
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic peptide macrocyclization via indole-N-acylation†
Hiroto
Maruyama
a,
Yuito
Yamada
a,
Yasuhiro
Igarashi
b,
Kenichi
Matsuda
 *a and
Toshiyuki
Wakimoto
*a
*a and
Toshiyuki
Wakimoto
*a
aFaculty of Pharmaceutical Sciences, Hokkaido University, Kita 12, Kita-ku, Sapporo, 060-0812, Japan. E-mail: kematsuda@pharm.hokudai.ac.jp; wakimoto@pharm.hokudai.ac.jp
bBiotechnology Research Center and Department of Biotechnology, Toyama Prefectural University, 5180 Kurokawa, Imizu, Toyama 939-0398, Japan
First published on 28th January 2025
Abstract
Indole N-acylation is chemically challenging, due to the low nucleophilicity of the indole nitrogen. Although a few similar transformations have been proposed in the biosynthesis of indole-containing natural products, their enzymatic basis remains elusive. Here, we show that BulbE TE is an N-acylindole-forming macrocyclase involved in the biosynthesis of the non-ribosomal cyclopeptide bulbiferamide. BulbE catalyzed macrocyclization not only via the indole nitrogen, but also via a primary amine and an alcohol. The uncommon catalytic residue Cys731 in BulbE TE was indispensable for the nucleophilic attack from the indole nitrogen. While the C731S variant failed to utilize the indole nitrogen and primary alcohol as nucleophiles, it retained the ability to employ the amine nucleophile, showing a clear correlation between the catalytic residues and the nucleophile scope. A model of the acyl–enzyme complex revealed how the substrate is recognized, including interactions involving a unique second lid-like structural motif in BulbE TE. This study provides an enzymatic basis for indole N-acylation and offers important insights into the nucleophile specificity in TE-mediated macrocyclization.
Introduction
N-Acylindole moieties are found in biologically active molecules, including synthetic drugs like indomethacin,1 as well as indole alkaloids derived from bacteria, fungi and plants.2 Indole N-acylation is a challenging chemical transformation due to the inherently lower nucleophilicity of the indole nitrogen, arising from the participation of its lone pair electrons in the aromatic system. Consequently, various chemical methodologies have been intensively developed to address this challenge.3–10The occurrence of indole N-acylation in nature remains largely elusive. It is hypothesized in the biosynthesis of plant-derived indole alkaloids such as correantoside11 and strynuxlines (Fig. 1 and S1†).12 Indole N-acylation is also proposed in the biosynthesis of fungal peptides, including the tetracyclic-fused peptide “compound 14”13 and the rare N-acylindole linkage-containing tripeptide psychrophilin (Fig. 1 and S1†).14 In 2016, Zhao et al. reported the biosynthetic gene cluster (BGC) of psychrophilin in Penicillium rivulum.15 Although macrocyclization via indole N-acylation is hypothesized to be mediated by a C-terminal condensation-like domain, its molecular basis remains unclear.
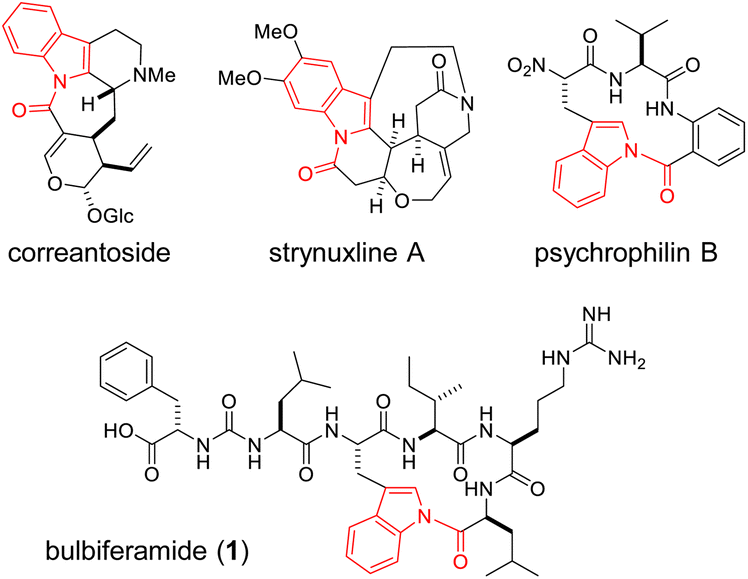 | ||
| Fig. 1 Structures of representative N-acylindole-containing natural products. | ||
Bulbiferamide (1) belongs to a recently discovered group of cyclic peptides produced by the marine obligate bacterium, Microbulbifer spp. (Fig. 1).16,17 It exhibits inhibitory activity against Trypanosoma cruzi, a protozoan that causes Chagas disease.16 Bulbiferamide has a ureido-containing pseudo-C-terminal tail, where the terminal Phe1 is inverted to expose its carboxylic acid due to the presence of the ureido backbone, and a C-terminal tetrapeptidyl cyclic head group, bridged by a very rare N-acylindole linkage. In 2023, Zhong et al. independently discovered 1 and its analogs from Microbulbifer sp., and identified their BGC (bulb), which contains six open reading frames, bulbA–F.17 While bulbF encodes a putative transporter, bulbA–E encode non-ribosomal peptide synthetases (NRPSs) to assemble the bulbiferamide sequence, including the backbone ureido moiety (Fig. S2†). Therefore, the N-acylindole is proposed to be installed by the C-terminal thioesterase domain of BulbE (BulbE TE),17 an α/β hydrolase fold domain generally responsible for product release via cyclization or hydrolysis in thio-templated biosynthesis,18–21 even though the detailed mechanism of the intriguing N-acylindole formation in bulbiferamide biosynthesis remains elusive, as in the case of psychrophilin.
TE catalysis involves a two-step mechanism: substrate loading and release. In the loading step, a catalytic Ser, which is activated by a His–Asp dyad, attacks the thioester intermediate tethered on a cognate carrier protein domain, leading to the formation of the acyl-O-enzyme complex. The succeeding release step can vary: attacks by exogenous nucleophiles generate linear products, whereas attacks by intramolecular nucleophiles generate cyclic products. Various nucleophiles—including primary and secondary amines, primary, secondary, and phenolic alcohols, thiols, and activated methylene groups—can be selectively incorporated in the TE-mediated cyclization step, even though the nucleophile selectivity mechanism remains to be elucidated.20,21 Furthermore, the use of the indole nitrogen for nucleophilic addition/substitution is unprecedented—not only in TE-mediated cyclization but, to our knowledge, in any experimentally validated enzymatic transformation.
Results and discussion
To assess the enzymatic basis of the N-acylindole linkage, the acyclic precursor 2, seco-bulbiferamide with a C-terminal pantetheine surrogate SNAC (N-acetyl cysteamine), was synthesized via the scheme shown in Fig. 2a. Briefly, Fmoc-L-Leu-OH, which was loaded on Trt(2-Cl) resin, was subjected to four rounds of piperidine-mediated deprotection and DIC/Oxyma-mediated coupling to generate the resin-bound pentapeptide 5. The isocyanate 7 was coupled with 5 to construct the ureido moiety, and after the resultant hexapeptide was cleaved from the resin, it was coupled with SNAC. Finally, the protecting groups were removed to afford 2. The BulbE TE from Microbulbifer sp. MLAF003 was expressed in E. coli as a PCP/TE didomain, and then 20 µM of enzyme was incubated with 200 µM of 2 at 25 °C for 12 hours. HPLC analysis of enzymatic reaction mixtures showed that 2 was converted to 1 and its linear counterpart hyd-2, with a cyclization![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hydrolysis ratio of 1:1.6 (Fig. 2b, trace i). This result demonstrates that BulbE TE is the first macrocyclase that catalyses peptide macrocyclization via indole-N-acylation. The high flux of hydrolysis may be due to the unnatural conditions surrounding BulbE TE in vitro, including the use of SNAC, a low-molecular-weight pantetheine surrogate, and the excision of BulbE TE from its native modular architecture.
:hydrolysis ratio of 1:1.6 (Fig. 2b, trace i). This result demonstrates that BulbE TE is the first macrocyclase that catalyses peptide macrocyclization via indole-N-acylation. The high flux of hydrolysis may be due to the unnatural conditions surrounding BulbE TE in vitro, including the use of SNAC, a low-molecular-weight pantetheine surrogate, and the excision of BulbE TE from its native modular architecture.
 | ||
| Fig. 2 In vitro characterization of BulbE TE. (a) Synthetic scheme of seco-bulbiferamide-SNAC (2). (b) HPLC analysis of the in vitro enzymatic reaction of BulbE TE (analytical conditions 2 in Table S5†). The structure of the pseudo-C-terminal region is omitted (R = Phe1-ureido-Leu2) (c–e). In vitro enzymatic reactions using variant substrates 9–11 (9 and 10: analytical conditions 2 in Table S5,†11: analytical conditions 3 in Table S5†). The nucleophile residues are highlighted in red. The structure of the pseudo-C-terminal region is omitted (R = Phe1-ureido-Leu2). The inset in panel c represents a 10× magnified view of the region enclosed by the rectangle highlighted with an asterisk (*). These reaction mixtures were also analysed by LC-MS (Fig. S3–S5†). | ||
The use of enzymes is a promising approach for site-selective peptide macrocyclization, which has otherwise remained challenging through chemical methodologies that often require multiple protecting groups and are accompanied by yield-decreasing side reactions such as epimerization and oligomerization.22,23 Among the known peptide cyclase families, NRPS TEs are particularly notable for the wide variety of nucleophiles they utilize; however, the nucleophile scopes of TEs and the molecular basis underlying their nucleophile specificities remain largely unknown. Since the Trp-utilizing BulbE TE is functionally distinct from other TEs in terms of nucleophile selection, we investigated the scope of BulbE TE toward nucleophiles. To this end, we synthesized compound 9, a variant of 2 in which Trp was substituted with Orn. BulbE TE macrolactamized 9 to afford cyc-9, with a cyc:hyd ratio of 1:0.9 (Fig. 2c trace i, S3†). Variant 10, in which Trp was substituted with Ser, was also cyclized to generate cyc-10, a 13-membered macrolactone, although the amount of cyclic product substantially decreased (cyc:hyd ratio of 1:6.2) (Fig. 2d trace i, S4†). As the ring size of cyc-10 is considerably smaller than that of 1, with a 15-member ring, we tested a substrate variant that would afford a product with a ring size identical to 1. To this end, variant 11 with 5-OH Nva (Norvaline) was synthesized and subjected to BulbE TE cyclization. As a result, 11 was cyclized with improved conversion (cyc:hyd ratio of 2:1) (Fig. 2e trace i, S5†). We further assessed the tolerance of BulbE TE for other potentially nucleophilic residues, including Tyr (12), His (13), Arg (14) and Asn (15). These variants equally underwent hydrolysis, but not cyclization (Fig. S6†). Overall, these results show that BulbE TE accepts not only the native indole nitrogen, but also tolerates primary amines and alcohols, showing its broad tolerance for nucleophile species in macrocyclization.
BulbE TE features the less common Cys as the catalytic residue, instead of the canonical Ser. Enzymes with catalytic Cys residues often catalyse unusual chemical transformations. For instance, ObiF in obafluorin biosynthesis catalyses β-lactone formation,24 while SulM in sulfazecin biosynthesis catalyses β-lactam formation,25 and both require catalytic Cys residues for their catalysis. PMB TE in polymyxin biosynthesis catalyses macrolactamization with a catalytic Cys, and the Cys-to-Ser mutation decreased the activity 60-fold.26 Conversely, the opposite Ser-to-Cys mutation has occasionally been reported to enhance catalytic properties. For example, the Ser-to-Cys mutation of Pik TE in pikromycin biosynthesis improved the catalytic efficiency and broadened the stereochemical configuration scope of the alcohol nucleophile.27 Similarly, the Ser-to-Cys mutant of MycC TE in microcystin biosynthesis improved the catalytic efficiency.28 These studies collectively demonstrate the profound impact of the type of catalytic residue (i.e. Ser or Cys) on the TE function.
