
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Injectable biocompatible hydrogels with tunable strength based on crosslinked supramolecular polymer nanofibers†
Hans F.
Ulrich
ab,
Ceren C.
Pihlamagi
ab,
Tobias
Klein
 ab,
Camille
Bakkali-Hassani
c,
Sylvain
Catrouillet
c and
Johannes C.
Brendel
*abde
ab,
Camille
Bakkali-Hassani
c,
Sylvain
Catrouillet
c and
Johannes C.
Brendel
*abde
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: johannes.brendel@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
cICGM, Univ. Montpellier, CNRS, ENSCM, 34095 Montpellier, France
dMacromolecular Chemistry I, University of Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany
eInstitute of Macromolecular Research (BIMF) and Bavarian Polymer Institute (BPI), University of Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany
First published on 20th December 2024
Abstract
Hydrogels based on supramolecular assemblies offer attractive features for biomedical applications including injectability or versatile combinations of various building blocks. We here investigate a system combining benzenetrispeptides (BTP), which forms supramolecular fibers, with polymer polyethylene oxide (PEO) forming a dense hydrophilic shell around the fibers. Hydrogels are created through the addition of a bifunctional crosslinker (CL). Rheological studies revealed that shorter hydrophobic n-hexyl spacers (BTP-C6) lead to stronger hydrogels than BTP-C12 comprising n-dodecyl chains. All hydrogels recovered rapidly (<5 s) after deformation in step-strain-measurements. We varied the crosslinker content between 0.1, 1 and 10 mol% and the overall concentration of the gelator. While the shear storage modulus of all BTP-C12 hydrogels remains below 1 kPa independent of the variations, the shear storage modulus of BTP-C6 hydrogels can be tuned from around 0.2 kPa up to almost 8 kPa. Shear rate dependent viscosity measurements further revealed similar shear thinning behavior of all hydrogels, and the calculation of extrusion parameters confirmed that the hydrogels can be easily injected even through thin cannulae. Accordingly, we injected a fluorescein-containing BTP-C6 sample into chicken breast demonstrating the potential for application as an injectable drug depot. Furthermore, BTP-C6 hydrogels prevent the adherence of L929 mouse fibroblasts but preserve their relative metabolic activity (>87%) during incubation on the gel when compared to cells growing on adherent surfaces. Our investigations overall reveal that the BTP-C6 system in particular has attractive features for applications in tissue engineering or as an injectable and biocompatible drug depot.
Introduction
A plethora of examples regarding self-assembly processes of biomacromolecules in nature (e.g., nucleic acids, proteins, and polysaccharides) has inspired the research on supramolecular hydrogels and their biomedical applications such as tissue regeneration, drug delivery, and artificial cell scaffolds.1–3 Unlike conventional synthetic hydrogels, which are made of three-dimensional networks of polymer chains linked by permanent covalent bonds, supramolecular hydrogels are formed through the self-assembly of small organic molecules (hydrogelators) via noncovalent interactions.4–9 These intermolecular forces often result in a dynamic system at room temperature, where liquification (viscous flow) of the supramolecular hydrogels can occur given a sufficient shear force (shear thinning) and the system recovers once the applied force is removed (self-healing).3 The peculiar viscoelastic properties and self-healing ability of supramolecular hydrogels make them promising candidates for applications, such as injectability for tissue engineering or drug delivery.1,2,10–14Ureido-pyrimidinone, peptides, benzenetrisamides, and benzenetrisurea are common examples of supramolecular motifs of hydrogels displaying shear thinning behavior.15–22 These systems mainly rely on H-bond interactions and on hydrophilic polymers, mostly polyethylene oxide (PEO), which are functionalized at their end groups with the corresponding supramolecular motifs. There are different ways these motifs can be utilized. One approach uses the interactions of the end groups in star or branched polymers to form a large crosslinked network in which the end groups serve as junction sides that act as linkage between the PEO chains.23–26 A different approach utilizes hydrophobic supramolecular motifs such as ureidopyrimidinones, benzenetrisamides, and benzenetrisureas (BTU) decorated with hydrophilic groups to obtain amphiphilic molecules, which are able to form fibers through self-assembly.16,22,27,28 An advantage of fiber formation is that through overlapping of the fibers or defects in the structure, junction sides are formed that lead to gel formation.29 Even though some of these systems form hydrogels without a special crosslinker, the gels become stronger when a homotelechelic polymer with a motif on both end groups is utilized as a crosslinker. Here the crosslinker is integrated during the assembly step to ensure even distribution throughout the fiber. For biological applications, fast recovery kinetics and optimum shear force are crucial parameters when designing supramolecular hydrogels.30,31 For instance, fast recovery rates were observed in hydrogels based on peptides.19,32,33
Previously, our group demonstrated that the BTU motif is able to form hydrogels with a strong shear thinning character, enabling 3D printing.28 Although these results are promising, BTUs allow almost no modification of their hydrophobic interactions by varying the spacers or changing the hydrogen bonding units without disturbing the assembly process.34 These aspects limit the adaptability of the hydrogels in terms of mechanical or rheological properties, which are crucial for cell and tissue interactions. Therefore, we explored benzenetrispeptide (BTP)-based supramolecular motifs, which form self-assembled fibers when conjugated with PEO chains that can be varied in their length by adjusting the kinetics of the assembly process.35,36 The strong interaction of the BTP allows for more variations of the hydrophobic groups as for the benzenetrisurea motif, for example, n-hexyl (C6) or n-dodecyl (C12) chains can both be employed as hydrophobic spacers.35,37 Moreover, recent investigations revealed that the BTP system shows a stronger tendency to form supramolecular fibers compared to the BTU system, even when more complex hydrophilic polymers are utilised.38 The robustness during the fiber assembly and versatility make the BTP system a promising candidate for future applications. We conducted multiple rheological measurements on both BTP-C6- and BTP-C12-based hydrogels, varying conditions such as the crosslinker content from 0.1 to 10 mol% and overall concentration from 25 to 50 mg mL−1. Furthermore, we examined the rheological behavior of the hydrogels at temperatures in the range of 0–80 °C. To reveal the biomedical applicability for future studies, the injectability and recovery of the gel in a biological environment were qualitatively assessed by the injection of a fluorescein containing BTP-C6 hydrogels into muscle tissue (chicken breast sample). In addition, the viability of L929 mouse fibroblast cells growing on the BTP-C6 hydrogels was determined.
Results and discussion
The synthesis of BTP-monomer and crosslinker (CL) units (Fig. 1) was performed according to the procedures established by Gruschwitz and Klein.28,35 For the confirmation of the successful PEO attachment, 1H-NMR (see ESI†) and size exclusion chromatography (SEC) measurements (Fig. 2) were performed. The SEC measurements of the monomer units showed monodisperse distribution in their elugrams (Fig. 2(a)). Interestingly, a shoulder towards higher molar masses was observed for CL12 (Fig. 2(b)), although no evidence of side reactions was observed in the NMR spectra. Therefore, we assume that this occurrence is a result of dimerization of CL12 in the SEC solvent (DMAc + 0.21 wt% LiCl), which indicates poor solubility and strong interaction. | ||
| Fig. 1 Overview of the chemical structures of the prepared benzenetrispeptide-based monomers and crosslinkers. | ||
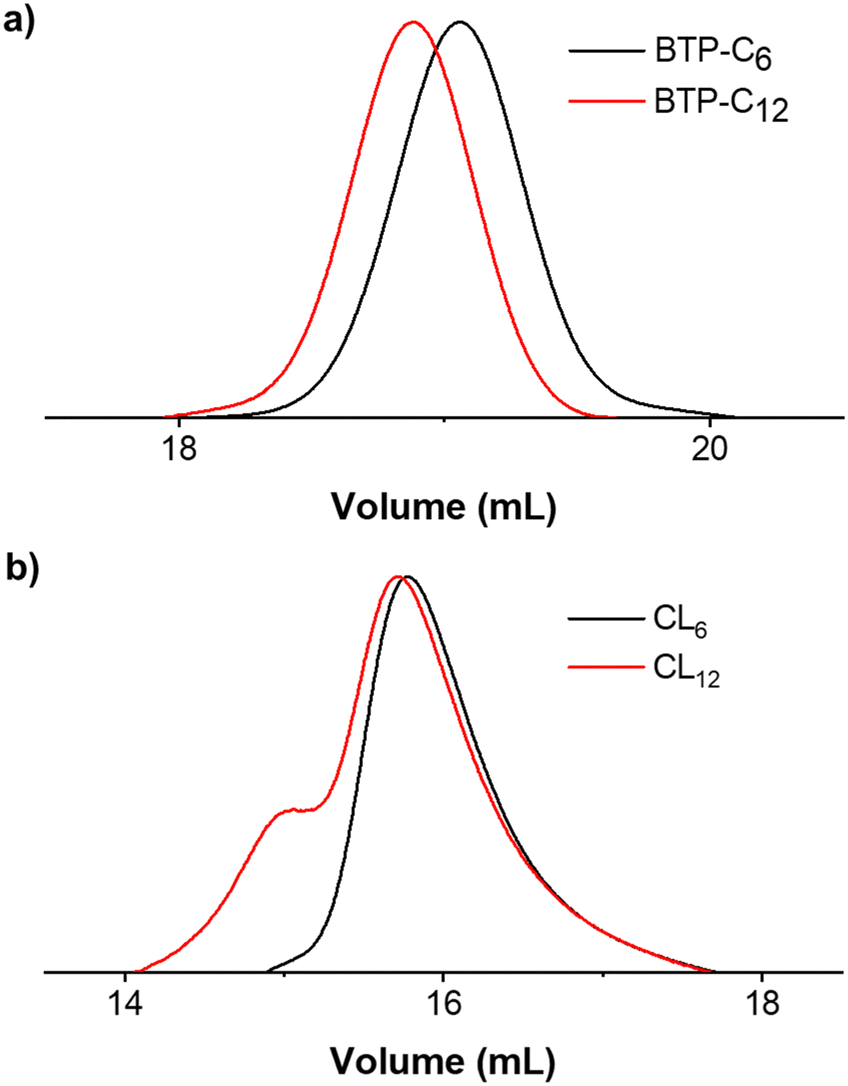 | ||
| Fig. 2 Overview of SEC measurements, showing elugrams of the BTP-monomer (a) and BTP-crosslinker units (b). Measurements were conducted using DMAc (+0.21 wt% LiCl) as the eluent and a flow rate of 1 mL min−1. | ||
Multiple parameters were considered for the rheological investigation to assess the potential of the BTP-based hydrogels for biomedical applications.39 Based on previous experience, the conditions for the first experiment were set at a sample concentration of 25 mg mL−1 and a crosslinker concentration of 1 mol%.28 Maintaining comparable weight contents of the supramolecular building blocks, the difference of molar mass between BTP-C6 and BTP-C12 causes slight discrepancies in their final molar concentrations (approximately 0.7 μmol mL−1). For the general hydrogel preparation procedure, we mixed the BTP-monomer and crosslinker and dissolved them in THF. To ensure that the system was fully dissolved, the solutions were heated to ∼40 °C. In the case of the BTP-C6 system, a small amount of water (<50 μL) was necessary for complete dissolution, as the system gelled in the pure organic solvent (THF) after cooling to room temperature. A gradual solvent switch from THF to water was then performed by slowly adding water (syringe pump at 1 mL h−1) to the organic mixture reaching a content of 67% water, before the organic solvent was finally removed by evaporation. This procedure results in free standing hydrogels for both samples BTP-C6 and BTP-C12 (Fig. 3(a)). Additionally, we also assessed the capability of the systems to form hydrogels in the absence of a crosslinker at the given concentrations, but we did not observe hydrogel formation for any of the samples (Fig. 3(b)).
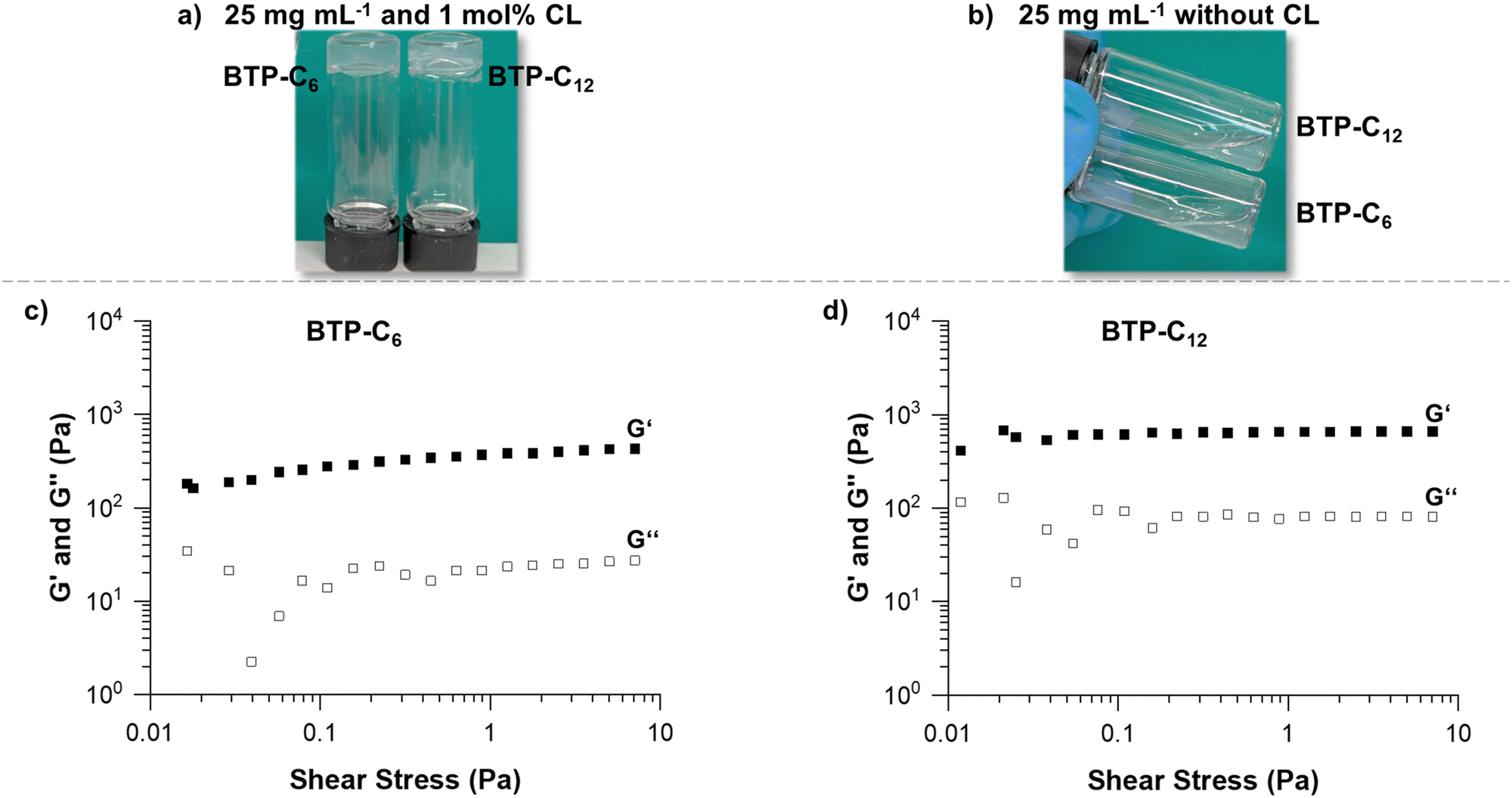 | ||
| Fig. 3 Images of the hydrogels (c = 25 mg mL−1) with 1 mol% crosslinker (a) and without crosslinker (b). Stress sweep of the BTP-C6 (c) and BTP-C12 hydrogels (d) at 20 °C to determine the stress strength usable for further tests. | ||
As the first step of rheological studies, we examined the linear viscoelastic domain of our samples using stress sweep measurement to determine how much stress we can apply to our sample without breaking the gel structure (Fig. 3(c) and (d), see the ESI† Section 2.2 stress sweep measurements of all samples). Shear storage modulus (G′) and loss modulus (G′′) are stable when subjected to a shear stress from 0.4 to 10 Pa for both samples. Therefore, we choose a shear stress of 1 Pa for further measurements of both samples.
As the first major investigation, we conducted step-strain measurements. During this measurement, the sample is repeatedly deformed by applying a large strain (200% deformation) and allowed to recover, which yields information on the self-healing properties of the hydrogels, i.e. whether the sample recovers and how long it takes to regain its initial strength.40 Surprisingly, the BTP-C6 system with 1 mol% crosslinking formed a significantly stronger hydrogel with an average G′ value of ∼1.1 kPa (Fig. 4(a), red squares), whereas the BTP-C12 system resulted in a lower G′ value of ∼500 Pa (Fig. 4(b), red squares) at similar concentrations. We initially anticipated the formation of stronger hydrogels by BTP-C12 motifs in comparison to BTP-C6 counterparts due to the longer alkyl chains present in BTP-C12. A longer aliphatic chain is expected to enhance the hydrophobic interaction and thus should induce a higher stability of the self-assembled fibers in the gel. The lower modulus of the BTP-C12 hydrogel might be related to a preaggregation of CL12 in THF (already observed in SEC experiments) and, subsequently, an insufficient incorporation into the fiber network or clustering of the crosslinker during the assembly process (Fig. 4(c) and (d)).
