activity
To monitor the formation of H2O2 during the ferroxidase/mineralization activity of Mtb BfrA variants, the Amplex Red/HRP assay was employed, as reported earlier.64 Briefly, the formation of resorufin (ε571 = 5.4 × 104 M−1 cm−1), the oxidized product of Amplex Red, was tracked using UV-visible absorption spectrophotometry to estimate the in situ generated H2O2 (resorufin:H2O2 = 1:1). 50 μM Amplex Red and 0.5 μM HRP were added to 1 μM cage/cage equivalent of protein solutions in 100 mM MOPS (pH 7.0), prior to the addition of iron. The absorption spectra were recorded ∼3 min after the addition of the desired amounts of FeSO4 solutions (0–480 μM; for 0–480 Fe/cage).
Manual kinetic measurements of Fe2+ oxidation and mineralization reaction
Kinetic measurements for the formation of iron biomineral within the nanocage of WT BfrA and its variants over extended time period was monitored by manually mixing desired amounts of freshly prepared FeSO4 with protein solutions (in 100 mM MOPS buffer, pH 7.0) using a UV-visible spectrophotometer. Additionally, sequential iron oxidation/mineralization in Mtb BfrA variants was also monitored by subsequent addition of 48 Fe/cage, manually, to 1.04 μM of protein to achieve 480 Fe/cage after ten consecutive additions.Kinetics of dissolved O2 consumption
The oxidoreductase activity of the Mtb BfrA variants was tracked by monitoring the real-time dissolved O2 levels during the ferroxidase/mineralization reaction by amperometry using a Clark-type microelectrode (Hansatech Instruments). Prior to the kinetic experiments, the current response of the oximeter was calibrated to concentration and the reaction kinetics were monitored as reported earlier.19 Additionally, pH dependent O2 consumption during ferroxidase/mineralization reaction was also recorded using two different buffers: 100 mM MOPS (pH 7.0) and 100 mM MES (pH 6.0).Similar to the sequential iron oxidation/mineralization, O2 consumption was also measured for ten consecutive additions of 48 Fe/cage to 0.5 μM cage of protein solutions (to achieve 480 Fe/cage).
DNA protection assay
The DNA protection ability of both self-assembled and assembly-defective Mtb BfrA variants was assessed in vitro, against the Fe2+/H2O2-induced Fenton reaction, using the reported method.53 The BfrA variants (∼2 μM cage or cage equivalent) were incubated separately with plasmid DNA (pET21c, ∼190 ng, 30 min), in 100 mM MOPS buffer (pH 7.0). 100 μM of FeSO4 solution was added to the above samples, followed by incubation for an additional 10 min. Following this, 4.5 mM H2O2 was added to initiate the DNA-cleavage by the Fenton reaction and the reaction was allowed to proceed for 25 min. For control experiments (in the absence of protein), 100 μM iron and DNA were separately incubated with and without H2O2. The reaction mixtures were run on a 0.8% (w/v) agarose gel at 70 V for 50 min in tris-acetate-EDTA (TAE) buffer. The DNA bands in the ethidium bromide (EtBr) stained gels were visualized using a ChemiDoc MP imaging system (Bio-Rad).Chemical and thermal unfolding analysis
Chemical unfolding was performed by increasing guanidine hydrochloride (GdnHCl) concentration from 0–8 M to investigate the impact of self-assembly on the unfolding behavior and conformational stability of self-assembled and assembly-defective Mtb BfrA variants using a Jasco J-1500 CD spectrophotometer. Similarly, temperature dependent far-UV CD spectra were monitored to investigate their thermal stability, by increasing the temperature from 20 to 95 °C at 1 °C min−1. The loss of α-helicity at 222 nm in the far-UV CD spectra was used to analyze their chemical/thermal stability and to obtain various thermodynamic parameters (eqn (3)–(6)), as per earlier reports.63 A subunit concentration of 6 μM was maintained in 10 mM Tris buffer (pH 7.0) for both chemical and thermal unfolding analyses.A two-state model, where folded (F) and unfolded (U) states of the protein remain in equilibrium, was employed to analyze the chemical/thermal denaturation parameters of WT BfrA and its variants.
F ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) U U | (3) |
The free energy change (ΔGU) is given as:
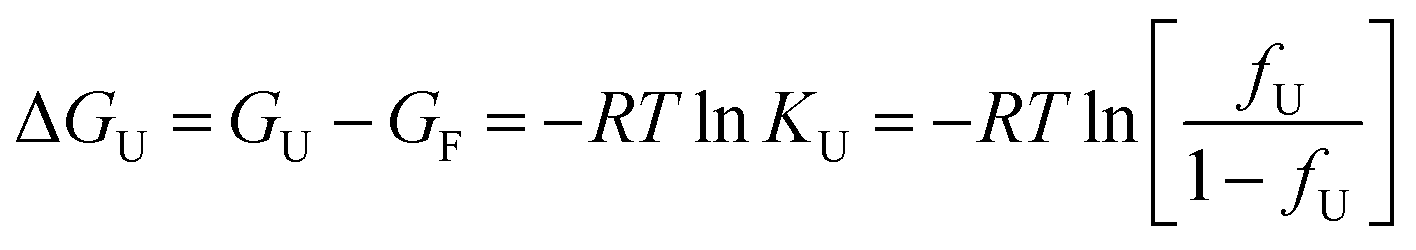 | (4) |
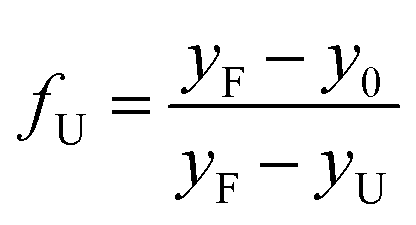 | (5) |
As reported earlier, WT BfrA was found to be too stable to completely unfold, even at 8 M GdnHCl.19 Hence, apparent thermodynamic parameters were calculated by considering yU (as 0, at 222 nm) for E121K, which exhibited complete unfolding.
Linear fitting of the unfolding data (ΔGU) at different concentrations of GdnHCl (in the transition region) gives the values for ‘Cm’, ‘m’ and the free energy of unfolding in aqueous solution, ΔGAq, based on the linear extrapolation method (LEM) using eqn (6).
| ΔGU = ΔGAq − m[GdnHCl] | (6) |
Results
Substitution of E121, at the 3-fold pore, resulted in self-assembled and assembly-defective Mtb BfrA variants
The rationally designed 3-fold pore variants of Mtb BfrA were synthesized (over-expressed and purified) and used for structural and functional analyses. Single bands observed in the denaturing gel (12.5% SDS-PAGE) indicated the purity of the recombinant bacterioferritins (Fig. 2A). However, E121K and E121F exhibited a different migration profile compared to WT, E121A, and E121Q; possibly due to differential binding of SDS, an anomalous phenomenon reported earlier in some cases including Mtb BfrA.19 The cage integrity and iron loading ability were examined using a 7% non-denaturing gel (Fig. 2B). Distinct self-assembled nanocages were observed in E121A and E121Q, along with the formation of comparable amounts of Prussian blue precipitate (iron mineral), similar to WT BfrA. In contrast, the faint Coomassie bands observed in the case of E121K and E121F with no Prussian blue precipitate (Fig. 2B, left), indicated the possible formation of only a small fraction of cage like structures, which was further supported by the appearance of weak Prussian blue bands when a 5× higher protein concentration was used (Fig. 2B, right). Therefore, a major fraction of these E121K and E121F variants possibly exist in undefined lower oligomeric states and lack well-defined self-assembled nanocages.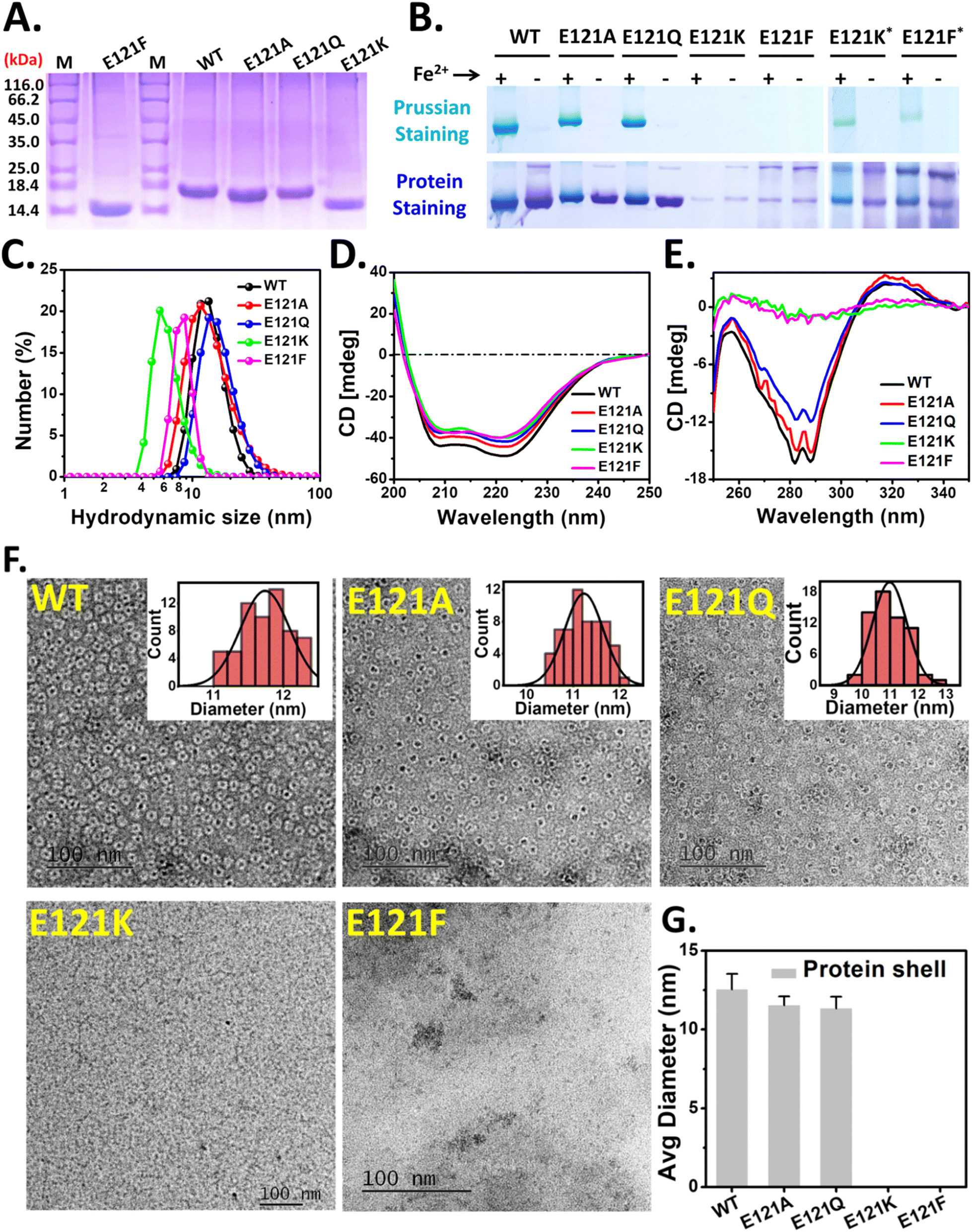 | ||
| Fig. 2 Cage-assembly, iron loading ability and structural analyses of 3-fold pore variants of Mtb BfrA. The purity of as-isolated Mtb BfrA and its variants was checked by SDS-PAGE (A). The first lane corresponds to the protein ladder/marker and their molecular weights are mentioned on the left. Protein samples with ∼1250 Fe/cage and without iron mineral cores, were run on a 7% (w/v) non-denaturing gel (native-PAGE) for checking the iron loading ability (top) and cage integrity (bottom) (B) (*five times concentrated protein samples to visualize the cage component). DLS profile (C), Far-UV (D) and near-UV (E) CD spectra of WT BfrA and its variants. Negatively stained TEM images of WT BfrA and its variants (1 mM Fe2+ added to 1 mg ml−1 protein solution; ∼480 Fe/cage) (F). The average diameters of protein shells of self-assembled variants analyzed using ImageJ (G). The average diameters for assembly-defective variants were not determined. p value ∼0.04, for the assembled variants (E121A and E121Q) with respect to WT for the size analysis data. The averages represent the results of at least two independent experiments, using two different protein batches. | ||
Dynamic light scattering (DLS) studies were performed to measure the hydrodynamic diameters of WT BfrA and its variants (Fig. 2C and Table S3†). E121A and E121Q displayed a diameter of ∼11–13 nm, similar to WT BfrA. However, E121K and E121F exhibited particle sizes of ∼5.6 ± 0.6 and ∼8.7 ± 0.8 nm, respectively, which are evidently smaller than the WT BfrA (Table S3†). The SEC elution profiles of E121K and E121F further corroborated the native PAGE and DLS data (Fig. S1†). Far-UV and near-UV CD spectra were recorded to investigate the impact of E121 substitution on the secondary and tertiary structure of the protein (Fig. 2D and E). While no significant changes in the secondary structure of the variants were observed, the characteristic features of the tertiary structure were missing in E121K and E121F, indicating the lack of a compact cage-like structure and/or exposure to an isotropic environment. The dips (at 285–290 nm) in the near-UV CD spectra for WT, E121A and E121Q are characteristic of the tertiary structure, arising from the anisotropic/compact environment around the aromatic amino acid residues.63
Although the λmax values for both the Soret and Q-bands are similar across these variants (in both the oxidized and reduced forms), they bind varying amounts of heme in their as-purified forms. The assembly-defective variants, E121K and E121F, bind similar amounts of heme (∼3–4 heme/cage equivalent) as WT, whereas E121A and E121Q bind ∼4–5 heme/cage (Fig. S2†).
Mineralized Mtb BfrA protein samples (∼480 Fe/cage) were negatively stained prior to TEM imaging for investigating the impact of E121 substitutions on the cage forming ability (self-assembly) of the synthesized BfrA variants. Core–shell architectures represent the encapsulated iron mineral inside the nanocavity of the self-assembled ferritin protein cage. While this feature was distinctly observed in the case of self-assembled variants, WT, E121A, and E121Q (∼11–12 nm), no such discrete mineral core encapsulated by the protein shell was observed in E121K and E121F. This supports our previous observation (PAGE and DLS data; Fig. 2B and C) that E121K and E121F, do not self-assemble to form intact nanocage structures and lack characteristics pores/channels and are referred to as the “assembly-defective variants” throughout the text (Fig. 2).
To promote cage-assembly of the E121K and E121F variants, alternative over-expression protocols were also implemented: supplementing the LB media with betaine/sorbitol or slowing down the expression with lower IPTG concentrations, lower incubation temperatures and longer incubation periods (details of modified protocols are listed in Table S2†). Interestingly, some degree of assembly was achieved for the E121K variant, as observed in native-PAGE (Fig. S3†). While an increased population of the cage was observed, additional bands were also evident that indicate the presence of E121K in other undefined states. The rehydration ability of betaine/sorbitol possibly helps in this regard. However, for the E121F variants, no distinct ferritin cage-like bands were seen and the yield (protein-expression) was comparatively low. The modified protocols possibly allow the re-orientation of the K121 side-chain (to form new interactions with the nearby residues) leading to improved cage assembly.65,66 F121, being a bulkier residue, possibly imparts steric obstruction at the pore during subunit–subunit interaction thereby inhibiting the self-assembly phenomena.
Self-assembly (electrostatic guiding and nanoconfinement) is crucial for rapid ferroxidase/mineralization reaction
Stopped-flow rapid kinetic measurements were performed at 350 and 650 nm to study the iron oxidation by monitoring the formation of [Fe3+–O]x species and DFP-like species, respectively, in both the self-assembled (E121A and E121Q) and assembly-defective variants (E121K and E121F). This rapid-kinetics experiment aims to assess the importance of 3-fold pore electrostatics and the role of self-assembly (nanoconfinement effect and electrostatic guiding) in rapid Fe2+ uptake/ferroxidase activity.The self-assembled variants exhibited similar kinetic profiles for the formation of [Fe3+–O]x species, as that of WT BfrA (Fig. 3), which implies that the alteration of 3-fold pore electrostatics (substitution of Glu121 by Ala or Gln) does not affect the rapid Fe2+ uptake/oxidation process significantly in Mtb BfrA. This indicates that the 3-fold pores are not the primary Fe2+ uptake routes in Mtb BfrA and validates the involvement of other pathways such as B-pores and 4-fold pores, as reported earlier.19 This was further supported by the similar initial rates and rapid phase rate constant values (Fig. 3D and Table 1). The initial rates of iron oxidation (ΔA/Δt) were determined by linear fitting of the initial data points (up to ∼0.03 s). Rate constants for the rapid phase, obtained from the non-linear fitting (eqn (1)) of the A350nm time courses, were found to be similar for all the assembled variants: ∼23 ± 2.3 s−1 (WT), ∼22.6 ± 2.2 s−1 (E121A) and ∼21.6 ± 2.3 s−1 (E121Q) (Table 1). Therefore, it can be inferred that the major iron entry routes i.e. the B-pores and 4-fold pores are un-affected/unaltered by these modifications. Despite the higher heme content (Fig. S2†), the E121A/Q variants exhibit minimal changes in their overall activities in comparison to WT, suggesting that heme plays a less critical role in ferroxidase activity or self-assembly properties, which is consistent with our earlier report.57
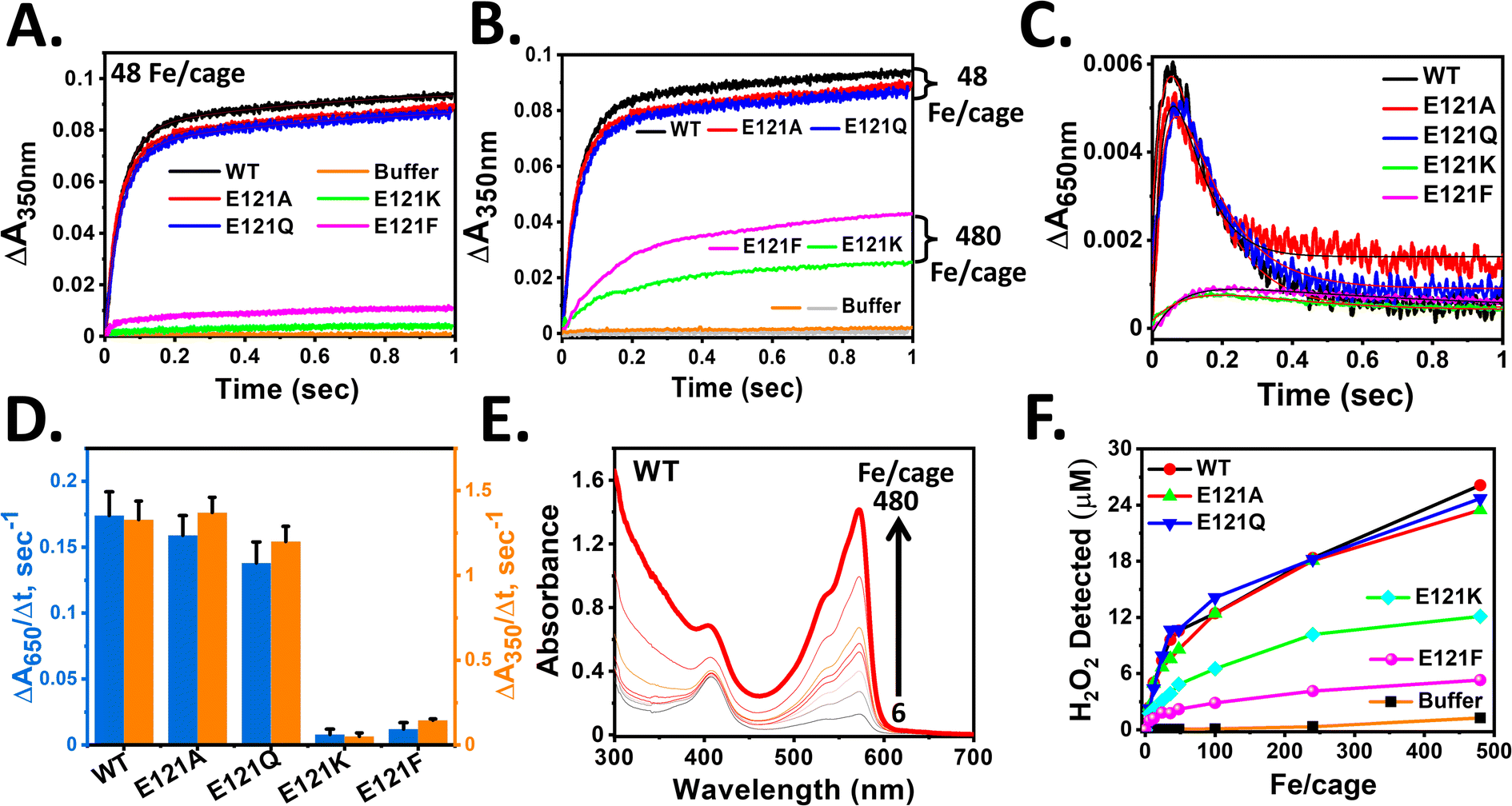 | ||
| Fig. 3 Impact of self-assembly on rapid oxidoreductase activity in Mtb BfrA. Progress curves for the formation of [Fe3+–O]x species at 350 nm in self-assembled and assembly-defective variants (A). Equal volume of 4.16 μM cage or cage equivalent of ferritin in 100 mM MOPS (pH 7.0) was mixed with a freshly prepared FeSO4 solution (in 1 mM HCl) in a stopped-flow rapid mixing spectrophotometer to achieve 48 Fe/cage at 25 °C. Buffer: autoxidation of iron in the absence of protein. (B) Self-assembled variants with lower substrate concentration (100 μM Fe2+) exhibit faster iron oxidation than the assembly-defective variants with 10-fold higher substrate concentration (1 mM Fe2+). Grey and orange lines: autoxidation in buffer with 48 and 480 Fe/cage. (C) Progress curves for the formation of DFP-like transient intermediates at 650 nm under identical reaction conditions as described in ‘A’. The initial rates obtained by linear fitting of the initial data points (up to ∼0.03 s) from ‘A’ and ‘C’ plotted in (D). The nonlinear kinetic profiles of ‘A’ and ‘C’ were fitted with double exponential equation and the modified Bateman equation, respectively, to obtain the rate constants listed in Table 1. Detection of H2O2 generated in situ (E and F). 50 μM Amplex Red and 0.5 μM HRP were added to 1.04 μM cage in 100 mM MOPS (pH 7.0). The absorption spectra were recorded ∼3 min after the addition of desired amounts of FeSO4 solutions (0–480 μM) (see Methods). (E) Representative figure of H2O2 detection using HRP assay for WT BfrA. (F) H2O2 detected at different Fe/cage (0–480) for all variants. For [Fe3+–O]x species at 350 nm and DFP-like species at 650 nm, p value ∼0.045 and <0.001 for the assembled variant (E121A) and assembly-defective variants (E121K and E121F) respectively, calculated with respect to WT. For DFP-like species at 650 nm, the p value is ∼0.03 for E121Q, determined with respect to WT. The averages are of results of at least four independent experiments, using two different protein batches. | ||
| Ferritin | ΔA350nmvs. time (s) | ΔA650nmvs. time (s) | ||
|---|---|---|---|---|
| (Rapid phase) k1 (s−1) | (Slow phase) k2 (s−1) | (Formation) k1 (s−1) | (Decay) k2 (s−1) | |
| a Rate constants were obtained by fitting the non-linear time courses with the double exponential (from Fig. 3A) and modified Bateman equations (from Fig. 3C). b N.D.: not determined, due to insufficient accumulation of iron mineral. | ||||
| WT | 23.6 ± 2.3 | 1.9 ± 0.3 | 24.1 ± 1.8 | 9.2 ± 1.2 |
| E121A | 22.6 ± 2.2 | 1.9 ± 0.6 | 23.4 ± 1.6 | 10.1 ± 1.5 |
| E121Q | 21.6 ± 2.3 | 1.5 ± 0.4 | 23.1 ± 1.7 | 8.9 ± 1.4 |
| E121K | N.D. | N.D. | 8.2 ± 1.2 | 2.1 ± 1.1 |
| E121F | N.D. | N.D. | 7.9 ± 1.1 | 2.6 ± 0.9 |
The slower formation of [Fe3+–O]x species in the case of assembly-defective variants suggested either repressed Fe2+ uptake or slower ferroxidase activity (iron oxidation at di-iron catalytic centers) (Fig. 3). The radical decrease in the initial rates of assembly-defective variants is possibly due to the absence of B-pores and 4-fold pores to sequester Fe2+ ions, as a consequence of the inhibited self-assembly process. Additionally, the absence of a well-defined protein cage and possibly the loss of its associated cooperativity further contribute to this effect.