To assess the importance of the catalytic Cys in BulbE TE-mediated indole-N-acylation, it was mutated to Ser. As a result, the mutant BulbE TEC731S lost the ability to convert 2 to 1, but converted 2 to hyd-2 (Fig. 2b trace ii). The hydrolytic flux indicated that BulbE TEC731S retained the ability to form the peptide-O-enzyme complex but the subsequent cyclization was impaired, highlighting the importance of the catalytic Cys residue for macrocyclizing indole-N-acylation. Furthermore, to determine whether the type of catalytic residue impacts the nucleophile scope of BulbE TE, the BulbE TEC731S mutant was tested with the nucleophile variants 9–15. The C731S mutant generated a small yet clearly detectable amount of cyc-9 (Fig. 2c trace ii, S3†), indicating that the catalytic Cys of BulbE TE is dispensable for macrocyclization via a primary amine. In contrast, the C731S mutant abolished the ability to macrolactonize 10 and 11 (trace ii in Fig. 2d, e, S4 and S5†), demonstrating the narrowed nucleophile scope of the C731S mutant. To our knowledge, this is the first demonstration of a catalytic residue being a determinant for the nucleophile scope in TE-mediated macrocyclization.
To gain insight into the structural basis of the macrocyclization via indole N-acylation, the structure model of BulbE TE was generated by AlphaFold2.29 BulbE TE has an α/β hydrolase fold30 with seven β-sheets (β2–β8), with only β2 being antiparallel, sandwiched by six α-helixes (Fig. 3a). As typical for TEs, BulbE TE possesses a so-called ‘lid region’, a large insertion between β6 and β7, which consists of three helixes (αL1–αL3). Notably, BulbE TE features an unusually long insertion between β6 and αE, which we refer to as the ‘second lid’ (αL4–αL5), as it plays a significant role in shaping the substrate binding pocket. A multiple structure alignment by FoldMason31 using nine crystal structures of NRPS TEs24,25,32–37 clearly indicated that the second lid region is unique to BulbE TE (Fig. S7 and S8†). The nucleophilic elbow with the catalytic Cys is located at the entrance of a putative, bowl-shaped substrate binding pocket. The Cys interacted with a second catalytic residue, the His in the loop after β8. A Foldseek server search38 of the model showed that BulbE TE is most structurally similar to Pks13 TE (aaid = 27%) in Mycobacterium tuberculosis, an essential component of mycolic acid biosynthesis that is thought to be a promising anti-tuberculosis drug target.39 BulbE TE is also structurally similar to NocB TE (aaid = 28%), a unique bifunctional TE in nocardicin biosynthesis that catalyses epimerization/hydrolysis.32 BulbE TE was less superimposable on other macrocyclizing NRPS TEs, such as Srf TE in surfactin biosynthesis33 and Fen TE in fengycin biosynthesis,34 underlining the divergence of structure and function in the TE family.
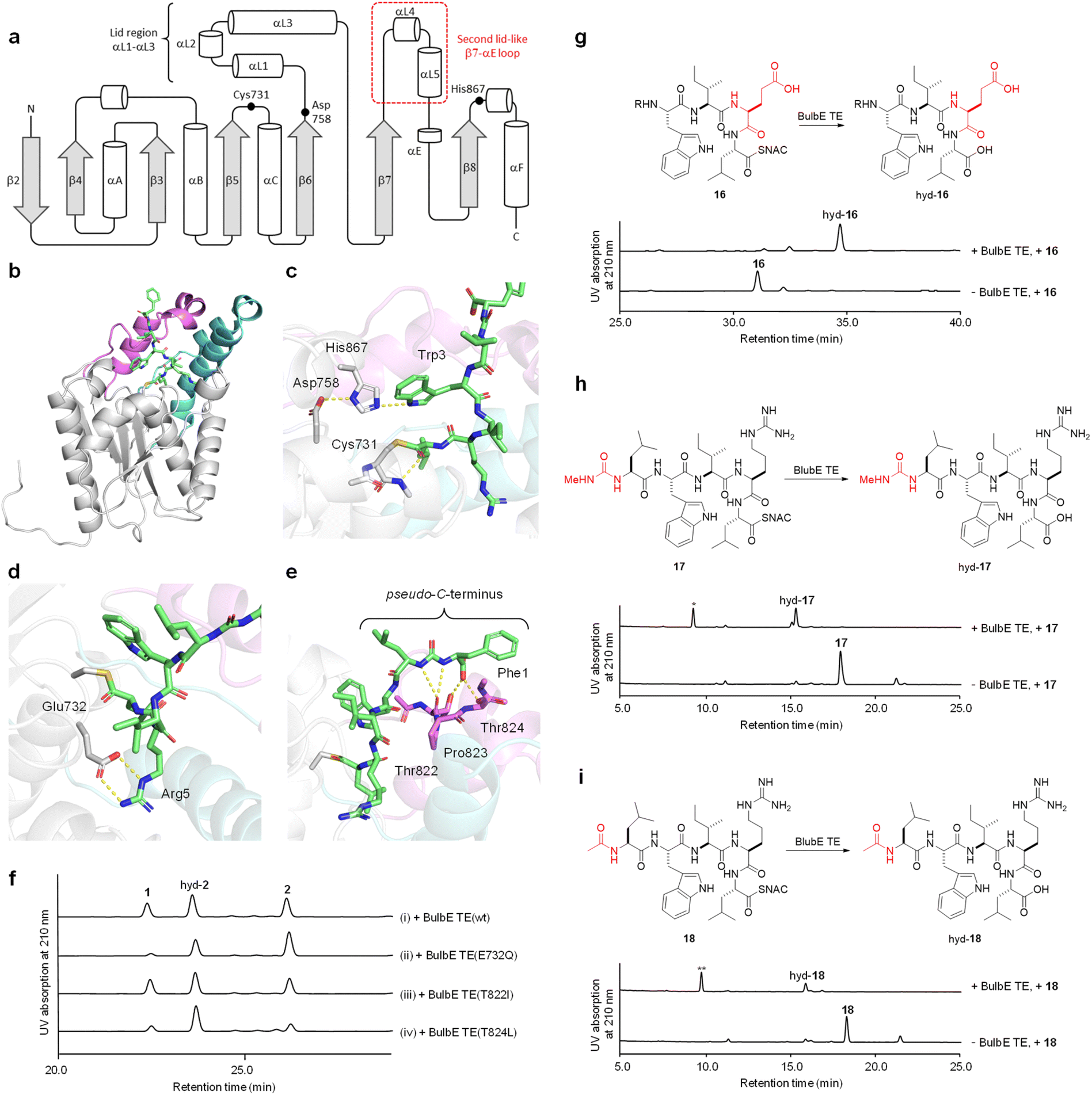 | ||
| Fig. 3 Substrate recognition of BulbE TE. (a) Topology diagram of BulbE TE secondary structure elements. (b) Acyl–enzyme complex model of BulbE TE. The structure of BulbE TE was predicted by AlphaFold2. The lid region (between β6 and β7) and the second lid-like loop (between β7 and αE) are colored cyan and magenta, respectively. (c) The active site of BulbE TE. The catalytic triad and tethered peptide are shown as sticks. The hydrogen-bond network is depicted as yellow dots. (d) The salt bridge between Arg5 of seco-bulbiferamide and Glu732 of BulbE TE. (e) Interaction between the pseudo-C-terminus of tethered seco-bulbiferamide and the second lid of BulbE TE. (f) HPLC analysis of in vitro enzymatic reactions of BulbE TE mutants (analytical conditions 2 in Table S5†). (g–i) In vitro enzymatic reactions using variant substrates 16–18 (analytical conditions 2 in Table S5†). The portions that differ from 2 are highlighted in red. *, **: these byproducts were likely generated by trace amounts of contaminating proteases from the E. coli host. MS2 analysis of these byproducts revealed that they are N-terminal fragments of 17/18, resulting from peptide bond cleavage between Trp and Ile. The MS2 spectra of these byproducts are shown in Fig. S28 and S29.† | ||
To further clarify the molecular basis of BulbE TE catalysis, we constructed a model of the acyl–enzyme complex based on the AlphaFold model using AutoDock Vina,40 and then relaxed it by a 20 ns molecular dynamics simulation using GROMACS41 (Fig. 3b and S9–S11†). Several interactions between BulbE TE and the tethered peptide were observed in the trajectory (Fig. S10†). The tethered peptide was tightly bent and accommodated within the bowl-shaped binding pocket. The pocket was occupied by the C-terminal Leu6 (counting pseudo-C-terminal Phe as residue 1) and Arg5, which formed a salt-bridge with Glu732 in the binding pocket (Fig. 3d). Ile4 was directed toward the solvent, and the nucleophile indole nitrogen of Trp3 was captured by the His of the catalytic triad (Fig. 3c). This interaction suggests the key role of His in activating a non-reactive indole nitrogen during indole N-acylation. While Leu2 is directed toward the solvent, the ureido moiety interacted with the backbone oxygen of Thr822. Notably, the carboxylic group of the pseudo-C-terminus Phe1 hydrogen-bonded with the β-OHs of two Thr residues, Thr822 and Thr824, located in the unique second lid of BulbE TE (Fig. 3e). Based on this model, we assessed the importance of the salt-bridge-forming Glu732 and the two Thr residues in the second lid, which are putatively involved in the recognition of the pseudo-C-terminus.
First, Glu732, which was thought to interact with the intermediate Arg5, was mutated to Gln. As a result, the cyc:hyd ratio shifted from 1:1.6 (wt) to 1:6.6 (E732Q) (Fig. 3f trace ii). The increased hydrolytic flux indicates that the salt bridge between Arg5 and Glu732 plays an important role in the cyclization step. To further assess the importance of the Arg5–Glu732 interaction, the variant substrate 16, in which Arg5 was substituted with Glu, was incubated with wild type BulbE TE. As a result, no cyclized product of 16 was observed (Fig. 3g), further supporting the importance of the Arg5–Glu732 interaction in BulbE TE catalysis.
Next, we investigated the role of the two residues Thr822 and Thr824 in the second lid, which were suggested to be involved in the recognition of the pseudo-C-terminus. To this end, both Thr residues were individually mutated to β-OH-lacking aliphatic residues, Ile and Leu, to generate BulbE TET822I and BulbE TET824L, respectively. The enzymatic reactions revealed that the T822I mutation had a negligible impact on the conversion (cyc:hyd ratio 1:1.5) (Fig. 3f trace iii), while the T824L mutation decreased the amount of 1 (cyc:hyd ratio 1:4.6) (Fig. 3f trace iv). This indicated that Thr824, rather than Thr822, plays an important role in the recognition on the pseudo-C-terminus. To further evaluate the importance of this interaction, the substrate variant 17, in which the pseudo-C-terminal Phe1 was substituted with a methyl group, and another variant 18, in which the Phe1-ureido portion was substituted with an acetyl group, were subjected to the BulbE TE reaction. As a result, both substrates were not cyclized, but hydrolysed (Fig. 3h and i), showing the crucial role of the pseudo-C-terminus of 2 for BulbE TE recognition. Notably, 18 was recently synthesized independently by Zhong et al. and subsequently cyclized by BulbE TE with 31% conversion.42 The inconsistent results may be attributed to variations in assay conditions; however, this will be the focus of future investigations.
Conclusions
In conclusion, BulbE TE catalyses an unprecedented indole N-acylation, using an uncommon Cys catalytic residue. The use of a more reactive acyl donor (i.e. thioester) compared to conventional oxoesters closely resembles strategies in organic synthesis, where the thioester acyl donor facilitates the attack from a weak nucleophile, the indole nitrogen.8 BulbE TE exhibits a broad nucleophile scope, tolerating not only the native indole nitrogen but also primary alcohols and amines. The Cys-to-Ser mutation renders BulbE TE specific to primary amine nucleophiles, which are more nucleophilic than the indole nitrogen, clearly highlighting the impact of this catalytic residue on the nucleophile scope. Overall, this study provides an enzymatic basis for indole N-acylation in nature and sets the stage for further explorations of the molecular basis of substrate specificity in TEs, as well as their potential biocatalytic applications for the synthesis of N-acylindole-containing macrocycles.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. M. and T. W. designed the whole experiment. H. M., Y. Y. and K. M. performed the experiments and data analysis. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by Hokkaido University, Global Facility Center (GFC), Pharma Science Open Unit (PSOU), funded by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) under “Support Program for Implementation of New Equipment Sharing System”, Global Station for Biosurfaces and Drug Discovery, a project of Global Institution for Collaborative Research and Education in Hokkaido University, Japan Foundation for Applied Enzymology, TERUMO Life Science Foundation, the Japan Agency for Medical Research and Development (JP24gm1610007, and JP24ama121039), Grants-in-Aid from MEXT, the Japan Science and Technology Agency (JST Grant Number ACT-X JPMJAX201F, FOREST JPMJFR233U, A-STEP JPMJTR24U6 and SPRING JPMJSP2119), and JSPS KAKENHI (Grant Numbers JP21H02635, JP22K15302, JP22H05128, JP23K17410, and JP24K01659).Notes and references
- T. Y. Shen, T. B. Windholz, A. Rosegay, B. E. Witzel, A. N. Wilson, J. D. Willett, W. J. Holtz, R. L. Ellis, A. R. Matzuk, S. Lucas, C. H. Stammer, F. W. Holly, L. H. Sarett, E. A. Risley, G. W. Nuss and C. A. Winter, J. Am. Chem. Soc., 1963, 85, 488–489 CrossRef CAS.
- Dictionary of Natural Products, 31.1, CRC Press, https://www.dnp.chemnetbase.com/faces/chemical/ChemicalSearch.xhtml, accessed 11 October 2024 Search PubMed.
- M. Terashima and M. Fujioka, Heterocycles, 1982, 19, 91–92 CrossRef CAS.
- B. B. Snider and H. Zeng, J. Org. Chem., 2003, 68, 545–563 CrossRef CAS PubMed.
- S. T. Heller, E. E. Schultz and R. Sarpong, Angew. Chem., 2012, 51, 8304–8308 CrossRef CAS PubMed.
- J. Woo, A. H. Christian, S. A. Burgess, Y. Jiang, U. F. Mansoor and M. D. Levin, Science, 2022, 376, 527–532 CrossRef CAS PubMed.
- A. Umehara, H. Ueda and H. Tokuyama, J. Org. Chem., 2016, 81, 11444–11453 CrossRef CAS PubMed.
- T. Du, X. Wei, H. Xu, X. Zhang, R. Fang, Z. Yuan, Z. Liang and Y. Li, J. Org. Chem., 2022, 18, 89–94 CAS.
- A. Umehara, S. Shimizu and M. Sasaki, ChemCatChem, 2023, 15, e202201596 CrossRef CAS.
- A. Umehara, S. Shimizu and M. Sasaki, Adv. Synth. Catal., 2023, 365, 2367–2376 CrossRef CAS.
- A. Berger, K. Valant-Vetschera, J. Schinnerl and L. Brecker, Phytochem. Rev., 2022, 21, 915–939 CrossRef CAS.
- Y. Fu, Y. Zhang, H. He, L. Hou, Y. Di, S. Li, X. Luo and X. Hao, J. Nat. Prod., 2012, 75, 1987–1990 CrossRef CAS PubMed.
- W. L. Liang, X. Le, H. J. Li, X. L. Yang, J. X. Chen, J. Xu, H. L. Liu, L. Y. Wang, K. T. Wang, K. C. Hu, D. P. Yang and W. J. Lan, Mar. Drugs, 2014, 12, 5657–5676 CrossRef CAS PubMed.
- P. W. Dalsgaard, T. O. Larsen, K. Frydenvang and C. Christophersen, J. Nat. Prod., 2004, 67, 878–881 CrossRef CAS PubMed.
- M. Zhao, H. C. Lin and Y. Tang, J. Antibiot., 2016, 69, 571–573 CrossRef CAS PubMed.
- S. Lu, Z. Zhang, A. R. Sharma, J. Nakajima-Shimada, E. Harunari, N. Oku, A. Trianto and Y. Igarashi, J. Nat. Prod., 2023, 86, 1081–1086 CrossRef CAS PubMed.
- W. Zhong, J. M. Deutsch, D. Yi, N. H. Abrahamse, I. Mohanty, S. G. Moore, A. C. McShan, N. Garg and V. Agarwal, ChemBioChem, 2023, 24, e202300190 CrossRef CAS PubMed.
- M. E. Horsman, T. P. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- R. E. Little and C. Hertweck, Nat. Prod. Rep., 2022, 39, 163–205 RSC.
- M. L. Adrover-Castellano, J. J. Schmidt and D. H. Sherman, ChemCatChem, 2021, 13, 2095–2116 CrossRef CAS PubMed.
- K. Matsuda, J. Nat. Med., 2025, 79, 1–14 CrossRef PubMed.
- R. Wills, V. Adebomi and M. Raj, ChemBioChem, 2021, 22, 52–62 CrossRef CAS PubMed.
- S. Qiao, Z. Cheng and F. Li, Beilstein J. Org. Chem., 2024, 20, 721–733 CrossRef CAS PubMed.
- D. F. Kreitler, E. M. Gemmell, J. E. Schaffer, T. A. Wencewicz and A. M. Gulick, Nat. Commun., 2019, 10, 3432 CrossRef PubMed.
- K. D. Patel, R. A. Oliver, M. S. Lichstrahl, R. Li, C. A. Townsend and A. M. Gulick, J. Biol. Chem., 2024, 300, 107489 CrossRef CAS PubMed.
- C. A. Galea, K. D. Roberts, Y. Zhu, P. E. Thompson, J. Li and T. Velkov, Biochemistry, 2017, 56, 657–668 CrossRef CAS PubMed.
- A. A. Koch, D. A. Hansen, V. V. Shende, L. R. Furan, K. N. Houk, G. Jiménez-Osés and D. H. Sherman, J. Am. Chem. Soc., 2017, 139, 13456–13465 CrossRef CAS PubMed.
- L. Qiao, J. Fang, P. Zhu, H. Huang, C. Dang, J. Pang, W. Gao, X. Qiu, L. Huang and Y. Li, Protein J., 2019, 38, 658–666 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- D. L. Ollis, E. Cheah, M. Cygler, B. Dijkstra, F. Frolow, S. M. Franken, M. Harel, S. J. Remington, I. Silman and J. Schrag, Protein Eng., 1992, 5, 197–211 CrossRef CAS PubMed.
- C. L. M. Gilchrist, M. Mirdita and M. Steinegger, bioRxiv, 2024, preprint, DOI: DOI:10.1101/2024.08.01.606130.
- K. D. Patel, F. B. d'Andrea, N. M. Gaudelli, A. R. Buller, C. A. Townsend and A. M. Gulick, Nat. Commun., 2019, 10, 3868 CrossRef PubMed.
- S. D. Bruner, T. Weber, R. M. Kohli, D. Schwarzer, M. A. Marahiel, S. T. Walsh and M. T. Stubbs, Structure, 2002, 10, 301–310 CrossRef CAS PubMed.
- S. A. Samel, B. Wagner, M. A. Marahiel and L. O. Essen, J. Mol. Biol., 2006, 359, 876–889 CrossRef CAS PubMed.
- N. Huguenin-Dezot, D. A. Alonzo, G. W. Heberlig, M. Mahesh, D. P. Nguyen, M. H. Dornan, C. N. Boddy, T. M. Schmeing and J. W. Chin, Nature, 2019, 565, 112–117 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. Leigh Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- J. Yu, J. Song, C. Chi, T. Liu, T. Geng, Z. Cai, W. Dong, C. Shi, X. Ma, Z. Zhang, X. Ma, B. Xing, H. Jin, L. Zhang, S. Dong, D. Yang and M. Ma, ACS Catal., 2021, 11, 11733–11741 CrossRef CAS.
- M. van Kempen, S. S. Kim, C. Tumescheit, M. Mirdita, J. Lee, C. L. M. Gilchrist, J. Söding and M. Steinegger, Nat. Biotechnol., 2024, 42, 243–246 CrossRef CAS PubMed.
- A. Aggarwal, M. K. Parai, N. Shetty, D. Wallis, L. Woolhiser, C. Hastings, N. K. Dutta, S. Galaviz, R. C. Dhakal, R. Shrestha, S. Wakabayashi, C. Walpole, D. Matthews, D. Floyd, P. Scullion, J. Riley, O. Epemolu, S. Norval, T. Snavely, G. T. Robertson, E. J. Rubin, T. R. Ioerger, F. A. Sirgel, R. van der Merwe, P. D. van Helden, P. Keller, E. C. Böttger, P. C. Karakousis, A. J. Lenaerts and J. C. Sacchettini, Cell, 2017, 170, 249–259 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- W. Zhong, Z. L. Budimir, L. O. Johnson, E. I. Parkinson and V. Agarwal, Org. Lett., 2024, 26, 9378–9382 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07839j |
| This journal is © The Royal Society of Chemistry 2025 |