
Metallosupramolecules of pillar[5]-bis-trithiacrown including a mercury(II) iodide ion-triplet complex†
Mingyeong
Shin
 a,
Sujin
Seo
a,
In-Hyeok
Park
ab,
Eunji
Lee
ac,
Yoichi
Habata
c and
Shim Sung
Lee
*a
a,
Sujin
Seo
a,
In-Hyeok
Park
ab,
Eunji
Lee
ac,
Yoichi
Habata
c and
Shim Sung
Lee
*a
aDepartment of Chemistry and Research Institute of Natural Science, Gyeongsang National University, Jinju 52828, South Korea. E-mail: sslee@gnu.ac.kr
bGraduate School of Analytical Science and Technology (GRAST), Chungnam National University, Daejeon 34134, South Korea
cDepartment of Chemistry, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan
First published on 24th July 2020
Abstract
A combination of pillar[5]-bis-trithiacrown (L) and mercury(II) halides afforded a monomer complex (Cl−-form), a 1-D coordination polymer (Br−-form) and a supramolecular ion-triplet complex [(I·Hg·I)@L] (I−-form). In the ion-triplet complex, the host encapsulates the (I−–Hg2+–I−) entity via Hg2+⋯π and C–H⋯I− interactions, reflecting geometrical complementarity.
Macrocyclic hosts have played a pivotal role in supramolecular chemistry especially because of their ability to exhibit selective recognition of particular cation (C+)1,2 or anion (A−)2,3 guests. The latter recognition is typically more challenging due to the intrinsic nature of many anions including their larger sizes, different shapes and often higher polarizabilities. A number of hetero-di or poly-topic receptors incorporating one or more macrocycle units have been shown to exhibit concurrent cation and anion recognition to form inclusive or endocyclic (guest-in-cavity) ion-pair complexes.4 Common motifs of such ion-pair complexes involve, as shown in Fig. 1, (a) contact and (b) solvent-separated arrangement of types [C+·A−@receptor] and [C+·solvent·A−@receptor], respectively.3 In principle, the formation of the corresponding ion-triplet complexes exhibiting [A−·C2+·A−@receptor] or [C+·A2−·C+@receptor] arrangements appeared possible under appropriate conditions (Fig. 1c).
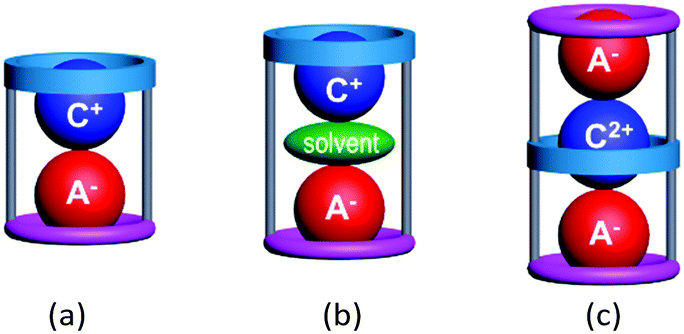 | ||
| Fig. 1 Schematic representation of (a) a contact ion-pair complex, (b) a solvent-separated ion-pair complex and (c) a contact ion-triplet complex with poly-topic macrocycles. | ||
Indeed, the Lüning group has shown the formation of a supramolecular ion-triplet complex of calcium(II) ions, [Cl−·Ca2+·Cl−@macrocycle], with a 30-membered [2+2] amide-macrocycle by ESI-mass in solution.5 However, no further examples of the formation or isolation on supramolecular ion-triplet complexes of metal ions both in solution and the solid state have been reported so far.
In our experience, endocyclic and exocyclic coordination modes of some thiamacrocyclic complexes are known to be controlled by several factors that include their anion coordination abilities.6,7 Pillar[n]arenes are a relatively new category of macrocyclic receptors and their inclusion complexes with organic guests such as diamines and dinitriles to yield polyrotaxane and pseudo-rotaxane structures have been widely studied in recent years.8
In marked contrast to the big advances in pillar[n]arene chemistry involving organic guests,9 their metallosupramolecular derivatives are rare except for studies from our10 and two other groups.11 In a prior study our modification of a pillar[5]arene to form a pseudo[1]catenane-type pillar[5]-mono-thiacrown showed a chiral inversion upon mercury(II) complexation under anion-coordination control.10a We have also prepared a di-armed pillar[5]arene-based two-dimensional silver(I) poly-pseudo-rotaxane in which the same dinitrile guest both threads and crosslinks.10b The Huang group has reported ion-pair recognition of pyridinium iodide12 and silver trifluoroacetate11b using a peralkylated pillar[5]arene. In this work, we have shown that it is possible to isolate an ion-triplet complex by employing pillar[5]-bis-trithiacrown (L).
In our results, the coordination modes of the mercury(II) halide complexes of L are anion-dependent. For example, L forms a mononuclear complex and an infinite type complex via an exo-coordination with chloride and bromide, respectively. Surprisingly, mercury(II) iodide led to the formation of an ion-triplet complex (Fig. 1c) as the first example isolated in the solid state to the best of our knowledge. The details are discussed below.
The pillar[5]-bis-trithiacrown L was synthesised by bicyclisation employing pillar[5]arene tetrabromide and the required dithiol (yield 30%, Scheme S1 and Fig. S1 and S2, ESI†). The tetrabromide precursor was obtained employing a mixture of dibromide, 1,4-dimethoxybenzene, paraformaldehyde and BF3·O(C2H5)2.10b The crystal structure of L was determined and shown to include one n-hexane molecule in its central cavity (Fig. S3, S4 and Table S1, ESI†). Both trithiacrown loops (O–S–S–S–O) in L show t–t–t–t (t = trans) torsion arrangements reflecting the repulsive interaction between adjacent S donors.
The reaction of L with HgCl2 in dichloromethane/methanol afforded a colourless crystalline product 1. The X-ray analysis revealed that 1 crystallises in the monoclinic space group Cc with Z = 4 (Tables S1 and S2, ESI†). The structure features an exocyclic mononuclear arrangement of type [HgCl2(CH2Cl2@L)] (Fig. 2 and Fig. S5, ESI†). The Hg1 atom, with a distorted tetrahedral geometry [82.41(12)–126.39(14)°], is bonded to two S atoms [Hg1–S2 2.611(4), Hg1–S3 2.714(3) Å], with its coordination environment completed by two Cl atoms [Hg1–Cl1 2.363(4), Hg1–Cl2 2.378(4) Å]. Four S donors remain uncoordinated, with a dichloromethane molecule occupying the cavity of L.
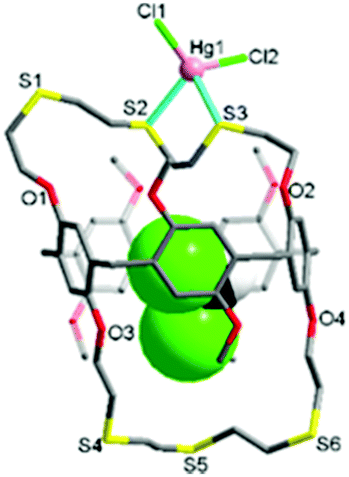 | ||
| Fig. 2 The exo-coordinated mercury(II) chloride complex [HgCl2(CH2Cl2@L)] (1). | ||
In 1, the nonsymmetrical binding of the Hg1 atom induces a large conformational change in the O1–S1–S2–S3–O2 loop segment from t–t–t–t to g–t–g–g (g = gauche, t = trans) torsion arrangements, with that of the O3–S4–S5–S6–O4 loop segment (t–t–t–) remaining unchanged. Unlike oxygen-bearing crown ethers, thiamacrocycles so often show a repulsive interaction between adjacent sulfur donors, stabilizing a trans torsion arrangement which tends to lead to the adoption of an exo-coordination mode.6,7,13
The reaction of L with HgBr2 in chloroform/acetonitrile yielded the colourless complex 2 which crystallizes in the monoclinic space group Pc with Z = 2 (Tables S1 and S3, ESI†). This product features an exocyclic one-dimensional (1-D) polymeric arrangement of formula [Hg6Br12(CH3CN@L)2]n (Fig. 3a). The asymmetric unit contains one formula unit. The crystallographically independent six Hg atoms (Hg1–Hg6) that lie outside the cavity have different coordination environments. First, the terminal Hg1 atom is three-coordinate, being bound to one S atom from one trithiacrown loop and two Br atoms. Second, each of the Hg2, Hg3, Hg5 and Hg6A atoms is four-coordinated by two S atoms from one trithiacrown loop and two Br atoms to form a distorted tetrahedral coordination geometry. The Hg4 atom is five-coordinate, being bound to four Br atoms and one S atom to yield a three-way node. An acetonitrile molecule occupies the cavity of each L.
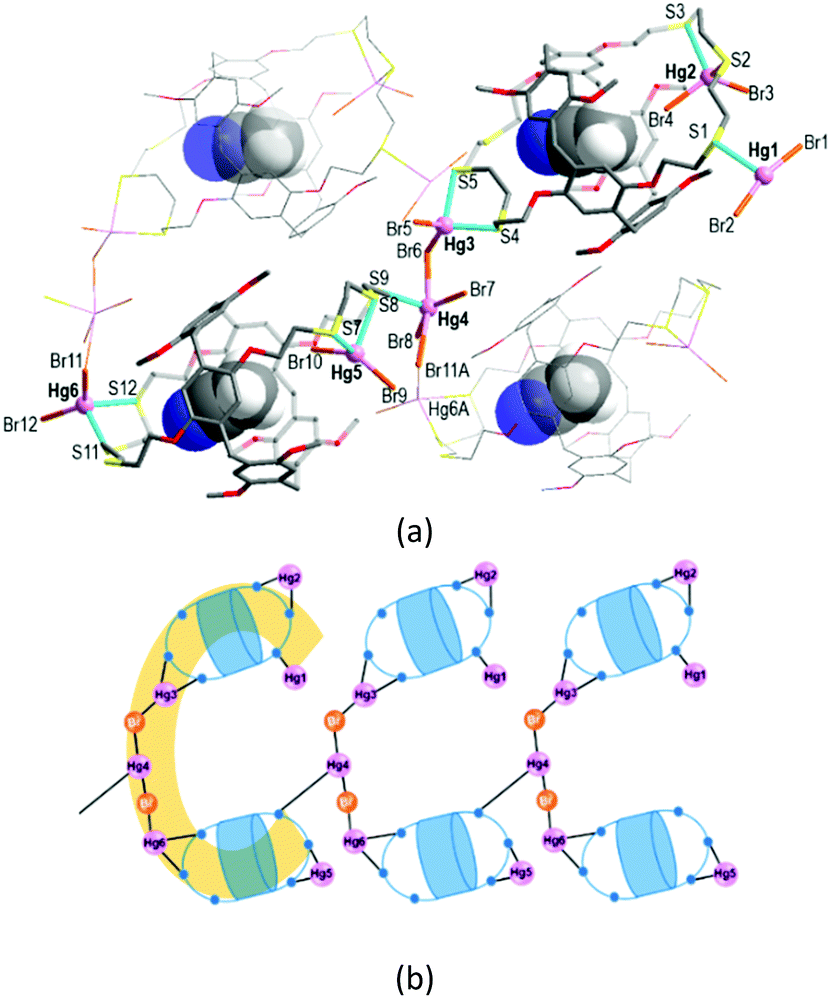 | ||
| Fig. 3 The exo-coordinated mercury(II) bromide complex [Hg6Br12(CH3CN@L)2]n (2): (a) the 1-D polymeric structure (the asymmetric unit is shown with the labelling) and (b) the connectivity pattern showing the linkage of the C-shaped dimers (terminal Br atoms are omitted). Symmetry operation A: −1 + x, −y, −0.5 + z. | ||
In 2, two (CH3CN@L) units which locate up and down are linked by a linear Hg3–Br–Hg4–Br–Hg6A segment via Hg–S bonds [2.577(5)–2.763(5) Å] to form a C-shaped dimer arrangement (Fig. 3b). Individual dimers are further linked via a Hg4–S9 bond [2.734(4) Å] to give a 1-D polymeric structure.
When HgI2 was reacted with L in dichloromethane/methanol, a pale yellow product 3 was isolated. In marked contrast to the chlorido and bromido complexes, 3 is an endocyclic ion-triplet complex with the formula [I·Hg·I@L] (3) which crystallises in the monoclinic space group P21/c with Z = 4 (Fig. 4a and Fig. S6, S7, Tables S1, S4, ESI†). In 3, surprisingly, one (I−–Hg2+–I−) entity locates inside the cavity of L. Although some ion-triplet organic cation complexes of types [(R1R2-NH2+)2·SO42−@bis(calix[6]arene)] and [(Cl−·R4N+·Cl−)@bis(calix[4]pyrrole)] have been reported,14 no solid ion-triplet metal complexes of such type have been reported so far.
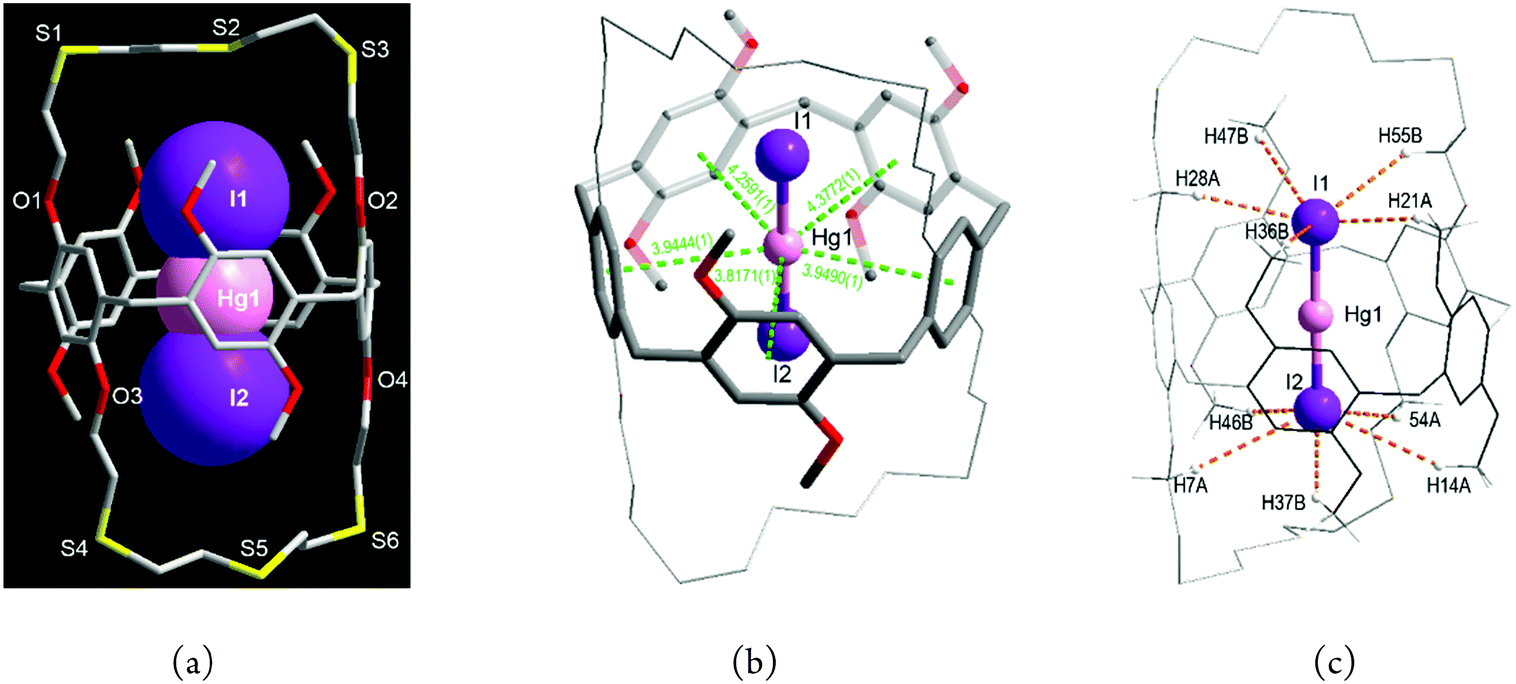 | ||
| Fig. 4 The contact ion-triplet complex with mercury(II) iodide [(I·Hg·I)@L] (3): (a) general view showing an (I−–Hg2+–I−) entity (space-filling) in the central cavity, (b) a pentagonal bipyramidal geometry with five Hg2+⋯π interactions (green dashed lines) and an I–Hg–I axis and (c) a mercury-shared double pentagonal pyramidal geometry with five C–H⋯I− H-bonds (orange dashed lines) and I–Hg–I axis (only H-bonded H atoms are shown). | ||
As mentioned above, L prefers to form exo-coordinated products with mercury(II) salts. Thus the formation of 3 raises some questions. (i) What is the driving force to stabilize the mercury(II) ion in the centre of the cavity? (ii) How is the iodide ion stabilized inside the crown loop? (iii) How is the linear (I−–Hg2+–I−) entity formed? Each of these aspects is now discussed.
First, some metal ions including K+, Cs+, Ag+ and Hg2+ interact with aromatic carbons via cation–π interactions, with such interactions being well established for calixarenes.15,16 With respect to this, the Hg2+ centre in 3 is stabilized by five such cation⋯π interactions (3.82–4.38 Å, dashed lines in Fig. 4b) to form a pentagonal planar array. Additionally, two I− ions occupy axial sites to yield an overall pentagonal bipyramidal geometry [Hg1–I1 2.5642(6), Hg1–I2 2.5555(6) Å, I1–Hg1–I2 178.30(2)°].
Second, the larger size and softer nature of the I− ion may play an advantageous role in the confined space. When large numbers of weak H-bonds act cooperatively, quite often they play an important role in stabilizing supramolecular structures. Thus, each I− ion in 3 is found to interact with five H atoms from different O–CH2 and O–CH3 groups via C–H⋯I− H-bonds,17 with the distances and angles being 3.2132(0)–3.5277(0) Å and 133.193(2)–157.987(2)° (Fig. 4c and Table S4, ESI†).
Third is the question of how the linear (I−–Hg2+–I−) entity might form. Mercury(II) iodide is known to crystallize in several polymorphs including linear, tetrahedral and tetragonal structures depending on the preparation process.18 In principle, the interaction of solvent-separated ion-pairs is primarily electrostatic, but mercury(II) iodide is quite soluble in common organic solvents including methanol19 and is present partially as a non-dissociated ion-triplet due to the covalent character.20 As mentioned, each I− ion is bound to a bridging μ-Hg2+ centre to yield a pentagonal pyramidal geometry (Fig. 4c). Since synthetic hosts for halide ions favour two to six binding interactions,2,3 each I− ion in Fig. 4c represents a good example of such a condition being met with six interactions present for each I− on forming the ion-triplet complex. The linear (I−–Hg2+–I−) entity in 3 is intimately associated with the peanut-shaped cavity of L giving rise to an excellent geometric and electronic complementary via the multiple cation–π interactions and H-bonds. Thus it not surprising how the linear arrays of HgI2 form because the solid structure of the yellow polymorph of HgI2, as mentioned, is almost linear (178.3°)18b and the linear HgI2 guest from [I–Hg–I⋯I–Hg–I] columns found in the CuII/HgII-MOF (MOF = metal–organic framework) is also stabilised by weak coordination of four iodide ions in the 3-D framework.21
Based on the UV-vis titration experiments in chloroform/methanol, the formation constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K) for the complexations of L with mercury(II) halides were obtained (Table 1 and Fig. S8–S10, ESI†). The logK values (for 1:1 complexes) tend to be smaller for larger anions because the larger halide ions as a softer base can bind to the mercury(II) centre (soft acid) more tightly and hence reduce the complex stability. The logK value for the iodide system is, however, larger than expected because the extra stabilization of the ion-triplet via the supramolecular interactions probably compensates this effect in part. In the bromide system, the ligand L binds to two mercury(II) ions via two steps but the 1:1 (ML) complex is more stable than the 2:1 complex (M2L) which is formed above the 1 equivalent of HgBr2.
K) for the complexations of L with mercury(II) halides were obtained (Table 1 and Fig. S8–S10, ESI†). The logK values (for 1:1 complexes) tend to be smaller for larger anions because the larger halide ions as a softer base can bind to the mercury(II) centre (soft acid) more tightly and hence reduce the complex stability. The logK value for the iodide system is, however, larger than expected because the extra stabilization of the ion-triplet via the supramolecular interactions probably compensates this effect in part. In the bromide system, the ligand L binds to two mercury(II) ions via two steps but the 1:1 (ML) complex is more stable than the 2:1 complex (M2L) which is formed above the 1 equivalent of HgBr2.
K) for the complexations of L with mercury(II) halides in chloroform/methanol (1:2.5, v/v) at 298 Ka
In summary, we report here the isolation of the first solid state example of a supramolecular ion-triplet complex incorporating a hetero-tritopic macrocycle. To achieve this, the pillar[5]-bis-trithiacrown L was synthesised and its anion-dependent mercury(II) halide complexes exhibiting different topological arrangements were isolated. In the chlorido and bromido complexes, exo-coordination via Hg–S bonds occurs, with the corresponding anions binding only to the cation (in different mutual configurations). In the iodido complex, however, the mode of interaction of this anion is totally different; it not only interacts with the metal cation but also with the ligand L to form the unique contact ion-triplet complex. Consequently, the formation of the ion-triplet complex reflects that the multiple Hg2+⋯π interactions stabilize the overall structure, with the concerted C–H⋯I− H-bonds being geometrically and electronically well matched to satisfy the mutual complementarity required for binding in the confined central space in L. A further investigation of the potential application of triplet complexes of the present type for the detection and removal of toxic heavy metal species is in progress.
This work was supported by the National Research Foundation (NRF) of South Korea (2017R1A4A1014595 and 2019R1A2C1002075). We are thankful to Prof. Naoki Hirayama (Toho University) for the helpful discussions on fitting data.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) L. F. Lindoy, The Chemistry of Macrocyclic Complexes, Cambridge University Press, Cambridge, U.K., 1989 CrossRef; (b) J.-M. Lehn, Supramolecular Chemistry, Concept and Perspectives, VCH, Weinheim, Germany, 1995 CrossRef.
- P. A. Gale and J. E. Haynes, Anion Receptors Containing Heterocyclic Rings. in Supramolecular Chemistry, ed. P. A. Gale and J. W. Steed, John Wiley & Sons Ltd, Chichester, U.K., 2012, vol. 3, pp. 1125–1151 Search PubMed.
- (a) N. Busschaert, C. Caltagirone, W. V. Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed; (b) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry; Monographs. in Supramolecular Chemistry, ed. J. F. Stoddart, RSC Publishing, Cambridge, U.K., 2006 Search PubMed.
- (a) Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed; (b) A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef CAS PubMed; (c) B. D. Smith, Ion-pair Recognition by Ditopic Receptors. in Macrocyclic Chemistry: Current Trends and Future, ed. K. Gloe and B. Antonioli, London, U.K., 2005 Search PubMed; (d) J.-M. Lü, S. V. Rosokha, S. V. Lindeman, I. S. Neretin and J. K. Kochi, J. Am. Chem. Soc., 2005, 127, 1797–1809 CrossRef PubMed; (e) J. M. Mahoney, A. M. Beatty and B. D. Smith, J. Am. Chem. Soc., 2001, 123, 5847–5848 CrossRef CAS PubMed; (f) S. Moerkerkr, S. L. Gac, F. Topić, K. Rissanen and I. Jabin, Eur. J. Org. Chem., 2013, 5315–5322 CrossRef.
- J. Eckelmann, V. Saggiomo, F. D. Sönnichsen and U. Lüning, New J. Chem., 2010, 34, 1247–1250 RSC.
- H. Kim and S. S. Lee, Inorg. Chem., 2008, 47, 10807–10809 CrossRef CAS PubMed.
- S. Park, S. Y. Lee, K.-M. Park and S. S. Lee, Acc. Chem. Res., 2012, 45, 394–403 Search PubMed.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) X. Shu, S. Chen, J. Li, Z. Chen, L. Weng, X. Jia and C. Li, Chem. Commun., 2012, 48, 2967–2969 RSC.
- (a) T. Ogoshi, Pillararenes, The Royal Society of Chemistry, Cambridge, U.K., 2016 Search PubMed; (b) M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed; (c) T. Ogoshi, T.-A. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed; (d) J. Chen, H. Ni, Z. Meng, J. Wang, X. Huang, Y. Dong, C. Sun, Y. Zhang, L. Cui, J. Li, X. Jia, Q. Meng and C. Li, Nat. Commun., 2019, 10, 3546–3553 CrossRef PubMed; (e) Y. Wang, K. Xu, B. Li, L. Cui, X. Jia, H. Zhao, J. Fang and C. Li, Angew. Chem., Int. Ed., 2019, 131, 10387–10390 CrossRef; (f) M. Guo, X. Wang, C. Zhen, P. Demay-Drouhard, W. Li, K. Du, M. A. Olson, H. Zhilhof and A. C.-H. Sue, J. Am. Chem. Soc., 2018, 140, 74–77 CrossRef CAS PubMed.
- (a) E. Lee, H. Ju, I.-H. Park, J. H. Jung, M. Ikeda, S. Kuwahara, Y. Habata and S. S. Lee, J. Am. Chem. Soc., 2018, 140, 9669–9677 CrossRef CAS PubMed; (b) E. Lee, H. Ryu, H. Ju, S. Kim, J.-E. Lee, J. H. Jung, S. Kuwahara, M. Ikeda, Y. Habata and S. S. Lee, Chem. Eur. J., 2019, 25, 949–953 CrossRef CAS PubMed; (c) E. Lee, I.-H. Park, H. Ju, S. Kim, J. H. Jung, Y. Habata and S. S. Lee, Angew. Chem., Int. Ed., 2019, 58, 11296–11300 CrossRef CAS PubMed.
- (a) N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed; (b) B. Hua, Z. Zhang, J. Liu and F. Huang, J. Am. Chem. Soc., 2019, 141, 15008–15012 CrossRef CAS PubMed.
- L. Shao, B. Hua, J. Liu and F. Huang, Chem. Commun., 2019, 55, 4527–4530 RSC.
- (a) R. E. Wolf Jr, J. R. Hartman, J. M. E. Storey, B. M. Foxman and S. R. Cooper, J. Am. Chem. Soc., 1987, 109, 4328–4335 CrossRef; (b) J. Buter, R. M. Kellog and F. van Bolhuis, J. Chem. Soc., Chem. Commun., 1991, 910–912 RSC; (c) G. H. Robinson and S. A. Sangokoya, J. Am. Chem. Soc., 1988, 110, 1494–1497 CrossRef CAS.
- (a) S. Moerkerke, M. Mén and I. Jabin, Chem. Eur. J., 2010, 16, 11712–11719 CrossRef CAS PubMed; (b) S. Moerkerke, S. L. Gac, F. Topić, K. Rissanen and I. Jabin, Eur. J. Org. Chem., 2013, 5315–5322 CrossRef CAS; (c) V. Valderrey, E. C. Escudero-Adán and P. Ballester, Angew. Chem., Int. Ed., 2013, 52, 6898–6902 CrossRef CAS PubMed; (d) R. Molina-Muriel, G. Aragay, E. C. Escudero-Adán and P. Ballester, J. Org. Chem., 2018, 83, 13507–13514 CrossRef CAS PubMed.
- (a) W. Lau and J. K. Kochi, J. Org. Chem., 1986, 51, 1801–1811 CrossRef CAS; (b) E. S. Meadows, S. L. De Wall, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 2001, 123, 3092–3107 CrossRef CAS PubMed; (c) Y. Habata, A. Taniguchi, M. Ikeda, T. Hiraoka, N. Matsuyama, S. Otsuka and S. Kuwahara, Inorg. Chem., 2013, 52, 2542–2549 CrossRef CAS PubMed.
- (a) A. T. Macias, J. E. Norton and J. D. Evanseck, J. Am. Chem. Soc., 2003, 125, 2351–2360 CrossRef CAS PubMed; (b) S. K. Kim, J. L. Sessler, D. E. Gross, C.-H. Lee, J. S. Kim, V. M. Lynch, L. H. Delmau and B. P. Hay, J. Am. Chem. Soc., 2010, 132, 5827–5836 CrossRef CAS PubMed; (c) H. Ju, C. Kim, K.-S. Choi, E. Lee, S. Kim, J. H. Jung, Y. Habata, L. F. Lindoy and S. S. Lee, Eur. J. Inorg. Chem., 2018, 3587–3594 CrossRef CAS.
- H. S. El-Sheshtawy, B. S. Bassil, K. I. Assaf, U. Kortz and W. M. Nau, J. Am. Chem. Soc., 2012, 134, 19935–19941 CrossRef CAS PubMed.
- (a) M. Hostettler and D. Schwarzenbach, Acta Crystallogr., 2002, B58, 914–920 CrossRef CAS PubMed; (b) M. Hostettler, H. Birkedal and D. Schwarzenbach, Helv. Chim. Acta, 2003, 86, 1410–1422 CrossRef CAS.
- I. Persson, M. Landgren and A. Marton, Inorg. Chim. Acta, 1986, 116, 135–144 CrossRef CAS.
- D. M. Considine and G. D. Considine, Mercury. Van Nostrand's scientific encyclopedia, Springer, US, Boston, 1995, pp. 2024–2025 Search PubMed.
- Y.-B. Dong, M. D. Smith and H.-C. Z. Loye, Angew. Chem., Int. Ed., 2000, 39, 4271–4273 CrossRef CAS PubMed.
- Online at http://www.hyperquad.co.uk/HypSpec2014.htm.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2004262–2004265. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03902k |
| This journal is © The Royal Society of Chemistry 2020 |