Graphene oxide dispersions: tuning rheology to enable fabrication†
Sina
Naficy‡
a,
Rouhollah
Jalili‡
a,
Seyed Hamed
Aboutalebi‡
b,
Robert A.
Gorkin III
a,
Konstantin
Konstantinov
b,
Peter C.
Innis
a,
Geoffrey M.
Spinks
a,
Philippe
Poulin
c and
Gordon G.
Wallace
*a
aIntelligent Polymer Research Institute and ARC Centre of Excellence for Electromaterials Science, AIIM Facility, Innovation Campus, University of Wollongong, North Wollongong, NSW 2522, Australia. E-mail: gwallace@uow.edu.au
bInstitute for Superconducting and Electronic Materials, AIIM Facility, Innovation Campus, University of Wollongong, North Wollongong, NSW 2522, Australia
cCentre de Recherche Paul Pascal, Université Bordeaux I, UPR CNRS 8641, France
First published on 14th January 2014
Abstract
Here, we show that graphene oxide (GO) dispersions exhibit unique viscoelastic properties, making them a new class of soft materials. The fundamental insights accrued here provide the basis for the development of fabrication protocols for these two-dimensional soft materials, in a diverse array of processing techniques.
The recent discovery of liquid crystalline behaviour in graphene oxide (GO) dispersions in water,1–5 and various organic solvents,6,7 can be instrumental in guiding material assembly at the atomic level through π–π stacking and hydrogen bonding interactions.1,5,6,8 Original rheological behaviour is expected, since monolayer GO dispersions constitute a new class of soft materials with rich unique properties in between rigid 2D nanoplatelets and 1D polymers.9 Probing and quantifying this dynamic behaviour will be instrumental in generating novel applications, such as coatings, fillers and molecular electronics by advancing our fundamental knowledge of soft materials.10 Despite the importance for fundamental, practical, and industrial applications, the rheological behaviour of GO monolayer dispersions and its origin remains an unexplored area. This, in turn, hinders the development of fabrication protocols for two-dimensional soft materials, including GO dispersions. Moreover, fabricating practical devices in large-scale with advanced architectural design is still an unresolved challenge.
Likewise, formulation of functional composite inks for industrially scalable fabrication methods such as electrospraying, spray coating, and printing techniques, is always hindered by the high concentration of supporting media (such as graphene, GO, liquid crystals (LCs) and polymers). Therefore, low concentration of the supporting media is desirable to simultaneously permit processing while making the preparation of composite inks with high concentrations of multi-functional materials possible. This challenge, if resolved, can be used to create geometrically complex multi-functional 3D architectures fabricated for use in areas such as printed electronics, organic field effective transistors and 3D bionic scaffolds.
Here, with the aim of investigating the rheological behaviour of GO to enable further processing, fabrication and integration of GO into complex architectures, we employed ultra-large GO sheets as a model material with a high aspect ratio (∼45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Fig. S1†) to study the phase transitions from the so-called isotropic phase to the liquid crystal (LC) phase and finally to a LC gel phase at higher loadings. We demonstrate that GO dispersions exhibit unique viscoelastic behaviour, wherein the rheological behaviour varies considerably with dispersion concentration. We show that finite yield stress in ultra-large GO dispersions occurs at a critical concentration approximately three orders of magnitude lower than the theoretical value for colloidal suspensions (ϕthis study ≈ 2.2 × 10−4vs. ϕc ≈ 0.5) enabling the processing of this 2D material at the lowest concentration ever reported for any dispersion, which allows for the self-assembly of multi-functional architectures. Our experiments reveal that there are four distinct regions: viscoelastic liquid, a transition state consisting of viscoelastic liquid and viscoelastic soft solid, viscoelastic soft solid (behaving like a solid below yield stress yet flow readily above a yield stress)11 and viscoelastic gel. Each of these unbinding regions were found amenable to unique processing techniques, some of which never before were shown possible in the case of GO, warranting the processing of this fascinating material based on its inherent complex flow properties.
000, Fig. S1†) to study the phase transitions from the so-called isotropic phase to the liquid crystal (LC) phase and finally to a LC gel phase at higher loadings. We demonstrate that GO dispersions exhibit unique viscoelastic behaviour, wherein the rheological behaviour varies considerably with dispersion concentration. We show that finite yield stress in ultra-large GO dispersions occurs at a critical concentration approximately three orders of magnitude lower than the theoretical value for colloidal suspensions (ϕthis study ≈ 2.2 × 10−4vs. ϕc ≈ 0.5) enabling the processing of this 2D material at the lowest concentration ever reported for any dispersion, which allows for the self-assembly of multi-functional architectures. Our experiments reveal that there are four distinct regions: viscoelastic liquid, a transition state consisting of viscoelastic liquid and viscoelastic soft solid, viscoelastic soft solid (behaving like a solid below yield stress yet flow readily above a yield stress)11 and viscoelastic gel. Each of these unbinding regions were found amenable to unique processing techniques, some of which never before were shown possible in the case of GO, warranting the processing of this fascinating material based on its inherent complex flow properties.
Control over the rheological properties can be achieved by adjusting the volume fraction (concentration) of GO particles. To this end, we probed, as plotted in Fig. 1 and S2,† the flow behaviour of a series of GO dispersions by the cone–plate method (see the Experimental section of the ESI†). The elastic G′ (storage) and viscous G′′ (loss) moduli of GO dispersions were determined as a function of frequency at a constant strain amplitude of 0.01.
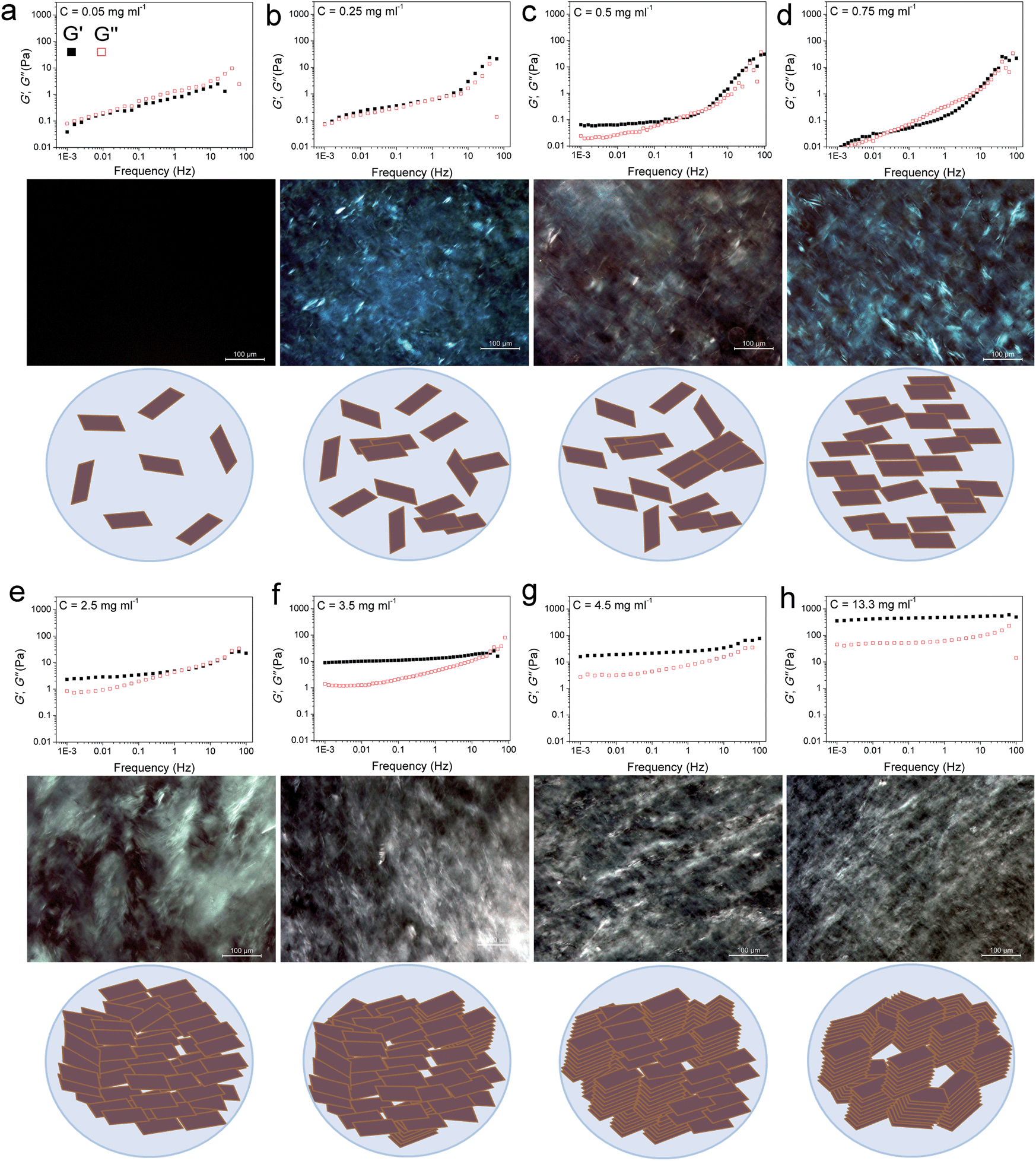 | ||
| Fig. 1 Storage and loss moduli (filled squares and open squares, respectively) of GO suspensions as a function of frequency accompanied by their polarized optical micrographs (POMs) and the schematic illustrations of the proposed model for the evolution of LC phases in GO dispersions upon increasing concentration. (a) At this extremely low concentration, GO sheets are randomly dispersed in the solution. (b and c) Upon increasing the concentration to 0.25 mg ml−1 some nematic ordering starts to appear. In this region, the storage modulus increases and overtakes the loss modulus, while the loss modulus remains almost constant with frequency. This can be attributed to the increase in the volume fraction of colloidal particles imparting elasticity to the system. However, this increase in concentration is very negligible to impart any serious effects on the loss modulus, as the dominating part is still water. (d) The dispersion forms a single phase nematic liquid crystal. This phase transformation frees up some additional space in the dispersion resulting in subsequent gain in entropy as well as a drop in elastic modulus. (e) Further increase in concentration results in higher packing of the nematic phase. An increase in volume fraction of GO sheets results in a frequency dependent plateau-like behaviour which is the direct consequence of sheets being trapped by their neighbours preventing the stress relaxation on the longest time-scale of the measurement. (Jammed systems such as weak polymer gels, entangled polymer networks, concentrated emulsions or biological cells exhibit this generic behaviour.)10,12,13 (f) In addition to long-range orientation in the nematic phase, some parts in GO exhibit long-range positional order. However, above this critical concentration, as the formation of the nematic LC phase is completed, further increase in volume fraction results in the simultaneous increase in both moduli, with the storage modulus increasing much faster than the loss modulus. (g and h) Smaller monodomains are formed associated with exceptional increase in elastic modulus. | ||
Quite interestingly, even at very low concentrations (as low as 0.05 and up to 0.25 mg ml−1) previously thought to be a completely isotropic and viscous phase,1,5 a considerable elastic component still exists. In overall time-scales, in the low concentration region (<0.25 mg ml−1), the dispersions exhibited a liquid-like response with G′′ being slightly higher than G′ suggesting a viscoelastic liquid-like behaviour similar to that of colloidal suspensions near their crystallization point at volume fractions ϕc ∼ 0.5.10 The dominance of G′′ along with a large G′ suggests the existence of so-called crowding or jamming.10 This implies that the configurational rearrangement necessary for equilibration cannot be achieved within the time frame studied here. In our system, jamming resulted in the development of a finite yield stress12 at concentrations approximately three orders of magnitude lower than the theoretical value for spherical colloidal suspensions (ϕthis study ≈ 2.2 × 10−4vs. ϕc ≈ 0.5).10 Jamming at such low concentrations is due to the extremely large aspect ratio of ultra-large wrinkled GO sheets used in this study (Fig. S1†). The dominance of the viscous part in all time scales (liquid-like behaviour) while having considerable elastic and viscosity components, in such low concentrations, has a direct impact on processability and fabrication. Specifically this behaviour enables us to easily electrospray and spray-coat with a high level of control (Table S1† and Fig. 2), while eliminating the drift perturbations typically associated with spraying of viscous fluids with no elastic components. These rheological features also allow us to spray at extremely low concentrations leading to low mass loading levels deposited at the substrate surface and subsequently enabling the fabrication of ultra-transparent GO thin films (transparency of 98.04% and 95.4% at 633 nm for single and twice coated glass slides after partial reduction by heat-treatment at 220 °C corresponding to approximately 1 layer of GO per run (Fig. 2d)). It should be noted that such a level of control is usually achieved by employing the highly time consuming Langmuir–Blodgett method.14,15 Spray coating of GO dispersions has great potential in many industrial processes such as electronics, painting, microencapsulation, electroemulsification, fine powder production, or micro- and nano-thin film deposition.16
 | ||
| Fig. 2 A correlation between rheological properties and the key prerequisites for various manufacturing techniques enabled us to process and fabricate GO via a wide range of industrial techniques. (a) Ratio of elastic and storage moduli for various GO concentrations measured over a range of testing frequencies. Overlaid are the approximate processing regimes for a number of industrial fabrication techniques. When the viscous modulus (G′′) dominates, the GO dispersion is suitable for high rate processing methods where the dispersion must spread on contact with the substrate. However, when the elastic modulus (G′) is high the rheological properties suit fabrication methods requiring the dispersion to keep its given shape, such as extrusion printing and fibre spinning. (b) Photograph of electrospraying of a viscoelastic liquid of a GO dispersion at a concentration of 0.05 mg ml−1. (c) Photograph of a GO thin film that was spray coated and thermally reduced (overnight at 220 °C) utilizing a transitional state to a viscoelastic liquid GO dispersion of 0.25 mg ml−1. (d) Transparency of the spray coated reduced GO thin films as a function of coating layers; the numbers show the number of coating layers. (e) Ink-jet printed ACES logo using a LC GO viscoelastic soft solid at a concentration of 0.75 mg ml−1. (f) As-prepared wet-spun fibers from a LC GO viscoelastic soft solid at a concentration of 2.5 mg ml−1. (g) Cross-section of the wet-spun LC GO fiber, showing that GO sheets are stacked in layers with some degree of folding and are ordered due to the formation of nematic liquid crystals. (h) Extrusion printed pattern using a LC GO viscoelastic gel of 4.5 mg ml−1. (i) Multi level extrusion printed 3D architecture using a LC GO viscoelastic gel of 13.3 mg ml−1. (j) Dry-spinning of LC GO fibers utilizing a LC GO viscoelastic gel of 13.3 mg ml−1. | ||
In the second region (GO concentration: ∼0.25 up to 0.75 mg ml−1), GO dispersions show biphasic behaviour (coexistence of both isotropic and nematic phases). Here a viscoelastic behaviour can be observed which is in contrast to normal lyotropic liquid crystal phases.17 In typical lyotropic liquid crystals, the viscous part (G′′) is often dominant at large time-scales or towards the lower frequencies.17 On the other hand, here G′ appears to be higher than G′′ for GO dispersions falling in this region at large time-scales. This behaviour suggests the crowding of particles and consequently gelation of the system as a result of repulsive interactions experienced by neighbouring GO sheets which is also consistent with the yield point observed in this region (Fig. 3a). Therefore, over long-time scales, GO dispersions respond more like a viscoelastic soft solid as the “long-range” rearrangements (convolutions) are very slow. However, above the G′–G′′ crossover point, in an intermediate time scale, G′ was found to be lower than G′′ suggesting a dominant viscous behaviour (liquid-like) implying that “short-range” rearrangements rapidly occur. Nevertheless, up to the concentration of 0.75 mg ml−1, the difference is not considerable and the zones are not yet well-defined. These fluid properties are ideal for inkjet printing as the ink formulation should be designed to quickly regain viscosity and hence shapes (rearrange the mesogens) once they are printed on the surface to preserve the printed structure for accurate reproduction.18 Importantly, this new finding enables us to formulate stable inks from pure GO dispersions without the need of any binders or additives to impart the dominant elastic components required for ink-jet printing (Fig. 2e). It should also be noted that although the size of our GO sheets (average diameter: 37 μm) was much larger than the safe zone19 criterion for our inkjet printer nozzles (1 μm for a 50 μm nozzle), we were able to successfully print GO without any clogging side effects as a consequence of the highly flexible nature of GO sheets (Fig. S1†). The inkjet printing of binder free GO will enable the fabrication of high quality electrode materials that are critical for use in electronic applications such as organic field effect transistors (OFETs).
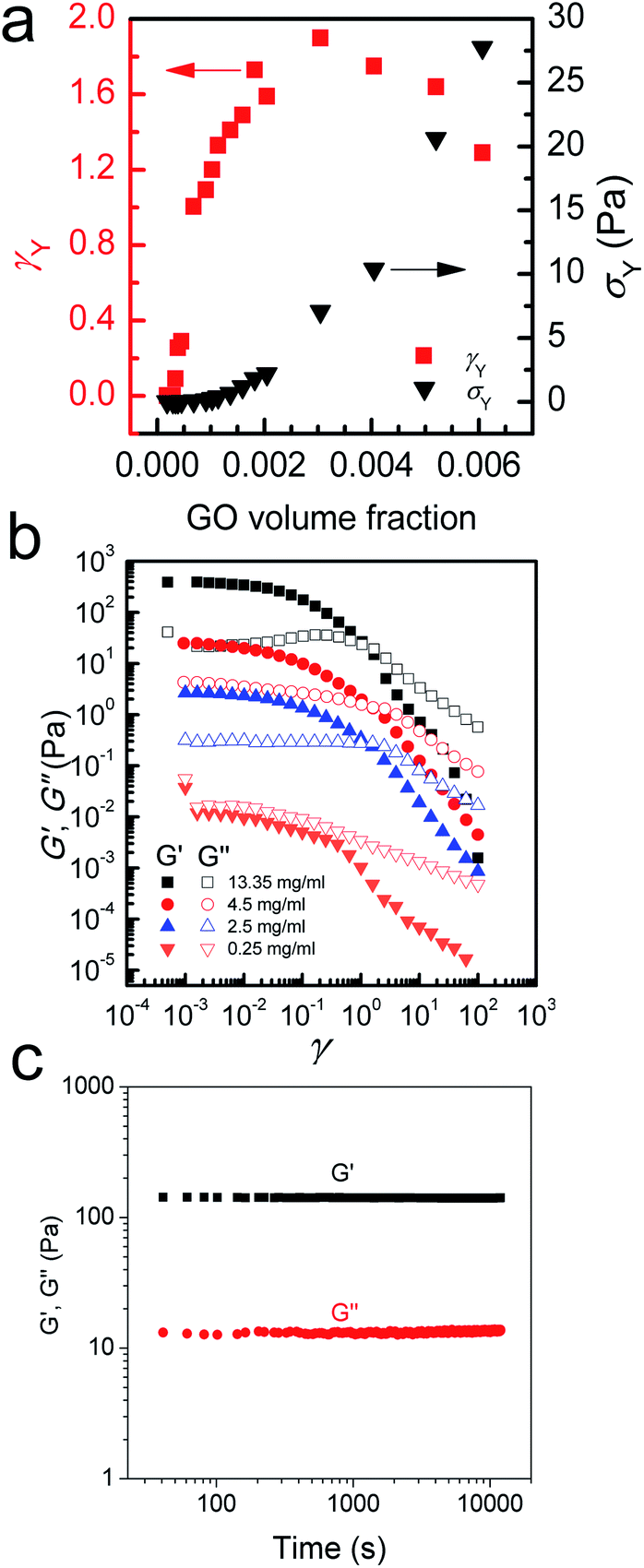 | ||
| Fig. 3 Interpretation of rheological behaviour of LC GO dispersions. (a) Yield strain (γY) and yield stress (σY) of various GO suspensions determined at different GO volume fractions. Yield point is considered as the point where the storage and loss moduli intercept when measured as a function of frequency. (b) Storage (elastic) and loss (viscous) moduli of GO suspensions at the frequency of 0.01 Hz at different strains. No strain-stiffening can be observed even at very high concentrations which is consistent with SGMs rheology generally exhibiting yielding and plasticity. However in contrast to soft glassy rheology (SGR), (c) no aging after shear rejuvenation/fluidization can be observed. | ||
At higher concentrations (above 0.75 up to 2.5 mg ml−1), the storage and loss moduli are completely and clearly well-separated from each other (Fig. 1d and e). This is the concentration range at which GO dispersions form single phase nematic liquid crystals. The two-dimensional GO sheets form a tenuous network architecture with a very dominant elastic part in spite of the considerably low concentration. The G′–G′′ crossover point begins to shift towards higher frequencies. The rheological behaviour of GO dispersions at concentrations as high as 2.5 mg ml−1 resembles that of soft glassy materials (SGMs), liquid crystals, and/or weak gels.10,20 This rheological behaviour suggests that the processing of GO dispersions is possible with established fabrication techniques usually reserved for processing weak gel-like materials (such as the wet-spinning approach).1,21–24 However, in contrast to cross-linked biopolymers10 and biological gels13 no strain stiffening is observed (Fig. 3b), and unlike gels of other disk-like colloids a fully frequency-dependent elastic modulus is not observed either (Fig. 1e).25 Another interesting aspect is the ability of these dispersions to retain their structure at very short time scales.
As concentration increases up to 4.5 mg ml−1G′ and G′′ become fully distinct with G′ reaching 15–77 Pa at 4.5 mg ml−1 (depending on frequency, Fig. 1f). This behaviour is slightly different from that of polymer networks and resembles the rheological characteristics of cells or SGMs.10,26 Purely elastic polymer networks exhibit a completely frequency-independent storage modulus plateau even at low frequencies.10,26 Similar to cells however,10 GO dispersions in this region show power-law rheology with a weak non-universal exponent (i.e. the storage modulus increases slowly with frequency). In this region, it is only at very short time scales that the viscous part dominates. The magnitude of storage modulus increases with increasing concentration (volume fraction) and the crossover point (G′ = G′′) shifts to time scales as short as 0.01–0.1 s as the network becomes more robust and exhibits a gel-like behaviour (Fig. 1g). This behaviour might suggest structural changes even though the rheological properties of liquid crystals of disk-like particles are not simply correlated with the phase behaviour of the materials.20,25,27 Upon increasing the concentration above 4.5 mg ml−1, the nematic domains (with same orientation and brightness) decrease in size; this in turn, could contribute to substantial increase in the elastic modulus associated with a greater density of defects.28
At even higher concentrations (up to 13.35 mg ml−1), no G′–G′′ crossover was observed in the frequency range studied here, resembling a gel-like (cells or SGMs) behaviour with an extraordinarily high elastic modulus of 350–490 Pa. The measured storage modulus at 13.35 mg ml−1 was considerably higher than the calculated elastic modulus of SWNT suspensions at the same concentration (∼60 Pa).29 GO dispersions at this concentration range are, therefore, viscoelastic liquid crystal gels that can flow after a yield point. Furthermore, there are some unique characteristics that our as-prepared LC GO viscoelastic gels exhibit such as the anisotropy arising from having a liquid crystal network, and the exceptional uniformity of the network structure. Therefore, GO dispersions prepared in this region enabled us to process GO for the first time in a range of industrial processes such as gel-extrusion printing and dry spinning that are most beneficial for many industrial applications (Fig. 2h–j). During gel-extrusion through a nozzle (i.e. extrusion printer nozzle), the loss modulus will become dominant (Fig. 3b, strain thinning effect), allowing for the dispersion to be easily extruded. However, when the LC GO viscoelastic gels leave the nozzle (at low strain), the physical sheet entanglement forces them to regain their high elasticity. The high elasticity of LC GO viscoelastic gels retains the printed structure and makes it possible to print fine lines or multi-level 3D architectures (Fig. 2h and i).
To provide further understanding of the phase transition behaviour of the GO dispersion, we measured the viscosity and calculated the ratio of elastic and loss moduli of the system with increasing GO concentration (Fig. 4). As expected, both viscosity and G′/G′′ increase with GO concentration in the low concentration range until a peak is reached at 0.5 mg ml−1; then a sudden drop in both viscosity and G′/G′′ is observed. As we showed before,5 entropy plays an important role in the ordering of GO sheets. Upon becoming concentrated, the isotropic fluid of two-dimensional disk-like GO sheets must undergo a transition to a nematic phase in which GO sheets encompass a preferred orientation. This orientational ordering consequently results in loss of orientational entropy. However, this loss of entropy is compensated with an increase in free volume. Therefore, there will be a net gain of entropy, as a result of the increase in free volume (packing) entropy. This sudden free volume expansion accompanied by ordering in the direction of shear, because of the spontaneous formation of LC domains, results in a sudden drop of viscosity and the ratio of elastic to viscous moduli.1,5 By increasing GO concentration furthermore, both viscosity and the moduli ratio gradually increase again until around 2.5 mg ml−1. However, in the concentration range of 2.5 mg ml−1 to 4 mg ml−1, a sudden drop of G′/G′′ can be observed suggesting a disturbance in the system. The latter might be due to an underlying phase transition with a tendency of the flakes to stack more regularly. Indeed, it has been observed that clay platelets can form columnar and hexagonal phases at high concentration.25 The formation of such phases can be prevented in the present case by the polydispersity of the system or by dynamical arrest; but the tendency of the flakes to pack more regularly could explain changes in rheological properties. At the critical concentration (4.5 mg ml−1), the increase in viscosity levels out and G′/G′′ ratio starts to increase again. Stacking of the GO mesogens results in further free volume expansion30 and subsequently hinders any increase in the viscosity at this critical concentration (Fig. 4a).