For autoxidation, the added Fe2+ (substrate) is distributed homogenously in the reaction medium and a similar scenario can be assumed for the assembly-defective variants, where the ferroxidase centers are exposed to the same concentration of substrate as in the bulk. However, for the self-assembled variants, >100-fold excess of Fe2+ is available at the catalytic sites prior and/or during the ferroxidase reaction, owing to the rapid Fe2+ sequestration and accumulation within the ferritin cavity. Therefore, a set of experiments were also designed to investigate the contribution of nanoconfinement towards rapid sequestration and oxidation of Fe2+ ions by recording iron oxidation kinetics of assembly-defective variants in the presence of excess concentrations of the substrate (to mimic the effective concentration in the self-assembled variants). Fig. 3B shows faster oxidation kinetics for the self-assembled variants even at lower substrate concentrations. Furthermore, a comparison of iron-oxidation profiles was made at low protein concentrations and significantly higher iron additions (Fig. S4†). Even at a 100-fold higher Fe2+ concentration, the assembly-defective variants could not match the initial rates of iron oxidation observed in the assembled variants (Fig. S4†).
In comparison to the autoxidation of Fe2+ in buffer, the assembly-defective variants exhibited significantly faster kinetics of ferroxidase activity (Fig. 3). This difference becomes more prominent with higher iron addition and at high pH (Fig. S5†) and is a clear depiction of the role of the catalytic nature of the intact di-iron sites, despite the loss of a well-defined nanocage. Kinetics of iron mineralization at different pH values (MES: pH 6.0 and MOPS: pH 7.0) was recorded to assess the contribution of autoxidation. At pH 6.0, the mineralization process was found to be comparatively slower and autoxidation reaction was relatively suppressed (Fig. S5†).
Similar to the slow formation of [Fe3+–O]x species, the accumulation of DFP-like transient intermediates was less in the case of assembly-defective variants. This transient intermediate involves two sequential processes, i.e., formation and decay, where the relative rates can dictate the accumulation and measured initial rates. Therefore, in addition to the initial rates (ΔA650/Δt), the rate constants for both the formation (k1) and decay (k2) were determined by fitting the A650nm time courses to the modified Bateman equation (eqn (2)). While WT BfrA and the self-assembled variants (E121A and E121Q) exhibited similar formation rate constant values, k1 ∼ 23–24 s−1, the assembly-defective variants exhibited a drastic decrease in the values, k1 ∼ 7–9 s−1 (Table 1). Similarly, the variations in the decay rate constants for the self-assembled (k2 ∼ 8–11 s−1) and assembly-defective (k2 ∼ 1–3 s−1) variants (Table 1), possibly indicates the existence of differential mechanisms for decay of DFP-like transient intermediate. These findings further highlight the consequences of the absence of a confined nanocompartment and the lack of major iron uptake pathways to sequester and deliver Fe2+ ions to the catalytic sites in the assembly-defective variants.
The significance of cage assembly towards ferroxidase activity was further highlighted by the enhanced iron-oxidation profiles observed in E121K samples with improved cage-assembly, over-expressed in modified LB medium. Compared to its assembly-defective counterparts, the assembled E121K proteins demonstrated an efficient restoration of iron oxidation (Fig. S6†).
Detection of in situ generated H2O2 during ferroxidase/mineralization activity
The in situ generation of H2O2 through the oxidoreductase activity of the synthesized variants was investigated using a peroxidase assay with Amplex Red (substrate) and HRP (catalyst) (Fig. 3E, F and S7†). The assay involves the spectrophotometric assessment of the amount of resorufin generated at 571 nm (ε = 5.4 × 104 M−1 cm−1) to estimate the in situ generated H2O2 (resorufin:H2O2 = 1:1).14 Similar amounts of H2O2 were detected for the self-assembled variants (WT, E121A, and E121Q), indicating a similar mechanism during the ferroxidase reaction. In contrast, for the assembly-defective variants (E121K and E121F), minimal amounts of H2O2 were detected, even at higher substrate concentrations. This is possibly a result of a slower ferroxidase reaction due to the slower occupancy of Fe2+ ions at their Fox centers, resulting from the absence of electrostatic guiding and confinement effect. All in all, the detection of in situ generated H2O2 levels corelates with the trends observed for the ferroxidase/mineralization activity of the assembled and assembly-defective variants. Previous work on frog M ferritin also reported the generation of H2O2 during ferroxidase activity by decay of di-ferric peroxo species and is stoichiometrically linked to its formation.67
Interestingly, in the case of assembled variants, although the detection of H2O2 increased with increasing Fe2+ concentrations (0–480 μM), the amount remained significantly lower than the value anticipated from stoichiometry at the first turnover (i.e., Fe2+:H2O2 ∼ 2:1 for <48 Fe/cage). This may possibly indicate the participation of the generated H2O2 in Fe2+ oxidation, which competes with the HRP present in the reaction mixture. Additionally, the lower amount of H2O2 detected may also be due to the inherent catalase activity of Mtb BfrA, as seen in earlier reports.53
Loss of cooperativity in assembly-defective variants
To investigate the iron occupancy and assess the presence of allosteric interactions (cooperativity) during ferroxidase activity at the di-iron oxidoreductase centers, the formation of [Fe3+–O]x species was recorded with increasing iron per cage (6–96) using a stopped-flow rapid kinetics system (Fig. S8†). The initial rate (ΔA350/Δt) increases with increasing Fe/cage and saturates at ∼40 Fe/cage, as reported for E. coli Bfr.68 Further addition of iron had a nominal impact on the initial rate, indicating saturation of the “stable di-Fe3+ catalytic sites”. The Hill coefficients (n) were determined by fitting the initial rates vs. the respective iron concentration in the Hill equation (see Methods) (Fig. S8F†). The sigmoidal plots for the self-assembled variants indicated the existence of positive cooperativity (Hill coefficient; n > 1) (Table S4†), which in turn possibly depicts the rapid and precise delivery of Fe2+ ions to the Fox centers via electrostatic guiding generated along the pores by self-assembly. However, Hill coefficient values close to unity (n ∼1), in the case of assembly-defective variants indicate that the di-iron centers of each sub-unit behave independently towards binding/oxidation of Fe2+. Furthermore, this implies that the affinity of their Fox centers towards Fe2+ ions possibly does not depend on whether or not the di-iron sites are occupied. The slower iron oxidation kinetics and the lack of self-assembly (multi-subunit interactions) in these variants further support this observation. Therefore, the loss of cooperativity may be a reason behind their slower iron oxidation in assembly-defective variants.Self-assembly facilitates the biomineralization reaction
Kinetic measurements for iron oxidation and subsequent biomineral formation in WT BfrA and its variants were performed by manual addition of FeSO4 solutions to 1.04 μM protein solution, to achieve 48 and 480 Fe/cage using a UV-visible spectrophotometer (Fig. S9A and B†). Manual-mixing kinetic trends over longer time (Fig. S9A–D†) were similar to stopped-flow kinetics findings and also support the role of self-assembly towards iron oxidation/mineralization. The self-assembled variants exhibited similar kinetic profiles for [Fe3+–O]x species formation as the WT BfrA both under single and multiple catalytic turnovers, clearly depicting the role of self-assembly in the rapid uptake of Fe2+ ions and also corroborating that the 3-fold pores do not participate in Fe2+ uptake in Mtb BfrA. However, in the assembly-defective variants, rapid iron oxidation was inhibited, yet the rate of oxidation was higher than that of background autoxidation in buffer, which indicates a combined effect of the Fox centers and the autoxidation phenomena.Similarly, to understand the differences in the iron mineralization mechanism for the self-assembled and the assembly-defective variants, sequential iron oxidation kinetics was monitored by manual addition of iron aliquots (48 Fe/cage) at regular time intervals. A rapid increase in each cycle followed by a steady plateau after the addition of each aliquot in the sequential Fe2+ oxidation data (Fig. S9E†) indicates that the Fox centers are possibly vacated at the end of each cycle indicating the labile nature of the ‘di-Fe3+’ species formed at the Fox centers, thereby behaving as P. aeruginosa Bfr.69 However, in E121K and E121F, the lack of a protein cage/self-assembled structure slows down the rapid iron oxidation despite having functional Fox centers. Therefore, instead of attaining a rapid plateau, a gradual increase in absorbance was seen during consecutive additions (Fig. S9E†). The slower iron oxidation at ferroxidase centers and the inhibition of iron sequestration in the assembly-defective variants may also lead to higher background oxidation in buffer.
Self-assembly aids in rapid consumption of dissolved O2 during ferroxidase/mineralization activity
Amperometry based oximetry is a useful technique to monitor the alterations in real time O2 concentration for a reaction that involves O2 either as a substrate or as a product. Ferroxidase activity of ferritin involves O2 as a substrate along with Fe2+. Here, a Clark-type electrode was used to compare the dissolved O2 consumption kinetics of self-assembled and assembly-defective variants of Mtb BfrA during their ferroxidase/mineralization activity to investigate the impact of pore-electrostatics and the role of nanoconfinement/self-assembly.In contrast to the assembly-defective variants (E121K and E121F), the self-assembled variants (E121A and E121Q) exhibited a rapid decrease in O2 concentration as observed in the case of WT BfrA (Fig. 4A), corroborating the stopped-flow kinetic trend for iron oxidation (Fig. 3A). The faster O2 consumption profiles depict rapid ferroxidase activity and mineralization in these assembled variants, thereby indicating the role of self-assembly. In comparison to autoxidation in buffer, the assembly-defective variants exhibited a faster O2 consumption kinetics (Fig. 4A). This difference can be attributed to the functional ferroxidase centers in the assembly-defective variants. Similar to the stopped-flow data, a comparison was made between the O2 consumption profiles for assembled (48 Fe/cage) and assembly-defective variants (480 Fe/cage) at different substrate concentrations (48 vs. 480 Fe/cage). Despite a 10-fold lower substrate (Fe2+) concentration, faster O2 consumption kinetics was observed for the self-assembled variants, indicating the role of nanoconfinement towards higher efficiency of oxidoreductase activity and biomineralization (Fig. 4B). pH dependent O2 consumption kinetics (carried out in MES: pH 6.0 and MOPS: pH 7.0) also support the above observations (Fig. S10†). Autoxidation (Fe2+ oxidation in buffer, outside the protein cage) contributes significantly to the overall O2 consumption kinetic profile in assembly-defective variants, particularly at pH 7.0. However, catalytic iron oxidation predominates in the self-assembled variants while the mineralization process is pH dependent but less impacted in comparison to assembly-defective variants. The initial rates of O2 consumption for different protein samples (2.08 μM: Fig. 5A; 0.5 μM: Fig. S10A†) with 480 Fe/cage also support the observation that the self-assembled variants exhibit faster O2 consumption (Fig. 4C).
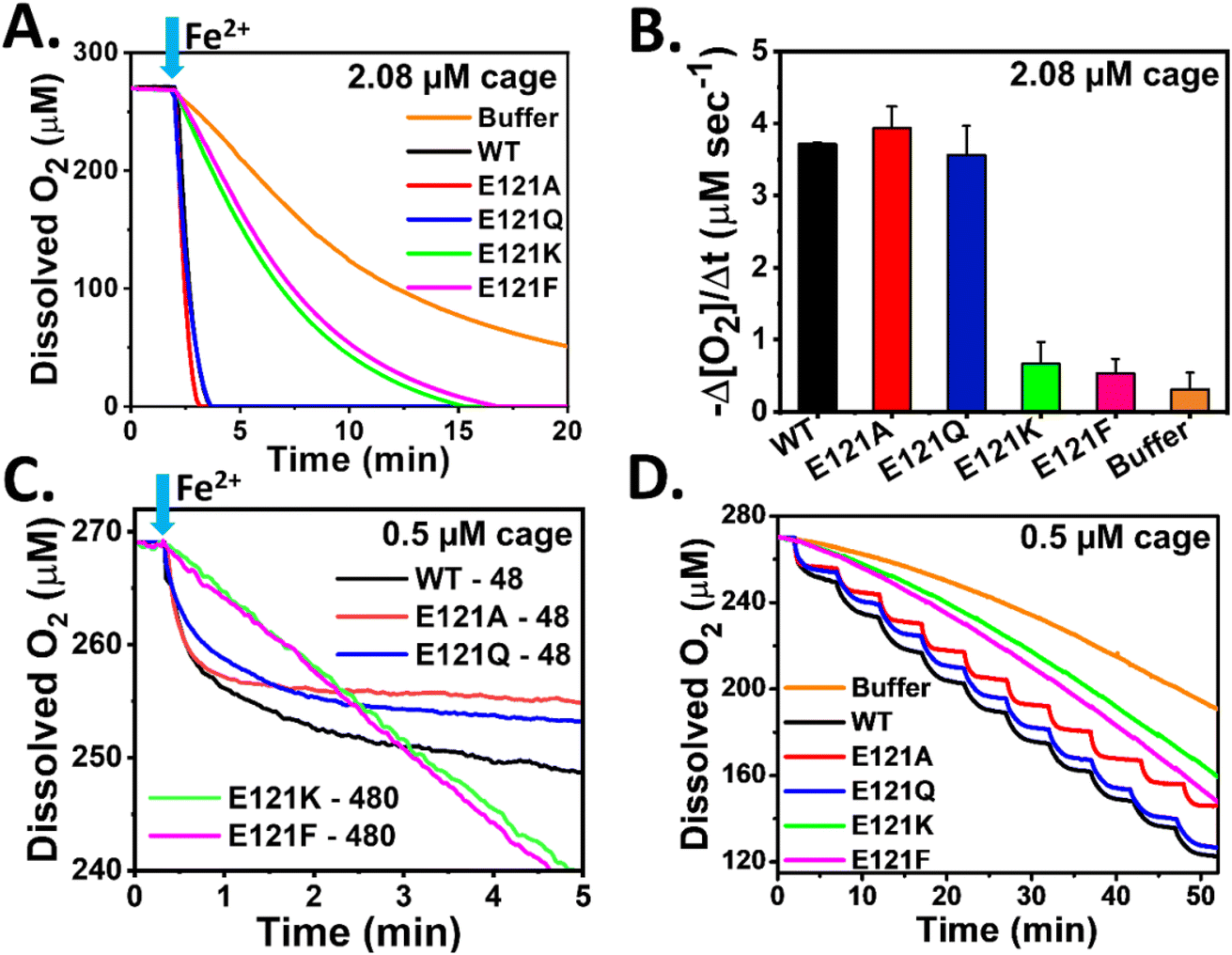 | ||
| Fig. 4 O2 consumption kinetics during the ferroxidase/mineralization activity of self-assembled and assembly-defective Mtb BfrA variants. (A) To begin the ferroxidase reaction, freshly prepared FeSO4 solutions (1 mM) were added to 2.08 μM protein cage using a Hamilton syringe to achieve 480 Fe/cage. The initial rates of O2 consumption obtained from (A) were compared in (B); the self-assembled variants exhibit faster O2 consumption. Error bars indicate the standard deviation. (C) Assembled variants with lower substrate concentrations (24 μM Fe2+) consume O2 faster than assembly-defective variants with 10-fold higher substrate concentrations (240 μM Fe2+). 48 and 480 indicate Fe/cage in respective protein samples (0.5 μM cage). (D) Dissolved O2 consumption during sequential addition of FeSO4 solutions (48 Fe/cage per injection) to protein solutions (0.5 μM cage) to achieve 480 Fe/cage. Here, all the experiments were carried out in 100 mM MOPS buffer (pH 7.0) at 25 °C. p value >0.06 and <0.001 for the assembled and assembly-defective variants respectively, calculated with respect to WT. The averages are derived from the results of at least three independent experiments, using two different protein batches. | ||
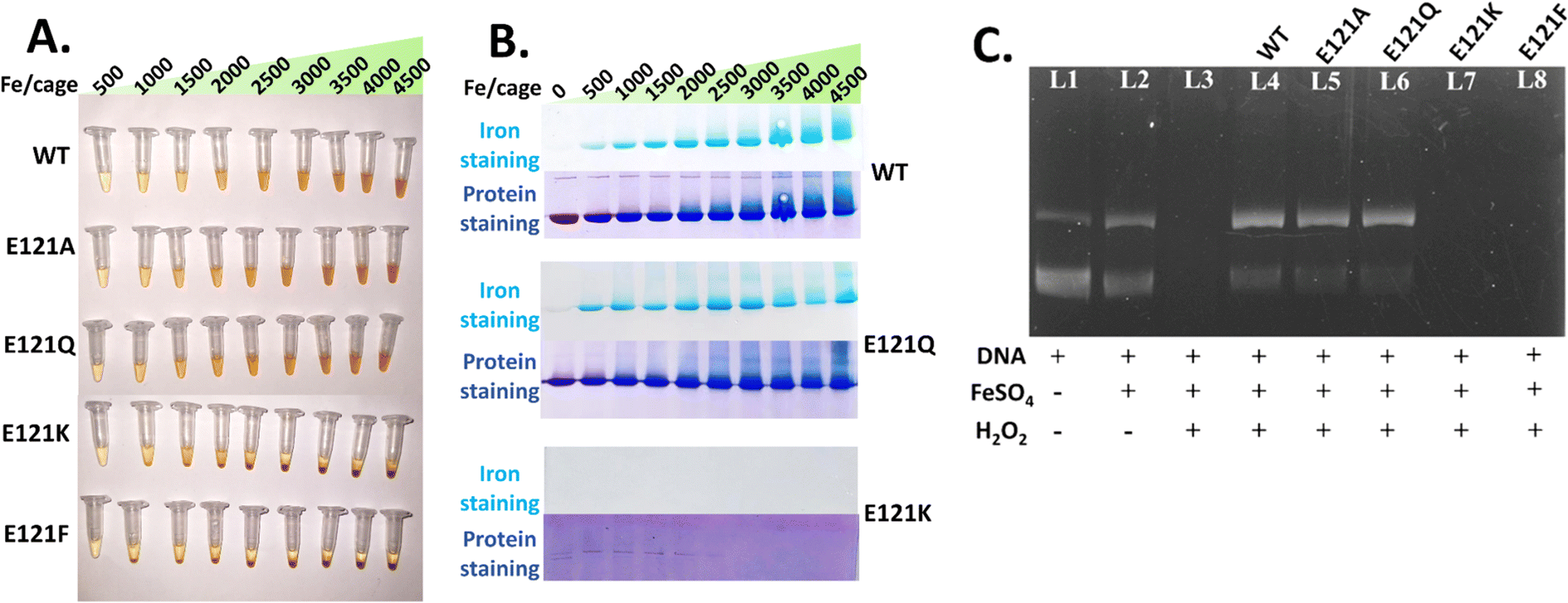 | ||
| Fig. 5 Analysis of iron incorporation ability and DNA protection activity of Mtb BfrA variants. (A) The self-assembled variants hold iron in a soluble form almost up to the addition of 4500 Fe/cage whereas for the assembly-defective variants distinct precipitation was seen beyond 500 Fe/cage. (B) Native-PAGE analysis of iron loading efficiency/accumulation in assembled and assembly-defective variants. WT and E121Q exhibited similar behaviour with higher iron loading, but E121K showed a complete absence of Prussian Blue precipitate. The gradual disappearance of protein bands on increasing iron for E121K indicates iron-induced protein precipitation. Protein samples, with and without iron mineral incorporation, were run in a 6% (w/v) non-denaturing gel (native-PAGE) for checking the iron loading ability (top) and cage integrity (bottom). (C) DNA protection activity of WT Mtb BfrA and its 3-fold assembled variants against oxidative cleavage induced by the Fenton reaction. Agarose gel (0.8% w/v) electrophoresis was carried out to assess the DNA protection activity of Mtb BfrA and its self-assembled/assembly-defective variants. DNA protection was observed only for self-assembled variants (L4 to L6) whereas the assembly-defective variants (L7 and L8) could not provide protection. For the control experiment: L1: only DNA; L2: iron and DNA; L3: DNA, iron and H2O2 (the absence of DNA bands indicates oxidative cleavage). ± signs represent the presence/absence of DNA, H2O2 and Fe2+ ions. | ||
To investigate the catalytic turnover of the Fox centers of the BfrA variants, 24 μM Fe2+ was added sequentially to 0.5 μM protein solution (48 Fe/cage per injection) at 5 min intervals to achieve 480 Fe/cage after 10 injections (Fig. 4D). WT BfrA and the self-assembled variants exhibited rapid consumption of dissolved O2 followed by a distinct plateau during each addition, possibly due to the rapid generation of ‘di-Fe3+’ at the Fox centers that enables the subsequent catalytic cycle to oxidize the incoming Fe2+ ions. On the other hand, the lack of a protein cage/self-assembled structure slows down the rapid dissolved O2 consumption in the assembly-defective variants despite having functional Fox centers. Therefore, instead of a distinct plateau, a gradual decrease in O2 levels was seen in E121K and E121F (Fig. 4D). This further indicates the synergistic effect of electrostatic guiding and nanoconfinement, responsible for rapid Fe2+ uptake and its increased effective concentration inside the nanocavity, which aids in rapid ferroxidase activity and O2 consumption.
Self-assembly is a necessity for iron incorporation and biomineral solubility
WT Mtb BfrA and the self-assembled variants (E121A and E121Q) were capable of maintaining iron in a soluble form up to the addition of ∼4000–4500 Fe/cage, without any notable precipitation (Fig. 5A). The native-PAGE profiles of self-assembled variants show distinct, gradually increasing Prussian blue bands, indicating that a major portion of the iron is incorporated and localized inside the nanoconfined ferritin cages (Fig. 5B). Smeared bands above ∼3000 Fe/cage, possibly indicate iron induced cage-aggregation.For the assembly-defective variants, visible precipitation was absent up to addition of 480 Fe/cage (Fig. 5A). However, no Prussian blue precipitate was seen in the native-PAGE profiles, possibly indicating that the iron mineral in these variants is not localized (owing to the absence of a nanoconfined cage) and rather distributed/bound in a non-specific manner. Upon increasing the iron addition beyond 480 Fe/cage, ranging from 1000–4500 Fe/cage, clear precipitation was seen. Moreover, the absence of Coomassie/protein bands in E121K under higher iron loading is an indication of iron-induced protein precipitation. These observations suggest that self-assembly is a rudimentary factor not only for faster iron oxidation but also for its higher iron-incorporation/mineralization/storage ability in Mtb BfrA.
DLS-based analysis was performed for the mineralized samples within the soluble window of all the variants i.e. up to 480 Fe/cage, to investigate if iron incorporation/mineral formation alters their hydrodynamic diameters. WT and the self-assembled variants exhibited only an ∼1 nm change in their respective sizes (Fig. S11 and Table S3†) of the apo-form, suggesting the ability of these variants to store iron efficiently and control the nucleation/growth of the iron mineral within the confined nanocompartment. In contrast, the assembly-defective variants exhibited increased particle size upon increasing the addition of iron. This could either be due to the possible formation of soluble iron-oxide/oxy-hydroxide clusters on the surface of the protein and/or iron-induced higher order oligomeric assemblies.
Furthermore, to analyze the amount of iron that the assembly-defective variants can retain without precipitation, E121K protein samples were mineralized (480 Fe/cage equivalent) with two different protein concentrations: ∼0.5 mg ml−1 (initially used for DLS) and ∼11 mg ml−1. Prior to iron analysis, the low protein mineralized samples (∼0.5 mg ml−1 with 540 μM Fe2+) were run through a PD-10 desalting column. However, iron/protein ratios (determined from ferrozine assay15) were found to be similar for the WT (450 ± 30) and E121K (400 ± 20) variants, possibly due to low resolution of the PD-10/desalting column, leading to poor separation of products. However, upon mineralizing high concentrations of E121K protein (∼11 mg ml−1), significant precipitation occurred. The solution was centrifuged and the supernatant was collected, syringe filtered and loaded into the SEC column (Sephacryl S-300). The resulting chromatogram showed three distinct fractions, indicating the presence of multiple species (Fig S12†). Interestingly, fraction 3 overlaps with the apo-protein and the additional fractions 1 and 2 correspond to some relatively larger sized species, with higher iron content.