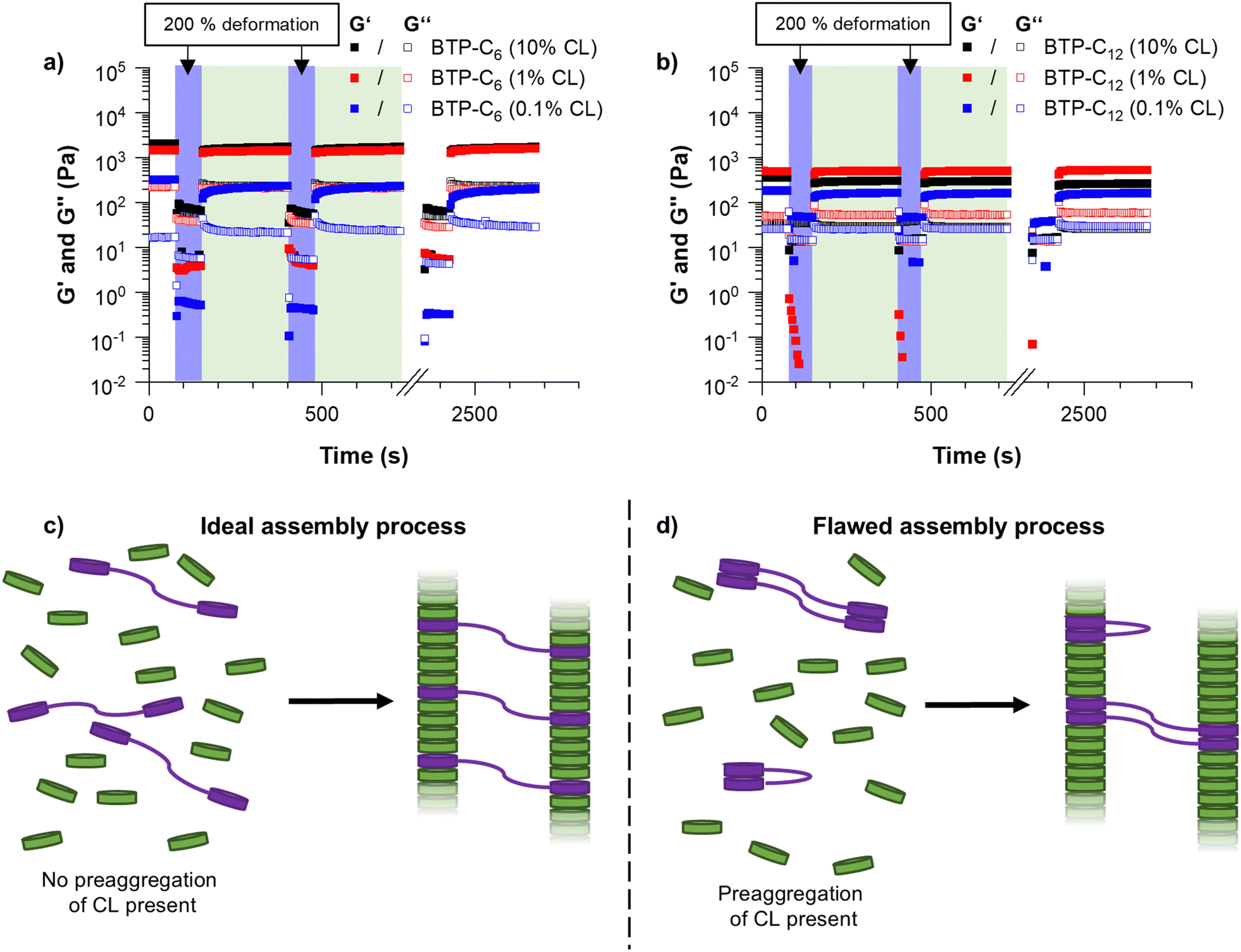 | ||
| Fig. 4 Step-strain measurements of BTP-C6 (a) and BTP-C12 (b) hydrogels with varying crosslinker concentrations of 0.1, 1 and 10 mol% at 20 °C. Schematic representation of assembly formation without (c) and with (d) preaggregation of the crosslinker. | ||
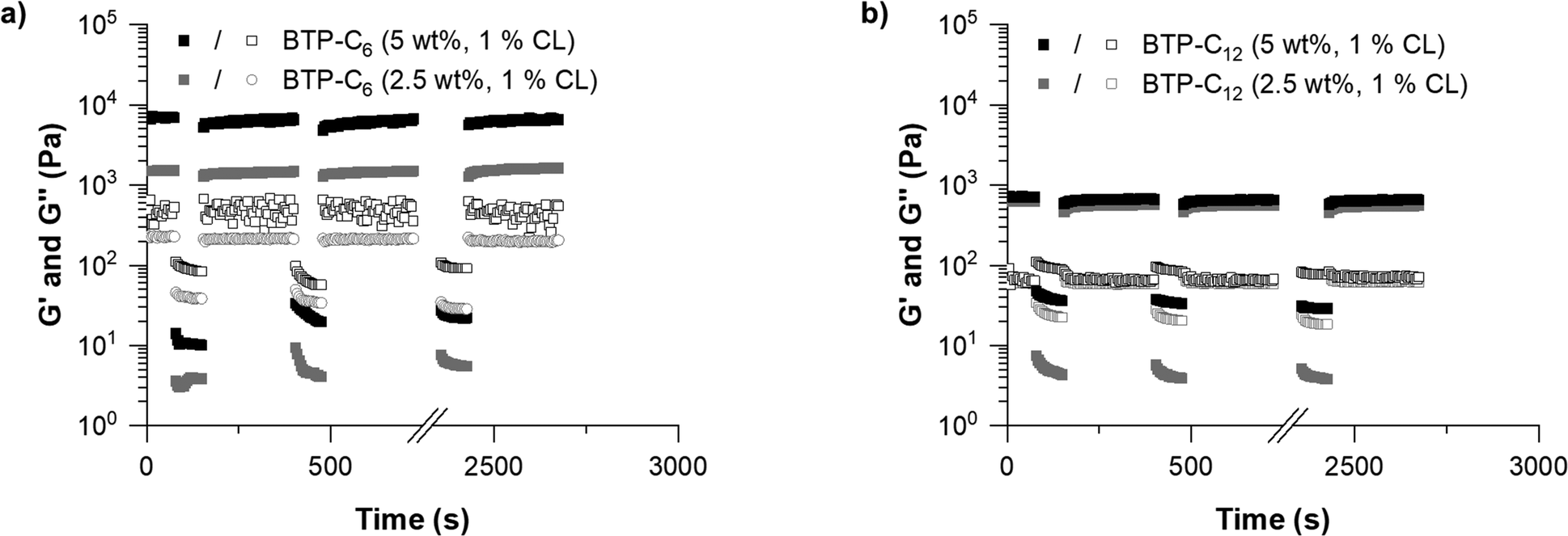
Concerning the capability of both systems to reform a gel after shear deformation, the rheological measurements revealed a rapid recovery within less than 5 seconds over several cycles of strain/recovery. Only in the case of BTP-C6, a slight decrease of shear storage modulus was observed after the first strain step, followed by a gradual increase over time during each recovery step. To further verify the robustness of the systems, we performed frequency sweep experiments before and after the step strain measurement, which show only little variation, indicating a full recovery of the systems (Fig. S3 and S7, ESI†).
Following these initial investigations on the BTP-C6 and BTP-C12 systems, we explored the influence of various crosslinker concentrations (0.1, 1, and 10 mol% CL) to evaluate their impact on the mechanical properties of the hydrogels. We examined both systems similar to the previous measurements. For the BTP-C6 system, we observed a slight increase in the G′ value of the hydrogels from ∼1.4 kPa to ∼1.7 kPa with 10 mol% CL and a significant decrease in the G′ value to ∼0.2 kPa for 0.1 mol% CL (Fig. 4(a); for individual measurements, see Fig. S3–S5, ESI†). These findings were in the line with our expectation because a higher crosslinking density leads to a higher stability of the hydrogel and thus a higher G′ value.41 The 10 mol% CL sample featured a similar decreased strength after the first step and gradual improvement as the initially tested sample with 1 mol% CL. In the case of the 0.1 mol% CL sample, however, the recovery occurs more slowly and requires several minutes to reach a plateau level again. Surprisingly, the BTP-C12 system revealed lower G′ values at higher (10 mol%,) CL content compared to the initially tested sample with 1 mol% CL (∼0.5 kPa) (Fig. 4(b); for individual measurements see Fig. S7–S9, ESI†). This weakening of the gel with increasing CL content supports our hypothesis that the limited solubility of CL12 causes a decrease in strength compared to the CL6 system. Furthermore, decreasing the CL content to 0.1 mol% has a less pronounced effect on gel strength (∼0.15 kPa compared to ∼0.3 kPa for 1 mol% CL) than that observed for the BTP-C6 system. All gels based on BTP-C12 recovered quickly (gel formation <5 s) and a plateau is rapidly reached for 10 mol% and 1 mol% CL, while the low CL content of 0.1 mol% again causes a slight delay.
We further conducted shear viscosity measurements to reveal the effect of the crosslinker concentration on yield stress and shear-dependent viscosity of BTP-C6 and BTP-C12 systems (Fig. 5). These are critical parameters to determine the pressure required to induce the flow of the gel and evaluate the injectability of the hydrogels. Therefore, we applied a continuously increasing shear rate ![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) to determine its influence on viscosity. We evaluated the results using a power law fit on the linear regions at higher shear rates according to
to determine its influence on viscosity. We evaluated the results using a power law fit on the linear regions at higher shear rates according to
| η = Kn−1 | (1) |
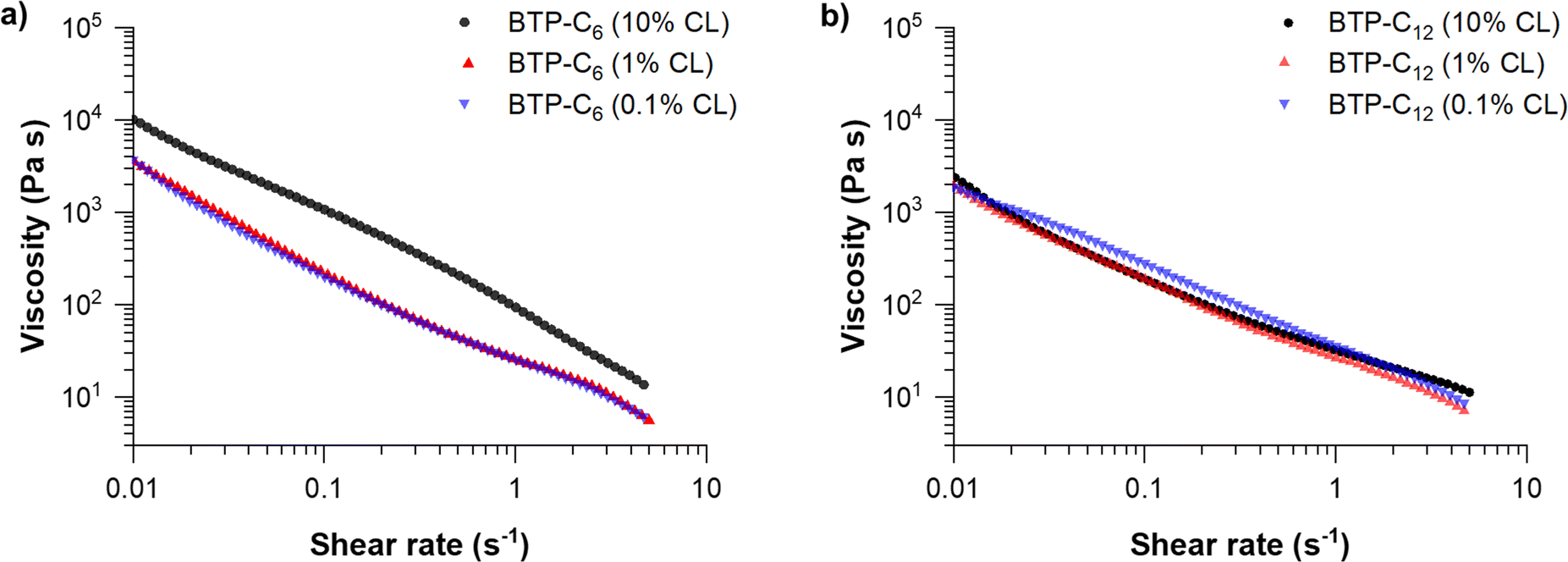 | ||
| Fig. 5 Viscosity measurements of BTP-C6 (a) and BTP-C12 (b) hydrogels with varying crosslinker concentrations of 0.1, 1 and 10 mol% at 20 °C. For individual measurements with measuring points below 0.01 s−1, see Fig. S3–S5 and S7, S8 (ESI†). | ||
These fits allowed us to determine the shear-thinning parameter n and the consistency index K for the tested samples (Table 1).30,42 In addition, we investigated the reversibility of the BTP-C12 system (1 mol% CL) to assess whether the viscosity of our system remains consistent based on multiple measurements. Therefore, we performed three consecutive runs by alternating between increasing and decreasing the shear rate (Fig. S11, ESI†). These results revealed minimal variations demonstrating the robustness of our measurement setup and the stability of our system.
| Sample | Crosslinker content (mol%) | Yield stress (Pa) | n | K (Pa s−1) |
|---|---|---|---|---|
| a Due to the abnormal curve progression of the measurement, the fit was applied to lower sheer rates between 0.02 and 0.3 s−1. | ||||
| BTP-C6 | 10 | 70.1 | 0.08 ± 0.01a | 122.95 ± 1.86a |
| 1 | 32.3 | 0.14 ± 0.01 | 25.12 ± 0.36 | |
| 0.1 | 28.22 | 0.14 ± 0.02 | 24.31 ± 0.70 | |
| BTP-C12 | 10 | 17.7 | 0.30 ± 0.02 | 33.35 ± 0.93 |
| 1 | 13.6 | 0.16 ± 0.02 | 25.48 ± 1.31 | |
| 0.1 | 10.3 | 0.10 ± 0.01 | 33.89 ± 0.33 | |
For all samples, the calculated values for n are well below 1 which corresponds to a strongly shear-thinning fluid, as the viscosity decreases significantly with increasing shear-rate. Comparing the previously reported benzenetrisurea systems, the obtained values for n and K are in a similar range (n = 0.15; K = 22.97), proving that this shear-thinning behavior is characteristic for these types of hydrogelators.28 In addition, both systems indicate significantly lower values compared to state-of-the-art printable polymer hydrogels based on poloxamer polymers (n = 0.13; K = 406) or alginates (n = 0.31; K = 254).42 Among the results obtained (Table 1), lower shear rates between 0.02 and 0.3 s−1 were applied for BTP-C6 with 10 mol% CL due to the distinct behavior of the material with higher shear rates (Fig. 5(a)). At higher shear rates >0.1 s−1, the value of n is negative (n = −0.15 ± 0.01; K = 91.36 ± 0.59), which is related to a rather steep slope of the viscosity change with increasing shear rate. Although such values are unusual, negative n values were also observed in other studies.43,44 A plausible explanation for this anomaly could be that the sample becomes more fragile with increasing crosslinker content.
The obtained values of n and K can also be used to calculate the pressure and force needed to extrude the hydrogel through a cannula, and so their injectability can be determined.30 Using the predetermined parameters, we calculated the force needed to extrude the hydrogel through a cannula of specific gauges (see the ESI,† Chapter 3 and Table S2). For a gauge 21 cannula with a length of 50 mm, commonly used for intramuscular injections, and a 1 mL syringe, BTP-C6 and BTP-C12 hydrogels with 1 mol% CL would require 0.62 N and 0.76 N force, respectively, to be extruded with a flowrate of 0.1 mL s−1. A gauge 23 cannula, with a length of 12 mm, commonly used for subcutaneous injection, on the other hand, would require a force of 0.33 N for BTP-C6 and 0.42 N for BTP-C12 under the same conditions.45 In all cases the required forces are considerably lower than the maximum forces considered for manual (40 N) or automatic injection (20 N) of 1 mL with an injection rate of 0.1 mL s−1.46 It is noteworthy that the injection into tissue also affects the necessary force due to the tissues counterpressure, which leads to an increase in the force needed for, for instance, subcutaneous injection.47 As a proof of concept for the injectability of the hydrogels into muscular tissue, we used a piece of chicken breast and injected the BTP-C6 gel using a syringe. This test not only enables a practical evaluation of the required injection forces but also assesses if the hydrogel recovers in such an environment (Fig. 6). We used a 1 mol% crosslinked BTP-C6 hydrogel soaked with 1 mg mL−1 fluorescein, which resembled an easy traceable model compound. For the injection, a gauge 23 cannula with a length of 80 mm and a 2 mL syringe were used, which still allowed an easy and effortless manual injection of the hydrogel. The resistance observed was similar to the extrusion of a viscous oil. After sample injection (∼0.2 mL), we gently “massaged” the sample to simulate muscle movement and cut the tissue to reveal the injection site. The hydrogel appears to be distributed between different muscle, cords but remains localized in the injection area. It is noteworthy that neither massaging nor cutting the chicken breast had a strong impact on the distribution, highlighting the stability of the gel even when injected into tissue.
 | ||
| Fig. 6 Injection of a BTP-C6 1 mol% CL sample (using a G27 cannula), loaded with 1 mg mL−1 fluorescein as the modal system, into chickenbreast (a) and a crosssection of the injectionside under UV light (b). | ||
In order to explore the further potential of the materials, we analyzed the influence of the total gelator concentration on the G′ value. A higher concentration should lead to a higher density of fibers in the hydrogel, which in return should lead to increased entanglement and thus a strengthening of the gel.48,49 We therefore kept the CL content at 1 mol% and increased the overall weight concentration of the BTP-system in the hydrogel from 2.5 wt% to 5 wt%, which in previous investigations on the benzenetrisurea system resulted in a noticeable (∼three-fold) increase of the gel strength.28 The G′ value of the BTP-C6 system increased to ∼8 kPa (Fig. 7(a)), which is almost a sixfold increase compared to the sample with 2.5 wt%. To our surprise, the increase in concentration caused only an increase in strength from 0.3 kPa to ∼0.8 kPa for the BTP-C12 system (Fig. 7(b)). We assume that in the latter case, the higher concentration of the building block and crosslinker further impedes the homogeneous distribution of the crosslinker throughout the fibers, which is in accordance with the observations made for increased crosslinker contents.
 | ||
| Fig. 7 Comparison between the hydrogels with concentrations of 2.5 wt% (grey) and 5 wt% (black) based on BTP-C6 (a) and BTP-C12 (b) with the 1 mol% crosslinker at 20 °C. | ||
Following these tests, we further evaluated the impact of increasing temperature on the hydrogels. Supramolecular interactions typically weaken at elevated temperatures.50 In our tests, the BTP-C12 samples demonstrated such weakening as the gel strength decreased from ∼0.4 kPa at 0 °C to ∼0.15 kPa at 80 °C (Fig. 8), which was reversible upon cooling of the sample (see Fig. S12, ESI†). In contrast, the BTP-C6 sample showed an increase in G′ from ∼1.4 kPa to ∼2.7 kPa (Fig. 8), which indicates an unexpected thermal stiffening behavior. Such effects are less common for supramolecular assemblies and could be due to changes in the aggregation structure.51,52 For example, Chakravarthy et al. proposed the formation of a double network at elevated temperatures for their system, which leads to thermostiffening behavior at 37 °C.53 Further investigations into the cause of this phenomenon are currently ongoing, but considering the limited reversibility of the BTP-C6 sample, we refrained from further temperature-dependent measurements.
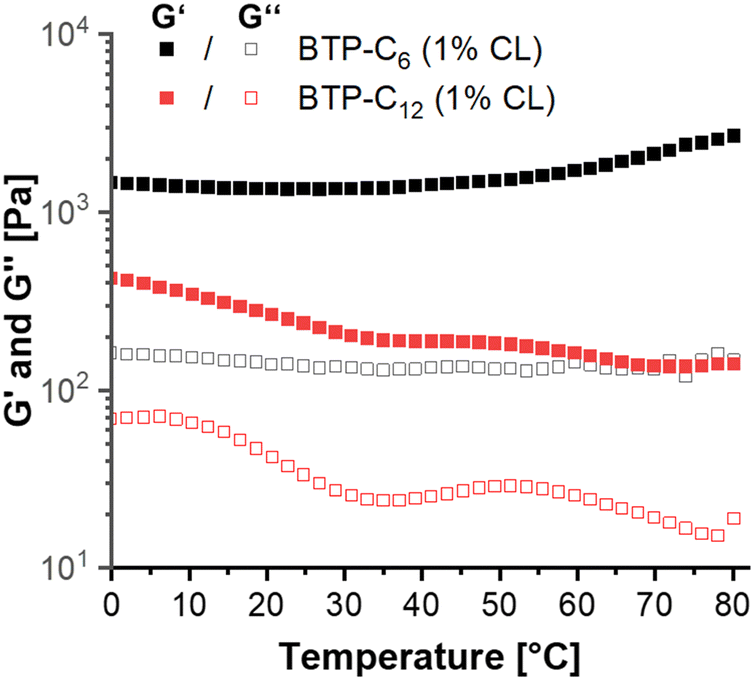 | ||
| Fig. 8 Temperature sweep measurements from 0 to 80 °C of BTP-C6 (black) and BTP-C12 (red) samples containing 25 mg mL−1 monomer and 1 wt% crosslinker. | ||
Since the BTP-C12 system was more reliable with temperature changes, we further investigated the temperature-dependent stress relaxation behavior of the corresponding hydrogels. Isothermal stress relaxation experiments were performed at 5 °C, 40 °C, 60 °C and 80 °C (Fig. 9(a)) on the BTP-C12 system (containing 1 mol% of crosslinker). The obtained data were normalized after 20 s of applied strain (1%, within the linear viscoelastic domain). This delay was necessary to reach a stable strain and to reduce the signal noise observed at the early stage of the experiments. As expected, the stress decay was faster at high temperatures, which we attribute to a faster exchange of supramolecular crosslinker or network reorganization. By analogy with covalent adaptable networks, i.e. thermosets containing covalent exchangeable bonds, the stress relaxation behavior of the BTP-C12 system can be described using a multimode Maxwell model (or KWW model), in which multiple relaxation modes coexist instead of a simple mode.54,55 The data obtained from Fig. 9(a) were accurately fitted (0.993 < R2 < 0.998) using the following equation:
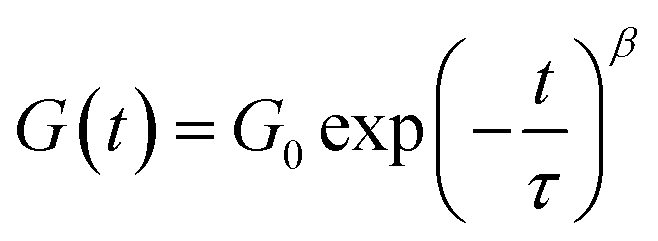 | (2) |
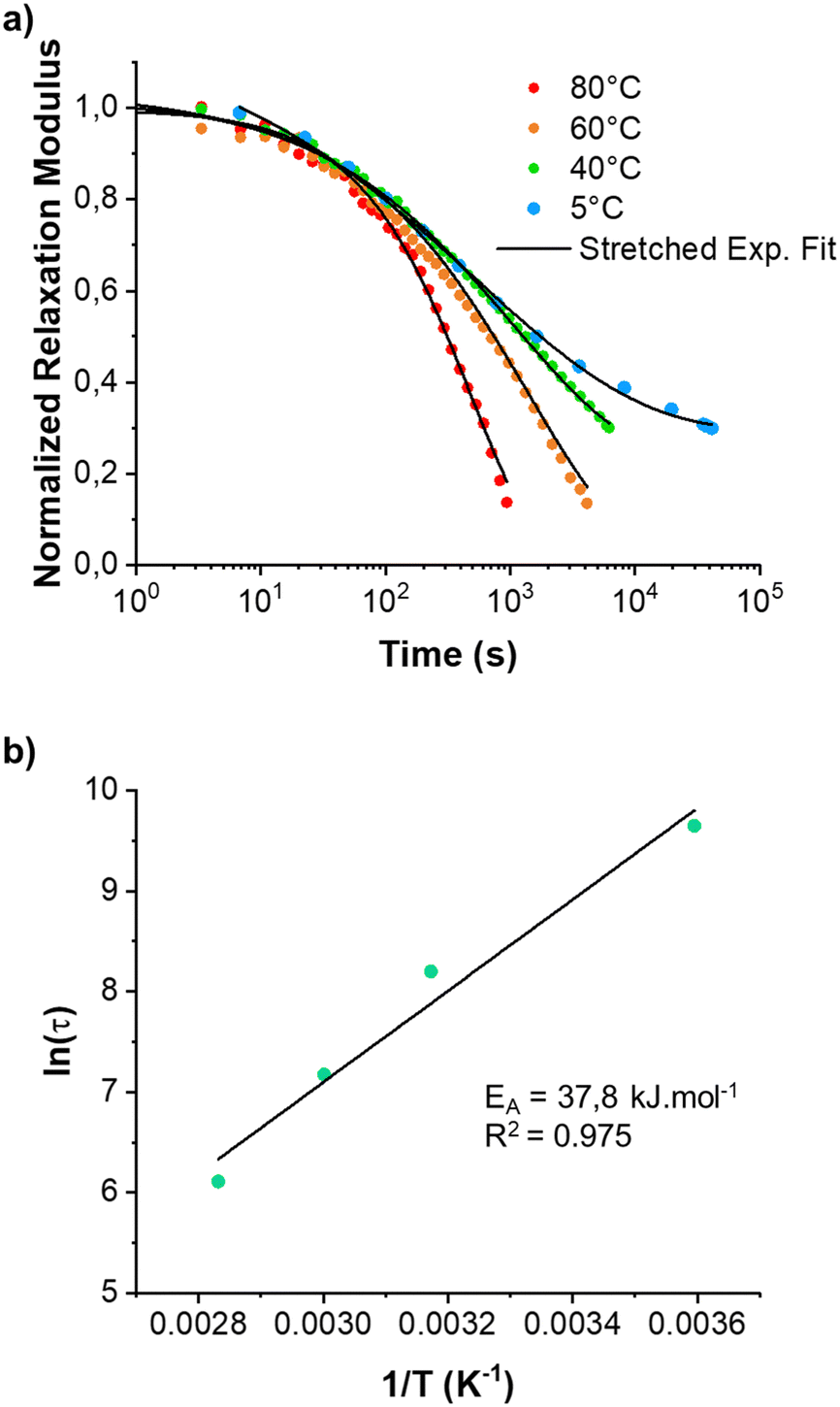 | ||
| Fig. 9 Normalized stress relaxation modulus (shear mode) of BTP-C12 (a) at 5, 40, 60 and 80 °C. The data were measured consecutively on the same sample and normalized 20 seconds after strain (1%) was applied. The characteristic relaxation times (in s) were plotted in a log plot (b) and fitted using a linear fit to obtain the activation energy (EA). | ||
Fitting the stress relaxation behavior with this stretched exponential function reveals that a broad relaxation time distribution is present at 5 °C (β(5 °C) = 0.32), which becomes “narrower” at higher temperatures (β(80 °C) = 0.81). The activation energy EA = 37.8 kJ mol−1 (Fig. 9(b)) was extracted from the Arrhenius plot of ln(τ) vs. 1/T, where τ is the average relaxation time determined from the stretched exponential fitting. These results indicate that the system becomes more dynamic with increasing temperature, which is in accordance with the behavior of other supramolecular systems. Further studies with regard to the general fiber dynamics are currently underway.
We finally aimed to assess the biocompatibility of the hydrogels. It is noteworthy to state that instead of using hydrogel extracts, we designed an experimental system incubating L929 mouse fibroblasts directly on a hydrogel layer, which allowed us to further investigate cell-hydrogel interactions. Therefore, we tried to cover the bottom of wells in standard cell culture plates with the hydrogel and subsequently seed cells on the surface. However, following the same procedure for preparing the hydrogels used for the rheological measurements, the BTP-C12 system started to gel at 20 to 30 vol% water content, which impeded a homogenous coverage of the culture plate surface (Fig. S13, ESI†). Therefore, we focused on the BTP-C6 hydrogel with 2.5 wt% and 1 mol% of CL as the exemplary system to evaluate cell interactions and biocompatibility. We added a predetermined amount of the BTP-C6 hydrogel stock solution to a tissue culture (TC)-treated 96-well culture plate aiming for a final hydrogel thickness of 1.1 mm after evaporation of the solvent. The aimed thickness ensured that the cells were only in contact with the hydrogel but not the bottom surface of the well. Upon THF evaporation, we obtained intact BTP-C6 hydrogels in the wells (Fig. S14, ESI†). To ensure the full removal of the solvent and create conditions suitable for cell seeding, we washed the surface of the hydrogels several times with PBS. Finally, we incubated the hydrogels treated with Dulbecco's modified Eagle medium (D10) overnight at 37 °C under a humidified 5% (v/v) CO2 atmosphere. It is noteworthy that the thickness and consistency of the hydrogels did not change over the course of this procedure even though they were constantly covered by 100 μL of cell culture medium.
Following the preparation of the culture plate with hydrogel, L929 cells (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per mL per well) were seeded in blank wells without hydrogel (control) and in wells covered with the hydrogels (gel) in triplicate and the experiment was repeated for four different cell passages (P1, P2, P3, and P4) (Fig. 10).
000 cells per mL per well) were seeded in blank wells without hydrogel (control) and in wells covered with the hydrogels (gel) in triplicate and the experiment was repeated for four different cell passages (P1, P2, P3, and P4) (Fig. 10).
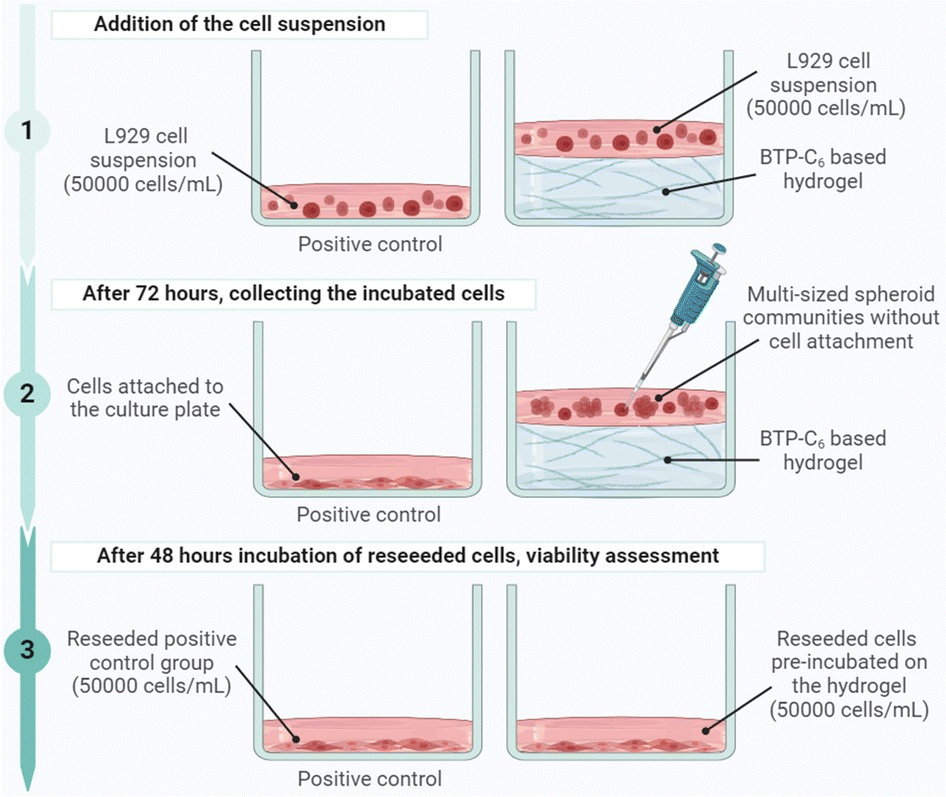 | ||
| Fig. 10 Graphical representation of the experimental setup for the biocompatibility assessment of the BTP-C6 hydrogel (created with https://www.BioRender.com). | ||
The cells were incubated for 72 hours and then imaged by light microscopy (Fig. 11). The cells did not attach to the surface of the hydrogel (Fig. 11(a)) but rather formed multi-sized communities in spheroid shapes based on cell-to-cell aggregation. This is a quite common behavior for the cells growing on inert surfaces and indicates that the hydration shell around the dense PEO chains on the fibers prevents protein adsorption and further cell interactions.56–58 After 72 hours of incubation, the cells were collected and reseeded into a TC treated 96-well culture plate to understand whether the gels had any negative effect on the cell viability, morphology, and attachment ability. After 48 hours of incubation, the cells pre-incubated on the hydrogels were able to reattach to the culture plate. (Fig. 11(b)). In addition, there was no significant change in their morphology compared to a control, so we can conclude that the BTP-C6 hydrogel did not have a detrimental effect on L929 cells.
 | ||
| Fig. 11 Transmitted light microscopy images of L929 mouse fibroblast cells incubated on the BTP-C6 hydrogel for 72 hours: (a) image taken on the hydrogel, and (b) image of cells reseeded after 48 hours of incubation, collected from the top of the hydrogel. Optical magnification is 10× and the scale bar is 100 μm. | ||
The viability of the reseeded cells was assessed quantitatively using the PrestoBlue™ assay. For all passages, the mean relative metabolic activity values of the control groups were set as 100%. A cell viability ≤70% is considered toxic according to DIN EN ISO 10993-5.59 Compared to the control groups, the cells previously incubated on the hydrogels showed relative metabolic activities around 90%, indicating that the BTP-C6 hydrogel had no toxic effect on L929 cells (Fig. 12). Irrespective of the passage number, the mean relative metabolic activity was higher than 87% for all gel groups compared to the control groups, and the mean values did not show any significant variation from passage to passage (Table S1, ESI†). In light of both qualitative and quantitative observations, the BTP-C6 hydrogel can be considered biocompatible and the formation of a multi-sized spheroid on the hydrogel is currently under further investigation.
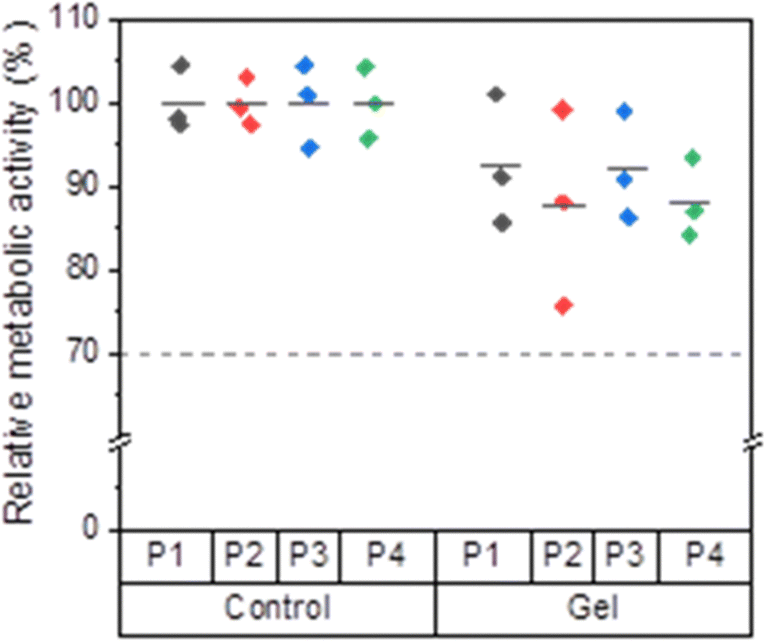 | ||
| Fig. 12 Relative metabolic activity (%) of L929 mouse fibroblast cells growing on the BTP-C6 hydrogel was determined using the PrestoBlue™ assay. The experiments were conducted as triplicate for four different passages represented as P1, P2, P3, and P4. The calculated mean values were depicted using lines. | ||
Conclusion
In conclusion, we investigated the rheological properties of hydrogels formed from the supramolecular assembly of benzenetrispeptides with C6 and C12 spacers and a conjugated PEO chain. The introduction of a bifunctional crosslinker induced the formation of the hydrogel and we found that both systems display fast recovery rates during step strain measurements. Varying the crosslinker content had a limited effect on the BTP-C12 system, which was related to an inefficient integration of the crosslinker. However, the BTP-C6 system could be modified in its shear modulus over a range from 0.2 kPa up to nearly 2 kPa when increasing the crosslinker content from 0.1 to 10 mol%. Increasing the total concentration of the materials from 2.5 w% to 5 wt% caused another significant increase of G′ for the BTP-C6 system up to 8 kPa, while only a minor increase in G′ (from 0.5 to 0.8 kPa) is observed for BTP-C12, which is most likely again due to a limited integration of the crosslinker. Temperature-dependent measurements further revealed a thermo-stiffening behavior of the BTP-C6 system, which cannot be clarified at present, while the BTP-C12 hydrogel becomes weaker with increasing temperature. Stress relaxation measurements of the latter sample revealed an Arrhenius behavior, indicating increasing dynamics at higher temperatures. Analyzing the shear-dependent viscosity further revealed a strong shear-thinning effect for all tested samples, which facilitates effortless injections even through thinnest needles. Thus, a fluorescein-containing BTP-C6 hydrogel could be easily injected into chicken breast as a muscle tissue sample applying only a small amount of force. The hydrogel distributes between the muscle cords but more importantly, it is able to reform even within this tissue and remains at the injection side even after massaging the muscle. Direct incubation of L929 mouse fibroblast cells on the hydrogel confirmed that the dense PEO shell on the fibers prevents an attachment of the cells and spheroid-like communities are formed. Nevertheless, reseeding of these cells and comparison with a control group resulted in a relative metabolic activity of over 87% independent of passage number, and no signs of changes in cell morphology or behavior were observed. Overall, these experiments demonstrate a high biocompatibility of these injectable supramolecular hydrogels, and the tunable shear modulus of the BTP-C6 system allows an adaption of the hydrogel to the specific strength of different soft tissues such as brain, pancreas, or lung making it an attractive material for applications as a drug depot or in tissue engineering.60–63Methods and materials
All reagents and solvents were commercial products purchased from Merck, ABCR, Iris BioTech, Rapp Polymere or TCI and were used without further purification. The BTP building blocks were prepared according to procedures described in the literature.28,35 Details on the synthesis of the crosslinker are provided in the ESI.†Gel preparation
The procedure for the preparation of hydrogels is based on the solvent switch and gel preparation method described as follows.28,36 The gels were prepared by weighing in the lyophilized building blocks and crosslinkers in a 20 mL vial with a wide neck. 0.75 mL of THF was added, the vial was closed, and the solids were dissolved using sonication and light heating. Afterwards, the samples were mixed using a magnetic stirrer for 30 min. To the closed vial, 1.5 mL of water was added at a speed of 1 mL h−1 using a syringe pump. Following this, the cap was then removed to slowly evaporate THF until the weight of the gel was 1.5 g. Lastly, the gel was transferred to the rheometer.Rheology measurements
The oscillatory dynamic measurements were performed using a Physica Modular Compact MCR102 Rheometer obtained from Anton Paar (Germany). Mechanical properties of the viscoelastic hydrogel material were assayed using a parallel (PP 25 sandblasted)-plate geometry. The linear viscoelastic regime of the samples was obtained from amplitude sweep experiments that were performed from 0.1 to 200% strain at 6.36 rad s−1 angular frequency. Frequency sweep measurements were recorded from 0.1 to 100 rad s−1 at 1% strain. Measurements were performed at 20 °C unless stated otherwise. For measurements above 20 °C, paraffin oil was added around the sample and geometries to hinder the sample from drying out due to water evaporation.Biological investigation
L929 cells (CLS Cell Lines Service GmbH, Eppelheim, Germany) were cultured in TC-treated cell culture flasks (Greiner Bio-One International GmbH and Labsolute Th. Geyer GmbH & Co. KG) in the presence of Dulbecco's modified Eagle medium (D10) (2 mM L-glutamine (Biochrom, Germany) supplemented with 10% fetal calf serum (FCS, Capricorn Scientific, Germany), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin (Biochrom)) at 37 °C under a humidified 5% (v/v) CO2 atmosphere. Dulbecco's phosphate-buffered saline (1×) (DPBS) and trypsin-EDTA (Capricorn Scientific GmbH, Germany) were used for each cell passaging. The cells with the passage numbers 22 (P1), 23 (P2), 24 (P3), and 25 (P5) showing ≥97% viability were used.For the cell viability assessment, a hydrogel stock solution was prepared strictly following the above-mentioned preparation methodology. Under a sterile environment, predetermined amount of hydrogel stock solution (57 μL) was added to each well of TC treated 96-well cell culture plates (VWR International GmbH) by aiming 1 to 1.2 mm gel thickness (Fig. S16, ESI†). After evaporation of THF from the system in a sterile environment, hydrogels were formed and a series of washes with PBS were conducted to adjust the pH of the system. Afterwards, 100 μL of DMEM was added in each well containing hydrogel. The plate was incubated overnight at 37 °C under a humidified 5% (v/v) CO2 atmosphere for the next day's cell seeding. The following day, L929 cells were seeded (5000 cells per well) both in the wells without gels (positive control) and with the gels in triplicate. The cells were incubated for three days at 37 °C under a humidified 5% (v/v) CO2 atmosphere.
After 72 hours, control groups were collected by trypsinization. However, the cells growing on the gels were directly collected since the cells did not attach to the surface of the hydrogel. For each sample, the viability and number of the cells were determined using a cell counter (Fluidlab R-300, Anvajo GmbH, Dresden, Germany). 5000 cells from each sample were reseeded in a TC treated 96-well cell culture plate and the cells were incubated for two days at 37 °C under a humidified 5% (v/v) CO2 atmosphere. After 48 hours, the viability assessment was conducted.
Relative metabolic activities of viable L929 mouse fibroblast cells were determined using the PrestoBlue™ assay (Thermo Fisher Scientific) following DIN EN ISO 10993-5. 10% (v/v) PrestoBlue™ solution, which was prepared according to the manufacturer's instructions, was added to each well. Afterwards, the cells were further incubated at 37 °C for 45 min and finally the fluorescence was measured with a multi-plate reader (Infinite M200 PRO Microplate Reader, Tecan, Switzerland) at λEx = 560 nm/λEm = 590 nm. The relative viability (%) of the cells was calculated as in eqn (3):
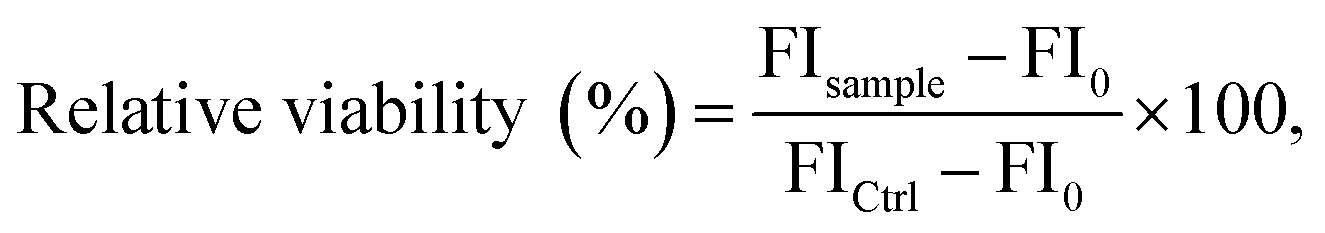 | (3) |
In addition to the viability assessment, cells were observed with a transmitted light microscope (Axio Oberserver Vert.A1, Zeiss, Germany) with a 10× objective using brightfield imaging. Images were recorded using ZEN lite software (2012, Zeiss).
For the statistical analysis, OriginPro2022b software was used. The mean relative metabolic activity of each group was calculated as mean ± SD. In addition, a one-way analysis of variance (ANOVA) was conducted. At level 0.05, there was no significant difference in the mean relative metabolic activity of the passages.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the German Science Foundation (DFG) for generous funding within the Emmy-Noether Programme (project-ID: 358263073) and the Heisenberg-Programme (project-ID: 517761335). The work was further supported by an Exploration Grant of the Boehringer Ingelheim Foundation (BIS). We acknowledge Sandra Henk and Carolin Kellner for maintaining the L929 cell line and Elisabeth Moek for the assistance in mycoplasma detection testing. Prof. U. S. Schubert is furthermore acknowledged for his continuous support and access to excellent research facilities at the FSU Jena.References
- R. Dong, Y. Pang, Y. Su and X. Zhu, Biomater. Sci., 2015, 3, 937–954 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Mater. Today Adv., 2019, 3, 100021 CrossRef.
- S. Khan, A. Ullah, K. Ullah and N.-U. Rehman, Des. Monomers Polym., 2016, 19, 456–478 CrossRef CAS.
- M. Santin, S. J. Huang, S. Iannace, L. Ambrosio, L. Nicolais and G. Peluso, Biomaterials, 1996, 17, 1459–1467 CrossRef CAS PubMed.
- K. Kowalczuk, P. Mons, H. Ulrich, V. Wegner, J. Brendel, A. Mosig and F. Schacher, Macromol. Biosci., 2023, 24, e2300230 CrossRef PubMed.
- R. Yoshida, K. Uchida, Y. Kaneko, K. Sakai, A. Kikuchi, Y. Sakurai and T. Okano, Nature, 1995, 374, 240–242 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- X. Wang and C. Feng, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2023, 15, e1847 CAS.
- R. F. Q. Grenfell, L. M. Shollenberger, E. F. Samli and D. A. Harn, Clin. Vaccine Immunol., 2015, 22, 336–343 CrossRef CAS PubMed.
- Y. Tian, H. Wang, Y. Liu, L. Mao, W. Chen, Z. Zhu, W. Liu, W. Zheng, Y. Zhao, D. Kong, Z. Yang, W. Zhang, Y. Shao and X. Jiang, Nano Lett., 2014, 14, 1439–1445 CrossRef CAS PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- Q. Wang, J. Wang, Q. Lu, M. S. Detamore and C. Berkland, Biomaterials, 2010, 31, 4980–4986 CrossRef CAS PubMed.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- M. M. C. Bastings, S. Koudstaal, R. E. Kieltyka, Y. Nakano, A. C. H. Pape, D. A. M. Feyen, F. J. van Slochteren, P. A. Doevendans, J. P. G. Sluijter, E. W. Meijer, S. A. J. Chamuleau and P. Y. W. Dankers, Adv. Healthcare Mater., 2014, 3, 70–78 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. J. A. van Luyn, A. Huizinga-van der Vlag, G. M. L. van Gemert, A. H. Petersen, E. W. Meijer, H. M. Janssen, A. W. Bosman and E. R. Popa, Biomaterials, 2012, 33, 5144–5155 CrossRef CAS PubMed.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024–2027 RSC.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- J. J. Van Gorp, J. A. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS PubMed.
- M. de Loos, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron, 2007, 63, 7285–7301 CrossRef CAS.
- C. M. A. Leenders, T. Mes, M. B. Baker, M. M. E. Koenigs, P. Besenius, A. R. A. Palmans and E. W. Meijer, Mater. Horiz., 2014, 1, 116–120 RSC.
- S. Bernhard and M. W. Tibbitt, Adv. Drug Delivery Rev., 2021, 171, 240–256 CrossRef CAS PubMed.
- G. Zhang, Y. Chen, Y. Deng, T. Ngai and C. Wang, ACS Macro Lett., 2017, 6, 641–646 CrossRef CAS PubMed.
- J. Li, X. Li, X. Ni, X. Wang, H. Li and K. W. Leong, Biomaterials, 2006, 27, 4132–4140 CrossRef CAS PubMed.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS PubMed.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- F. V. Gruschwitz, F. Hausig, P. Schüler, J. Kimmig, S. Hoeppener, D. Pretzel, U. S. Schubert, S. Catrouillet and J. C. Brendel, Chem. Mater., 2022, 34, 2206–2217 CrossRef CAS.
- J.-L. Li and X.-Y. Liu, Adv. Funct. Mater., 2010, 20, 3196–3216 CrossRef CAS.
- S. Correa, A. K. Grosskopf, H. Lopez Hernandez, D. Chan, A. C. Yu, L. M. Stapleton and E. A. Appel, Chem. Rev., 2021, 121, 11385–11457 CrossRef CAS PubMed.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- L. A. Haines-Butterick, D. A. Salick, D. J. Pochan and J. P. Schneider, Biomaterials, 2008, 29, 4164–4169 CrossRef CAS PubMed.
- F. V. Gruschwitz, M.-C. Fu, T. Klein, R. Takahashi, T. Higashihara, S. Hoeppener, I. Nischang, K. Sakurai and J. C. Brendel, Macromolecules, 2020, 53, 7552–7560 CrossRef CAS.
- T. Klein, H. F. Ulrich, F. V. Gruschwitz, M. T. Kuchenbrod, R. Takahashi, S. Fujii, S. Hoeppener, I. Nischang, K. Sakurai and J. C. Brendel, Polym. Chem., 2020, 11, 6763–6771 RSC.
- F. V. Gruschwitz, T. Klein, M. T. Kuchenbrod, N. Moriyama, S. Fujii, I. Nischang, S. Hoeppener, K. Sakurai, U. S. Schubert and J. C. Brendel, ACS Macro Lett., 2021, 10, 837–843 CrossRef CAS PubMed.
- T. Klein, H. F. Ulrich, F. V. Gruschwitz, M. T. Kuchenbrod, R. Takahashi, S. Hoeppener, I. Nischang, K. Sakurai and J. C. Brendel, Macromol. Rapid Commun., 2021, 42, e2000585 CrossRef PubMed.
- H. F. Ulrich, F. V. Gruschwitz, T. Klein, N. Ziegenbalg, D. T. N. Anh, S. Fujii, S. Hoeppener, K. Sakurai and J. C. C. Brendel, Chem. – Eur. J., 2024, 30, e202400160 CrossRef CAS PubMed.
- P. Bertsch, M. Diba, D. J. Mooney and S. C. G. Leeuwenburgh, Chem. Rev., 2023, 123, 834–873 CrossRef CAS PubMed.
- M. H. Chen, L. L. Wang, J. J. Chung, Y.-H. Kim, P. Atluri and J. A. Burdick, ACS Biomater. Sci. Eng., 2017, 3, 3146–3160 CrossRef CAS PubMed.
- R. S. H. Wong, M. Ashton and K. Dodou, Pharmaceutics, 2015, 7, 305–319 CrossRef CAS PubMed.
- N. Paxton, W. Smolan, T. Böck, F. Melchels, J. Groll and T. Jungst, Biofabrication, 2017, 9, 044107 CrossRef PubMed.
- J. S. Weston, J. H. Harwell and B. P. Grady, Soft Matter, 2017, 13, 6743–6755 RSC.
- J. C. P. Hollister, A. C. Wang, W. Kim, C. C. Giza, M. L. Prins and H. P. Kavehpour, Front. Phys., 2023, 11, 1308136 CrossRef.
- https://www.immunize.org/wp-content/uploads/catg.d/p2020a.pdf, (accessed 11.08.2024).
- R. P. Watt, H. Khatri and A. R. G. Dibble, Int. J. Pharm., 2019, 554, 376–386 CrossRef CAS PubMed.
- L. Wu, H. Li, Y. Wang, C. Liu, Z. Zhao, G. Zhuang, Q. Chen, W. Zhou and J. Guo, Eur. J. Pharm. Biopharm., 2024, 197, 114221 CrossRef PubMed.
- J. Cheng, D. Amin, J. Latona, E. Heber-Katz and P. B. Messersmith, ACS Nano, 2019, 13, 5493–5501 CrossRef CAS PubMed.
- R. E. Kieltyka, A. C. H. Pape, L. Albertazzi, Y. Nakano, M. M. C. Bastings, I. K. Voets, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 11159–11164 CrossRef CAS PubMed.
- S. Xian and M. J. Webber, J. Mater. Chem. B, 2020, 8, 9197–9211 RSC.
- G. Chaudhary, A. Ghosh, N. A. Bharadwaj, J. G. Kang, P. V. Braun, K. S. Schweizer and R. H. Ewoldt, Macromolecules, 2019, 52, 3029–3041 CrossRef CAS.
- P. de Almeida, M. Jaspers, S. Vaessen, O. Tagit, G. Portale, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2019, 10, 609 CrossRef CAS PubMed.
- R. D. Chakravarthy, I. Sahroni, C. W. Wang, M. Mohammed and H. C. Lin, ACS Nano, 2023, 17, 11805–11816 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- R. G. Ricarte and S. Shanbhag, Polym. Chem., 2024, 15, 815–846 RSC.
- L. Chen, C. Yan and Z. Zheng, Mater. Today, 2018, 21, 38–59 CrossRef CAS.
- B. D. Ippel, H. M. Keizer and P. Y. W. Dankers, Adv. Funct. Mater., 2019, 29, 1805375 CrossRef.
- H. Shen, S. Cai, C. Wu, W. Yang, H. Yu and L. Liu, Micromachines, 2021, 12, 96 CrossRef PubMed.
- DIN EN ISO 10993-5:2009-10, Biological evaluation of medical devices-Part5: Test for in vitro cytotoxicity, European Standard EN ISO 10993-5, Brussels.
- D. M. Tremmel, S. D. Sackett, A. K. Feeney, S. A. Mitchell, M. D. Schaid, E. Polyak, P. J. Chlebeck, S. Gupta, M. E. Kimple, L. A. Fernandez and J. S. Odorico, Sci. Rep., 2022, 12, 7188 CrossRef CAS PubMed.
- G. Burgstaller, B. Oehrle, M. Gerckens, E. S. White, H. B. Schiller and O. Eickelberg, Eur. Respir. J., 2017, 50, 1601805 CrossRef PubMed.
- L. Glorieux, L. Vandooren, S. Derclaye, S. Pyr dit Ruys, P. Oncina-Gil, A. Salowka, G. Herinckx, E. Aajja, P. Lemoine, C. Spourquet, H. Lefort, P. Henriet, D. Tyteca, F. M. Spagnoli, D. Alsteens, D. Vertommen and C. E. Pierreux, Int. J. Mol. Sci., 2023, 24, 10268 CrossRef CAS PubMed.
- S. Budday, T. C. Ovaert, G. A. Holzapfel, P. Steinmann and E. Kuhl, Arch. Comput. Methods Eng., 2020, 27, 1187–1230 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01873g |
| This journal is © The Royal Society of Chemistry 2025 |