
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeyond scaling relations in electrocatalysis: unifying concepts from molecular systems and metallic surfaces†
Kirstine Nygaard
Kolding
 ab,
Kristian
Torbensen
c and
Alonso
Rosas-Hernández
*abc
ab,
Kristian
Torbensen
c and
Alonso
Rosas-Hernández
*abc
aDepartment of Chemistry, Aarhus University, Langelandsgade 140, 8000 Aarhus C, Denmark. E-mail: arosas@chem.au.dk
bCarbon Dioxide Activation Center (CADIAC), Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Gustav Wieds Vej 14, 8000 Aarhus C, Denmark
cNovo Nordisk Foundation (NNF) CO2 Research Center, Aarhus University, Gustav Wieds Vej 10C, 8000 Aarhus C, Denmark
First published on 26th February 2025
Abstract
Transitioning the chemical industry away from fossil fuels is a critical goal that requires the adoption of alternative, non-fossil carbon feedstocks. The electrochemical CO2 reduction reaction, driven via renewable-derived electricity, represents an unparalleled technology that uses CO2 as a C1-building block to generate industrially relevant products. Although many electrocatalytic systems have demonstrated promising activities in producing a wide range of products, challenges remain in controlling the product selectivity and reducing the operating overpotential for large-scale applications. This Perspective outlines recent efforts in designing tailored microenvironments in electrocatalytic systems to boost their selectivity and energy efficiency. We review examples from homogeneous and heterogeneous systems, emphasizing mechanistic studies that elucidate how the modulation of the space surrounding catalytic active sites can control the outcome of electrocatalysis. Lastly, we carry out a thermodynamic–kinetic analysis to identify existing scaling relationships that govern the electrocatalytic performance of molecular catalysts, and we highlight examples of catalysts that circumvent these relations through the functionalization of their secondary coordination sphere.
Introduction
The chemical industry has significantly contributed to the rapid societal development over the past century by providing solutions in areas as diverse as energy, agriculture, health, construction, and information technology.1 However, due to its dependence on fossil fuels, this sector has a substantial carbon footprint, emitting 1.33 gigatonnes (Gt) of CO2 in 2022, which accounted for about 4% of total global CO2 emissions.2 According to various scenarios modeled by the Intergovernmental Panel on Climate Change (IPCC), global net-zero carbon emissions must be achieved by around 2050 to limit global warming to 1.5 °C above pre-industrial levels.3 Therefore, reducing the reliance of the chemical industry on fossil fuels is crucial for transitioning toward a stable and sustainable future. It is important to note that although this industry subsector is the largest consumer of oil and gas, it is only the third largest industrial emitter of CO2 because approximately half of the fossil fuel input is used as feedstock.4 This indicates that electrifying energy sources or deploying carbon capture and storage technologies alone would not completely eliminate the dependence of the chemical sector on fossil carbon. Accordingly, using renewable carbon as a raw input material is critical to curbing CO2 emissions during the large-scale production of primary chemicals.5,6When powered by green electricity, the electrochemical CO2 reduction reaction (CO2RR) is a unique technology capable of a near de-fossilization of the chemical industry by producing several of its essential building blocks entirely from renewable feedstocks, such as water and CO2.7,8 Whilst the feasibility of electrochemically upgrading CO2 has been fully demonstrated, the primary challenge for the market deployment of this technology lies in developing catalytic systems with improved activity and selectivity toward specific products. Such improvements are essential to increase the overall energy efficiency and reduce production costs.9 In recent years, the use of sophisticated in situ and operando techniques,10 theoretical models,11 and electrochemical methods12 has enabled a deeper understanding of the underlying principles governing the overall kinetics of this reaction. These comprehensive mechanistic studies have highlighted the limitations in developing novel electrocatalytic systems with tailored reactivity to accelerate specific CO2RR pathways.
For example, the upper limit catalytic performance of any novel material is ultimately determined by scaling relations. Based on empirical observations, Brønsted, Evans, and Polanyi were the first to identify correlations between the activation energies of a series of reactions with structurally similar reactants and the overall reaction energies.13,14 These scaling relations between kinetic and thermodynamic parameters provide a conceptual framework for understanding the typical volcano-type reactivity of catalytic materials. Within this framework, a unique thermodynamic descriptor (e.g., the binding energy of a reaction intermediate) can explain the displayed efficiency of distinct catalysts.15,16 Most recently, Nørskov and coworkers extended this framework by identifying scaling relations among the binding energies of reaction intermediates on transition metal surfaces.17 These linear correlations are found among adsorbates bonded to the metal surface through the same type of atoms, i.e., C, H, O, N, or S.18
In complex reaction networks like CO2RR, the overall reaction rate is dictated by both the energy barrier and the reaction energy of the distinct elementary steps along the reaction coordinate. Interestingly, since the activation energies are linearly correlated with the overall reaction energies and the adsorption energies of different intermediates are interrelated, the dimensionality of the system can be simplified. These energy correlations often lead to significantly fewer independent energies than expected in a non-correlated system. As a result, the kinetics of the catalyzed reaction can be expressed using a smaller set of fundamental independent descriptors. Identifying these descriptors and understanding how they control the reaction kinetics is central to catalysis science. This approach enables us to dissect the trends observed across various catalytic systems, paving the way for a descriptor-driven design of enhanced catalytic materials. Such materials may exhibit descriptors close to maximum activity or, crucially, bypass current correlations to advance the catalytic performance even further.
The chemical architecture of the space surrounding the active sites of enzymes, also called the microenvironment, has commonly served as a template for designing homogeneous19 and, more recently, heterogeneous electrocatalysts.20 The main feature of the microenvironments is the existence of organic residues precisely positioned to interact with reaction intermediates bound to the metal active sites (Fig. 1). For instance, the structure of the active site of the [FeFe] hydrogenase enzyme displays a pendant amine group functioning as a proton relay, thus facilitating both the formation and cleavage of H–H bonds at the iron active center (Fig. 1c).21 This proton relay has been proposed to be the origin of reported rate constants as high as 9000 s−1 during the catalytic formation of H2 from water.22
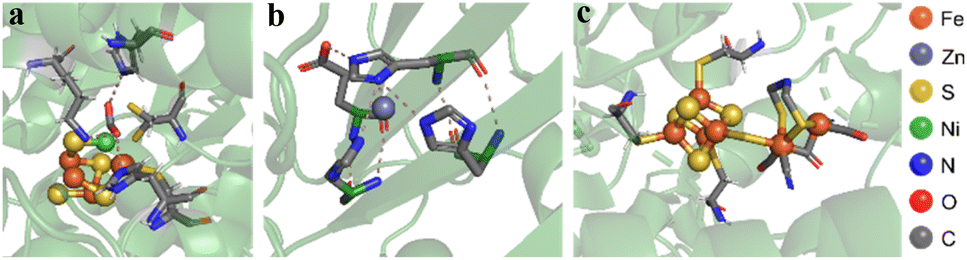 | ||
| Fig. 1 Microenvironments of distinct enzymes. (a) [NiFe]–CO dehydrogenase displays a [3Fe–4S] cluster bonded to the Ni–Fe moiety. The Ni apex binds the C atom of the CO2 substrate, while the acidic Fe center binds to the O atom. (Protein Data Bank identifier: 4UDX). (b) Carbonic anhydrase features a Zn(II) atom coordinated to three histidine side-chains and a fourth histidine shuttling protons in and out of the active site via conformational switching (Protein Data Bank identifier: 1CA2). (c) [FeFe] hydrogenase contains a cubane-shaped [4Fe4S] cluster coupled to a low valent di-iron center connected by a bridging thiolate ligand. The pendant secondary amine of this ligand can relay protons, facilitating H2 activation at one of the Fe centers (Protein Data Bank identifier: 4XDC). | ||
This Perspective focuses on the use of synthetic tailored microenvironments as a strategy to circumvent scaling relations in CO2RR. We describe selected examples of molecular and heterogeneous electrocatalysts in which mechanistic investigations have established the role of organic functionalities in accelerating catalytic reaction rates for specific products. In the last section, we identify existing scaling relations in the CO2RR through quantitative data and show which molecular systems escape from such correlations. Based on this analysis, we draw mechanistic parallels between molecular and heterogeneous electrocatalysts to circumvent scaling relations using tailored microenvironments.
CO2RR with homogeneous electrocatalysts
This section describes two major groups of molecular catalysts applied in the field of electrochemical CO2 reduction, namely iron porphyrins and rhenium and manganese bipyridine tricarbonyl complexes. Before discussing the effects of secondary coordination sphere interactions on the catalytic performance of these catalysts, the general reduction pathways of the unfunctionalized catalysts are outlined.The CO2-to-CO reduction mechanism for iron tetraphenylporphyrin (Fe1) reported by Savéant and coworkers is shown in Scheme 1.23 The cycle starts with a one-electron reduction of the [Fe(I)]− precursor to generate the [Fe(0)]2− species, followed by the nucleophilic attack onto CO2, leading to the formation of the [Fe(II)COO]2− intermediate. Protonation of [Fe(II)COO]2− forms the carboxyl intermediate [Fe(II)COOH]−, which undergoes a second protonation and a C–O bond cleavage event to produce the [Fe(II)CO] species. Finally, the reduction of [Fe(II)CO] liberates CO and regenerates the initial [Fe(I)]− catalytic precursor. This last step is suggested to occur via a homogeneous electron transfer from [Fe(0)]2−, thus generating two equivalents of the [Fe(I)]− species.
 | ||
| Scheme 1 Proposed mechanism for CO2-to-CO reduction by Fe1. | ||
Savéant and coworkers suggested that the rate-determining step (RDS) in CO2RR with Fe1 proceeds as a concerted proton–electron transfer-bond cleavage reaction.24 In this key step, an electron transfer from an [Fe(I)COO˙−]− center co-occurs with the second protonation of the carboxylate intermediate, [Fe(II)COO]2−, and with the C–O bond cleavage, resulting in the formation of [Fe(II)CO]. This mechanism was convincingly supported by foot-of-the-wave analysis of the reaction kinetics and kinetic isotope effect studies. Computational investigations carried out by Neese and coworkers found this mechanism implausible, and point instead to a slightly altered mechanism.25 In their mechanism, [Fe(II)COO]2− is initially protonated by the Brønsted acid (HA) to form the carboxylic acid intermediate, [Fe(II)COOH]−. This intermediate is stabilized by one HA molecule, followed by a subsequent protonation and C–O bond cleavage step to release [Fe(II)CO], A−, and water. Another computational study on the reduction mechanism of Fe1 carried out by de Visser and Davethu points, however, to a completely different RDS.26 Using comprehensive density functional theory (DFT) calculations, they found that the binding of CO2 to the iron center constitutes the RDS rather than the second protonation and C–O bond cleavage. Specifically, when using phenol as the proton donor, they found that the first and second proton transfers have low-energy transition states and, hence, should proceed rapidly, making CO2-binding the RDS. Furthermore, they suggest that the first protonation is concerted with an electron transfer rather than the second protonation, making this latter step a pure proton transfer process.
The rhenium and manganese bipyridine tricarbonyl complexes, Re1 and Mn1, share similar reduction mechanisms as well as several intermediates. There are, however, also fundamental differences between the two catalysts that are reflected in the catalysts' properties as well as in their preferred reduction pathways. Carter and coworkers published a detailed study comparing the reduction mechanisms of Re1 and Mn1, where they elucidate many differences between the two electrocatalysts, some of which will be discussed here.27 The overall reduction mechanism for these two catalysts is seen in Scheme 2. Starting with the characteristic six-coordinate species 1, the initial one-electron reduction can occur alone or simultaneously with the dissociation of the halide ligand, X−. Based on comparisons between experimental and computed reduction potentials, Re1 is predicted to favor the formation of 2-X followed by a second reduction where the halide ligand is expelled to give the catalytically active complex 3. On the other hand, Mn1 preferentially forms the five-coordinate intermediate 2, which can undergo a second reduction to give intermediate 3. 2-X can also undergo ligand exchange with a solvent molecule such as acetonitrile when electrochemical experiments are carried out in this solvent. However, this variant is omitted here for simplicity. An essential difference between Re1 and Mn1 in the initial reduction steps is the tendency of Mn1 to dimerize and form the inactive dimer, 2-D. This dimer requires a large overpotential to form the active complex 3, which deteriorates the catalytic properties of Mn1.28,29 Several studies have suggested that dimerization occurs from the five-coordinate intermediate 2 by the combination of two radicals as shown in Scheme 2.30 Grills and coworkers demonstrated this dimerization experimentally using pulse radiolysis combined with time-resolved infrared spectroscopy and determined a dimerization rate of 1.3 × 109 M−1 s−1.31 However, a different mechanism involving a parent–child reaction between intermediate 1 and the catalytically active intermediate 3 has also recently been proposed as an alternative pathway for the formation of 2-D.32Re1 does not show this tendency, and calculations confirm that the rate of dimerization for Re1 is approximately 109 times slower than that of Mn1.27 The catalytically active species 3 binds CO2, leading to the formation of intermediate 4 followed by its protonation to afford the carboxylic acid intermediate 5. Such an intermediate can pursue either of two pathways depending on the order of protonation and reduction.33 Using microkinetic simulations, Carter and coworkers showed the accumulation of intermediate 6a in the reduction of CO2 by Re1 at potentials greater than −1.7 V vs. SCE, indicating that CO formation proceeds via the reduction-first pathway with a proton-coupled C–O cleavage as the RDS.27 Depending on the applied reduction potential, Mn1 exhibits a duality in its behavior. Mn1 was found to behave similarly to Re1 at high overpotentials, thus favoring the reduction-first pathway, as demonstrated by the accumulation of intermediate 6a. At low overpotentials, the accumulation of 6b suggests that in such a potential range, the protonation-first pathway is operative. Importantly, independent of the applied potential, the RDS was again found to be the proton-coupled C–O bond cleavage.
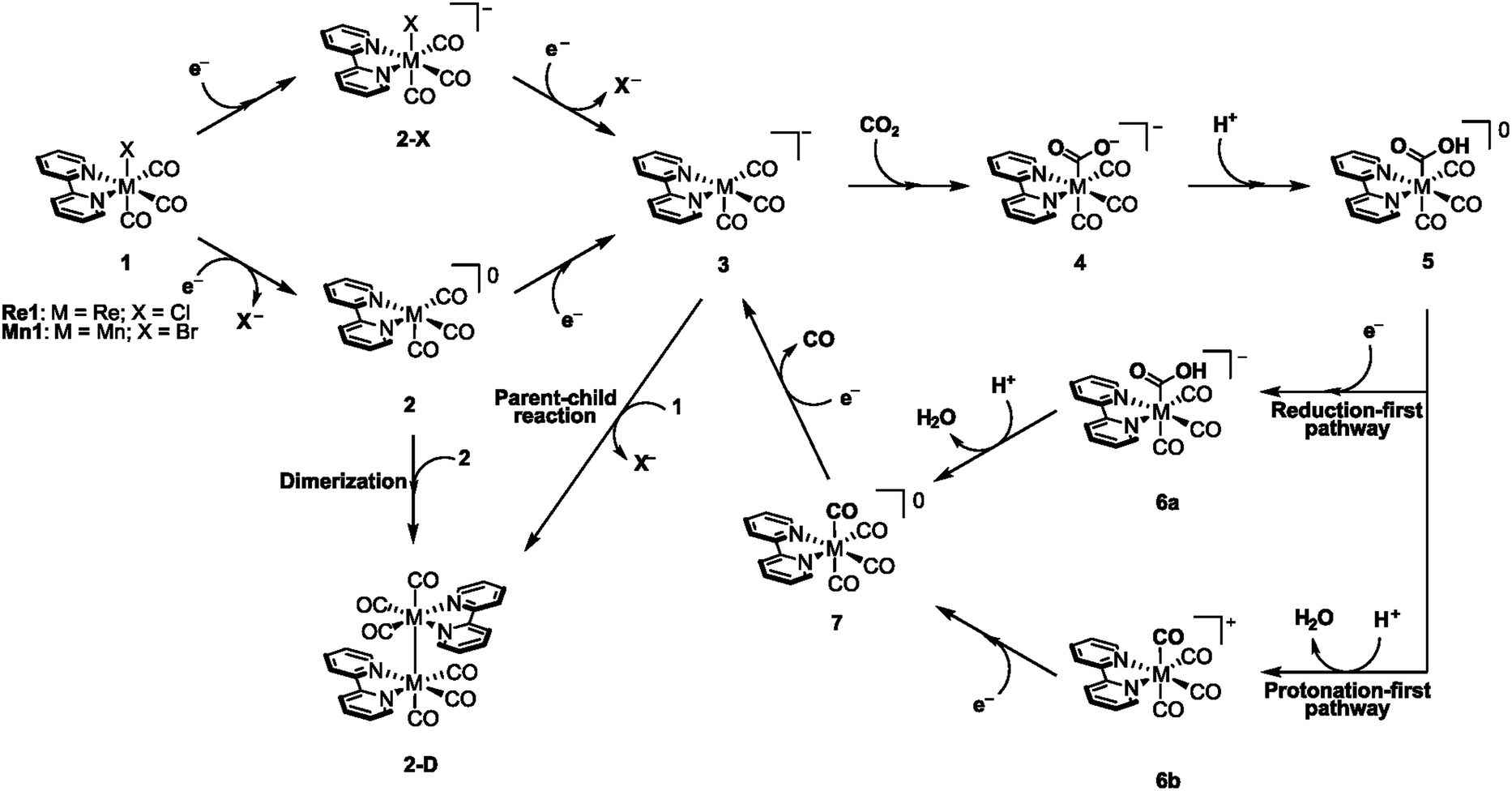 | ||
| Scheme 2 Proposed CO2-to-CO reduction mechanism by rhenium and manganese bipyridine tricarbonyl catalysts, Re1 and Mn1. | ||
Iron porphyrin complexes featuring secondary coordination sphere interactions
Molecular electrocatalysts for CO2RR have been developed for decades, and early works focused mainly on optimizing the first coordination sphere of the catalysts, which led to the discovery of several molecular systems that varied in d-block transition metal and ligand type.34–36 While these efforts did result in useful electrocatalysts for CO and formate production, modulation of the secondary coordination sphere has increasingly proved to be an effective means of optimizing the catalytic activity and selectivity of molecular catalysts.19 This strategy has provided an extensive toolbox for rationally designing the environment near the metal active site of various molecular electrocatalysts. While this work highlights selected examples of the use of secondary coordination sphere interactions in CO2RR molecular catalysts, several comprehensive reviews published in recent years provide a broader overview of the achievements in this field and offer additional examples for interested readers.37–40This section introduces various concepts of secondary coordination sphere modifications in Fe porphyrins. Several research groups have utilized Fe porphyrins as benchmark catalysts for studying second coordination sphere effects due to their near unity selectivity toward CO production. The examples described below cover two substituent effects categories: intramolecular hydrogen bonding and tuning of proton inventories, and electrostatic effects exerted by substituents.
The report of Savéant and coworkers on the electrocatalytic reduction of CO2 to CO mediated by Fe1 in the presence of a Brønsted acid represents a seminal point for the ongoing development of CO-producing molecular catalysts.23 Based on the dramatic increase in the catalytic activity of Fe1 upon the addition of an external proton source, Savéant and coworkers reasoned that the installment of acidic organic residues on the phenyl rings of the porphyrin ligand would further enhance the CO2-to-CO electrocatalytic activity due to the anticipated increased local concentration of available protons.41 Indeed, tailoring the second coordination sphere to obtain a hydroxyl-functionalized tetraphenyl iron porphyrin (Fe2, Scheme 3) resulted in a highly active and selective catalyst for CO production, exhibiting turnover frequencies (TOFs) > 3000 s−1, which is two orders of magnitude higher than those obtained for the unsubstituted porphyrin Fe1.
 | ||
| Scheme 3 Iron porphyrin catalysts Fe1–Fe16. The anionic axial ligand coordinated to the Fe center has been omitted for clarity. | ||
According to the suggested mechanism, the prepositioned phenol groups strongly stabilize the [Fe–CO2] adduct through hydrogen bonding. The second proton-coupled electron transfer (PCET) step, in conjunction with a C–O bond cleavage, occurs upon the participation of another phenolic proton. Cyclic voltammetry (CV) studies of Fe2 at various scan rates and phenol concentrations confirmed the dual role of the pendant phenol groups as both hydrogen bond promotors and proton relays.42 Here, a one-electron pre-catalytic wave is observed prior to catalysis for Fe2, indicating the reductive transformation of [Fe(I)(HO–)8]− to [Fe(0)(HO–)8]2− as seen in Scheme 4. The active catalyst [Fe(0)(HO–)8]2− coordinates with CO2 forming the adduct [Fe(II)(COO)(HO–)8]2−. A pre-catalytic wave, which is absent in the case of Fe1, indicates a strong hydrogen bonding that stabilizes the [Fe(II)(COO)(HO–)8]2− adduct. The close follow-up of the second catalytic electron transfer to this adduct is hence indicative of a reversible intramolecular protonation step forming the [Fe(II)(COOH)(O–)(HO–)7]2− intermediate. This step is then followed by re-protonation of the resulting internal phenoxide group, generating [Fe(II)(COOH)(HO–)8]−. The second electron–proton transfer to the protonated intermediate and loss of CO completes the catalytic cycle. Inspection of the catalytic wave revealed that for Fe2, the kinetics were governed by the rate of the internal protonation step. Evaluation of the charge transfer parameter for the second PCET showed that the C–O bond breaking co-occurs with this step.
 | ||
| Scheme 4 The proposed CO2-to-CO electroreduction mechanism by iron porphyrin catalyst Fe2. | ||
The methylated analog to Fe2 bearing methoxy substituents instead of hydroxy groups, Fe3, proved to be a poor catalyst with its kinetics being dependent on the phenol concentration in solution,41 underlining the positive effect of proton availability in the secondary coordination sphere. The internal phenol groups play the role of high-concentration and mass transport-independent proton donors. In fact, when the foot-of-the-wave TOF values of catalysts Fe1 and Fe2 were compared, it was concluded that the effect of the eight intramolecular phenolic groups was equivalent to an effective concentration in solution of added phenol of 150 M.41,43,44
Chang and coworkers investigated the impact of the spatial configuration of the second coordination sphere interactions on the electrocatalytic activity of molecular complexes featuring amide functionalities.45 They found, by comparing Fe porphyrin Fe4–Fe7, that the stabilizing hydrogen bonding effect of an NH group on the CO2-adduct was most beneficial if positioned in the ortho-position and two carbon atoms away from the phenyl ring (Fe5). This spatial arrangement was inspired by the configuration of the CODH active site shown in Fig. 1a. CV performed in aprotic conditions was used to probe the CO2 binding affinity at the iron centers of Fe4–Fe7. The most significant anodic shift of the Fe(I)/Fe(0) reduction wave in the presence of CO2 was observed for Fe5, showing that a properly positioned amide functionality increases the affinity for CO2via hydrogen bonding (Scheme 5). The equivalent para-substituted structures Fe6 and Fe7, with the NH moieties positioned further away from the iron atom, did not show the same positive effect.
 | ||
| Scheme 5 Iron porphyrin catalysts Fe4 and Fe5, showing how the differently positioned amide functionality coordinates to the [Fe–CO2] adduct via hydrogen bonding. | ||
For catalysts like Fe4 and Fe5, not only the positioning but also the acidity of the amide affects catalysis. This was demonstrated by Nichols and coworkers, who prepared Fe catalysts similar to Fe5 but with different electronic groups in the para-position of the most distant phenyl group (Fe8–Fe11).46 They found that the catalysts with the more acidic amides (OCF3- and CF3-substituted, Fe10–Fe11) exhibited the highest activity toward CO production at roughly the same potential for all tested catalysts. Furthermore, they investigated the interplay between the endogenous and exogenous proton donor and found that the highest rate increase was obtained when using the catalyst with the most acidic amide (Fe11) together with the most acidic of the tested exogeneous proton donors (para-trifluoromethyl phenol). The authors also discovered a greater sensitivity in terms of activity toward the pKa of the exogenous proton donor for Fe11 compared to the less acidic complexes, Fe8–Fe10.
To further optimize the effect of a prepositioned hydrogen bonding moiety, Aukauloo and coworkers took inspiration from the multipoint hydrogen-bonding interactions between the [Fe–CO2] adduct and the protein matrix, specifically, lysine and histidine residues in the CO dehydrogenase enzyme (CODH, Fig. 1a).47–49 This biological model guided their design and synthesis of the iron porphyrin Fe12.50 CV of Fe12 with urea functionalities positioned on all four porphyrin phenyl rings, was performed in an aprotic solvent where no catalytic reduction of CO2 is possible. An anodic shift in the Fe(I)/Fe(0) reduction potential and the absence of irreversibility in the presence of CO2 indicated a strong CO2 binding rate constant favored by hydrogen bonding via the urea moieties. DFT calculations further confirmed the existence of hydrogen bonds between the N–H hydrogen atoms of the urea groups and the oxygen atoms of the [Fe–CO2] species, resulting in the stabilization of such an intermediate. In addition, the use of water as the proton donor enhanced the catalytic activity for CO production despite its much higher pKa value compared to phenol or trifluoroethanol. X-ray structure analysis revealed that a water network exists around the urea moiety of the catalyst. Hence, it was concluded that water works in synergy with the urea moiety to supply protons, unlike when organic proton donors were used. The possibility that the substantial anodic shift in the reduction potential could arise from an inductive effect exerted by the fluorinated substituents was ruled out by synthesizing Fe13. With the absence of the urea moiety in the favorable position, Fe13 showed only a minor anodic potential shift.
Nocera and coworkers carried out experimental and computational analysis of the so-called hangman porphyrins, in which the functional groups dangle above the [Fe–CO2] adduct.51 The investigated Fe complexes display intramolecular proton donors with distinct pKa values, Fe14–Fe16 (Scheme 3), and were electrocatalytically tested in the presence of various concentrations of an exogenous Brønsted acid (phenol). Catalysis was corroborated by the sigmoidal-shaped voltammograms for all three porphyrins, with increased activity in the presence of phenol. The catalytic efficiency in the order Fe14 > Fe15 > Fe16 reflects the interplay between the endogenous and exogenous proton donors. With a reaction order of one with respect to phenol, the authors proposed a reaction mechanism where the concerted binding of CO2 at the active Fe(0) site and endogenous hydrogen coordination/exogenous re-protonation comprise a first chemical RDS.
Hence, the pKa values of the endogenous and exogenous proton donors must be of such different values that a fast re-protonation occurs to maintain the proton shuttle mechanism. If the pKa of the hanging proton donor was lower than that of phenol, the former could not become re-protonated, and the proton shuttle process would cease to operate. Computational analysis showed that the intramolecular hydrogen bonding to CO2 within the hangman moiety was thermodynamically favorable only in the case of Fe14 and Fe15 when compared to CO2 coordination external to the hangman residue (i.e., at the unmodified side of the porphyrin ring). For Fe16, the sulfonic acid group was deprotonated under the pH conditions examined. Due to coulombic repulsion between the sulfonate group and the reduced iron center, it was not possible to computationally optimize a structure in which the sulfonate group could assist in CO2 binding. Hence, it was found that CO2 could only bind at the unmodified side of the porphyrin ring.
Dey and coworkers reported the electrocatalytic reduction of CO2 to formate using the partly saturated iron porphyrin Fe17 (Scheme 6), which features four electron-withdrawing groups and a pendant tertiary amine.52 CV studies showed that this catalyst does not bind CO2 in the absence of a proton donor, as indicated by the superposition of cyclic voltammograms recorded under nitrogen and CO2 in aprotic conditions. However, the addition of increasing amounts of water as a proton donor led to a rising catalytic current in the Fe(II)/Fe(I) redox potential peak. Typically, Fe porphyrins bind CO2 in their formal Fe(0) state, but Fe17 was active toward CO2RR in the Fe(I) state, resulting in an exceptionally low overpotential of only 50 mV in CV measurements. Cyclic voltammograms of a similar Fe catalyst lacking the tertiary amine did not exhibit a comparable catalytic response, highlighting the crucial role of the tertiary amine in the secondary coordination sphere for the activity of Fe17. Controlled potential electrolysis (CPE) experiments demonstrated a formate selectivity up to 97% with a TOF of 50 s−1 at an overpotential of only 100 mV (−1.55 V vs. Fc/Fc+).
 | ||
| Scheme 6 The mechanistic pathway showing the formation of formate by Fe17 as proposed by Dey and coworkers.52 | ||
The reduction mechanism proposed for such an unusual product selectivity for a Fe porphyrin was investigated using a combination of in situ Fourier transform infrared spectroelectrochemistry (FTIR-SEC), resonance Raman spectroscopy, and in situ Mössbauer spectroscopy, leading to the mechanistic proposal in Scheme 6. The Fe(III) pre-catalyst (Fe17) is initially reduced to the [Fe(II)(–N–)] intermediate, which undergoes a second, proton-coupled 1e− reduction to afford [Fe(I)(–NH+–)] in the presence of water, where the tertiary amine gets protonated. [Fe(I)(–NH+–)] binds and activates CO2, resulting in the carboxylic acid intermediate, [Fe(III)(COOH)(–N–)], through an intramolecular proton transfer. Under electrochemical conditions, this intermediate is further reduced to [Fe(II)(COOH)(–N–)]−, which was detected by FTIR-SEC measurements (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration at 1682 cm−1). Finally, formate is released in a hydrolysis step to reform [Fe(II)(–N–)]. The accumulation of the [Fe(II)(COOH)(–N–)]− species led the authors to suggest that the hydrolysis step to release formate constitutes the RDS. This mechanism demands that the reduction potential of [Fe(III)(COOH)(–N–)] is more positive than that where catalysis occurs. Dey and coworkers52 confirmed this by recording cyclic voltammograms of a chemically prepared [Fe(III)(COOH)(–N–)] species, detecting its 1e−-reduction at −0.64 V vs. Fc/Fc+. DFT calculations were used to confirm the importance of the tertiary amine in the secondary coordination sphere. The calculations showed that CO2 would only bind to [Fe(I)(–NH+–)] when the amine was protonated, in agreement with the cyclic voltammograms recorded in the absence and presence of water. Other formate-producing electrocatalysts, such as Mn bipyridine tricarbonyl catalysts, depend on the formation of a metal hydride intermediate (vide infra).53 Yet, in the work presented by Dey and coworkers,52 formate is generated from the carboxylic acid intermediate, which in all previous reports on Fe porphyrins undergoes protonation and C–O bond breakage to form CO and water in the last step of the mechanism (vide supra).24 The peculiar protonation of the carboxylic carbon atom instead of the O–H oxygen atom in [Fe(II)(COOH)(–N–)]− is central to the observed selectivity toward formate instead of CO. A possible explanation points to the lower reducing power of the Fe center in Fe17 compared to the previously discussed Fe porphyrins. This results from the catalytically active Fe(I) center, which facilitates the transformation at extremely low overpotentials. The greater Lewis acidity of the Fe center in the [Fe(II)(COOH)(–N–)]− species decreases the amount of π backbonding that takes place between the d-orbitals of the Fe center and the π* orbital of the COOH group. This, in turn, makes the C–OH bond less susceptible to cleavage, which may allow for the C-centered protonation to take place instead. Taken together, this work demonstrated how the modulation of the secondary coordination sphere can completely shift the product selectivity and reduction behavior for this class of catalysts, leading to selective formate production at unprecedently low overpotentials.
O vibration at 1682 cm−1). Finally, formate is released in a hydrolysis step to reform [Fe(II)(–N–)]. The accumulation of the [Fe(II)(COOH)(–N–)]− species led the authors to suggest that the hydrolysis step to release formate constitutes the RDS. This mechanism demands that the reduction potential of [Fe(III)(COOH)(–N–)] is more positive than that where catalysis occurs. Dey and coworkers52 confirmed this by recording cyclic voltammograms of a chemically prepared [Fe(III)(COOH)(–N–)] species, detecting its 1e−-reduction at −0.64 V vs. Fc/Fc+. DFT calculations were used to confirm the importance of the tertiary amine in the secondary coordination sphere. The calculations showed that CO2 would only bind to [Fe(I)(–NH+–)] when the amine was protonated, in agreement with the cyclic voltammograms recorded in the absence and presence of water. Other formate-producing electrocatalysts, such as Mn bipyridine tricarbonyl catalysts, depend on the formation of a metal hydride intermediate (vide infra).53 Yet, in the work presented by Dey and coworkers,52 formate is generated from the carboxylic acid intermediate, which in all previous reports on Fe porphyrins undergoes protonation and C–O bond breakage to form CO and water in the last step of the mechanism (vide supra).24 The peculiar protonation of the carboxylic carbon atom instead of the O–H oxygen atom in [Fe(II)(COOH)(–N–)]− is central to the observed selectivity toward formate instead of CO. A possible explanation points to the lower reducing power of the Fe center in Fe17 compared to the previously discussed Fe porphyrins. This results from the catalytically active Fe(I) center, which facilitates the transformation at extremely low overpotentials. The greater Lewis acidity of the Fe center in the [Fe(II)(COOH)(–N–)]− species decreases the amount of π backbonding that takes place between the d-orbitals of the Fe center and the π* orbital of the COOH group. This, in turn, makes the C–OH bond less susceptible to cleavage, which may allow for the C-centered protonation to take place instead. Taken together, this work demonstrated how the modulation of the secondary coordination sphere can completely shift the product selectivity and reduction behavior for this class of catalysts, leading to selective formate production at unprecedently low overpotentials.
Savéant and coworkers investigated the effect of having charged substituents at the pendant phenyl rings (Scheme 7).54 Positively charged trimethylanilinium (TMA) groups in the para-position (Fe18) showed a similar through-structure inductive effect as observed for electron-withdrawing groups, leading to decreased TOFs along with lower operating potentials.55 When the charged groups were moved to the ortho-position (Fe19), a dramatic improvement in the performance was observed in terms of both overpotential and catalytic activity. Such an increased performance was ascribed to the through-space coulombic stabilization of the [Fe–CO2] adduct, in which the negative charge residing at the oxygen of the bound CO2 interacts with the positive charge borne by the TMA substituents. This effect is only achievable with the positive charges positioned close to the iron active site of the adduct, i.e., in the ortho-position of the pendant phenyl ring. An unprecedented catalytic activity with a TOF of 106 s−1 at only 220 mV of overpotential and with selectivity for CO close to 100% was reported.
 | ||
| Scheme 7 Iron porphyrin catalysts Fe18–Fe20. The anionic axial ligand coordinated to the Fe center has been omitted for clarity. | ||
The positional effect of the TMA groups on the porphyrin ring of Fe19 was further investigated by Mayer and coworkers,56 who tested the four different atropisomers of this catalyst (αααα, αααβ, ααββ, αβαβ) under the same conditions as those used by Savéant and coworkers.54 They found that all four atropisomers are excellent CO2RR electrocatalysts, producing CO with fast rates and at low overpotentials. The αααα atropisomer exhibited the highest TOFmax (2 × 105 s−1), yet the largest difference in TOFmax between the atropisomers was less than a factor of 5. This led the authors to conclude that the orientation of the TMA groups is not decisive for the catalysts' performance, but rather the total charge near the metal center is essential.
Another example of a through-space electrostatic interaction was reported by Aukauloo and coworkers, who demonstrated a similar positive effect on the catalytic activity by incorporating methylimidazolium functionalities (Fe20, Scheme 7) in the porphyrin second coordination sphere.57 The positively charged methylimidazolium groups did not affect the onset potential of the Fe(III)/Fe(II) reduction wave under non-catalytic conditions, as expected from the lack of conjugation between the positive charge and the porphyrin ring. Upon further reduction of Fe20, anodic shifts of the Fe(II)/Fe(I) and Fe(I)/Fe(0) waves were observed and ascribed to a through-space charge interaction between the iron center and the positively charged methylimidazolium groups. Albeit CO2 has a lower solubility, an increase in the TOF was observed when switching solvent from DMF to water. This was attributed to a higher degree of dissociation of the methylimidazolium+Cl− units in water, leading to a better intermediate stabilization. CPE experiments conducted in water resulted in a TOF of 1.5 × 104 s−1 for CO production at an overpotential of 418 mV.
Head-Gordon and coworkers also investigated the stabilization of the [Fe–CO2] adduct in Fe18–Fe20 using an energy decomposition analysis of DFT calculations based on absolutely localized molecular orbitals (ALMO-EDA).58 For Fe19, their calculations suggested that the observed adduct stabilization is mainly due to reduced Pauli repulsion, meaning that the through-structure electronic effects of the positively charged groups are dominating. Furthermore, they found that the through-space coulombic interactions between the ortho-TMA nitrogens in Fe19 and the negatively charged oxygens of the [Fe–CO2] species are significantly attenuated by solvent screening. Thus, the TMA groups are found to operate mainly via inductive through-structure effects, yet the co-existence of the through-space and through-structure effects leads to the greater stabilization of [Fe–CO2] by Fe19 compared to Fe18. The authors also conducted this analysis for a model catalyst resembling Fe20 but containing only two methyl imidazolium-substituted groups instead of four. Here, they found that the through-space stabilization of [Fe–CO2] was dominating and that through-structure inductive effects from the charged groups were basically nonexistent. The authors argued that the bulky chain to which the imidazolium group is attached creates a pocket where the solvent is excluded, allowing the through-space interactions to occur. These results suggest a greater overall stabilization of the [Fe–CO2] adduct in Fe20 compared to Fe19 due to the stronger through-space coulombic interactions exerted by the four imidazolium moieties.
Rhenium tricarbonyl bipyridine electrocatalysts decorated with secondary coordination sphere groups
Rhenium(I) bipyridine (bpy) complexes of the form Re(bpy)(CO)3X (X = halogen) have been known as active electrocatalysts since the 1980s when Lehn and coworkers first reported their use in the CO2-to-CO electroconversion.59 Under CO2RR conditions at −1.25 V vs. the normal hydrogen electrode (NHE), this catalyst afforded Faradaic efficiencies (FEs) for CO up to 98%. Subsequent research has focused on modifying the steric and electronic properties of the catalyst by introducing electron-donating and electron-withdrawing functional groups at the 4,4′-positions of the bpy ligand.60,61 These studies collectively suggest that electron-rich substituents enhance the electrocatalytic activity of these molecular complexes, while electron-withdrawing groups have the opposite effect. This trend has been attributed to the fact that a highly nucleophilic metal center promotes CO2 activation, leading to the formation of intermediate 4 in Scheme 2. As expected, this increased activity comes with a trade-off of larger overpotentials, as the formation of intermediate 3 requires more energy due to the presence of the electron-donating groups in the ligand structure.Through functionalization of the 6,6′-positions of the bpy ligand, the effect of secondary coordination sphere interactions in Re bpy catalysts has also been investigated. As for iron porphyrins, a widely applied strategy entails the utilization of pendant proton donors within the secondary coordination sphere. When positioned near the rhenium center, they can engage in beneficial hydrogen bond interactions, leading to enhanced catalytic activity. For example, Marinescu and coworkers investigated the role of pendant amines in creating an intermolecular network of hydrogen bond donors.62 CV analysis of rhenium bipyridine catalysts modified with primary (Re2), secondary (Re3), or tertiary amines (Re4) in the 6,6′-positions showed that Re2 and Re3 exhibited an additional reduction peak (Scheme 8). Since Re4 did not give rise to such a peak, the additional reduction event was attributed to the formation of an intermediate stabilized by hydrogen bonding.63 This was further supported by switching the solvent from MeCN to DMF, where the enhanced hydrogen bonding ability of DMF appeared to interrupt the formation of the intermediate displaying intramolecular hydrogen bonding interactions. This effect was corroborated by the disappearance of the extra reduction peak in the cyclic voltammograms of Re2 and Re3. CPE experiments of the complexes showed that Re2 exhibited the greatest FE for CO (83%) of all three complexes at a potential of −2.30 V vs. Fc/Fc+.
 | ||
| Scheme 8 Re tricarbonyl bipyridine catalysts. | ||
The position of primary amines on the bpy ligand has also been deemed important for electrocatalysis, and comparative studies of the substituted complexes Re2, Re5, and Re6 showed that a more negative catalytic onset potential was required for Re5 and Re6 than for Re2.64 Unlike Re2, the former two complexes do not exhibit the aforementioned additional reduction peak, indicating the lack of intermolecular hydrogen bonding interactions. CPE experiments revealed that all three complexes exhibited excellent FE for CO (83–99%), however, Re5 and Re6 required more negative electrolysis potentials, and the CO production of Re6 was half of that of Re2. DFT calculations indicated a larger electron density at the metal center of Re2 than Re5 and Re6. Combined with the hydrogen bonding ability of this complex, this substantiates that the proximity of the proton donor to the metal center is a significant parameter to enhance CO2-to-CO electroconversion.
Surprisingly, the insertion of hydroxyl groups in the second coordination sphere of a Re bipyridine catalyst (Re7) led to a severely diminished catalytic activity despite the structural similarity with Re3.65 In optimized CPE experiments, Re7 afforded a FE for CO of 70% at −1.7 V vs. Ag/AgCl, however, the turnover number (TON) was less than 1. This result indicated that Re7 did not function as an electrocatalyst, likely due to slow product release in conjunction with the decomposition of unstable intermediates after CO release. This observation differs markedly from the dramatic improvement in catalytic performance observed with the similarly modified iron porphyrin Fe2. This result furthermore shows that the nature and acidity of the proton donor play important roles in the ability of the ligand to facilitate beneficial secondary coordination sphere interactions.
Neumann and coworkers also investigated the effect of a local proton source in the second coordination sphere by asymmetrically inserting a thiourea tether in the 6-position of the bipyridine ligand (Re8).66 To enhance the proton donating ability of the thiourea moiety, it was bonded to an electron-withdrawing –PhCF3 group through the most distant nitrogen atom. This electrocatalyst exhibited significantly improved catalytic properties compared to Re1, resulting in a TOF of 3040 s−1 as determined by CV measurements. In comparison, Re1 yielded a TOF of 355 s−1. CPE carried out at −1.9 V vs. Ag/AgNO3 resulted in the production of 12.4 μmol of CO over 12 hours with a FE of 89%. Using 1H nuclear magnetic resonance (NMR) experiments, the authors showed that the thiourea tether interacts with CO2 through hydrogen bonding. After observing a downfield shift for the hydrogen atom (NH) adjacent to the bipyridine ring upon changing the environment from N2 to CO2 gas, they confirmed the existence of a hydrogen-bonded CO2-adduct via an interaction with the pendant thiourea. Complementary 13C NMR measurements showed an additional peak under CO2 conditions, which was assigned to CO2 bound to the thiourea tether. In addition to being a hydrogen bond promoter, the thiourea also acted as the proton donor in the C–O bond cleavage step of the catalytic cycle in Scheme 2. In fact, DFT calculations were unable to locate a minimum where the NH proton adjacent to the bipyridine would reside on the nitrogen atom. Instead, this proton would transfer to the nearest O atom of CO2 and stabilize the carboxylate intermediate. This was further supported by electrochemical experiments in which an external proton source was added, leading to diminished catalytic activity due to the interruption of the favorable internal proton donation.
In a different approach toward enhanced electrocatalysis, Nippe and coworkers inserted charged imidazolium moieties with and without a C2 methyl group on the imidazolium unit in the 6-position of the bpy ligands (Re9 and Re10).67 CV measurements conducted under Ar revealed a positive shift of approximately 100 mV in the half-wave potential (E1/2) of the first quasi-reversible peak for Re9 and Re10 compared to the unfunctionalized Re1. This redox peak has been previously assigned to the initial 1e− reduction to generate the [Re(I)(bpy˙−)(CO)3Cl]− species,60 which is analogous to 2-X in Scheme 2. Additionally, the quasi-reversibility of such a redox peak is decreased with the functionalized Re complexes. Using DFT calculations, the authors suggested that the decreased reversibility originates from accelerated Cl− dissociation rates in Re9 and Re10 compared to Re1. In Re9, the intramolecular C2–H⋯Cl− interaction may allow for faster dissociation of Cl− from the [Re(I)(bpy˙−)(CO)3Cl]− species.60 In the case of Re10, it was suggested that the electrostatic and C4,5-H⋯Cl− interactions may contribute to the faster Cl− dissociation. Accordingly, increased reversibility of the initial reduction peak was observed at higher scan rates, where the Cl− dissociation rate is outrun by the scan rate.
Functionalized Re9 and Re10 catalysts exhibited enhanced CO2RR activity compared to Re1. CV measurements under CO2 conditions showed that Re9 produced a larger reduction current at potentials approximately 170 mV less negative than Re1. With the addition of small amounts of H2O, reaching up to 0.28 M, the ratio of the catalytic peak current (icat) to the initial peak current of the 1e− redox process under Ar (ip) increased. After this point, the catalytic current decreased up to approximately 1 M H2O. Beyond 1 M, the catalytic current rose again, reaching its highest icat/ip values at 2–3 M H2O, followed by a gradual decrease. CPE experiments conducted with Re9 yielded an FE of 73% for CO at a potential of −1.74 V vs. Fc/Fc+ in the presence of 9.4 M H2O as the proton source. The non-linear dependence of the electrocatalytic activity of Re9 and Re10 on [H2O] contrasts with the classical linear increase of icat/ip observed for Re1 as [H2O] increases. These results suggest that the CO2RR mechanism for the functionalized catalysts differs from that of Re1. The authors hypothesized that, in the case of Re9, the mechanism likely involves stabilizing hydrogen bonding interactions, either in the form of C2–H⋯−OOC–Re or through interactions with water molecules, such as C2–H⋯OH(O)C–Re. For Re10, these interactions could only involve the C4–H and C5–H groups, which are expected to result in weaker hydrogen bonding compared to the C2–H interactions in Re9. This difference in stabilization strength is reflected in the estimated catalytic rate constants, with Re9 exhibiting rates more than twice those of Re10 and approximately ten times higher than those of the unfunctionalized Re1.
Manganese tricarbonyl bipyridine electrocatalysts decorated with secondary coordination sphere interfering groups
The bpy ligand in Mn-based complexes has been subject to extensive modifications to probe its effect on electrocatalysis. Studies have covered not only steric effects, as described by Kubiak and coworkers,29 but also electronic effects in terms of the insertion of electron-donating or electron-withdrawing groups on the bpy ligand.68 Generally, electron-withdrawing groups on the bpy ligand shift the reduction potential to more positive values yet reduce the electron density on the metal center. This results in the inhibition of the reaction between the metal center and the weakly electrophilic CO2 substrate. In contrast, electron-donating substituents show the opposite trend, pushing the reduction potential toward more negative values while maintaining catalytic activity. Opposite to the Re catalysts, Mn bpy tricarbonyl catalysts such as the simple Mn1 (Scheme 9) are only active in the CO2RR in the presence of a proton donor.30 Hence, a question arises as to whether a similar catalytic activity could be achieved by inserting local proton donors in the secondary coordination sphere. Substitution of the bpy ligand with a 4-phenyl-6-(1,3-dihydroxybenzen-2-yl)-2,2′-bipyridine ligand (Mn2) yielded a CO2RR catalyst that exhibited similar activity as Mn1 in the absence of an external proton donor.69 Specifically, the catalyst achieved an FE of 70% for CO as well as 22% for HCOOH during a four-hour CPE at −1.8 V vs. SCE. The generation of CO from CO2 is enhanced by intramolecular hydrogen bonding stabilization of the CO2-adduct by the phenolic hydroxyls.70 The generation of HCOOH was suggested to proceed via a different mechanistic pathway than that for CO generation. It was attributed to the formation of a Mn hydride anion intermediate which was observed as an additional reversible reduction wave at −1.66 V vs. SCE in the cyclic voltammogram of the catalyst.69 The hydride anion is proposed to form via an intramolecular proton transfer from the local OH group of the bipyridine ligand to the Mn atom in [Mn(bpy-OH)(CO)3]−, which is reversibly reduced into the corresponding dianion. Notably, the additional reversible reduction wave was absent from cyclic voltammograms recorded of the methoxylated control complex, Mn3, indicating the crucial role of the spatially close phenolic protons in the generation of the Mn hydride anion and, hence, the formation of the HCOOH product. DFT calculations also supported this mechanism. Additionally, Mn3 exhibited no catalytic activity toward CO2 reduction in the absence of an external proton source. Furthermore, the spatial closeness and correct orientation of the local proton source relative to the metal center affect catalysis significantly. Indeed, the Mn complex functionalized with a 3,4,5-trihydroxybenzene group on the bpy ring (Mn4), in which the OH groups are placed at a slightly larger distance from the metal center, exhibited a different reduction mechanism (reminiscing that of the original Mn1), as well as substantially reduced formate production and impaired catalytic activity.71 | ||
| Scheme 9 Mn tricarbonyl bipyridine catalysts. | ||
High activity and selectivity for HCOOH production have since been observed for Mn complexes carrying tertiary amines in the second coordination sphere. This result constitutes an interesting case of how second coordination sphere interactions can effectively alter the preferred reaction mechanism and, hence, the product selectivity of a catalyst. With a series of amine-functionalized Mn tricarbonyl bipyridine complexes (Mn5–Mn7), Daasbjerg and coworkers obtained FEs of 63–72% for HCOOH at a potential of −2.17 V vs. Fc/Fc+ and could be increased to 90% for Mn5 at −1.77 V.53 TOF values for Mn5–Mn7 were estimated ≥4000 s−1, placing them as some of the most active electrocatalysts for HCOOH production. The position and basicity of the amine moiety affected the selectivity, leading to a substantially diminished formate production when the amine groups were relocated from the ortho to the para position of the bipyridine ligand (Mn8) and when the tertiary amines were substituted with a less basic morpholine group (Mn9) or secondary amine (Mn10).
Mechanistic insights obtained using both theoretical and experimental methods point to the key role of a Mn hydride intermediate for the observed HCOOH selectivity. The authors suggested that the [MnIBr] complexes Mn5–Mn7 are initially reduced via a two-electron reaction to generate a [Mn0]− intermediate (corresponding to intermediate 3 in Scheme 2), which undergoes a “parent-child” reaction with the initial [MnIBr] catalyst, affording [Mn0Br]− and [Mn0]. The [Mn0] complex is then reduced in a one-electron reduction step to reform the [Mn0]− intermediate, which is subsequently protonated via assistance from the amine ligand to yield the Mn hydride, [Mn–H] (Scheme 10). In this process, the tertiary amine acts as a Brønsted base co-catalyst, reacting with the added acid (R′OH) and CO2 to generate the ammonium intermediate and the corresponding alkyl carbonate (R′OCOO−). The role of the ammonium intermediate is two-fold; firstly, it acts as a proton shuttle that effectively delivers a proton to the Mn center, leading to the formation of [Mn–H]. Secondly, before it transfers the proton to the Mn center, it forms a strong (∼10 kcal mol−1) hydrogen bond to the Mn center, inhibiting CO2 from attacking the metal center and forming the [Mn–COO]− adduct known to be involved in CO formation. The generation of the [Mn–H] intermediate steers the selectivity toward HCOOH formation via either a low overpotential or a high overpotential pathway. Both scenarios lead to the generation of the [Mn–OCHO]− intermediate upon CO2 insertion, which releases HCOOH upon subsequent protonation and reduction. The initial protonation of the amine ligand was found to be the RDS of the reaction, explaining the decreased HCOOH selectivity observed for Mn9 containing an amine with lower basicity.
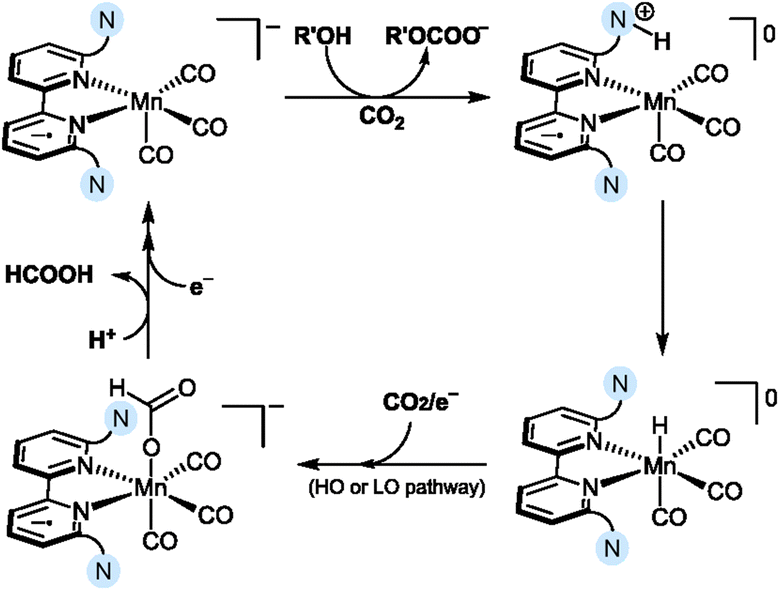 | ||
| Scheme 10 Proposed reduction mechanism showing the generation of the Mn hydride for amine-substituted Mn complexes, Mn5–Mn7.53 The nitrogen atom enclosed in blue denotes a tertiary amine in the secondary coordination sphere. | ||
As already outlined for the iron porphyrin and rhenium tricarbonyl bpy complexes, electrostatic hydrogen bonding effects facilitate proton transfer steps. The resulting enhanced hydrogen bond stabilization of key reaction intermediates is also a promising strategy for the CO2-to-CO conversion by Mn tricarbonyl bipyridine complexes. With a series of imidazolium-functionalized Mn complexes (Mn11–Mn14), CO2 reduction was conducted, yielding FEs >70% for CO at a reduction potential of −1.56 V vs. Fc/Fc+.72 Although the FEs are somewhat lower than those reported for other systems that convert CO2 to CO (vide supra), a significantly more positive reduction potential was used than those needed for such systems. The activity of Mn11–Mn14 follows the order Mn11 ≈ Mn12 > Mn13 > Mn14, indicating that the C2–H functionality of the imidazolium moiety is essential for catalysis, since Mn13 and Mn14 are devoid of this hydrogen atom. DFT calculations have shown that the C2 hydrogen participates in stabilizing hydrogen bond interactions at various points during electroreduction, allowing for a feasible reduction of CO2 to CO at a potential of −1.56 V vs. Fc/Fc+.72 Initially, a C2–H⋯OCO interaction locates the CO2 molecule close to the manganese center and increases the electrophilicity of CO2, thereby enhancing nucleophilic attack by the Mn atom. Once bonded, the C2–H hydrogen participates in synergistic interactions with various reaction intermediates as well as water molecules, reminiscing the behavior of the imidazolium functionalized Re catalyst (Re9, vide supra). This type of reactivity facilitates the protonation of key intermediates by creating a hydration shell around the active site via a network of hydrogen bonds. Combined with the previous examples of charged functionalities in the secondary coordination sphere of molecular catalysts, incorporating cationic moieties with hydrogen bonding abilities is a promising strategy to combine low reduction potentials with high selectivity for CO. Although restricted to exclusively promoting the catalytic 2e−/2H+ conversion of CO2 into CO and HCOOH, the examples of secondary coordination sphere interactions in homogeneous catalysis described in this section have inspired the design of the microenvironment around the catalytic sites in heterogeneous CO2 reduction. As described in the next section, such modifications can enhance the selectivity for further reduced products in the CO2RR.
Tailored microenvironments in heterogeneous Cu catalysts
In this section, we summarize recent efforts to tailor the microenvironment of Cu electrodes to enhance their electrocatalytic performance in the CO2RR. While other metal electrodes have demonstrated promising reactivity in activating and converting CO2 into value-added products,73 Cu stands out for its unique ability to produce hydrocarbon products, particularly multicarbon compounds such as ethylene and ethanol, which are crucial building blocks in the chemical industry.7Mechanistic overview of CO2RR on polycrystalline Cu surfaces leading to C1 and C2+ products
The general reduction pathways for CO2RR on polycrystalline Cu surfaces are illustrated in Fig. 2. Such pathways have been proposed through the results of spectroscopic and computational works.74–76 The initial step involves the electroadsorption of CO2, forming the surface-bound *CO2− intermediate (* indicates a surface-bound species). The binding configuration of CO2 is crucial in determining the specific product pathway. For instance, coordination of CO2via the oxygen atoms followed by a PCET step results in the η2(O,O)–CO2H intermediate, which can subsequently yield HCOOH after an additional PCET step. Alternatively, CO2 binding through the carbon atom yields the *CO2− intermediate, which can undergo two PCET steps to form the *CO intermediate. The desorption of *CO can lead to CO production; however, the binding energy of *CO on Cu favors further reactions on this intermediate that generate further reduced products.77 Specifically, *CO can undergo PCET reactions to form *CHO and *COH intermediates, which are key for methane production. In this pathway, the hydrogenation of *CO to *CHO/*COH is rate-determining, followed by several PCET steps that lead to the release of methane from the Cu surface.74,75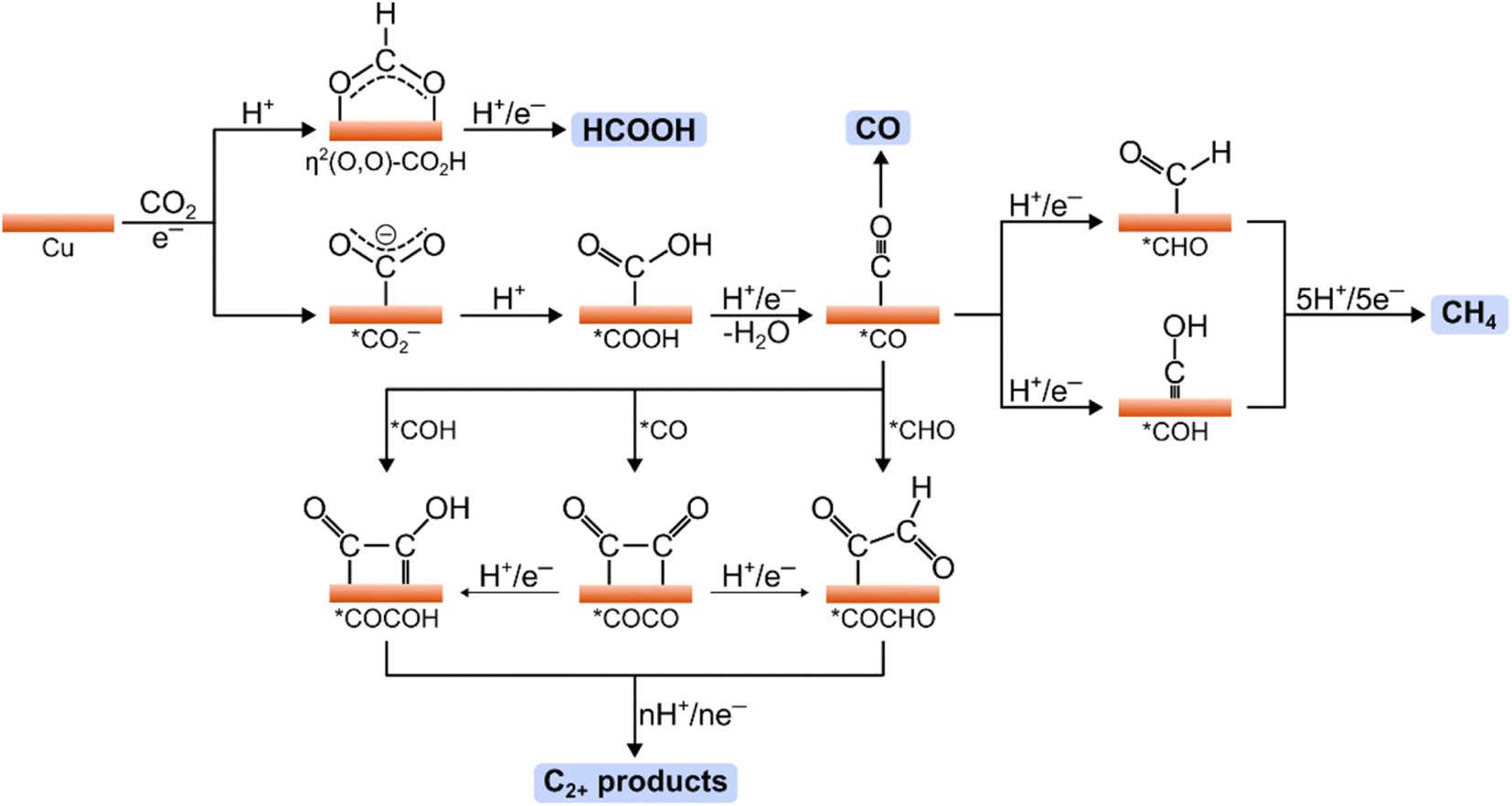 | ||
| Fig. 2 Mechanistic pathways in the CO2RR on Cu electrodes showing key intermediates toward HCOOH, CO, CH4, and C2+ products. | ||
*CO is also crucial for producing multicarbon products such as ethylene and ethanol. The rate-limiting step in these pathways is the C–C coupling reaction, which may occur either between two *CO intermediates or between *CO and its hydrogenated derivatives (*CHO or *COH).78–81 The preferred C–C coupling pathway is influenced by numerous factors, including surface facets, oxidation states, applied potential, and the pH and composition of the electrolyte.75 Notably, the microenvironment around the surface active sites can significantly affect selectivity for various multicarbon products (e.g., ethylene, ethanol, or acetate). The following section will address strategies to optimize these microenvironments for specific product generation.
Molecularly enhanced Cu surfaces as electrocatalysts in CO2RR
A preliminary indication that a metal electrode functionalized with surface organic residues successfully surpasses scaling relations is the enhancement of the intrinsic activity for a specific reduction product. This metric can be determined by normalizing the partial current densities with the electrochemically active surface area (ECSA) of the catalyst, obtainable through various methods such as the capacitance method82 or lead underpotential deposition.83,84 Normalizing the partial current density solely by the geometric area of the electrode does not accurately reflect the catalyst's intrinsic activity, as surface reconstruction or nano-structuring effects may artificially skew its performance.In previous work, we found that the intrinsic activity toward multicarbon products could be enhanced by modifying Cu substrates with N-heterocyclic carbene–carbodiimide (NHC–CDI) ligands.85 The catalysts were prepared via electrodeposition of Cu nanoparticles in the presence of different NHC–CDI ligands, resulting in hybrid Cu–NHC–CDI electrodes (Cu-1–5). The catalysts were tested in the CO2RR in an H-cell electrolyzer configured with a continuous CO2 flow using 0.1 M KHCO3 as electrolyte. The best-performing hybrid catalyst (Cu-3) achieved a >10-fold increase in the ECSA-normalized partial current density for multicarbon products, compared to a reference Cu sample, with ethylene as the main multicarbon product. Interestingly, we found that the generation of CO was suppressed as the only C1 product for all hybrid catalysts compared to the Cu reference sample. This indicated that the enhanced activity for multicarbon products was due to increased *CO dimerization. Using a flow cell electrolyzer, Cu-3 was also tested in a gas-diffusion electrode configuration, leading to an increase in the FE for multicarbon products from 33% to 58% and more than a doubling of the partial current density compared to a reference Cu sample (Fig. 3a).
 | ||
| Fig. 3 (a) Electrocatalytic performance of the best-performing NHC–CDI-modified Cu catalyst (CuGDE-3) and a reference Cu catalyst (CuGDE) in a GDE-based electrolysis setup at −1.1 V vs. RHE, and (b) partial current density for multicarbon products, |jC2+,ECSA|, as a function of the accumulated surface charge, Qan,ECSA, at ΔE = 1.6 V. Adapted with permission from J. Am. Chem. Soc. 2024, 146, 13034–13045. Copyright 2024 American Chemical Society. | ||
X-ray photoelectron spectroscopy (XPS) was employed to investigate the nature of the Cu species present at the catalyst's surface. We discovered that Cu-3 exhibited a higher ratio of Cu0/Cu+ surface states than the poorest-performing hybrid catalyst (Cu-5) and the Cu reference sample. We proposed that the enhanced activity toward multicarbon products observed with Cu-3 resulted from the more electron-rich surface, which stabilized *CO to a greater extent due to enhanced π back-bonding between the d-band from Cu and the CO 2π* orbital. To further link the electronic tuning effects of the NHC–CDI ligands to the catalytic activity, we carried out modified pulsed voltammetry (mPV) to determine the surface charge that accumulates under electrocatalytic conditions (Qan,ECSA). We found that the surface charge linearly correlates with the intrinsic activity for multicarbon products, in which the best-performing catalysts exhibited the highest accumulated surface charge (Fig. 3b). This result aligns with the increased stabilization of *CO due to a more reduced surface in Cu-3, i.e., a higher density of surface Cu(0) states, and hence an increased accumulation of *CO.
In situ Raman spectroscopy confirmed *CO as a key accumulating intermediate and probed its specific behavior under catalytic conditions. We found that *CO adsorbs in different binding configurations on the hybrid catalysts (Cu-3 and Cu-5) and on the reference Cu sample. On Cu-3 and Cu-5, *CO binds in the atop configuration, whereas in the reference Cu sample, *CO binds in a bridge configuration, which has previously been reported to exhibit poor kinetics for C–C bond formation.86 This result explained the enhanced multicarbon product activity observed across all hybrid catalysts. By deconvolution of the Raman spectra of Cu-3 and Cu-5 into high- and low-frequency bands, we discovered that *CO accumulated at Cu step sites known to be highly active toward C–C coupling (high-frequency band) on Cu-3. Accordingly, the binding of *CO at the less active Cu terrace sites (low-frequency band) on Cu-5 explained the difference in performance between these catalysts. Collectively, this study showed that the molecular modification of Cu catalysts with additives that shift the surface electronic Cu states can facilitate the breaking of scaling relations, enhancing the intrinsic activity toward multicarbon products. This is achieved due to an increased stabilization of *CO, particularly at Cu sites with a high C–C coupling activity.
Acetic acid is a highly desirable multicarbon product with a worldwide production of more than 10 million tonnes annually.87 Industrial production of acetic acid is currently achieved through the carbonylation of methanol using rhodium or iridium organometallic catalysts under high pressure and temperature conditions.88 The electrochemical conversion of CO2 into acetate offers the possibility of producing this bulk chemical at ambient conditions. Zeng and coworkers reported using amino-functionalized copper surfaces (Cu@NH2) for the electrocatalytic reduction of CO to acetate.89 The hybrid Cu@NH2 catalysts were prepared by the electroreduction of the molecular precursor [Cu(NH3)4]Cl2·H2O previously spray-coated on carbon fiber papers.
Upon reduction at −0.6 V vs. RHE in 1.0 M KOH for 600 s, the formation of copper nanoparticles with a mean diameter of 100 nm was observed with the use of high-resolution transmission electron microscopy (HRTEM), and the common Cu(100) and Cu(111) facets were detected. Energy-dispersive X-ray spectroscopy (EDX) showed a uniform distribution of Cu and N on the surface of the hybrid Cu@NH2 catalyst. XPS, in situ Cu K-edge X-ray absorption near-edge structure (XANES), and Cu LMM Auger electron spectroscopy revealed that the surface valence states of Cu@NH2 consisted of a mixture of Cu0 and Cu+ with a significant contribution from the zero-valent state. Furthermore, the nature of the surface amino group, –NH2, was corroborated by XPS and ex situ XANES, where the corresponding Cu–N and N–H transitions were observed at 401 eV and 407 eV, respectively.
The Cu@NH2 catalysts were tested for the electrochemical reduction of CO in 1 M KOH electrolyte, displaying a CO-to-acetate FE of 51.5% and a partial current density of −150 mA cm−2 at −0.75 V vs. RHE (Fig. 4a). For comparison, commercial copper nanoparticles (60–80 nm diameter) only reached a partial current density for acetate of −50 mA cm−2 under the same electrocatalytic conditions, suggesting that the surface amino functionalization on Cu can significantly enhance its catalytic activity for the generation of acetate. Comparing the ECSA-normalized acetate evolution activities of Cu@NH2 and bare Cu catalysts, a four-fold increase in acetate production was observed at −0.85 V vs. RHE, excluding the possibility of a higher surface area to be responsible for the enhanced catalytic activity.
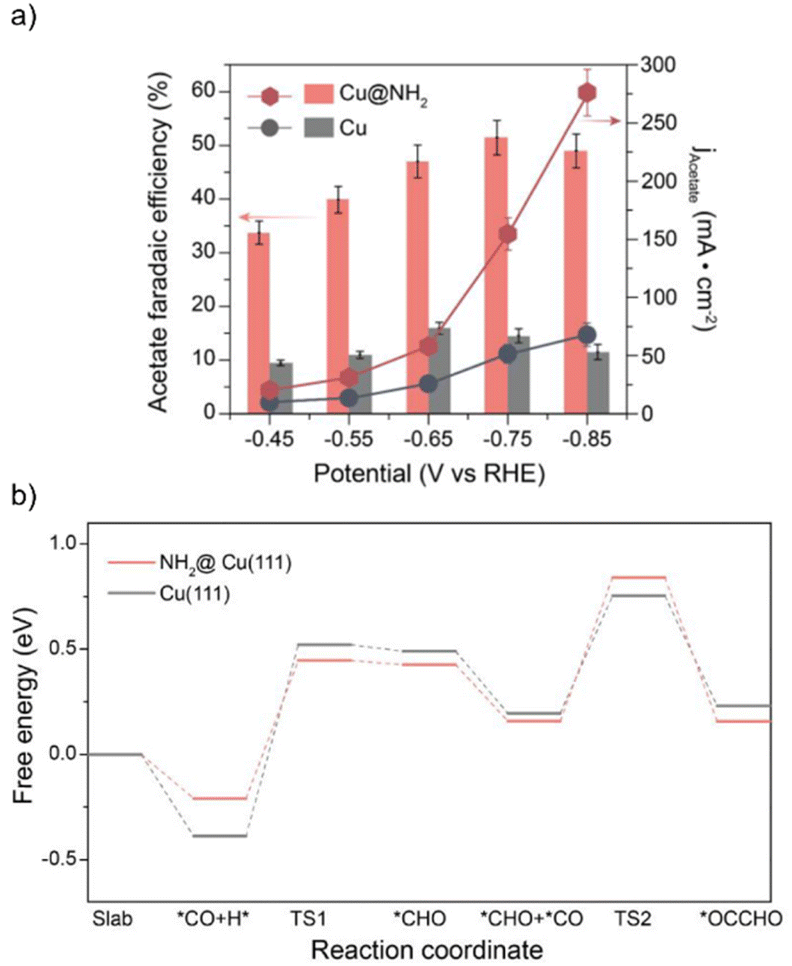 | ||
| Fig. 4 (a) Acetate FE and partial current density for CO reduction on Cu@NH2 and Cu recorded in CO-saturated 1.0 M KOH solution. (b) Free energy diagram of the CO reduction mechanism showing *CO hydrogenation to *CHO and the subsequent coupling with another *CO to *OCCHO on Cu@NH2(111) and Cu(111). Adapted with permission from ACS Catal. 2023, 13, 3532–3540. Copyright 2023 American Chemical Society. | ||
In situ Raman spectroscopy and in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) revealed different surface speciation of reaction intermediates on Cu@NH2 and bare Cu catalysts during CO electroreduction. Compared to bare Cu, the surface coverage of key intermediates *CO and *CHO was higher on the hybrid surface at potentials more negative than −0.4 V vs. RHE. Additionally, it was found that the absorption bands for *COatop in the Raman spectra of Cu@NH2 are red-shifted compared with the bare Cu samples, indicating that the C–O bond in *COatop on Cu@NH2 is weaker than that on bare Cu.
DFT calculations showed that in the presence of the surface amino groups, the energy barrier for the hydrogenation of *CO on a Cu(111) slab to form *CHO is reduced by 0.3 eV (Fig. 4b). Such hydrogenated intermediates have been demonstrated to trigger the C–C bond formation step to form multicarbon products since the *CO–*CHO coupling reaction has a lower energy barrier than the dimerization reactions of *CO and *CHO.90 The electron-donating properties of the surface amino groups were evidenced by the influence on the electronic structure of the hybrid catalyst by shifting the center of the Cu d-band from −2.19 eV in bare Cu to −2.23 eV in Cu@NH2, resulting in increased π backbonding from the Cu surface to *CO. Consequently, the C–O bond is weakened, as reflected in the red-shifted Raman spectra of Cu@NH2, and more labile toward protonation reactions. Most importantly, charge density difference plots showed an electronic interaction between the surface NH2 residues and the *CHO intermediate, corroborating the existence of a hydrogen bonding effect. Such a hydrogen bonding interaction selectively enhances the adsorption of *CHO and facilitates the reduction of *CO into *CHO, thus overcoming the existing scaling relationship in bare Cu. Overall, the amino groups on the surface of the hybrid electrocatalyst are responsible for maintaining the low valence state of Cu. The Hδ+ of the organic residues can stabilize the *CHO intermediates, enhancing the intrinsic activity of Cu sites for acetate generation.
Lim and coworkers reported histidine-functionalized Cu electrodes for the electrochemical CO2RR to multicarbon products with a combined FE of up to 76.6% at −2.0 V vs. RHE and excellent stability for 48 h.91 The histidine-functionalized Cu2O catalysts (Cu-Hist) were prepared by reducing copper chloride with glucose in basic pH and the presence of 1.5 mol% of histidine. Histidine was selected due to its ability to bind strongly to Cu while containing an imidazole group that can bind and activate CO2.92 The obtained Cu-Hist nanoparticles exhibited the same surface morphology and lattice spacing compared to samples prepared in the absence of an organic modifier, as revealed in scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, respectively.
Cu-Hist was tested in the electrochemical CO2RR using 0.1 M KHCO3 solutions as electrolytes, obtaining a peak FE of 76.6% at −2.0 V vs. RHE. These results contrast with a bare Cu catalyst for which, at potentials more negative than −1.2 V vs. RHE, the FE sharply decreased from 50% to less than 10% at −2.0 V vs. RHE. Additionally, partial current densities for the production of multicarbon products were ten times higher than for bare Cu at −2.0 V vs. RHE. The ECSAs of Cu-Hist and bare Cu catalysts were similar, as expected from the lack of a new metal component in the hybrid catalyst. These results suggested an enhancement of the intrinsic activity of the copper active sites in the Cu-Hist system. The product distribution during the electrocatalytic evaluations proved sensitive to the organic modifier's molecular structure. Similar Cu2O nanoparticles were prepared in the presence of imidazole (Cu-Im), imidazolepropionic acid (Cu-ImPA), and 2-methylimidazole (Cu-2mIm). Although increased selectivities for multicarbon products were obtained with Cu-Im and Cu-ImPA compared to bare Cu, their electrocatalytic performance was poorer than Cu-Hist. On the other hand, Cu-2mIm displayed similar electrocatalytic activity as bare Cu, suggesting that an unblocked C2 imidazole position is crucial for altering CO2RR selectivity. Masel and coworkers have suggested that imidazole-based cations can lower the activation energy for the formation of the CO2− intermediate by facilitating the conformational change from the linear CO2 molecule to the bent CO2− anion.92 Furthermore, the authors argue that the presence of carboxylic acid and imidazole groups results in a more stable anchoring of Hist and ImPa to the Cu surface compared to the other tested organic additives. This, in turn, may be reflected in the remarkable stability of Cu-Hist and higher sustained performance throughout the electrolysis. Finally, the extra α-amino functionality in the histidine molecule affords an additional interaction site with the surface-bound intermediates during the CO2RR, rendering the Cu-Hist system the most selective electrocatalyst for multicarbon products among all tested hybrid catalysts.
In situ Raman spectroscopy of Cu-Hist showed three distinct bands under electrochemical bias at 1009, 1640, and 2079 cm−1, which are absent for a bare Cu electrode, indicating that they correspond to new interactions between histidine and either reduced Cu or CO2RR intermediates. Surprisingly, the expected bands at 276–290 cm−1 for the Cu–CO frustrated rotation and 354–370 cm−1 for the Cu–CO stretching93 were absent in the hybrid catalyst, even after ramping the cathodic potential to −1.10 V.
Based on DFT calculations, the authors proposed an alternative reaction mechanism for the Cu-Hist electrocatalyst where the amino group of histidine functions as a binding site for CO2, remaining attached to the nitrogen atom throughout its reduction to multicarbon products (Fig. 5). In this mechanistic proposal, only one equivalent of *CO must be formed on the Cu surface to be hydrogenated to *CHO and coupled with a histidine-bound *CO. Such a C–C coupling step was calculated to be kinetically and thermodynamically more favorable than a *CO–*CO and *CO–*CHO coupling on a Cu(100) surface. This alternative mechanism for CO2RR via histidine-assisted transformations helped to rationalize the absence of the Cu–CO frustrated rotation and stretching bands in the Raman spectra. Since CO2 might be transformed into CO while bound to the histidine molecule, the interaction of *CO with the Cu surface sites is limited, thus affecting the intensity of the characteristic Raman bands. In situ Raman and DFT calculations hence revealed a direct interaction between reaction intermediates and the adsorbed histidine.
 | ||
| Fig. 5 (a) Gibbs free energy diagram and (b) snapshots of the first few surface intermediates in the histidine-assisted CO2RR mechanism. Adapted with permission from Nat. Commun., 2023, 14, 335. | ||
A recent investigation on the oxygen evolution reaction (OER) showed the interplay between the applied voltage, accumulated surface charge, catalyst oxidation state change, and reaction rate using a pulsed voltammetry-based technique.94 Employing a similar approach, Lim and coworkers also determined the surface charge's role in controlling CO2RR selectivity with Cu catalysts functionalized with organic additives.91 A linear correlation was observed between surface charge and the multicarbon product selectivity, where higher selectivities are linked to higher rates for the build-up of charged species on the catalyst surface and slower rates for desorption. Although the mPV method does not directly identify the accumulated species, the observed correlations suggest that the increased surface charge in the best-performing catalysts originates from the enhanced stabilization of CO2RR intermediates (vide supra). These results offer a novel approach for quantifying the effectiveness of organic functionalities in stabilizing key CO2RR intermediates by measuring the population of charged species on the catalytic surface. Although such a surface charge modulation effect may be similar to the cation effect,95–97 surface-tethered organic groups have the advantage of being specifically adsorbed through their unique combination of functional groups, providing a tailored strategy to boost CO2RR selectivity by selectively stabilizing surface intermediates. Overall, the results of Lim and coworkers showed how introducing organic residues on metallic surfaces can enable alternative reaction pathways by regulating the population density of crucial reaction intermediates, overcoming scaling relationships, and rerouting the electrocatalytic conversion of CO2 toward multicarbon products.
The microenvironment around Cu sites can also be adjusted by incorporating local proton sources and hydrogen-bond donors, leading to a shift in CO2RR selectivity toward multicarbon products. Zhao and colleagues utilized a Cu-based metal–organic framework (MOF) as a well-defined crystalline model electrocatalyst to explore the influence of local proton availability on the rate of the C–C coupling step. This hybrid catalyst was also used to elucidate mechanistic aspects of the C–C coupling step in the pathway toward multicarbon products.98Cu-HITP (HITP = 2,3,6,7,10,11-hexaiminotriphenylene) was synthesized through a solvothermal reaction involving the ligand HITP and hydrated Cu(NO3)2, resulting in the formation of the Cu MOF as a microcrystalline powder. Cu-HITP comprises a two-dimensional MOF structure constructed from HITP linkers and square-planar CuN4 nodes. The two-dimensional layers are stacked via π–π interactions along the [001] direction, forming hexagonal channels.99 HRTEM images depicted the characteristic rod-like structure of the microcrystals and revealed a distance of 3.4 Å between adjacent interlayer Cu sites along the c-axis. Considering that the surface of Cu-HITP is terminated with copper ions, numerous dual copper sites are present along the c-axis on the surface of the MOF crystal, potentially serving as active sites for the CO2RR.
The electrocatalytic performance of Cu-HITP was evaluated using a 0.1 M KHCO3 solution as the electrolyte in a gastight H-type cell. FEs toward multicarbon products and H2 reached 42% and 17% at −1.2 V vs. RHE, respectively, with a total current density of −5.2 mA cm−2 (Fig. 6a). Electrochemical experiments conducted with a nanosheet Cu-HITP sample, where CuN4 sites are predominantly abundant, exhibited lower selectivity for multicarbon products (FE = 14%). This finding supports the notion that the dual-atomic Cu sites in the rod-like sample serve as the active sites, thus increasing the selectivity for multicarbon products. DFT calculations have shown that the energy barrier for the coupling between *CO and *COH (0.87 eV at U = 0 V) is much lower than the barrier for the direct *CO dimerization (1.15 eV at U = 0 V), which was attributed to the ability of *COH to acquire a radical character.81 The resulting *COCOH intermediate shows a C–C distance of 1.55 Å and a C–C site angle of about 108–120°, indicating that the ideal distance between two Cu sites for forming the *COCOH species is within 2.78–3.56 Å. Accordingly, the value of the interlayer Cu–Cu distance in Cu-HITP (3.4 Å) is suitable to favor the low-energy path of *CO coupling to *COH for generating multicarbon products (Fig. 6b).
 | ||
| Fig. 6 (a) Comparison of CO2RR performances of Cu-HITP with different PDA coating amounts at −1.2 V vs. RHE. (b) Free energy diagram for forming the C–C bond on the Cu-HITP model. Adapted with permission from ACS Catal., 2022, 12, 7986–7993. Copyright 2022 American Chemical Society. | ||
Local proton sources and hydrogen-bond donors were introduced around the bimetallic copper sites by coating the surface of Cu-HITP with polydopamine (PDA), a conductive polymer featuring amino and phenolic hydroxyl groups. HRTEM and SEM images showed that the hybrid Cu-HITP@PDA samples, prepared through the self-polymerization of dopamine in the presence of the MOF microcrystals, present a homogenous coating of PDA on the surface of Cu-HITP. Electrocatalytic evaluation of Cu-HITP@PDA indicated an optimal Cu-HITP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PDA ratio of 10:5, with a selectivity toward multicarbon products reaching 75% at −1.2 V vs. RHE and no loss of the total current density when compared to Cu-HITP (Fig. 6a). The enhanced selectivity toward multicarbon products for Cu-HITP@PDA indicates that the microenvironment at the dual-Cu sites formed by the PDA layer played an essential role in shifting the selectivity. Long-term bulk electrolysis experiments showed a maintained performance for up to 10 hours.
:PDA ratio of 10:5, with a selectivity toward multicarbon products reaching 75% at −1.2 V vs. RHE and no loss of the total current density when compared to Cu-HITP (Fig. 6a). The enhanced selectivity toward multicarbon products for Cu-HITP@PDA indicates that the microenvironment at the dual-Cu sites formed by the PDA layer played an essential role in shifting the selectivity. Long-term bulk electrolysis experiments showed a maintained performance for up to 10 hours.
Cu K-edge in situ XANES spectra of Cu-HITP@PDA measured at −1.2 V vs. RHE in 0.1 M KHCO3 after 1 hour of electrolysis showed no significant change in the oxidation state of CuII, ruling out the possible formation of Cu or Cu2O clusters during electrocatalysis.100 The spectra of extended X-ray absorption fine structure (EXAFS) of Cu-HITP@PDA showed that the first coordination center peak of 1.96 Å (corresponding to the Cu–N bond) remains unchanged, demonstrating that the coordination configuration of CuII ions was unchanged throughout the catalytic process.
Electrocatalytic control experiments employing two distinct types of polymeric surface additives elucidated the pivotal role of PDA in modulating proton delivery to enhance multicarbon product formation. Coating a Cu-HITP sample with poly(p-vinylphenol), a polymer solely comprising phenolic hydroxy groups, increased the selectivity for CH4 production and decreased the CO generation compared to uncoated Cu-HITP. This outcome suggests that phenolic hydroxyl groups facilitate the hydrogenation of *CO to *COH, a crucial intermediate in the methane pathway. Conversely, when Cu-HITP was coated with a polymer consisting solely of amino groups on phenyl rings (polyaniline, PANI), the resultant hybrid catalyst exhibited enhanced selectivity for multicarbon products relative to Cu-HITP, albeit not reaching the high values observed for Cu-HITP@PDA. Overall, these findings suggest that the presence of both functional groups in the Cu-HITP@PDA structure promotes the hydrogenation of *CO into *COH by the phenolic hydroxyls and the stabilization of critical multicarbon products intermediates (*COH and *COCOH) through hydrogen-bond interactions with the amino groups. Hence, tailoring the environment of Cu sites with local proton sources and hydrogen-bond donors facilitates the low-energy pathway for multicarbon product generation, thereby enhancing their selectivity.
Unifying concepts: scaling relations in molecular and heterogeneous electrocatalysts
In the previous sections, we discussed how second coordination sphere interactions in molecular electrocatalysts can accelerate the conversion of CO2 into CO and HCOOH. Similarly, functionalizing metallic electrodes with organic ligands that interact with surface intermediates can selectively promote the transformation of CO2 into multicarbon products. In both electrocatalytic systems, modifying the microenvironment with organic residues positively influences the reaction outcome, and understanding the origin of this enhanced activity is crucial for designing more advanced systems. Specifically, the improvement may result from established scaling relations between catalytic activity and an intrinsic property of the modified metal center, or it may stem from creating a new reaction pathway that bypasses this linear correlation. This type of analysis can also shed light on mechanistic parallels between successful molecular and heterogeneous electrocatalysts that do not adhere to typical scaling relations.For instance, the benchmarking of molecular electrocatalysts for the CO2-to-CO conversion using catalytic Tafel plots has revealed a scaling relationship between catalytic activity and the standard potential of the transition-metal complex (E0cat).43,54,55 Catalytic Tafel plots are constructed by plotting the measured TOF of the electrocatalyst against the overpotential (η), which is defined as the difference between the applied potential (E) and the equilibrium potential of the catalyzed reaction (ECO2/CO). The overpotential represents the driving force required to produce CO at a given TOF, and thus, efficient catalyst systems are expected to display high TOF values at low η. These plots typically show two regions: first, a linear increase in log(TOF) with increasing η, followed by a second region where the TOF reaches a maximum plateau value (TOFmax) as η increases further.
Analysis of log(TOF)–η diagrams for a series of structurally similar metal complexes, all tested under identical reaction conditions, has shown that introducing electron-donating substituents shifts E0cat to more cathodic values. This favorably increases TOFmax but raises the required overpotential.45 Conversely, electron-withdrawing substituents have the opposite effect. The origin of this trend for such through-structure effects lies in the existing scaling relationship between TOFmax and E0cat. In fact, for these particular cases, plotting ln(TOFmax) as a function of E0cat produces a linear correlation, indicating that any increase in the electrocatalytic activity is directly linked to the electron density at the metal center. Therefore, when a system effectively circumvents such a correlation, TOFmax increases without the compensating changes in E0cat that also increase η.
The primary drawback of using the classic definition of overpotential as a figure of merit in benchmarking molecular electrocatalysts is that it relies solely on thermodynamic parameters, which can obscure mechanistic insights. In contrast, previous research in the field of the oxygen reduction reaction (ORR) has introduced an alternative measure known as the effective overpotential (ηeff).101–106 This approach calculates the difference between the equilibrium potential of the catalyzed reaction and the reduction potential at which catalysis begins, typically defined as the potential at which half of the maximum catalytic current is achieved (Ecat/2) (see Section S1.6. in the ESI for more details†).107 Determining this type of catalytic potential provides a higher precision compared to other measures, such as the onset or peak potential. Furthermore, it is crucial to account for all factors influencing the equilibrium potential ECO2/CO to accurately compare electrocatalytic systems evaluated under different conditions. These include solvent type, the concentration and pKa of the proton donor, and the water content in the electrolyte.
For the case of the electrochemical conversion of CO2 into CO in a nonaqueous solvent (eqn (1)), the equilibrium potential of this reaction is given by the Nernst equation (eqn (2)):
| CO2 + 2H+ + 2e− ⇌ CO + H2O | (1) |
 | (2) |
 is the equilibrium potential under standard conditions. Therefore, ηeff takes the following form:
is the equilibrium potential under standard conditions. Therefore, ηeff takes the following form: | (3) |
In this section, we extend the analysis to molecular CO2RR catalysts and establish a framework for identifying any scaling relation that governs their performance. This enables pinpointing electrocatalysts whose enhanced activity results from overcoming a particular scaling relation. As a first step, we calculated ηeff according to eqn (3) for a comprehensive set of electrocatalysts, each evaluated under different reaction conditions (details are provided in the ESI†). To do this, we use a recently published method of Matsubara, allowing for a comprehensive determination of ECO2/CO.110,111 Subsequently, we constructed plots correlating TOFmax, measured by CV, with ηeff for various molecular CO2RR catalysts (a complete list of the analyzed catalyst structures can be found in the ESI†).
As anticipated from the variables in eqn (3), our analysis revealed a linear correlation between TOFmax and the concentration of the proton donor when all other experimental conditions were held constant. Fig. 7 illustrates these correlations for a range of iron-based hangman porphyrin catalysts (Fe14–Fe16) alongside the original non-substituted Fe1 catalyst, each tested at various concentrations of phenol. The linear trend observed for Fe1 indicates that higher TOFmax values are associated with increased concentrations of phenol, which correlates with a rise in ηeff. This result agrees with the well-established proton-dependency in the RDS for this catalyst,23,24 and argues against the computational work by de Visser and Davethu who found the CO2 binding event to be rate-determining (vide supra).26 The slope of this linear plot (mHA) is 15.6 dec V−1, closely aligning with the theoretical value of 16.9 dec V−1 (see ESI for details†). The derivation of the theoretical slope assumes an expression for TOFmax in which the reaction order of the proton donor concentration, [HA], and the CO2 partial pressure, pCO2, equal 1. The excellent compliance between the experimental and theoretical slope supports this assumption, which is also in agreement with the reaction order of 1 for [HA] identified by Nocera and coworkers for the Fe hangman porphyrins.51
 | ||
| Fig. 7 Scaling relationships for iron hangman porphyrins Fe14–Fe16 and Fe1 showing the effect of phenol (PhOH) concentration. For Fe14–Fe16, [PhOH] = 2, 4, 10, 20, and 40 mM were tested. For Fe1, [PhOH] = 0.1, 0.75, 3.0 M were used. In all cases, a higher phenol concentration led to a higher TOFmax. The slopes of the linear regions are 15.58, 11.65, and 18.08 dec V−1 for Fe1/Fe14, Fe15, and Fe16, respectively. | ||
Furthermore, the scaling slopes for the four catalysts in Fig. 7 are very similar (15.58, 11.65, and 18.08 dec V−1 for Fe1/Fe14, Fe15, and Fe16, respectively). This could indicate that the general mechanism for these catalysts is the same, including the RDS. Notably, catalysts Fe15 and Fe16, which contain intramolecular proton-donating groups like an amidine or a benzenesulfonic acid, respectively, deviate from the scaling relationship observed with Fe1. This is demonstrated by the differing TOFmax values at the same ηeff. However, these modified iron complexes are less effective electrocatalysts compared to Fe1, indicating that introducing secondary coordination effects does not always improve catalytic performance over unmodified systems. On the other hand, Fe14 follows the same scaling relationship as Fe1, suggesting that the presence of the dangling phenol group near the iron center does not significantly alter the reaction mechanism compared to Fe1. Instead, this functional group likely modifies the local proton inventory around the metal center, with any increase in activity being accompanied by a simultaneous increase in the effective overpotential.
This type of analysis can also identify scaling relations arising from through-structure electronic effects, similar to those observed in the catalytic Tafel plots. When molecular complexes do not undergo side phenomena during electrocatalysis, such as decomposition, Ecat/2 is essentially equivalent to the standard potential of the catalyst couple E0cat.112 This is generally true for most iron porphyrin catalysts, where E0cat corresponds to the Fe(I/0) redox couple. For these catalysts, a clear scaling relationship can be established between TOFmax and ηeff when E0cat is modulated by incorporating electron-withdrawing or electron-donating groups into the ligand structure. Fig. 8 illustrates this by presenting linear correlations for a series of iron porphyrin catalysts (Fe21–Fe23), which incorporate one, two, and four perfluorinated phenyl rings, respectively, along with the unmodified Fe1.
 | ||
| Fig. 8 Scaling relationships for Fe21–Fe23 and Fe1 at different concentrations of phenol. ◊ = Fe1, ○ = Fe21, Δ = Fe22, □ = Fe23. The slopes of the lines are purple = 10.3 dec V−1, green = 28.2 dec V−1, and orange = 24.10 dec V−1. | ||
The plot shows these correlations for three different concentrations of the proton donor (phenol). As the level of fluorination increases, the iron center becomes more Lewis-acidic, resulting in a positive shift in E0cat. This shift decreases the Lewis basicity of the reduced form of the catalyst, thereby lowering its reactivity in the product formation process. As shown in Fig. 8, Fe1 achieves the highest TOFmax across all phenol concentrations, followed by Fe21, Fe22, and Fe23, in that order. The slopes of the lines are 24.1, 28.2, and 10.2 dec V−1 for phenol concentrations of 0.1, 1.0, and 3.0 M, respectively. While the slopes at 0.1 M and 1.0 M are similar, indicating that the same mechanism prevails and hence the same proton dependency, the slope at 3.0 M is significantly lower, consistent with the reported inhibitory effect of phenol at very high concentrations.44 These findings demonstrate the utility of log(TOFmax)–ηeff plots for straightforwardly extracting mechanistic insights from analyzing a series of electrocatalysts.
Similar linear correlations were observed for structurally related iron porphyrin complexes, Fe24 and Fe25, which are tetra-substituted with CF3 and OMe groups, respectively. Along with Fe1, a slope of 29.7 dec V−1 was obtained, as shown in Fig. 9. As expected, the most electron-rich iron center in Fe25 exhibited the highest TOFmax. The slope is similar to those obtained in Fig. 8 for phenol concentrations of 0.1 M and 1.0 M, which agrees with an unchanged reduction mechanism for Fe24 and Fe25 compared to Fe21–Fe23. Interestingly, a similar plot constructed for the Re bipyridine catalysts Re1 and Re11–Re13 showed the same varying effect of Ecat/2 on the correlation between ηeff and TOFmax, yet with a significantly smaller slope of 1.54 dec V−1 (Fig. S3 in the ESI†). The substantial difference in slopes between the Re and Fe catalysts likely stems from their distinct reduction mechanisms. As outlined in Scheme 2, the Re-based catalysts generate CO predominantly via the reduction-first pathway, with proton-coupled C–O cleavage serving as RDS.27 Because this RDS is a purely chemical step, it remains largely independent of Ecat/2. Conversely, the RDS for the traditional Fe porphyrins is proposed to proceed via a concerted proton-electron transfer-bond cleavage reaction, in which the electron transfer originates from the Fe center to the substrate.24 This step is highly sensitive to electronic effects, as the porphyrin substituents tune the electron density at the Fe center, thereby influencing the rate of the rate-determining electron transfer. As a result, TOFmax increases sharply when electron-donating substituents enhance the electron density of the Fe center, explaining the much steeper slopes observed for the Fe catalysts in Fig. 8 and 9.
 | ||
| Fig. 9 Breaking of the scaling relation set by Fe24, Fe1, and Fe25 (green) by functionalized catalysts Fe2, Fe5, and Fe12 (purple), all containing hydrogen-bonding/proton-donating organic groups in the second coordination sphere. The slope of the scaling relation set by Fe24, Fe1, and Fe25 is 29.7 dec V−1. Fe1, Fe24, Fe25, and Fe5 were tested in the presence of 0.1 M PhOH, while Fe2 and Fe12 were tested in the presence of 2.0 M and 5.5 M of water, respectively. | ||
The plots in Fig. 8 and 9 illustrate that, in most cases, tuning the electronic density at the metal center via synthetic modification of the supporting ligand does not enable the electrocatalytic systems to escape the scaling relations that govern these catalysts. However, several catalysts were identified that successfully deviated from such linear correlations. The five best-performing catalysts – Fe2, Fe5, Fe12, Fe19, and Fe20 – all feature a functional group in the second coordination sphere capable of selectively interacting with reaction intermediates at the metal center (Fig. S4 in the ESI†). For example, the microenvironments of Fe2, Fe5, and Fe12 contain organic residues with hydrogen-bonding and proton-donating groups, such as hydroxyl, amide, and urea (Fig. 9). Aukauloo and coworkers investigated the origin of the enhanced activity of Fe12 through DFT modeling.50 They found that the urea moieties can selectively interact with the [Fe–CO2] adduct, resulting in a stabilization of 29 kcal mol−1. This type of through-space interaction offers an orthogonal approach to stabilizing key reduction intermediates via hydrogen-bonding interactions, independently of Ecat/2. As a result, a different correlation between TOFmax and ηeff emerges, leading to the deviation of functionalized catalysts from the scaling relation governed by Ecat/2. Consequently, these catalysts can enter a new scaling relationship that allows for higher activity without the corresponding increase in effective overpotential.
Our analysis also highlighted the impact of the spatial arrangement of second-sphere functional groups within a series of structurally related electrocatalysts. As shown in Fig. 10, complexes Fe4–Fe7, which feature pendant amide groups, successfully deviate from the scaling relations indicated in yellow and discussed previously. Based on their position in the log(TOFmax)–ηeff plot, the electrocatalytic efficiency follows the order: Fe5 > Fe4 > Fe7 > Fe6. Notably, the arrangement of the pendant amide group relative to the metal center plays a significant role in determining how much they deviate from the linear correlation. The superior performance of Fe5 is attributed to the ortho-positioning of the amide, which leads to a shorter hydrogen bond between the amide and the [Fe–CO2] intermediate.45 This configuration enhances the through-space interaction compared to other positional isomers. As previously discussed for Fe12, the independent through-space stabilization of specific reduction intermediates introduces an additional degree of freedom. When this interaction is favorable, as seen in Fe5 and Fe4, it enables the catalysts to enter a superior scaling relationship. In contrast, Fe6 and Fe7, where the amide groups are positioned para to the porphyrin phenyl rings, exhibit this effect to a much lesser extent and, therefore, adhere more closely to the established scaling relations.
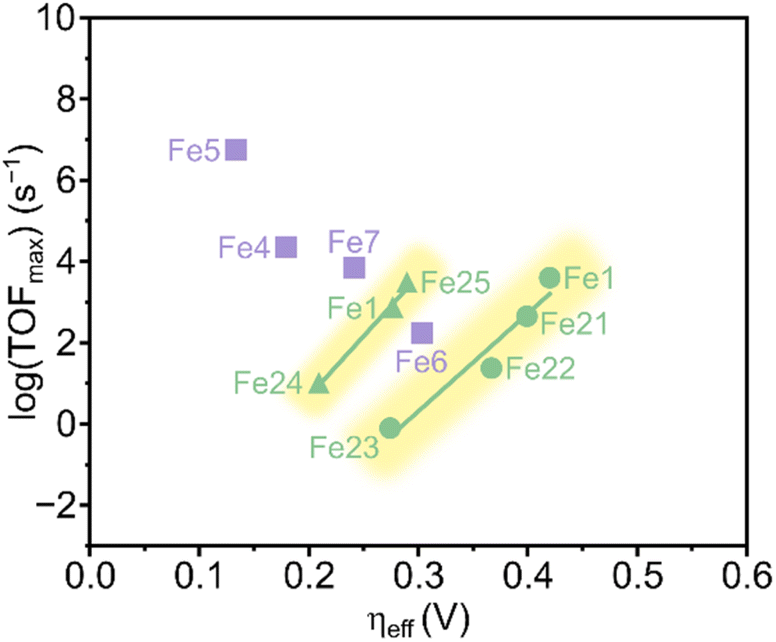 | ||
| Fig. 10 Overcoming linear scaling relations by amide-functionalized catalysts Fe4–Fe7. All catalysts included in this plot were tested in the presence of 0.1 M PhOH. | ||
We also found that attaching cationic groups in the second coordination sphere is an effective strategy for developing highly efficient molecular electrocatalysts that can bypass established scaling relations (Fig. 11). For example, Fe19 and Fe20, which feature positively charged ortho-TMA and imidazolium groups, respectively, exhibit the lowest ηeff among the studied catalysts while maintaining a high activity. In the case of Fe20, Nippe and colleagues suggested that the C2–H moiety of the intramolecular imidazolium group selectively stabilizes the [Fe–CO2] intermediate through hydrogen bonding, thus facilitating its protonation.72 Similarly, Savéant and coworkers proposed that the enhanced electrocatalytic performance of Fe19 stems from coulombic interactions between the four positively charged TMA groups and the negative charge on the initial [Fe–CO2] adduct.54 When these TMA groups are placed at the para positions of the phenyl rings, the stabilization effect diminishes due to the weaker coulombic interaction from the increased distance between the charged fragments. Consequently, Fe18 deviates less from the linear trend shown in Fig. 11. This stabilization mechanism is further supported by the poor catalytic performance of Fe26, which appears in the lower right of the linear correlation plot. The unfavorable effect in Fe26 is due to the replacement of the four positive charges with four negative charges provided by sulfonate groups at the para positions of the phenyl rings.
 | ||
| Fig. 11 Iron porphyrins with cationic (Fe18, Fe19, Fe20) and anionic (Fe26) groups in the second coordination sphere circumventing the linear scaling relationship set by Fe1 and Fe21–Fe23. The slope of the scaling relation is 28.2 dec V−1. Fe1 and Fe21–Fe23 were tested in the presence of 1.0 M PhOH, Fe18, Fe19, Fe26 were tested in the presence of 0.1 M H2O and 3.0 M PhOH, and Fe20 was tested in the presence of 5.55 M H2O. | ||
The free energies of all intermediates and transition states along a reaction pathway determine the overall rate of a catalyzed reaction. However, due to the linear correlations among the energies of these intermediates and the relationships between the transition states and intermediates, the number of independent descriptors that influence the reaction rate is reduced. Our analysis highlights that incorporating strategically positioned functional groups within the microenvironment of the metal center can selectively stabilize specific intermediates, providing an additional degree of freedom. This extra flexibility enables the system to overcome the linear correlations that constrain the reaction rate of the catalyzed process. The significance of selectively tuning the energy of a reaction intermediate is well-established in heterogeneous electrocatalysis. In this field, the binding energy of key surface intermediates plays a critical role in determining the catalytic selectivity and activity, especially in CO2RR, where multiple reaction pathways coexist. In fact, the adsorption energies of surface intermediates are similarly correlated through thermodynamic scaling relations that govern the overall performance of the catalysts.17 The hybrid electrodes discussed in the previous section, which feature surface organic films, operate on the same principle as the functionalized molecular catalysts. Here, the interactions between the organic residues at the electrode surface and a specific intermediate serve as an independent descriptor, resulting in a more efficient electrocatalytic system that escapes previously established scaling relations.
For instance, in the previous section, we highlighted examples of hybrid electrocatalytic systems exhibiting enhanced intrinsic reaction rates for multicarbon product generation. In each case, the metal–organic interfaces selectively stabilized a particular CO2RR intermediate. By favoring multicarbon products, these molecular films stabilize intermediates such as *CO, *CHO, or *COH, effectively lowering the energy barrier for the crucial C–C coupling step.
Outlook
The analysis in the previous section allowed us to compare the intrinsic activities of various homogeneous electrocatalysts tested under different experimental conditions using a universal descriptor such as ηeff. Extending this quantitative approach to heterogeneous systems would be highly beneficial, as it could enable the systematic investigation of the effects of molecular additives on hybrid organic–metal interfaces and establish a rational framework for the design of future catalytic systems. For homogeneous catalysts, calculating ηeff relied on the ability to determine Ecat/2—a metric specific to each electrocatalyst. Moreover, this metric relates solely to the formation of a single product, specifically CO, in the cases analyzed here. However, a similarly straightforward parameter capturing the catalyst performance akin to Ecat/2 is not yet well established for heterogeneous systems. The onset potential at which specific products are generated at the electrode surface shows a promising candidate for such a metric. Yet, for catalytic materials like Cu, multiple products are frequently generated at similar potentials, complicating this type of analysis. The challenge thus lies in disentangling the potential effects on the formation of all the possible CO2RR products.For example, Koper and coworkers demonstrated the application of online electrochemical mass spectrometry (OLEMS) to determine the onset potentials for different products during CO reduction on Cu(111) and Cu(100) electrodes.113 This approach enabled the monitoring of mass spectrometry signals for C2H4 and CH4 as a function of the applied potential. Extending this type of in situ technique to heterogeneous catalysts with altered microenvironments would offer valuable insights into the catalyst's performance. By plotting the ECSA-normalized partial current density for a specific product as a function of its measured onset potential, it may be possible to identify scaling relations on unfunctionalized metal surfaces. Comparing these plots with those obtained for functionalized metal surfaces could reveal whether surface organic functionalities enhance electrocatalytic performance by overcoming the identified scaling relationships.
Experimentally measuring the adsorption energies of the key CO2RR intermediates could also enable a more systematic benchmarking of hybrid electrodes and yield quantitative insights into how tailored microenvironments influence the electrocatalytic performance. Theoretical studies have previously identified a linear correlation between *CO adsorption energy and electrocatalytic activity for generating multicarbon products.74,75 While DFT calculations can readily calculate these adsorption energies, they frequently demonstrate considerable inaccuracies, leading to overpotential errors reaching up to 200 mV.114 This underscores the necessity to substantiate such calculations with experimental measurements. Techniques such as ambient pressure XPS (AP-XPS),115in situ/operando XANES, and EXAFS116 can provide these critical experimental values. Additionally, Liu and coworkers recently developed a thermodynamic–kinetic model to extract adsorption energies of reactive intermediates in the OER by integrating DFT calculations with electrochemical probing methods.117,118 This model could potentially be adapted for CO2RR with hybrid metal–organic electrodes, enabling the determination of the binding energies of important intermediates.
The electrocatalysts discussed in this Perspective highlight how tuning the microenvironment is a powerful strategy to enhance the electrocatalytic performance in both homogeneous and heterogeneous systems. The selective stabilization of key CO2RR intermediates accelerates critical mechanistic steps, resulting in highly active and selective electrocatalysts. While these trends are well-supported by experimental data for the molecular catalysts examined here, similar insights remain sparse for heterogeneous systems. Identifying accessible experimental descriptors of catalyst performance would enable meaningful analyses, particularly for complex reactions like the CO2RR. Such advancements could provide valuable guidance for designing the next generation of electrocatalysts for the CO2RR.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization & supervision, A. R.-H.; literature data collection and calculations for the thermodynamic–kinetic analysis of scaling relations, K. N. K.; writing – original draft, K. N. K.; writing – review & editing, K. N. K., K. T., and A. R.-H. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Danish National Research Foundation (Grant No. DNRF118). This work was supported by the Novo Nordisk Foundation CO2 Research Center (CORC) with grant number NNF21SA0072700 and is published under the number CORC_24_35. We would like to thank Prof. Kim Daasbjerg and Assoc. Prof. Steen U. Pedersen for their valuable discussions during the preparation of this manuscript.References
- IEA, ICCA, and DECHEMA Technology Roadmap, Energy and GHG Reductions in the Chemical Industry via Catalytic Processes, 2013 Search PubMed.
- IEA, Direct CO2 emissions from industry in the Net Zero Scenario, pp. 2000–2030, 2023 Search PubMed.
- IPCC, Global Warming of 1.5 °C: An IPCC Special Report on the Impacts of Global Warming of 1.5 °C above Pre-industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, 2018 Search PubMed.
- IEA, Chemicals, https://www.iea.org/energy-system/industry/chemicals, accessed June 4, 2024.
- G. Lopez, D. Keiner, M. Fasihi, T. Koiranen and C. Breyer, Energy Environ. Sci., 2023, 16, 2879–2909 RSC.
- Á. Galán-Martín, V. Tulus, I. Díaz, C. Pozo, J. Pérez-Ramírez and G. Guillén-Gosálbez, One Earth, 2021, 4, 565–583 CrossRef.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 Search PubMed.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 Search PubMed.
- E. Pastor, Z. Lian, L. Xia, D. Ecija, J. R. Galán-Mascarós, S. Barja, S. Giménez, J. Arbiol, N. López and F. P. García de Arquer, Nat. Rev. Chem, 2024, 8, 159–178 CrossRef PubMed.
- A. Rendón-Calle, S. Builes and F. Calle-Vallejo, Curr. Opin. Electrochem., 2018, 9, 158–165 CrossRef.
- J. Resasco, F. Abild-Pedersen, C. Hahn, Z. Bao, M. T. M. Koper and T. F. Jaramillo, Nat. Catal., 2022, 5, 374–381 CrossRef.
- J. N. Bronsted, Chem. Rev., 1928, 5, 231–338 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1936, 32, 1333 RSC.
- V. Pallassana and M. Neurock, J. Catal., 2000, 191, 301–317 CrossRef CAS.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS.
- M. Frey, ChemBioChem, 2002, 3, 153–160 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1994, 116, 5015–5016 CrossRef CAS.
- C. Costentin, S. Drouet, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2013, 135, 9023–9031 CrossRef CAS PubMed.
- M. Tarrago, S. Ye and F. Neese, Chem. Sci., 2022, 13, 10029–10047 RSC.
- P. A. Davethu and S. P. de Visser, J. Phys. Chem. A, 2019, 123, 6527–6535 CrossRef CAS PubMed.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2014, 136, 16285–16298 Search PubMed.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 Search PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- D. C. Grills, J. A. Farrington, B. H. Layne, S. V. Lymar, B. A. Mello, J. M. Preses and J. F. Wishart, J. Am. Chem. Soc., 2014, 136, 5563–5566 CrossRef CAS PubMed.
- M. H. Rønne, M. R. Madsen, T. Skrydstrup, S. U. Pedersen and K. Daasbjerg, ChemElectroChem, 2021, 8, 2108–2114 CrossRef.
- K. T. Ngo, M. McKinnon, B. Mahanti, R. Narayanan, D. C. Grills, M. Z. Ertem and J. Rochford, J. Am. Chem. Soc., 2017, 139, 2604–2618 CrossRef CAS PubMed.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- A. Briš and D. Margetić, Organics, 2023, 4, 277–288 CrossRef.
- X.-M. Liang, Z.-J. Ruan, G.-Q. Guo, J.-Q. Lin and D.-C. Zhong, ChemCatChem, 2025, 17, e202401394 CrossRef CAS.
- S. Amanullah, P. Saha, A. Nayek, M. E. Ahmed and A. Dey, Chem. Soc. Rev., 2021, 50, 3755–3823 RSC.
- A. W. Nichols and C. W. Machan, Front. Chem., 2019, 7, 397 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2014, 136, 11821–11829 Search PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, ChemElectroChem, 2014, 1, 1226–1236 CrossRef CAS.
- E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Chem. Sci., 2018, 9, 2952–2960 RSC.
- K. Teindl, B. O. Patrick and E. M. Nichols, J. Am. Chem. Soc., 2023, 145, 17176–17186 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 Search PubMed.
- E. J. Kim, J. Feng, M. R. Bramlett and P. A. Lindahl, Biochemistry, 2004, 43, 5728–5734 CrossRef CAS PubMed.
- M. D. Wodrich and X. Hu, Nat. Rev. Chem, 2018, 2, 0099 CrossRef CAS.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504–4509 CrossRef CAS PubMed.
- C. G. Margarit, C. Schnedermann, N. G. Asimow and D. G. Nocera, Organometallics, 2019, 38, 1219–1223 Search PubMed.
- S. Amanullah, P. Saha and A. Dey, J. Am. Chem. Soc., 2021, 143, 13579–13592 CrossRef CAS PubMed.
- M. H. Rønne, D. Cho, M. R. Madsen, J. B. Jakobsen, S. Eom, É. Escoudé, H. C. D. Hammershøj, D. U. Nielsen, S. U. Pedersen, M.-H. Baik, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2020, 142, 4265–4275 CrossRef PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- D. J. Martin and J. M. Mayer, J. Am. Chem. Soc., 2021, 143, 11423–11434 CrossRef CAS PubMed.
- A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Chem. Commun., 2018, 54, 11630–11633 Search PubMed.
- Y. Mao, M. Loipersberger, K. J. Kron, J. S. Derrick, C. J. Chang, S. Mallikarjun Sharada and M. Head-Gordon, Chem. Sci., 2021, 12, 1398–1414 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc. Chem. Commun., 1984, 328–330 RSC.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, ACS Catal., 2018, 8, 2021–2029 CrossRef CAS.
- A. N. Hellman, R. Haiges and S. C. Marinescu, ChemElectroChem, 2021, 8, 1864–1872 CrossRef CAS.
- C. W. Machan, S. A. Chabolla, J. Yin, M. K. Gilson, F. A. Tezcan and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 14598–14607 CrossRef CAS PubMed.
- A. N. Hellman, J. A. Intrator, J. C. Choate, D. A. Velazquez and S. C. Marinescu, Polyhedron, 2022, 223, 115933 CrossRef CAS.
- G. F. Manbeck, J. T. Muckerman, D. J. Szalda, Y. Himeda and E. Fujita, J. Phys. Chem. B, 2015, 119, 7457–7466 CrossRef CAS PubMed.
- E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2018, 140, 12451–12456 Search PubMed.
- S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef CAS PubMed.
- L. Rotundo, E. Azzi, A. Deagostino, C. Garino, L. Nencini, E. Priola, P. Quagliotto, R. Rocca, R. Gobetto and C. Nervi, Front. Chem., 2019, 7, 417 CrossRef CAS PubMed.
- F. Franco, C. Cometto, F. Ferrero Vallana, F. Sordello, E. Priola, C. Minero, C. Nervi and R. Gobetto, Chem. Commun., 2014, 50, 14670–14673 RSC.
- J. Agarwal, T. W. Shaw, H. F. Schaefer and A. B. Bocarsly, Inorg. Chem., 2015, 54, 5285–5294 Search PubMed.
- F. Franco, C. Cometto, L. Nencini, C. Barolo, F. Sordello, C. Minero, J. Fiedler, M. Robert, R. Gobetto and C. Nervi, Chem.–Eur. J., 2017, 23, 4782–4793 CrossRef CAS PubMed.
- S. Sung, X. Li, L. M. Wolf, J. R. Meeder, N. S. Bhuvanesh, K. A. Grice, J. A. Panetier and M. Nippe, J. Am. Chem. Soc., 2019, 141, 6569–6582 CrossRef CAS PubMed.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, New York, 2008, vol. 42, pp. 89–189 Search PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 Search PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 Search PubMed.
- Z.-Z. Wu, X.-L. Zhang, Z.-Z. Niu, F.-Y. Gao, P.-P. Yang, L.-P. Chi, L. Shi, W.-S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M.-R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 Search PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 Search PubMed.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 Search PubMed.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 Search PubMed.
- H. Xiao, T. Cheng, W. A. I. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353–376 CrossRef CAS.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- E. Plaza-Mayoral, P. Sebastián-Pascual, K. N. Dalby, K. D. Jensen, I. Chorkendorff, H. Falsig and M. Escudero-Escribano, Electrochim. Acta, 2021, 398, 139309 CrossRef CAS.
- K. N. Kolding, M. Bretlau, S. Zhao, M. Ceccato, K. Torbensen, K. Daasbjerg and A. Rosas-Hernández, J. Am. Chem. Soc., 2024, 146, 13034–13045 CrossRef CAS PubMed.
- C. M. Gunathunge, V. J. Ovalle, Y. Li, M. J. Janik and M. M. Waegele, ACS Catal., 2018, 8, 7507–7516 CrossRef CAS.
- Y. Ni, L. Shi, H. Liu, W. Zhang, Y. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2017, 7, 4818–4822 RSC.
- A. Haynes, in Advances in Catalysis, ed. B. C. Gates and H. Knözinger, Academic Press, 2010, vol. 53, pp. 1–45 Search PubMed.
- Y. Wang, J. Zhao, C. Cao, J. Ding, R. Wang, J. Zeng, J. Bao and B. Liu, ACS Catal., 2023, 3532–3540 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- C. Y. J. Lim, M. Yilmaz, J. M. Arce-Ramos, A. D. Handoko, W. J. Teh, Y. Zheng, Z. H. J. Khoo, M. Lin, M. Isaacs, T. L. D. Tam, Y. Bai, C. K. Ng, B. S. Yeo, G. Sankar, I. P. Parkin, K. Hippalgaonkar, M. B. Sullivan, J. Zhang and Y.-F. Lim, Nat. Commun., 2023, 14, 335 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- H. Li, P. Wei, D. Gao and G. Wang, Curr. Opin. Green Sustainable Chem., 2022, 34, 100589 CrossRef CAS.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 Search PubMed.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- Z.-H. Zhao, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 7986–7993 Search PubMed.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- Z.-H. Zhao, K. Zheng, N.-Y. Huang, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, Chem. Commun., 2021, 57, 12764–12767 Search PubMed.
- G. Passard, A. M. Ullman, C. N. Brodsky and D. G. Nocera, J. Am. Chem. Soc., 2016, 138, 2925–2928 CrossRef CAS PubMed.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- D. J. Martin, B. Q. Mercado and J. M. Mayer, Sci. Adv., 2020, 6, eaaz3318 CrossRef CAS PubMed.
- Y.-H. Wang, B. Mondal and S. S. Stahl, ACS Catal., 2020, 10, 12031–12039 CrossRef CAS.
- Y.-H. Wang, M. L. Pegis, J. M. Mayer and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 16458–16461 CrossRef CAS PubMed.
- T. Marshall-Roth, L. Liu, V. Mannava, D. M. Harraz, B. J. Cook, R. M. Bullock and Y. Surendranath, ACS Catal., 2024, 14, 18590–18602 CrossRef CAS.
- A. M. Appel and M. L. Helm, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
- M. L. Pegis, C. F. Wise, B. Koronkiewicz and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 11000–11003 CrossRef CAS PubMed.
- D. J. Martin, C. F. Wise, M. L. Pegis and J. M. Mayer, Acc. Chem. Res., 2020, 53, 1056–1065 CrossRef CAS PubMed.
- Y. Matsubara, ACS Energy Lett., 2019, 4, 1999–2004 CrossRef CAS.
- Y. Matsubara, ACS Energy Lett., 2017, 2, 1886–1891 CrossRef CAS.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- J. Greeley, J. K. Nørskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef CAS PubMed.
- Y. Han, H. Zhang, Y. Yu and Z. Liu, ACS Catal., 2021, 11, 1464–1484 CrossRef CAS.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
- J. Zhang, H. B. Tao, M. Kuang, H. B. Yang, W. Cai, Q. Yan, Q. Mao and B. Liu, ACS Catal., 2020, 10, 8597–8610 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for the calculation of ηeff, derivation of ceq,CO, Dcat, and DCO2, compilation of catalyst data extracted from literature and calculation results, structures of analyzed catalysts, and additional figures. See DOI: https://doi.org/10.1039/d4sc07864k |
| This journal is © The Royal Society of Chemistry 2025 |