
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extending the accessibility of catalytic active sites through L-cysteine assisted sulfidation for promoting the hydrogen evolution reaction†
Prince J. J.
Sagayaraj
a,
Kavinkumar
S.
a,
Keishi
Oyama
b,
Naoko
Okibe
*b,
Hyoung-il
Kim
 *c and
Karthikeyan
Sekar
*ac
*c and
Karthikeyan
Sekar
*ac
aSustainable Energy and Environmental Research Laboratory, Department of Chemistry, Faculty of Engineering and Technology, SRM Institute of Science and Technology, Kattankulathur 603203, Tamil Nadu, India. E-mail: karthiks13@srmist.edu.in
bDepartment of Earth Resources Engineering, Kyushu University, Fukuoka 819-0395, Japan. E-mail: okibe@mine.kyushu-u.ac.jp
cDepartment of Civil & Environmental Engineering, Yonsei University, Seoul 03722, Republic of Korea. E-mail: hi.kim@yonsei.ac.kr; karthiks@yonsei.ac.kr
First published on 18th December 2024
Abstract
Green hydrogen production has been a particular focus in recent times for implementing sustainable fuels in the future energy economy. One of the most effective ways to produce clean and green hydrogen is electrocatalytic overall water splitting. Various researchers with their persistent explorations have made this topic, the research hotspot in understanding the catalysis mechanism and developing new novel materials. As the hydrogen evolution reaction (HER) kinetically limits the overall water splitting reaction, this work demonstrates the L-cysteine assisted synthesis of millerite nickel sulfide dispersed as particles on nickel foam (NS/NF) by a simple one-step hydrothermal process as a self-supported working electrode. The controlled phase of NiS is confirmed by XRD and TEM analysis and the size and morphology of the catalyst are characterised by SEM analysis. XAS analysis further explores the bulk structure and chemical coordination within the crystal system according to the XANES and EXAFS findings. The HER performance of the NS/NF catalyst exhibits superior activity to bare NF, requiring an overpotential of 140 mV to deliver a current density of −10 mA cm−2 with a Tafel slope of 112.3 mV dec−1. The catalyst demonstrated excellent durability for 50 h with further electro-activation of NS/NF under reduction conditions. In a two-electrode system, NS/NF||RuO2 required only 1.79 V as the overall cell voltage to generate a current density of 10 mA cm−2. This study illustrates a simple and facile route for NiS synthesis with extendable electrochemical surface area (ECSA), demonstrating superior HER activity over time, under alkaline conditions.
Introduction
The need to focus on the production of renewable energy is inevitable due to the rapid increase in pollution caused by non-renewably sourced fuels and the consequent demand for energy. Hydrogen (H2) is considered to be an effective alternative to conventional fossil-based fuels as a clean sustainable energy source with high energy density.1–3 Although various methods like coal gasification, steam methane reforming, thermochemical methods and hydrocarbon splitting have been in use for H2 production, the problem associated in reaching higher efficiency often means, seeking an effective alternative method.4–6 Various electrochemical devices, like electrolyzers, fuel cells, and metal–air batteries, have been the prime focus for research as they are highly eco-friendly, producing higher energy conversion efficiencies.7–10 Electrochemical water splitting (EWS)11,12 is a green way to produce H2, which primarily involves the 2e− hydrogen evolution reaction (HER) at the cathode and the 4e− oxygen evolution reaction (OER) at the anode. The whole EWS occurs at a cell voltage of 1.23V vs. RHE (theoretical value) while the practical cell voltage is 1.48 V vs. RHE, considering the thermodynamics of the electrolytic system.13–15 However, noble metals like Pt (for HER) or RuO2/IrO2 (for OER) are available with high catalytic conversion efficiencies and optimum adsorption and desorption energy,16,17 but in an alkaline medium these catalysts suffer from poor durability, and due to their high cost their large-scale development is still being negotiated.18–20 Hence, the development of earth-abundant and cost-effective catalysts is imperative.Transition metal (TM)-based catalysts are optimum for alkaline EWS due to their stability, earth-abundance, relative non-toxicity, and high durability and could be suitably modified to ease the complex 4e− OER process of forming *O–OH bonds, thereby controlling the overall kinetics of EWS. However, it is highly challenging to catalyse alkaline HER, which involves an additional H2O dissociation step.18,21–25 Several studies on earth-abundant TM-based chalcogenides, like sulfides,26–29 selenides,30–32 tellurides,33–35 nitrides,36 and phosphides,37–40 have been reported for catalysing HER via alkaline overall water splitting and promoting the kinetics of the water dissociation step. Among them, metal sulfides have indeed been explored for HER due to their excellent stability and they could be synthesized without any difficulty, resulting in different phases, crystal systems and varying stoichiometries.41,42 Also the metal centres show a greater tendency for H− attraction and sulfides for proton adsorption due to the difference in electronegativity between the metal and S.43
One of the most commonly studied forms of metal sulfides are nickel sulfides, as Ni sites could afford abundant water dissociation sites and the S atoms chemically react to form stoichiometric or non-stoichiometric polymorphs (NiS, NiS2, Ni3S2, Ni3S4 and Ni9S8). Over the last few decades, Ni3S2 has been more evident in alkaline HER by confining the sizes and shapes, varying synthetic routes and the controlled use of different precursors.44,45 However other polymorphs have been less explored and are expected to be less HER active. Jiang et al.46 reported a case study comparing the HER activity of NiS, NiS2 and Ni3S2 and concluded that Ni3S2 performed better. But these materials are synthesized via a microwave-assisted solvothermal route and the catalyst is drop-cast on a glassy-carbon electrode (GCE). Efforts have been made to adapt NiS and NiS2 catalysts to access the sites for HER activity. Cimen et al.47 synthesized NiSx microspheres via the atomic layer deposition (ALD) method and presumed that high-area supports and template-free synthesis could be advantageous in creating more spaces for proton adsorption and hydrogen evolution. Wang et al.48 theoretically and experimentally analysed the mesoporous NiS and inferred that S atoms in NiS is highly resistive to *OH poisoning, leaving a rational avenue for alkaline electrolysis.
To avoid all these pitfalls, we synthesized NiS in a self-supported manner via a simple hydrothermal treatment. The 3-dimensional Ni foam (NF) is adapted as a substrate with high-area support and L-cysteine is employed to be the S source. During hydrothermal synthesis, the L-cysteine solvates with Ni ions and hydrolyses to form NiS in the millerite phase and pyrite phase in a highly dispersed manner. The resulting NiS grown on NF (NS/NF) has reduced solution resistance and the durability of the electrode is enhanced. Under alkaline HER conditions, NS/NF exhibits a low overpotential of 140 mV at −10 mA cm−2 with a Tafel slope value of 112.3 mV dec−1. On coupling with the OER catalyst RuO2 in a two-electrode system, NS/NF||RuO2 required an overall cell voltage of 1.79 V at 10 mA cm−2. The durability of the catalyst is well maintained with noticeably voltage-induced improved HER activity and wetness-assisted activity in both half-cell and full-cell reactions.
Results and discussion
Scheme 1 illustrates the synthetic scheme for preparing the nickel sulfide catalysts. Under hydrothermal conditions at 120 °C, Ni2+ ions are slowly liberated from the Ni foam, forming a complex with L-cysteine molecules. Under redox conditions, L-cysteine hydrolyses to HS− anions, liberating NH3, which then nucleates with Ni2+ ions, forming nickel sulfide particles on nickel foam (NS/NF). As the reaction proceeds, clusters of millerite NiS grow and aggregate through an Ostwald ripening process and Ni2+ ions are swiftly released into the solution over time, thereby increasing the Ni concentration.49–51 In addition, the pressure inside the autoclave increases, and the excess of Ni ions slightly distorts the millerite lattice, forming pyrite nuclei within the NS/NF catalyst. Since the hydrothermal reaction is arrested within 6 h, complete transformation into the pyrite phase is avoided. To confirm the crystallinity and phase purity of NS/NF, powder X-ray diffraction (PXRD) analysis was performed. The XRD pattern (Fig. 1) reveals the formation of highly crystalline nickel sulfide in the millerite phase (NiS).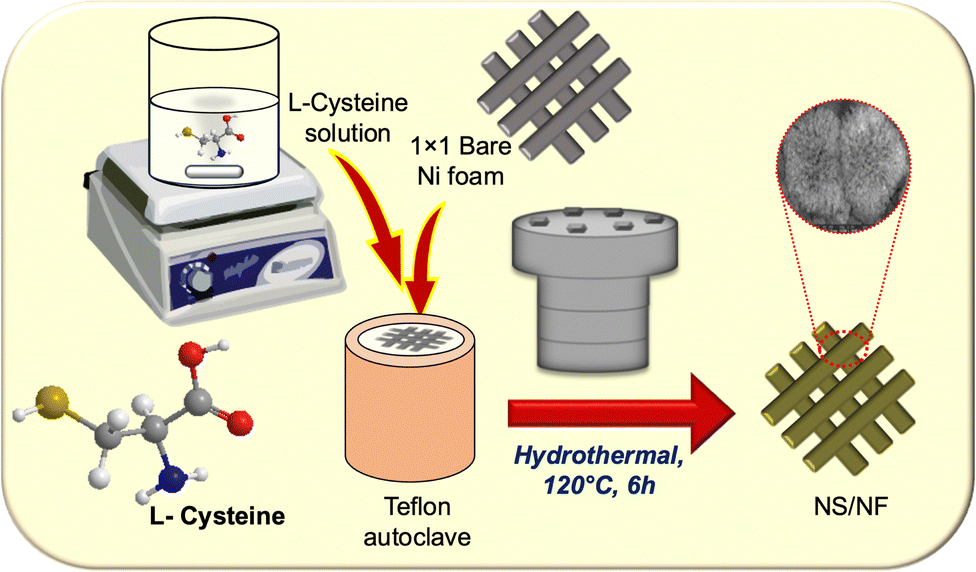 | ||
| Scheme 1 Schematic representation of the synthetic strategy for an NS/NF catalyst. | ||
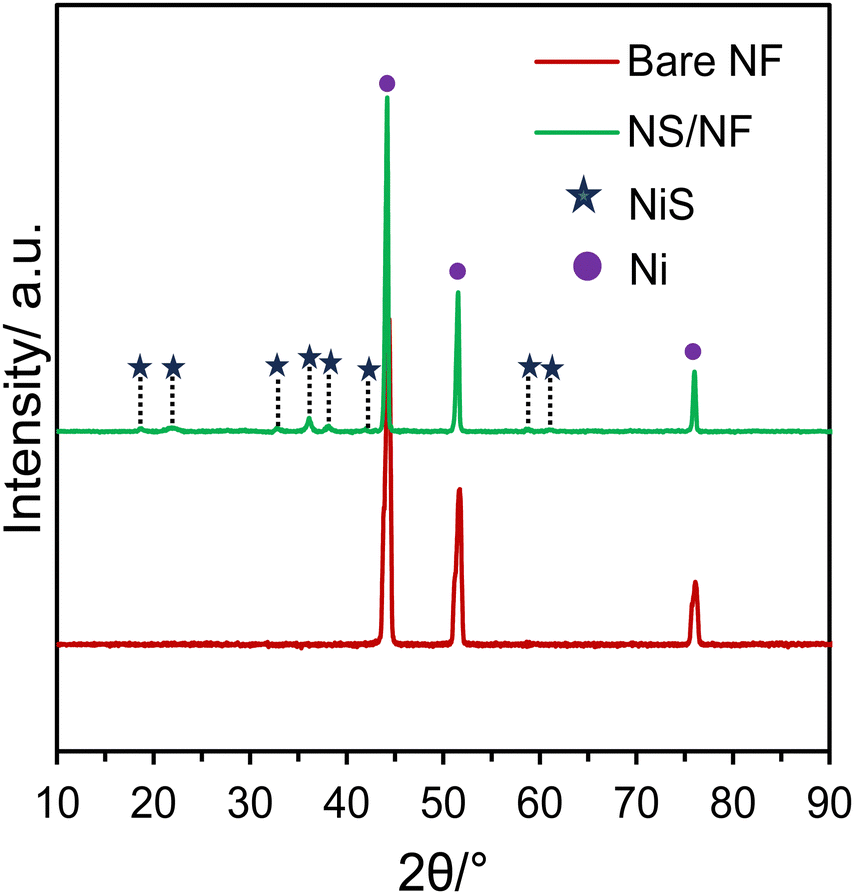 | ||
| Fig. 1 Comparison of the XRD patterns for the electrocatalysts, bare NF and NS/NF. | ||
The diffraction peaks at 2θ = 18.7°, 32.7°, 35.9°, 37.9° and 58.3° correspond to the planes (110), (300), (021), (220) and (330), respectively, following the JCPDS 03-065-3867 pattern (NiS; crystal system – rhombohedral; space group – R3M; space number – 160). The peaks at 21.7 and 61.2 follow hexagonal pyrite NiS from JCPDS 01-1286 and 01-075-0613, respectively, since the pH of the hydrothermal reaction mixture is neutral, which favours the formation of NiS in millerite and pyrite phases. The NS/NF also shows the characteristic Ni foam peaks at 44.4°, 51.6°, and 76.1°, matching the pattern JCPDS 03-065-0380 with planes (111), (200) and (222) planes, respectively.
The morphology and dimensions of the NS/NF catalyst were demonstrated using field-emission scanning electron microscopy (FE-SEM). As can be seen from the FESEM (Fig. 2a and b), sea-urchin like microspheres of NiS with an average size of 7 μm are formed on the NF. Fig. S1 (ESI†) shows the FESEM images of bare NF under different magnifications. On capturing them with higher magnification (Fig. 2c), each microsphere consists of smaller aggregated NiS particles, as expected.51 The uniform distribution of Ni and S atoms as seen in the elemental mapping (Fig. S2a–c, ESI†) and energy dispersive X-ray (EDX) spectra (Fig. S2d, ESI†) confirms the presence of Ni and S atoms and confirms the formation of NiS with a relatively high Ni concentration, as the catalyst is self-supported on NF.
 | ||
| Fig. 2 (a)–(c) FESEM images under different magnifications, (d) TEM, (e) HR-TEM showing lattice fringes and (f) SAED pattern for the catalyst NS/NF. | ||
To further explore the morphology, high resolution transmission electron microscopy (HR-TEM) images are demonstrated. The TEM and HR-TEM images in Fig. 2d–f reveal the formation of thin wrinkled sheets with dispersed NiS particles. The high-resolution images clearly depict the presence of lattice fringes with interplanar spacing d = 0.27 nm for the (300) plane, as confirmed by the XRD analysis, supporting the formation of crystalline NiS. The selected area electron diffraction (SAED) pattern (Fig. 2e) implies that NiS is polycrystalline in nature with hkl planes calculated for the NiS millerite and pyrite phases.
To provide more insight into the structural transformation and chemical composition in bulk, NS/NF was subjected to X-ray absorption spectroscopy (XAS) analysis. The X-ray absorption near edge spectra (XANES) for the Ni-K edge are displayed in Fig. 3a. The pre-edge region NS/NF closely resembles standard Ni(0) and the rising part matches Ni in the +2 oxidation state, which supports the presence of NiS formed on Ni foam.52 Linear combination fitting (LCF) was performed, showing that 20% of Ni is in the +2 state while the remaining Ni is in the zero oxidation state, which supports the slow release of Ni2+ ions involved in Ni–S formation. The extended X-ray absorption fine structure (EXAFS) was ascertained to infer the radial distance and coordination number of Ni and S atoms. Fig. 3b shows the Fourier transform (FT-EXAFS) spectra of the normalized Ni-K edge fitted in R space with the NiS millerite model. NS/NF has the first shell with a 2.44 Å radial distance corresponding to Ni–S and the second coordination shell corresponds to Ni–Ni with a radial distance of 2.19 Å while Reff is 2.24 Å. The presence of shortened bonds reveals lattice distortion arising from pyrite nucleation in millerite NiS.
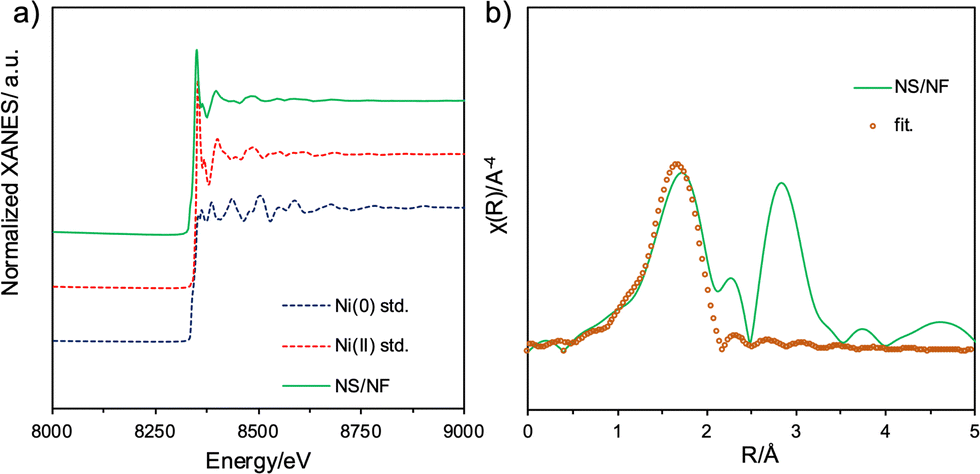 | ||
| Fig. 3 Ni-K edge (a) XANES spectra and (b) FT-EXAFS spectra fitted in R-space for the catalyst NS/NF. | ||
Electrochemical studies
Electrochemical measurements were undertaken in a single-compartment three-electrode cell-setup in 1.0 M KOH (pH = 14) utilising Hg/HgO as the reference electrode, Ni foam with a large surface area as the counter electrode and the synthesized catalysts as the working electrode. All the voltammogram measurements were compared with the benchmark HER catalyst Pt/C coated on Ni foam under similar operating conditions.The linear sweep voltammogram (LSV) studies provided in Fig. S3 (ESI†) confirm that NS/NF shows higher catalytic activity than bare NF. To further exclude the contributions from charging current, sampled current voltammetry (SCV) was performed by extracting the values from the chronoamperometry (CA) curves (Fig. S4, ESI†) in a region where the double-layer charging current ceases. Fig. 4a depicts the SCV curves for the catalyst in comparison with bare NF and Pt/C. After iR compensation, the plot implies enhanced HER activity for NS/NF. The overpotential required by NS/NF to deliver −10 mA cm−2 (η10) was only 140 mV, whereas bare NF required 210 mV. Pt/C required 95 mV to deliver same amount of current density. This is attributed to the polycrystalline nature of NS/NF, where the extended grain boundaries can facilitate the exposure and accessibility of more electro-active sites with greater dispersion, reducing the diffusion path for charge-carriers (H atoms and electrons), which enabled it to reach a maximum current density of −287 mA cm−2 at −0.318 V vs. RHE. On increasing the cathodic potentials, the activity increased with overpotentials of 220, 260 and 295 mV required to deliver current densities of 50, 100 and 200 mA cm−2, while bare NF hardly reached 150 mA cm−2. Fig. 4b shows a comparative plot of overpotential η10 and onset overpotential for all the catalytic materials. NS/NF exhibits significant HER activity with lowered overpotential and HER onset overpotential of only 50 mV. This is attributed to the increased electrochemical surface area (ECSA) of NS/NF, which activates a greater number of H atoms in generating a sufficient amount of H2 at the requirement of low overpotential, as evidenced in Fig. S5 (ESI†). To further explore the kinetics of NS/NF, the Tafel plot is demonstrated from SCV in the catalytic-turnover region. In general, the kinetics of HER is limited by the electrochemical discharge of a proton accompanied either by another electrochemically discharged proton to evolve H2 or by another proton that is yet to discharge.53 However, under alkaline conditions, the prospect of finding a free proton is unlikely; hence each step has to rely on the H2O splitting step following eqn (1)–(3).54
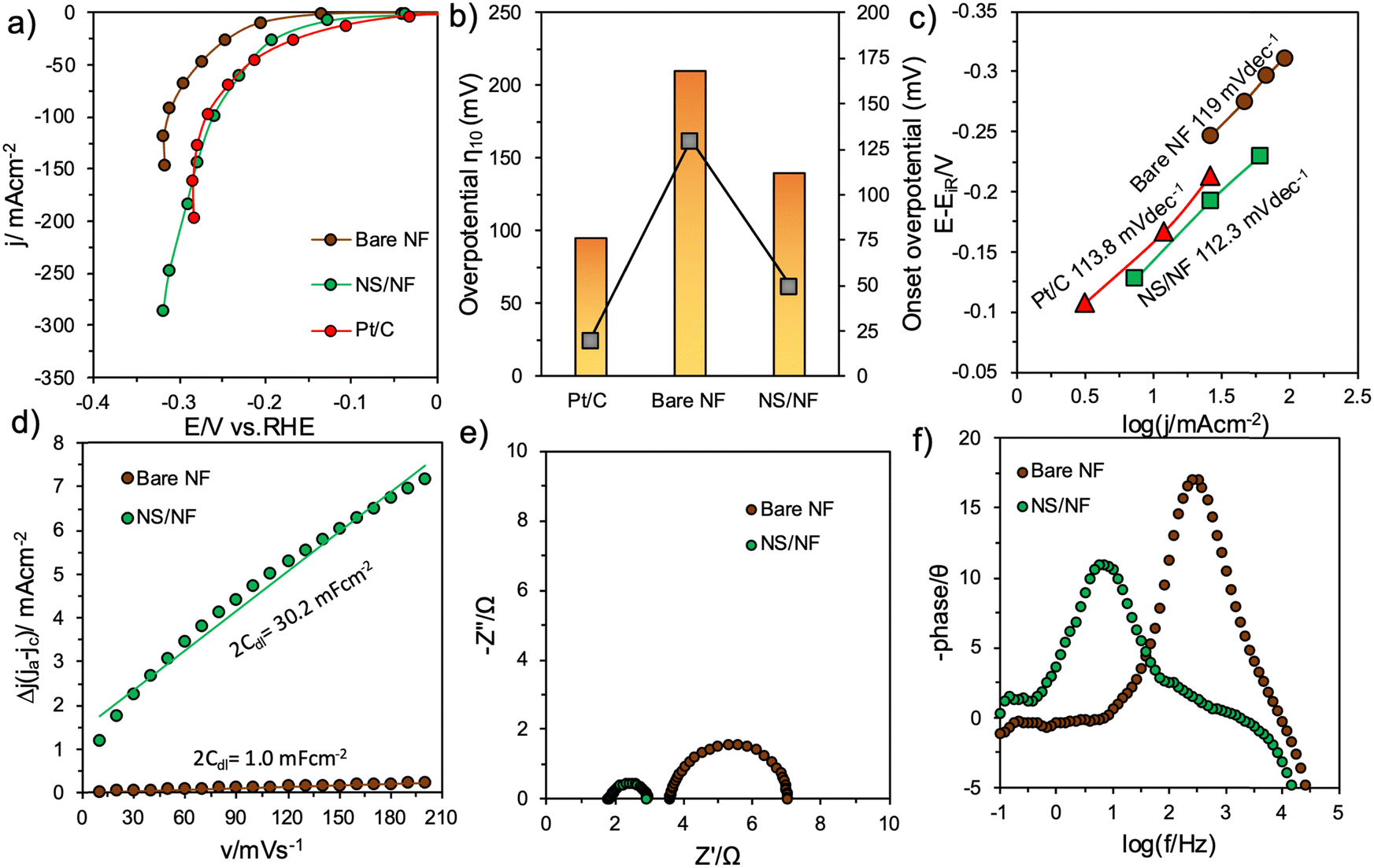 | ||
| Fig. 4 Electrochemical HER studies in 1.0 M KOH showing (a) an SCV plot derived from CA studies with 100% iR compensation, (b) a comparative plot of overpotential values at −10 mA cm−2 and onset overpotential, (c) a Tafel plot under catalytic-turnover conditions extracted from the SCV plots for bare NF, NS/NF and Pt/C, respectively, (d) a linear plot of current density against scan-rates showing 2Cdl values recorded in a non-faradaic region, (e) a Nyquist plot and (f) a Bode-phase angle plots for bare NF and NS/NF catalysts, respectively. | ||
Volmer–H2O dissociation step
| H2O + e− → HO− + Hads | (1) |
Heyrovsky–H2O dissociation step
| Hads + H2O + e− → HO− + H2 | (2) |
Tafel step
| Hads + Hads → H2 | (3) |
The NS/NF catalyst exhibited a lower Tafel slope value of 112.3 mV dec−1, which implies that the kinetics of HER is limited by the Volmer–Heyrovsky step with the associated water splitting step (Fig. 4c). Although the reaction proceeds through the slowest step, the kinetics are even fast at maximum current densities relative to Pt/C (113.8 mV dec−1), as there is an increase in the ECSA of NS/NF. This also suggests that the adsorption of H atoms is mainly responsible for pushing the onset of HER closer to 0 V vs. RHE and that the evolution of H2 is greater due to the higher packing efficiency in the rhombohedral NiS system, which favours the formation of activated H atoms in closer proximity.55 The improved accessibility of active sites promoting the HER kinetics in NS/NF is assessed from the linear plot (Fig. 4d) of current density versus scan-rate by the Cdl method. The current densities are deciphered from the difference between the anodic and cathodic current densities of the cyclic voltammograms (CVs) recorded in the non-faradaic region from their respective scan-rates (Fig. S6, ESI†). The slope gives the double-layer capacitance value 2Cdl whose value is extremely high for NS/NF at 30.2 mF cm−2 due to the high dispersibility of NiS particles compared to bare NF (2Cdl = 30.2 mF cm−2). Supporting the aforementioned results of improved activity and kinetics of NS/NF, the quickness of electron transit during the electrochemical reaction is analysed from electrochemical impedance spectroscopy (EIS) studies. From the Nyquist plot (Fig. 4e), it is calculated that NS/NF has a low charge-transfer resistance at (Rct) of 1.33 Ω, whereas bare NF has an Rct value of 3.12 Ω. This supports the improved electrochemical conductivity with an easiness in the charge transfer rate at the electrode–electrolyte interface of NiS due to the increased creation of active sites. The uncompensated resistances Ru for NS/NF are 1.587 Ω and 3.987 Ω for bare NF. The reduction in Ru reveals that the roughness of the catalyst increases the wettability for water adsorption. The resistance values agree with the Bode-absolute impedance plot (Fig. S7a, ESI†) and from the calculated admittance (Y) values for NS/NF (Y = 0.24 Ω−1) and bare NF (Y = 0.067 Ω−1) the lifetime of electrons in the NS/NF catalyst is long, which facilitates the charge-transfer kinetics in HER. The Bode-phase angle indicates the nature of an electrochemical reaction. All the catalysts show a phase shift of <45°, which is distinctive for a charge-transfer reaction (HER is a 2e− process), as can be seen from Fig. 4f. NS/NF exhibits a phase shift of θ = 10.8°, whereas bare NF shows 17°, supporting the improved HER activity in NS/NF.
The RC time constant (τ) is also decreased for NS/NF, as the time required to charge 63.2% of the electro-active material is reduced, which is turn implies an increased ECSA. The trend observed from the EIS results seems to be in good agreement with all the other results (Fig. S7b, ESI†).
To test the durability and robustness of the material, NS/NF was subjected to prolonged stability testing for 50 h at a fixed potential. The material was able to deliver a current density of −50 mA cm−2, as the highly dispersed polycrystalline NS/NF exhibits grain boundary pinning, which can improve stability and durability by reducing grain growth and coarsening whose activity initially increases with time (Fig. 5a). This could be due to the improving wettability56 of the electrode material as the S atom in NiS is highly resistant to hydroxide poisoning, Ni promotes water dissociation and the millerite phase of NiS is electrochemically activated under reduction potential over time under high alkaline conditions, which enables the material NS/NF to attain a constant current density for 50 h.57 To physically characterise the NS/NF catalyst, the XRD pattern (Fig. S8, ESI†) was obtained after stability testing. Since, it is bulk-responsive, NS/NF showed the XRD pattern for pure NF and an additional small peak with reduced intensity at 2θ = 35.9° corresponding to the (021) plane of millerite NiS. The lowered intensity of the 2θ peaks for NS/NF after stability testing compared to pure NS/NF is a clear indication of reduced crystallinity due to the utilisation of surface-active sites introducing disorderliness into the long-range order of the crystal planes. To check the activity after stability (AS) testing, SCV was recorded, which clearly supported the increased HER activity (Fig. 5b) with a reduced overpotential value η10 of 92 mV. But, the onset of HER remains the same as before stability testing, retaining the intrinsic activity of NiS. The EIS measurements after stability testing (Fig. S9a and b, ESI†) also prove the improved catalytic activity of NS/NF during prolonged electrolysis while maintaining its intrinsic activity and extending the accessibility of active sites over time. The linear plot derived from the non-faradaic CV curves (Fig. S9c and d, ESI†) also shows an improved capacitance 2Cdl value of 39.4 mF cm−2, revealing an enhanced ECSA. All the results suggest that NS/NF could be used as a cathode material in alkaline electrolyzers for large-scale H2 production.
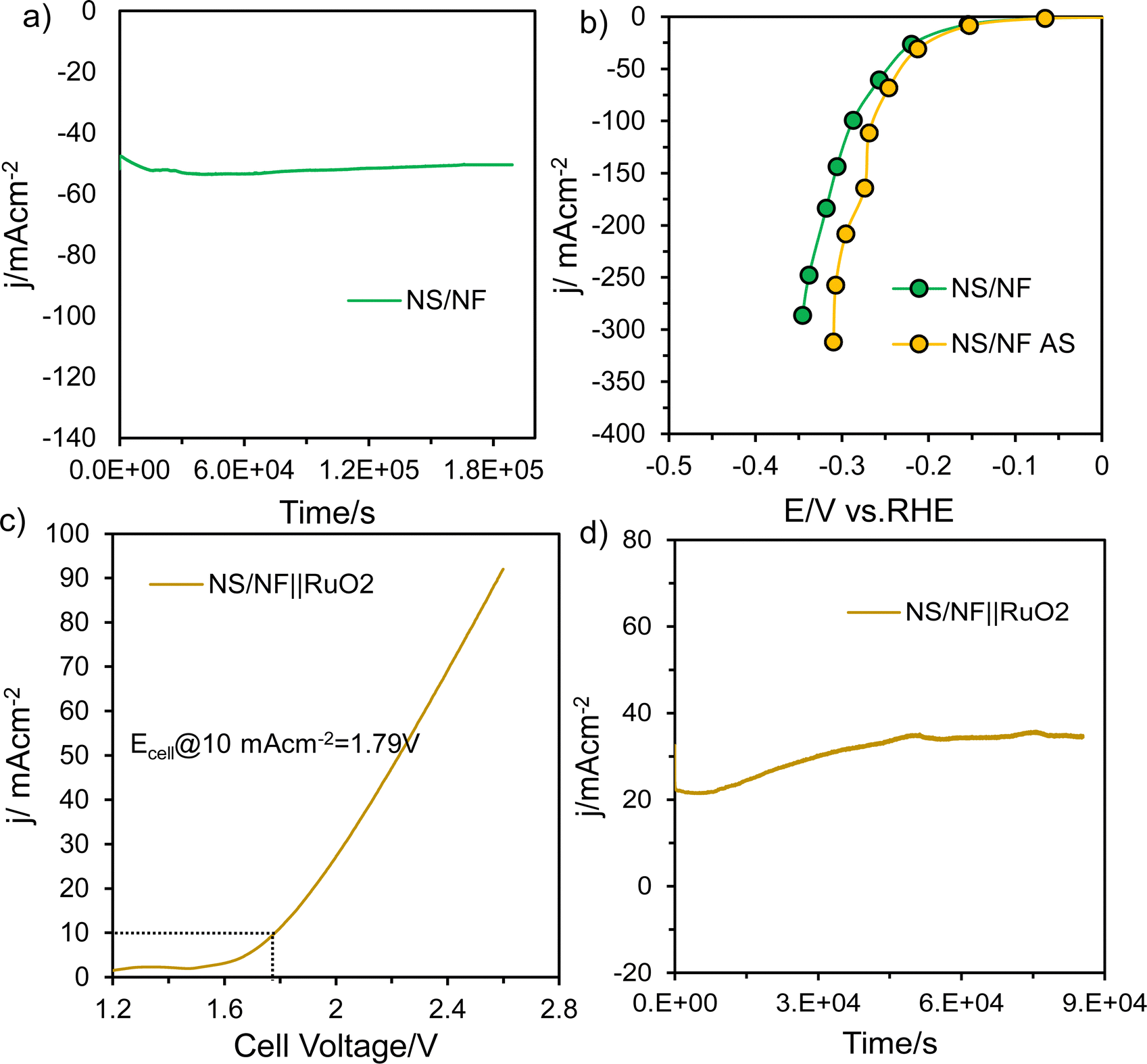 | ||
| Fig. 5 (a) i–t curve demonstrating the durability of NS/NF catalyst for HER at a constant potential in 1.0 M KOH, (b) SCV curves of NS/NF before and after HER stability, (c) polarisation curves showing overall cell voltage and (d) chronopotentiometry curves for NS/NF||RuO2 in 1.0 M KOH. | ||
To check its overall performance, the NS/NF catalyst was evaluated for overall water splitting in a two-electrode system, where NS/NF served as the cathode and RuO2 coated on NF was connected to the anode for the OER reaction. The full cell NS/NF||RuO2 was tested for alkaline electrolysis (Fig. 5c) in 1.0M KOH and the cell required only 1.79 V to reach a current density of 10 mA cm−2. Since NiS impedes hydroxide poisoning and Ni promotes H2O dissociation, the kinetics of the HER is improved synergistically in a two-electrode system. The full cell NS/NF||RuO2 exhibited massive robustness (Fig. 5d) for about 24 h showing similar activating trends to those observed in half-cell conditions for overall water splitting.
Conclusions
In this work, we successfully synthesized a self-supported NiS catalyst on Ni foam (NS/NF) using a simple single-step hydrothermal process. L-Cysteine was used as a sulfur source for the slow release of S atoms into the crystal lattice, forming the millerite phase and the nucleated pyrite phase of NiS, which is unique and confirmed by XRD analysis. The existence of polycrystallinity and the highly dispersed NiS particles improved the ECSA and the accessibility of surface-active sites to promote the HER mechanism. The NS/NF catalyst required only 140 mV overpotential to deliver a current density of −10 mA cm−2 with a reduced Tafel slope of 112.3 mV dec−1. The durability of the catalyst was checked and it was found to be highly active for 20 h with increased electrochemical activity due to its improved wettability and voltage-induced activation, as the overpotential was reduced to 92 mV after stability testing. Impressively, the two-electrode overall alkaline water splitting exhibited a cell voltage of only 1.79 V for j = 10 mA cm−2. Moreover, this work improved the HER activity of simple NiS and proposed a new synthetic strategy for controlling the phase of the crystal in a single-step hydrothermal method.Author contributions
P. J. J. S. conceptualisation, data Curation, methodology, validation, visualization, formal analysis, investigation, writing – original draft, and writing – review & editing. K. K. S. methodology, data curation. O. K. &O. N. XAS analysis, funding acquisition, methodology, validation, visualization, review & editing. Hyoung-il Kim, review & editing. S. K. formal analysis, funding acquisition, methodology, validation, visualization, supervision, review & editing – original draft.Data availability
The data supporting this article have been included as part of the manuscript and ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
K. S. would like to thank the Royal Society-Newton International Fellowship Alumni follow-on funding support AL\211016 and AL\221024, SERB Start-up Research Grant (SRG/2023/000658) and Department of Chemistry at SRMIST. The authors also acknowledge the SRM Institute of Science and Technology (SRM IST) for providing all the research facilities, including SRM-SCIF and NRC.References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed
.
- D. H. Marin, J. T. Perryman, M. A. Hubert, G. A. Lindquist, L. Chen, A. M. Aleman, G. A. Kamat, V. A. Niemann, M. B. Stevens, Y. N. Regmi, S. W. Boettcher, A. C. Nielander and T. F. Jaramillo, Joule, 2023, 7, 765–781 CrossRef CAS
- P. J. J. Sagayaraj and K. Sekar, Chem. Commun., 2024, 60, 6817–6820 RSC
- A. Hassan, O. Abdel-Rahim, M. Bajaj and I. Zaitsev, Sci. Rep., 2024, 14, 24767 CrossRef CAS PubMed
- D. Çelik and M. Yıldız, Int. J. Hydrogen Energy, 2017, 42, 23395–23401 CrossRef
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
- L. Li, X. Tang, B. Wu, B. Huang, K. Yuan and Y. Chen, Adv. Mater., 2024, 36, 2308326 CrossRef CAS
- J. Tang, R. Xie, P. Pishva, X. Shen, Y. Zhu and Z. Peng, J. Mater. Chem. A, 2024, 12, 15580–15591 RSC
- Y. Guo, Y. Cao, Q. Tan, D. Yang, Y. Che, C. Zhang, P. Ming and Q. Xiao, RSC Adv., 2024, 14, 19636–19647 RSC
- P. R. Santos, D. Pérez-Coll, M. T. Azcondo, G. C. Mather, Á. Muñoz-Noval, E. Salas-Colera, U. Amador, K. Boulahya and D. Muñoz-Gil, J. Mater. Chem. A, 2024, 12, 7631–7644 RSC
- Q. Qian, Y. Zhu, N. Ahmad, Y. Feng, H. Zhang, M. Cheng, H. Liu, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2024, 36, 2306108 CrossRef CAS
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2021, 14, 4740–4759 RSC
- A. Sengeni, Catal. Sci. Technol., 2024, 14, 2025–2039 RSC
- W. Li, H. Tian, L. Ma, Y. Wang, X. Liu and X. Gao, Mater. Adv., 2022, 3, 5598–5644 RSC
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef
- H. Sun, X. Xu, H. Kim, W. Jung, W. Zhou and Z. Shao, Energy Environ. Mater., 2023, 6, e12441 CrossRef CAS
- M. N. Lakhan, A. Hanan, A. Hussain, I. Ali Soomro, Y. Wang, M. Ahmed, U. Aftab, H. Sun and H. Arandiyan, Chem. Commun., 2024, 60, 5104–5135 RSC
- Y. Zhang, R. Yan, X. Xu, C. He, M. Zhou, Z. Zeng, T. Ma, M. Wang and S. Li, Adv. Funct. Mater., 2024, 34, 2308813 CrossRef CAS
- H. Kamaruddin, Z. Jianghong, L. Yu, W. Yuefan and H. Yizhong, J. Mater. Chem. A, 2024, 12, 9933–9961 RSC
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC
- X. Yang, R. Guo, R. Cai, W. Shi, W. Liu, J. Guo and J. Xiao, Dalton Trans., 2022, 51, 4590–4607 RSC
- M. D. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421–14461 RSC
- M. Wang, L. Zhang, Y. He and H. Zhu, J. Mater. Chem. A, 2021, 9, 5320–5363 RSC
- H. G. Shiraz, X. Crispin and M. Berggren, Int. J. Hydrogen Energy, 2021, 46, 24060–24077 CrossRef CAS
- X. Liu, Y. Li, Z. Cao, Z. Yin, T. Ma and S. Chen, J. Mater. Chem. A, 2022, 10, 1617–1641 RSC
- H. J. Liu, S. Zhang, Y. M. Chai and B. Dong, Angew. Chem., 2023, 135, e202313845 CrossRef
- W. Feng, W. Pang, Y. Xu, A. Guo, X. Gao, X. Qiu and W. Chen, ChemElectroChem, 2020, 7, 31–54 CrossRef CAS
- C. Zeng, L. Dai, Y. Jin, J. Liu, Q. Zhang and H. Wang, Sustainable Energy Fuels, 2021, 5, 1347–1365 RSC
- S. Anantharaj and S. Noda, Int. J. Hydrogen Energy, 2020, 45, 15763–15784 Search PubMed
- M. Ahmad, T. Nawaz, I. Hussain, F. Meharban, X. Chen, S. A. Khan, S. Iqbal, P. Rosaiah, M. Z. Ansari, W. A. Zoubi and K. Zhang, Small, 2024, 20, 2310099 CrossRef CAS PubMed
- D. Siegmund, N. Blanc, M. Smialkowski, K. Tschulik and U.-P. Apfel, ChemElectroChem, 2020, 7, 1514–1527 CrossRef CAS
- S. Anantharaj, K. Karthick and S. Kundu, Inorg. Chem., 2018, 57, 3082–3096 CrossRef CAS PubMed
- H. Hu, X. Wang, J. P. Attfield and M. Yang, Chem. Soc. Rev., 2024, 53, 163–203 RSC
- Z.-Z. Liu, N. Yu, R.-Y. Fan, B. Dong and Z.-F. Yan, Nanoscale, 2024, 16, 1080–1101 RSC
- K.-L. Yan, J.-F. Qin, Z.-Z. Liu, B. Dong, J.-Q. Chi, W.-K. Gao, J.-H. Lin, Y.-M. Chai and C.-G. Liu, Chem. Eng. J., 2018, 334, 922–931 CrossRef CAS
- J. Zhang, X. Shang, H. Ren, J. Chi, H. Fu, B. Dong, C. Liu and Y. Chai, Adv. Mater., 2019, 31, 1905107 CrossRef CAS PubMed
- X.-Y. Zhang, Y.-R. Zhu, Y. Chen, S.-Y. Dou, X.-Y. Chen, B. Dong, B.-Y. Guo, D.-P. Liu, C.-G. Liu and Y.-M. Chai, Chem. Eng. J., 2020, 399, 125831 CrossRef CAS
- S. Anantharaj, S. Kundu and S. Noda, J. Mater. Chem. A, 2020, 8, 4174–4192 RSC
- Y. Chen, Y. Fan, Z. Cui, H. Huang, D. Cai, J. Zhang, Y. Zhou, M. Xu and R. Tong, Int. J. Hydrogen Energy, 2023, 48, 27992–28017 CrossRef CAS
- W. Du, Y. Shi, W. Zhou, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 7051–7055 CrossRef CAS PubMed
- S. Wang, Z. Geng, S. Bi, Y. Wang, Z. Gao, L. Jin and C. Zhang, Green Energy Environment, 2024, 9, 659–683 CrossRef CAS
- A. Gaur, J. Sharma and V. Bagchi, ChemCatChem, 2024, 16, e202301438 CrossRef CAS
- N. Jiang, Q. Tang, M. Sheng, B. You, D.-E. Jiang and Y. Sun, Catal. Sci. Technol., 2016, 6, 1077–1084 RSC
- Y. Çimen, A. W. Peters, J. R. Avila, W. L. Hoffeditz, S. Goswami, O. K. Farha and J. T. Hupp, Langmuir, 2016, 32, 12005–12012 CrossRef PubMed
- A. Wang, H. Li, J. Xiao, Y. Lu, M. Zhang, K. Hu and K. Yan, ACS Sustainable Chem. Eng., 2018, 6, 15995–16000 CrossRef CAS
- A. Roffey, N. Hollingsworth, H.-U. Islam, M. Mercy, G. Sankar, C. R. A. Catlow, G. Hogarth and N. H. de Leeuw, Nanoscale, 2016, 8, 11067–11075 RSC
- X. Zheng, X. Han, Y. Zhang, J. Wang, C. Zhong, Y. Deng and W. Hu, Nanoscale, 2019, 11, 5646–5654 RSC
- G. Barim, S. R. Smock, P. D. Antunez, D. Glaser and R. L. Brutchey, Nanoscale, 2018, 10, 16298–16306 Search PubMed
- N. Jiang, L. Bogoev, M. Popova, S. Gul, J. Yano and Y. Sun, J. Mater. Chem. A, 2014, 2, 19407–19414 Search PubMed
- O. van der Heijden, S. Park, R. E. Vos, J. J. J. Eggebeen and M. T. M. Koper, ACS Energy Lett., 2024, 9, 1871–1879 CrossRef CAS PubMed
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed
- V. Pundir, A. Gaur, R. Kaur, Aashi and V. Bagchi, Energy Adv., 2023, 2, 321–327 RSC
- X. Shan, J. Liu, H. Mu, Y. Xiao, B. Mei, W. Liu, G. Lin, Z. Jiang, L. Wen and L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 1659–1665 CrossRef CAS PubMed
- X. Ding, D. Liu, P. Zhao, X. Chen, H. Wang, F. E. Oropeza, G. Gorni, M. Barawi, M. García-Tecedor, V. A. de la Peña O’Shea, J. P. Hofmann, J. Li, J. Kim, S. Cho, R. Wu and K. H. L. Zhang, Nat. Commun., 2024, 15, 5336 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00578c |
| This journal is © The Royal Society of Chemistry 2025 |