Self-assembled Mtb BfrA variants protect DNA against Fe2+/H2O2-induced Fenton reaction
To further understand the consequences of self-assembly in ferritin on its anti-oxidative properties, in vitro DNA protection activity against Fe2+/H2O2-induced Fenton reaction was carried out by agarose gel electrophoresis (see Methods) using both assembled and assembly-defective Mtb BfrA variants. Similar to the WT, the assembled variants (E121A and E121Q) exhibited DNA protection activity, indicated by intact DNA bands (Fig. 5C, L4 to L6). In contrast, the assembly-defective variants (E121K and E121F) were unable to protect the plasmid DNA as evidenced by the absence of the DNA bands (Fig. 5C, L7 and L8). This further supports that ferritin self-assembly is essential/crucial for its anti-oxidative properties. The WT and its self-assembled variants having intact primary iron entry pathways (B-pores and 4-fold pores) and a confined nanocage possibly inhibit/minimize the generation of OH˙ radicals by rapidly scavenging Fe2+ (Fenton substrate). This is evidenced by faster iron oxidation and increased iron incorporation ability of the self-assembled Mtb BfrA variants.For the in vitro DNA protection assay, 4.5 mM H2O2 was externally added to induce oxidative stress conditions, which is significantly higher than the in situ concentration of H2O2 detected during ferroxidase activity (up to ∼28 μM, Fig. S7†). Despite the high concentration of externally added H2O2, self-assembled variants effectively safeguarded the DNA, underscoring their remarkable protective capabilities under oxidative stress.
Self-assembly (cage formation) imparts stability to the protein structure
E121K and E121F retained their secondary structure similar to WT and its self-assembled variants but exhibited a significant loss of tertiary structure and inter-subunit interactions, due to the absence of self-assembly. To analyze the impact of self-assembly (cage formation) on the overall conformational/thermal stability of the bacterioferritins, GdnHCl and temperature induced unfolding of their secondary structures was monitored by far-UV CD. The loss of α-helicity at 222 nm (Fig. 6 and S13†) was used as an indicator to interpret their chemical/thermal stability and obtain various thermodynamic parameters (eqn (3)–(7)).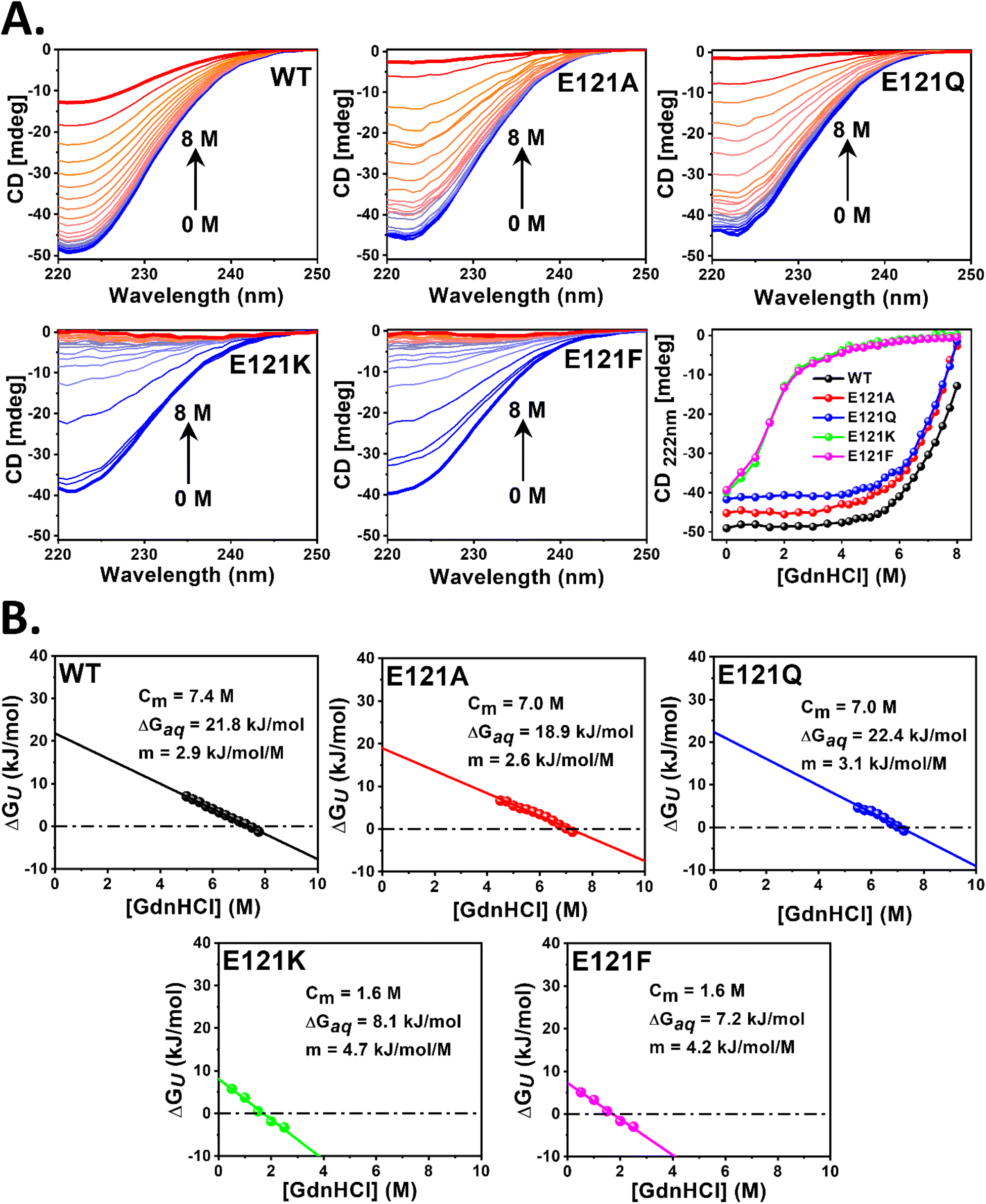 | ||
| Fig. 6 GdnHCl induced unfolding of self-assembled and assembly-defective Mtb BfrA variants. Far-UV CD spectra and changes in the CD signal at 222 nm of WT BfrA and its variants at increasing concentrations of GdnHCl i.e. from 0 to 8 M (A). Protein concentration was maintained at 0.25 μM cage or cage equivalent (6 μM subunit). Conformational stability parameters of WT BfrA and its variants determined from the linear extrapolation method (i.e., ΔGUvs. [GdnHCl]) at 25 °C (B). | ||
The linear extrapolation method (eqn (6)) was used to determine the free energies of unfolding in aqueous solution (ΔGAq) and Cm values (Fig. 6B and Table S5†) from the GdnHCl-induced unfolding data (Fig. 6A and Table S5†). The Cm value indicates the GdnHCl concentration at which the protein is 50% unfolded (fU = fF = 0.5) and ΔGU = 0. While WT BfrA exhibited a Cm of ∼7.4 ± 0.2 M, the self-assembled variants (E121A and E121Q) exhibited slightly lower values of Cm of ∼7.0 ± 0.2 M. However, the assembly-defective variants (E121K and E121F) unfolded at drastically lower concentrations of GdnHCl (Cm ∼1.6 ± 0.1 M), which clearly indicates their lower conformational stability, possibly due to the loss of various inter-subunit non-covalent interactions that arise as a consequence of self-assembly. The self-assembled variants exhibited ΔGAq values similar to those of WT BfrA (∼19–22 kJ mol−1), whereas the assembly-defective variants exhibited much lower values, ∼7–8 kJ mol−1, indicating the contribution of self-assembly in the overall conformational stability of Mtb BfrA.
The thermal stability of the self-assembled variant, E121A, was found to be similar as that of WT (Tm ∼90 °C); however E121Q was relatively less thermostable (Tm ∼84 °C) (Fig. S13 and Table S5†). In contrast, the assembly-defective variants, E121K and E121F, exhibited lower Tm values of ∼78 and 80 °C, respectively (Fig. S13 and Table S5†), possibly due to the loss of the compact cage structure (Fig. 2), further corroborating the importance of self-assembly towards the overall cage stability. The differences in the thermal stability of self-assembled and assembly-defective variants can also be correlated with the revival of intact, self-assembled nanocage architectures after cooling (Fig. S14†). While the assembled variants exhibited almost complete revival of their secondary structures, the assembly-defective variants exhibited an irreversible loss in their helical content, as indicated by the disappearance of two α-helix signature peaks at 208 and 222 nm (Fig. S14†). To examine if the irreversibility of assembly-defective variants was influenced by the speed at which they were brought back to 25 °C, the samples were cooled, at different speeds, under three different conditions (Fig. S14D and E†). However, complete reversibility of unfolding of the assembly-defective variants was not observed i.e. the CD signal was not restored to the original value at 25 °C. This further emphasizes that cage formation imparts extra stability to the overall protein structure.
Discussion
The formation of ferritin protein nanocages, by self-assembly phenomena, is driven by non-covalent interactions at the subunit–subunit interfaces, ultimately generating a central hollow nanocavity (∼30% of total cage volume) and multiple symmetric/asymmetric pores/channels (Fig. 1A). The amino acid residues present on the external/internal surface and along the pore linings impart necessary electrostatics that decipher the ability to sequester/oxidize Fe2+ ions and store them in the form of a soluble ferric oxyhydroxide mineral.70 The relatively higher negative charge density on the internal surface (Fig. 1E) stabilizes and safeguards the iron mineral within its hollow protein nanocompartment until cellular requirements.15,71Iron biomineralization inside the ferritin nanocages involves a complex series of events/reactions.72–74 This cascade of events leading to iron storage commences by rapid uptake of Fe2+ (via electrostatic guiding/focusing22,32) and its migration to the Fox centers (enzymatic sites responsible for rapid iron oxidation), through dedicated pathways involving different pores/channels in different ferritins.19,75 The oxidoreductase and mineralization activity of ferritin involves consumption of Fe2+ and O2; their ratio varies with the amount of Fe2+ and the type of ferritin.11,64 The in situ generated H2O2 is consumed during the mineralization process.
| 2Fe2+ + O2 + 4H2O → 2FeOOHCore + H2O2 + 4H+ |
While eukaryotic ferritins mostly utilize the 3-fold pores/channels as their primary Fe2+ entry pathways,76 recent reports on a few bacterioferritins suggest the involvement of the asymmetric B-pores.19,21,69 These studies established the importance of negatively charged Glu and Asp residues lined along these hydrophilic pores. In addition to Fe2+ sequestration, specific pores are possibly involved in Fe2+ mobilization77 and the selective translocation of phosphate/O2/H+78 to and from the protein cavity and its surrounding environment (cytosol, mitochondria). Cytosolic chaperones are reported to possibly assist in the transportation of iron to and from ferritins and other target sites.79–81
The protein cage acts as a selective barrier and reinforces the establishment of a concentration gradient of Fe2+, similar to the ion gradient across lipid membranes. This electrostatically driven accumulation of Fe2+ inside the ferritin nanocompartment increases its effective concentration in the vicinity of the Fox centers. As a consequence of this augmented proximity between Fe2+ and the Fox center, the probability of collision increases; as demonstrated by the rapid ferroxidase activity occurring within milliseconds to seconds (Fig. 3). However, it is difficult to segregate the relative contributions of the catalytic sites and self-assembly towards the rapid ferroxidase/mineralization activity in bacterioferritins owing to their inherent self-assembly ability. In addition, these protein cages are exceptionally stable60 and thus, isolation of single/oligomeric assembly units in folded form is difficult, despite substitution of multiple amino acids.19 In our recent report on Mtb BfrA, site-directed mutagenesis was employed to ascertain the role of B-pores as primary Fe2+ uptake routes; the robustness of the nanocage was evidenced through one of its B-pore variants, where the introduction of multiple Lys on a single helix (D132K/E135K/T136K/E139K: 4 MUT) failed to disassemble the protein nanocage.19 Therefore, in the current work, a ‘hot-spot’ residue at the 3-fold pores was identified and rationally perturbed by site-directed mutagenesis to investigate the role of these pores in the interplay between the formation of self-assembled protein nanocage and its functional aspects. The 3-fold pores of Mtb BfrA consist of alternate layers of negative and positive amino acid residues, where E121 is located in between two positively charged residues, R109 and K122 (Fig. 1), and is possibly involved in essential interactions (salt-bridge and/or hydrogen bond) that constitute the pore and enable the self-assembly process and hence the formation of other pores (Fig. S15A†).
In the case of buffer alone and for the assembly-defective variants, the added Fe2+ is expected to be distributed homogenously in the reaction medium (say, 1 ml = 1021 nm3) (Fig. 7A). Thus, the effective substrate concentration does not change in the vicinity of the enzymatic sites and is similar to that of the bulk. By virtue of the rapid Fe2+ sequestration and its accumulation within the ferritin nanocavity (say, for 2 μM ferritin cage, effective volume in 1 ml = no of ferritin × effective volume offered by each ferritin cavity i.e. 1.2 × 1015 × 268 nm3 = 3.21 × 1017 nm3), the self-assembled variants gain access to an effective concentration of up to ∼3115-fold excess of Fe2+ at the catalytic sites (Fig. 7A). This phenomenon is expected to drastically enhance the reactivity of the ferroxidase centers.
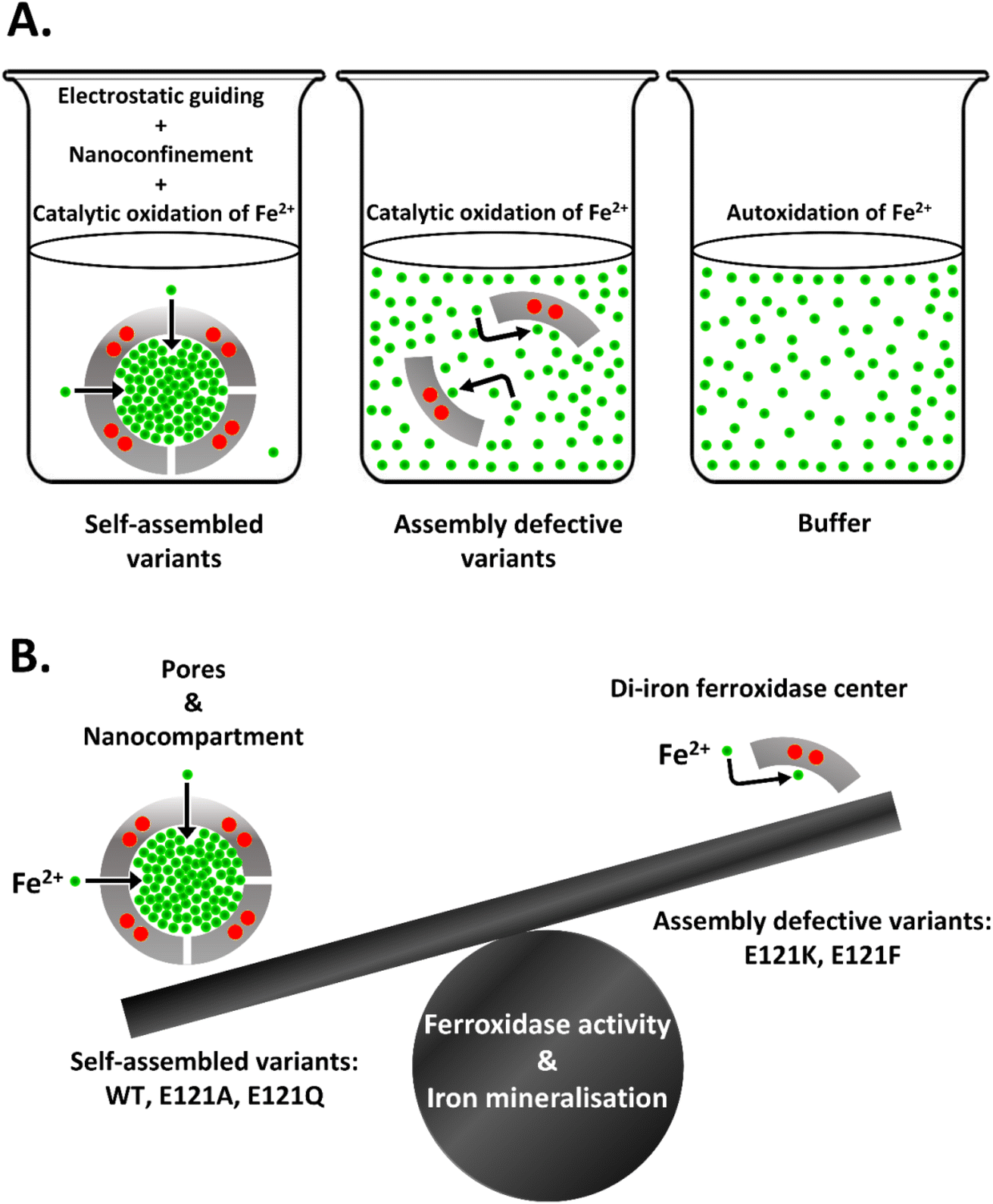 | ||
| Fig. 7 Impact of electrostatic guiding and cage-confinement on rapid iron oxidation/mineralisation and establishment of relative contribution of enzymatic sites and self-assembly. Self-assembly generates favorable electrostatics to guide the ingress of Fe2+ ions via electrostatic guiding and enhance the local substrate concentration in the vicinity of the di-iron catalytic sites (highlighted in red) by cage-confinement that is absent in the case of assembly-defective variants and buffer (A). Pore electrostatics and nanoconfinement provide major assistance to the di-iron catalytic centers in facilitating rapid ferroxidase activity and iron mineralization (B). | ||
The electrostatics/dynamics of pore residues (breathing motion) and faster water exchange rates (k ∼106 s−1)3 collectively favour the translocation of hydrated Fe2+ (∼6 Å) through the narrow channels (∼3–4 Å, primary entry pathways: B-pores).19 Hydrated Fe2+ traverses along the channel by shedding/exchanging its inner-sphere water molecules with the amino acids or water molecules present in its ingress route and reach the catalytic Fox centers possibly via the transit residues (such as the “bucket brigade” in eukaryotic ferritins).13 However, when the electrostatics of these entry routes are perturbed, though rapid Fe2+ uptake and oxidation are inhibited, yet similar mineral accumulation inside the cage is reported over longer time durations.19 This indicates that Fe2+ possibly enters the cage through other secondary ‘slower’ routes and/or the Fe3+ species generated as a consequence of slower autoxidation of Fe2+ ions are also sequestered by the ferritin nanocages. This Fe3+ uptake process probably occurs over longer time owing to its larger hydration sphere (∼9 Å), higher dehydration energy and slower water exchange rate (k ∼102 s−1).3 However, this delayed iron uptake and oxidation may contribute to Fe2+ induced oxidative stress/cellular toxicity and infection. This situation gets worse in the case of assembly-defective variants, where most of the iron is freely distributed in the buffer.
Free iron in its ferrous (Fe2+) form, can be highly toxic to cells and tissues due to its ability to catalyze the formation of harmful reactive oxygen species such as OH˙ radicals through the Fenton reaction.46,47,53 The OH˙ radical is a highly potent oxidizing agent that reacts indiscriminately with proteins, DNA, membranes and other biomolecules at rates that are nearly diffusion-controlled.5 Ferritin checks the Fenton reaction in vivo by rapidly sequestering free Fe2+, thereby limiting the generation of harmful OH˙ radicals. This regulation accounts for the anti-oxidative activity of ferritin.
Naturally, the mini-ferritins (Dps proteins) safeguard DNA by capturing the Fenton substrates and/or by binding directly to DNA to form a protective complex.82,83 Although, Mtb lacks the Dps gene, it still manages to survive oxidative stress and continue proliferating.53,84 Our earlier reports established that WT Mtb BfrA exhibited DNA protection activity, possibly by forming a BfrA–DNA complex.53 To elucidate the effect of cage-assembly on anti-oxidative properties of ferritin, DNA cleavage assay was performed in the presence of 3-fold pore-variants of Mtb BfrA, which exhibit differences in their cage integrity. Interestingly, only the self-assembled variants showed DNA-protection activity, whereas the DNA was completely degraded in the presence of assembly-defective variants. As the modifications in the variants are only limited to their pores, the external-surface electrostatics are possibly unaltered. Thus, it is anticipated that all the variants would bind DNA to a similar extent and protect it against the Fenton reaction. The difference in the DNA protection ability, likely indicates that DNA binding may not be the only mechanism by which it protects DNA. The protective mechanism maybe somewhat related to its cage-assembly and pore electrostatics and it ultimately minimizes the accumulation of free Fe2+ in the reaction medium.
The inherent stability of the ferritin nanocage along with its natural ability to internalize via cellular receptors makes it an excellent choice for numerous nanobiotechnology applications viz. diagnosis (MRI contrast agents), targeted drug delivery, theranostics (for cancer therapy and diagnosis), catalysis, etc.10,85,86 Additionally, the ferritin compartments serve as confined reaction chambers (nanoreactor), where non-natural ions/substrates can be incorporated into their cavity via electrostatic guiding to carry out nanoparticle synthesis.7,85,87 However, the encapsulation of neutral or larger cargo/drugs requires ferritin cage-disassembly under harsh conditions,42,60,85 often resulting in the loss of both the protein and cargo. The current work presents/demonstrates the identification and alteration of a single “hot-spot” residue that can be targeted to disassemble the stable bacterioferritin cage, bypassing the use of any extreme reaction conditions. These findings can be further extended towards the encapsulation of cargo, following its disassembly and subsequent reassembly by making adjustments in the ionic strength (screening repulsive forces).88Mtb, like any other pathogen, requires iron for its virulence but still proliferates under iron-restricted conditions inside human macrophages and lungs. Furthermore, this study can also be extended towards combating Mtb proliferation by hindering its Fe2+ storage and sequestration abilities through the strategic manipulation of its iron uptake routes.
Conclusion
In the current report, we assess the relative contribution of enzymatic sites and self-assembly towards rapid ferroxidase activity and mineralization reaction in bacterioferritins. To evaluate the role of self-assembly, 3-fold pore electrostatics were perturbed by rational substitution of the E121 residue, generating two sets of variants that either maintained self-assembly (E121A, E121Q) or were assembly-defective (E121K and E121F). In the case of the self-assembled variants, the altered pore electrostatics did not impact the formation of the primary iron uptake routes (i.e., the B-pores), nor did it affect the iron oxidation/mineralization reaction, suggesting that the 3-fold pores are not involved in Fe2+ uptake by Mtb BfrA. However, E121-substitution with positively charged Lys (E121K; electrostatic repulsions due to lysine crowding) or with bulkier/hydrophobic Phe (E121F; steric hindrance) abolished the self-assembly phenomena highlighting the importance of key interfacial interactions that control the formation of nanocage/pores and their functions (Fig. S15B and Table S6†). Moreover, self-assembly was also seen to impart extra stability to the protein nanocage. Assembly-defective variants, bearing active catalytic sites, exhibited notably faster ferroxidase/mineralization activities and O2 consumption as compared to the autoxidation of Fe2+ in buffer, but they still failed to reach the efficiency of the self-assembled variants, even at 100 fold higher Fe2+ concentrations. Positive cooperativity, the capability to incorporate iron in its soluble form and to protect DNA against the Fenton reaction was exclusive to the self-assembled variants. The absence of a defined nanocompartment and loss of its associated electrostatic guiding accounts for the poor enzymatic activity of the assembly-defective variants. In summary, this report highlights the concerted action of self-assembly and ferroxidase sites that facilitates swift iron uptake, its oxidation, and subsequent biomineralization in bacterioferritins. Our findings comprehend that ferritin self-assembly, which electrostatically directs the ingress of Fe2+ ions via pores/channels and enhances local substrate concentration via cage-confinement, is the major contributor towards ferroxidase/mineralization reaction and its anti-oxidative behaviour (Fig. 7B). Additionally, the current work identifies a key “hot-spot” residue (E121) in Mtb bacterioferritin, rational engineering of which enables the disassembly of the stable bacterioferritin cage, without the need for harsh reaction conditions. A similar strategy can be adopted towards encapsulation of cargo in ferritins, following their disassembly through alterations of specific “hot-spot” residues and subsequent reassembly by making adjustments to the ionic strength. The study holds significance in exploiting ferritin nanocages as a nanosink and nanoreactor for sequestration, encapsulation and nanomaterial synthesis for a multitude of nano-biomedical applications.Abbreviations
| Mtb | Mycobacterium tuberculosis |
| Bfr | bacterioferritin |
| BfrA | bacterioferritin A |
| ROS | reactive oxygen species |
| Fox | ferroxidase center |
| DFP | di-ferric peroxo |
| PDB | protein data bank |
| IPTG | isopropyl-1-thio-β-D-galactopyranoside |
| MOPS | 3-(N-morpholino) propane sulfonic acid |
| PAGE | poly-acrylamide gel electrophoresis |
| TEM | transmission electron microscopy |
| DLS | dynamic light scattering |
| HRP | horseradish peroxidase |
| CD | circular dichroism |
| GdnHCl | guanidine hydrochloride |
| C m | midpoint of unfolding concentration of GdnHCl |
| T m | midpoint unfolding (melting) temperature |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Rabindra Kumar Behera, Akankshika Parida and Gargee Bhattacharyya designed the study and drafted the manuscript. Akankshika Parida, Swagatika Mallik and Gargee Bhattacharyya performed the experiments and revised the document. All the authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Engineering Research Board (SERB), India (CRG/2020/005332), the Science and Technology Department, Odisha, India (ST-SCST-MISC-0036-2023) to R. K. B. G. B. and S. M. thank DST INSPIRE (2021/IF210168) and UGC, India respectively for their research fellowship. We are thankful to Dr S. Mazumdar and Mr B. T. Kansara (TIFR, Mumbai) for the collection of stopped flow kinetics data. We are thankful to Dr Anil K. Tyagi and Dr Garima Khare (University of Delhi, South Campus) for their generous support in providing the Mtb Bfr clone.References
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 4th edn, 2002 Search PubMed.
- L. Bar-Peled and N. Kory, Principles and functions of metabolic compartmentalization, Nat. Metab., 2022, 4, 1232–1244 CrossRef PubMed.
- Biological Inorganic Chemistry: Structure and Reactivity, ed. H. Gray, Stiefel, E. I., Valentine, J. S. and Bertini, I., University Science Books, 2007 Search PubMed.
- E. Gouaux and R. Mackinnon, Principles of selective ion transport in channels and pumps, Science, 2005, 310, 1461–1465 CrossRef CAS PubMed.
- C. C. Winterbourn, Reconciling the chemistry and biology of reactive oxygen species, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Chemical reactivity under nanoconfinement, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed.
- M. Taher, B. Maity, T. Nakane, S. Abe, T. Ueno and S. Mazumdar, Controlled Uptake of an Iridium Complex inside Engineered apo-Ferritin Nanocages: Study of Structure and Catalysis, Angew Chem. Int. Ed. Engl., 2022, 61, e202116623 CrossRef CAS PubMed.
- B. K. John Kuriyan and D. Wemmer, The Molecules of Life: Physical and Chemical Principles, W.W. Norton & Company, 2012 Search PubMed.
- E. C. Theil, T. Tosha and R. K. Behera, Solving Biology's Iron Chemistry Problem with Ferritin Protein Nanocages, Acc. Chem. Res., 2016, 49, 784–791 CrossRef CAS PubMed.
- E. C. Theil, R. K. Behera and T. Tosha, Ferritins for Chemistry and for Life, Coord. Chem. Rev., 2013, 257, 579–586 CrossRef CAS PubMed.
- F. Bou-Abdallah, The iron redox and hydrolysis chemistry of the ferritins, Biochim. Biophys. Acta, 2010, 1800, 719–731 CrossRef CAS PubMed.
- W. M. Aumiller, M. Uchida and T. Douglas, Protein cage assembly across multiple length scales, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- R. K. Behera and E. C. Theil, Moving Fe2+ from ferritin ion channels to catalytic OH centers depends on conserved protein cage carboxylates, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7925–7930 CrossRef CAS PubMed.
- N. Behera, G. Bhattacharyya, S. Behera and R. K. Behera, Iron mobilization from intact ferritin: effect of differential redox activity of quinone derivatives with NADH/O(2) and in situ-generated ROS, J. Biol. Inorg. Chem., 2024, 29, 455–475 CrossRef CAS PubMed.
- A. Parida and R. K. Behera, Iron Accumulation in Ferritin, Methods Mol. Biol., 2023, 2671, 121–134 CrossRef CAS PubMed.
- E. C. Theil and R. K. Behera, in Coordination Chemistry in Protein Cages, ed. T. Ueno and Watanabe, Y., John Wiley & Sons, Inc., 2013, ch. 1, p. 3, DOI:10.1002/9781118571811.ch1.
- Y. Zhang and B. P. Orner, Self-assembly in the ferritin nano-cage protein superfamily, Int. J. Mol. Sci., 2011, 12, 5406–5421 CrossRef CAS PubMed.
- Y. Kwak, J. K. Schwartz, S. Haldar, R. K. Behera, T. Tosha, E. C. Theil and E. I. Solomon, Spectroscopic studies of single and double variants of M ferritin: lack of conversion of a biferrous substrate site into a cofactor site for O2 activation, Biochemistry, 2014, 53, 473–482 CrossRef CAS PubMed.
- A. Parida, A. Mohanty, R. K. Raut, I. Padhy and R. K. Behera, Modification of 4-Fold and B-Pores in Bacterioferritin from Mycobacterium tuberculosis Reveals Their Role in Fe2+ Entry and Oxidoreductase Activity, Inorg. Chem., 2023, 62, 178–191 CrossRef CAS PubMed.
- M. Rivera, Bacterioferritin: Structure, Dynamics, and Protein-Protein Interactions at Play in Iron Storage and Mobilization, Acc. Chem. Res., 2017, 50, 331–340 CrossRef CAS PubMed.
- S. G. Wong, J. C. Grigg, N. E. Le Brun, G. R. Moore, M. E. Murphy and A. G. Mauk, The B-type channel is a major route for iron entry into the ferroxidase center and central cavity of bacterioferritin, J. Biol. Chem., 2015, 290, 3732–3739 CrossRef CAS PubMed.
- R. K. Behera, R. Torres, T. Tosha, J. M. Bradley, C. W. Goulding and E. C. Theil, Fe(2+) substrate transport through ferritin protein cage ion channels influences enzyme activity and biomineralization, J. Biol. Inorg. Chem., 2015, 20, 957–969 CrossRef CAS PubMed.
- C. Pozzi, F. Di Pisa, D. Lalli, C. Rosa, E. Theil, P. Turano and S. Mangani, Time-lapse anomalous X-ray diffraction shows how Fe(2+) substrate ions move through ferritin protein nanocages to oxidoreductase sites, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 941–953 CrossRef CAS PubMed.
- M. Mehlenbacher, M. Poli, P. Arosio, P. Santambrogio, S. Levi, N. D. Chasteen and F. Bou-Abdallah, Iron Oxidation and Core Formation in Recombinant Heteropolymeric Human Ferritins, Biochemistry, 2017, 56, 3900–3912 CrossRef CAS PubMed.
- F. Bou-Abdallah, J. Fish, G. Terashi, Y. Zhang, D. Kihara and P. Arosio, Unveiling the stochastic nature of human heteropolymer ferritin self-assembly mechanism, Protein Sci., 2024, 33, e5104 CrossRef CAS PubMed.
- H. Yao, A. Soldano, L. Fontenot, F. Donnarumma, S. Lovell, J. R. Chandler and M. Rivera, Pseudomonas aeruginosa Bacterioferritin Is Assembled from FtnA and BfrB Subunits with the Relative Proportions Dependent on the Environmental Oxygen Availability, Biomolecules, 2022, 12, 366 CrossRef CAS PubMed.
- K. Pantopoulos, Iron Metabolism and the IRE/IRP Regulatory System: An Update, Ann. N. Y. Acad. Sci., 2004, 1012, 1–13 CrossRef CAS PubMed.
- D. B. Kell and E. Pretorius, Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells, Metallomics, 2014, 6, 748–773 CrossRef CAS PubMed.
- G. W. H. Organization, WHO guideline on use of ferritin concentrations to assess iron status in individuals and populations, CC BY-NC-SA 3.0 IGO, 2020.
- T. Douglas and D. R. Ripoll, Calculated electrostatic gradients in recombinant human H-chain ferritin, Protein Sci., 1998, 7, 1083–1091 CrossRef CAS PubMed.
- P. Ceci, G. Di Cecca, M. Falconi, F. Oteri, C. Zamparelli and E. Chiancone, Effect of the charge distribution along the "ferritin-like" pores of the proteins from the Dps family on the iron incorporation process, J. Biol. Inorg. Chem., 2011, 16, 869–880 CrossRef CAS PubMed.
- B. Chandramouli, C. Bernacchioni, D. Di Maio, P. Turano and G. Brancato, Electrostatic and Structural Bases of Fe2+ Translocation through Ferritin Channels, J. Biol. Chem., 2016, 291, 25617–25628 CrossRef CAS PubMed.
- R. Laghaei, W. Kowallis, D. G. Evans and R. D. Coalson, Calculation of iron transport through human H-chain ferritin, J. Phys. Chem. A, 2014, 118, 7442–7453 CrossRef CAS PubMed.
- K. H. Ebrahimi, P. L. Hagedoorn and W. R. Hagen, Self-assembly is prerequisite for catalysis of Fe(II) oxidation by catalytically active subunits of ferritin, J. Biol. Chem., 2015, 290, 26801–26810 CrossRef CAS PubMed.
- H. Sies, V. V. Belousov, N. S. Chandel, M. J. Davies, D. P. Jones, G. E. Mann, M. P. Murphy, M. Yamamoto and C. Winterbourn, Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology, Nat. Rev. Mol. Cell Biol., 2022, 23, 499–515 CrossRef CAS PubMed.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A. F. Miller, M. Teixeira and J. S. Valentine, Superoxide dismutases and superoxide reductases, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- D. R. Ripoll, C. H. Faerman, P. H. Axelsen, I. Silman and J. L. Sussman, An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5128–5132 CrossRef CAS PubMed.
- R. H. Meltzer, E. Thompson, K. V. Soman, X.-Z. Song, J. O. Ebalunode, T. G. Wensel, J. M. Briggs and S. E. Pedersen, Electrostatic Steering at Acetylcholine Binding Sites, Biophys. J., 2006, 91, 1302–1314 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. Olsson, Electrostatic basis for enzyme catalysis, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- F. Wegerich, A. Giachetti, M. Allegrozzi, F. Lisdat and P. Turano, Mechanistic insights into the superoxide-cytochrome c reaction by lysine surface scanning, J. Biol. Inorg. Chem., 2013, 18, 429–440 CrossRef CAS PubMed.
- T. Tosha, M. R. Hasan and E. C. Theil, The ferritin Fe2 site at the diiron catalytic center controls the reaction with O2 in the rapid mineralization pathway, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18182–18187 CrossRef CAS PubMed.
- A. Mohanty, M. K., S. S. Jena and R. K. Behera, Kinetics of Ferritin Self-Assembly by Laser Light Scattering: Impact of Subunit Concentration, pH, and Ionic Strength, Biomacromolecules, 2021, 22, 1389–1398 CrossRef CAS PubMed.
- A. D. Sheftel, A. B. Mason and P. Ponka, The long history of iron in the Universe and in health and disease, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 161–187 CrossRef CAS PubMed.
- X. Liu and E. C. Theil, Ferritins: dynamic management of biological iron and oxygen chemistry, Acc. Chem. Res., 2005, 38, 167–175 CrossRef CAS PubMed.
- P. A. Frey and G. H. Reed, The ubiquity of iron, ACS Chem. Biol., 2012, 7, 1477–1481 CrossRef CAS PubMed.
- W. H. Koppenol, Ferryl for real. The Fenton reaction near neutral pH, Dalton Trans., 2022, 51, 17496–17502 RSC.
- K. O. Muranov, Fenton Reaction in vivo and in vitro. Possibilities and Limitations, Biochemistry, 2024, 89, S112–S126 CAS.
- P. Arosio, R. Ingrassia and P. Cavadini, Ferritins: a family of molecules for iron storage, antioxidation and more, Biochim. Biophys. Acta, 2009, 1790, 589–599 CrossRef CAS PubMed.
- J. E. Cassat and E. P. Skaar, Iron in infection and immunity, Cell Host Microbe, 2013, 13, 509–519 CrossRef CAS PubMed.
- A. Chao, P. J. Sieminski, C. P. Owens and C. W. Goulding, Iron Acquisition in Mycobacterium tuberculosis, Chem. Rev., 2019, 119, 1193–1220 CrossRef CAS PubMed.
- J. M. Bradley, D. A. Svistunenko, M. T. Wilson, A. M. Hemmings, G. R. Moore and N. E. Le Brun, Bacterial iron detoxification at the molecular level, J. Biol. Chem., 2020, 295, 17602–17623 CrossRef CAS PubMed.
- A. Soldano, H. Yao, A. N. D. Punchi Hewage, K. Meraz, J. K. Annor-Gyamfi, R. A. Bunce, K. P. Battaile, S. Lovell and M. Rivera, Small Molecule Inhibitors of the Bacterioferritin (BfrB)-Ferredoxin (Bfd) Complex Kill Biofilm-Embedded Pseudomonas aeruginosa Cells, ACS Infect. Dis., 2021, 7, 123–140 CrossRef CAS PubMed.
- A. Mohanty, B. Subhadarshanee, P. Barman, C. Mahapatra, B. Aishwarya and R. K. Behera, Iron Mineralizing Bacterioferritin A from Mycobacterium tuberculosis Exhibits Unique Catalase-Dps-like Dual Activities, Inorg. Chem., 2019, 58, 4741–4752 CrossRef CAS PubMed.
- G. Khare, P. Nangpal and A. K. Tyagi, Differential Roles of Iron Storage Proteins in Maintaining the Iron Homeostasis in Mycobacterium tuberculosis, PLoS One, 2017, 12, e0169545 CrossRef PubMed.
- D. Schnappinger, S. Ehrt, M. I. Voskuil, Y. Liu, J. A. Mangan, I. M. Monahan, G. Dolganov, B. Efron, P. D. Butcher, C. Nathan and G. K. Schoolnik, Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment, J. Exp. Med., 2003, 198, 693–704 CrossRef CAS PubMed.
- K. Borah, T. A. Mendum, N. D. Hawkins, J. L. Ward, M. H. Beale, G. Larrouy-Maumus, A. Bhatt, M. Moulin, M. Haertlein, G. Strohmeier, H. Pichler, V. T. Forsyth, S. Noack, C. W. Goulding, J. McFadden and D. J. V. Beste, Metabolic fluxes for nutritional flexibility of Mycobacterium tuberculosis, Mol. Syst. Biol., 2021, 17, e10280 CrossRef CAS PubMed.
- A. Mohanty, A. Parida, B. Subhadarshanee, N. Behera, T. Subudhi, P. K. Koochana and R. K. Behera, Alteration of Coaxial Heme Ligands Reveals the Role of Heme in Bacterioferritin from Mycobacterium tuberculosis, Inorg. Chem., 2021, 60, 16937–16952 CrossRef CAS PubMed.
- P. K. Koochana, A. Mohanty, A. Parida, N. Behera, P. M. Behera, A. Dixit and R. K. Behera, Flavin-mediated reductive iron mobilization from frog M and Mycobacterial ferritins: impact of their size, charge and reactivities with NADH/O2, J. Biol. Inorg. Chem., 2021, 26, 265–281 CrossRef CAS PubMed.
- R. K. Raut, G. Bhattacharyya and R. K. Behera, Gastric stability of bare and chitosan-fabricated ferritin and its bio-mineral: implication for potential dietary iron supplements, Dalton Trans., 2024, 53, 13815–13830 RSC.
- B. Subhadarshanee, A. Mohanty, M. K. Jagdev, D. Vasudevan and R. K. Behera, Surface charge dependent separation of modified and hybrid ferritin in native PAGE: Impact of lysine 104, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 1267–1273 CrossRef CAS PubMed.
- J. R. Blackwell and R. Horgan, A novel strategy for production of a highly expressed recombinant protein in an active form, FEBS Lett., 1991, 295, 10–12 CrossRef CAS PubMed.
- E. A. Berry and B. L. Trumpower, Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra, Anal. Biochem., 1987, 161, 1–15 CrossRef CAS PubMed.
- R. K. Behera, H. Nakajima, J. Rajbongshi, Y. Watanabe and S. Mazumdar, Thermodynamic Effects of the Alteration of the Axial Ligand on the Unfolding of Thermostable Cytochrome c, Biochemistry, 2013, 52, 1373–1384 CrossRef CAS PubMed.
- A. Parida, A. Mohanty, B. T. Kansara and R. K. Behera, Impact of Phosphate on Iron Mineralization and Mobilization in Nonheme Bacterioferritin B from Mycobacterium tuberculosis, Inorg. Chem., 2020, 59, 629–641 CrossRef CAS PubMed.
- D. J. Huard, K. M. Kane and F. A. Tezcan, Re-engineering protein interfaces yields copper-inducible ferritin cage assembly, Nat. Chem. Biol., 2013, 9, 169–176 CrossRef CAS PubMed.
- C. Bernacchioni, V. Ghini, C. Pozzi, F. Di Pisa, E. C. Theil and P. Turano, Loop electrostatics modulates the intersubunit interactions in ferritin, ACS Chem. Biol., 2014, 9, 2517–2525 CrossRef CAS PubMed.
- G. N. Jameson, W. Jin, C. Krebs, A. S. Perreira, P. Tavares, X. Liu, E. C. Theil and B. H. Huynh, Stoichiometric production of hydrogen peroxide and parallel formation of ferric multimers through decay of the diferric-peroxo complex, the first detectable intermediate in ferritin mineralization, Biochemistry, 2002, 41, 13435–13443 CrossRef CAS PubMed.
- A. Crow, T. L. Lawson, A. Lewin, G. R. Moore and N. E. L. Brun, Structural Basis for Iron Mineralization by Bacterioferritin, J. Am. Chem. Soc., 2009, 131, 6808–6813 CrossRef CAS PubMed.
- H. Yao, H. Rui, R. Kumar, K. Eshelman, S. Lovell, K. P. Battaile, W. Im and M. Rivera, Concerted motions networking pores and distant ferroxidase centers enable bacterioferritin function and iron traffic, Biochemistry, 2015, 54, 1611–1627 CrossRef CAS PubMed.
- F. Bou-Abdallah, G. Zhao, G. Biasiotto, M. Poli, P. Arosio and N. D. Chasteen, Facilitated diffusion of iron(II) and dioxygen substrates into human H-chain ferritin. A fluorescence and absorbance study employing the ferroxidase center substitution Y34W, J. Am. Chem. Soc., 2008, 130, 17801–17811 CrossRef CAS PubMed.
- P. K. Koochana, A. Mohanty, B. Subhadarshanee, S. Satpati, R. Naskar, A. Dixit and R. K. Behera, Phenothiazines and phenoxazines: as electron transfer mediators for ferritin iron release, Dalton Trans., 2019, 48, 3314–3326 RSC.
- N. D. Chasteen and P. M. Harrison, Mineralization in Ferritin: An Efficient Means of Iron Storage, J. Struct. Biol., 1999, 126, 182–194 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P. L. Hagedoorn and W. R. Hagen, Unity in the biochemistry of the iron-storage proteins ferritin and bacterioferritin, Chem. Rev., 2015, 115, 295–326 CrossRef CAS PubMed.
- E. C. Theil, Ferritin protein nanocages-the story, Nanotechnol. Perceptions, 2012, 8, 7–16 CrossRef CAS PubMed.
- J. M. Bradley, G. R. Moore and N. E. Le Brun, Diversity of Fe2+ entry and oxidation in ferritins, Curr. Opin. Chem. Biol., 2017, 37, 122–128 CrossRef CAS PubMed.
- E. C. Theil, Ferritin protein nanocages use ion channels, catalytic sites, and nucleation channels to manage iron/oxygen chemistry, Curr. Opin. Chem. Biol., 2011, 15, 304–311 CrossRef CAS PubMed.
- G. Bellapadrona, S. Stefanini, C. Zamparelli, E. C. Theil and E. Chiancone, Iron translocation into and out of Listeria innocua Dps and size distribution of the protein-enclosed nanomineral are modulated by the electrostatic gradient at the 3-fold "ferritin-like" pores, J. Biol. Chem., 2009, 284, 19101–19109 CrossRef CAS PubMed.
- M. Uchida, S. Kang, C. Reichhardt, K. Harlen and T. Douglas, The ferritin superfamily: Supramolecular templates for materials synthesis, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 834–845 CrossRef CAS PubMed.
- C. E. Outten, Checks and balances for the iron bank, J. Biol. Chem., 2017, 292, 15990–15991 CrossRef CAS PubMed.
- C. C. Philpott, M. S. Ryu, A. Frey and S. Patel, Cytosolic iron chaperones: Proteins delivering iron cofactors in the cytosol of mammalian cells, J. Biol. Chem., 2017, 292, 12764–12771 CrossRef CAS PubMed.
- H. Shi, K. Z. Bencze, T. L. Stemmler and C. C. Philpott, A cytosolic iron chaperone that delivers iron to ferritin, Science, 2008, 320, 1207–1210 CrossRef CAS PubMed.
- S. M. Williams, A. V. Chandran, M. S. Vijayabaskar, S. Roy, H. Balaram, S. Vishveshwara, M. Vijayan and D. Chatterji, A histidine aspartate ionic lock gates the iron passage in miniferritins from Mycobacterium smegmatis, J. Biol. Chem., 2014, 289, 11042–11058 CrossRef CAS PubMed.
- E. Chiancone and P. Ceci, The multifaceted capacity of Dps proteins to combat bacterial stress conditions: Detoxification of iron and hydrogen peroxide and DNA binding, Biochim. Biophys. Acta, 2010, 1800, 798–805 CrossRef CAS PubMed.
- G. Khare, P. Nangpal and A. K. Tyagi, in Frontiers in Protein Structure, Function, and Dynamics, ed. D. B. Singh and T. Tripathi, Springer Singapore, Singapore, 2020, pp. 425–452, DOI:10.1007/978-981-15-5530-5_17.
- A. Mohanty, A. Parida, R. K. Raut and R. K. Behera, Ferritin: A Promising Nanoreactor and Nanocarrier for Bionanotechnology, ACS Bio & Med Chem Au, 2022, 2, 258–281 Search PubMed.
- M. Uchida, M. L. Flenniken, M. Allen, D. A. Willits, B. E. Crowley, S. Brumfield, A. F. Willis, L. Jackiw, M. Jutila, M. J. Young and T. Douglas, Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles, J. Am. Chem. Soc., 2006, 128, 16626–16633 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, T. Sho, S. Ozeki, S. Abe, T. Hikage, T. Kuchimaru, S. Kizaka-Kondoh and T. Ueno, Intracellular CO release from composite of ferritin and ruthenium carbonyl complexes, J. Am. Chem. Soc., 2014, 136, 16902–16908 CrossRef CAS PubMed.
- J. L. Prelesnik, R. G. Alberstein, S. Zhang, H. Pyles, D. Baker, J. Pfaendtner, J. J. De Yoreo, F. A. Tezcan, R. C. Remsing and C. J. Mundy, Ion-dependent protein-surface interactions from intrinsic solvent response, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(26), e2025121118 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: DLS based size distribution analysis, kinetic parameters of iron oxidation kinetics, thermodynamic parameters of chemical and thermal unfolding, elution profile after size exclusion chromatography, UV-vis absorption spectra of oxidized and reduced WT BfrA and its variants, kinetics of sequential and manual iron oxidation, H2O2 quantification by peroxidase assay, and thermal unfolding data. See DOI: https://doi.org/10.1039/d4sc07021f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |