
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the anticancer activity of platinum(IV) prodrugs using a dual-targeting strategy with a dichloroacetate axial ligand†
Fengfan Liua,
Xiaomei Donga,
Qiwen Shia,
Jianli Chena and
Weike Su *ab
*ab
aNational Engineering Research Center for Process Decelopment of Active Pharmaceutial Ingredients, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, P. R. China. E-mail: pharmlab@zjut.edu.cn
bCollege of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou, P. R. China
First published on 17th July 2019
Abstract
Four novel platinum(IV) complexes, characteristic of DCA/TFA and with chloride ions as axial ligands, were designed and synthesized. This type of platinum(IV) complexes 1a–2b exhibited significant cytotoxic activity, and the cytotoxicity of 1b was the greatest among these four complexes, which was 20.61 fold and 7.65 fold higher than that of cisplatin against HepG-2 and NCI-H460 cancer cells, respectively. The result from the apoptosis assay of 1b was consistent with the result from the cytotoxicity assay. In addition, complexes 1a and 1b induced cell cycle arrest at the S phase on HepG-2 cells. Taken together, our data showed that Pt(IV) complex 1b released the corresponding Pt(II) complex and DCA, and induced apoptosis as well as disruption of the mitochondrial membrane potential, establishing Pt(IV) complex 1b as a potential dual-targeting anticancer agent.
1. Introduction
The United States Food and Drug Administration (FDA) has approved cisplatin, carboplatin, and oxaliplatin for the treatment of cancer.1,2 According to previous reports, platinum drugs were administered to 50–70% of cancer patients, either as single drugs or in combination with other drugs.3 However, side effects and drug resistance have greatly limited the therapeutic application of platinum drugs.3–6 Therefore, a variety of functional platinum complexes with different strategies have been studied to improve cancer therapy, including receptor targeting of platinum complexes, nano-delivery of platinum complexes and dual-threat platinum prodrugs.7–12 In addition, the FDA has approved an immunotherapy for patients with a specific type of lung cancer in 2018, which needs adjuvant treatment with platinum-containing chemotherapy.13,14 Therefore, through a process of continuous design for improvement, these drugs will still play an important role in the future.15–18 In our previous research, complexes A and B exhibited similar activity compared with cisplatin and oxaliplatin, but their toxicity against normal cells was lower than that of cisplatin and oxaliplatin.19,20 The results indicated that both of them were promising anticancer lead compounds for further study (Fig. 1).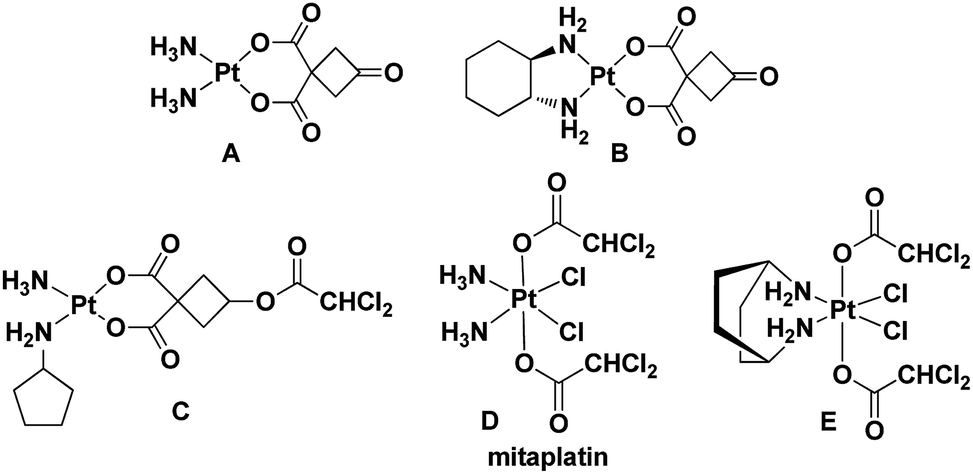 | ||
| Fig. 1 Leading compounds (A and B) and known platinum complexes of ligands with dichloroacetate (C–E). | ||
Dichloroacetate (DCA), an enzyme pyruvate dehydrogenase kinase inhibitor, can reverse the Warburg effect by improving the way pyruvate enters mitochondria.21 It can reduce tumor cell proliferation and promote cancer cell death via inducing apoptosis and mitochondrial dysfunction. In fact, Pt(II)/(IV) complexes could effectively increase the anticancer activity by introducing one or two DCA into the axial ligands (Fig. 1).22–24 On the other hand, taking the trifluoroacetate (TFA) group as the ligand can increase the lipophilicity and possess good ability to be dissociated.25,26 Therefore, we designed and synthesized Pt(IV) complexes 2a and 2b with TFA as the axial ligand.
Since platinum(IV) prodrugs have kinetic inertia and contain functional groups in the axial position, their characteristics include minimizing unwanted side reactions with biomolecules prior to activation of Pt(IV) prodrugs, enhancing the cytotoxicity of platinum complexes, regulating the reduction rate and altering the lipophilicity of resulting complexes. The bioactive molecule can ultimately combine with platinum(II) complex dissociated from the platinum(IV) prodrug under the reductive condition to improve anti-tumor effects. Until now, cisplatin-based and oxaliplatin-based Pt(IV) complexes have been used to act as leading compounds. Herein, we designed and synthesized four novel Pt(IV) complexes 1a–2b with a new structure to reduce side effects prior to activation of Pt(IV) prodrugs and target nuclear DNA and mitochondria (Scheme 1). This work was motivated idea from Mitaplatin analogue, which firstly displayed a dual-targeting strategy with dichloroacetate axial ligand.22
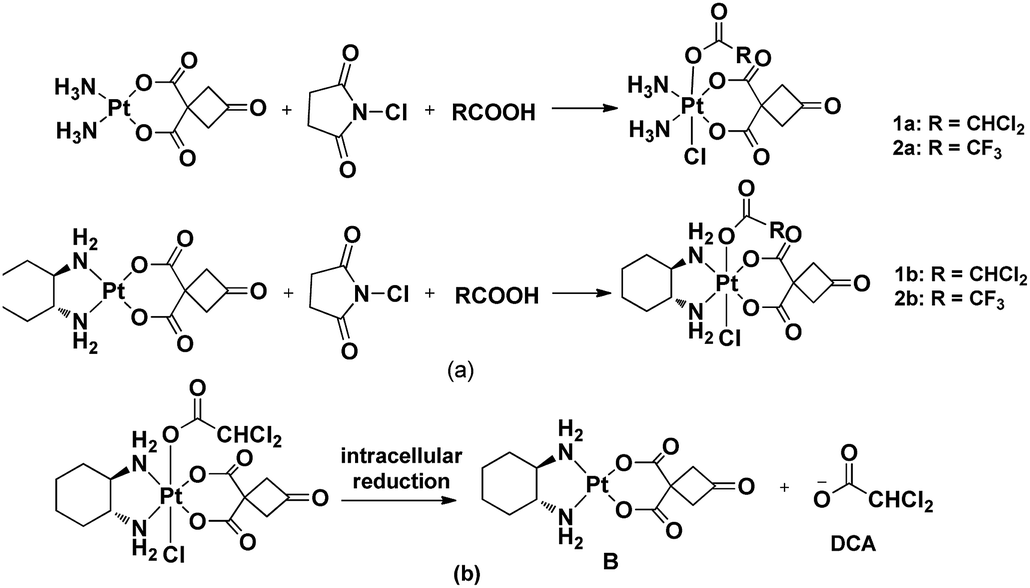 | ||
| Scheme 1 (a) Synthetic route of target complexes 1a–2b; (b) proposed mechanism of activation of 1b. | ||
2. Results and discussion
2.1 Chemistry
Notably, Pt(IV) complexes 1a–2b were synthesized by the oxidative chlorination of the corresponding platinum(II) complexes A and B with N-chlorosuccinimide in the acetone solution of dichloroacetic acid or trifluoroacetic acid, respectively. This procedure obtained Pt(IV) complexes adopting a one-pot synthesis (Scheme 1). All complexes were characterized by using IR, 1H NMR, 13C NMR and ESI-MS and elemental analysis. N–H stretching vibrations were found in the IR spectra of the platinum complexes ranging from 3061–3574 cm−1. The coordination of the amino group to platinum(II) caused the difference between the vibrations. The presence of stretches between 2936 and 2939 cm−1 confirmed the presence of C–H bonds. In the 1H NMR spectra, the cyclobutyl proton of complexes A and B appeared at a single peak in 3.71 ppm. These signals were completely different from those of the corresponding platinum(IV) complexes 1a–2b appearing the split by the introduction ligands into the axial position. The CHCl2 signal of the complexes 1a and 1b appeared at a single peak in 6.52 ppm and 6.49 ppm, respectively. In the 13C NMR spectrum, the peaks at δ 67.1, 67.7 assignable to CHCl2 carbon, at δ(62.4, 58.6), δ(62.6, 58.3) for C–N carbons (1b and 2b), respectively. The ESI-MS showed [M + Na]+ peaks that were in agreement with the proposed molecular formulas of the metal complexes.2.2 Stability of complexes 1a–2b in PBS
The stability of complexes 1a and 1b in a solution of deuterated aqueous and phosphate buffered solution (PBS 7.87 mM, pH 7.4 in DMSO-d6/D2O ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 (v/v)) at 37 °C was investigated by 1H NMR technique at different time. As shown in Fig. 2, the peak of coordinated DCA in complex 1a located at 6.18 ppm. After 50 minutes incubation under the physiological condition, we observed the decay of the CHCl2 signal at 6.18 ppm (marked with
:90 (v/v)) at 37 °C was investigated by 1H NMR technique at different time. As shown in Fig. 2, the peak of coordinated DCA in complex 1a located at 6.18 ppm. After 50 minutes incubation under the physiological condition, we observed the decay of the CHCl2 signal at 6.18 ppm (marked with  in Fig. 2) and the concomitant increase of a signal resonating at 5.86 ppm (
in Fig. 2) and the concomitant increase of a signal resonating at 5.86 ppm ( indicates the peak of free DCA). The half-life of 1a was approximately 50 minutes, which had a value quite similar to that reported for kiteplatin (60 min).20 Meanwhile, 1b (60 min) has the same half-life as 1a and kiteplatin in the same experimental conditions. As shown in Fig. S32 and S33,† the half-lives of complexes 1a and 1b were similar to those obtained by 1H NMR. On the other hand, the characteristic peaks of complexes 2a and 2b are not so obvious in 1H NMR. The stability of 2a and 2b in the solution of MeOH/PBS (v/v = 1:9) was examined by HPLC at different times. As shown in Fig. S34 and S35† (ESI†), the half-life of 2a and 2b was approximately 50–60 minutes under the physiological condition, which were lower than that found for ct-[Pt(NH3)2(TFA)2(CBCDA)] (5) with TFA as axial ligands (PBS, 37 °C, pH 7.0, t1/2 = 142 min).27 Furthermore, the complexes 1a–2b were stable in methanol for 48 hours (S36–S39†).
indicates the peak of free DCA). The half-life of 1a was approximately 50 minutes, which had a value quite similar to that reported for kiteplatin (60 min).20 Meanwhile, 1b (60 min) has the same half-life as 1a and kiteplatin in the same experimental conditions. As shown in Fig. S32 and S33,† the half-lives of complexes 1a and 1b were similar to those obtained by 1H NMR. On the other hand, the characteristic peaks of complexes 2a and 2b are not so obvious in 1H NMR. The stability of 2a and 2b in the solution of MeOH/PBS (v/v = 1:9) was examined by HPLC at different times. As shown in Fig. S34 and S35† (ESI†), the half-life of 2a and 2b was approximately 50–60 minutes under the physiological condition, which were lower than that found for ct-[Pt(NH3)2(TFA)2(CBCDA)] (5) with TFA as axial ligands (PBS, 37 °C, pH 7.0, t1/2 = 142 min).27 Furthermore, the complexes 1a–2b were stable in methanol for 48 hours (S36–S39†).
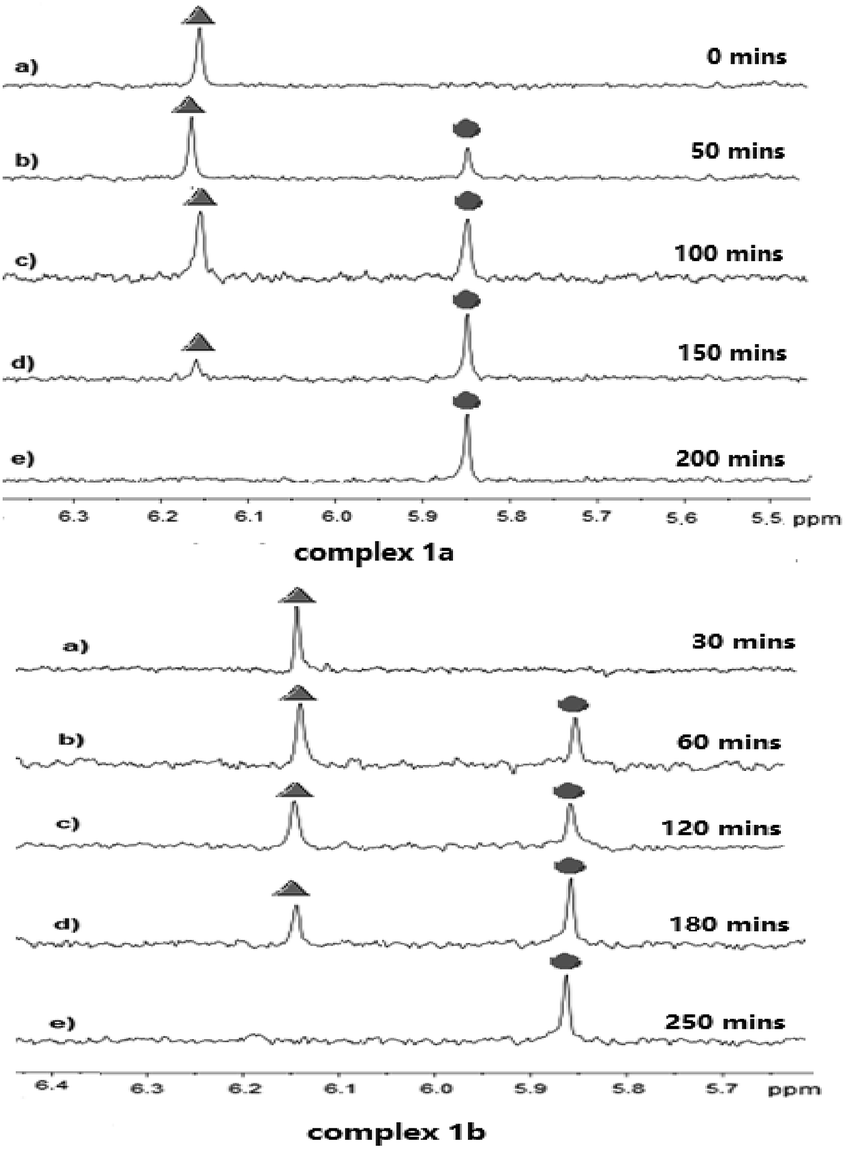 | ||
Fig. 2 Portion of the 1H NMR (400 MHz) spectra of complexes 1a and 1b in deuterated aqueous and phosphate buffered solution (PBS 7.87 mM, pH 7.4; at 37 °C; 10% v/v DMSO-d6).  indicates the peak of coordinated DCA in complex 1a and 1b. indicates the peak of coordinated DCA in complex 1a and 1b.  indicates the peak of free DCA. indicates the peak of free DCA. | ||
2.3 In vitro cytotoxicity assay
The cytotoxicity of complexes 1a–2b was first tested in vitro by MTT assay in BGC803, HepG-2, MCF-7 and NCI-H460 cancer cell lines with cisplatin, oxaliplatin, and carboplatin as positive controls. The IC50 values of them were reported in Table 1.| Complex | IC50a (μM) | |||
|---|---|---|---|---|
| BGC803b | HepG-2c | MCF-7d | NCI-H460e | |
| a IC50 is the drug concentration effective in inhibiting 50% of the cell growth measured by the MTT assay after 48 h drug exposure.b BGC803: human gastric cancer cell line.c HepG-2: human hepatocellular carcinoma cell line.d MCF-7: human breast carcinoma cell line.e NCI-H460: large cell lung cancer cell line. | ||||
| Cisplatin | 21.58 ± 1.35 | 27.82 ± 2.56 | 29.19 ± 1.38 | 14.85 ± 1.35 |
| Oxaliplatin | 29.10 ± 1.65 | 26.49 ± 2.55 | 41.27 ± 3.43 | 13.43 ± 1.13 |
| Carboplatin | 77.14 ± 5.35 | 42.32 ± 2.55 | 179.40 ± 10.56 | 52.93 ± 4.98 |
| DCA | >100 | >100 | >100 | >100 |
| A:DCA (1:1) |
431.11 ± 15.56 | 99.47 ± 2.89 | 11.36 ± 1.56 | 10.22 ± 0.99 |
| B:DCA (1:1) |
190.06 ± 13.51 | 41.09 ± 3.02 | 60.46 ± 4.85 | 18.86 ± 1.19 |
| 1a | 57.73 ± 4.34 | 10.36 ± 1.24 | 27.08 ± 2.25 | >200 |
| 1b | 7.63 ± 0.56 | 1.35 ± 0.15 | 7.96 ± 0.76 | 1.94 ± 0.13 |
| 2a | 14.09 ± 0.98 | 15.00 ± 1.42 | 22.63 ± 2.05 | 5.46 ± 0.43 |
| 2b | 15.01 ± 1.05 | 17.54 ± 1.65 | 22.20 ± 2.17 | 7.66 ± 0.57 |
These four complexes displayed potent antitumor activity against BGC803, HepG-2, MCF7 and NCI-H460 cancer cell lines with the IC50 values in the range of 7.63–57.73 μM, 1.35–17.54 μM, 7.96–27.08 μM and 1.94–200 μM, respectively. Among them, complex 1b showed the highest activity against four cancer cell lines, which was superior to positive controls (cisplatin, oxaliplatin and carboplatin). DCA had no obvious cytotoxic activity on cancer cell lines with the IC50 values exceeding 100 μM. It was noted that 1b exhibited higher cytotoxic activity than an equivalent mixture: DCA:A (1:1) and DCA:B (1:1), implying that the special structure of complex 1b resulted in better cytotoxic activity. Notably, complex 1b exhibited 20.61 fold and 7.65 fold higher cytotoxicity toward HepG-2 and NCI-H460 cancer cells than that of cisplatin. Meanwhile, the cytotoxic activity of complex 1b was 2.82 fold and 3.66 fold more potent than that of cisplatin against BGC803 and MCF-7, respectively.
By the analysis of the structure–activity relationship and cytotoxic activity, the design of novel Pt(IV) 1a and 1b effectively improved the anti-tumor activity. Moreover, taking this type of complexes as prodrugs could reduce the attack of small sulfur-containing molecules (e.g., glutathione, methionine and metallothionein) and maintain a relatively stable structure before reaching tumor cells. After that, complexes 1a and 1b could release B and DCA, playing a dual-targeting anti-tumor effect against cancer cells, targeting nuclear DNA and mitochondria, respectively. In addition, complexes 2a and 2b exhibited good effects against BGC803, HepG-2, MCF-7 and NCI-H460 cancer cell lines, demonstrating that the axial ligand can be modified to alter the reductive rate and the lipophilicity of Pt(IV) complexes and consequently enhance the anti-tumor activity of Pt(IV) complexes.
Based on the above results, complexes 1a–2b were selected for further investigation against SGC7901, SGC7901/CDDP and HUVEC cells lines by MTT assay. As shown in Table 2, complexes 1a and 1b showed the same cytotoxicity as oxaliplatin against SGC7901. However, SGC7901/CDDP cell line was not sensitive to all the tested compounds, indicating that these compounds were not able to overcome cisplatin resistance. In HUVEC, the cytotoxicity of complexes 1a and 1b (IC50 = 59.11 μM and IC50 = 56.92 μM) was lower than that of cisplatin (IC50 = 36.92 μM). Meanwhile, neither compounds 2a nor 2b had detectable cytotoxic activity on HUVEC with IC50 values exceeding 200 μM. This type of Pt(IV) complexes was more selective to tumor cells compared with HUVEC. Consequently, both complexes 1a and 1b were promising anticancer candidates for further research.
| Complex | IC50a (μM) | ||
|---|---|---|---|
| SGC7901b | SGC7901/CDDPc | HUVECd | |
| a IC50 is the drug concentration effective in inhibiting 50% of the cell growth measured by the MTT assay after 48 h drug exposure.b SGC7901: gastric cancer cell line.c SGC7901/CDDP: cisplatin-resistant gastric cancer cell line.d HUVEC: human umbilical vein endothelial cell line. | |||
| Cisplatin | 9.96 ± 0.87 | 35.65 ± 2.55 | 36.92 ± 2.23 |
| Oxaliplatin | 13.47 ± 1.55 | 99.44 ± 5.23 | 51.03 ± 4.42 |
| 1a | 16.79 ± 1.45 | 92.45 ± 2.56 | 59.11 ± 2.54 |
| 1b | 16.76 ± 1.78 | 120.16 ± 6.74 | 56.47 ± 1.65 |
| 2a | 20.15 ± 1.44 | 92.65 ± 1.56 | >200 |
| 2b | 8.73 ± 1.87 | 97.50 ± 3.67 | >200 |
2.4 Cellular uptake
Given that complexes 1a and 1b exhibited better cytotoxicity, they were chosen to conduct a cellular uptake test on HepG-2 and NCI-H460 cells by using ICP-MS. As for HepG-2 cell line, platinum accumulation of 1b was 1.28 times more potent than that of cisplatin. Meanwhile, the intracellular Pt levels of 1a and 1b were higher than that of cisplatin due to the lipophilicity, while complex 1a had the same Pt content as 1b in HepG-2 cells. Interestingly, the uptake of complex 1b by NCI-H460 cells was 2.32 times higher than that of 1a.According to Tables 1 and 3, it seems that enhanced cellular uptake is associated with increased cytotoxicity of complexes. However, the intracellular platinum level is not the only factor deciding the cytotoxicity of platinum complexes. In fact, many other factors also affect the cytotoxic activity of platinum complexes, such as the effective amounts of the compounds that finally act on DNA. Actually, we have already found similar results in previous studies.28–30
| Complex | Pt contentb (nanograms per 106 cells) | |
|---|---|---|
| HepG-2 | NCI-H460 | |
| a HepG-2 and HCI-H460 cells were treated with 20 μM of cisplatin, oxaliplatin, complexes 1a and 1b for 12 h.b Results are expressed as the mean ± SD for four independent experiments. | ||
| Cisplatin | 176 ± 15 | 189 ± 20 |
| Oxaliplatin | 188 ± 12 | 196 ± 19 |
| 1a | 213 ± 17 | 106 ± 17 |
| 1b | 227 ± 26 | 246 ± 31 |
2.5 Complex 1a and 1b induced apoptotic cell death
Since complexes 1a and 1b showed better cytotoxicity, they were selected to carry out the apoptotic study against HepG-2 cells by Annexin V-FITC/PI assay. HepG-2 cells were incubated with 50 μM of indicated complexes for 24 hours. The quadrants Q1–Q4 represented four different cancer cell states: necrotic cells, late apoptotic or necrotic cells, living cells and early apoptotic cells, respectively.As shown in Fig. 3, complexes 1a and 1b (28.75% and 28.97%) had nearly the same apoptotic rate as cisplatin (28.79%) against HepG-2 cells while showing a higher apoptotic rate than oxaliplatin (21.74%; Fig. 2). As for HepG-2 cell line, after treated with 1b for 24 hours, the early apoptotic rate rose strikingly from 2.14% to 24.97% and the late apoptotic rate was increased from 4.17% to 4.99%. Similarly, the early apoptotic rate of HepG-2 cell for 1a surged from 2.14% to 21.61% and the late apoptotic rate was increased from 4.17% to 7.14%. Complex 1b showed a slightly higher population of apoptotic cells than complex 1a did. Based on the results from the cytotoxicity tests and the apoptotic assays, it was noted that there might be a positive correlation between these two assays. The order of apoptotic rates against HepG-2 cells from high to low was 1b, 1a, cisplatin, and oxaliplatin. Complexes 1a and 1b were able to cause cancer cell death through apoptotic pathways.
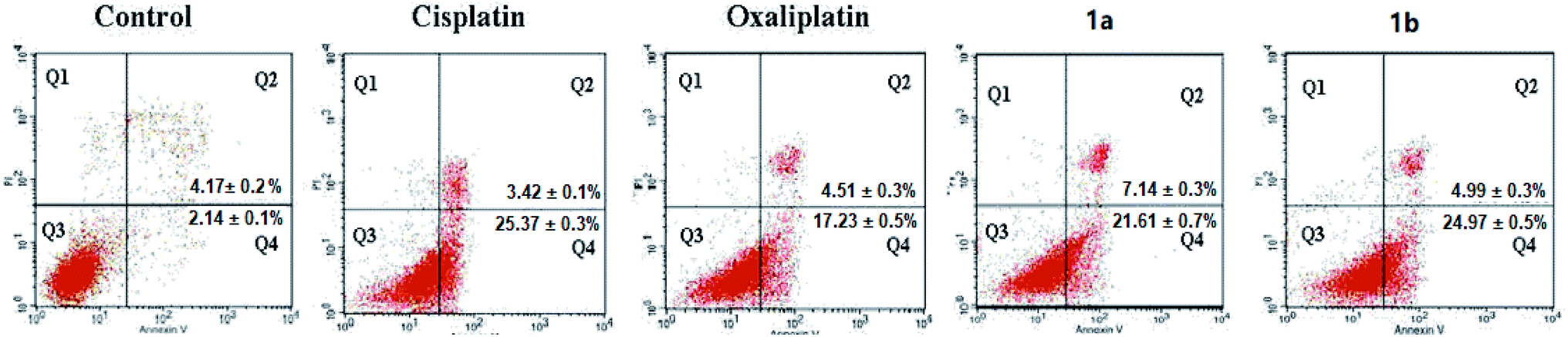 | ||
| Fig. 3 Flow cytometric analysis of the distribution of HepG-2 cells treated with 50 μM of cisplatin, oxaliplatin, complexes 1a and 1b for 24 hours (DMF final concentration < 0.4%). Cells were stained with 5 mL of Annexin V-FITC and incubated in the dark at 25 °C for 10 min. The fluorescence was measured by using a flow cytometer. The results were expressed as the percentage of normal and apoptotic cells at various stages by FCS Express software. | ||
2.6 Effect on cell cycle arrest
It is confirmed that cell cycle contains four different phases, including G1, G2, S, and M stages. Pt(IV) complexes could inhibit cancer cell growth by cell cycle arrest. As shown in Fig. 4, HepG-2 cells were treated with cisplatin, oxaliplatin, complex 1a or 1b for 12 hours, and the cell cycle distribution was examined by PI staining and following flow cytometry analysis.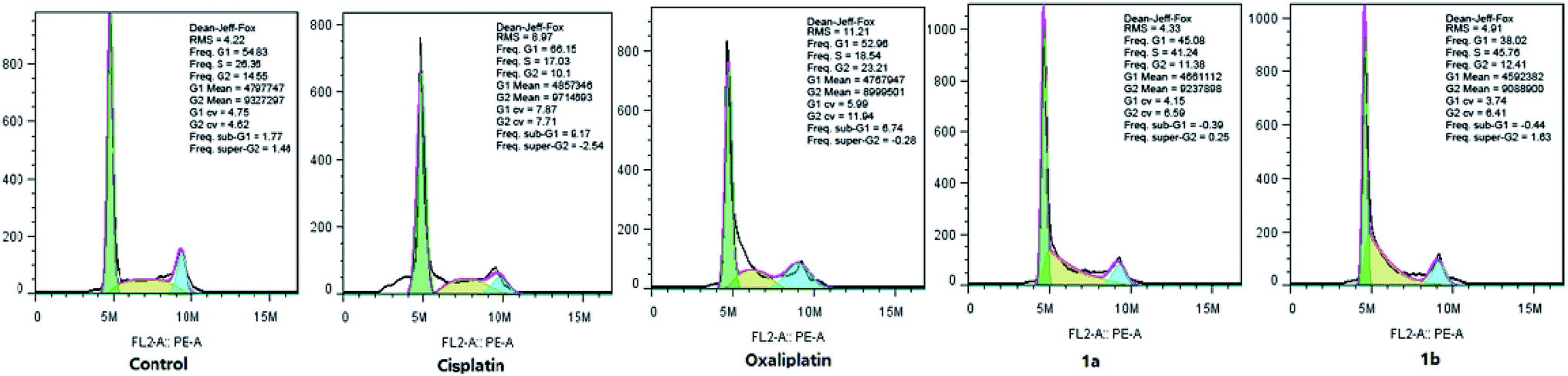 | ||
| Fig. 4 Cell cycle distribution of HepG-2 cells cultured in the presence of 30 μM of cisplatin, oxaliplatin, complexes 1a and 1b (DMF final concentration < 0.4%). The cells were seeded in 6-well plates for 12 hours at 37 °C, and then were treated with cisplatin, oxaliplatin, complex 1a or 1b. After 12 hours of treatment, cells were harvested with trypsin and washed twice with PBS. | ||
The percentage of cells at G1 phase was increased significantly upon cisplatin treatment (from 54.83% to 66.15%), while oxaliplatin arrested HepG-2 cells at G2 phase (from 14.55% to 23.21%). Notably, the cell cycle arrest caused by complex 1a and 1b mainly occurred at S phase (from 26.36% to 41.24% and from 26.36% to 45.76%). Overall, it was confirmed that different from cisplatin and oxaliplatin, complex 1a and 1b induced cell cycle arrest at S phase by inhibiting DNA replication in the nucleus. The different feature of cell cycle arrest is probably due to distinct DNA binding modes.
2.7 Mitochondrial membrane potential
Platinum(IV) complexes 1a and 1b can release platinum(II) complexes and DCA under the intracellular reductive condition, targeting nuclear DNA and mitochondria, respectively. In addition, DCA can result in mitochondrial dysfunction, and the change of mitochondrial function is manifested as the decrease in membrane potential and stability. The change of transmembrane potential causes the release of cytochrome C and a variety of apoptosis inducing factors (AIFs), which activate a cascade of apoptosis and eventually lead to apoptosis. In order to confirm that the change of mitochondria membrane potential was caused by Pt(IV) complexes, HepG-2 cells were incubated with the tested complexes for 48 hours and examined by JC-1 staining method.Complexes 1a and 1b were selected to test their effects on mitochondrial membrane potential with carbonylcyanide-m-chlorophenylhydrazone (CCCP), DCA, cisplatin, carboplatin and oxaliplatin as positive controls. As shown in Table 4, compared with the negative control, red-green fluorescence ratio of each group was dropped. In fact, complexes 1a and 1b notably reduced the mitochondrial membrane potential (FL2/FL1 = 0.98 and FL2/FL1 = 0.86). However, the decrease caused by complexes 1a and 1b was less than that induced by 1a:DCA (1:1) and 1b:DCA (1:1) (FL2/FL1 = 0.59 and FL2/FL1 = 0.80). The decline of mitochondrial membrane potential was a landmark event in the early stage of apoptosis, and the IC50 values of complexes 1a and 1b were lower than an equivalent mixture: DCA:A (1:1) and DCA:B (1:1). Besides, complexes 1a and 1b could release the corresponding platinum(II) and DCA by reduction, which could target mitochondria.
| Complex | Ratio (FL2/FL1) |
|---|---|
| a HepG-2 cells cultured in the presence of 30 μM of cisplatin, carboplatin, oxaliplatin, complex 1a or 1b for 48 hours at 37 °C (DMF final concentration < 0.4%). Then the cells were harvested by centrifugation and incubated with JC-1 solution for 30 min. After briefly washing, the proportions of red and green fluorescence intensity were immediately detected and analyzed by flow cytometry (Ex = 488 nm; Em = 530 nm). | |
| Negative control | 5.42 |
| CCCP | 0.04 |
| DCA | 4.17 |
| Cisplatin | 0.54 |
| Carboplatin | 2.56 |
| Oxaliplatin | 0.94 |
| 1a:DCA (1:1) |
0.59 |
| 1b:DCA (1:1) |
0.80 |
| 1a | 0.98 |
| 1b | 0.86 |
3. Conclusions
In this manuscript, we explored two types of Pt(IV) complexes 1a–1b and 2a–2b by taking complexes A and B as the lead compounds, and investigated dual-threat platinum prodrugs and the lipophilicity of the TFA as the axial ligand, respectively. Complexes 1a and 1b could rapidly transform in PBS (pH = 7.4) with the hydrolysis of the axial DCA ligands (t1/2 = 60 min). According to the IC50 values, 1a and 1b had better cytotoxic activity than cisplatin, oxaliplatin and carboplatin against HepG-2 and MCF-7 cell lines. Since Pt(IV) prodrugs 1a–2b had relatively kinetic inertia, they possessed less toxicity than cisplatin and oxaliplatin against HUVEC cell line. In addition, it was confirmed that complexes 1a and 1b induced cell cycle arrest at S phase (41.24% and 45.76%) by inhibiting DNA replication in the nucleus. Moreover, both of them significantly reduced the mitochondrial membrane potential. Above all, platinum(IV) prodrugs could improve the anticancer activity using a dual-targeting strategy with dichloroacetate axial ligand, which released platinum(II) complexes and DCA under the intracellular reductive condition, targeting nuclear DNA and mitochondria, respectively.Furthermore, a series of novel platinum complexes with ethylenediamine-N,N′-di-2/3-propionate ester as ligands has been reported to induce caspase-dependent apoptosis.31 Therefore, it is still a reasonable way to design antitumor platinum complexes targeting nuclear DNA and mitochondria. It may be an interesting approach to combine such complexes with drug delivery systems in the future.
4. Experimental
4.1 Chemistry
:C, 17.51; H, 2.02; N, 5.11; found: C, 17.47; H, 2.14; N, 5.13.| Molecular formula | [M + Na]+ experimental | [M + Na]+ theoretical | Error [ppm] | |
|---|---|---|---|---|
| 1a | C8H11Cl3N2O7Pt | 569.9196 | 569.9172 | −4.0 |
| 1b | C14H19Cl3N2O7Pt | 649.9818 | 649.9798 | −2.9 |
| 2a | C8H10ClF3N2O7Pt | 555.9687 | 555.9669 | −3.2 |
| 2b | C14H18ClF3N2O7Pt | 636.0323 | 636.0295 | −4.2 |
:90 (v/v)). 1H NMR spectra recorded at different time intervals.Complexes 2a and 2b (0.786 μmol) were dissolved in MeOH (1 mL). The stability of 2a and 2b in a MeOH/PBS (1:9) was investigated by HPLC (PBS 7.87 mM, pH 7.4; at 37 °C). HPLC recorded at different time intervals. Reversed phase HPLC was implemented on a 250 × 4.6 mm ODS column and the HPLC profiles were recorded on UV detection at 226 nm. Mobile phase consisted of MeCN:H2O = 25:75 (v/v), and flow rate was 1.0 mL min−1. The samples were taken for HPLC analysis after filtration by 0.5 μm filter.
4.2 Bioactivity study
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the China Postdoctoral Science Foundation (no. 2016M600473), the Postdoctoral Science Foundation of Zhejiang Province (no. zj20160107), and the National Natural Science Foundation of China (Project 21808205) for financial aids to this work. We are grateful to the Zhejiang Provincial Key R&D Project (No. 2018C03074). We thank the Jiangsu Province Hi-Tech Key Laboratory for Biomedical Research at Southeast University for completing the in vitro tests.Notes and references
- D. M. Cheff and M. D. Hall, J. Med. Chem., 2017, 60, 4517–4532 CrossRef CAS.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- J. D. White, M. F. Osborn, A. D. Moghaddam, L. E. Guzman, M. M. Haley and V. J. DeRose, J. Am. Chem. Soc., 2013, 135, 11680–11683 CrossRef CAS.
- R. A. Hazlitt, J. Min and J. Zuo, J. Med. Chem., 2018, 61, 5512–5524 CrossRef CAS.
- R. Qun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- E. Martinez-Balibrea, A. Martinez-Cardus, A. Gines, V. R. Porras, C. Moutinbo, L. Layos, J. L. Manzano, C. Buges, S. Bystrup, M. Esteller and A. Abad, Mol. Cancer Ther., 2015, 14, 1767–1776 CrossRef CAS PubMed.
- X. Y. Wang, X. H. Wang and Z. J. Guo, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS.
- M. Yang, H. Z. Wu, J. L. Chu, L. A. Gabriel, K. S. Anderson, C. M. Furdui and U. Bierbach, Chem. Commun., 2018, 54, 7479–7482 RSC.
- A. Abu Ammar, R. Raveendran, D. Gibson, T. Nassar and S. Benita, J. Med. Chem., 2016, 59, 9035–9046 CrossRef CAS PubMed.
- N. Wang, Z. G. Wang, Z. F. Xu, X. F. Chen and G. Y. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3426–3430 CrossRef CAS PubMed.
- D. Y. Q. Wong, J. Y. Lau and W. H. Ang, Dalton Trans., 2012, 41, 6104–6111 RSC.
- K. Mitra, C. E. Lyons and M. C. T. Hartman, Angew. Chem., Int. Ed., 2018, 57, 10263–10267 CrossRef CAS.
- G. De Lima Lopes, Y. L. Wu, S. Sadowski, J. Zhang, R. Rangwala, D. Kush and T. Mok, J. Thorac. Oncol., 2016, 11, S244–S245 CrossRef.
- C. A. Garcia, S. Dacic and L. C. Villaruz, J. Thorac. Oncol., 2018, 13, e135–e136 CrossRef.
- Z. Z. Zhu, Z. H. Wang, C. L. Zhang, Y. J. Wang, H. M. Zhang, Z. J. Gan and Z. J. Guo, Chem. Sci., 2019, 10, 3089–3095 RSC.
- F. F. Chen, G. Xu, X. D. Qin, X. F. Jin and S. H. Gou, J. Pharmacol. Exp. Ther., 2017, 263, 221–239 CrossRef.
- E. Gabano, M. Ravera, E. Perin, I. Zanellato, B. Rangone, M. J. McGlinchey and D. Osella, Dalton Trans., 2019, 48, 435–445 RSC.
- X. Y. Yao, C. M. Tracy and U. Bierbach, Inorg. Chem., 2019, 58, 43–46 CrossRef CAS.
- J. Zhao, S. H. Gou and F. F. Liu, Chem.–Eur. J., 2014, 20, 15216–15225 CrossRef CAS PubMed.
- F. H. Chen, X. F. Jing, J. Zhao and S. H. Gou, Exp. Cell Res., 2018, 364, 68–83 CrossRef CAS.
- E. D. Michelakis, L. Webster and J. R. Mackey, Cancer, 2008, 99, 989–994 CrossRef CAS.
- S. Dhar and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef CAS.
- W. P. Liu, J. Su, J. Jiang, X. Y. Li, Q. S. Ye, H. Y. Zhou, J. L. Chen and Y. Li, Sci. Rep., 2013, 3, 2464 CrossRef.
- S. Savino, V. Gandin, J. D. Hoeschele, C. Marzano, G. Natile and N. Margiotta, Dalton Trans., 2018, 47, 7144–7158 RSC.
- N. Margiotta, P. Papadia, F. Lazzaro, M. Crucianelli, F. De Angelis, C. Pisano, L. Vesci and G. Natile, J. Med. Chem., 2005, 48, 7821–7828 CrossRef CAS.
- J. J. Wilson and S. J. Lippard, J. Med. Chem., 2012, 55, 5326–5336 CrossRef CAS PubMed.
- E. Wexselblatt, R. Raveendran, S. Salameh, A. Friedman-Ezra, E. Yavin and D. Gibson, Chem.–Eur. J., 2015, 21, 3108–3114 CrossRef CAS.
- F. F. Liu, S. H. Gou, F. H. Chen, L. Fang and J. Zhao, J. Med. Chem., 2015, 58, 6368–6377 CrossRef CAS PubMed.
- X. C. Huang, R. Z. Huang, S. H. Gou, Z. M. Wang, Z. X. Liao and H. S. Wang, Bioconjugate Chem., 2016, 27, 2132–2148 CrossRef CAS.
- J. Pracharova, T. Saltarella, T. R. Radosova Muchova, S. Scintilla, V. Novohradsky, O. Novakova, F. P. Intini, C. Pacifico, G. Natile, P. Ilik, V. Brabec and J. Kasparkova, J. Med. Chem., 2015, 58, 847–859 CrossRef CAS.
- G. N. Kaluderović, S. A. Mijatović, B. B. Zmejkovski, M. Z. Bulatović, S. Gómez-Ruiz, M. K. Mojić, D. Steinborn, D. M. Miljković, H. Schmidt, S. D. Stošić-Grujičić, T. J. Saboe and D. D. Maksimović-Ivanić, Metallomics, 2012, 4, 979–987 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03690c |
| This journal is © The Royal Society of Chemistry 2019 |