
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-bridged secondary building units in a titanium metal–organic framework catalyze cascade reduction of N-heteroarenes†
Xuanyu
Feng‡
 a,
Yang
Song‡
a,
Justin S.
Chen
a,
Zhe
Li
ab,
Emily Y.
Chen
a,
Michael
Kaufmann
a,
Cheng
Wang
b and
Wenbin
Lin
*ab
a,
Yang
Song‡
a,
Justin S.
Chen
a,
Zhe
Li
ab,
Emily Y.
Chen
a,
Michael
Kaufmann
a,
Cheng
Wang
b and
Wenbin
Lin
*ab
aDepartment of Chemistry, University of Chicago, 929 E. 57th St., Chicago, Illinois 60637, USA. E-mail: wenbinlin@uchicago.edu
bCollege of Chemistry and Chemical Engineering, iCHEM, State Key Laboratory of Physical Chemistry of Solid Surface, Xiamen University, Xiamen 361005, PR China
First published on 19th December 2018
Abstract
We report here a novel Ti3–BPDC metal–organic framework (MOF) constructed from biphenyl-4,4′-dicarboxylate (BPDC) linkers and Ti3(OH)2 secondary building units (SBUs) with permanent porosity and large 1D channels. Ti–OH groups from neighboring SBUs point toward each other with an O–O distance of 2 Å, and upon deprotonation, act as the first bidentate SBU-based ligands to support CoII-hydride species for effective cascade reduction of N-heteroarenes (such as pyridines and quinolines) via sequential dearomative hydroboration and hydrogenation, affording piperidine and 1,2,3,4-tetrahydroquinoline derivatives with excellent activity (turnover number ∼ 1980) and chemoselectivity.
Introduction
The development and functionalization of Ti-oxo materials is a fertile research area due to their high crust abundance, low toxicity, excellent chemical and thermal stability, and unique photophysical properties. The introduction of permanent porosity into bulk TiO2 allows efficient utilization of all surface functionalizable sites to ensure sustainable resource utilization.1–3 For example, surface modifications of mesoporous titania have generated a series of Ti–OH anchored metal species displaying impressive catalytic or photocatalytic activities.4–6Ti metal–organic frameworks (MOFs) provide a highly tunable platform to realize molecular-level design and construction of single-crystalline and porous Ti-oxo materials that can be further functionalized for catalytic applications.7–10 However, only a few Ti-carboxylate MOFs have been synthesized and characterized since the report of the first Ti-carboxylate MOF (MIL-125) with Ti8O8(OH)4 secondary building units (SBUs) in 2009 (Table S1, ESI†).11–17 Ti MOFs with permanent porosity, large channels, and sterically open functionable sites are even rarer.
Herein we report the rational synthesis of a novel porous Ti-carboxylate MOF, Ti3–BPDC (BPDC = biphenyl-4,4′-dicarboxylate) with unprecedented Ti3(OH)2 SBUs. Two terminal TiIV–OH groups in the neighboring SBUs of Ti3–BPDC point at each other with a 2 Å distance and act as excellent bidentate ligands to complex CoII-hydride species to provide a sterically open and electronically-rich Earth-abundant metal catalyst for the cascade reduction of N-heteroarenes to piperidine and 1,2,3,4-tetrahydroquinoline derivatives (Fig. 1).
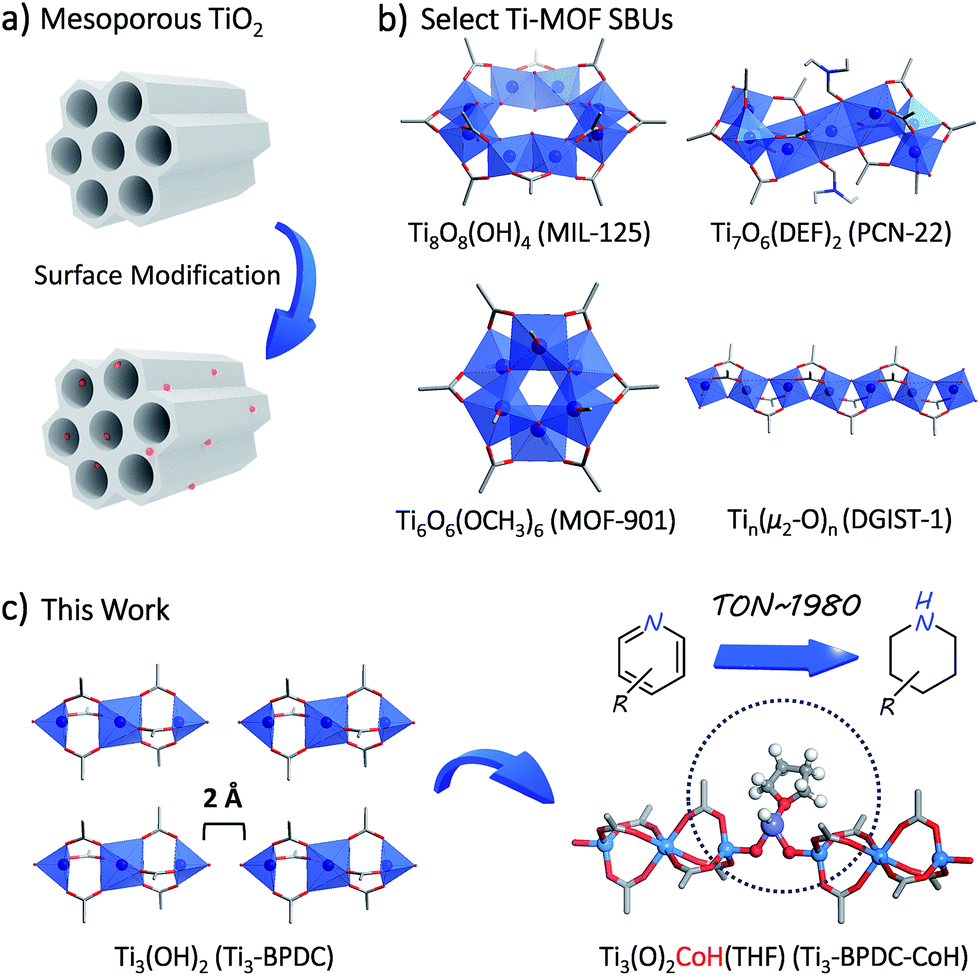 | ||
| Fig. 1 (a) Surface modification of mesoporous TiO2 generates functional materials. (b) Select Ti-MOF SBU structures (MIL-125, PCN-22, MOF-901, and DGIST-1). (c) Schematic illustration of novel Ti3(OH)2 SBUs in Ti3–BPDC and their metalation to generate anchored CoII-H species for cascade reduction of N-heteroarenes. | ||
Results and discussion
Synthesis and postsynthetic metalation of Ti3–BPDC
To prevent the formation of amorphous TiO2, we used Ti6O6(OiPr)6(abz)6 (abz = 4-aminobenzoate) cluster18 as the Ti source for MOF growth.12 Colorless rhombic Ti3–BPDC crystals were obtained in 31% yield via a solvothermal reaction between Ti6O6(OiPr)6(abz)6 and H2BPDC in N,N-dimethylformamide at 120 °C for 3 days with acetic acid (HOAc) as modulator. Single-crystal X-ray diffraction (SXRD) revealed that Ti3–BPDC crystallizes in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. Every three Ti atoms are linked by carboxylate groups to form Ti3(OH)2 SBUs, with the central Ti atom in octahedral coordination with six carboxylate groups and each of the two terminal Ti atoms in tetrahedral coordination with three carboxylate groups and one OH group. Ti3(OH)2 SBUs are connected by six BPDC linkers to afford a 3D framework of dia topology with 1D triangle channel of 11 Å in length running along the c axis (Fig. 2a). The void space was calculated to be 71.9% by PLATON. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) revealed the presence of νO–H stretching band at 3678 cm−1 for the terminal TiIV–OH.191H NMR analysis of digested Ti3–BPDC (in D3PO4/HF) showed the presence of 1.38 equiv. of HOAc w.r.t. H2BPDC (Fig. S6, ESI†), affording a formula of [Ti3(BPDC)3(OH)2](OAc)4. Powder X-ray diffraction (PXRD) showed that bulk phase and single crystals of Ti3–BPDC have identical structure (Fig. 2b). Due to relatively stable Ti3-cluster SBUs and strong coordination of carboxylate to TiIV, Ti3–BPDC was thermally stable up to 274 °C (Fig. S7, ESI†) and maintained its crystalline structure in a range of organic solvents and bases (Fig. S15, ESI†). Ti3–BPDC exhibits a type I isotherm with a Brunauer–Emmett–Teller (BET) surface area of 636 m2 g−1 and a pore size of 11.8 Å. To the best of our knowledge, Ti3–BPDC is the first Ti-MOF based on the biphenyldicarboxylate ligand, which provides a potential for further functionalization.20,21
m space group. Every three Ti atoms are linked by carboxylate groups to form Ti3(OH)2 SBUs, with the central Ti atom in octahedral coordination with six carboxylate groups and each of the two terminal Ti atoms in tetrahedral coordination with three carboxylate groups and one OH group. Ti3(OH)2 SBUs are connected by six BPDC linkers to afford a 3D framework of dia topology with 1D triangle channel of 11 Å in length running along the c axis (Fig. 2a). The void space was calculated to be 71.9% by PLATON. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) revealed the presence of νO–H stretching band at 3678 cm−1 for the terminal TiIV–OH.191H NMR analysis of digested Ti3–BPDC (in D3PO4/HF) showed the presence of 1.38 equiv. of HOAc w.r.t. H2BPDC (Fig. S6, ESI†), affording a formula of [Ti3(BPDC)3(OH)2](OAc)4. Powder X-ray diffraction (PXRD) showed that bulk phase and single crystals of Ti3–BPDC have identical structure (Fig. 2b). Due to relatively stable Ti3-cluster SBUs and strong coordination of carboxylate to TiIV, Ti3–BPDC was thermally stable up to 274 °C (Fig. S7, ESI†) and maintained its crystalline structure in a range of organic solvents and bases (Fig. S15, ESI†). Ti3–BPDC exhibits a type I isotherm with a Brunauer–Emmett–Teller (BET) surface area of 636 m2 g−1 and a pore size of 11.8 Å. To the best of our knowledge, Ti3–BPDC is the first Ti-MOF based on the biphenyldicarboxylate ligand, which provides a potential for further functionalization.20,21
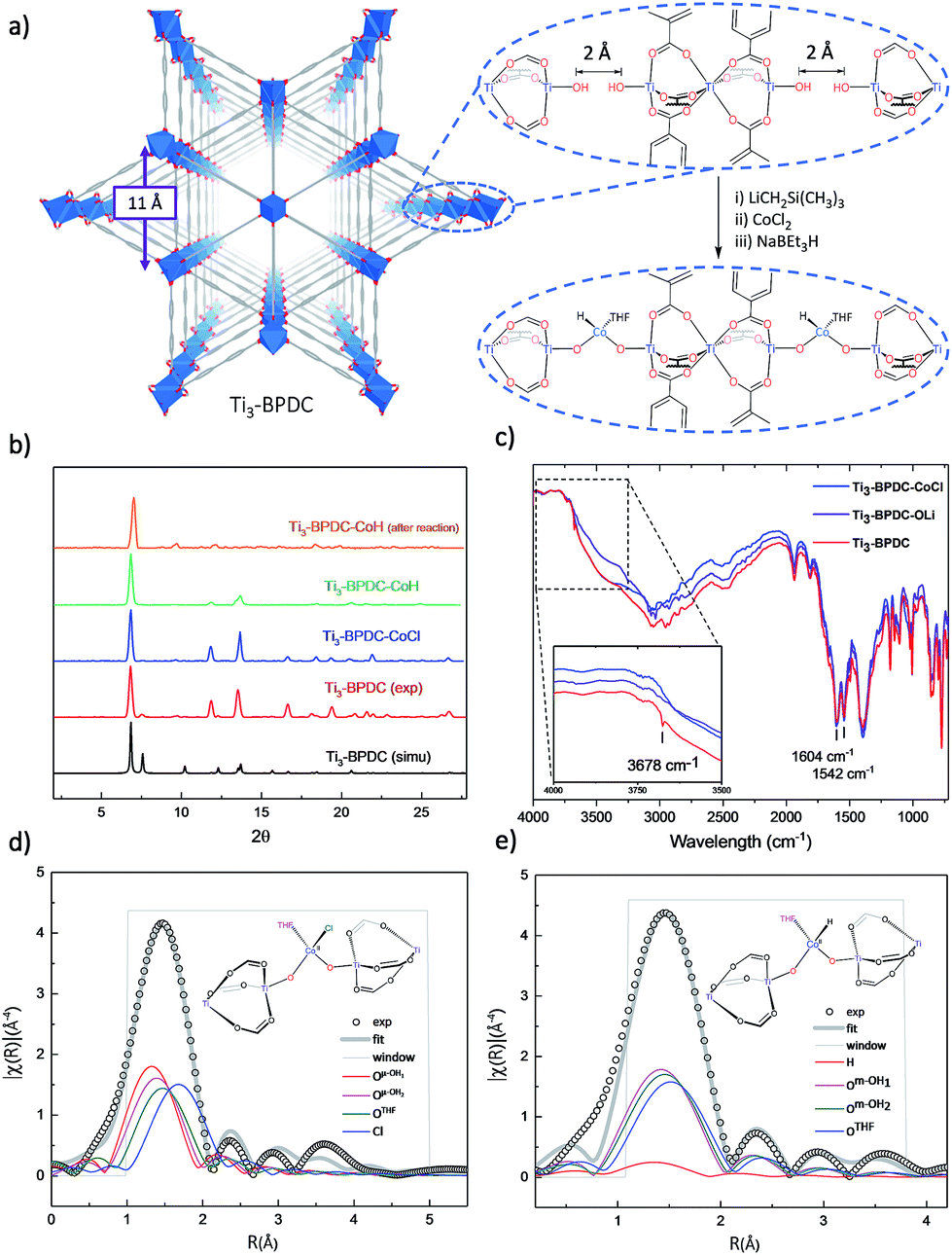 | ||
| Fig. 2 (a) Structure and SBU arrangement of Ti3–BPDC and its conversion to Ti3–BPDC–CoH. (b) PXRD patterns of Ti3–BPDC (red), Ti3–BPDC–CoCl (navy), and Ti3–BPDC–CoH (violet) and after-reaction (orange) along with simulated PXRD based on the SXRD structure (black). (c) DRIFT spectrum of Ti3–BPDC showing stretching vibration of Ti–OH at 3678 cm−1 from Ti3(OH)2 SBUs. (d, e) EXAFS spectra (black circles) and fits (grey solid line) in R-space at the Co K-edge adsorption of (d) Ti3–BPDC–CoCl and (e) Ti3–BPDC–CoH. | ||
The unique structure of Ti–BPDC, particularly the proximity of the neighboring TiIV–OH groups, inspired us to use the SBUs to support Earth-abundant metal catalysts.22 Although individual SBUs of MOFs have been previously used as structural and functional mimics of metal oxide catalyst supports,23–28 there has been no study of using neighboring SBUs as bidentate ligands to support Earth-abundant metal catalysts. We posited that chelation by neighboring SBUs affords several advantages including stronger coordination from two or more M–O− groups, more electron-rich active metal sites, and more sterically open coordination environment around catalytic centers.
Ti3–BPDC was deprotonated with 5 equiv. of LiCH2Si(CH3)3 and then metalated with 1 equiv. of CoCl2 in THF to afford Ti3–BPDC–CoCl as the purple solid. Both the carboxylate groups and linkers remained intact during the lithiation and metalation as evidenced by unchanged carbonyl stretching vibrations in the IR spectra. The disappearance of terminal OH stretching band (Fig. 2c) indicated complete deprotonation of TiIV–H groups. Inductively coupled plasma-mass spectrometry (ICP-MS) analysis of digested Ti3–BPDC–CoCl revealed the presence of 0.92 ± 0.04 Co per Ti3 SBU, indicating nearly complete metalation of all Ti–OH pairs. Crystallinity maintained after metalation as evidenced in the PXRD patterns, and SXRD of Ti3–BPDC–CoCl displayed a nearly identical unit cell parameter to Ti3–BPDC. Unfortunately, after numerous trials, the coordination environment of Co could not be established through SXRD due to the highly disordered nature of coordinated cobalt atoms. Instead, extended X-ray absorption fine structure (EXAFS) spectroscopy and density functional theory (DFT) were used to determine the Co coordination environment in Ti3–BPDC–CoCl. DFT calculation at the B3LYP level of theory converged at a distorted tetrahedral [(Ti–O−)2CoCl(THF)] coordination with two oxo groups from the deprotonation of Ti–OH (Co–O− distances of 1.89 and 1.95 Å), one chloride ion (Co–Cl distance of 2.33 Å), and one THF (Co–OTHF distance of 2.06 Å). The calculated model fitted well to the experimental EXAFS data of Ti3–BPDC–CoCl (Fig. 2d, Table S5, ESI†), which gave Co–(OTi) distances of 1.86 and 1.93 Å, respectively, with a Co–Cl distance of 2.25 Å and a Co–OTHF distance of 2.03 Å.
Treatment of Ti3–BPDC–CoCl with 10 equiv. of NaBEt3H in toluene generated Ti3–BPDC–CoH as a dark blue solid through Cl/H exchange. MOF crystallinity was maintained based on similar PXRD patterns before and after NaBEt3H treatment (Fig. 2b). 96.5% of chloride was exchanged by hydride as determined by X-ray Fluorescence analysis (Fig. S19, ESI†). X-ray photoelectron spectroscopy (XPS) and X-ray absorption near-edge spectroscopy (XANES) were used to determine the oxidation states of Ti and Co centers in Ti3–BPDC–CoH. The Co centers exhibited strong 2p3/2 and 2p1/2 peaks at 781.1 and 796.8 eV along with 2p3/2 and 2p1/2 shake-up peaks at 785.9 and 802.6 eV, consistent with CoII species (Fig. 3a). Ti centers showed strong 2p3/2 and 2p1/2 peaks at 458.3 and 464.1 eV, which are characteristic of TiIV species (Fig. 3b). The Co pre-edge features of Ti3–BPDC–CoCl and Ti3–BPDC–CoH are identical to that of CoCl2 (Fig. 3c), confirming the CoII oxidation state before and after hydride activation. Similar Ti pre-edge features were observed for Ti3–BPDC–CoCl and Ti3–BPDC–CoH (Fig. 3d), indicating the identical Ti oxidation states. Treatment of Ti3–BPDC–CoH with excess formic acid generated 0.96 ± 0.04 equiv. of H2 w.r.t. Co, indicating the presence of Co–H species. EXAFS fitting on Ti3–BPDC–CoH showed that Co centers adopt (Ti–O−)2Co bridging mode with Co–(OTi) distances of 1.93 Å and 1.96 Å (Fig. 2e) which are similar to those of Ti3–BPDC–CoCl. Due to the unique site-isolation effect within MOF structure,29,30 such monomeric Co–H species with oxo-based chelating ligands cannot be prepared in homogeneous systems due to their tendency to oligomerize.
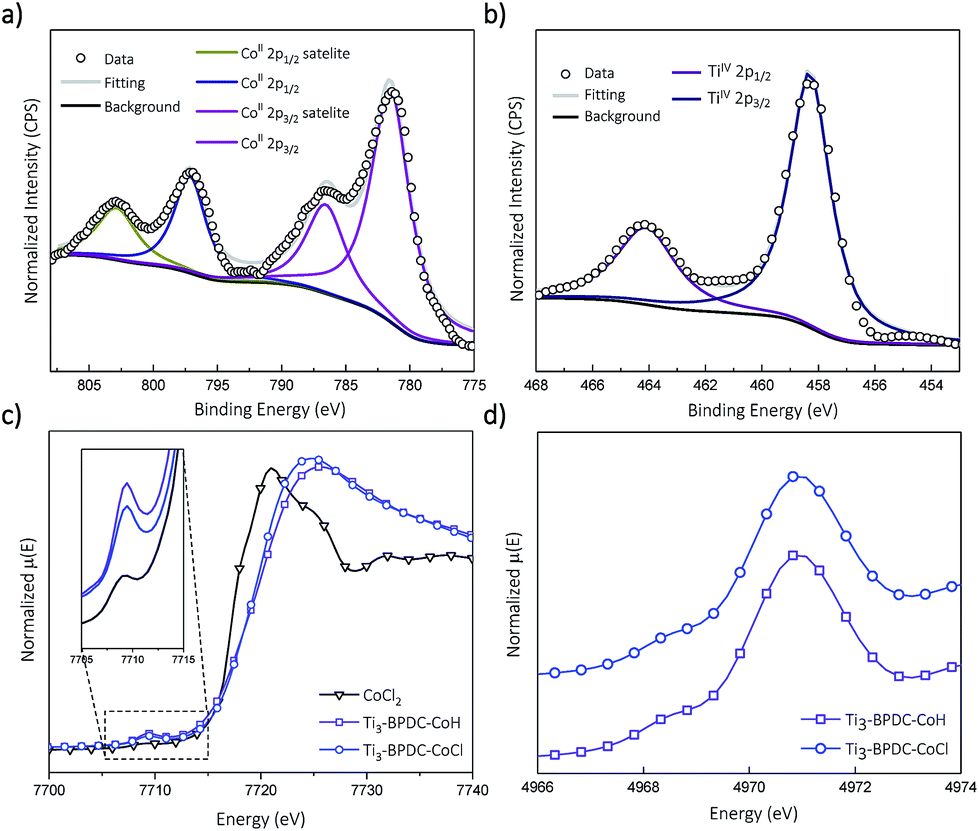 | ||
| Fig. 3 (a) Co 2p XPS spectra of Ti3–BPDC–CoH and the fitted result indicates the Co2+ oxidation state after NaBEt3H treatment. (b) Ti 2p XPS spectra of Ti3–BPDC–CoH and the fitted result indicates the Ti4+ oxidation state after NaBEt3H treatment. (c) Co pre-edge XANES features of Ti3–BPDC–CoCl (navy) and Ti3–BPDC–CoH (violet) in comparison to that of CoCl2 (black). (d) Ti pre-edge XANES features of Ti3–BPDC–CoCl (navy) and Ti3–BPDC–CoH (violet). | ||
Ti–BPDC–CoH catalyzed cascade reduction of pyridines
Ti3–BPDC–CoH displayed high catalytic activities in the cascade reduction of pyridines to piperidine derivatives. As one of the most important building blocks in drugs and bioactive alkaloids,31,32 the synthesis of piperidines has drawn significant interest in the past few decades. Compared to traditional ring-closing pathways, catalytic hydrogenation of pyridines provides an atom-/step-efficient strategy to construct piperidines.33 However, there has been only a few examples of hydrogenation of pyridines using homogeneous Rh34 and Ir,35 heterogeneous Rh,36 Pd,37 and Co,38 and metal-free systems,39 as well as the semi-reduction of pyridines,40,41 likely due to strong poisoning effect of pyridines on the catalytic sites. Inspired by our previous success in hydroboration and hydrogenation reactions catalyzed by SBU-supported Co-hydrides,23,42 we envisaged a one-pot cascade reduction pathway to prepare piperidines from pyridines using Ti3–BPDC–CoH. Pinacolborane (HBpin) was added to the reactions to first dearomatize N-heteroarenes via hydroboration, followed by the hydrogenation of the remaining unsaturated bonds of the hydroborated intermediates.At 0.2 mol% Ti3–BPDC–CoH, treatment of 3-picoline with 1.05 equiv. of HBpin in n-octane under 20 bar H2 at 100 °C for 22 h gave 3-methylpiperidine in 97% yield. No obvious change was observed in the MOF's PXRD pattern after catalysis (Fig. 2b), indicating the stability of the MOF framework under reaction condition. Changing n-octane to coordinating solvents (e.g., THF) or solvent-free condition led to a dramatic drop in yields (Table S8, ESI†), suggesting the inhibition effect from coordination of the solvent or pyridine substrate. No piperidine product was observed in the absence of MOF, HBpin, or H2 (Table S9, ESI†). An unprecedentedly high turnover number (TON) of 1980 was achieved at 0.05 mol% catalyst loading in 3 days. At 0.2–0.5 mol% of Co loading, a wide range of pyridines with electron donating or withdrawing groups on 3-, 4-, or 5-positions of the pyridine rings, were all reduced to piperidines in high yields (73–100%). This cascade reduction reaction also exhibits good functional group tolerance and outstanding product selectivity. Alkoxy, ester, dialkyl amide, and silyl groups all remained intact during the reduction process. For pyridine substrates containing aromatic rings, e.g., 3/4-phenylpridines and 4-benzylpyridines, the pyridyl rings were selectively reduced without affecting the phenyl rings (Table 1).
| a 0.50 mmol pyridine, 0.525 mmol HBpin, 20 bar H2, 0.2–0.5 mol% of Ti3–BPDC–CoH, 100–140 °C, 22 h; yield determined by GC-MS using mesitylene as internal standard. b Reaction time: 40 h. c 1.05 equiv. HBpin, 35 bar H2. d 3 equiv. HBpin, 50 bar H2. e 3 equiv. HBpin, 35 bar H2. f 3-((Trimethylsilyl)ethynyl)pyridine as substrate, the triple bond was hydrogenated. |
|---|
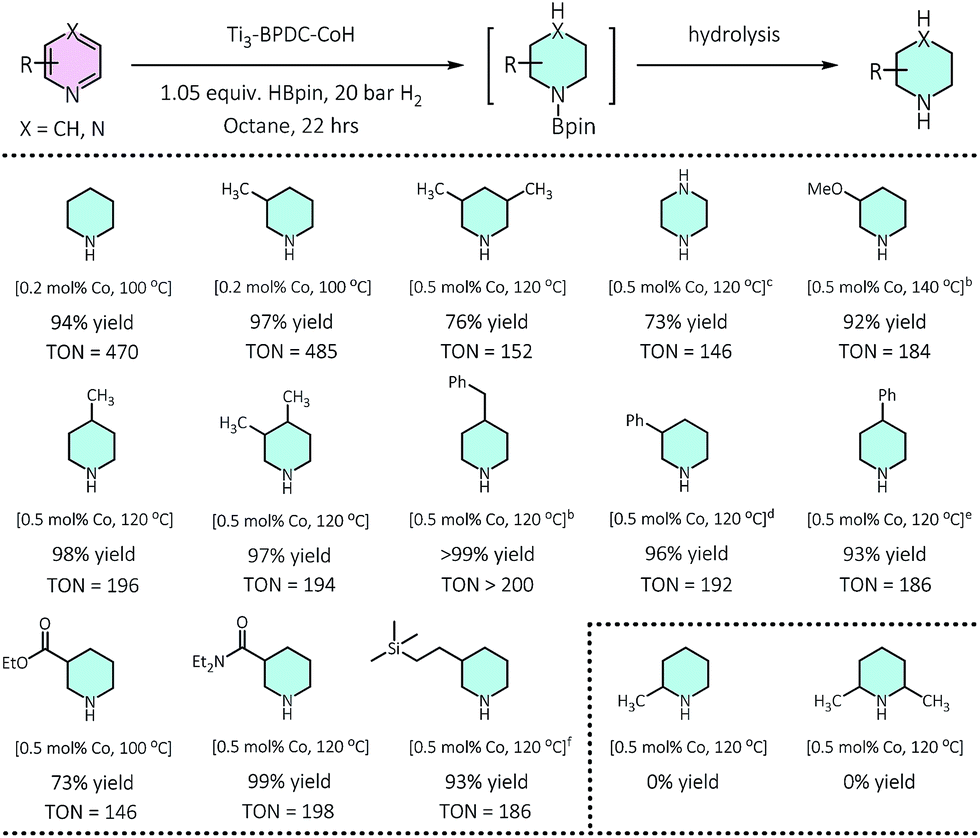
|
Several lines of evidences support the cascade process (Tables 1, S8, Fig. S30†). First, 2-picoline and 2,6-lutidine showed dramatic activity decrease compared to 3-picoline and 3,5-lutidine, respectively, likely due to the blocking of Co coordination by the methyl group(s) to inhibit the hydroboration step. Second, we detected 25% hydroborated 3-picoline by Ti3–BPDC–CoH in the absence of H2, which demonstrates the Co centers could catalyze the hydroboration step. Third, isolated hydroborated pyridines41 were quantitatively converted to piperidine at 0.5 mol% of Ti3–BPDC–CoH under 20 bar H2, while no piperidine was observed without the MOF catalyst. Cascade reduction was thus initiated by dearomative hydroboration of pyridines followed by hydrogenation of the remaining unsaturated bonds (Fig. 4a). Both steps are catalyzed by the Co–H species.
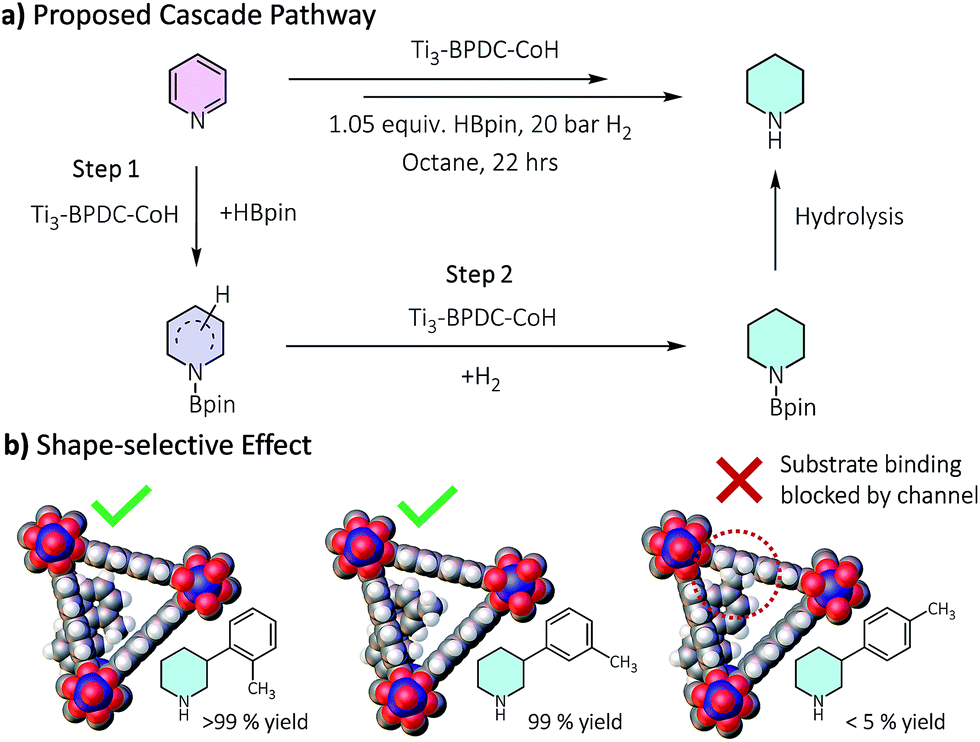 | ||
| Fig. 4 (a) Proposed cascade reduction pathway. (b) Shape selectivity of the cascade reduction of 3-(m/p/o-toly)pyridine. | ||
Ti3–BPDC–CoH was recovered and reused at least 6 times without significant decrease in yields (93–100%, Fig. S32, ESI†). ICP-MS showed minimal leaching of Co and Ti (0.4% and 0.6%, respectively). Hot filtration experiment ruled out the possibility of leached Co species contributing to the cascade reduction reactivity (Fig. S34, ESI†). Additionally, neither Co nanoparticles nor NaBEt3H afforded any product in the cascade reduction of 3-picoline (Table S10, ESI†). Notably, different regioisomers of 3-tolylpyridines showed remarkable shape selectivity in Ti3–BPDC–CoH catalyzed cascade reduction (Fig. 4b). Under the same conditions, 3-(o-tolyl)pyridine and 3-(m-tolyl)pyridine were completely reduced to piperidines, while most of 3-(p-tolyl)pyridine remained unreacted, likely caused by unfavorable steric repulsion between the methyl group of Co-coordinated 3-(p-tolyl)pyridine and BPDC ligands from far side of the channel wall (Fig. 4b) to inhibit the hydroboration step.
Ti–BPDC–CoH catalyzed cascade reduction of quinolines
We then applied this cascade protocol to dearomatize other N-heteroarenes. Semi-hydrogenation of quinolines is the most straightforward and convenient way to synthesize 1,2,3,4-tetrahydroquinolines (1,2,3,4-4HQLs), which have broad applications in pharmaceuticals and agrochemicals.43 Although several catalysts have been reported for such semi-hydrogenation reactions,44–46 challenges still exist in terms of product selectivity and functional group tolerance. Ti3–BPDC–CoH catalyzed semi-hydrogenation of quinolines to generate 1,2,3,4-4HQLs with excellent selectivity and functional group tolerance via the cascade reduction process. At 0.2 mol% catalyst loading and 100 °C, 1,2,3,4-4HQL was generated in >99% yield with <1% of 5,6,7,8-4HQL byproduct. Quinolines with methyl groups on different positions (3-,4-,6-,7-) and with different functional groups (Cl, OMe, COOMe) as well as quinoxaline were all semi-hydrogenated to 1,2,3,4-4HQLs in >84% yields and good selectivities (Table 2). No conversion was detected for 2,6-dimethylquinoline due to the inhibition of substrate coordination to the Co center.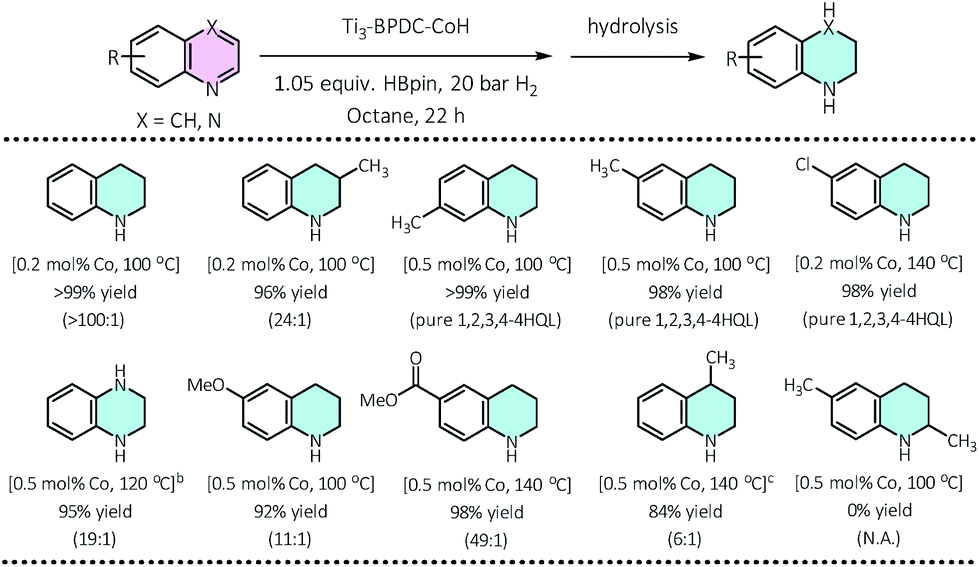
Conclusions
We have synthesized a novel single-crystalline Ti-carboxylate MOF with permanent porosity and large 1D channels based on unique Ti3(OH)2 SBUs and BPDC linkers. Each pair of closely spaced TiIV–OH groups from neighboring SBUs are deprotonated and then chelate to CoII centers. Such SBU-supported CoII-hydride species is highly active for selective cascade reduction of N-heterocyclic rings of pyridines and quinolines: heteroarenes first undergo dearomative hydroboration followed by hydrogenation of the remaining unsaturated bonds to afford synthetically useful piperidines and 1,2,3,4-tetrahydroquinolines with excellent activity and chemoselectivity. This work expands the applications of MOFs in developing single-site solid catalysts by using neighboring SBUs to support Earth-abundant metal complexes and highlights the great potential of MOF catalysts in fine chemical synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSF (CHE-1464941). We thank Dr Yuanyuan Zhu, Dr Zekai Lin, and Vlad Kamysbayev for experimental help. XAS analysis was performed at Beamline 10-BM, supported by the Materials Research Collaborative Access Team (MRCAT). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. DOE Office of Science by ANL, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Z. Li acknowledges financial support from the China Scholarship Council and the National Science Foundation of China (21671162).Notes and references
- R. Zhang, A. A. Elzatahry, S. S. Al-Deyab and D. Zhao, Nano Today, 2012, 7, 344–366 CrossRef CAS.
- W. Zhou and H. Fu, ChemCatChem, 2013, 5, 885–894 CrossRef CAS.
- W. Li, Z. Wu, J. Wang, A. A. Elzatahry and D. Zhao, Chem. Mater., 2014, 26, 287–298 CrossRef CAS.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS.
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed.
- G. Kennedy, L. R. Baker and G. A. Somorjai, Angew. Chem., Int. Ed., 2014, 53, 3405–3408 CrossRef CAS PubMed.
- C. Serre, J. A. Groves, P. Lightfoot, A. M. Z. Slawin, P. A. Wright, N. Stock, T. Bein, M. Haouas, F. Taulelle and G. Férey, Chem. Mater., 2006, 18, 1451–1457 CrossRef CAS.
- H. Assi, G. Mouchaham, N. Steunou, T. Devic and C. Serre, Chem. Soc. Rev., 2017, 46, 3431–3452 RSC.
- H. L. Nguyen, New J. Chem., 2017, 41, 14030–14043 RSC.
- S. Yuan, J.-S. Qin, C. T. Lollar and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- S. Yuan, T.-F. Liu, D. Feng, J. Tian, K. Wang, J. Qin, Q. Zhang, Y.-P. Chen, M. Bosch, L. Zou, S. J. Teat, S. J. Dalgarno and H.-C. Zhou, Chem. Sci., 2015, 6, 3926–3930 RSC.
- J. A. Mason, L. E. Darago, W. W. Lukens and J. R. Long, Inorg. Chem., 2015, 54, 10096–10104 CrossRef CAS PubMed.
- H. L. Nguyen, F. Gándara, H. Furukawa, T. L. H. Doan, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 4330–4333 CrossRef CAS PubMed.
- H. L. Nguyen, T. T. Vu, D. Le, T. L. H. Doan, V. Q. Nguyen and N. T. S. Phan, ACS Catal., 2017, 7, 338–342 CrossRef CAS.
- S. Wang, T. Kitao, N. Guillou, M. Wahiduzzaman, C. Martineau-Corcos, F. Nouar, A. Tissot, L. Binet, N. Ramsahye, S. Devautour-Vinot, S. Kitagawa, S. Seki, Y. Tsutsui, V. Briois, N. Steunou, G. Maurin, T. Uemura and C. Serre, Nat. Commun., 2018, 9, 1660 CrossRef PubMed.
- Y. Keum, S. Park, Y.-P. Chen and J. Park, Angew. Chem., Int. Ed., 2018, 57, 14852 CrossRef CAS PubMed.
- K. Hong and H. Chun, Inorg. Chem., 2013, 52, 9705–9707 CrossRef CAS.
- N.-Y. Topsøe, J. Catal., 1991, 128, 499–511 CrossRef.
- D. Sun, Y. Gao, J. Fu, X. Zeng, Z. Chen and Z. Li, Chem. Commun., 2015, 51, 2645–2648 RSC.
- D. Kim, D. R. Whang and S. Y. Park, J. Am. Chem. Soc., 2016, 138, 8698–8701 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 12610 CrossRef CAS PubMed.
- P. Ji, Y. Song, T. Drake, S. S. Veroneau, Z. Lin, X. Pan and W. Lin, J. Am. Chem. Soc., 2018, 140, 433–440 CrossRef CAS PubMed.
- I. S. Kim, Z. Li, J. Zheng, A. E. Platero-Prats, A. Mavrandonakis, S. Pellizzeri, M. Ferrandon, A. Vjunov, L. C. Gallington, T. E. Webber, N. A. Vermeulen, R. L. Penn, R. B. Getman, C. J. Cramer, K. W. Chapman, D. M. Camaioni, J. L. Fulton, J. A. Lercher, O. K. Farha, J. T. Hupp and A. B. F. Martinson, Angew. Chem., Int. Ed., 2018, 57, 909–913 CrossRef CAS PubMed.
- S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed.
- H. G. T. Nguyen, N. M. Schweitzer, C.-Y. Chang, T. L. Drake, M. C. So, P. C. Stair, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2014, 4, 2496–2500 CrossRef CAS.
- P. J. Larson, J. L. Cheney, A. D. French, D. M. Klein, B. J. Wylie and A. F. Cozzolino, Inorg. Chem., 2018, 57, 6825–6832 CrossRef CAS PubMed.
- T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed.
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS PubMed.
- S. Källström and R. Leino, Bioorg. Med. Chem., 2008, 16, 601–635 CrossRef PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- R. Vardanyan, in Piperidine-Based Drug Discovery, ed. R. Vardanyan, Elsevier, 2017, pp. 1–82, DOI:10.1016/B978-0-12-805157-3.00001-6.
- E. Baralt, S. J. Smith, J. Hurwitz, I. T. Horvath and R. H. Fish, J. Am. Chem. Soc., 1992, 114, 5187–5196 CrossRef CAS.
- X.-B. Wang, W. Zeng and Y.-G. Zhou, Tetrahedron Lett., 2008, 49, 4922–4924 CrossRef CAS.
- M. Freifelder, R. M. Robinson and G. R. Stone, J. Org. Chem., 1962, 27, 284–286 CrossRef CAS.
- F. Glorius, N. Spielkamp, S. Holle, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2004, 43, 2850–2852 CrossRef CAS PubMed.
- F. Chen, W. Li, B. Sahoo, C. Kreyenschulte, G. Agostini, H. Lund, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2018, 57, 14488 CrossRef CAS.
- Z.-Y. Liu, Z.-H. Wen and X.-C. Wang, Angew. Chem., Int. Ed., 2017, 56, 5817–5820 CrossRef CAS PubMed.
- A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100 CrossRef CAS PubMed.
- P. Ji, X. Feng, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 15600–15603 CrossRef CAS PubMed.
- P. Ji, K. Manna, Z. Lin, X. Feng, A. Urban, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 7004–7011 CrossRef CAS.
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed.
- N. A. Beckers, S. Huynh, X. Zhang, E. J. Luber and J. M. Buriak, ACS Catal., 2012, 2, 1524–1534 CrossRef CAS.
- F. Chen, A.-E. Surkus, L. He, M.-M. Pohl, J. Radnik, C. Topf, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 11718–11724 CrossRef CAS PubMed.
- I. Sorribes, L. Liu, A. Doménech-Carbó and A. Corma, ACS Catal., 2018, 8, 4545–4557 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1859034. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04610g |
| ‡ X. F. and Y. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |