
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA minireview on the perturbation effects of polar groups to direct nanoscale hydrophobic interaction and amphiphilic peptide assembly
Feiyi Zhang†
a,
Lanlan Yu†b,
Wenbo Zhang†b,
Lei Liu *a and
Chenxuan Wang*b
*a and
Chenxuan Wang*b
aInstitute for Advanced Materials, Jiangsu University, Zhenjiang, Jiangsu 212013, China. E-mail: liul@ujs.edu.cn
bState Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences Chinese Academy of Medical Sciences, School of Basic Medicine Peking Union Medical College, Beijing 100005, China. E-mail: wangcx@ibms.pumc.edu.cn
First published on 25th August 2021
Abstract
Hydrophobic interaction provides the essential driving force for creating diverse native and artificial supramolecular architectures. Accumulating evidence leads to a hypothesis that the hydrophobicity of a nonpolar patch of a molecule is non-additive and susceptible to the chemical context of a judicious polar patch. However, the quantification of the hydrophobic interaction at the nanoscale remains a central challenge to validate the hypothesis. In this review, we aim to outline the recent efforts made to determine the hydrophobic interaction at a nanoscopic length scale. The advances achieved in the understanding of proximal polar groups perturbing the magnitude of hydrophobic interaction generated by the nonpolar patch are introduced. We will also discuss the influence of chemical heterogeneity on the modulation of amphiphilic peptide/protein assembly and molecular recognition.
1 Introduction
Hydrophobic interactions underlying the water-mediated organization of nonpolar molecules provide the key driving force for a broad range of biological phenomena, such as protein folding and aggregation, cell membrane and vesicle formation, and phase separation of specific proteins and nucleic acids.1–3 A profound understanding of hydrophobic interactions is essential to disentangle the evolutionary drivers of assembly in the enormous protein structures, unraveling the causes of chemical selection in the rational design of molecular machines.4–6 Most biological systems do not retain a macroscopic nonpolar surface.7,8 The nonpolar domain within a protein is typically nanometer-sized and flanked by a variety of polar functional groups. The nanoscopic length scale of a nonpolar patch produces tremendous difficulty to characterize hydrophobicity. First, the hydrophobicity of a solid surface at the macroscopic length scale can be manifested by the optically measured contact angle of a macro-sized aqueous droplet with the surface. But such an approach does not apply to the characterization of the nanometer-sized hydrophobicity due to the limitation of resolution in the microscope.9 Second, the potential for assembly can be described as ΔG = γΔA, in which γ is the excess free energy density of the interfaces of a protein nonpolar patch in water, and ΔA is the area of a nonpolar patch to provide the driving force for assembly.8,10–12 However, the magnitude of γ for a protein nonpolar domain is usually smaller than the empirical magnitude, such as the nonpolar oil/water interfacial tension, resulting in a failure in the translation of hydrophobicity from macroscopic scale to nanoscale.8,13 Thus, the determination of nanoscale hydrophobicity remains a challenging task. The development of experimental and theoretical techniques is of genuine interest and could help study the mechanism underlying hydrophobically driven biomolecular folding, assembly, and intermolecular recognition.Substantial efforts have been made to improve the understanding of hydrophobic interactions based on conceptual models, in which a protein is usually decomposed into a sum of its constituent amino acids and different types of intermolecular interactions are conventionally regarded as independent and pairwise-additive. But such an approach seems to be inadequate. Many attempts to predict the protein folding structure via an energetic summation over the constituent parts have encountered difficulty in practice.7,14,15 For example, the A domain and B domain of G protein, a multidomain cell wall protein, share 88% sequence identity and are calculated to be similar with each other in structure and functions from the perspective of the additive energetics of protein folding.16 However, these two domains adopt distinct folding structures, α-helical versus α/β heterozygous conformation. They also differ in molecular recognition (i.e., the A domain of G protein binds to human serum albumin, whereas the B domain binds to the Fc region of IgG).16 Collective experimental and theoretical studies lead to the fact that the intermolecular interactions that govern the folding and assembly of a protein/peptide present the non-additive characteristics and hydrophobic interaction displays a dependence on the nanoscale chemical heterogeneity of a biological molecule.
In this review, we aim to introduce the recent approaches to reveal the chemical heterogeneity-dependence of hydrophobic interactions and present the advances of efforts to characterize the hydrophobicity at a nanoscopic length scale. We will also elucidate the outcomes of the proximal polar group altering hydrophobic interactions on the modulation of peptide assembly and molecular recognition. We envision that the study with the chemical heterogeneity-dependence of hydrophobic interactions provides a fresh vision for engineering the hydrophobically driven protein/peptide assemblies and reconsidering the structure–activity correlations for protein mutations.
2. The perturbation effect of polar group on hydrophobic interactions
Conventionally, the hydrophobicity of a molecule is approximately regarded as additive.17,18 For instance, the hydrophobic energy corresponding to an alkyl chain transferred from water into micelle is usually considered to be linearly proportional to the number of –CH2– group with an increment of 3.8 kJ mol−1 per –CH2– group at room temperature.17,18 However, collective evidence from different chemical and biological systems suggests that there exists a correlation between the polar group adjacent to the nonpolar patch and the hydrophobicity of a molecule, i.e., the hydrophobic interaction is non-additive. For example, asparagine and glutamine have different numbers of –CH2– groups in their side chains, but there is no appreciable difference in the solubility.19 Similarly, the hydrophobic contribution of abundant –CH2– groups in polyethylene glycol (PEG) is missing and PEG is infinitely soluble in water since the –CH2– groups in PEG are nearby polar oxygen.20 These facts lead to an important hypothesis that the contribution of the nonpolar –CH2– group to molecular solubility/hydrophobicity is influenced by the judicious polar groups.19Quantifying the nanometer-scale hydrophobicity of a chemically heterogeneous system remains a key challenge for improving our understanding of hydrophobic interaction. Ma et al. designed a conformationally stable β3-amino acid oligomer GA-KKK that generated a globally amphiphilic (GA) helix and quantified the adhesive forces arising from hydrophobic interactions of the GA-KKK with an atomic force microscope (AFM) tip that was made non-polar by adsorbing a monolayer of dodecanethiol (Fig. 1a and b). β-Peptide GA-KKK contains 14-atom hydrogen-bonded rings and presents a 1 nm2-sized non-polar domain comprised of six cyclohexyl groups from (1S,2S)-2-aminocyclohexane carboxylic acid (ACHC) residues.21 On the opposing helical face of GA-KKK, a charged domain comprised of three β3-homolysine (β3-hLys) immobilized ∼1 nm from the non-polar domain (Fig. 1b). At pH 7, where the side chains of β3-hLys are protonated, the hydrophobic interactions between the non-polar ACHC-rich domain of GA-KKK and the AFM tip were determined to be 0.90 ± 0.05 nN (Fig. 1c and d). Strikingly, the replacement of β3-hLys by another cationic β3-amino acid, β3-homoarginine (β3-hArg), in the GA peptide sequence (i.e., GA-RRR), led to a substantial decrease in the strength of hydrophobic interactions between the GA peptide and the AFM tip (Fig. 1b–d). This result demonstrated that the hydrophobic interactions generated by the nonpolar domain (∼1 nm2 in size) are modulated by the proximally immobilized cations and such modulation effects depend on the charge identity.21
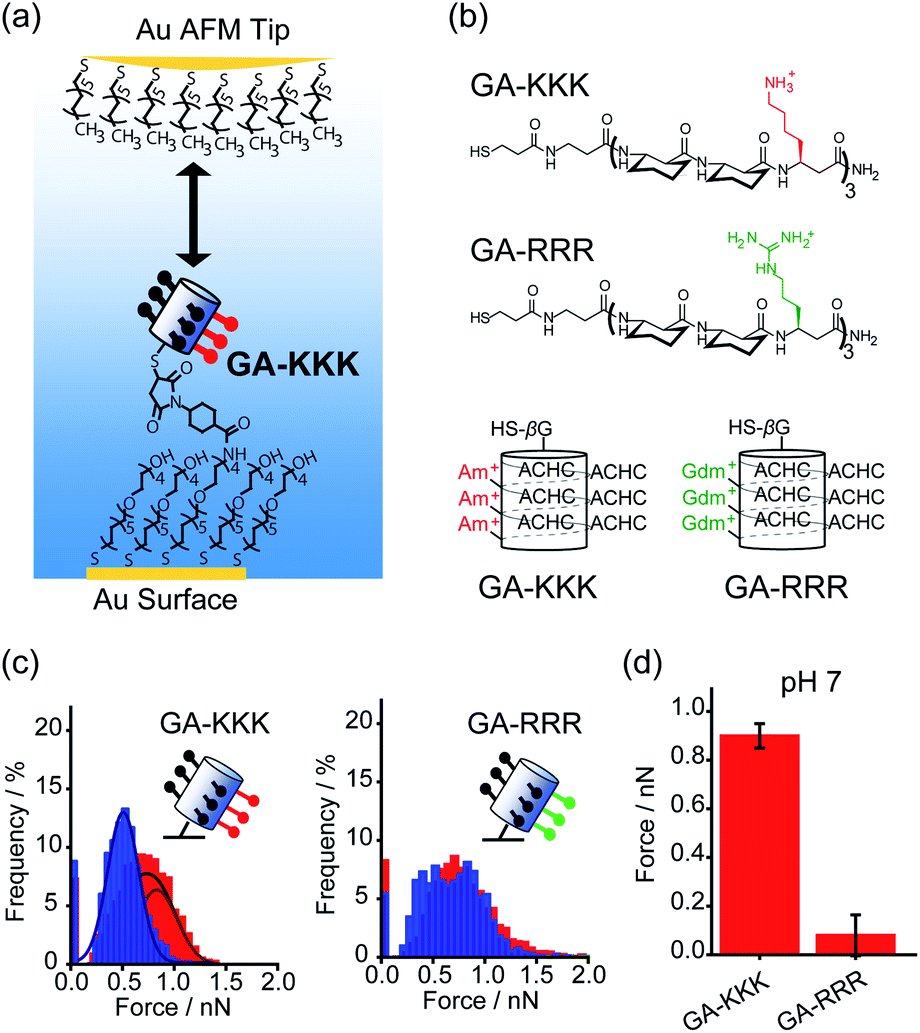 | ||
| Fig. 1 Influence of proximal cations on the hydrophobic interactions of globally amphiphilic β-peptides. (a) Schematic illustration of an alkyl-terminated AFM tip interacting with an immobilized GA-KKK.21 Reproduced with permission from ref. 21. Copyright 2015 Springer Nature. (b) The chemical structures (top) and helical representations (bottom) of β-peptides GA-KKK and GA-RRR at pH 7. (c) Histograms of adhesion forces were measured between a nonpolar AFM tip and immobilized GA-KKK (left) or GA-RRR (right) in either aqueous solution at pH 7 (red) or 60 vol% MeOH aqueous solution at pH 7 (blue).21,37 Reproduced with permission from ref. 37. Copyright 2021 American Chemical Society. (d) The magnitudes of mean hydrophobic interactions are determined from (c).21,37 Data showed mean ± SEM. | ||
Other than cations, anions and neutral-polar groups also participate in the modulation of adjacent hydrophobicity. For example, the immobilized univalent anions are observed to weaken the hydrophobic interactions generated by the neighboring nonpolar domain.22 The potential of mean force (PMF) simulation of a coarse-grained GA β-peptide nearby a nonpolar surface (Fig. 2a) revealed that the immobilization of F−, Cl−, Br−, and I− into the proximity of the nonpolar domain of a GA β-peptide substantially decreased the PMF strength of hydrophobic interactions. The PMFs of hydrophobic interactions measured from the GA β-peptides with negative charges followed the rank as I− < F− < Cl− ≈ Br− (Fig. 2b).22 This result also provided instructive information that immobilized ions versus free ions exert divergent impacts on hydrophobic interaction. The free F−, Cl−, and I− in bulk solution were found to strengthen hydrophobic interaction, contrasting with the influence of immobilized anions on the GA β-peptide surface to weaken hydrophobic interaction (Fig. 2c).22
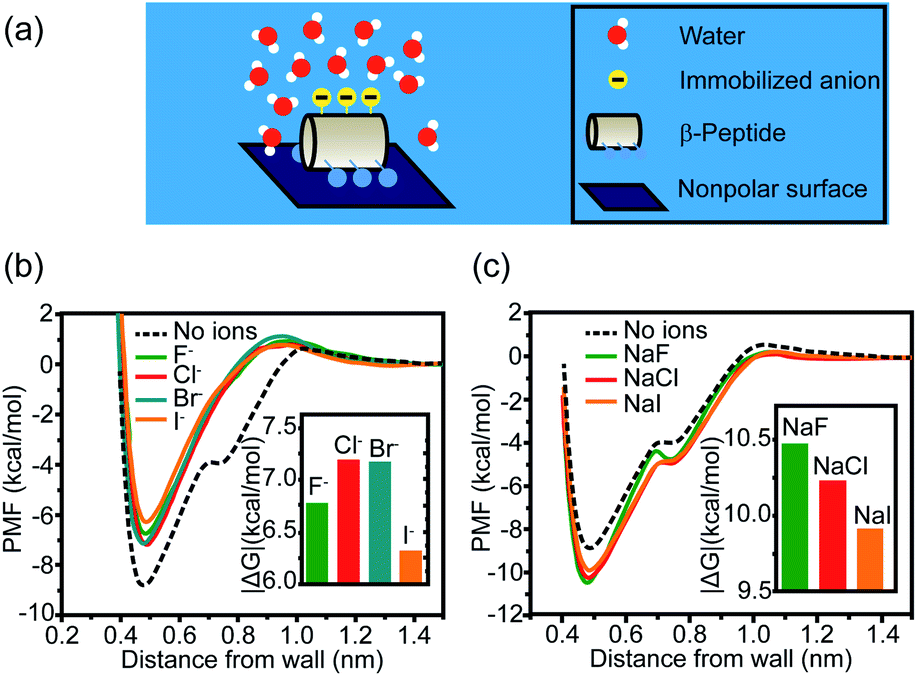 | ||
| Fig. 2 Influence of proximal anions on the hydrophobic interactions. (a) Schematic illustration of a globally amphiphilic β-peptide interacting with a nonpolar surface in water. (b) The PMFs of hydrophobic interactions between a nonpolar surface and the β-peptide with proximally immobilized anions. (c) The PMFs of hydrophobic interactions between a nonpolar surface and the β-peptide in the presence of dissolved anions. The inset represents the absolute value of the difference in PMF between the depth of contact minimum and the reference state.22 Reproduced with permission from ref. 22. Copyright 2015 American Chemical Society. | ||
The perturbation effect of the neutral-polar group on hydrophobic interaction is manifested by the molecular dynamics simulation of the hydration feature of heterogeneous self-assembled monolayer (SAM) surface.14 As revealed by the local compressibility of interfacial water in the vicinity of the SAM surface, the addition of a single hydroxyl-terminated group into the methyl-terminated SAM surface led to a delocalized effect on the local density and dynamic structuring of interfacial water (Fig. 3a). The perturbation effect propagates from the single hydroxyl group site with an extending area of ∼4 nm2 (Fig. 3b).14 For the uniformly mixed hydroxyl/methyl-terminated SAM surfaces composing of 25%, 50%, and 75% hydroxyl groups, the interfacial water compressibility decreased nonlinearly with the increased surface concentration of hydroxyl-terminated groups (Fig. 3c and d).14 This nonlinearity in how the polar group affects the wetting property highlights the non-additive nature of hydrophobicity at the nanoscale.
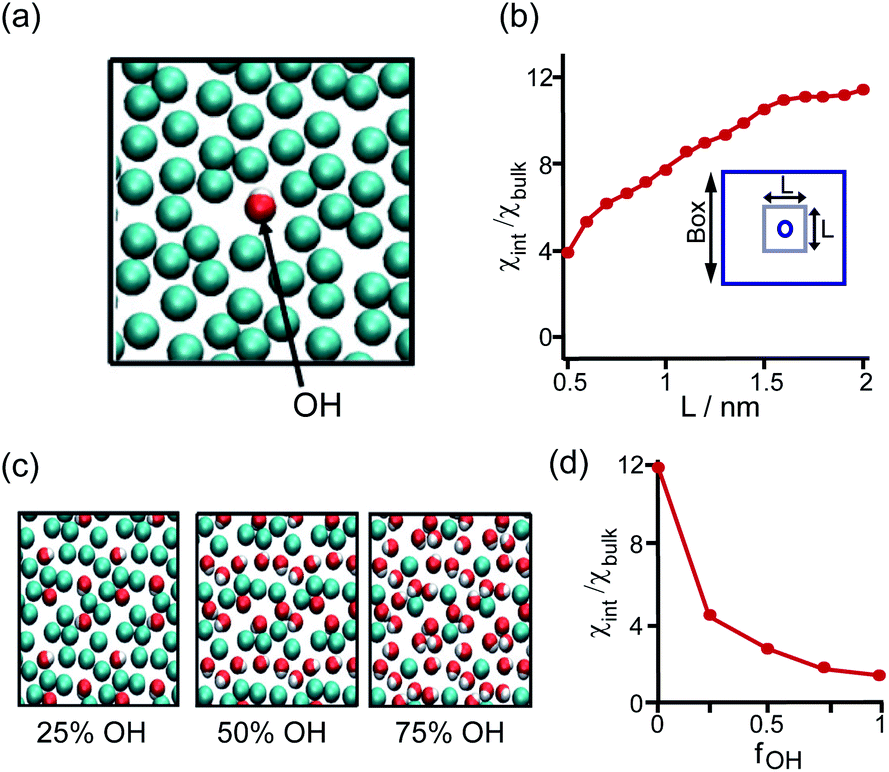 | ||
| Fig. 3 Influence of the proximal neutral-polar group on the hydrophobic interactions. (a) Schematic illustration of the top view of the heterogeneous methyl-terminated SAM surface containing one hydroxyl group. (b) Dependence of normalized interfacial compressibility (χint/χbulk) of the perturbation caused by a single hydroxyl group on the length scale (L). (c) Top view of the heterogeneous methyl-terminated SAM surface containing 25%, 50%, and 75% hydroxyl groups. (d) Dependence of the normalized interfacial compressibility of perturbation on the surface fraction of hydroxyl group (fOH).14 Reproduced with permission from ref. 14. Copyright 2010 Springer Nature. | ||
3 Interfacial water and the mechanism underlying the perturbation effect of polar group
The effect of the proximally immobilized polar group on the hydrophobic interaction generated by the nonpolar patch motivates consequent studies to reveal the modulation mechanism of how the immobilized polar group contributes to hydrophobic interaction. Hydrophobic interaction is defined as the attractive interaction with a magnitude exceeding van der Waals interaction between two nonpolar particles in an aqueous solution.23,24 Hydrophobic interaction is originated from the assembly structure of interfacial water molecules adjacent to the nonpolar patch (Fig. 4).23,24 The interfacial water assembly structure around the nonpolar patch is historically depicted by the iceberg model, in which interfacial water molecules are thought to form frozen patches or microscopic icebergs around the nonpolar parts of solute molecules in an aqueous solution.23,25 Different from the disordered structure of water molecules in bulk solution, interfacial waters are suspected to adopt tetrahedral configuration and assemble into a continuous hydrogen-bonded interfacial water network, constructing polyhedral cage-shaped hydration shell around hydrophobic solute (Fig. 4).25,26 The ordered hydrogen-bonded water network gives rise to hydrophobic free energy and thus a fluctuation of the structure and density of interfacial water nearby the nonpolar patch will cause a variation in the magnitude of hydrophobic interaction.24 | ||
| Fig. 4 The assembly structure of interfacial water molecules near hydrophobic surface. The blue and white particles represent the oxygen and hydrogen atoms of water. The dashed lines indicate hydrogen bonds. The red particles represent the hydrophobic species.26 Reproduced with permission from ref. 26. Copyright 2005 Springer Nature. | ||
By using the iceberg model, the perturbation effect of polar group observed with the comparison of GA-KKK versus GA-RRR is attributed to the correlation between the proximally immobilized cation and the interfacial water structure. Two possible mechanisms might participate in the interaction between the immobilized cation and interfacial water (Fig. 5a).22,27 First, the protonated cationic side chain can interact with interfacial water adjacent to the ACHC-rich nonpolar domain via a charge–dipole interaction (Fig. 5a). Considering the side chains of β3-hLys and β3-hArg possess similar net charge density, the charge–dipole interaction between the immobilized cation and interfacial water is not appreciable to cause the perturbing effect on hydrophobic interaction. In contrast, a second mechanism has been raised that the fluctuation of interfacial water organization propagates from charged group to the site of water nearby the nonpolar domain via the hydrogen-bonded interfacial water network (Fig. 5a).22,27 Such a mechanism can be leveraged to formulate a hypothesis that two polar but neutral groups with divergent hydration free energy possess the group-specific potential to modulate adjacent hydrophobic interaction. Wang et al. substituted the homolysine side chains in β-peptide GA-KKK by using polar but nonionic amino acid homoglutamine (β3-hGln) to generate GA-QKK and GA-QQK and reported chemical force microscopy measurements on single globally amphiphilic β-peptides (Fig. 5b).27 At pH 10.5, where the side chain of β3-hLys is deprotonated, a weaker hydrophobic interaction was measured using GA-QKK (0.54 ± 0.01 nN) and GA-QQK (0 ± 0.01 nN) relative to GA-KKK (0.61 ± 0.04 nN) with the AFM tips (Fig. 5c and d).27 The distinct modulation effects displayed by neutral β3-hLys versus β3-hGln on hydrophobic interaction reflect the different ability of amine versus amide, the polar groups carried by the side chains of β3-hLys versus β3-hGln, to interact with water. The hydration free energy of amine is −18.8 kJ mol−1 (ethylamine), whereas the hydration free energy of amide is −40.8 kJ mol−1 (acetamide).27–29 Thus, the side chain of β3-hGln is more favorable to interact with water and perturb the interfacial water structure nearby the nonpolar domain of β-peptide in energy relative to the side chain of deprotonated β3-hLys.27
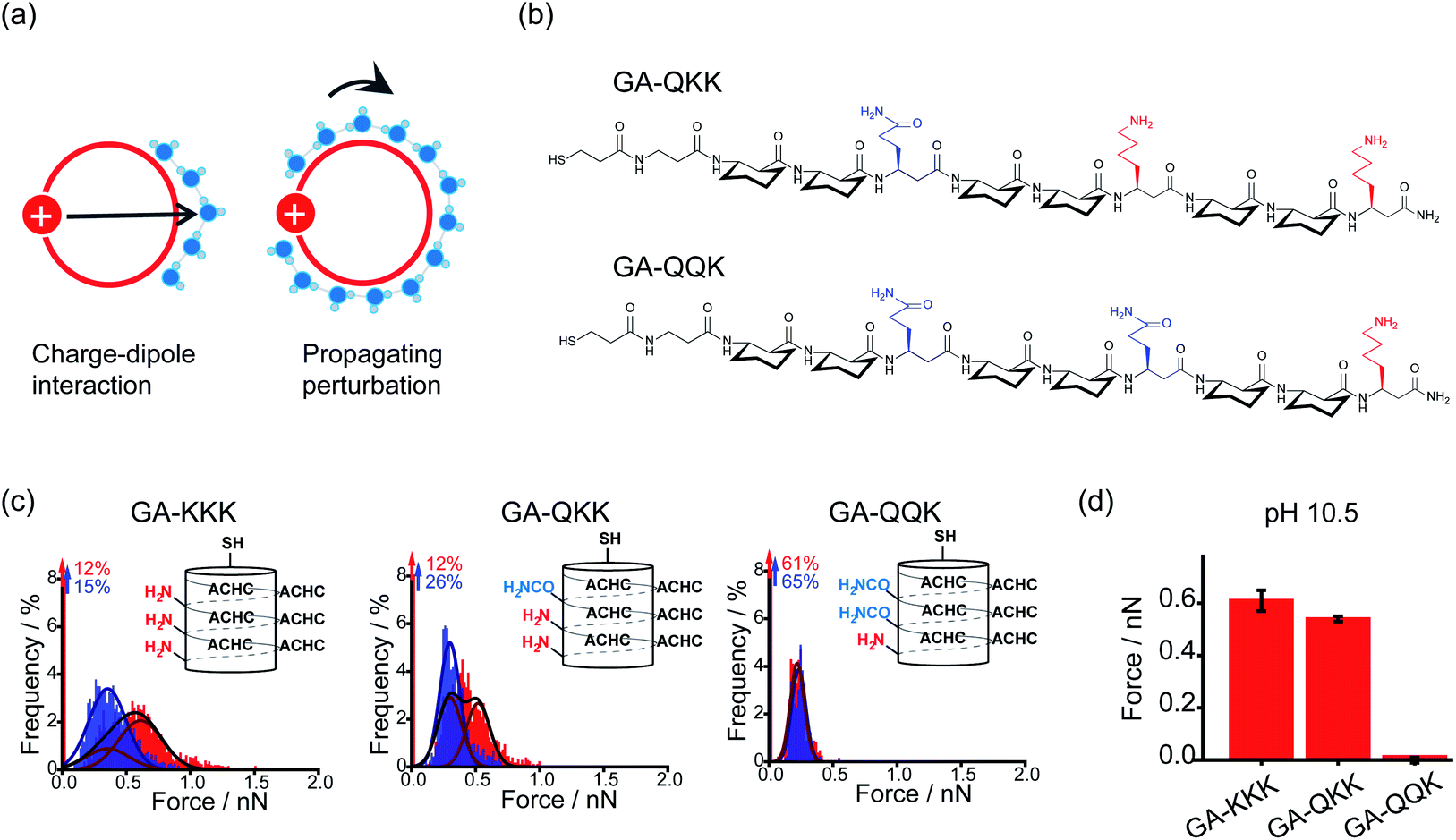 | ||
| Fig. 5 Mechanisms underlying the perturbation effect of polar groups. (a) Schematic illustrations of two possible mechanisms by which cationic groups influence the organization of water molecules nearby the nonpolar surface of β-peptide. The red circles represent cross-sections of the helical β-peptides. The solid red disk with a white plus sign represents the cationic side chain. Blue spheres are indicative of interfacial water molecules. (b) The chemical structures of GA-QKK and GA-QQK at pH 10.5. (c) Histograms of adhesion forces were measured between a nonpolar AFM tip and immobilized GA-KKK, GA-QKK, and GA-QQK in either aqueous solution at pH 7 (red) or 60 vol% MeOH aqueous solution at pH7 (blue).27 Reproduced with permission from ref. 27. Copyright 2017 American Chemical Society. (d) The magnitudes of mean hydrophobic interactions are determined from (c).27 Data showed mean ± SEM. | ||
The perturbation effect of polar group on hydrophobic interaction can also be understood from the view of interfacial water dynamics. The liquid/solid interface strongly influences the dynamic features of interfacial water nearby the solid surface, including the structural ordering, density, and diffusion.30–32 The perturbation imposed by the interface to the interfacial water dynamics is a function of distance to the interface, typically decaying within 1 nm of the interface (Fig. 6a).31,32 Molecular simulations revealed that the density and the dipole orientation distribution of interfacial water molecules next to the hydrated surface were modulated by the chemically functional groups on the surface (Fig. 6b and c).8,33 Such modulation effect is non-additive, in which the adhesive free energy of the surface deviates from the linear dependence on the surface chemical composition.33 The diffusion of interfacial waters within the hydration shell of the surface is also heterogeneous and affected by the chemical identity presenting on the surface.34–36 As manifested by the Overhauser dynamic nuclear polarization relaxometry experiment, significant differences emerged from the water motions within a distance of ∼1 nm from the interface, with fast-diffusing water molecules in the vicinity of hydrophobic patches and slow-moving water molecules at the surface of hydrophilic patches (Fig. 6d).36
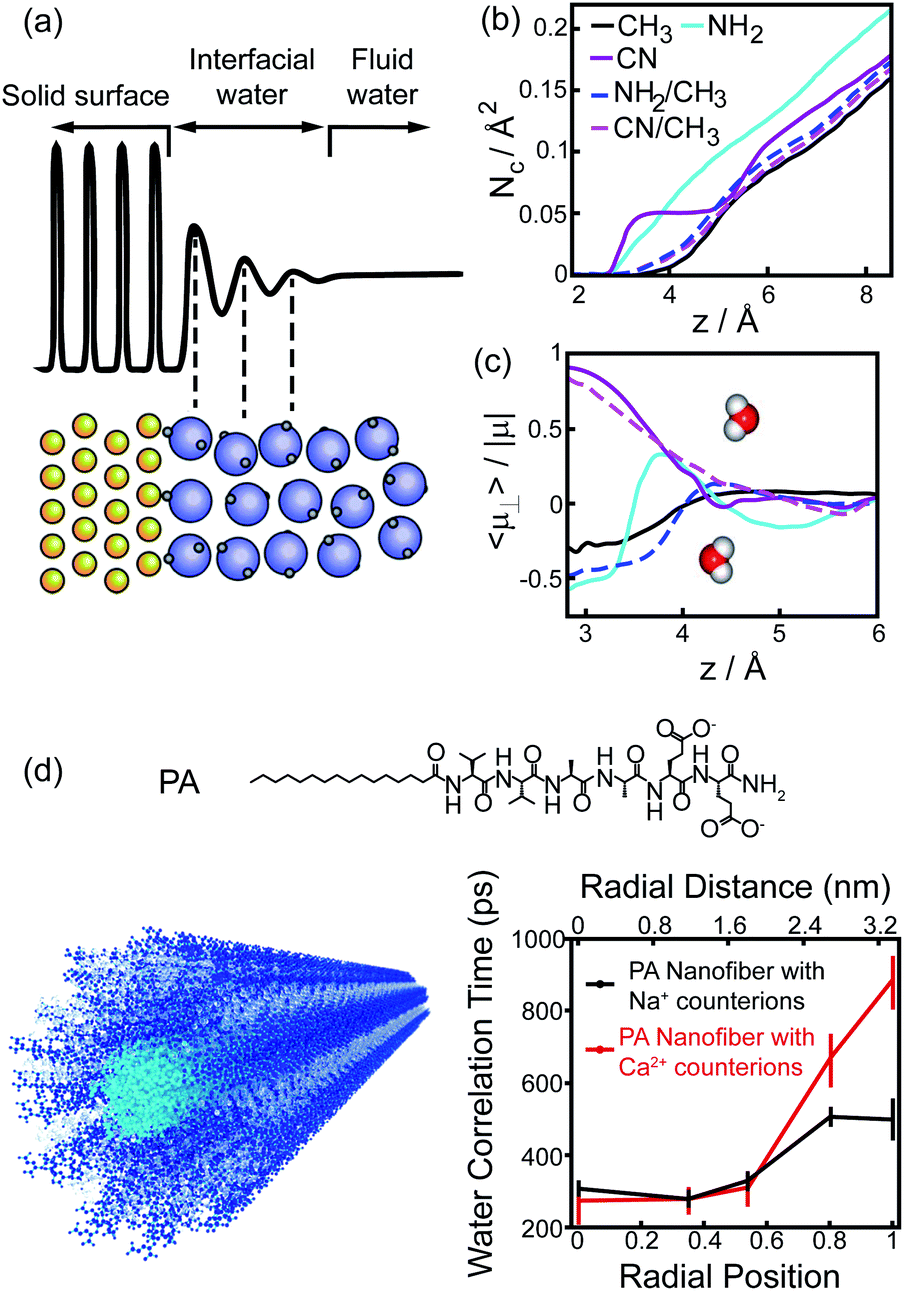 | ||
| Fig. 6 Interfacial water dynamics surrounding a chemically heterogeneous surface. (a) The interfacial water density oscillates nearby an interface.32 Reprinted with permission from ref. 32, Copyright 2016 American Chemical Society. (b), (c) The coordination numbers (b) and dipole orientations (c) of interfacial water molecules close to the surfaces terminated with different functionalized groups.33 Reprinted with permission from ref. 33, Copyright 2011 National Academy of Sciences. (d) The chemical structure of PA peptide and the schematic image of PA assembled nanofiber (left). Blue, PA peptide; cyan, hydrophobic tail; grey, interfacial water. Water-correlation times measured by the Overhauser dynamic nuclear polarization depend on the radial position within the cross-section of PA nanofibers.36 Reproduced with permission from ref. 36. Copyright 2017 American Chemical Society. | ||
4 The perturbation effect of polar group is manifested in the hydrophobically-driven self-assembly of peptide
The judicious placing of polar groups near hydrophobic domains dramatically tunes hydrophobic interaction, providing an invaluable handle for manipulating and designing peptide–peptide interactions.5 Conventionally, the self-assembled structure of an amphiphile is theoretically dependent on its packing parameter, which is defined relative to the area of the amphiphile head-group, the nonpolar chain volume, and the nonpolar chain length.17 Different from the conventional opinion, the interplay between polar group and nanoscale hydrophobicity can be employed as a harness to control the hydrophobically driven self-assembly by manipulating the chemical identity of the polar group instead of changing the molecular geometry of amphiphile. For example, the globally amphiphilic β-peptides, GA-KKK and GA-RRR, possess the same packing parameter but exhibit divergent assembled structures in aqueous 10 mM TEA solution at pH 7 (Fig. 7a).37 GA-KKK preserves a strong tendency to hydrophobically assemble into sheet-like aggregates of about 100 nm in hydrodynamic diameter. In contrast, GA-RRR displays substantially weak assembly propensity with association states ranging from monomer to nonamer (Fig. 7a).37 The phenomenon that lysine and arginine are not interchangeable in protein structure has been occasionally observed in some past studies, such as cell-penetrating peptides preferring arginine-rich domain for membrane permeation or strong nucleic acid binding potency.38–42 However, these past studies attribute the structural variations correlated with the lysine-to-arginine substitution to be their difference in hydrogen bonding or mediating coulombic effect. The result obtained from the cation-directed β-peptide assembly experiment provided the first clear demonstration that it is a feasible strategy to toggle the hydrophobically-driven self-assembly behaviors of peptides by a rational design of the chemical identity of a judicious polar group.37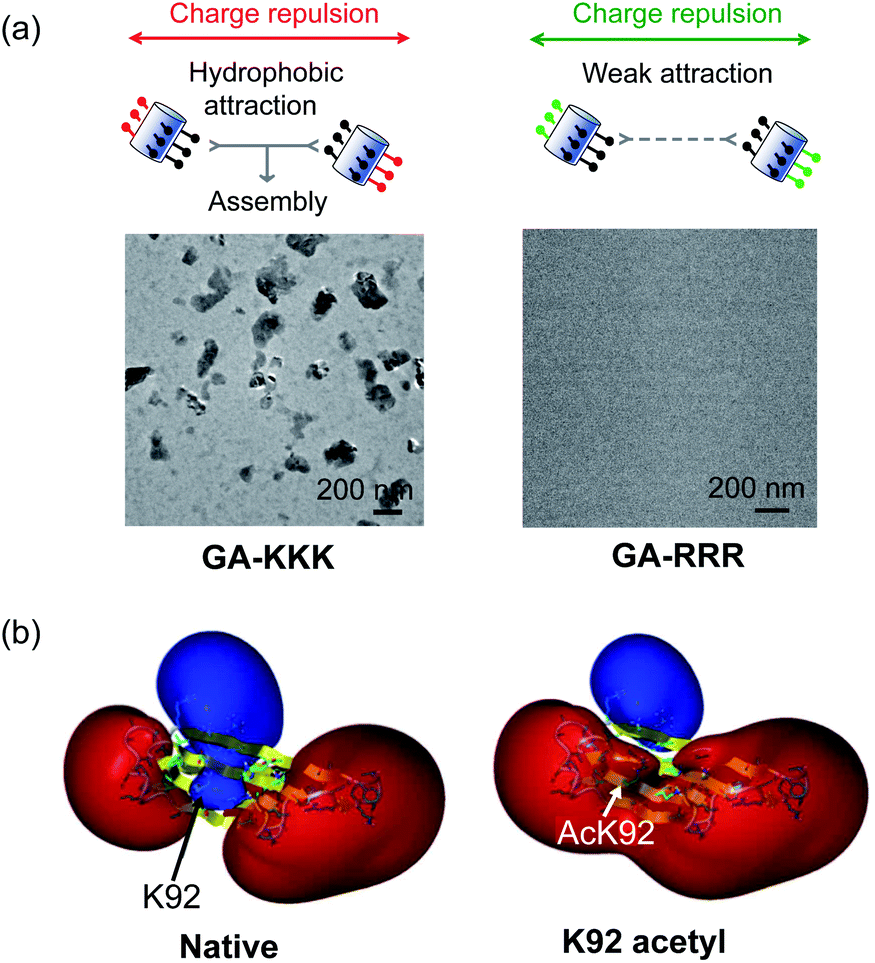 | ||
| Fig. 7 The perturbation effect of polar group and peptide/protein assembly. (a) Schematic illustrations of GA-KKK (left) and GA-RRR (right) assembly in bulk aqueous solution (top). Cryo-TEM images of 2.0 mM GA-KKK and GA-RRR in 10 mM aqueous TEA, pH 7 (bottom).37 Reproduced with permission from ref. 37. Copyright 2021 American Chemical Society. (b) Acetylation of Lys92 converts a part of αB-crystallin from hydrophilic to hydrophobic.45 The electrostatic potentials for native (left) and acetyl αB-crystallin (right) are shown as isosurfaces at a level of +25 kcal mol−1 (blue) and −25 kcal mol−1 (red). Reproduced with permission from ref. 45. Copyright 2013 American Chemical Society. | ||
The scope of hydrophobicity modulation by locally immobilized polar groups in the complicated biologically relevant system is a crucial issue to be explored. An experimental approach is required to probe the generality of the trends that were identified from artificial β-peptide systems in the realm of native α-peptide and protein folding and assembling. Native peptides and proteins differ from β-peptides in the complexity of chemical context and some important physical properties, including molecular topology, nanoscale curvature, and backbone flexibility, which substantially affect the hydrophobicity of a molecule.8 Recent studies with a poly-α-peptide in coiled-coil conformation highlighted the profound influence of the perturbation effect of polar groups in the construction of native polypeptide supramolecular structures.43,44 In the field of protein folding, the perturbation effect of polar groups on hydrophobic interaction is relevant to a series of intriguing examples as follows. The loss of a positive charge by acetylation with the side chain of Lys92 residue in αB-crystallin, a 573 kDa protein, led to a delocalized and long-distance perturbation to the protein structure, improving the hydrophobicity of the whole crystallin domain (Fig. 7b).45 The substituting Arg18 for histidine in the non-amyloidogenic rat islet amyloid polypeptide (rIAPP) increased the tendency to hydrophobically driven amyloidogenesis.46 The number of neutral-polar glutamine/asparagine residues surrounding the nonpolar patch of helices governs the hydrophobic aggregation tendency of coiled-coil proteins.47
5 Conclusion and future perspectives
Recent advances have been made towards understanding the nanoscale hydrophobicity present on the peptide chemically heterogeneous surface. The adjacent polar group plays a profound influence on the hydrophobic interaction generated by the nonpolar patch in a group-specific manner. This modulation effect reflects the differential potency of polar groups to interact with water and perturb the assembly structure of interfacial water molecules nearby the nonpolar patch. More importantly, the efforts to uncover the mystery of proximal groups on hydrophobic interaction benefit the self-assembly in a wide range of biological and chemical systems with broad ramifications.5 These findings open perspectives for the design of self-assembled supramolecular architectures beyond current design rules.Theoretical challenges remain in the efforts to define the non-additivity of intermolecular interactions. The interplays among van der Waals interaction, hydrogen bond, electrostatic interaction, and hydrophobic interaction that provide efficient revenues to fold and organize polypeptide should be rigorously pursued in future efforts for designing peptide-assembled systems.21,48 Additionally, a fine characterization of the assembly structure and the dynamic feature of interfacial water molecules within the local space around the nonpolar patch with the single-molecule resolution is also needed for sketching the detailed molecular mechanisms underlying the perturbation effect of polar group. The employment of the atomic force microscope equipped with a qPlus sensor or the Raman scattering measurements with multivariate curve resolution (Raman-MCR) would be the future direction.49,50 The efforts to address the mentioned challenge will be beneficial for advancing our knowledge of directing biomolecular assembly and recognition by designing molecularly chemical heterogeneity.
Author contributions
F. Z., L. Y., W. Z., L. L., and C. W. discussed and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the National Natural Science Foundation of China (31901007), China Postdoctoral Science Foundation (2020T130006ZX), Fundamental Research Funds for the Central Universities (3332021039), the Open Project Fund provided by Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS (NSKF202019), and State Key Laboratory Special Fund 2060204.Notes and references
- A. J. Patel, P. Varilly, S. N. Jamadagni, M. F. Hagan, D. Chandler and S. Garde, J. Phys. Chem. B, 2012, 116, 2498–2503 CrossRef CAS PubMed.
- W. Zhang, S. Mo, M. Liu, L. Liu, L. Yu and C. Wang, Front. Chem., 2020, 8, 587975–587998 CrossRef CAS PubMed.
- L. Xie, D. Yang, Q. Lu, H. Zhang and H. Zeng, Curr. Opin. Colloid Interface Sci., 2020, 47, 58–69 CrossRef CAS.
- W. M. Park and J. A. Champion, J. Am. Chem. Soc., 2014, 136, 17906–17909 CrossRef CAS.
- S. Garde, Nature, 2015, 517, 277–279 CrossRef CAS PubMed.
- D. N. Woolfson, G. J. Bartlett, M. Bruning and A. R. Thomson, Curr. Opin. Struct. Biol., 2012, 22, 432–441 CrossRef CAS PubMed.
- N. Giovambattista, C. F. Lopez, P. J. Rossky and P. G. Debenedetti, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2274–2279 CrossRef CAS.
- E. Xi, V. Venkateshwaran, L. Li, N. Rego, A. J. Patel and S. Garde, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13345–13350 CrossRef CAS PubMed.
- A. V. Lukyanov and A. E. Likhtman, ACS Nano, 2016, 10, 6045–6053 CrossRef CAS PubMed.
- Y. K. Kang, K. D. Gibson, G. Nemethy and H. A. Scheraga, J. Phys. Chem., 1988, 92, 4739–4742 CrossRef CAS.
- B. Roux and T. Simonson, Biophys. Chem., 1999, 78, 1–20 CrossRef CAS.
- D. Eisenberg and A. D. McLachlan, Nature, 1986, 319, 199–203 CrossRef CAS.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- H. Acharya, S. Vembanur, S. N. Jamadagni and S. Garde, Faraday Discuss., 2010, 146, 353–365 RSC.
- L. Li, C. J. Fennell and K. A. Dill, J. Phys. Chem. B, 2014, 118, 6431–6437 CrossRef CAS.
- A. E. Kister and J. C. Phillips, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9233–9237 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and surface forces, Elsevier Inc, USA, 3rd edition, 2013 Search PubMed.
- C. Tanford, The hydrophobic effect: Formation of micelles and biological membranes, New York, 2nd, 1980 Search PubMed.
- G. A. Greathouse and N. W. Stuart, Plant Physiol., 1936, 11, 873–880 CrossRef CAS.
- B. Ensing, A. Tiwari, M. Tros, J. Hunger, S. R. Domingos, C. Perez, G. Smits, M. Bonn, D. Bonn and S. Woutersen, Nat. Commun., 2019, 10, 2893–2901 CrossRef.
- C. D. Ma, C. Wang, C. Acevedo-Velez, S. H. Gellman and N. L. Abbott, Nature, 2015, 517, 347–350 CrossRef CAS PubMed.
- K. Huang, S. Gast, C. D. Ma, N. L. Abbott and I. Szlufarska, J. Phys. Chem. B, 2015, 119, 13152–13159 CrossRef CAS.
- G. Hummer, S. Garde, A. E. Garcia, A. Pohorille and L. R. Pratt, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8951–8955 CrossRef CAS PubMed.
- R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7310–7313 CrossRef CAS PubMed.
- H. S. Frank and M. W. Evans, J. Chem. Phys., 1945, 13, 507–532 CrossRef CAS.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- C. Wang, C. D. Ma, H. Yeon, X. Wang, S. H. Gellman and N. L. Abbott, J. Am. Chem. Soc., 2017, 139, 18536–18544 CrossRef CAS PubMed.
- C. Sergio, G. Paolo, M. Vincenzo and L. Luciano, J. Solution Chem., 1981, 10, 563–595 CrossRef.
- K. T. No, S. G. Kim, K. H. Cho and H. A. Scheraga, Biophys. Chem., 1999, 78, 127–145 CrossRef CAS PubMed.
- F. F. Abraham, J. Chem. Phys., 1978, 68, 3713–3716 CrossRef CAS.
- L. Cheng, P. Fenter, K. L. Nagy, M. L. Schlegel and N. C. Sturchio, Phys. Rev. Lett., 2001, 87, 156103–156107 CrossRef CAS PubMed.
- O. Bjorneholm, M. H. Hansen, A. Hodgson, L. M. Liu, D. T. Limmer, A. Michaelides, P. Pedevilla, J. Rossmeisl, H. Shen, G. Tocci, E. Tyrode, M. M. Walz, J. Werner and H. Bluhm, Chem. Rev., 2016, 116, 7698–7726 CrossRef.
- J. Wang, D. Bratko and A. Luzar, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6374–6379 CrossRef CAS.
- J. H. Ortony, C.-Y. Cheng, J. M. Franck, R. Kausik, A. Pavlova, J. Hunt and S. Han, New J. Phys., 2011, 13, 015006–015022 CrossRef.
- Y. Itoh and T. Aida, Nat. Chem., 2017, 9, 934–936 CrossRef CAS.
- J. H. Ortony, B. Qiao, C. J. Newcomb, T. J. Keller, L. C. Palmer, E. Deiss-Yehiely, M. Olvera de la Cruz, S. Han and S. I. Stupp, J. Am. Chem. Soc., 2017, 139, 8915–8921 CrossRef CAS PubMed.
- C. Wang, N. A. Biok, K. Nayani, X. Wang, H. Yeon, C. K. Derek Ma, S. H. Gellman and N. L. Abbott, Langmuir, 2021, 37, 3288–3298 CrossRef CAS.
- K. E. Tiller, L. Li, S. Kumar, M. C. Julian, S. Garde and P. M. Tessier, J. Biol. Chem., 2017, 292, 16638–16652 CrossRef CAS.
- D. Kalafatovic and E. Giralt, Molecules, 2017, 22, 1929–1967 CrossRef.
- E. H. Egelman, C. Xu, F. DiMaio, E. Magnotti, C. Modlin, X. Yu, E. Wright, D. Baker and V. P. Conticello, Structure, 2015, 23, 280–289 CrossRef CAS.
- B. J. Calnan, B. Tidor, S. Biancalana, D. Hudson and A. D. Frankel, Science, 1991, 252, 1167–1171 CrossRef CAS PubMed.
- J. I. Austerberry, A. Thistlethwaite, K. Fisher, A. P. Golovanov, A. Pluen, R. Esfandiary, C. F. van der Walle, J. Warwicker, J. P. Derrick and R. Curtis, Biochemistry, 2019, 58, 3413–3421 CrossRef CAS PubMed.
- N. A. Biok, A. D. Passow, C. Wang, C. A. Bingman, N. L. Abbott and S. H. Gellman, Biochemistry, 2019, 58, 4821–4826 CrossRef CAS PubMed.
- L. Yu, Y. Zheng, X. Fang, Y. Zou, C. Wang, Y. Yang and C. Wang, J. Pept. Sci., 2021, 27, e3310 CrossRef CAS PubMed.
- R. B. Nahomi, R. Huang, S. K. Nandi, B. Wang, S. Padmanabha, P. Santhoshkumar, S. Filipek, A. Biswas and R. H. Nagaraj, Biochemistry, 2013, 52, 8126–8138 CrossRef CAS.
- X. B. Mao, C. X. Wang, X. K. Wu, X. J. Ma, L. Liu, L. Zhang, L. Niu, Y. Y. Guo, D. H. Li, Y. L. Yang and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19605–19610 CrossRef CAS.
- F. Fiumara, L. Fioriti, E. R. Kandel and W. A. Hendrickson, Cell, 2010, 143, 1121–1135 CrossRef CAS.
- S. Chen, Y. Itoh, T. Masuda, S. Shimizu, J. Zhao, J. Ma, S. Nakamura, K. Okuro, H. Noguchi, K. Uosaki and T. Aida, Science, 2015, 348, 555–559 CrossRef CAS.
- J. G. Davis, K. P. Gierszal, P. Wang and D. Ben-Amotz, Nature, 2012, 491, 582–585 CrossRef CAS PubMed.
- J. Peng, J. Guo, P. Hapala, D. Cao, R. Ma, B. Cheng, L. Xu, M. Ondracek, P. Jelinek, E. Wang and Y. Jiang, Nat. Commun., 2018, 9, 122–129 CrossRef.
Footnote |
| † These authors share the first authorship. |
| This journal is © The Royal Society of Chemistry 2021 |