
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of the product-spectrum during the biocatalytic hydrolysis of PEF (poly(ethylene furanoate)) with various esterases†
Tobias
Heinks
 a,
Katrin
Hofmann
b,
Lennard
Zimmermann
c,
Igor
Gamm
d,
Alexandra
Lieb
e,
Luise
Blach
d,
Ren
Wei
f,
Uwe T.
Bornscheuer
f,
Julian
Thiele
c,
Christof
Hamel
bd and
Jan
von Langermann
*a
a,
Katrin
Hofmann
b,
Lennard
Zimmermann
c,
Igor
Gamm
d,
Alexandra
Lieb
e,
Luise
Blach
d,
Ren
Wei
f,
Uwe T.
Bornscheuer
f,
Julian
Thiele
c,
Christof
Hamel
bd and
Jan
von Langermann
*a
aFaculty of Process and Systems Engineering, Institute of Chemistry, Biocatalytic Synthesis, Otto von Guericke University of Magdeburg, Germany. E-mail: Jan.Langermann@ovgu.de
bDepartment of Applied Biosciences and Process Engineering, Anhalt University of Applied Sciences, Germany
cFaculty of Process and Systems Engineering, Institute of Chemistry, Organic Chemistry, Otto von Guericke University of Magdeburg, Germany
dFaculty of Process and Systems Engineering, Institute of Process Engineering, Chemical Process Engineering, Otto von Guericke University of Magdeburg, Germany
eFaculty of Process and Systems Engineering, Institute of Chemistry, Industrial Chemistry, Otto von Guericke University of Magdeburg, Germany
fInstitute of Biochemistry, Dept. of Biotechnology & Enzyme Catalysis, University of Greifswald, Germany
First published on 21st November 2024
Abstract
Poly(ethylene furanoate) (PEF) is considered the greener alternative to poly(ethylene terephthalate) (PET) and other plastics, as it can be produced 100% biobased from renewable resources based on the building blocks 2,5-furandicarboxylic acid (FDCA) and ethylene glycol (EG). So far, most of the literature has dealt with the synthesis and detailed characterization of this synthetic polymer, but very few articles deal with enzymatic depolymerization, which is increasingly favored due to environmental reasons. This study therefore aimed to perform hydrolysis of Nano-PEF using 12 different esterases, which have been shown to depolymerize PET very efficiently. All enzymes were compared in terms of their hydrolysis efficiency, showing very different hydrolysis rates and different product profiles over time. A wide variety of hydrolysis products were identified using ESI-TOF including FDCA, (mono(2-hydroxyethyl)-furanoate) (MHEF), (bis(2-hydroxyethyl)-furanoate) (BHEF), dimers, and trimers. Among the tested enzymes, LCCICCG was the most efficient one performing best at pH 8–9 and elevated temperatures (>70 °C). Finally, all hydrolysis intermediates were hydrolyzed to the final building block FDCA (>99% with almost complete depolymerization of Nano PEF), and higher Nano-PEF-concentrations (up to about 1.4 mg mL−1) were depolymerized equally efficient.
Sustainability spotlightPolymers are an indispensable chemical component in almost all areas of the modern world and fulfill numerous functions, e.g. chemical production and food packaging. PEF (polyethylene furan-2,5-dicarboxylate) is a bio-based polymer with an excellent low CO2 footprint. This novel polymer has excellent properties, some of which are even better than the petrochemical-based classic variant PET (polyethylene terephthalate). However, the use of bio-based PEF still requires efficient recycling strategies, whereby biocatalytic reaction systems are essentially preferred due to the low energy input. This work addresses this issue and examines the product spectrum of PEF with various enzymes in preparation for reuse. This work contributes to the following UN sustainable development goals: industry, innovation and infrastructure (SDG 9) and responsible consumption and production (SDG 12). |
Introduction
By 2030, it is estimated that 700 million tons of synthetic plastics will be produced annually, which corresponds to around 80 kg per human,1 demonstrating their widespread use and importance. Their benefits are versatile (e.g., high resistance to influences and lightness),2 justifying their wide use in various fields. However, one of their greatest benefits is also their greatest disadvantage, namely their durability. Without proper waste management, they often remain as synthetic polymers in the environment for decades, leading to significant accumulation especially in marine ecosystems, as about half of the plastic waste is currently released into nature.1,3,4 One of the most commonly used plastics is poly(ethylene terephthalate) (PET),3,5 which is comparatively easy to depolymerize due to its reactive ester bonds. Besides well-established chemical depolymerization methods (in particular solvolysis),5,6 enzymatic depolymerization became feasible using hydrolases.3,7,8PET is composed of the building blocks terephthalic acid (TPA) and ethylene glycol (EG) (Scheme 1), while TPA is currently still produced using petroleum-derived xylenes.9 An alternative to petroleum-based TPA is FDCA, which is chemically similar to TPA (i.e., furan instead of phenyl ring) and derivable from renewable sources via environmentally friendly syntheses.10 Using FDCA and EG as the basic building blocks for polymerization, poly(ethylene furanoate) (PEF) is produced (Scheme 1). The synthesis of PEF has been extensively researched and is well established, and the properties of this polymer have been characterized in detail as reported in several publications.11,12 PEF provides several advantages compared to PET such as (i) a higher thermal resistance due to a higher glass transition temperature (Tg) (85–88 °C vs. 70–76 °C, respectively)13–15 caused by a higher chain rigidity,13 (ii) superior barrier properties (e.g. for oxygen and carbon dioxide),13,16 (iii) a lower melting temperature,13,14 and (iv) enhanced degradability (e.g., when exposed to UV light).12,17 Consequently, when both FDCA and EG are produced from renewable resources and used as feedstock for synthesis, PEF can be considered as a fully biobased and therefore ‘green’ alternative to PET.
 | ||
| Scheme 1 Structure of PEF vs. PET and principle of enzymatic hydrolysis. The enzyme structure is IsPETasewt (PDB-ID: 6ILW). | ||
Analogous to PET, PEF can be depolymerized by chemical techniques.18 However, enzymatic degradation is generally favored for depolymerization mainly due to lower process temperatures and less (harmful) chemicals. To our knowledge, the biocatalytic depolymerization of PEF has only been investigated as reported in a limited set of publications, applying a total of only five ester hydrolases with different PEF-preparations as substrates.19–23 Most of the work was done by Guebitz and co-workers, who used the bacterial cutinase from Thermobifida cellulosilytica Thc_Cut1, and the commercially available (from Novozyms/Novonesis) fungal cutinase HiC from Humicola insolens for depolymerization of PEF films and powders with different crystallinities, molecular weights and particle sizes.19–21 Similar to PET, PEF films with a lower crystallinity (0–10%) degraded significantly faster. However, PEF films with higher crystallinity (≥20%) were better depolymerized than PET films, which were almost not hydrolyzed at a crystallinity higher than 10%.19 If the PEF sample had a low crystallinity (<1%), higher molecular weights led to a faster degradation,20 and the particle size of PEF powders showed a minor impact.21 In contrast, powders with a high crystallinity were degraded faster when the particle size was low.21 Both has also been observed with PET.24 As expected, higher temperatures (i.e., 65 °C instead of 50 °C) led to a higher efficiency, and a phosphate buffer was better than Tris buffer.21 Another short study was published by Kawai and co-workers, who engineered the PET hydrolyzing Cut190**SS/L136F, which was also proven to depolymerize PEF.22 More recently, Kumar and co-workers published the enzymatic depolymerization of different PEF substrates at high loadings using the esterases LCC and Fast-PETase.23
This study aimed to extend the knowledge on the enzymatic hydrolysis of PEF and to comprehensively compare a wide range of 12 esterases with respect to their PEF hydrolysis efficiency. In this study, Nano-PEF was used as a model system as the basic analysis of this polymer was targeted independent of the crystallinity of the polymer applied. All enzymes have already been shown to successfully hydrolyze PET, and most of them are recent and improved enzymes in terms of their PET hydrolysis activity and/or stability at elevated temperatures. The most efficient hydrolase was further analyzed regarding the depolymerization temperature and pH, and was applied in reactions with higher concentrations of Nano-PEF (see Materials and methods for details). The product profile (i.e., all hydrolysis products/intermediates) was analyzed at specific time points to gain insight into the proportion of the product profile for the individual enzyme over time.
Results and discussion
Pre-screening of various PET-hydrolyzing enzymes
The similarity between PEF and PET is very high, as they only differ in the core group (i.e., FDCA with the furan ring vs. TPA with the phenyl ring) and both are analogously linked by EG units via ester bonds (Scheme 1). Consequently, 12 literature-known PET-hydrolyzing enzymes, which were recently compared using PET as substrate,25 were investigated for their ability to hydrolyze PEF with Nano-PEF as a model substrate. Consistent with the literature,26 the Nano-PEF produced and used in this study proved to be predominantly amorphous in XRD measurements (Fig. S9†). The particle size of the Nano-PEF suspension used was about 78 nm (Fig. S10†). All 12 enzymes were applied to Nano-PEF (0.2 mg mL−1) under standard conditions (0.1 M phosphate buffer, pH 7.5, 60 °C, 100 nM (≈3.3 μg mL−1) enzyme) and the resulting concentrations of the products were monitored after 1, 8 and 24 h. A variety of different products were determined (see next section), while Fig. 1 represents the total concentration of all products (sum product concentration) and the proportion of FDCA therein. All enzymes showed activity towards Nano-PEF albeit to varying extents. For PET2-7M,27 PESH1L92F/Q94Y,28 PESH1wt,28 IsPETasewt,29 LCCICCG,30,31 and TurboPETase,32 conversion of the total polymer was already maximal after 1 h. This indicates that the PEF polymer was hydrolyzed very quickly to smaller units (e.g., FDCA-dimers) whereafter these intermediates were further hydrolyzed to the final product FDCA over time. LCCICCG was the most efficient enzyme with the highest FDCA concentration at all time points (84% after 24 h), while the other enzymes were close to that. In contrast, for HotPETase,33 MoPE,34 DuraPETaseN233C/S282C,29 TfCut2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 and Cut190**SS/L136F22 increasing total product concentrations and of FDCA were detected throughout the 24 h, but not reaching as high as the more efficient enzymes. Consequently, these enzymes exhibit a slower PEF hydrolysis rate, which was similar in the case of PET hydrolysis,25 probably due to a lower esterase activity or a higher product inhibition. Interestingly, DmPETase36 showed no activity as in the PET study, whereas MoPE, DuraPETaseN233C/S282C and TfCUT2 showed significant activity here, while in the case of PET hydrolysis there was no activity or only at reduced temperatures (40 °C).25 The hydrolysis of PEF generally appeared to be faster than that of PET under equal conditions (Fig. S1†), which was also supported by another study showing that the hydrolysis of PEF-films was faster than that of PET-films.19 Overall, the term ‘PETase’ should be reconsidered and ‘esterase’ or ‘carboxylesterase’ generally preferred instead, which was also recently discussed by a review about the diverse nomenclature in literature.37 LCCICCG proved to be the most efficient enzyme, so it was selected for further investigations.
35 and Cut190**SS/L136F22 increasing total product concentrations and of FDCA were detected throughout the 24 h, but not reaching as high as the more efficient enzymes. Consequently, these enzymes exhibit a slower PEF hydrolysis rate, which was similar in the case of PET hydrolysis,25 probably due to a lower esterase activity or a higher product inhibition. Interestingly, DmPETase36 showed no activity as in the PET study, whereas MoPE, DuraPETaseN233C/S282C and TfCUT2 showed significant activity here, while in the case of PET hydrolysis there was no activity or only at reduced temperatures (40 °C).25 The hydrolysis of PEF generally appeared to be faster than that of PET under equal conditions (Fig. S1†), which was also supported by another study showing that the hydrolysis of PEF-films was faster than that of PET-films.19 Overall, the term ‘PETase’ should be reconsidered and ‘esterase’ or ‘carboxylesterase’ generally preferred instead, which was also recently discussed by a review about the diverse nomenclature in literature.37 LCCICCG proved to be the most efficient enzyme, so it was selected for further investigations.
 | ||
| Fig. 1 Screening of various esterases for Nano-PEF-hydrolysis. 12 enzymes that were shown to hydrolyze PET were analyzed regarding their potential to depolymerize the alternative plastic PEF. All reactions were performed using standard conditions. FDCA is depicted in green and the total product concentration in grey, visualized as cumulative bars. The reactions were performed in triplicates, and the error bars represent the standard deviation. The background due to self-hydrolysis of Nano-PEF (see ‘control’) was always subtracted. | ||
Product range of biocatalytic Nano-PEF hydrolysis
Various hydrolysis products after Nano-PEF depolymerization were observed in the chromatograms (Fig. S2†). To identify the product spectrum of biocatalytically hydrolyzed Nano-PEF, the samples were additional analyzed using an HPLC equipped with mass spectrometry (ESI-TOF). Consequently, the wide variety of products could be identified and assigned to an intermediate of Nano-PEF-hydrolysis (Table 1 and Fig. S3, S4†). In particular, FDCA, FDCA coupled with one or two EG (i.e., MHEF (mono(2-hydroxyethyl)-furanoate) and BHEF (bis(2-hydroxyethyl)-furanoate); analogous to MHET and BHET in PET hydrolysis, respectively), dimer and trimers coupled to one or two EG could be verified. Most of these products were also found in other studies analyzing the biocatalytic hydrolysis of PEF.20,21,23 Additionally, unexpected structures were also found that were coupled to di- or tri- rather than mono-EG (i.e., entries 7–9, 13–15 in Table 1). However, all structures were finally hydrolyzed to FDCA and mono-(MEG), di- and tri-EG was detected by ESI-TOF. These impurities may occur in the structure of PEF, when MEG-feedstocks are used for synthesis that are not completely pure and contain some degree of di- and tri-EG. In some cases, polyethylene glycols (PEG) are also incorporated on purpose into PEF- and PET-polymers, as they can positively modify its behavior, like improving degradability or lowering its melting temperature.38 Consequently, latter compounds (carrying PEG) are not found in PEF synthesized with pure MEG-feedstocks. Notably, oligomers with PEG structures exhibited a lower solubility, while other insolubilities were not detected.| # | Chemical structure | Abbr. | MWtheo [g mol−1] | MWdetectedb [g mol−1] | rtc [min] |
|---|---|---|---|---|---|
| a Several products were detected by HPLC during Nano-PEF-hydrolysis. To identify the products generated by the biocatalysis, another HPLC equipped with mass spectrometry (ESI-TOF) was used and Nano-PEF hydrolysis was catalyzed by Turbo-PETase and LCCICCG for 1 h under standard conditions to generate a high distribution of the hydrolysis products. As the analytes were separated by different HPLC methods, the results are not directly transferable between both HPLC systems and thus chromatograms. The respective chromatogram can be found in Fig. S3 and S4. Notably, MEG was not found in this setup for unknown reason. b The ammonium (+18.03 g mol−1) and sodium adducts (+22.99 g mol−1) result from the use of different buffer systems (ammonium acetate in the eluent and sodium phosphate during hydrolysis). c The masses listed were determined on the Waters LC/MS system (see Experimental section) and therefore have different retention times than the analytical HPLC runs. rt: retention time; MWtheo: theoretical molecular weight; abbr.: abbreviation. | |||||
