
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-chain synergy in ionic liquid electrolytes for lithium batteries†
Feng
Wu
 ab,
Qizhen
Zhu
a,
Renjie
Chen
*ab,
Nan
Chen
a,
Yan
Chen
a and
Li
Li
ab
ab,
Qizhen
Zhu
a,
Renjie
Chen
*ab,
Nan
Chen
a,
Yan
Chen
a and
Li
Li
ab
aBeijing Key Laboratory of Environmental Science and Engineering, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing, 100081, China. E-mail: chenrj@bit.edu.cn; Fax: +86-10-68451429
bCollaborative Innovation Center of Electric Vehicles in Beijing, Beijing, 100081, China
First published on 18th September 2015
Abstract
Lithium-ion batteries have been attracting much attention which enables the revolution of wireless global communication. Ionic liquids are regarded as promising candidates for lithium-ion battery electrolytes because they can overcome the limitations of high operating temperatures and flammability concerns of traditional electrolytes. However, at low temperatures they suffer from low ionic conductivity and phase transition. In this paper mixed electrolyte systems are described based on N-methoxyethyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)-imide (Pyr1,2O1TFSI) and lithium difluoro(oxalate)borate (LiODFB) lithium salt, with ethylene sulphite (ES) or dimethyl sulphite (DMS) as a cosolvent. The mixed electrolyte system exhibits good ion transport properties (a conductivity of 8.163 mS cm−1), a wide electrochemical window (5.2 V), non-flammability, the ability to form films to protect the anode and a large operating temperature range (−40 °C to 60 °C). We compare the performance and function of the new mixed electrolyte system with a variety of ionic liquid/cosolvent electrolyte systems developed in previous studies. The ring-chain synergy takes advantage of the availability of both high permittivities based on the ring-like components and low viscosities based on the chain-like components in the mixed electrolyte system and causes the electrolyte to exhibit a good overall performance in safety, ion transport and compatibility with electrodes.
Introduction
With increasing global concerns about climate change and the environment and a growing scarcity of traditional fossil fuels, substantial efforts have been focused on the development of new and renewable energy sources.1,2 Lithium-ion batteries are important as storage and conversion devices, which have a high energy density, long lifecycle, and significant environmental benefits.3,4 However, the adoption of lithium-ion battery technology still faces major challenges because the batteries can only operate within a narrow temperature range and because they present safety issues, namely inflammability.5–7 As a key component of a battery, the electrolyte has a potent influence on both of these issues, so the solution lies in improving/tailoring the electrolyte.8,9In lithium-ion batteries, a variety of liquid solvents can be used as electrolytes to produce batteries with excellent ionic conductivity, and the use of organic carbonates as solvents for electrochemical applications is well established.10 However, these carbonates are highly flammable, posing a safety problem for large-scale applications. Ionic liquids, which have been the focus of significant research,11–19 are regarded as one of the most promising alternatives to the carbonates used as electrolytes in lithium batteries because of their high electrochemical and thermal stability. One challenge with ionic liquids, however, is that they tend to have a high viscosity, which can lead to decreased ionic conductivity in batteries. To overcome this problem, ionic liquid/organic solvent binary electrolyte systems, which combine the nonflammability of the ionic liquid and the low viscosity of the organic solvent, have been prepared.14,15,20,21 The addition of a small amount of carbonate in alkyl ammonium significantly improves the conductivity, reaching 12 mS cm−1 at 40 °C.22 By introducing 20% diethyl carbonate as a co-solvent into a pure ionic liquid electrolyte, the discharge capacity of the LiCoO2/Li cells reaches up to 102 mA h g−1 at 10 °C with negligible damage to the safety characteristics.23
In this study, we report the preparation and electro-chemical evaluation of a mixed electrolyte system that takes advantage of the ring-like ionic liquid N-methoxyethyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)-imide (Pyr1,2O1TFSI). The ether-functionalised cation (Fig. 1) effectively decreases the viscosity and provides an amorphous system below room temperature.24–27 Pyr1,2O1TFSI and EC/DEC binary mixtures with a volume ratio of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 with LiTFSI demonstrated remarkably reduced flammability, ionic conductivity exceeding 7 mS cm−1 at 20 °C and an overall electrochemical window of 4.5 V.15 Sulphites are chosen as the cosolvents for their low viscosity, low melting point and outstanding ability to form effective solid electrolyte interface (SEI) layers to protect the electrodes.28–32 LiODFB is used as the lithium salt because of its good passivation toward Al33,34 to avoid corrosion of the Al current collector from the TFSI− anion. The Pyr1,2O1TFSI/sulphite binary electrolytes were combined with LiODFB as the lithium salt.
:40 with LiTFSI demonstrated remarkably reduced flammability, ionic conductivity exceeding 7 mS cm−1 at 20 °C and an overall electrochemical window of 4.5 V.15 Sulphites are chosen as the cosolvents for their low viscosity, low melting point and outstanding ability to form effective solid electrolyte interface (SEI) layers to protect the electrodes.28–32 LiODFB is used as the lithium salt because of its good passivation toward Al33,34 to avoid corrosion of the Al current collector from the TFSI− anion. The Pyr1,2O1TFSI/sulphite binary electrolytes were combined with LiODFB as the lithium salt.
 | ||
| Fig. 1 Molecular structures of the mixture components: dimethyl sulphite (DMS), ethylene sulphite (ES), N-methoxyethyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)-imide (Pyr1,2O1TFSI) and lithium difluoro(oxalate)borate (LiODFB). | ||
So far, there has been no suitable single solvent available that simultaneously possesses a high dielectric permittivity, low viscosity and good compatibility with both an anode and cathode. As a compromise, the majority of the most common electrolyte systems used in lithium-ion batteries is a mixture of the ring-like ethylene carbonate with high dielectric permittivity or the chain-like carbonate with low viscosity. The combination of the beneficial effects of the carbonate species with atoms arranged in both rings and chains can improve the performance of the bulk electrolyte systems, which is designated as ring-chain synergy. However, key questions about the electrolyte performance remain unanswered. Can ring-chain synergy also be applicable for ionic liquid electrolytes? What is the optimal composition of an ionic liquid/organic cosolvent-based electrolyte that meets the demands in terms of safety, ion transport, and compatibility with the both cathodes and anodes in a lithium-ion battery? In order to answer these questions, this paper compares the improvement offered by ring-like ethylene sulphite (ES) and chain-like dimethyl sulphite (DMS) in electrochemical and physicochemical performance for the electrolyte based on Pyr1,2O1TFSI. Furthermore, we summarise and discuss the properties and performance of ionic liquid/cosolvent electrolyte systems derived both in our new research outlined here and in previous studies.
Results, discussion and experimental
Electrolyte properties
Solvation of Li+ in the electrolyte depends upon the interaction of Li+ and the solvent molecules.35 The Fourier transform infrared spectroscopy (FTIR) spectra of the electrolyte systems in which LiODFB and sulphites are added into Pyr1,2O1TFSI (Fig. S1†) are shown to investigate the Li+ solvation which depends on the intensity of interaction between LiODFB and the solvent molecules. The electron cloud density of –SO3 in DMS is more negative than that in ES. Affected by the coordination between Li+ and the electronegative O atom, the stretching vibrations of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O become weaker. Consequently, the bond energy of O–S–O becomes stronger and the peak is red shifted to a lower frequency. The O–S–O stretching bands varying in ES (910 cm−1 and 654 cm−1) and DMS (953 cm−1 and 692 cm−1) indicates that the enhancement of Li+ solvation is more obvious in DMS.
O become weaker. Consequently, the bond energy of O–S–O becomes stronger and the peak is red shifted to a lower frequency. The O–S–O stretching bands varying in ES (910 cm−1 and 654 cm−1) and DMS (953 cm−1 and 692 cm−1) indicates that the enhancement of Li+ solvation is more obvious in DMS.
The ionic conductivity and the Arrhenius plots of the ionic conductivity of the LiODFB–Pyr1,2O1TFSI electrolyte with and without sulphites over a temperature range of −40 °C to 80 °C are given in Fig. 2(a) and Table 1. Although the ether group, as a lateral substituent, decreases the viscosity of the ionic liquid, the conductivity of the ionic liquid (1.9 mS cm−1) is not yet comparable to that of traditional carbonate electrolytes (5–10 mS cm−1).33 However, when Pyr1,2O1TFSI is mixed with sulphites, a significant increase (3–4 times) in the ionic conductivity is observed. LiODFB–Pyr1,2O1TFSI/DMS possesses an ionic conductivity at room temperature of 8.163 mS cm−1 with a relative standard departure of 4.14% among permitted error bars after testing 5 times (Fig. S2†). The conductivity value is close to that of carbonate-based ionic liquid electrolytes (8 mS cm−1).15 Fig. S3† shows the differential scanning calorimetry (DSC) curves of the electrolytes. LiODFB–Pyr1,2O1TFSI exhibits a large exothermal crystallisation peak (at −43 °C) due to incomplete crystallization of the sample prior to the DSC heating scan. It becomes lower (−51 °C) when DMS is introduced, which suggests that the addition of DMS makes the electrolyte useful at low temperatures.
 | ||
| Fig. 2 (a) VTF plots of the conductivity of LiODFB–Pyr1,2O1TFSI electrolytes with and without sulphites at various temperatures. Burning tests of (b) LiODFB–Pyr1,2O1TFSI/DMS and (c) LiPF6–EC/DEC. (d) CVs of LiODFB–Pyr1,2O1TFSI electrolytes with and without sulphite. (e) The energy level diagram of frontier molecular orbitals relative to the energy in a vacuum (Evac) and HOMO and LUMO plots of Pyr1,2O1TFSI, ES and DMS. | ||
| Electrolyte system | LiODFB–Pyr1,2O1TFSI | LiODFB–Pyr1,2O1TFSI/ES | LiODFB–Pyr1,2O1TFSI/DMS | |
|---|---|---|---|---|
| Ionic conductivity/mS cm−1 | −40 °C | 5.26 × 10−3 | 2.06 × 10−1 | 4.63 × 10−1 |
| −20 °C | 8.40 × 10−2 | 9.63 × 10−1 | 1.50 | |
| 0 °C | 4.91 × 10−1 | 2.69 | 3.80 | |
| 25 °C | 1.91 | 6.06 | 8.16 | |
| 60 °C | 6.66 | 14.0 | 16.6 | |
| VTF parameters | A/S cm−1 K−1/2 | 17.1 | 12.7 | 8.1 |
| B/K | 835 | 748 | 609 | |
| T 0/°C | −109 | −131 | −125 | |
The relationship between temperature and ionic conductivity is described by the Vogel–Tamman–Fulcher (VTF) equation,36,37
 | (1) |
To clarify the influence of the sulphites on electrochemical stability, the cyclic voltammograms (CVs) determining the oxidation/reduction potentials of the electrolytes are shown in Fig. 2(d). When the potential is increased to 5.2 V, the decomposition of LiODFB–Pyr1,2O1TFSI becomes significant, suggesting oxidative decomposition at a potential of 5.2 V (as compared to Li/Li+). The addition of sulphites only has a small influence on the oxidation potentials of the electrolytes; for all cases the oxidation potential exceeded 5 V (vs. Li/Li+). On the basis of molecular orbital theory, the difference between the calculated energies of the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) determine the vertical electron affinity (VEA) and the vertical ionisation potential (VIP).38,39 As shown in Fig. 2(e), the LUMO and HOMO of ES have lower energies than those of DMS, indicating that ES has higher oxidation and reduction potentials.
The flammability of the electrolytes based on either ionic liquids or carbonates was tested, and the results are shown in Fig. 2(b) and (c) and S4.† The traditional carbonate electrolyte ignited immediately, while the ionic liquid electrolytes, including those with sulphites, did not ignite when exposed to a flame of 1300 °C for 60 s. This excellent flame resistance makes ionic liquid electrolytes a promising safe choice for use in lithium-ion batteries.
Electrochemical compatibility with electrodes
A mesocarbon microbead (MCMB) is chosen as the electrode to evaluate the electrochemical properties of cells with the electrolytes because it is abundant, inexpensive and has a high specific energy. In Fig. 3(a)–(c), the galvanostatic charge–discharge profiles of Li/MCMB cells with LiODFB–Pyr1,2O1TFSI do not feature a stable plateau, and the CV (Fig. S5†) without symmetrical oxidation/reduction peaks implies irreversible Li+ insertion/extraction. Li/MCMB cells containing electrolytes with sulphites show two discharge voltage plateaus in the initial cycle. The small plateau at 1.8 V in the first cycle disappearing in the following cycle may be mainly assigned to the formation of a SEI, leading to loss of capacity, and the smooth plateau at 0.005–0.4 V indicates a good reversibility in the Li+ insertion/extraction performance. The CVs in Fig. S5(b) and (c)† show reduction peaks at about 1.7 V indicating SEI forming in the first cycle and symmetrical oxidation/reduction peaks below 0.5 V which are in good agreement with the galvanostatic charge–discharge results. | ||
| Fig. 3 First and subsequent charge and discharge curves of Li/MCMB cells with (a) LiODFB–Pyr1,2O1TFSI, (b) LiODFB–Pyr1,2O1TFSI/ES or (c) LiODFB–Pyr1,2O1TFSI/DMS electrolytes. (d) Cycling performance and coulombic efficiency of the cells with the three electrolyte samples. | ||
The cycling performances of the Li/MCMB cells containing the electrolytes are demonstrated in Fig. 3(d). The cells containing LiODFB–Pyr1,2O1TFSI exhibit a limited reversible charge capacity below 100 mA h g−1. The initial discharge of cells with LiODFB–Pyr1,2O1TFSI/ES are about 313.9 mA h g−1. Compared with ES, the cells upon addition of DMS acquire a higher charge capacity of 376.2 mA h g−1 in the first cycle and retain a value of 345.0 mA h g−1 after 50 cycles. The coulombic efficiency is 99.6%, which is higher than that of cells containing electrolytes of disiloxane-functionalised phosphonium,40 allyl-functionalised piperidinium41 and imidazolium, pyrrolidinium, and piperidinium with film-forming additives.42,43 The results above mean an outstanding cycle stability for Li/MCMB cells with LiODFB–Pyr1,2O1TFSI/DMS because DMS is beneficial for SEI forming on the surface of the MCMB.
The Nyquist plots of AC impedance spectra of cells containing different electrolytes are shown in Fig. S6.† In general, the total cell resistance mainly consists of the bulk resistance (Rb), the resistance to Li+ migration through the SEI layer (Rsei), and the charge transfer resistance (Rct).38 It is revealed that both the Rb and Rct values are much smaller for electrolytes with sulphites than for those of LiODFB–Pyr1,2O1TFSI. The Rct value of cells containing LiODFB–Pyr1,2O1TFSI/ES is higher than that of cells containing LiODFB–Pyr1,2O1TFSI/DMS, which might be attributed to the excessively thick SEI layer formed in ES and the consequent difficulty for Li+ to transfer through the electrode–electrolyte interface.
A combination of density functional theory (DFT) modelling, electrochemical testing and scanning electron microscopy (SEM) analysis reveals the film-forming mechanisms of the mixed electrolytes. LiODFB can facilitate the formation of a SEI by electrochemical redox reactions.44 The reduction of the F2B[ox]− anions may contribute to the formation of the inorganic inner layer of the SEI:45
| F2B[ox]− + e−˙ → [ox]B˙ + 2F− | (2) |
The [ox]B˙ radical can react with sulphite molecules, and the reactions with the energy change obtained by DFT calculations are shown in Fig. 4. According to the principle of lowest energy, the prime reactions for ES and DMS are
| [ox]B˙ + ES → [ox]BOCH2CH2OS˙O | (3) |
| [ox]B˙ + DMS → [ox]BOCH3 + CH3OS˙O | (4) |
 | ||
| Fig. 4 Reactions of [ox]B˙ radicals with (a) ES and (b) DMS. | ||
To validate the calculation results, the SEM, energy dispersive spectrometry (EDS) and schematic illustration of the synthesis route to the SEI layer with prime reactions in the electrolytes are shown in Fig. 5. The spherically shaped particles of the original MCMB electrode are obscure, as a film covers their surface after cycling. The films formed in the electrolyte containing sulphites are more compact than those formed in LiODFB–Pyr1,2O1TFSI. Moreover, the film grown in the electrolyte containing ES is thicker and has a lower S content than in DMS (Table S1†), which is consistent with the calculation results.
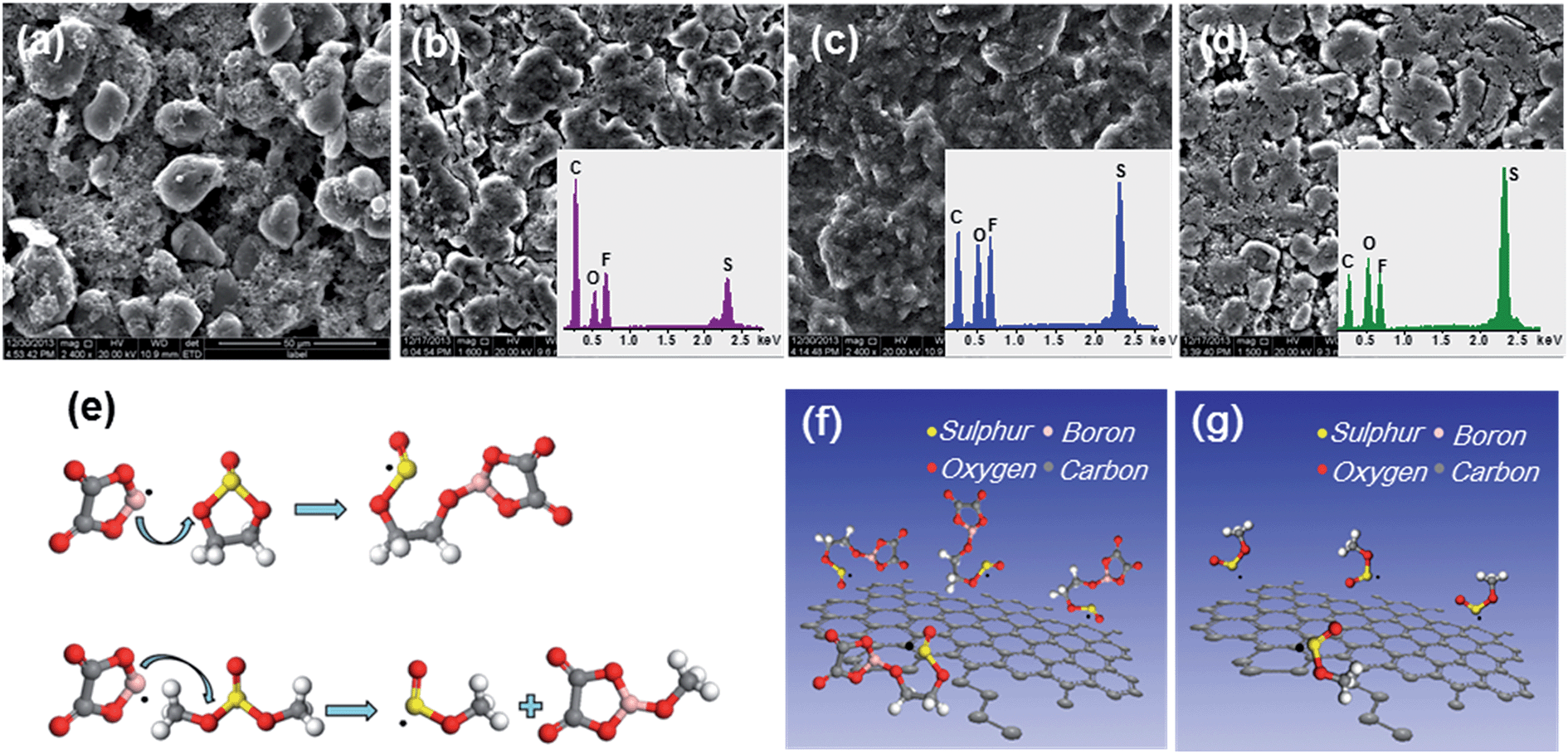 | ||
| Fig. 5 (a) SEM image of the MCMB anode before cycling. SEM and EDS of the SEI layer on the MCMB anode in (b) LiODFB–Pyr1,2O1TFSI, (c) LiODFB–Pyr1,2O1TFSI/ES, and (d) LiODFB–Pyr1,2O1TFSI/DMS electrolytes after 10 cycles. Schematic illustration of the synthesis route to SEI: (f) ES; (g) DMS, with the prime reactions (e). | ||
The olivine LiFePO4 has been developed with a low toxicity, low cost and high safety46 as an electrode material for lithium-ion batteries. However, Li/LiFePO4 containing the traditional electrolyte, cannot operate at low temperatures below −20 °C according to observations reported by Wang Y.46Fig. 6 and S7† show the electrochemical performance of the Li/MCMB cells and Li/LiFePO4 cells in LiODFB–Pyr1,2O1TFSI/ES, LiODFB–Pyr1,2O1TFSI/DMS, the traditional electrolyte (1 M LiPF6–EC/DEC (1:1 by volume)) and the 1 M LiODFB–EC/DEC (1:1 by volume) at temperatures ranging from −40 °C to 60 °C. The cells with the traditional electrolyte exhibit poor low temperature performance. LiODFB provides some improvement in the electrochemical performance of the lithium-ion cells at low temperatures,47 but not great breakthroughs when the carbonate species is solidified below −30 °C. The electrolytes based on ionic liquids operate well at temperatures higher than 25 °C because of their thermal stability,48,49 but suffer from the same problem as traditional electrolytes at low temperatures. Improvement by adding sulphites in ether-functional ionic liquid electrolytes enables operation at lower temperatures. The capacities of Li/MCMB and Li/LiFePO4 cells containing LiODFB–Pyr1,2O1TFSI/DMS are 354.2 mA h g−1 and 130.3 mA h g−1 at −10 °C with average voltages of 0.306 V and 3.283 V, respectively. Additionally, these cells are able to operate with acceptable electrochemical performance at temperatures as low as −40 °C.
 | ||
| Fig. 6 Performance of Li/MCMB (a) and Li/LiFePO4 cells (b) containing LiODFB–Pyr1,2O1TFSI/ES, LiODFB–Pyr1,2O1TFSI/DMS, LiODFB–EC/DEC or LiPF6–EC/DEC electrolytes at various temperatures with a current density of 0.1C. | ||
LiCoO2 has been widely used in lithium-ion batteries as a cathode material. To ensure the effect of the developed electrolyte on the electrochemical performance of lithium-ion cells, the rate capability of the LiODFB–Pyr1,2O1TFSI/DMS electrolyte in Li/LiFePO4, Li/LiCoO2 and Li/MCMB cells are shown in Fig. 7. The discharge capacities for the Li/LiFePO4 and Li/LiCoO2 cells containing LiODFB–Pyr1,2O1TFSI/DMS at 1C (150 mA g−1) are around 129 and 106.7 mA h g−1, respectively, and the charge capacity for the Li/MCMB cells at 0.5C (150 mA g−1) is about 206.5 mA h g−1. A good rate performance depends on both satisfactory ion transport and acceptable interfacial reaction resistance in the cells.
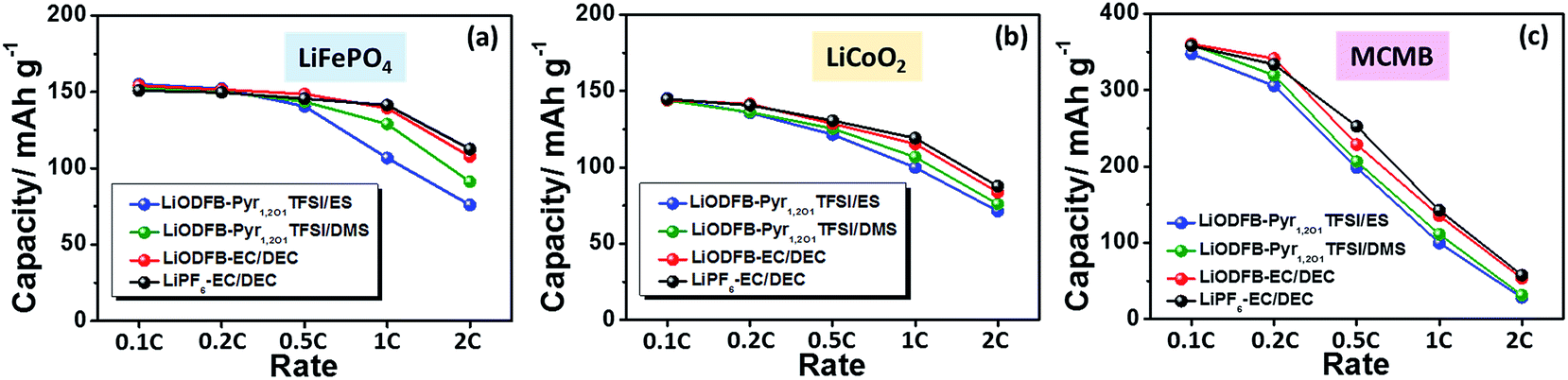 | ||
| Fig. 7 Rate capability of the Li/LiFePO4 (a), Li/LiCoO2 (b) and Li/MCMB (c) cells with LiODFB–Pyr1,2O1TFSI/ES, LiODFB–Pyr1,2O1TFSI/DMS, LiODFB–EC/DEC or LiPF6–EC/DEC electrolytes. | ||
Ring-chain synergy
To clarify if the ring-chain synergy in the carbonate electrolytes used in lithium-ion batteries is also applicable for the ionic liquid electrolytes, a summary of representative electrolyte systems of ionic liquids with organic cosolvents for lithium-ion batteries and the corresponding characteristics and properties are given in Fig. 8.11,13–17,19,24,34,46–78 The relationship among the structures of electrolyte components and their properties is shown to provide a good starting point for strategically designing new electrolyte systems. Furthermore, the comparisons of the improvements offered by the ring-like ES and chain-like DMS in the performance of the electrolytes based on Pyr1,2O1TFSI offer some support for this relationship. | ||
| Fig. 8 (a) Illustration of representative electrolyte systems based on ionic liquids for lithium batteries with the corresponding characteristics. Semi-quantitative threshold of the properties of the electrolyte systems: (b) ring-like ionic liquid and ring-like cosolvent, (c) ring-like ionic liquid and chain-like cosolvent, (d) chain-like ionic liquid and ring-like cosolvent and (e) chain-like ionic liquid and chain-like cosolvent.11,13–17,19,24,34,46–78 | ||
Generally, a safe electrolyte should satisfy several requirements such as non-flammability, good ion transport and compatibility with both the cathode and anode in lithium-ion batteries. The electrolytes based on imidazolium are electrochemically unstable at high potential.79 Therefore we have focused on ammonium-based electrolytes.
General performances of the electrolytes based on ionic liquids with cosolvents are divided into four factors: safety, conductivity, oxidation potential and SEI forming ability. They depend on the nature of the ionic liquids and the cosolvents. We tried to suggest how the electrochemical performances of the electrolyte systems varied with the contents of the cosolvents through analyzing research data from this work and previous studies and the results are shown in Fig. 8(b)–(e). For example, Fig. 8(b) exhibits the general performance of a mixed electrolyte based upon a ring-like ionic liquid with ring-like cosolvents. The applicable content of the cosolvent should be less than 40 wt% for non-flammability of the bulk electrolyte (green zone), because the electrolyte with 40 wt% ring-like cosolvent and 60 wt% ring-like ionic liquid was just beginning to become non-flammable.19 Cosolvents were added to an ionic liquid to increase the room temperature ionic conductivity of the bulk electrolyte to over 1 mS cm−1. The amount of cosolvents added depends on the nature of the specific components. For systems exhibited in the present and previous studies, it should be more than 25–35 wt% (purple zone).19,49,53,54 The oxidation potentials of the investigated electrolyte systems containing a ring-like ionic liquid and a ring-like cosolvent are higher than 5 V (brown line), which can meet the requirements of the cathodes widely used in lithium-ion batteries such as LiFePO4, LiCoO2, LiNi1/3Mn1/3Co1/3O2, and Li1.2Ni0.2Mn0.6O2. Besides, the contents of the cosolvents should be chosen to form a suitable SEI layer to protect the carbonaceous anodes, which is about 20 wt% (blue zone) in accordance with the previous studies. The performances of the electrolyte systems containing “ring-like ionic liquid and chain-like cosolvent”, “chain-like ionic liquid and ring-like cosolvent” and “chain-like ionic liquid and chain-like cosolvent” are analogously summed up to search for the optimal proportion of the electrolyte components on the basis of this work and the reported studies.
Conclusions
Study of the electrolytes based on the ring-like Pyr1,2O1TFSI with ES and DMS reveals that the sulphites improve the electrolyte conductivity, compatibility with MCMB electrodes and cell performance at low temperatures. DMS with a lower viscosity and melting point can better enhance the cell performance than ES in the electrolyte with the main component of Pyr1,2O1TFSI. The conductivity of the LiODFB–Pyr1,2O1TFSI/DMS electrolyte was 8.163 × 10−3 S cm−1. The Li/MCMB and Li/LiFePO4 cells containing this electrolyte had good cycle performance and a normal charge curve in the temperature range of −40 °C to 60 °C.Furthermore, this research identifies the relationship between ionic liquid/cosolvent electrolyte performances and the component structures. According to this work and previous studies, ionic liquid electrolyte systems consisting of both a ring-like component and a chain-like component in a specific ratio can offer good overall battery performance – measured in terms of safety, ionic conductivity and compatibility with cathode and anode materials. The ring-chain synergy that takes advantage of the availability of the two structures might offer a useful starting point to design mixed electrolytes in lithium-ion batteries. In addition, electrolytes with both ring-like components and chain-like components might be developed for applications under particular conditions due to their outstanding electrochemical window and ionic conductivity, respectively. The insight into the ring-chain synergy will allow further anticipation of the ionic liquid/cosolvent electrolytes. It is noteworthy that only the chemical structures of the cosolvent and the cations of ionic liquid were taken into consideration here. In order to gain a better understanding of a broader range of systems and to achieve an even better performance, further study should explore functional modifications and the effect of anions, which might further improve the properties of the electrolyte systems.
Acknowledgements
The authors appreciate the support from the National Science Foundation of China (21373028), Major Achievements Transformation Project for Central University in Beijing, National Key Program for Basic Research of China (2015CB251100), and Beijing Science and Technology Project (D151100003015001).Notes and references
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS.
- Y. Shim and H. J. Kim, ACS Nano, 2010, 4, 2345–2355 CrossRef CAS PubMed.
- K. Byoungwoo and C. Gerbrand, Nature, 2009, 458, 190–193 CrossRef PubMed.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- H. Li, Y. Wang, H. Na, H. Liu and H. Zhou, J. Am. Chem. Soc., 2009, 131, 15098–15099 CrossRef CAS PubMed.
- J. Y. Luo, W. J. Cui, P. He and Y. Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- H. S. Jeong, J. H. Kim and S. Y. Lee, J. Mater. Chem., 2010, 20, 9180–9186 RSC.
- M. Hirayama, H. Ido, K. Kim, W. Cho, K. Tamura and J. Mizuki, J. Am. Chem. Soc., 2010, 132, 15268–15276 CrossRef CAS PubMed.
- B. Schffner, F. Schffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- A. Chagnes, M. Diaw, B. Carré, P. Willmann and D. Lemordant, J. Power Sources, 2005, 145, 82–88 CrossRef CAS PubMed.
- Q. Du, X. Fu, S. Liu and L. Niu, Polym. Int., 2012, 61, 222–227 CrossRef CAS PubMed.
- N. Madria, T. A. Arunkumar, N. G. Nair, A. Vadapalli, Y.-W. Huang and S. C. Jones, J. Power Sources, 2013, 234, 277–284 CrossRef CAS PubMed.
- S. Ferrari, E. Quartarone, C. Tomasi, D. Ravelli, S. Protti, M. Fagnoni and P. Mustarelli, J. Power Sources, 2013, 235, 142–147 CrossRef CAS PubMed.
- I. Quinzeni, S. Ferrari, E. Quartarone, C. Tomasi, M. Fagnoni and P. Mustarelli, J. Power Sources, 2013, 237, 204–209 CrossRef CAS PubMed.
- L. Lombardo, S. Brutti, M. A. Navarra, S. Panero and P. Reale, J. Power Sources, 2013, 227, 8–14 CrossRef CAS PubMed.
- F. Wu, G. Q. Tan, R. J. Chen, L. Li, J. Xiang and Y. L. Zheng, Adv. Mater., 2011, 23, 5081–5085 CrossRef CAS PubMed.
- J. Xiang, F. Wu, R. Chen, L. Li and H. Yu, J. Power Sources, 2013, 233, 115–120 CrossRef CAS PubMed.
- I. Stepniak, E. Andrzejewska, A. Dembna and M. Galinski, Electrochim. Acta, 2014, 121, 27–33 CrossRef CAS PubMed.
- K. Kim, Y. H. Cho and H. C. Shin, J. Power Sources, 2013, 225, 113–118 CrossRef CAS PubMed.
- X. Li, Z. Yin, X. Li and C. Wang, Ionics, 2014, 20, 795–801 CrossRef CAS.
- M. L. P. Le, L. Cointeaux, P. Strobel, J. C. Lepretre, P. Judeinstein and F. Alloin, J. Phys. Chem. C, 2012, 116, 7712–7718 CAS.
- H. F. Xiang, B. Yin, H. Wang, H. W. Lin, X. W. Ge, S. Xie and C. H. Chen, Electrochim. Acta, 2010, 55, 5204–5209 CrossRef CAS PubMed.
- Z. Chen, T. Xue and J. M. Lee, RSC Adv., 2012, 2, 10564–10574 RSC.
- S. Fang, Z. Zhang, Y. Jin, L. Yang, S. Hirano and K. Tachibana, J. Power Sources, 2011, 196, 5637–5644 CrossRef CAS PubMed.
- S. Fang, Y. Tang, X. Tai, L. Yang, K. Tachibana and K. Kamijima, J. Power Sources, 2011, 196, 1433–1441 CrossRef CAS PubMed.
- Y. Jin, S. Fang, L. Yang, S. Hiranob and K. Tachibana, J. Power Sources, 2011, 196, 10658–10666 CrossRef CAS PubMed.
- R. Chen, F. Wu, L. Li, Y. Guan, X. Qiu and S. Chen, J. Power Sources, 2007, 172, 395–403 CrossRef CAS PubMed.
- P. Ping, Q. Wang, D. Kong, C. Zhang, J. Sun and C. Chen, J. Electroanal. Chem., 2014, 731, 119–127 CrossRef CAS PubMed.
- R. Chen, L. Zhu, F. Wu, L. Li, R. Zhang and S. Chen, J. Power Sources, 2014, 245, 730–738 CrossRef CAS PubMed.
- E. G. Leggesse and J. C. Jiang, J. Phys. Chem. A, 2012, 116, 11025–11033 CrossRef CAS PubMed.
- S. Li, W. Zhao, X. Cui, Y. Zhao, B. Li and H. Zhang, Electrochim. Acta, 2013, 91, 282–292 CrossRef CAS PubMed.
- J. L. Allen, D. W. Mcowen, S. A. Delp, E. T. Fox, J. S. Dickmann and S.-D. Han, J. Power Sources, 2013, 237, 104–111 CrossRef CAS PubMed.
- J. Li, K. Xie, Y. Lai, Z. Zhang, F. Li and X. Hao, J. Power Sources, 2010, 195, 5344–5350 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS.
- J. Cruickshank, H. V. S. A. Hubbard, N. Boden and I. M. Ward, J. Power Sources, 1995, 36, 3779–3781 CAS.
- K. Hayamizu, Y. Aihara, H. Nakagawa, T. Nukuda and W. S. Price, J. Phys. Chem. B, 2004, 108, 19527–19532 CrossRef CAS.
- F. Wu, J. Xiang, L. Li, J. Chen, G. Tan and R. Chen, J. Power Sources, 2012, 202, 322–331 CrossRef CAS PubMed.
- J. Chen, S. Wang, L. Wang, K. Zhang, Z. Zhu and Z. Tao, Nano Lett., 2013, 13, 4404–4409 CrossRef PubMed.
- W. Weng, Z. Zhang, J. Lu and K. Amine, Chem. Commun., 2011, 47, 11969–11971 RSC.
- J. Reiter and M. Nadherna, Electrochim. Acta, 2012, 71, 22–26 CrossRef CAS PubMed.
- I. A. Profatilova, N. S. Choi, S. W. Roh and S. S. Kim, J. Power Sources, 2009, 192, 636–643 CrossRef CAS PubMed.
- A. Lewandowski and M. A. Świderska, J. Power Sources, 2009, 194, 502–507 CrossRef CAS PubMed.
- M. Xua, L. Zhou, L. Hao, L. Xing, W. Li and B. L. Lucht, J. Power Sources, 2011, 196, 6794–6801 CrossRef PubMed.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. P. Abraham, J. Phys. Chem. C, 2013, 117, 23750–23756 CAS.
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2011, 4, 805–817 CAS.
- S. S. Zhang, J. Power Sources, 2007, 163, 713–718 CrossRef CAS PubMed.
- N. Wongittharom, T. C. Lee, C. H. Hsu, G. T. K. Fey, K. P. Huang and J. K. Chang, J. Power Sources, 2013, 240, 676–682 CrossRef CAS PubMed.
- J. Li, S. Jeong, R. Kloepsch, M. Winter and S. Passerini, J. Power Sources, 2013, 239, 490–495 CrossRef CAS PubMed.
- M. Diaw, A. Chagnes, B. Carré, P. Willmann and D. Lemordant, J. Power Sources, 2005, 146, 682–684 CrossRef CAS PubMed.
- S. Y. Lee, H. H. Yong, Y. J. Lee, S. K. Kim and S. Ahn, J. Phys. Chem. B, 2005, 109, 13663–13667 CrossRef CAS PubMed.
- M. Holzapfel, C. Jost and P. Novák, Chem. Commun., 2004, 2098–2099 RSC.
- V. Borgel, E. Markevich, D. Aurbach, G. Semrau and M. Schmidt, J. Power Sources, 2009, 189, 331–336 CrossRef CAS PubMed.
- J. Jin, H. H. Li, J. P. Wei, X. K. Bian, Z. Zhou and J. Yan, Electrochem. Commun., 2009, 11, 1500–1503 CrossRef CAS PubMed.
- B. S. Lalia, N. Yoshimoto, M. Egashira and M. Morita, J. Power Sources, 2010, 195, 7426–7431 CrossRef CAS PubMed.
- H. Nakagawa, Y. Fujino, S. Kozono, Y. Katayama, T. Nukuda and H. Sakaebe, J. Power Sources, 2007, 174, 1021–1026 CrossRef CAS PubMed.
- H. Sakaebe, H. Matsumoto and K. Tatsumi, J. Power Sources, 2005, 146, 693–697 CrossRef CAS PubMed.
- C. Arbizzani, G. Gabrielli and M. Mastragostino, J. Power Sources, 2011, 196, 4801–4805 CrossRef CAS PubMed.
- I. A. Profatilova, N. S. Choi, S. W. Roh and S. S. Kim, J. Power Sources, 2009, 192, 636–643 CrossRef CAS PubMed.
- A. Lewandowski, I. Acznik and M. A. Swiderska, J. Appl. Electrochem., 2010, 40, 1619–1624 CrossRef CAS.
- H. Sakaebe and H. Matsumoto, Electrochem. Commun., 2003, 5, 594–598 CrossRef CAS.
- A. Lewandowski and M. A. Swiderska, J. Power Sources, 2007, 171, 938–943 CrossRef CAS PubMed.
- S. Ferrari, E. Quartarone, P. Mustarelli, A. Magistris, S. Protti, S. Lazzaroni, M. Fagnoni and A. Albini, J. Power Sources, 2009, 194, 45–50 CrossRef CAS PubMed.
- G. B. Appetecchi, M. Montanino, M. Carewska, M. Moreno, F. Alessandrini and S. Passerini, Electrochim. Acta, 2011, 56, 1300–1307 CrossRef CAS PubMed.
- G. B. Appetecchi, M. Montanino, A. Balducci, S. F. Lux, M. Winter and S. Passerini, J. Power Sources, 2009, 194, 599–605 CrossRef PubMed.
- S. F. Lux, M. Schmuck, G. B. Appetecchu, S. Passerini, M. Winter and A. Balducci, J. Power Sources, 2009, 192, 606–611 CrossRef CAS PubMed.
- M. Egashira, M. Nakagawa, I. Watanabe, S. Okada and J. Yamaki, J. Power Sources, 2005, 146, 685–688 CrossRef CAS PubMed.
- H. Zheng, K. Jiang, T. Abe and Z. Ogumi, Carbon, 2006, 44, 203–210 CrossRef CAS PubMed.
- H. Zheng, B. Li, Y. Fu, T. Abe and Z. Ogumi, Electrochim. Acta, 2006, 52, 1556–1562 CrossRef CAS PubMed.
- T. Sato, T. Maruo, S. Marukane and K. Takagi, J. Power Sources, 2004, 138, 253–261 CrossRef CAS PubMed.
- S. Seki, Y. Ohno, H. Miyashiro, Y. Kobayashi, A. Usami, Y. Mita, N. Terada, K. Hayamizu, S. Tsuzuki and M. Watanabe, J. Electrochem. Soc., 2008, 155, A421–A427 CrossRef CAS PubMed.
- K. Tsunashima, F. Yonekawa and M. Sugiya, Electrochem. Solid-State Lett., 2009, 12, A54–A57 CrossRef CAS PubMed.
- M. Taggougui, M. Diaw, B. Carré, P. Willmann and D. Lemordant, Electrochim. Acta, 2008, 53, 5496–5502 CrossRef CAS PubMed.
- Z. B. Zhou, H. Matsumoto and K. Tatsumi, Chem.–Eur. J., 2006, 8, 2196–2212 CrossRef PubMed.
- K. R. Harris, T. Makino and M. Kanakubo, Phys. Chem. Chem. Phys., 2014, 16, 9161–9170 RSC.
- N. Terasawa and K. Asaka, Sens. Actuators, B, 2014, 193, 851–856 CrossRef CAS PubMed.
- S. Fang, Y. Jin, L. Yang, S. Hirano, K. Tachibana and S. Katayama, J. Power Sources, 2014, 254, 137–147 CrossRef PubMed.
- M. Egashira, S. Okada, J. Yamaki, D. A. Dri, F. Bonadies and B. Scrosati, J. Power Sources, 2004, 138, 240–244 CrossRef CAS PubMed.
- M. Ishikawa, T. Sugimoto, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 162, 658–662 CrossRef CAS PubMed.
- G. Nagasubramanian and C. J. Orendorff, J. Power Sources, 2011, 196, 8604–8609 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. E. Gordon and G. N. S. Rao, J. Am. Chem. Soc., 1978, 100, 7445–7454 CrossRef CAS.
- Z. Gu and J. F. Brennecke, J. Chem. Eng. Data, 2002, 47(2), 339–345 CrossRef CAS.
- S. N. Baker, G. A. Baker, M. A. Kane and F. V. Bright, J. Phys. Chem. B, 2001, 105, 9663–9668 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, ATR-FTIR spectra and ionic conductivity with error bar of all the electrolyte systems. See DOI: 10.1039/c5sc02761f |
| This journal is © The Royal Society of Chemistry 2015 |