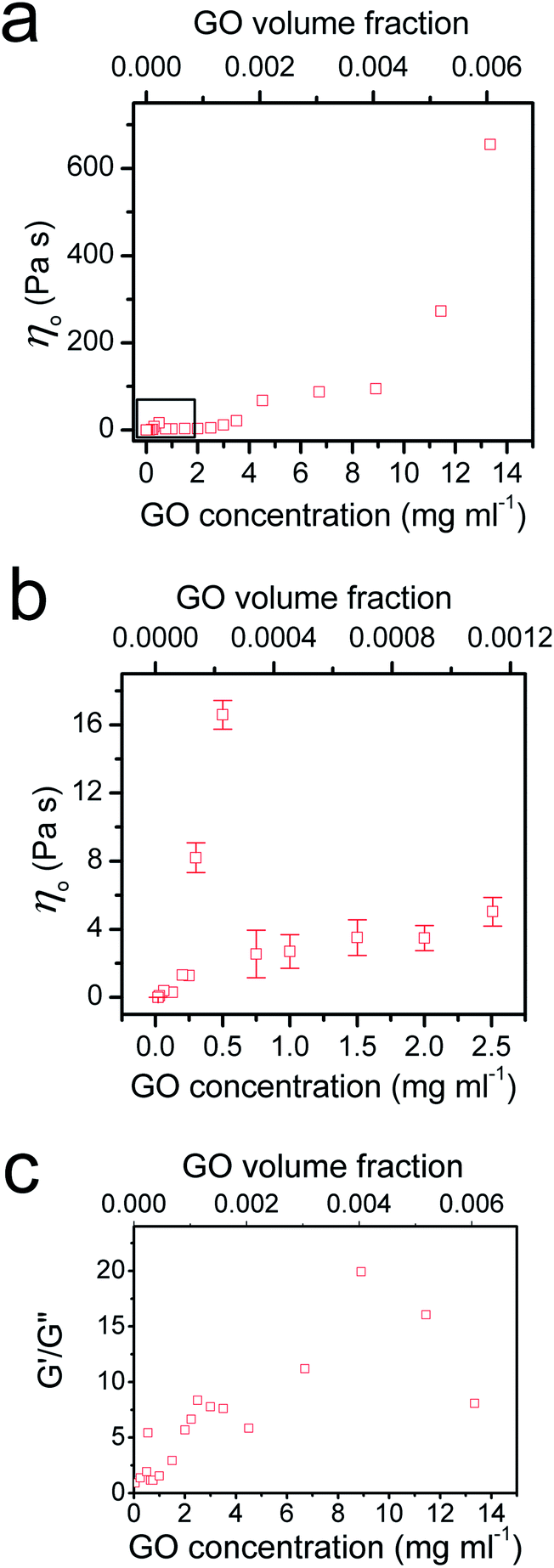 | ||
Fig. 4 Viscosity and the ratio of loss and storage moduli of GO dispersions at different GO concentrations. (a) Viscosity (![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) = 0.01 s−1) as a function of GO volume fraction. Increasing the concentration of GO results in an overall increase in viscosity until a peak at ϕ ∼ 2.3 × 10−4 (0.5 mg ml−1). However as depicted in (b) a sudden drop in viscosity as a result of ordering happens at higher concentrations. (c) The same general trend also occurs in the case of the ratio between loss and storage moduli. Moreover, at almost all concentrations, except in the very beginning, the ratio of G′ to G′′ is higher than 1 indicating a very dominant elastic behaviour. = 0.01 s−1) as a function of GO volume fraction. Increasing the concentration of GO results in an overall increase in viscosity until a peak at ϕ ∼ 2.3 × 10−4 (0.5 mg ml−1). However as depicted in (b) a sudden drop in viscosity as a result of ordering happens at higher concentrations. (c) The same general trend also occurs in the case of the ratio between loss and storage moduli. Moreover, at almost all concentrations, except in the very beginning, the ratio of G′ to G′′ is higher than 1 indicating a very dominant elastic behaviour. | ||
Conclusions
The viscoelastic behaviour of this fascinating material offers significant features that can prove to be useful for both fundamental research in two-dimensional materials and practical applications as demonstrated by a wide range of processing techniques employed in the present paper. We emphasize that the characteristic flow behaviour of LC GO is fundamentally different from those associated with usual viscoelastic materials including polymers. The generic properties reported here can be considered as a universal guideline to process different GO dispersions based on their rheological properties. Therefore, a simple rheological test and the comparison with the guidelines provided here can assist others in the field to decide on what processing techniques should be employed and why.Acknowledgements
The authors thank the Australian National Fabrication Facility (ANFF) Materials Node for their provision of research facilities. This work was supported by ARC Discovery Project DP1093952 (KK and SHA), ARC Federation Fellowship (GGW) and ARC Professorial Fellowship (GMS).Notes and references
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, L. Shepherd Roderick, C. Jun, A.-Y. Sima, K. Konstantinov, I. Minett Andrew, J. M. Razal and G. G. Wallace, Adv. Funct. Mater., 2013, 23, 5344 CrossRef.
- S. H. Aboutalebi, M. M. Gudarzi, Q. B. Zheng and J.-K. Kim, Adv. Funct. Mater., 2011, 21, 2978–2988 CrossRef CAS.
- Z. Xu and C. Gao, ACS Nano, 2011, 5, 2908–2915 CrossRef CAS PubMed.
- J. E. Kim, T. H. Han, S. H. Lee, J. Y. Kim, C. W. Ahn, J. M. Yun and S. O. Kim, Angew. Chem., Int. Ed., 2011, 50, 3043–3047 CrossRef CAS PubMed.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, J. M. Razal, S. E. Moulton and G. G. Wallace, Mater. Horiz., 2014, 1, 87–91 RSC.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, S. E. Moulton, J. M. Razal and G. G. Wallace, ACS Nano, 2013, 7, 3981–3990 CrossRef CAS PubMed.
- M. M. Gudarzi, M. H. M. Moghadam and F. Sharif, Carbon, 2013, 64, 403–415 CrossRef CAS PubMed.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571 CrossRef PubMed.
- C. Cheng and D. Li, Adv. Mater., 2013, 25, 13–30 CrossRef CAS PubMed.
- D. T. N. Chen, Q. Wen, P. A. Janmey, J. C. Crocker and A. G. Yodh, Annu. Rev. Condens. Matter Phys., 2010, 1, 301–322 CrossRef CAS.
- J. R. Seth, L. Mohan, C. Locatelli-Champagne, M. Cloitre and R. T. Bonnecaze, Nat. Mater., 2011, 10, 838–843 CrossRef CAS PubMed.
- G. Ovarlez, Q. Barral and P. Coussot, Nat. Mater., 2010, 9, 115–119 CrossRef CAS PubMed.
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS PubMed.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538–542 CrossRef CAS PubMed.
- Q. Zheng, W. H. Ip, X. Lin, N. Yousefi, K. K. Yeung, Z. Li and J.-K. Kim, ACS Nano, 2011, 5, 6039–6051 CrossRef CAS PubMed.
- A. Jaworek and A. T. Sobczyk, J. Electrost., 2008, 66, 197–219 CrossRef CAS PubMed.
- R. Mezzenga, C. Meyer, C. Servais, A. I. Romoscanu, L. Sagalowicz and R. C. Hayward, Langmuir, 2005, 21, 3322–3333 CrossRef CAS PubMed.
- B. Derby, Annu. Rev. Mater. Res., 2010, 40, 395–414 CrossRef CAS.
- F. Torrisi, T. Hasan, W. Wu, Z. Sun, A. Lombardo, T. S. Kulmala, G.-W. Hsieh, S. Jung, F. Bonaccorso, P. J. Paul, D. Chu and A. C. Ferrari, ACS Nano, 2012, 6, 2992–3006 CrossRef CAS PubMed.
- M. Kroon, W. L. Vos and G. H. Wegdam, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, 1962–1970 CrossRef CAS.
- R. Jalili, J. M. Razal and G. G. Wallace, J. Mater. Chem., 2012, 22, 25174–25182 RSC.
- R. Jalili, J. M. Razal, P. C. Innis and G. G. Wallace, Adv. Funct. Mater., 2011, 21, 3363–3370 CrossRef CAS.
- D. Esrafilzadeh, J. M. Razal, S. E. Moulton, E. M. Stewart and G. G. Wallace, J. Controlled Release, 2013, 169, 313–320 CrossRef CAS PubMed.
- R. Jalili, J. M. Razal and G. G. Wallace, Sci. Rep., 2013, 3, 3438 Search PubMed.
- M. C. D. Mourad, D. V. Byelov, A. V. Petukhov, D. A. Matthijs de Winter, A. J. Verkleij and H. N. W. Lekkerkerker, J. Phys. Chem. B, 2009, 113, 11604–11613 CrossRef CAS PubMed.
- P. Sollich, F. Lequeux, P. Hébraud and M. E. Cates, Phys. Rev. Lett., 1997, 78, 2020–2023 CrossRef CAS.
- L. J. Michot, C. Baravian, I. Bihannic, S. Maddi, C. Moyne, J. r. m. F. L. Duval, P. Levitz and P. Davidson, Langmuir, 2008, 25, 127–139 CrossRef PubMed.
- M. Zapotocky, L. Ramos, P. Poulin, T. C. Lubensky and D. A. Weitz, Science, 1999, 283, 209–212 CrossRef CAS.
- L. A. Hough, M. F. Islam, P. A. Janmey and A. G. Yodh, Phys. Rev. Lett., 2004, 93, 168102–168106 CrossRef CAS.
- H. H. Wensink and H. N. W. Lekkerkerker, Mol. Phys., 2009, 107, 2111–2118 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, supporting discussion, Fig. S1–S5 and Table S1. See DOI: 10.1039/c3mh00144j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |