
Palladium-catalyzed carbonylative transformation of aryl chlorides and aryl tosylates
Xiao-Feng
Wu
*ab
aDepartment of Chemistry, Zhejiang Sci-Tech University, Xiasha Campus, Hangzhou 310018, People's Republic of China. E-mail: xiao-feng.wu@catalysis.de
bLeibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Strasse 29a, 18059 Rostock, Germany
First published on 25th August 2016
Abstract
Developments in the carbonylative transformations of aryl chlorides and aryl tosylates have been collected and discussed.
Introduction
Palladium-catalyzed coupling reactions have become a powerful tool for advanced chemical synthesis, and are of interest in both academic and industrial settings.1 In particular, the palladium-catalyzed activation of Ar–X (X = I, Br, Cl, OTf, OTs, etc.) bonds allow for the selective and specific formation of new C–C, C–N and C–O bonds after coupling with the corresponding nucleophiles. The value of these reactions has been recognized by the 2010 Nobel Prize in Chemistry, which was shared by Ei-ichi Negishi, Akira Suzuki and Richard F. Heck.2 The reaction mechanism, taking the palladium-catalyzed Suzuki reaction as an example, consists of oxidative addition, transmetalation, and finishes with reductive elimination (Scheme 1). In more detail, the oxidative addition step can be favoured by electron rich ligands, while bulky ligands can assist the reductive elimination step. With respect to the bond dissociation energy of Ar–X bonds, the accepted sequence is I > OTf > Br > Cl > OTs > OMs > OAc.1j Until now, various Ar–X compounds, from ArI to ArOAc, have been successfully applied as electrophiles in palladium-catalyzed cross-coupling reactions. | ||
| Scheme 1 General reaction mechanism for the Suzuki cross coupling reaction. | ||
The palladium-catalyzed carbonylation reaction was first reported by Richard F. Heck and co-workers in the 1970s.3 Since then, carbonylation has experienced impressive progress over the past few decades.4 From alkoxycarbonylation to aminocarbonylation, from carbonylative Stille coupling to carbonylative C–H activation, and from intermolecular carbonylation to intramolecular carbonylation, numerous methodologies have been developed (Scheme 2).
 | ||
| Scheme 2 Palladium-catalyzed carbonylation reactions. | ||
Mechanistically, palladium-catalyzed carbonylation includes oxidative addition, coordination and insertion of carbon monoxide, transmetalation and reductive elimination (Scheme 3). In carbonylative coupling reactions, the oxidative addition step needs an electron rich metal center, but the CO coordination and insertion requires an electron deficient metal center. The reductive elimination can be promoted by bulky ligands, while the CO coordination and insertion step demands that the ligand is not too bulky. Elevated temperature can favor oxidative addition, but it also favors the decarbonylation of the acylpalladium complex (the reverse reaction of CO insertion). The key to develop a carbonylative coupling reaction is to find the appropriate combination of reaction temperature, ligand and other parameters.
 | ||
| Scheme 3 Reaction mechanism for palladium-catalyzed carbonylation reactions. | ||
At this stage it is interesting to discuss the relationship between palladium-catalyzed coupling reactions and their corresponding carbonylative coupling reactions. In principle, the reactions that work under carbonylative coupling conditions can also afford a noncarbonylation product under CO-free conditions. However, we should not be restricted by the idea that noncarbonylative coupling must be developed before the corresponding carbonylative coupling reaction. For example, aminocarbonylation was reported by Heck and co-workers in the 1970s, while Buchwald–Hartwig amination was established between 1994 and the late 2000s.5
Aryl chlorides are important starting materials in palladium-catalyzed coupling reactions.6a Compared with the corresponding aryl iodides or aryl bromides, the advantages of aryl chlorides are obvious, such as their low cost, ease of preparation and stability, etc. This is also the case if we compare aryl tosylates or aryl mesylates with their corresponding aryl triflates, as aryl tosylates and mesylates are stable, abundant and simple to prepare.6b,c Although aryl chlorides, aryl mesylates and also aryl acetates have been studied and have succeeded in cross-coupling reactions, their corresponding carbonylative coupling transformations are still rarely reported. Until now, no general methodology has been developed for the carbonylative coupling of aryl chlorides. The possible explanations for this situation are: (1) CO coordinated to the metal centre decreases its electron density, which inhibits the oxidative addition step in some way; (2) the high temperature required by oxidative addition, or the strong base needed for transmetalation can destroy the formed arylpalladium or acylpalladium species; (3) in order to avoid high temperature induced decarbonylation, CO at high pressure is normally applied to drive the reaction, but this may result in the formation of a palladium carbon monoxide complex which is inactive for cross-coupling reactions (Scheme 4).6d–i
 | ||
| Scheme 4 Carbonylation and decarbonylation equilibrium. | ||
In this review we summarize contributions involving the palladium-catalyzed carbonylative coupling of aryl chlorides (ArOTs and ArOMs are also included) and discuss their transformations, with the wish to encourage further development of this field and overcome remaining challenges.
Literature discussion
As early as 1989, Osborn and co-workers reported the catalytic carbonylation of chlorobenzene and dichloromethane.7 By using [Pd(PCy3)2(dba)] as a catalyst in the presence of additional PCy3, chlorobenzene was catalytically carbonylated at 180 °C in toluene under 15 bar of CO using NEt3 as a base (Scheme 5). In their report, instead of PCy3, PiPr3 was used as effective ligand for the activation of DCM at 25 °C to also give the ketene, but not P(CH2Ph)3, PPh2Cy or PPh3. In the case of chlorobenzene, Pd0 complexes with PtBu3, PtBu2Ph, PPh2Cy and P(m-tol)3 are completely non-active. Here, it is also interesting to point out that chlorobenzene can react with [Pd(PCy3)2(dba)] to give a phenylpalladium complex at 60 °C, and a benzoylpalladium complex can be prepared from the phenylpalladium complex at room temperature under 30 bar of CO (the reverse transformation can be realized easily at 60 °C under argon). However, the catalytic alkoxycarbonylation of chlorobenzene must be carried out at 180 °C. | ||
| Scheme 5 [Pd(PCy3)2(dba)]-catalyzed alkoxycarbonylation of chlorobenzene. | ||
Meanwhile, the group of Milstein succeeded in their breakthrough work on the carbonylation of aryl chlorides with their dippp (1,3-bis(di-isopropylphosphino)propane) ligand.8 In their studies, aryl chlorides were transformed into esters, amides, acids and aldehydes in good yields under relatively mild conditions (Scheme 6). Under the best conditions, the reactivity of ligands decrease in the sequence dippp > dippb > dippe ⋙ dppp > dppe. In the case of the formylation of aryl chlorides, the reaction is specific to the dippp ligand. Their mechanistic study demonstrated the importance of chelate stability, ligand basicity, concentration of the active 14e species and the effect of the P–Pd–P angle on its reactivity.9 The reactivity toward the oxidative addition of chlorobenzene follows the trend Pd(dippp)2 > Pd(PiPr2nBu)3 ≫ Pd(dippe)2 ≫ Pd(dppp)2. This catalyst system was applied in the synthesis of polyamides from aromatic dichlorides, diamines and CO.10
 | ||
| Scheme 6 Pd(dippp)2-catalyzed carbonylation of aryl chlorides. | ||
In 2001, the group of Beller developed another efficient phosphine ligand for the carbonylative transformation of aryl chlorides.11 Under the assistance of the 1-[2-(dicyclohexylphosphanyl)ferrocenyl]ethyldicycloheyxlphosphine ligand, both activated and non-activated aryl chlorides were carbonylated under mild reaction conditions and gave the corresponding products in good yields (Scheme 7). Compared with previous methodologies, the advantage of this procedure is that the ligand used is air stable and commercially available. Later, the same group performed a comprehensive study on the alkoxycarbonylation of various N-heteroaryl chlorides.12 Studies involving the butoxycarbonylation of 2- and 3-chloropyridine revealed the importance of selecting both the right phosphine ligand and ligand concentration in order to obtain efficient conversion and selectivity. Amongst the different ligands tested, 1,4-bis(diphenylphosphino)butane (dppb) and 1,1′-bis(diphenylphosphino)ferrocene (dppf) led to the most efficient palladium catalyst systems for the conversion of 2- and 4-chloropyridines and similar heteroaryl chlorides. The best catalytic systems for the alkoxycarbonylation of less activated substrates, such as 3-chloropyridines, were found to be those containing 1,4-bis(dicyclohexylphosphino)butane. Good to excellent yields of a number of N-heterocyclic carboxylic acid esters were realized by applying the appropriate ligand in the right concentration at low catalyst loadings (0.005–0.5 mol% Pd). For the first time, catalyst turnover numbers (TON) of up to 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 were obtained for the carbonylation of a (hetero)aryl chloride.
000 were obtained for the carbonylation of a (hetero)aryl chloride.
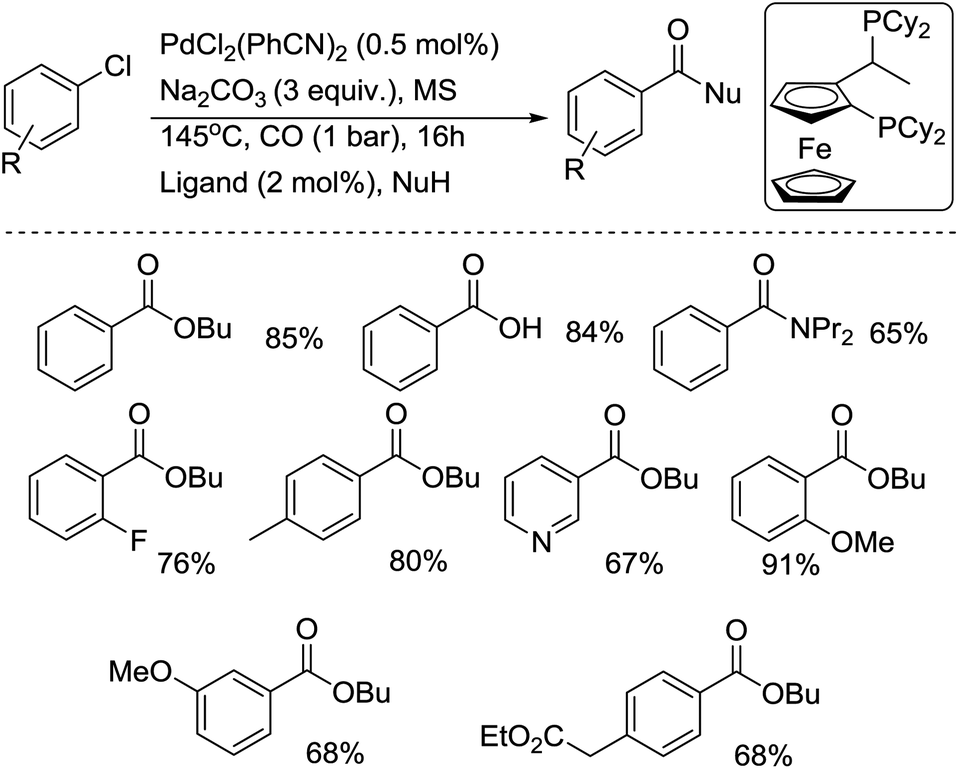 | ||
| Scheme 7 Pd–Josiphos-catalyzed carbonylation of aryl chlorides. | ||
Recently, another type of Josiphos ligand was discovered as an efficient ligand for the palladium-catalyted carbonylation of aryl sulfonates.13 The catalyst system is effective for the carbonylation of aryl p-fluorobenzenesulfonates and tosylates, with a general functional group tolerance under less severe reaction conditions. Both electron-rich and electron-poor aryl arenesulfonates were carbonylated and gave the corresponding esters in good to excellent yields (Scheme 8). This method provides an alternative route to aryl carboxylic acid derivatives from readily available and air-stable starting materials, and is therefore useful in industrial manufacturing processes. As the authors of this study indicated, the catalyst was also found to be effective for the carbonylation of aryl chlorides.
 | ||
| Scheme 8 Pd–Josiphos-catalyzed carbonylation of aryl arenesulfonates. | ||
In 2008, Buchwald and co-workers developed an efficient procedure for the carbonylation of aryl chlorides, aryl tosylates and aryl mesylates.14a,b Under their reaction conditions, carboxylic acid derivatives were prepared in good yields (Scheme 9). The advantages of this procedure are: (1) 1,3-bis(dicyclohexylphosphino)propane bis(tetrafluoroborate) as the ligand used is stable and easily available; (2) the reactions were carried out in a reaction tube under 1 bar of CO, avoiding the use of an autoclave. In this procedure, the aliphatic alcohols are better nucleophiles than phenol, and are suitable for the lower reaction temperatures of aryl tosylates. Recently, the catalyst system has been further studied and extended to aminocarbonylation with various amines (both aromatic amines and aliphatic amines).14d
 | ||
| Scheme 9 Pd(OAc)2–DCPP-catalyzed carbonylation of aryl chlorides and aryl tosylates. | ||
A catalyst system based on palladium-1,2-bis-(di-tert-butylphosphinomethyl)benzene (BDTBPMB) was reported by Cole-Hamilton and co-workers, which shows good activity for the methoxycarbonylation of strongly activated aryl chlorides, like methyl 4-chlorobenzoate or 4-chlorocyanobenzene (Scheme 10).15 In this system, the selective carbonylation of aromatic chlorides to carboxylic acid esters is catalyzed by Pd/BDTBPMB complexes in alcohol in the presence of base. For less activated aromatic rings such as that in 4-chloroacetophenone, selective carbonylation can only be achieved by using alcohols of low nucleophilicity, such as 2,2,2-trifluoroethanol. More nucleophilic alcohols give many side products arising from nucleophilic aromatic substitution, reductive dehalogenation and an unusual transformation of the methylketone into a methyl ester. Labelling studies show that this reaction formally occurs through the displacement of the methyl group by methoxide. Nitro and cyano groups on the ring also undergo severe side reactions.
 | ||
| Scheme 10 PdCl2–BDTBPMB-catalyzed carbonylation of activated aryl chlorides. | ||
In addition to the ligand-tuned carbonylative activation of aryl chlorides, several other catalyst systems have also been developed based on the use of nanoparticles, biphasic systems and Lewis acids.
Pd/C was found to act as an effective catalyst for the carbonylation of aryl chlorides at 200 °C.16 Both activated and non-activated aryl chlorides were transformed into the corresponding methyl esters [CO (3 bar), AcONa, MeOH], and the addition of K2Cr2O7 was found to enhance the catalytic activity. The likely role of K2Cr2O7 is to reoxidize large particles of metallic palladium into a highly dispersed palladium(II) species which can be reduced again under CO into an active zero-valent palladium complex.
Alper and Grushin found that bis(tricyclohexylphosphine)palladium dichloride is an active catalyst for the carbonylation of chloroarenes to carboxylic acids under biphasic conditions.17 The reactions were carried out in an aqueous solution of KOH under 1 bar of CO at 100 °C, and both activated and non-activated aryl chlorides were carbonylated in good yields. In 1997, a highly selective procedure for the hydrocarbonylation of chlorobenzene was developed, using a combination of PdCl2(PCy3)2–K2CO3 aqueous solution and triethylamine [CO (5 bar), 180 °C].18 Using this procedure, 4-chlorobenzophenone and/or benzoyl chloride was synthesized from chlorobenzene using PdCl2 in the presence of a Lewis acid (AlCl3 or GaCl3), under 10 bar of CO at 120 °C.19 In the absence of PdCl2, 4-chlorobenzaldehyde was produced as the product of the Gattermann–Koch reaction which is promoted by AlCl3.20 Activated aryl chlorides can also give the corresponding carbonylated products in low to moderate yields in ionic liquids using Pd-benzothiazole carbine as a catalyst under CO at atmospheric pressure at 140 °C.21
The group of Perry developed a palladium-catalyzed aminocarbonylation of activated aryl chlorides under CO at low pressure and in the presence of iodide salt (KI, NaI).22 Amides were prepared in moderate to excellent yields from activated chlorides, while electron-rich aryl chlorides were not effectively amidated, even after adding iodide (Scheme 11).
 | ||
| Scheme 11 PdCl2(PPh3)2–DPPE-catalyzed aminocarbonylation of activated aryl chlorides. | ||
Aside from electron-withdrawing activated aryl chlorides, heteroaryl chlorides are another family of activated chlorides that also exhibit interesting biological activity. In this context, there are many procedures that have been developed for the carbonylative transformation of heteroaryl chlorides. In general, the reaction conditions for these methodologies are much milder.
In 1984, Head and co-workers reported the palladium-catalyzed carbonylative synthesis of heterocyclic esters from their corresponding halides.23 The main substrates were heterocyclic bromides, and one example of a heterocyclic chloride was also described. Using PdCl2(PPh3)2 as a catalyst under 7.9 bar of CO at 100 °C in ethanol with NEt3 as a base, 5-chloroethylnicotinate was produced from 3,5-dichloropyridine.
The palladium-catalyzed carbonylation of chloroquinolines and chloropyridines was also developed.24 3-Subtituted 2-chloroquinolines were carbonylated with MeOH under the assistance of Pd(OAc)2 (2 mol%), DPPP (4 mol%), NaOAc (1 equiv.) and CO (100 bar), in DMF at 140 °C for 2 days. Esters were isolated in good to excellent yields. In comparison, 4,7-dichloroquinone was selectively carbonylated at the 4-position using 2 mol% of PdCl2(PPh3)2 and 10 mol% of PPh3 in methanol using NEt3 as a base, under 50 bar of CO at 150 °C for one hour. The corresponding aldehyde, ester and amide were produced in good yields with excellent selectivity. Under the same reaction conditions, 2,6-dichloropyridine was converted into dimethyl diester in 90% yield, while 2,3- and 2,5-dichloropyridines mainly afforded monoesters at the 2-position. 2,7-Dichloro-1,8-naphthyridine and 2,9-dichloro-1,10-phenanthroline were transformed into dibutyl 1,8-naphthyridine-2,7-dicarboxylate and dibutyl 1,10-phenanthroline-2,9-dicarboxylate in 40–60% yield.25 The reactions were carried out using PdCl2(PPh3)2 (2 mol%) as a catalyst, under 1 bar of CO at 120 °C.
The group of Takeuchi performed a comprehensive study on the palladium-catalyzed carbonylation of N-heteroaryl chlorides.26 Carbonylation of chloropyrazines in methanol or amines gave the corresponding esters or amides in good to excellent yields (Scheme 12). In the methoxylcarbonylation of 2-chloropyrazine, PPh3 and DPPE afforded esters in excellent yields, while the use of tri-n-butylphosphine or tricyclohexylphosphine was not effective. Using 1 mol% of Pd(dba)2 and 2 mol% of PPh3, at 120 °C under 39.24 bar of CO, chloropyrazines, including N-oxides, were easily carbonylated. The reactivity of the substrates increases in the following order: 2-chloropyrazine > 2-chloropyridine ≫ 3-chloropyridine. In the aminocarbonylation of 2-chloropyrizine, the use of tri-n-butylphosphine was effective. Using a high pressure of carbon monoxide decreased the selectivity towards the amides.
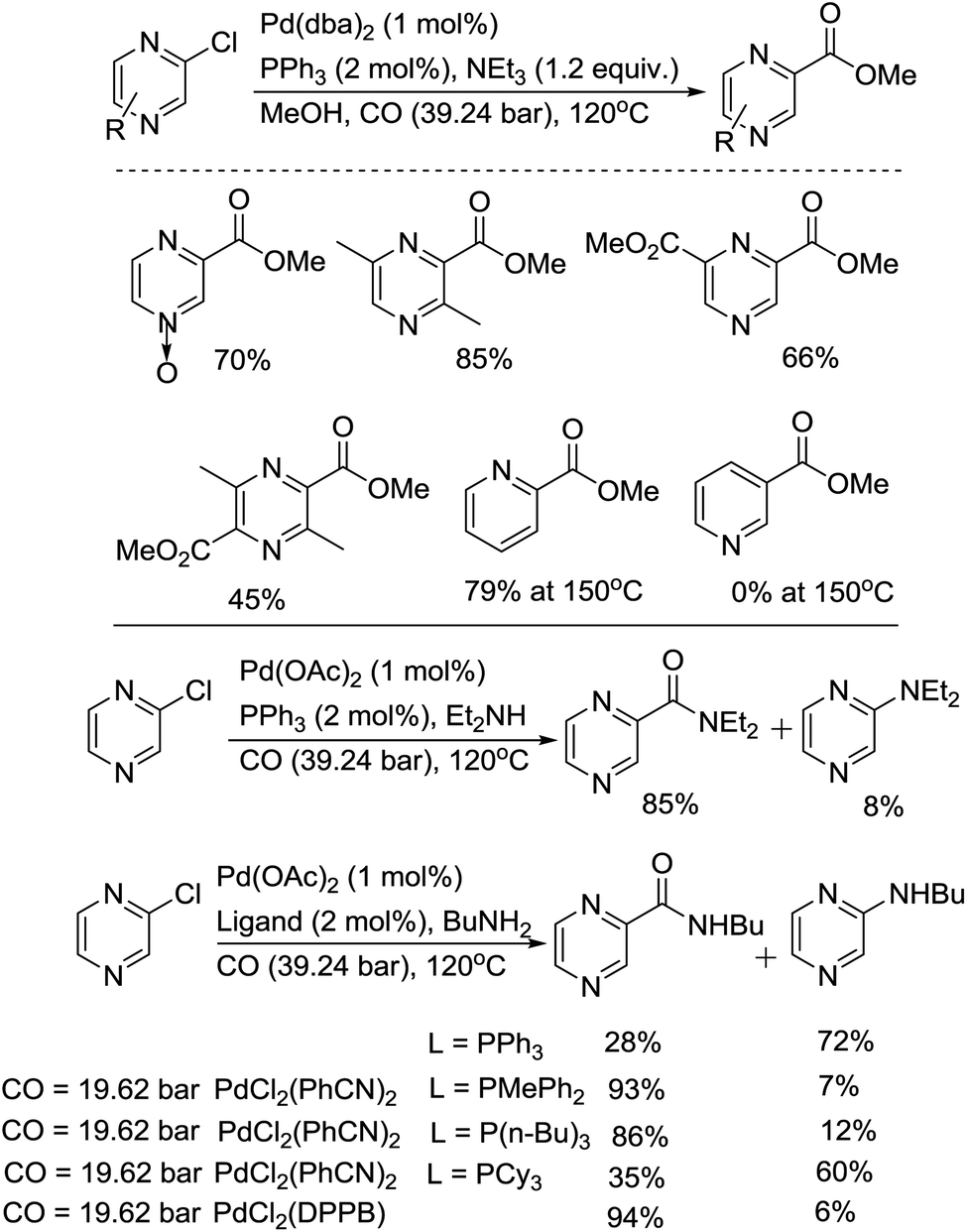 | ||
| Scheme 12 Pd-catalyzed carbonylation of 2-chloropyrizines. | ||
Bessard and co-workers developed a procedure for the selective mono- or di-carbonylation of 2,3-dichloropyridines (Scheme 13).27 In this procedure, the choice of base and ligand play a crucial role in the carbonylation. When Na2CO3 was used as a base, the major reaction pathway was substitution at the 2-position, giving a methoxy-substituted product. Recently, some other catalytic systems have been established for the carbonylation of 2-chloropyridines.28 DPPP and Binap were used as ligands for the alkoxycarbonylation of 2-chloropyridines, and the corresponding esters were prepared in good to excellent yields.
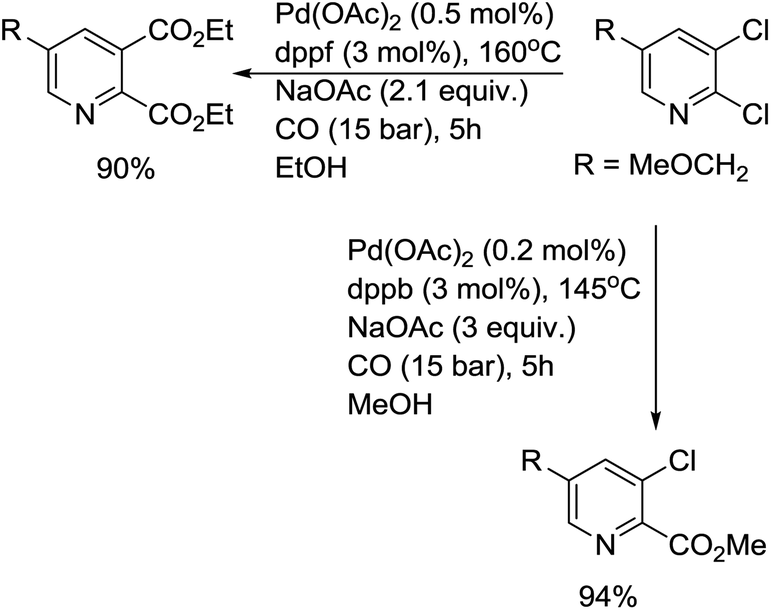 | ||
| Scheme 13 Pd-catalyzed carbonylation of 2,3-dichloropyridine. | ||
Moreover, several other CO sources were also discovered and applied in the carbonylation of aryl chlorides.
Jenner and Taleb developed a palladium-catalyzed carbonylation of aryl chlorides using methyl formate as a CO source, and benzoic acids were produced in good yields under aqueous conditions at 160 °C.29 PdCl2(PCy3)2 was found to be the only efficient catalyst. Ru3(CO)12 and ammonium formate improved the yield and selectivity.
More recently, an improved protocol for the palladium-catalyzed alkoxycarbonylation of aryl chlorides with alkyl formates was developed by Beller and co-workers.30 In the presence of palladium(II) acetate/n-butylbis(1-adamantyl)phosphine, and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base, non-activated chloroarenes could be conveniently carbonylated in good yields for the first time (Scheme 14). In this report, it was shown that the presented catalyst system does not require the presence of ruthenium co-catalysts.
 | ||
| Scheme 14 Pd(OAc)2/BuPAd2-catalyzed carbonylation of aryl chlorides. | ||
The group of Larhed developed the microwave-promoted aminocarbonylation of aryl chlorides using a palladacycle and Mo(CO)6 as a solid carbon monoxide source.31 This procedure offers the rapid transformation of aryl chlorides into a variety of benzamides (Scheme 15). Noteworthy features of this microwave method include the use of commercially available [(t-Bu)3PH]BF4 to activate the strong Ar–Cl bond, impressive results using sluggish aniline and tert-butylamine reactants, tolerance of air, short reaction times, and employment of Mo(CO)6 as a solid carbon monoxide source.
 | ||
| Scheme 15 Palladacycle-catalyzed carbonylation of aryl chlorides. | ||
Interestingly, Jun and co-workers recently reported the Pd/C-catalyzed alkoxycarbonlyation of aryl chlorides.32 Here, primary alcohols were applied as a carbonyl source and coupling partner. In addition, aryl chlorides were applied as the oxidants. Using this procedure, esters, arenes and alkanes can be obtained with NaF as the additive at 150 °C under solvent-free conditions.
Conclusions
The achievements in the palladium-catalyzed carbonylation of (hetero)aryl chlorides have been summarized. The developments involving the carbonylative transformation of aryl tosylates and aryl mesylates have also been included. It has been shown that ligands play a crucial role in the carbonylative activation of the C–Cl bond. They make the metal centre more electron rich and also stabilize the palladium catalyst at high temperature. The successful application of a palladacycle, due to its high stability, has been reported as well.Compared with aryl bromides, aryl chlorides are much less explored in carbonylation reactions. In the future, efforts should be made in this area to decrease the reaction temperature and CO pressure, and increase the reaction efficiency, etc. The development of new ligands or a more active palladacycle will certainly be good directions to take.
Acknowledgements
The author thanks the financial support from NSFC (21472174) and Zhejiang Natural Science Fund for Distinguished Young Scholars (LR16B020002). X.-F. Wu appreciates the general support from Professor Matthias Beller in LIKAT.Notes and references
- For selected reviews, see:
(a) F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2008, 64, 3047–3101 CrossRef CAS
; (b) P. Rollet, W. Kleist, V. Dufaud and L. Djakovitch, J. Mol. Catal. A: Chem., 2005, 241, 39–51 CrossRef CAS
- http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2010/ .
-
(a) A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326 CrossRef CAS
- For selected reviews on palladium-catalyzed carbonylation, see:
(a) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC
-
(a) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS
-
(a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS
- M. Huser, M.-T. Youinou and J. A. Osborn, Angew. Chem., Int. Ed., 1989, 28, 1386–1388 CrossRef
-
(a) Y. Ben-David, M. Portnoy and D. Milstein, J. Am. Chem. Soc., 1989, 111, 8742–8744 CrossRef CAS
-
(a) M. Portnoy and D. Milstein, Organometallics, 1993, 12, 1655–1664 CrossRef CAS
- J. S. Kim and A. Sen, J. Mol. Catal. A: Chem., 1999, 143, 197–201 CrossRef CAS
-
(a) W. Mägerlein, A. F. Indolese and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 2856–2859 CrossRef
- M. Beller, W. Mägerlein, A. F. Indolese and C. Fischer, Synthesis, 2001, 1098–1109 CAS
- C. Cai, N. R. Rivera, J. Balsells, R. R. Sidler, J. C. McWilliams, C. S. Shultz and Y. Sun, Org. Lett., 2006, 8, 5161–5164 CrossRef CAS PubMed
-
(a) D. A. Watson, X. Fan and S. L. Buchwald, J. Org. Chem., 2008, 73, 7096–7101 CrossRef CAS PubMed
- C. Jimenez-Rodriguez, G. R. Eastham and D. J. Cole-Hamilton, Dalton Trans., 2008, 1826–1830 Search PubMed
- V. Dufaud, J. Thivolle-Cazat and J.-M. Basset, J. Chem. Soc., Chem. Commun., 1990, 426–427 RSC
- V. V. Grushin and H. Alper, J. Chem. Soc., Chem. Commun., 1992, 611–612 RSC
- T. Miyawaki, K. Nomura, M. Hazama and G. Suzukamo, J. Mol. Catal. A: Chem., 1997, 120, L9–L11 CrossRef CAS
- Y. G. Noskov and E. S. Petrov, React. Kinet. Catal. Lett., 1998, 64, 359–363 CrossRef CAS
- L. Toniolo and M. Graziani, J. Organomet. Chem., 1980, 194, 221–228 CrossRef CAS
- V. Calò, P. Giannoccaro, A. Nacci and A. Monopoli, J. Organomet. Chem., 2002, 645, 152–157 CrossRef
- R. J. Perry and B. D. Wilson, J. Org. Chem., 1996, 61, 7482–7485 CrossRef CAS
- R. A. Head and A. Ibbotson, Tetrahedron Lett., 1984, 25, 5939–5942 CrossRef CAS
-
(a) M. A. Ciufolini, J. W. Michell and F. Roschangar, Tetrahedron Lett., 1996, 37, 8281–8284 CrossRef CAS
-
(a) A. El-ghayoury and R. Ziessel, Tetrahedron Lett., 1998, 39, 4473–4476 CrossRef CAS
- R. Takeuchi, K. Suzuki and N. Sato, J. Mol. Catal., 1991, 66, 277–288 CrossRef CAS
-
(a) Y. Bessard and J. P. Roduit, Tetrahedron, 1999, 55, 393–404 CrossRef CAS
-
(a) H.-U. Blaser, M. Diggelmann, H. Meier, F. Naud, E. Scheppach, A. Schnyder and M. Studer, J. Org. Chem., 2003, 68, 3725–3728 CrossRef CAS PubMed
- G. Jenner and A. B. Taleb, J. Organomet. Chem., 1994, 470, 257–261 CrossRef CAS
- T. Schareina, A. Zapf, A. Cotté, M. Gotta and M. Beller, Adv. Synth. Catal., 2010, 352, 1205–1209 CrossRef CAS
-
(a) O. Lagerlund and M. Larhed, J. Comb. Chem., 2006, 8, 4–6 CrossRef CAS PubMed
- H. S. Park, D. S. Kim and C. H. Jun, ACS Catal., 2015, 5, 397–401 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |