
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManifold dynamic non-covalent interactions for steering molecular assembly and cyclization†
Shaotang
Song‡
 a,
Lulu
Wang‡
a,
Jie
Su‡
a,
Zhen
Xu
b,
Chia-Hsiu
Hsu
cd,
Chenqiang
Hua
e,
Pin
Lyu
a,
Jing
Li
a,
Xinnan
Peng
a,
Takahiro
Kojima
b,
Shunpei
Nobusue
b,
Mykola
Telychko
a,
Yi
Zheng
e,
Feng-Chuan
Chuang
cd,
Hiroshi
Sakaguchi
*b,
Ming Wah
Wong
*a and
Jiong
Lu
*af
a,
Lulu
Wang‡
a,
Jie
Su‡
a,
Zhen
Xu
b,
Chia-Hsiu
Hsu
cd,
Chenqiang
Hua
e,
Pin
Lyu
a,
Jing
Li
a,
Xinnan
Peng
a,
Takahiro
Kojima
b,
Shunpei
Nobusue
b,
Mykola
Telychko
a,
Yi
Zheng
e,
Feng-Chuan
Chuang
cd,
Hiroshi
Sakaguchi
*b,
Ming Wah
Wong
*a and
Jiong
Lu
*af
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmwmw@nus.edu.sg
bInstitute of Advanced Energy, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: sakaguchi.hiroshi.7z@kyoto-u.ac.jp
cDepartment of Physics, National Sun Yat-sen University, Kaohsiung 80424, Taiwan
dPhysics Division, National Center for Theoretical Sciences, Taipei, 10617, Taiwan
eZhejiang Province Key Laboratory of Quantum Technology and Device, Department of Physics, Zhejiang University, Hangzhou, People's Republic of China
fCentre for Advanced 2D Materials (CA2DM), National University of Singapore, 6 Science Drive 2, Singapore 117546, Singapore. E-mail: chmluj@nus.edu.sg
First published on 5th August 2021
Abstract
Deciphering rich non-covalent interactions that govern many chemical and biological processes is crucial for the design of drugs and controlling molecular assemblies and their chemical transformations. However, real-space characterization of these weak interactions in complex molecular architectures at the single bond level has been a longstanding challenge. Here, we employed bond-resolved scanning probe microscopy combined with an exhaustive structural search algorithm and quantum chemistry calculations to elucidate multiple non-covalent interactions that control the cohesive molecular clustering of well-designed precursor molecules and their chemical reactions. The presence of two flexible bromo-triphenyl moieties in the precursor leads to the assembly of distinct non-planar dimer and trimer clusters by manifold non-covalent interactions, including hydrogen bonding, halogen bonding, C–H⋯π and lone pair⋯π interactions. The dynamic nature of weak interactions allows for transforming dimers into energetically more favourable trimers as molecular density increases. The formation of trimers also facilitates thermally-triggered intermolecular Ullmann coupling reactions, while the disassembly of dimers favours intramolecular cyclization, as evidenced by bond-resolved imaging of metalorganic intermediates and final products. The richness of manifold non-covalent interactions offers unprecedented opportunities for controlling the assembly of complex molecular architectures and steering on-surface synthesis of quantum nanostructures.
Introduction
Non-covalent interactions span a wide range of binding energies, and consist of hydrogen bonding, halogen bonding, dipole–dipole interaction, various types of aromatic interactions, steric repulsion and London dispersion,1 which govern a diversity of chemical and biochemical processes including chemical interactions within protein–drug, catalyst–substrate and molecular self-assemblies.2–4 Understanding these non-covalent interactions at the atomic bond level is therefore vital not only for identifying the driving forces that control the molecular arrangements in complex natural processes, but also for the design of drugs and new supramolecular architectures.One peculiar feature of non-covalent interactions lies in their dynamic nature exemplified by great flexibility and reversibility, which can be readily influenced by external factors such as molecular density,5 pH value,6 temperature,7 and light irradiation.8 Despite considerable interest in this field, microscopic knowledge of the interplay between manifold non-covalent interactions, particularly at the single-bond level, which controls the formation of elegant supramolecular architectures remains elusive.
Common strategies for the characterization of non-covalent interactions rely on the utilisation of sample-averaged techniques including nuclear magnetic resonance, mass spectroscopy and X-ray diffraction (XRD). These techniques allow us to gather information on the presence of certain non-covalent bonds in a molecular compound, but their spatial resolution is rather limited, thus making it challenging to visualize dynamic processes in supramolecular assemblies governed by multiple non-covalent interactions.