| 1 |

|
E | 62.04 | 63 (H+), 80 (NH4+), 85 (Na+) | — |
| 2 |

|
E-E | 106.06 | 107 (H+), 124 (NH4+), 129 (Na+) | 0.53–0.64 |
| 3 |
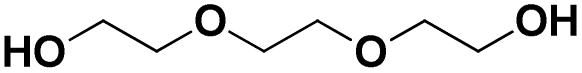
|
E-E-E | 150.09 | 151 (H+), 168 (NH4+), 173 (Na+) | 0.78 |
| 4 |
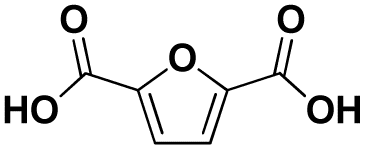
|
FDCA (F) | 156.01 | 157 (H+), 174 (NH4+), 179 (Na+) | 0.47–0.70 |
| 5 |
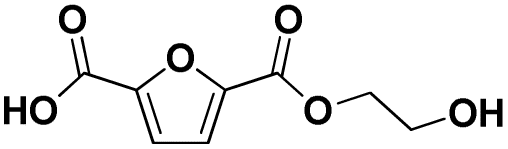
|
F-E | 200.03 | 201 (H+), 218 (NH4+), 223 (Na+) | 0.70–0.85 |
| 6 |

|
E-F-E | 244.06 | 245 (H+), 262 (NH4+), 267 (Na+) | 1.07 |
| 7 |
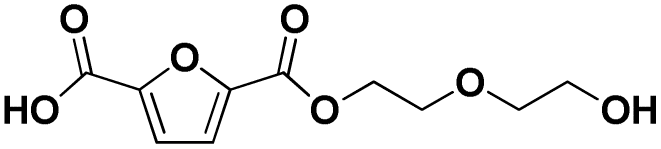
|
F-E-E | 244.06 | 245 (H+), 262 (NH4+), 267 (Na+) | 1.80 |
| 8 |

|
E-F-E-E | 288.08 | 289 (H+), 306 (NH4+), 311 (Na+) | 2.42 |
| 9 |

|
F-E-E-E | 288.08 | 289 (H+), 306 (NH4+), 311 (Na+) | 3.00 |
| 10 |

|
E-F-E-E-E | 332.11 | 350 (NH4+), 355 (Na+) | 3.25 |
| 11 |

|
F-E-F | 338.03 | 339 (H+), 356 (NH4+), 361 (Na+) | 0.93 |
| 12 |
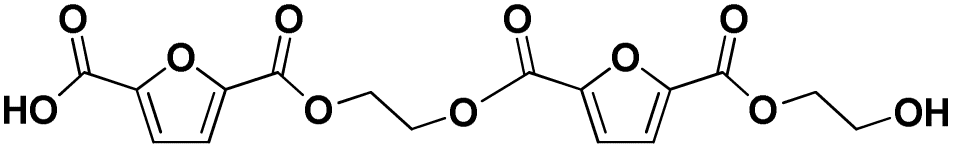
|
F-E-F-E | 382.05 | 400 (NH4+), 405 (Na+) | 2.67 |
| 13 |
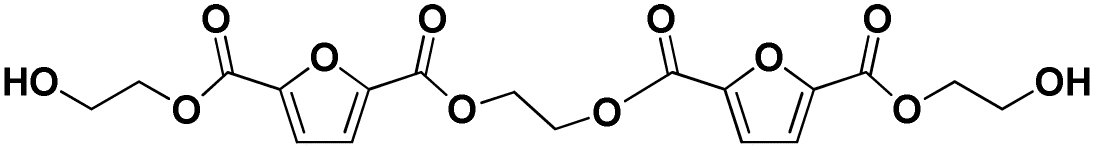
|
E-F-E-F-E | 426.08 | 444 (NH4+), 449 (Na+) | 2.94 |
| 14 |
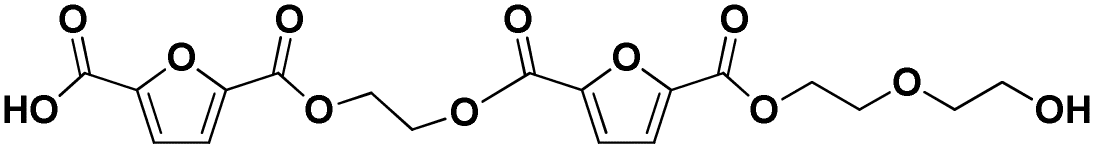
|
F-E-F-E-E | 426.08 | 444 (NH4+), 449 (Na+) | 3.49 |
| 15 |
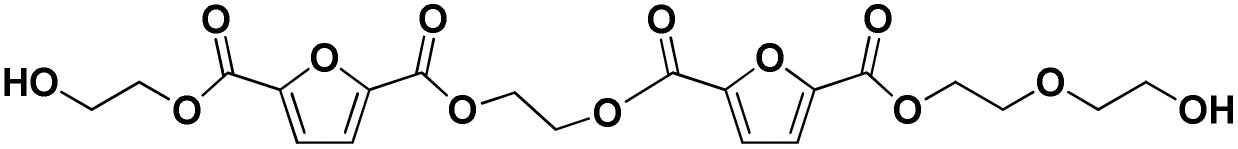
|
E-F-E-F-E-E | 470.11 | 488 (NH4+), 493 (Na+) | 3.66 |
| 16 |
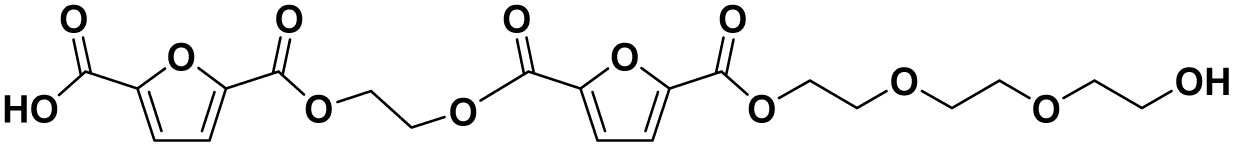
|
F-E-F-E-E-E | 470.11 | 488 (NH4+), 493 (Na+) | 4.01 |
| 17 |
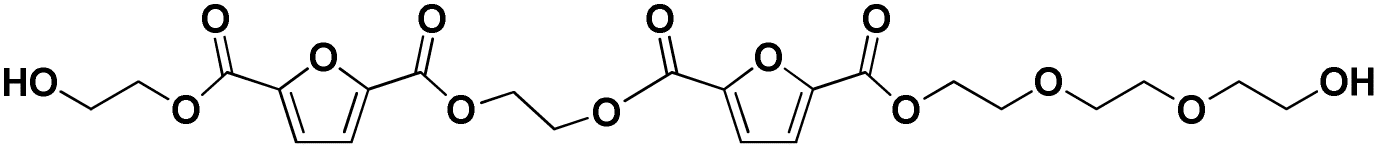
|
E-F-E-F-E-E-E | 514.13 | 532 (NH4+) | 5.51 |
| 18 |
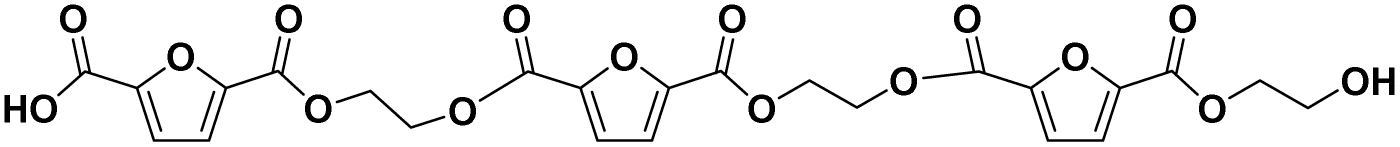
|
F-E-F-E-F-E | 564.08 | 626 (NH4+), 631 (Na+) | 5.25 |
| 19 |

|
F-E-F-E-F-E-E | 608.10 | 670 (NH4+) | 5.30 |
Several hydrolysis intermediates were formed very quickly at the beginning of the reaction (1 h), with monomers (i.e., FDCA, MHEF and BHEF) and dimers being the predominant products as shown for LCCICCG and IsPETasewt by ESI-TOF (Fig. S5†). In particular, for the shown LCCICCG reaction, MHEF had the highest proportion of all products at early stage (Fig. S5†). Furthermore, assuming that FDCA, MHEF and BHEF run similarly in analytical HPLC as in qualitative HPLC (LC/MS) as well as their analogs TPA, MHET and BHET (Fig. S6†), MHEF was present in high concentrations in reactions with all enzymes, especially at the beginning of the reactions. Conclusively, it is possible that the intermediate MHEF reduces the reaction rate due to inhibition of the enzyme, being thus the rate-limiting step. This was comprehensively proven for the analog MHET in the case of PET hydrolysis.39 The proportions of the intermediates were highly dependent on the enzyme used and, consequently, its hydrolysis efficiency. Enzymes with high catalytic activity rapidly produced substantial amounts of FDCA, whereas those with lower activity generated significant quantities of intermediates, which were gradually decomposed over time.
Further investigations of biocatalytic Nano-PEF-hydrolysis
The Tg of PEF is higher than of PET. However, under the degradation conditions, the polymers are totally soaked in water, which has a substantial plasticization impact and lowers the polymer's Tg.8,15,40 As a result, Tg of nano-sized PET or its surface layer can typically fall within the range of 40 °C.40 However, it is widely accepted that the most suitable temperature for PET hydrolysis is between 68 and 72 °C.8,41 This temperature range strikes a balance between enhancing the flexibility of the polymer chains beyond its actual Tg and preventing rapid recrystallization when exposed to water-plasticization conditions.31,42 Hence, based on our understanding of enzymatic depolymerization of (nanosized) PET, we anticipate that nanosized PEF will exhibit similar characteristics when subjected to water plasticization. Additionally, we predict that nanosized PEF will have a higher optimal temperature for enzymatic hydrolysis because PEF inherently has a higher Tg than PET (under dry conditions). Therefore, we investigated the efficiency of Nano-PEF hydrolysis at 60, 70, 80 and 90 °C with LCCICCG under standard conditions analyzed after 1, 8, and 24 h (Fig. 2). Increasing the temperature led to an increase in the total product concentration in the first hour. Moreover, the degradation of the hydrolysis intermediates to FDCA was faster at elevated temperatures, proving a higher reaction efficiency under these conditions. Interestingly, the conversion was most efficient at very high temperatures (i.e., 90 °C; (>99% vs. 84% FDCA under standard conditions after 24 h)), although other studies showed a decrease of activity at this temperature. Fig. 2 demonstrates that this effect is only noticeable during the initial hour of the reaction, suggesting that the thermoactivated enzyme41 and thermoactivated polymers contribute together to efficient depolymerization. However, as the reaction continued to 8 h (and much more remarkably to 24 h), it became evident that 70 °C was once again the most favorable temperature. The lack of long-term stability of LCCICCG (with a melting temperature of 94.2, as reported by Tournier et al. in 2020) may be the cause of this. Nonetheless, the impact of polymer recrystallization on the degradability of PEF in this temperature range during the lengthy course of reactions should not be dismissed, but rather further investigated in future research, as it presently remains unclear but very likely due to our previous knowledge of PET (and other polymers). For the wildtype LCC, a decrease of activity during Nano-PEF hydrolysis was observed above 70 °C,23 whereas LCCICCG as an engineered/improved enzyme was proven to exhibit a higher thermostability.31 However, at 70–90 °C, almost complete depolymerizations (95–97%) and high FDCA contents (97% and >99%, respectively) occur at the end of the reaction. | ||
| Fig. 2 Biocatalytic hydrolysis of Nano-PEF by LCCICCG at different temperatures and pH values. All reactions were performed under standard conditions except the varying temperatures (70, 80, 90 °C; constant pH of 7.5) and varying pH values (0.1 M buffer, buffers are mentioned in the methods; constant temperature of 60 °C) and analyzed by HPLC. The reactions were performed in triplicates, and the error bars represent the standard deviation. The background due to self-hydrolysis of Nano-PEF under the used conditions (Fig. S7†) was always subtracted. The conversion was calculated based on the concentration of FDCA after the chemical total hydrolysis of the same amount PEF substrate, which was set to 100% (see Experimental section). | ||
Previously, we25 and another study43 showed that the reaction pH has an impact on the hydrolysis efficiency, the auto-hydrolysis of the intermediates, and the product profile in the biocatalytic hydrolysis of PET. Therefore, the depolymerization of Nano-PEF was performed at different pH values (5.0, 6.0, 7.0, 7.5, 8.0, 9.0) under standard conditions analyzed after 1, 8, and 24 h (Fig. 2). The reaction catalyzed by LCCICCG was strongly affected by the different pH values. Acidic pH values led to significantly lower total product concentrations, as also observed for PET hydrolysis by several esterases.23,27,36,44 Furthermore, the hydrolysis of the intermediates to FDCA is very limited at this pH (i.e., 54% total conversion and 7% FDCA after 24 h). Increasing the pH between 6 and 9 resulted in faster reaction rates, and almost complete conversion was observed after 8 or 24 h in all cases. The hydrolysis rate of the intermediates to FDCA enhanced with increasing pH, reaching the highest conversion at pH 9 after 24 h (>99% vs. 84% FDCA under standard conditions after 24 h). This indicates that LCCICCG is significantly affected by pH and performing better at higher pH values. The latter was also recently demonstrated for the wild type.23 Additionally, Nano-PEF and the hydrolysis intermediates are chemically hydrolyzed under basic conditions (Fig. S7†) as also shown previously for PET and PET-intermediates,25 which further improves the depolymerization. The product profile is not significantly affected by pH (data not shown), suggesting that the overall reaction rate is equally affected for all intermediates and not the hydrolysis rate of a particular intermediate.
Concentration-dependent hydrolysis of Nano-PEF
In the next step, the hydrolysis of different Nano-PEF concentrations (0.2, 0.6, 1.0, 1.4 mg mL−1 Nano-PEF) was analyzed under standard conditions and the product concentrations were determined at specific time points (Fig. 3). Although the substrate concentration was increased up to 7-fold (i.e., from 0.2 to 1.4 mg mL−1), the reaction behavior was similar in all cases (Fig. 3). Equal rates of the hydrolysis of Nano-PEF to the intermediates (see total product concentrations) and of the intermediates to FDCA (see FDCA concentrations) were obtained for all substrate amounts applied to the reaction (Fig. 3B). Almost the highest total product concentration was already observed after 8 h for all substrate amounts applied, while the FDCA-concentration was about 40%. Subsequently, all hydrolysis intermediates were hydrolyzed to the end product FDCA over the course of the reaction, reaching >99% FDCA formation with almost complete depolymerization of Nano-PEF after the reaction in all cases. | ||
| Fig. 3 Biocatalytic hydrolysis of different Nano-PEF concentrations by LCCICCG. All reactions were performed under standard conditions except the varying concentrations of Nano-PEF: 0.2 (purple, circle), 0.6 (green, star), 1.0 (red, triangle), 1.4 (blue, square) mg mL−1. The total product concentrations are represented by filled and the FDCA-concentrations by open symbols. (A) Concentration of the products over the course of the reaction. (B) All concentrations were related to the final concentration after the reaction. (C) The conversion was calculated based on the concentration of FDCA after the chemical total hydrolysis of the same amount PEF substrate, which was set to 100% (see Experimental section). | ||
Conclusions
This study compared for the first time a wide range of esterases in terms of their depolymerization efficiency for the polymer PEF. Nano-PEF was used as a model substrate here, which was determined to be predominantly amorphous in XRD measurements with average particle size of 78 nm. Different hydrolysis rates were observed for all enzymes, while LCCICCG was the most efficient enzyme under the applied reaction conditions with a few enzymes having similar activity. The enzymatic Nano-PEF hydrolysis was enhanced at higher pH values (8–9) and higher temperatures (>70 °C) as also shown for PET hydrolysis, proving similar efficiencies for both polyesters. Both increased the FDCA production from about 84% to >99% in 24 h with almost complete depolymerization of the polymer. Higher concentrations of Nano-PEF (7-fold; up to 1.4 mg mL−1) were degraded equally efficiently over time to the basic monomer FDCA with very similar reaction rates. Several intermediates were observed during hydrolysis including MHEF, BHEF, dimers and trimers of FDCA, enabling a deeper kinetic analysis of both the hydrolysis of PEF to intermediates and of each intermediate to FDCA in further studies.Experimental section
General information
FDCA (purity: 98%) and PEF pellets (viscosity: 0.76 dL g−1, purity: 98%) were obtained from Alfa Chemical (Zhengzhou, China). Based on PXRD measurements and qualitative comparisons with literature data (see below), the used PEF is determined to be predominantly amorphous. All other chemicals were purchased at analytical grade from commercial providers and used as received: Merck (Darmstadt, Germany), Carl Roth (Karlsruhe, Germany), Thermo Fisher Scientific (Waltham, MA, USA), TCI Europe (Zwijndrecht, Belgium), Th.Geyer (Renningen, Germany), BLD Pharmatech (Reinbek, Germany). The plasmids were provided by other scientists as stated recently,25 except for TurboPETase and Cut190**SS/L136F, which were synthesized by BioCat GmbH (Heidelberg, Germany).Plasmid transformation, gene expression and protein purification
Transformation, expression and purification were performed analogously as recently reported.25 Briefly, the enzymes were expressed using E. coli Shuffle T7 or E. coli BL21(DE3) in LB medium at 20 °C for 16 h with the respective IPTG concentration. Enzyme purification was performed via His tag purification after cell disruption in sonication buffer (50 mM Tris pH 8.0, 150 mM NaCl, 15 mM imidazole, 0.015 mg mL−1 DNAseI, 0.15 mg mL−1 lysozyme) using sonication. The filtered supernatant was applied to HisTrap™ HP columns and enzymes were purified using a step gradient (15, 30, 45, 210, 300 mM elution buffer). After a buffer exchange (50 mM Tris pH 8.0, 50 mM NaCl), enzymes were concentrated (Amicon® Ultra-15 centrifuge filter units) and stored at −20 °C.Enzymatic reactions
The initial biocatalytic reactions were carried out in a total volume of 1.6 mL in 2 mL reaction vessels with 100 nM (≈3.3 μg mL−1) enzyme and 0.2 mg mL−1 Nano-PEF as substrate representing the standard conditions (0.1 M phosphate buffer, pH 7.5, 60 °C). For the reactions at varying pH values, different buffers were used (i.e., pH 5.0 (0.1 M sodium acetate buffer) or 9.0 (0.1 M bicine buffer)) instead of the phosphate buffer and for the reactions with varying temperatures, different temperatures were applied (i.e., 70, 80, 90 °C) instead of 60 °C. For the reactions with varying concentrations of Nano-PEF, the reactions were performed under standard conditions, with different Nano-PEF concentrations (i.e., 0.2, 0.6, 1.0, 1.4 mM). To analyze the reactions via HPLC, 500 μL were taken at each reaction time point stated, acidified with 10 μL 37% HCl to a final pH of 1–2, and heated at 95 °C for 10 min. After centrifugation, 450 μL of the supernatant was diluted with an equal volume of analytic solution (0.1 M phosphate buffer, 20% DMSO, pH 5.0) to obtain a final pH of 1.5–4. Importantly, the solutions had to be acidified to a pH value below 2.6, otherwise FDCA behaved differently in the HPLC runs (Fig. S8†). The initial and characterization reactions were always performed in triplicates.HPLC measurements
These were performed to analyze the PEF hydrolysis and quantify the product concentrations for all biocatalytic reactions. Therefore, 20 μL of each sample were injected into a Chromaster® HPLC system (VWR, Darmstadt, Germany) equipped with an Eclipse XDB-C18 (4.6 × 12.5 mm; 5 μm) guard column and a Zorbax Eclipse XDB-C18 (4.6 × 250 mm; 5 μm) column (Agilent Technologies, Waldbronn, Germany) maintained at 35 °C. To achieve efficient separation of the hydrolysis products, a gradient was applied, using as the mobile phase varying portions of the eluents comprising (A) 0.1% (v/v) formic acid in water and (B) acetonitrile. The detailed gradient steps are provided in the ESI (Table S2†). A flow rate of 0.8 mL min−1 was employed and the analytes were detected via UV detector at 260 nm. Using a commercial standard, the retention time of FDCA was determined and a calibration curve was established over the concentration range of 1.7–680 μM. Based on this dataset, all other detected peaks were quantified. An exemplary chromatogram after biocatalysis with the diverse product profile is shown in Fig. S2.†Determination of protein concentration
These were determined using the Pierce™ Coomassie Protein Assay Kit (ThermoFisher Scientific; Bradford Assay) with BSA as calibration standard.LC/MS
To identify the product spectrum of biocatalytic PEF hydrolysis qualitatively, a HPLC equipped with mass spectrometry (ESI-TOF) was used. Quantitative analyses were conducted using the alternative HPLC system. To separate the product mixture, 4 μL of each sample was injected onto an HPLC (Acquity UPLC H-Class, Waters, Milford, USA), equipped with an Acquity UPLC® BEH C18 column (50 mm length, 2.1 mm inner diameter, 1.7 μm pore size). A gradient of the eluents (A: 2 mM ammonium acetate buffer pH ≈ 4.7; B: acetonitrile) was used for the separation with a flow of 0.4 mL min−1. The detailed gradient steps are provided in Table S2.† The column temperature was held at 40 °C. The sum formulas of the products were determined using a Xevo G2 time-of-flight mass spectrometer (Waters, Milford, USA), utilizing electrospray ionization. The measurements were done with a positive polarity and a mass range of 40–1200 dalton. The chromatogram was searched specifically for the masses of the suspected adducts (Table 1). The relative abundance of the ions found was used to determine the product distribution.Preparation of Nano-PEF
Nano-PEF was produced based on already established methods for the production of Nano-PET.45 Briefly, 0.5 g of PEF was dissolved in 10 mL of hexafluoroisopropyl alcohol (HFP) (ratio of 1:20 (PEF:HFP)) until fully dissolved. To speed up the dissolving process, the PEF particles can be crushed under cooling using a cryomill. The resulting solution was then added dropwise to 100 mL water (ratio of 1:10 (PEF/HFP:H2O)), under vigorous stirring, to form a suspension. This suspension was subsequently filtered through a paper filter. Finally, HFP was removed under reduced pressure using a rotary evaporator. An additional step involved transferring the final solution to a graduated cylinder and allowing it to settle for 2 hours. At high suspension densities, two layers were formed, with the upper layer being more stable. This upper layer was decanted and remains stable for up to several months. After evaporating a fixed volume of Nano-PEF, a concentration of 2.1 mg mL−1 was detected.
Determination of the crystallinity of Nano-PEF
To determine the crystallinity of the PEF, the PEF particles were crushed under cooling using a cryomill resulting in pulverized PEF particles that were analyzed using PXRD. The PXRD measurements were performed using an Empyrean diffractometer (PANalytical, Almelo, The Netherlands) with copper K alpha1/alpha2 radiation in a reflection geometry setup. The diffractometer was equipped with a PIXcel(3D) detector and the sample was loaded using so-called backloading sample holders to avoid preferred orientation. Data was collected from 2–60° 2theta at 40 kV and 40 mA. The resulting measurements were then qualitatively compared with data from the literature,46 and the PEF used in this study was classified as predominantly amorphous (Fig. S9†). This is consistent with the literature, which shows that after the nanoparticles are produced, the crystallinity is largely removed and highly amorphous particles are present.26Dynamic light scattering (DLS) measurements
The particle size distribution of the Nano-PEF suspension was determined via DLS on a Zetasizer Nano ZS (Malvern Panalytical, Westborough, USA), equipped with a 4 mW, 633 nm laser. The detection angle was 173°. For the measurement, 2 mL of the Nano-PEF suspension was transferred in a polystyrene cuvette (DTS0012). An average particle size of 78.38 nm was determined (Fig. S10†).Total hydrolysis of Nano-PEF to determine the conversion
To obtain the maximum FDCA concentration releasable by the substrate, 1 mL of the Nano-PEF suspension was totally hydrolyzed using 1 mL of 2 M NaOH and incubated at 50 °C for 24 h. Afterwards, the solution was acidified using 2 mL 1 M HCl and diluted as required. Per mg of applied Nano-PEF, 4.74 μmol FDCA was obtained (or 5048 (μM mL) mg−1) without byproducts (Fig. S11†). This released FDCA concentration was set to 100% and the enzymatically produced product concentrations were related to it.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. H.: conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing – original draft; K. H.: data curation, formal analysis, investigation, methodology, validation, writing – review & editing; L. Z.: data curation, formal analysis, investigation, methodology, validation, writing – review & editing; I. G.: data curation, investigation, formal analysis, methodology, writing – review & editing; A. L.: data curation, formal analysis, investigation, methodology, writing – review & editing; L. B.: writing – review & editing; R. W.: writing – review & editing; U. B.: writing – review & editing; J. T.: writing – review & editing; C. H.: funding acquisition, project administration, supervision, writing – review & editing; J. v. L.: funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the research initiative “SmartProSys: Intelligent Process Systems for the Sustainable Production of Chemicals” funded by the Ministry for Science, Energy, Climate Protection and the Environment of the State of Saxony-Anhalt. Personal funding for J. v. L. was provided by Deutsche Forschungsgemeinschaft through the Heisenberg Programme (project number 450014604). The authors gratefully acknowledge Evangelos Topakas, Ryota Iino, and Anthony Green who kindly provided the plasmids encoding the enzymes used in this study, who are listed and cited in our recently published work (Table S1†).25References
- P. F. Britt, et al., Report of the basic energy sciences roundtable on chemical upcycling of polymers, 2019 Search PubMed.
- R. O. Ebewele, Polymer science and technology, CRC Press, 2000 Search PubMed.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- (a) J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed; (b) X. Zhu, C. Rochman and M. R. Mazloff, Marine animals as a global reservoir of plastic pollution: a case study on sea turtles, 2024 Search PubMed; (c) R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- A. McNeeley and Y. A. Liu, Ind. Eng. Chem. Res., 2024, 63, 3400–3424 CrossRef CAS.
- (a) A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Cleaner Prod., 2019, 225, 1052–1064 CrossRef CAS; (b) F. Cao, L. Wang, R. Zheng, L. Guo, Y. Chen and X. Qian, RSC Adv., 2022, 12, 31564–31576 RSC.
- J. Mican, D. M. Jaradat, W. Liu, G. Weber, S. Mazurenko, U. T. Bornscheuer, J. Damborsky, R. Wei and D. Bednar, Appl. Catal., B, 2024, 342, 123404 CrossRef CAS.
- R. Wei, et al. , ACS Catal., 2022, 12, 3382–3396 CrossRef CAS PubMed.
- (a) H. M. Lapa and L. M. D. R. S. Martins, Molecules, 2023, 28, 1922 CrossRef CAS PubMed; (b) F. Yuan, P. Cao, W. Sun and L. Zhao, Ind. Eng. Chem. Res., 2024, 63, 8208–8215 CrossRef CAS.
- (a) A. Karich, S. B. Kleeberg, R. Ullrich and M. Hofrichter, Microorganisms, 2018, 6, 5 CrossRef CAS; (b) T. Lechtenberg, B. Wynands and N. Wierckx, Metab. Eng., 2024, 81, 262–272 CrossRef CAS; (c) R. O. Rajesh, T. K. Godan, R. Sindhu, A. Pandey and P. Binod, Bioengineered, 2020, 11, 19–38 CrossRef CAS PubMed; (d) G. Totaro, L. Sisti, P. Marchese, M. Colonna, A. Romano, C. Gioia, M. Vannini and A. Celli, ChemSusChem, 2022, 15, e202200501 CrossRef CAS PubMed; (e) M. A. do Nascimento, B. Haber, M. R. B. P. Gomez, R. A. C. Leão, M. Pietrowski, M. Zieliński, R. O. M. A. de Souza, R. Wojcieszak and I. Itabaiana, Green Chem., 2024, 26, 8211–8219 RSC.
- K. Loos, R. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, N. Sbirrazzuoli, A. J. D. Silvestre, N. Guigo and A. F. Sousa, Front. Chem., 2020, 8, 585 CrossRef CAS.
- E. de Jong, H. R. A. Visser, A. S. Dias, C. Harvey and G.-J. M. Gruter, Polymers, 2022, 14, 943 CrossRef CAS PubMed.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- A. Koltsakidou, Z. Terzopoulou, G. Z. Kyzas, D. N. Bikiaris and D. A. Lambropoulou, Molecules, 2019, 24, 564 CrossRef PubMed.
- T. B. Thomsen, C. J. Hunt and A. S. Meyer, New Biotechnol., 2022, 69, 28–35 CrossRef CAS PubMed.
- S. K. Burgess, R. M. Kriegel and W. J. Koros, Macromolecules, 2015, 48, 2184–2193 CrossRef CAS.
- E. Maaskant and D. S. van Es, ACS Macro Lett., 2021, 10, 1616–1621 CrossRef CAS PubMed.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, M. Majdoub, G. Z. Papageorgiou, D. N. Bikiaris and J. Analy, Appl. Pyrolysis, 2017, 126, 357–370 CrossRef CAS.
- S. Weinberger, K. Haernvall, D. Scaini, G. Ghazaryan, M. T. Zumstein, M. Sander, A. Pellis and G. M. Guebitz, Green Chem., 2017, 19, 5381–5384 RSC.
- A. Pellis, K. Haernvall, C. M. Pichler, G. Ghazaryan, R. Breinbauer and G. M. Guebitz, J. Biotech., 2016, 235, 47–53 CrossRef CAS PubMed.
- S. Weinberger, J. Canadell, F. Quartinello, B. Yeniad, A. Arias, A. Pellis and G. Guebitz, Catalysts, 2017, 7, 318 CrossRef.
- F. Kawai, Y. Furushima, N. Mochizuki, N. Muraki, M. Yamashita, A. Iida, R. Mamoto, T. Tosha, R. Iizuka and S. Kitajima, AMB Express, 2022, 12, 134 CrossRef CAS PubMed.
- V. Kumar, A. Pellis, R. Wimmer, V. Popok, J. d. C. Christiansen and C. Varrone, ACS Sustain. Chem. Eng., 2024, 12(26), 9658–9668 CrossRef CAS PubMed.
- R. K. Brizendine, E. Erickson, S. J. Haugen, K. J. Ramirez, J. Miscall, D. Salvachúa, A. R. Pickford, M. J. Sobkowicz, J. E. McGeehan and G. T. Beckham, ACS Sustainable Chem. Eng., 2022, 10, 9131–9140 CrossRef CAS.
- T. Heinks, K. Hofmann, S. Last, I. Gamm, L. Blach, R. Wei, U. T. Bornscheuer, C. Hamel and J. von Langermann, Selective Modification of the Product Profile of Biocatalytic Hydrolyzed PET via Product-specific Medium Engineering, ChemSusChem, 2024, e202401759 CrossRef PubMed.
- R. Wei, T. Oeser, M. Barth, N. Weigl, A. Lübs, M. Schulz-Siegmund, M. C. Hacker and W. Zimmermann, J. Mol. Catal. B: Enzym., 2014, 103, 72–78 CrossRef CAS.
- A. Nakamura, N. Kobayashi, N. Koga and R. Iino, ACS Catal., 2021, 11, 8550–8564 CrossRef CAS.
- L. Pfaff, et al. , ACS Catal., 2022, 12, 9790–9800 CrossRef CAS.
- S. Brott, L. Pfaff, J. Schuricht, J.-N. Schwarz, D. Böttcher, C. P. S. Badenhorst, R. Wei and U. T. Bornscheuer, Eng. Life Sci., 2022, 22, 192–203 CrossRef CAS.
- G. Arnal, J. Anglade, S. Gavalda, V. Tournier, N. Chabot, U. T. Bornscheuer, G. Weber and A. Marty, ACS Catal., 2023, 13, 13156–13166 CrossRef CAS.
- V. Tournier, et al. , Nature, 2020, 580, 216–219 CrossRef CAS.
- B. Wu, Y. Cui, Y. Chen, J. Sun, T. Zhu, H. Pang, C. Li and W. Geng, Deep learning-aided redesign of a hydrolase for near 100% PET depolymerization under industrially relevant conditions, 2023 Search PubMed.
- E. L. Bell, et al. , Nat. Catal., 2022, 5, 673–681 CrossRef CAS.
- E. Nikolaivits, G. Taxeidis, C. Gkountela, S. Vouyiouka, V. Maslak, J. Nikodinovic-Runic and E. Topakas, J. Hazard. Mater., 2022, 434, 128900 CrossRef CAS PubMed.
- K. Vogel, et al. , Sci. Total Environ., 2021, 773, 145111 CrossRef CAS PubMed.
- K. Makryniotis, E. Nikolaivits, C. Gkountela, S. Vouyiouka and E. Topakas, J. Hazard. Mater., 2023, 455, 131574 CrossRef CAS.
- R. Amanna and S. K. Rakshit, Biodegradation, 2024, 35, 341–360 CrossRef.
- (a) P. Ji, D. Lu, S. Zhang, W. Zhang, C. Wang and H. Wang, Polymers, 2019, 11, 2105 CrossRef CAS PubMed; (b) M. B. Banella, J. Bonucci, M. Vannini, P. Marchese, C. Lorenzetti and A. Celli, Ind. Eng. Chem. Res., 2019, 58, 8955–8962 CrossRef CAS.
- (a) M. Barth, T. Oeser, R. Wei, J. Then, J. Schmidt and W. Zimmermann, Biochem. Eng. J., 2015, 93, 222–228 CrossRef CAS; (b) E. Erickson, et al. , ChemSusChem, 2022, 15, e202101932 CrossRef CAS; (c) R. Wei, T. Oeser, J. Schmidt, R. Meier, M. Barth, J. Then and W. Zimmermann, Biotechnol. Bioeng., 2016, 113, 1658–1665 CrossRef CAS PubMed.
- N. A. Tarazona, R. Wei, S. Brott, L. Pfaff, U. T. Bornscheuer, A. Lendlein and R. Machatschek, Chem Catal., 2022, 2, 3573–3589 CrossRef CAS.
- E. Akram, Y. Cao, H. Xing, Y. Ding, Y. Luo, R. Wei and Y. Zhang, Chin. J. Catal., 2024, 60, 284–293 CrossRef CAS.
- (a) R. Wei, D. Breite, C. Song, D. Gräsing, T. Ploss, P. Hille, R. Schwerdtfeger, J. Matysik, A. Schulze and W. Zimmermann, Adv. Sci., 2019, 6, 1900491 CrossRef PubMed; (b) A. Patel, et al. , Polymer, 2023, 285, 126370 CrossRef CAS.
- K. Świderek, S. Velasco-Lozano, M. À. Galmés, I. Olazabal, H. Sardon, F. López-Gallego and V. Moliner, Nat. Commun., 2023, 14, 3556 CrossRef PubMed.
- (a) M. Dimarogona, E. Nikolaivits, M. Kanelli, P. Christakopoulos, M. Sandgren and E. Topakas, Biochim. Biophys. Acta, 2015, 1850, 2308–2317 CrossRef CAS PubMed; (b) Y. L. Lee, N. R. Jaafar, J. G. Ling, F. Huyop, F. D. Abu Bakar, R. A. Rahman and R. M. Illias, Int. J. Biol. Macromol., 2024, 263, 130284 CrossRef; (c) C. Liu, C. Shi, S. Zhu, R. Wei and C.-C. Yin, Biochem. Biophys. Res. Commun., 2019, 508, 289–294 CrossRef CAS PubMed.
- K. Welzel, PhD thesis, Universitätsbibliothek Braunschweig, 2003.
- (a) L. Maini, M. Gigli, M. Gazzano, N. Lotti, D. N. Bikiaris and G. Z. Papageorgiou, Polymers, 2018, 10, 296 CrossRef PubMed; (b) V. Tsanaktsis, D. G. Papageorgiou, S. Exarhopoulos, D. N. Bikiaris and G. Z. Papageorgiou, Cryst. Growth Des., 2015, 15, 5505–5512 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00722k |
| This journal is © The Royal Society of Chemistry 2025 |