Recent experimental breakthroughs in tip-functionalised scanning probe microscopy (SPM) techniques demonstrate their remarkable capability for sub-angstrom real-space imaging of chemical bonds and elucidation of the intermolecular interactions in self-assembled molecular architectures.9–14 However, the majority of these studies have been focused on planar or nearly-planar molecular structures. In addition, on-surface molecular assemblies investigated to date mainly involve a limited category of non-covalent interactions, such as halogen bond5,15,16 or hydrogen bond interactions.17 Molecular-level insights into complex three-dimensional (3D)-like assemblies with manifold non-covalent interactions remain largely unexplored.18
Here, we exploited combined scanning tunnelling microscopy (STM) and non-contact atomic force microscopy (ncAFM) techniques, corroborated by an exhaustive structural search algorithm and quantum chemistry calculations to elucidate manifold non-covalent interactions in 3D molecular clusters. These clusters are self-assembled from non-planar precursor molecules with two flexible bromo-triphenyl moieties adopting a V-shaped conformation. Upon deposition of these precursor molecules on Au(111), they spontaneously organised into dimers and trimers (Fig. 1a). The dominant interactions in dimers are found to be hydrogen bonding coexisting with lone pair⋯π interactions. In contrast, despite the presence of hydrogen bonding, the trimer is mainly driven by halogen bonding with the formation of two Br3 synthons by type-II (Fig. S1†) synergistic interactions at two different vertical planes. Moreover, such manifold non-covalent interactions can be exploited to tune on-surface reaction pathways, leading to different desired products, as evidenced by atomic-scale visualization of both metalorganic intermediates and final products via bond-resolved ncAFM imaging.
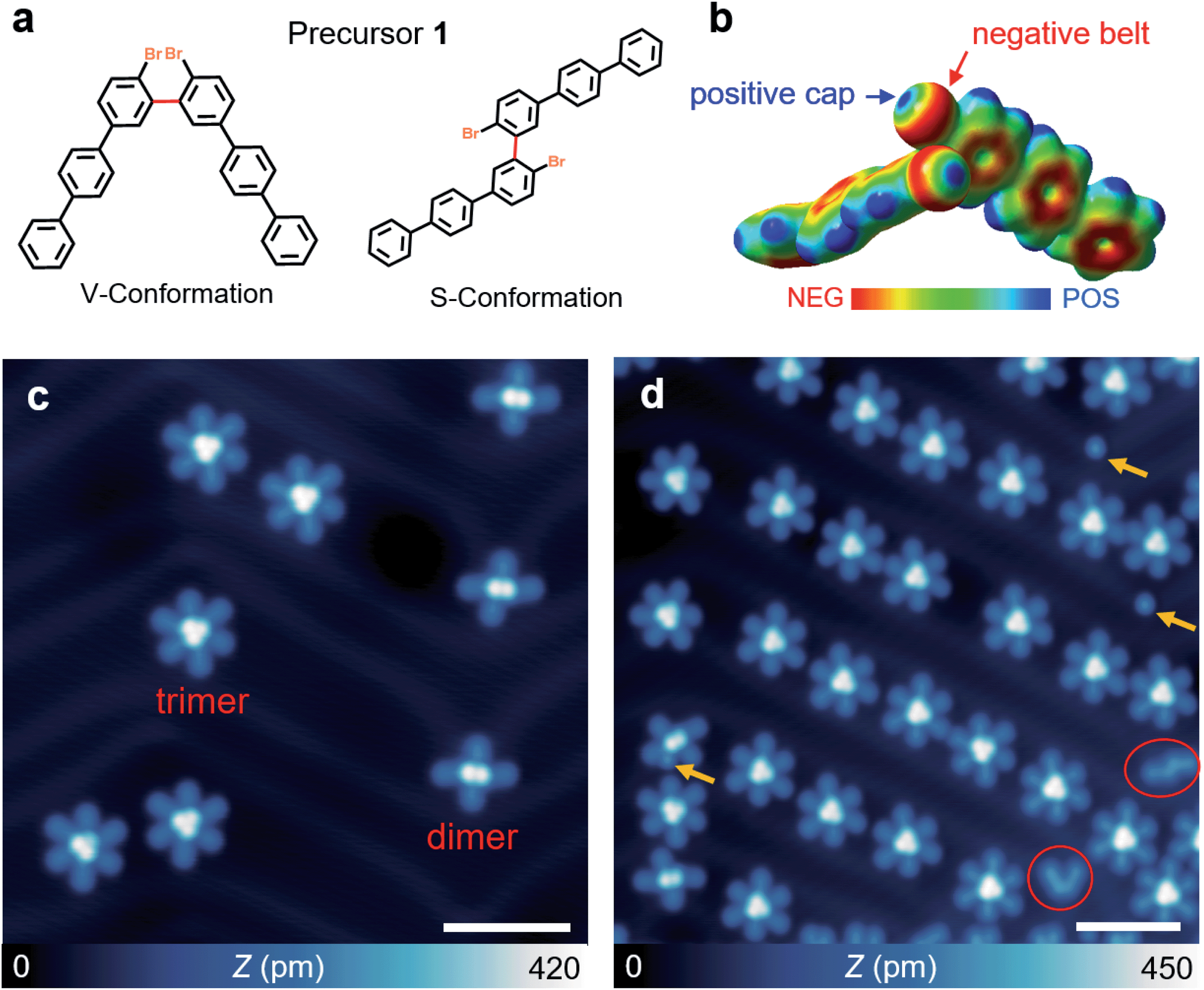 | ||
| Fig. 1 Self-assembly of precursor 1 into dimer and trimer clusters on Au(111). (a) The V- and S-conformations of the precursor upon its adsorption on Au(111) due to the C–C bond rotation (highlighted in red). (b) The electrostatic potential (ESP) map projected on an electron density isosurface (0.012 a.u.) of the electron density reveals the σ-hole (blue) and electron-rich belt (red) of the Br atoms. (c and d) STM images of the self-assembled molecular clusters of precursors including dimers and trimers at a low (c) and high (d) coverage, respectively. The red circles in (d) highlight the V- and S-conformations of debrominated precursor 1, respectively. The bright dots pointed by orange arrows are attributed to the disassociated Br atoms from the precursor monomers, which adsorb over elbow sites or are attached to dimer clusters. Scale bars for (c) and (d) are 5 nm. | ||
Results and discussion
STM/ncAFM measurements
Precursor 1 was synthesized via Suzuki coupling of 2,2′-dibromo-5,5′-diiodo-1,1′-biphenyl and biphenyl boronic acid (see synthetic details in the ESI†). Single-crystal XRD measurements of a bulk single crystal reveal the expected atomic structures of precursor 1 which is arranged in a 3D V-shaped conformation with two triphenyl branches intersected by an angle of 75° and packed in herringbone-like conformations (Fig. S2a†). A detailed analysis based on real-space non-covalent interaction (NCI) calculations reveals that the herringbone crystalline packing is governed by multiple non-covalent interactions including hydrogen bonding, π⋯π stacking, and lone-pair⋯π interaction (Fig. S2b†).To explore the packing geometries and non-covalent interactions in molecular assemblies in the two-dimensional regime at the single-molecule level, precursor 1 was sublimated onto Au(111) held at 300 K (see ESI†). Subsequent STM/ncAFM measurements were carried out at 4.5 K with a CO-functionalized tip. Distinct from the herringbone-like packing in 3D bulk crystals, precursor 1 forms two different molecular clusters on Au(111). At a low coverage of ∼0.1 ML, X-shaped (∼40%) and *-shaped (∼60%) molecular clusters are formed and predominantly located at face-centred cubic regions or elbow sites of the Au(111) herringbone reconstruction as evidenced by the large scale STM image (Fig. 1c and S3a†). X-shaped clusters exhibit two bright dots in the center surrounded by four “legs”, while *-shaped clusters show three bright dots in the center surrounded by six “legs” (Fig. 1c). Such unique features indicate that X- and *-shaped molecular clusters are likely formed by two (dimer) and three (trimer) monomers, respectively. Moreover, we found that an increase of molecular coverage to 0.3 ML yields a predominant presence of *-shaped clusters (94%) but with a low percentage of X-shaped ones (∼6%) (Fig. 1d and S3b†).
We then performed a controlled STM tip manipulation to disassemble the clusters for the confirmation of their composition (Fig. 2). Our manipulation approach relies on applying a high voltage pulse through the tip apex placed in the vicinity of the clusters (Fig. 2a). Upon disassembly of the molecular clusters, we observed two (for the X-shape) and three (for the *-shape) V-shaped structures which resemble the conformation of precursor 1 (Fig. 2f–i and b–e). This further confirms that X- and *-shaped structures indeed represent the self-assembled dimer and trimer clusters of V-shaped monomers. We would like to note that the bright dots attached to the V-shaped monomer are likely attributed to the dissociated Br atoms due to the cleavage of the C–Br bond triggered by the voltage pulse (as highlighted with yellow circles in Fig. 2b–i).19–21 Apart from the dominant dimer and trimer clusters, we also observed a low percentage of isolated monomers in its trans V-shape (<1%) and cis S-shape (<1%) configurations (Fig. 1d). The majority of molecules deposited on the surface adopt a V-shaped conformation at different molecular coverages, while a small amount of isolated molecules exhibits an S-shaped conformation. Molecules with a V-shape conformation favor head-to-head self-assemblies as revealed in our STM images. The absence of any bright dot suggests the dissociation of Br atoms from precursor 1 in these monomer-like structures upon its landing on the surface. These observations indicate that the self-assembly of monomers into molecular clusters tends to stabilize the precursor from surface-assisted debromination, crucial for tuning their subsequent intramolecular and intermolecular transformation.22,23
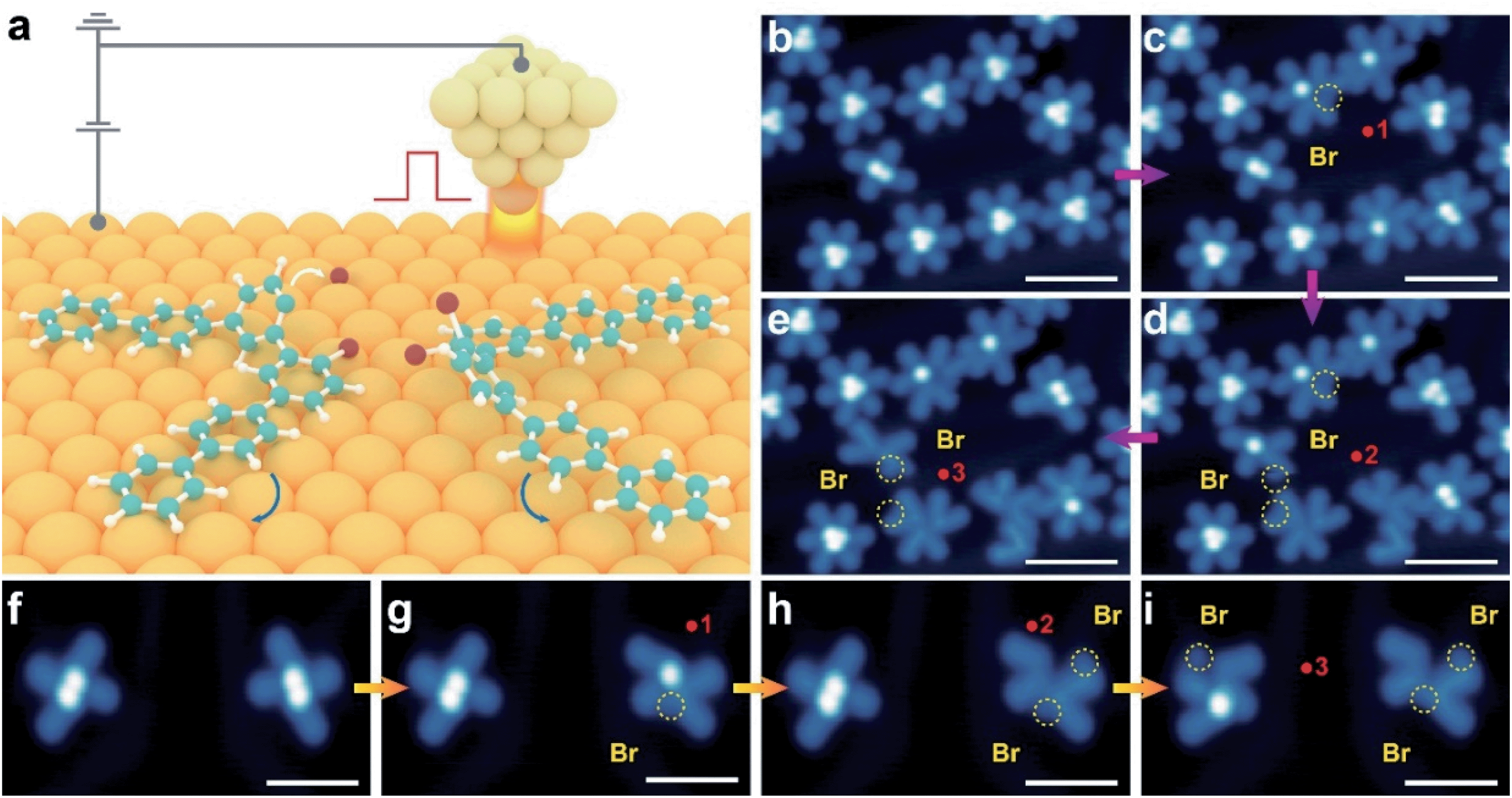 | ||
| Fig. 2 The disassembly of molecular clusters via STM tip manipulation. (a) Schematic illustration of a voltage pulse applied in the vicinity of the cluster. It triggers the disassembly of molecular clusters via breaking the weak non-covalent interactions between precursors. (b–i) The manipulation of the trimer (b–e) and dimer (f–i) clusters. Red dots refer to the positions where the voltage pulses (−8 V) are applied. The Arabic numbers indicate the sequence for applying voltage pulses. The yellow circles highlight the Br atoms disassociated from the precursors by the voltage pulse. Scale bars for (b)–(d) are 4 nm, and for (f)–(i) are 3 nm. | ||
To get further insight into the structural information of these molecular clusters, we performed constant height ncAFM imaging with a CO functionalized tip. The frequency shift (Δf) contrast in such ncAFM images reflects the magnitude of short-range forces acting between the CO-decorated tip and the sample in the Pauli repulsion regime.14,24 The bright areas in ncAFM images (i.e. higher Δf value) represent regions with higher height and electron density.25 The ncAFM images of the dimer (trimer) cluster (Fig. 3b and 4b) reveal four (three) dots interconnected via sharp lines to form a rhombus (triangular) shape. The surrounding “leg-like” molecular backbones are not visible due to a large tip–molecule separation. Such a unique ncAFM contrast indicates that these molecular clusters are composed of non-planar geometries, wherein the central bright regions are higher than the periphery.
 | ||
| Fig. 3 Characterization of multiple non-covalent interactions in the dimer. (a) STM image of the dimer (1 V, 100 pA). Scale bar is 1 nm. (b) A full-range ncAFM of the dimer with a CO functionalized tip overlaid with the dimer molecular model. Scale bar is 0.5 nm. (c) Zoom-in ncAFM image reveals four bright dots connected with sharp line features forming a rhombus shape. Scale bar is 0.3 nm. (d and e) Top- and side-views of the DFT-modelled dimer structure on Au(111). (f and g) Top- and side-views of the electrostatic potential maps of the dimer. (h) BCP calculation reveals the BCP of the hydrogen bonds and lone pair⋯π interactions revealed from AIM analysis of the dimer. (i) NCI plot of the dimer showing the presence of three types of non-covalent interactions including lone pair⋯π, π⋯π and C–H⋯Br (indicated with green discs). | ||
 | ||
| Fig. 4 Characterization of multiple noncovalent interactions in the trimer. (a) STM image of the trimer (1 V, 100 pA). Scale bar is 1 nm. (b) A full-range ncAFM image of the trimer with a CO functionalized tip overlaid with the trimer molecular model. Scale bar is 0.5 nm. (c) Zoom-in ncAFM image showing three bright dots connected with the sharp line features forming a triangular shape. Scale bar is 0.4 nm. (d and e) Top-view and side-view of the DFT-modelled trimer structure on Au(111); (f and g) top- and side-view of the electrostatic potential maps of the trimer cluster; (h) BCPs of the halogen bond, hydrogen bonds, CH⋯π and lone pair⋯π interactions revealed from AIM analysis of the trimer; and (i) NCI plot of the trimer revealing four types of non-covalent interactions including halogen, hydrogen, lone pair⋯π, and C–H⋯π (indicated with green discs). | ||
Intermolecular interactions
According to the structural information obtained by both STM and ncAFM imaging, we then carried out an exhaustive search coupled with a quasi-Newtonian algorithm (>200 initial atomic structures) using first-principles calculations to find the most possible structural configurations of dimers and trimers. The low-lying energy isomers were placed on Au(111) to further relax using VASP. As shown in Fig. 3c and 4c, strong steric hindrance between the two Br atoms of the precursor causes one bromophenyl motif to bend up with tilting angles of 57° and 40° for the dimer and trimer, respectively. As a result of such a dramatic structural geometric change, one Br and one H of each bromophenyl moiety protrude up, which are readily reflected as bright dots in the ncAFM images. Indeed, the ncAFM image of the dimer (Fig. 3c) reveals four bright protrusions associated with two Br and two H atoms from two interacting monomers. In contrast, the three bright dots revealed by the ncAFM image of the trimer (Fig. 4c) are attributed to three protruding Br atoms. In addition, the three protruding H atoms in the centre are almost invisible, due to a relatively small bending angle (40°) of the bromophenyl motif.In order to gain atomic-level insight into the intermolecular interactions in these intriguing molecular assemblies, we then performed atoms in molecules (AIM) and real-space non-covalent interaction (NCI) calculations to semi-quantitatively analyse various possible contributions of the intermolecular non-covalent interactions in the dimer and trimer, respectively.26 As shown in Fig. 3h, the computed numerical electron density [ρ(r)] and its Laplacian [∇2(r)] at the bond critical point (BCP) indicate the presence of multiple non-covalent interactions, including C–H⋯Br (denoted as a1 and a2) and Br lone pair⋯π (denoted as b1 and b2), featuring the interatomic distances of dC–H⋯Br ≈ 2.9 Å and dlone-pair⋯π ≈ 3.1 Å (Fig. S4 and Table S1†). The Br lone pair⋯π bonding represents attractive interaction between a lone pair electron of Br and a π-system of the up-tilted bromophenyl ring.27–29 This type of interaction was also reported to stabilize the formation of biological macromolecules30,31 and binding of inhibitors in the binding pocket of biochemical receptors.32 In addition, NCI analysis highlights1 the attractive interaction regions marked with green disks in the areas of the BCP (Fig. 3i and S6†). Interestingly, the lower Br atoms pointing towards the up-tilted bromophenyl ring lead to the emergence of lone pair⋯π interactions, which further increase the cohesive energy of the dimer.
Compared to the dimer, the trimer features a larger number (four types) of non-covalent interactions (Fig. 4). First, the three up-tilted and three lower Br atoms in the trimer participate in two sets of Br3 synthons with type-II X2 interactions (a1–a6 in Fig. 4h, S5a and b†), acting as a main stabilizing force for the trimer, as evidenced in the BCP by AIM analysis. Significantly stronger Br⋯Br halogen bonds (a1–a3, 3.51 Å) are observed for the interactions of the three up-tilted Br atoms (Fig. S5†). Electrostatic potential maps also reveal that the electropositive σ-hole points to the electronegative belt of the Br atoms in both of the Br3 synthons (Fig. 4f and g).
Moreover, the AIM analysis also identifies weak hydrogen bonding (C–H⋯Br, b1–b3) between the up-tilted Br and H atoms from the adjacent monomers at a separation distance of ∼3.4 Å (Fig. 4h and S5c†). In addition, up-tilted benzene rings are nearly perpendicular to their neighbouring bromophenyl motifs in close proximity (3.0 Å), which leads to the formation of strong C–H⋯π (c1–c3) and lone pair⋯π (d1–d3) interactions (Fig. 4h, and Table S2†). A combination of these attractive non-covalent interactions is further represented by green discs in the NCI plot (Fig. 4i and S7†).
To gain a more quantitative understanding of these non-covalent interactions, we set out to investigate the attractive interaction energies of these clusters by employing symmetry-adapted perturbation theory (SAPT) calculations.33,34 SAPT provides accurate NCI energies and decomposes the intermolecular interaction energies into physically meaningful components, namely electrostatic, exchange-repulsion, induction, and dispersion terms.33 The dimer and trimer reveal a total attractive stabilization energy of 30.4 kJ mol−1 and 55.9 kJ mol−1, respectively (Table S3†). It is worth noting that dispersion is the largest stabilizing force, which is consistent with the important roles of the C–H⋯π,35 lone pair⋯π,36 and halogen bonding37 interactions in the dimer and trimer. A higher ratio of the trimer than that of the dimer is attributed not only to a higher on-surface coverage of 1, but also to a larger energetic gain in the formation of trimers arising from a larger number of manifold non-covalent interactions. Notably, our DFT calculations reveal a rather negligible charge transfer between molecular precursors and Au(111), suggesting limited influence of molecule–substrate interactions on non-covalent interactions (Fig. S8†).
On-surface transformation of precursor 1
The pre-arrangement of molecular precursors in the self-assembled structures can dramatically impact on-surface reaction pathways triggered by thermal annealing.38 Here, the dimers with a relatively low stabilization energy may tend to dissociate, which thus favours intramolecular cyclization and suppresses intermolecular cyclization. In contrast, the trimers with a high stabilization energy are expected to facilitate an intermolecular coupling reaction of the debrominated biradicals.To explore such an effect, we conducted a mild thermal annealing of the samples decorated with dimer and trimer clusters with different ratios at 400 K. After the thermal activation of the samples, both V- and X-shaped products can be obtained (Fig. 5b). A statistical analysis of 3235 product molecules (at a precursor coverage of 0.35 ML) reveals the yields of 91.7% and 8.3% for products 2 and 4, respectively. The ratio of these products can be tuned by controlling the precursor coverage (Fig. 5g and h).
 | ||
| Fig. 5 Self-assembly steered intra- and intermolecular cyclization of precursor 1via STM and ncAFM imaging. (a) Schematic illustration of the debromination of precursor 1 leading to the formation of biradical intermediate 1′, followed by the radical cyclization to form product 2 (upper). The lower path illustrates the Ullmann coupling of 1′ followed by cyclodehydrogenation, yielding the products 3 and 4, respectively. The formation of 5 is prohibited, presumably due to a high steric hindrance. The red lines in products 5, 2, and 4 indicate newly formed chemical bonds. (b) Large-scale STM images (metal tip, I = 100 pA, V = 1.5 V) of the three products (2, 3, and 4) after thermal annealing (420 K) of the dimer and trimer clusters of precursor 1 pre-assembled on Au(111). Scale bar is 5 nm. (c–e) ncAFM images of products 2, 3, and 4, respectively. Magnified ncAFM images (the bottom panels) highlight the molecular backbone of the center region (marked with a dashed rectangle). Scale bars for (c)–(e) are 5 Å. (f–h) Distribution of reaction products at different precursor coverage ratios (0.02, 0.04, and 0.09 ML for (f), (g), and (h), respectively). Increasing the coverage yields a higher ratio of trimer clusters, therefore leading to a greater amount of product 4. Scale bars for (f)–(h) are 5 nm. | ||
The bond-resolved ncAFM image reveals a symmetric backbone structure of product 2, consisting of a biphenylene core connected with two biphenyl branches on both sides. Two of the inner benzene rings of the biphenyl motifs are tilted due to a relief of steric hindrance, consistent with DFT calculations (Fig. S10†). The magnified view of the biphenylene core unveils that the central four-membered ring is slightly brighter than the side six-membered rings (Fig. S11b†). This could probably originate from the cyclobutadiene anti-aromaticity or the tilting effect of the CO tip over a small size of the ring. As shown in Fig. S11d,† there are three possible contributing resonance structures including one cyclobutadiene and two radialene forms of the biphenylene core. Alternating single and double bonds give rise to the anti-aromatic cyclobutadiene form, while four single-bonds lead to the radialene counterpart. A detailed bond order analysis of the biphenylene motif (Fig. S11c†) reveals that the four bonds have nearly the same bond length, which suggests that the radialene form dominates the resonance structures.39
The magnified view of the X-shaped products resolves two distinct structures with a notably different contrast at the centre (denoted as products 3 and 4) (Fig. S12†). The ncAFM measurements also captured their structural difference, namely, product 3 has a dark contrast at the centre and product 4 adopts a planar geometry formed by a fused hexagonal ring. The magnified ncAFM view of the central region of product 3 shows a bright dot corresponding to an Au adatom (Fig. 5d), coordinated with the four dibrominated carbon atoms. The dark ncAFM contrast is attributed to the four benzene rings that bent downwards to the Au(111), consistent with previous reports of organometallic compounds.39 On the other hand, product 4 shows a dibenzo[e,l]pyrene core (Fig. 5e). These results allow us to deduce a possible reaction mechanism of the Ullmann coupling process from two precursors 1 (via debromination to the biradical 1′) to product 4, through the formation of metalorganic intermediate 3 (Fig. 5a).40,41 This intermediate provides mechanistic insight into aryl–aryl bond formation via a metal-catalysed Ullmann reaction, which may serve as a new path for synthesizing polycyclic aromatic hydrocarbons.
Competition between thermal diffusion and intermolecular reactions during thermal annealing of the self-assembled structures plays an important role in the on-surface synthesis.42 Random diffusion of the precursor molecules with multiple reaction pathways usually leads to the yield of undesired by-products, such as uncontrolled graphene nanoribbon junctions43 and unexpected molecular oligomers,44 which thus severely limits the yield of desired products. To this end, taking advantage of non-covalent interactions between the precursor reactants may provide a promising approach for steering the reaction pathways towards the desired products. Indeed, the thermal diffusion of the precursors causes the disassembly of dimers at a low coverage, which facilitates the intramolecular cyclization reaction leading to the biphenylene derivative 2. At an increased molecular coverage, the trimer with a larger cohesive energy dominates the assemblies. However, the trimerization reaction to a highly strained product 5 is prohibited due to a large steric hindrance. As a result, the intermolecular Ullmann coupling reaction occurs between two of the three strongly bonded monomers, while the third monomer undergoes the intramolecular reaction (Fig. 5a).
It is important to note that Cu-catalysed dimerization of the precursor 1 derivative (two tert-butyl groups installed at the 4,4′-sites) in solution yields a three-dimensional Greek cross dodecaphenylene with a tetra-o-phenylene core.45 In stark contrast to the planar dibenzo[e,l]pyrene core on the Au(111) surface, our results demonstrated that the surface-assisted reactions in the two-dimensional limit can be steered by tuning multiple intermolecular non-covalent interactions, and thus proceed under distinct pathways towards the formation of the different products.
Conclusion
Real-space characterization of dynamic non-covalent interactions in molecular assemblies at the atomic bond level, combined with quantum chemistry simulations, allows us to decode the role of multiple weak interactions in the molecular assemblies and their on-surface chemical reactions. The dynamic nature of these weak interactions allows for transformation of the arrangement of monomers in the assembled clusters as molecular density increases, which alters the reaction pathways in the subsequent on-surface synthesis of cyclized products. Our findings highlight a vital route for controlling on-surface supramolecular assemblies and steering their chemical transformations through manipulation of manifold dynamic non-covalent interactions.Data availability
Raw data are available upon request from the authors.Author contributions
J. L. supervised the project. S. S. and J. S. conducted the STM and ncAFM measurements. L. W. performed AIM, NCI, and DFT calculations under the supervision of M. W. W., C.-H. H, F.-C. C., C. Q. H., and Y. Z. performed DFT calculations. S. S., Z. X., T. K. and S. N. carried out the organic synthesis under the supervision of H. S.; P. L., J. L., and M. T. participated in scientific discussion. S. S. and J. L. wrote the manuscript with contribution from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Lu acknowledges the support from MOE grants (MOE2019-T2-2-044 and R-143-000-B58-114). M. Telychko acknowledges the support from A*STAR AME YIRG grant (Project No. A20E6c0098, R-143-000-B71-305) and J. Su acknowledges the support from A*STAR AME YIRG grant (A2084c0171, R-143-000-B92-305). F-C. C. acknowledges the support from the National Center for Theoretical Sciences and the Ministry of Science and Technology of Taiwan under grant no. MOST-107-2628-M-110-001-MY3 and the National Center for High-performance Computing for computer resources. Y. Zheng acknowledges the support from Zhejiang Provincial Natural Science Foundation (D19A040001). L. Wang acknowledges Applied Materials for the PhD scholarship.References
- E. R. Johnson, et al. Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- G. Cavallo, et al. The halogen bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- J. Teyssandier, K. S. Mali and S. De Feyter, Halogen bonding in two-dimensional crystal engineering, ChemistryOpen, 2020, 9, 225–241 CrossRef CAS.
- J. Shang, et al. Assembling molecular Sierpiński triangle fractals, Nat. Chem., 2015, 7, 389–393 CrossRef CAS PubMed.
- J. Lawrence, et al. Combining high-resolution scanning tunnelling microscopy and first-principles simulations to identify halogen bonding, Nat. Commun., 2020, 11, 1–7 Search PubMed.
- C. Le Bourvellec, S. Guyot and C. M. G. C. Renard, Non-covalent interaction between procyanidins and apple cell wall material: Part I. Effect of some environmental parameters, Biochim. Biophys. Acta, Gen. Subj., 2004, 1672, 192–202 CrossRef CAS PubMed.
- J. F. Jheng, et al. Influences of the non-covalent interaction strength on reaching high solid-state order and device performance of a low bandgap polymer with axisymmetrical structural units, Adv. Mater., 2013, 25, 2445–2451 CrossRef CAS.
- K. Ichimura, Photoalignment of liquid-crystal systems, Chem. Rev., 2000, 100, 1847–1874 CrossRef CAS PubMed.
- R. Temirov, S. Soubatch, O. Neucheva, A. C. Lassise and F. S. Tautz, A novel method achieving ultra-high geometrical resolution in scanning tunnelling microscopy, New J. Phys., 2008, 10, 053012 CrossRef.
- C. Weiss, et al. Imaging Pauli repulsion in scanning tunneling microscopy, Phys. Rev. Lett., 2010, 105, 086103 CrossRef CAS PubMed.
- G. Kichin, C. Weiss, C. Wagner, F. S. Tautz and R. Temirov, Single molecule and single atom sensors for atomic resolution imaging of chemically complex surfaces, J. Am. Chem. Soc., 2011, 133, 16847–16851 CrossRef CAS.
- F. Mohn, B. Schuler, L. Gross and G. Meyer, Different tips for high-resolution atomic force microscopy and scanning tunneling microscopy of single molecules, Appl. Phys. Lett., 2013, 102, 073109 CrossRef.
- H. Mönig, et al. Quantitative assessment of intermolecular interactions by atomic force microscopy imaging using copper oxide tips, Nat. Nanotechnol., 2018, 13, 371–375 CrossRef.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, The chemical structure of a molecule resolved by atomic force microscopy, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- Z. Han, et al. Imaging the halogen bond in self-assembled halogenbenzenes on silver, Science, 2017, 358, 206–210 CrossRef CAS.
- J. Tschakert, et al. Surface-controlled reversal of the selectivity of halogen bonds, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- J. Zhang, et al. Real-space identification of intermolecular bonding with atomic force microscopy, Science, 2013, 342, 611–614 CrossRef CAS PubMed.
- B. Alldritt, et al. Automated structure discovery in atomic force microscopy, Sci. Adv., 2020, 6, eaay6913 CrossRef CAS PubMed.
- N. Pavliček, et al. Synthesis and characterization of triangulene, Nat. Nanotechnol., 2017, 12, 308–311 CrossRef.
- N. Pavliček, et al. On-surface generation and imaging of arynes by atomic force microscopy, Nat. Chem., 2015, 7, 623–628 CrossRef.
- B. Schuler, et al. Reversible Bergman cyclization by atomic manipulation, Nat. Chem., 2016, 8, 220–224 CrossRef CAS.
- M. Bieri, et al. Two-dimensional polymer formation on surfaces: insight into the roles of precursor mobility and reactivity, J. Am. Chem. Soc., 2010, 132, 16669–16676 CrossRef CAS PubMed.
- M. Telychko, et al. Strain-induced isomerization in one-dimensional metal–organic chains, Angew. Chem., Int. Ed., 2019, 131, 18764–18770 CrossRef.
- P. Hapala, et al. Mechanism of high-resolution STM/AFM imaging with functionalized tips, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 085421 CrossRef CAS.
- S. Kawai,
et al. Direct quantitative measurement of the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–C bond by atomic force microscopy, Sci. Adv., 2017, 3, e1603258 CrossRef PubMed.
O⋯H–C bond by atomic force microscopy, Sci. Adv., 2017, 3, e1603258 CrossRef PubMed. - R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- M. Egli and S. Sarkhel, Lone pair-aromatic interactions: To stabilize or not to stabilize, Acc. Chem. Res., 2007, 40, 197–205 CrossRef CAS.
- T. J. Mooibroek, P. Gamez and J. Reedijk, Lone pair–π interactions: a new supramolecular bond?, CrystEngComm, 2008, 10, 1501–1515 RSC.
- S. R. Gadre and A. Kumar, Understanding lone pair-π interactions from electrostatic viewpoint, in Noncovalent forces, Springer, Cham, 2015, pp. 391–418 Search PubMed.
- J. C. Calabrese, D. B. Jordan, A. Boodhoo, S. Sariaslani and T. Vannelli, Crystal structure of phenylalanine ammonia lyase: multiple helix dipoles implicated in catalysis, Biochemistry, 2004, 43, 11403–11416 CrossRef CAS.
- E. J. Stollar, et al. Unconventional interactions between water and heterocyclic nitrogens in protein structures, Proteins: Struct., Funct., Bioinf., 2004, 57, 1–8 CrossRef CAS.
- J. Li, et al. Strategy for discovering chemical inhibitors of human cyclophilin A: focused library design, virtual screening, chemical synthesis and bioassay, J. Comb. Chem., 2006, 8, 326–337 CrossRef CAS.
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherrill, Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies, J. Chem. Phys., 2014, 140, 094106 CrossRef PubMed.
- K. Patkowski, Recent developments in symmetry-adapted perturbation theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1452 CAS.
- Y. Cao and M. W. Wong, Roles of electrostatic interaction and dispersion in CH⋯CH, C–H⋯π and π⋯π in ethylene dimers, J. Mol. Model., 2014, 20, 2185–2197 CrossRef.
- J. Ran and P. Hobza, On the nature of bonding in lone pair···π-electron complexes: CCSD(T)/complete basis set limit calculations, J. Chem. Theory Comput., 2009, 5, 1180–1185 CrossRef CAS PubMed.
- S. J. Ang, A. M. Mak and M. W. Wong, Nature of halogen bonding involving π-systems, nitroxide radicals and carbenes: A highlight on the importance of charge transfer, Phys. Chem. Chem. Phys., 2018, 20, 26463–26478 RSC.
- X. Zhou, et al. Steering surface reaction dynamics with a self-assembly strategy: Ullmann coupling on metal surfaces, Angew. Chem., Int. Ed., 2017, 129, 13032–13036 CrossRef.
- S. Kawai, et al. Competing annulene and radialene structures in a single anti-aromatic molecule studied by high-resolution atomic force microscopy, ACS Nano, 2017, 11, 8122–8130 CrossRef CAS.
- S. Clair and D. G. de Oteyza, Controlling a chemical coupling reaction on a surface: tools and strategies for on-surface synthesis, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- G. Galeotti, et al. An unexpected organometallic intermediate in surface-confined Ullmann coupling, Nanoscale, 2019, 11, 7682–7689 RSC.
- R. Otero, et al. Lock-and-key effect in the surface diffusion of large organic molecules probed by STM, Nat. Mater., 2004, 3, 779–782 CrossRef CAS PubMed.
- J. Li, et al. Single spin localization and manipulation in graphene open-shell nanostructures, Nat. Commun., 2019, 10, 1–7 CrossRef.
- J. Su, et al. On-surface synthesis and characterization of [7]Triangulene quantum ring, Nano Lett., 2021, 21, 861–867 CrossRef CAS.
- A. Rajca, H. Wang, P. Bolshov and S. Rajca, A Greek cross dodecaphenylene: sparteine-mediated asymmetric synthesis of chiral D2-symmetric π-conjugated tetra-o-phenylenes, Tetrahedron, 2001, 57, 3725–3735 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2091854. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03733a |
| ‡ Authors with equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |