
Porous polyelectrolyte frameworks: synthesis, post-ionization and advanced applications
Ting
Zhou
,
Xingye
Huang
,
Ning
Ding
,
Zheng
Lin
,
Ying
Yao
and
Jia
Guo
 *
*
State Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200438, China. E-mail: guojia@fudan.edu.cn
First published on 8th December 2021
Abstract
Porous organic polymers (POPs), which feature high surface areas, robust skeletons, tunable pores, adjustable functionality and versatile applicability, have constituted a designable platform to develop advanced organic materials. Endowing polyelectrolytes with the distinct characteristics of POPs will attract mounting interest as the structural diversity of polyelectrolytes will bring the new hope of intriguing applications and potential benefits. In this review, the striking progress in ionized POPs (i-POPs) has been systematically summarized with regard to their synthetic strategies and applications. In the synthesis of i-POPs, we illustrate the representative ionic building blocks and charged functional groups capable of constructing the polyelectrolyte frameworks. The synthetic methods, including direct synthesis and post-modification, are detailed for the i-POPs with amorphous or crystalline structures, respectively. Subsequently, we outline the distinctive performances of i-POPs in adsorption, separation, catalysis, sensing, ion conduction and biomedical applications. The survey concerns the interplay between the surface chemistry, ionic interaction and pore confinement that cooperatively promote the performance of i-POPs. Finally, we conclude with the remaining challenges and promising opportunities for the on-going development of i-POPs.
 Ting Zhou | Ting Zhou received her BS degree from the Nanjing University of Science and Technology in 2016, and obtained her PhD degree in 2021 from Fudan University under the supervision of Professor Jia Guo. Currently she is a postdoctoral researcher at Fudan University. Her research interests focus on exploiting functional porous organic frameworks for photo- and electro-catalysis. |
 Xingye Huang | Xingye Huang received her BS degree in polymer materials and engineering from Fudan University in 2020. Currently she is finishing her MS degree under the supervision of Prof. Jia Guo in the Department of Macromolecular Science at Fudan University. Her research is focused on covalent organic frameworks in the field of photocatalysis. |
 Ning Ding | Ning Ding received her BS degree in composite material and engineering from East China University of Science and Technology in 2019. Then she joined Prof. Jia Guo's group as a graduate student in the Department of Macromolecular Science at Fudan University. Her research is focused on the design, synthesis and application of ionic covalent organic frameworks. |
 Zheng Lin | Zheng Lin received her BS degree from East China University of Science and Technology in 2019. She is currently a PhD candidate under the supervision of Prof. Jia Guo in the Department of Macromolecular Science at Fudan University. Her research is focused on the design, synthesis, and biological application of COF-based photocatalysts. |
 Ying Yao | Ying Yao received her BS degree in new energy materials and devices from Nanjing Tech University in 2019. She is currently an MS candidate under the supervision of Prof. Jia Guo in the Department of Macromolecular Science at Fudan University. Her research is focused on covalent organic frameworks in the field of electrocatalysis. |
 Jia Guo | Jia Guo received his PhD in polymer chemistry and physics from Fudan University in 2007. Then, he joined the group of Prof. Donglin Jiang at the Institute for Molecular Science (IMS, Japan) as a JSPS postdoctoral fellow. At the end of 2009, he joined the faculty of the Department of Macromolecular Science at Fudan University, where he is now a full professor. His current research efforts are focused on exploring the structure–activity relationships of porous organic polymers in the fields of catalysis and biomedical applications. |
1. Introduction
Over decades, porous organic polymers (POPs) have emerged as a new class of porous materials in which atoms are covalently bonded into two-dimensional (2D) or three-dimensional (3D) polygons. POPs are well known not only for their adjustable porosity, robust skeletons and pre-designable structures, but also for their chemically adaptable functions and synergistically enhanced performance. From a structural point of view, there are two kinds of representative POPs reported so far: amorphous POPs, which include polymers of intrinsic microporosity (PIMs),1 hyper-crosslinking polymers (HCPs),2 conjugated microporous polymers (CMPs)3 and porous aromatic frameworks (PAFs);4 and crystalline POPs, which predominantly refer to covalent organic frameworks (COFs)5 and covalent triazine frameworks (CTFs)6 with a periodic atomic arrangement and hierarchical ordering. Beyond the traditional applications of porous materials, such as adsorption,7 separation8 and catalysis,9,10 the diverse functionality of POPs has expanded their application scope enormously due to their structurally enhanced redox, optical, electrical and magnetic properties, thereby underpinning the rapid development of POPs for sensing,11 energy storage,12–14 photo/electrocatalysis,15 ion conduction,16 biological applications17,18 and so on. Encouraged by these striking achievements, functionalized POPs offer great opportunities for the creation of advanced materials with unique characteristics.Polyelectrolytes are charged macromolecules that can bear cations, anions or zwitterions on their backbones or pendant groups. They can be categorized as traditional all-organic polyelectrolytes and metallo-polyelectrolytes, both of which are well suited for exploring functional materials. Porous polyelectrolytes have been actively pursued to promote applicability via the synergy between Coulombic charges and porous structures, while conventional routes such as phase separation and layer-by-layer assembly usually render the polyelectrolyte macropores. These polyelectrolytes are remarkably different from the porous framework materials like POPs conferring intrinsic microporosity and mesoporosity and in turn, resulting in specific confinement effects. Therefore, based on a reticular chemistry strategy, polyelectrolyte porous frameworks, namely ionized POPs (i-POPs), have been intensively studied to exploit the advantages that include but are not limited to the following. (1) Electrostatic interaction is superimposed with a size-sieving effect to strengthen the affinity and selectivity of pore walls towards guests, particularly in mass transfer such as ion exchange/conduction and molecular adsorption/separation. (2) Charged functional modules are immobilized onto the side groups of POP skeletons to ionize the pore channels and substantially expose the active sites at interfaces for catalysis and sensing. (3) The incorporation of ions into the π-extended skeletons of POPs will improve the electron/hole dissociation and free charge mobility for photochemical and electrochemical applications. Therefore, the incorporation of polyelectrolytes can tailor specific functions of POPs, afford charge-induced effects and provide new possibilities for task-specific applications.
This review deals with the synthesis and application of polyelectrolyte porous frameworks, of which amorphous and crystalline structures are both involved. The review starts with the design principles of i-POPs from anionic, cationic and zwitterionic building blocks to synthetic methods including bottom-up and post-modification strategies. Next, applications of i-POPs in adsorption/separation, ion conduction, catalysis, sensing and bio-applications are comprehensively introduced. To conclude, the promising potentials and remaining challenges of i-POPs regarding technological synthesis and performances are summarized, which might inspire further research in this specific field. It is noted that many comprehensive reviews on neutral POPs19 or porous polyelectrolytes20,21 have been well documented, while porous polyelectrolyte frameworks have rarely been reviewed so far.
2. Synthesis of i-POPs
To establish the ionic frameworks with covalent linkages and high surface areas, it is necessary to consider the geometries and properties of building units, the properties of counterions, and the mechanisms of reactions. There are two general strategies to construct i-POPs, i.e., direct synthesis and post-modification. For direct synthesis, i-POPs can be obtained via the self-polymerization of ionic building blocks, the co-polymerization of ionic building units and neutral building units and the co-polymerization of neutral building units with ionic linkages. For the post-modification strategy, ionic units can be covalently immobilized to predesigned reactive sites in the neutral networks. Based on the ordering degree of frameworks, i-POPs can be classified into amorphous i-POPs, which include i-HCPs, i-CMPs, i-PAFs and i-PIMs, and crystalline i-POPs, which include i-COFs and i-CTFs. Thus, synthetic strategies of various i-POPs are introduced according to the above structural classification.2.1 Synthesis of amorphous i-POPs
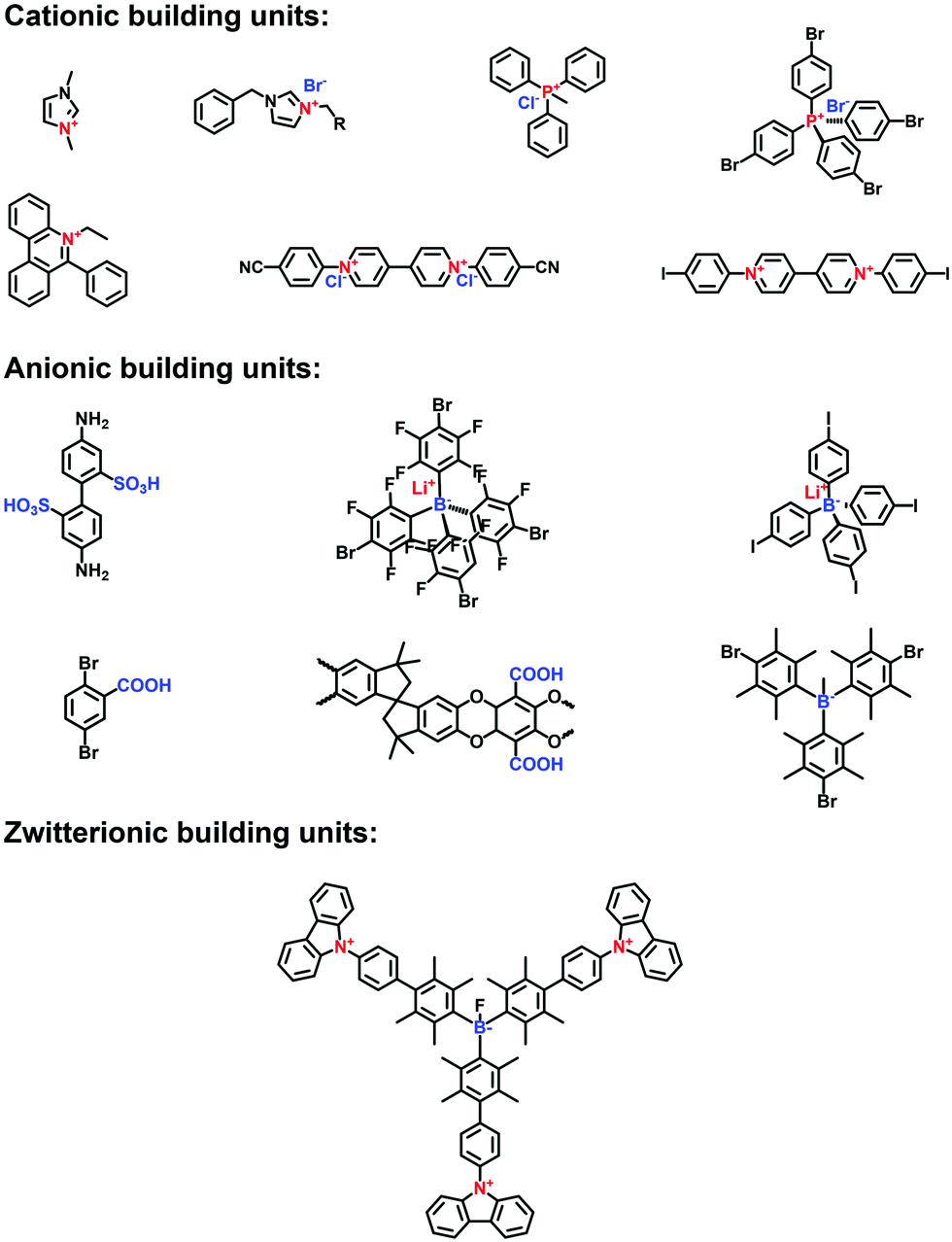 | ||
| Fig. 1 Building blocks for the construction of amorphous i-POPs. | ||
Imidazole derivatives are promising as a source of cationic building blocks. Wang et al.26 reported a series of imidazolium-functionalized i-HCPs prepared from the derivatives of 1-benzyl-imidazolium bromide through the hyper-crosslinking reaction. Also, imidazole-based ionic liquids (ILs) could be used to decorate PAFs via stepwise post-synthetic modification, using N-methylimidazole as a nucleophilic reagent that reacted with the chloromethyl side groups of the PAF frameworks.27 Song et al.28 adopted post-quaternization to incorporate methyl pyrimidinium salts into the frameworks, resulting in a pyrimidine-based i-CMP.
Carboxylic acid has often been applied to functionalize the side groups of POP skeletons, providing the chelating sites for metal ions. Zhu et al.29 synthesized a carboxyl-functionalized PAF via the Sonogashira–Hagihara reaction of 2,5-dibromobenzoic acid with tetrakis(4-ethynylphenyl) methane as a linker. A variety of metal counterions were tethered on the acidic sites of the frameworks, yielding enhanced adsorption properties for CO2 and CH4. Also, Meng et al.30 synthesized a PIM-1 via the condensation reaction of 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane and 2,3,5,6-tetrafluoroterephthalonitrile, using anhydrous K2CO3 as a catalyst. The PIM-1 with nitrile groups on the polymer backbone was hydrolyzed in NaOH solution to produce abundant –COONa side groups, which were then exchanged with divalent Zn2+ ions to form a physically crosslinked PIM-COONa-Zn.
Borane-based anions can be designed as building blocks to synthesize i-POPs. Typically, lithium tetrakis(4-iodophenyl)borate with a tetrahedral motif was applied to undergo the Sonogashira–Hagihara reaction with different alkyne monomers for anionic POPs.31 Borane can also be fluorinated to be negatively charged, which has been developed as a post-modification method for the ionization of POPs. For example, Feng et al.32 synthesized a stable anionic POP via a post-reaction between a boron-containing CMP and tetrabutylammonium fluoride, by which Lewis acid–base adducts were formed between tris(bromoduryl)borane and fluorine ions.
Zwitterionic POPs bear cationic and anionic sites, while both are anchored on the backbones of i-POPs. Compared with the cationic or anionic POPs, there have been only a few publications reporting zwitterionic POPs. Jiang et al.33 explored a zwitterion-containing POP film via a two-step sequence. The borane carbazole consisting of a tris(2,3,5,6-tetramethylphenyl) borane focal core and three N-substituted carbazole arms was prepared. It then underwent electro-polymerization to form a continuous and even film, which was subsequently fluorinated to form the borane anions. Finally, electro-oxidization of the N-substituted carbazole resulted in the quaternary ammonium cations. The zwitterionic POP films could offer the outstanding performances in various electronic devices.
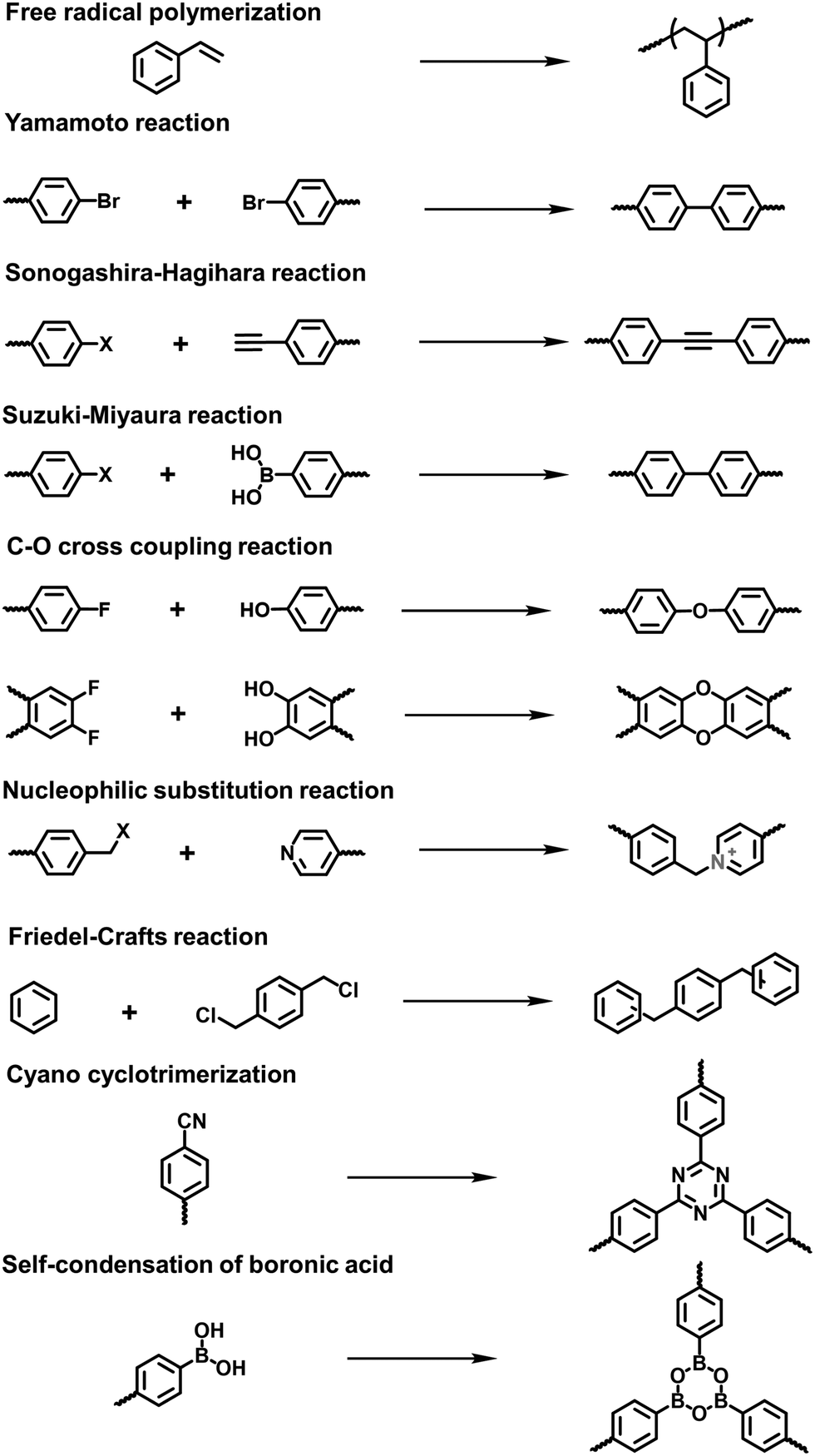 | ||
| Fig. 2 Reactions for the synthesis of amorphous i-POPs with different linkages. | ||
Xiao et al. reported the radical copolymerization of vinyl-functionalized quaternary phosphonium salts with tris-(4-vinylphenyl)-phosphine and 4-vinylbenzyl chloride for a hierarchical polymerized quaternary phosphonium (PQP) salt (Fig. 3a), using azobisisobutyronitrile (AIBN) as an initiator.34 The PQP salt afforded the N2 sorption profile with the type I plus type IV isotherm, indicative of the hierarchical pore character. Its BET surface area reached 758 m2 g−1. Also, Ding et al. applied a similar radical polymerization to prepare a series of P+X−@POPs (X = Cl, Br, I).35
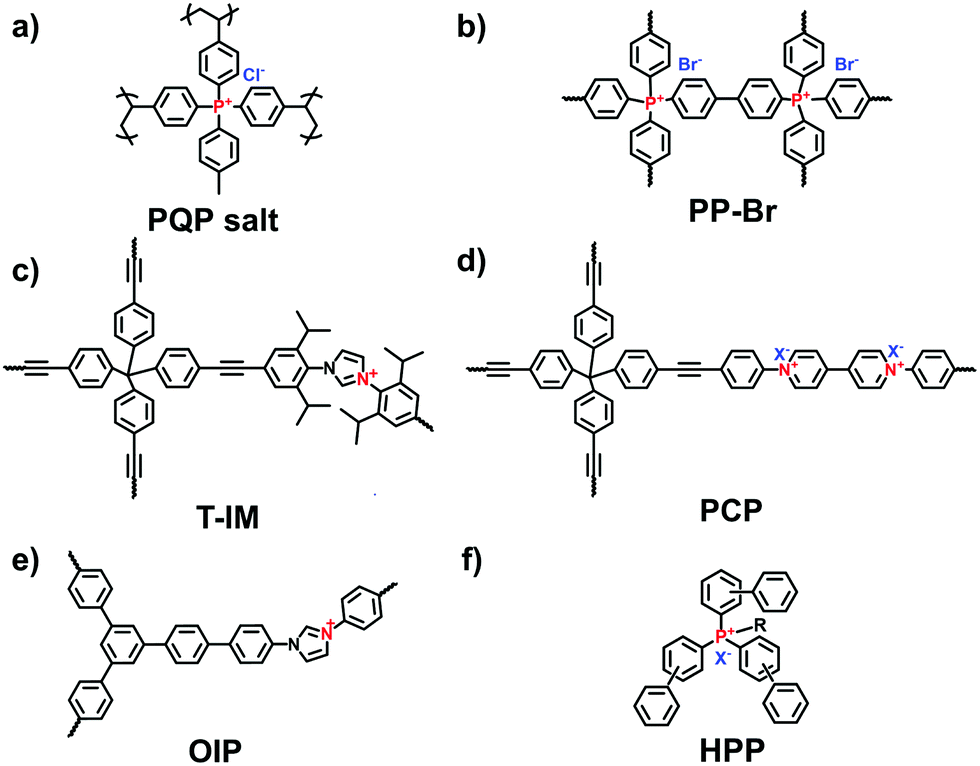 | ||
| Fig. 3 Structures of (a) PQP salt, (b) PP-Br, (c) T-IM, (d) PCP, (e) OIP and (f) HPP. | ||
The Yamamoto reaction brings about the dehalogenation condensation of aryl halides in the presence of a nickel(0) complex as the catalyst. Given its high efficiency and facile post-treatment, the Yamamoto reaction has been one of the commonly used coupling reactions for the synthesis of i-POPs. Zhang et al. reported the homo-polymerization of tetrakis(4-chlorophenyl)phosphonium bromide via the nickel(0)-catalyzed Yamamoto reaction,36 producing a cationic porous PP-Br network (Fig. 3b). The N2 uptake of PP-Br was remarkable at very low pressures, indicative of a micropore character, and afforded the BET surface area of 650 m2 g−1. However, the 31P NMR spectra of PP-Br revealed the decomposition of monomers into tertiary phosphine and quaternary phosphonium during the polymerization. To tackle the issue, Thomas et al. employed the copolymerization of tetrakis(4-bromophenyl)phosphonium bromide and tetraphenylmethane to discretely immobilize the charged units in the network.22 Such a cationic CPN-1-Br had no tertiary phosphine signals in the 31P NMR spectra, confirming that the addition of neutral building units successfully retained the charged phosphonium units. As a result, the BET surface area of CPN-1-Br was as high as 1455 m2 g−1.
The Sonogashira–Hagihara reaction has been intensively reported to synthesize i-POPs through the coupling of an aromatic halide and an aromatic alkyne in the presence of a palladium catalyst, a copper(I) co-catalyst and an amine base. Son et al. presented an imidazolium-based microporous organic network (T-IM) via the Sonogashira–Hagihara reaction of tetrakis(4-ethynylphenyl)methane with 1,3-bis(2,6-diisopropyl-4-iodophenyl)imidazolium chloride under solvothermal conditions (Fig. 3c).37 The cationic T-IM retained high porosity (620 m2 g−1) as well as exceptional catalytic activity for the cycloaddition of CO2 into cyclic carbonates. Thomas et al. designed a fluorinated tetraphenylborate anion, i.e., lithium tetrakis(4-bromo-2,3,5,6-tetrafluorophenyl)borate, (Li[B-(C6F4Br)4]), with a tetrahedral motif.31 The cross-coupling of Li[B-(C6F4Br)4] and 1,3,5-triethynylbenzene afforded an anionic microporous polymer network, showing a high surface area of 890 m2 g−1. Likewise, Zhu et al. built up a series of i-PAFs via the Sonogashira–Hagihara coupling of lithium tetrakis(4-iodophenyl)borate with different alkynes as linkers.38 Also, Coskun et al. used the same reaction to develop a series of cationic viologen-based POPs (PCPs) using 1,10-bis(4-iodophenyl)-[4,40-bipyridine]-1,10-diium salts and tetrakis(4-ethynylphenyl)methane (Fig. 3d). Their surface areas could be increased from 433 to 586 and to 755 m2 g−1 when the radius of that counterions of PF6−, BF4− and Cl− was decreased.24
The Suzuki–Miyaura reaction is another well-known C–C coupling reaction using aryl boronic acid and aryl halides for aryl–aryl linkages. Wang et al. reported the imidazolium-based i-POPs (OIPs) by following the Suzuki–Miyaura reaction of 1,3,5-tri(4-pinacholatoborolanephenyl)benzene and 1,3-bis(4-bromophenyl) imidazolium bromide (Fig. 3e).39 During the reaction, well-dispersed palladium nanoparticles could be generated in situ and stabilized by imidazolium groups in the confined pores, showing high catalytic activity towards the reduction of nitroarenes.
The Friedel–Crafts reaction catalysed by a Lewis acid under mild conditions has been used particularly for the synthesis of i-HCPs.26,40 Zhang et al. reported a series of phosphonium-based hypercrosslinked porous polymers (HPPs) via the Friedel–Crafts reaction of aldehyde dimethyl acetal with different triphenyl phosphonium salts (Fig. 3f), of which the BET surface areas could be controlled in the range from 770 to 1168 m2 g−1.23
With the rapid development of the design and synthesis of i-POPs, reactions for preparing i-POPs are not only limited to C–C formation but also include the covalent coupling of heteroatoms such as oxygen and nitrogen. Using the C–O cross-coupling reaction between fluorine and phenolic hydroxyl groups, Dai et al. synthesized a 1,4-dioxin-linked POP with fluorine-substituted ionic monomers and highly contorted 5,5,6,6-tetrahydroxy-3,3,3,3-tetramethyl-1,1′-spirobisindane (TTSBI).41 Through a solvent-free mechanochemical route, the cationic and anionic POPs were formed by the ball grinding of TTSBI with 1-methyl-3-(2,3,4,5,6-pentafluorobenzyl)-imidazolium hexafluorophosphate (ImPF6) and lithium tetrakis(2,3,4,5,6-pentfluorophenyl)borate (Li[B(C6F5)4]), respectively. Similarly, Ghosh et al. solvothermally prepared a sulfonate POP (PCF-1-SO3H) via the C–O cross-coupling of 1,1,1-tris(4-hydroxyphenyl)ethane and 2,4,6-tris(4-fluorophenyl)-1,3,5-triazine in the presence of K2CO3 in dimethylacetamide (Fig. 4a).42
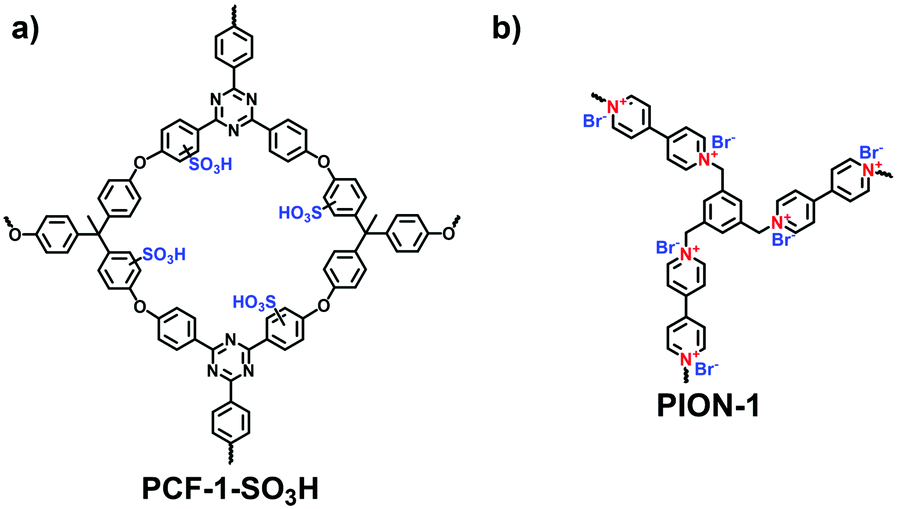 | ||
| Fig. 4 Structures of (a) PCF-1-SO3H and (b) PION-1. | ||
The nucleophilic substitution reaction is flexible and eco-friendly without using a metal catalyst for the formation of ionic linkages. Dai et al. reported the synthesis of a series i-POPs (PIONs) via the nucleophilic substitution of 1,3,5-tris(bromomethyl)benzene with 1,2-bis(4-pyridyl)ethylene, 1,2-di(4-pyridyl)diazene or 4,4′-dipyridyl, respectively, in the presence of a silica template (Fig. 4b).43 The formed quaternary ammonium salts provided the cationic linkages for the i-POPs. After removal of the SiO2 support, their BET surface areas were obtained in the range of 107–132 m2 g−1. With this method, the synthesized i-POPs often give moderate surface areas as the less rigid networks with abundant rotatable benzylic groups are prone to intermolecular packing.44,45
To improve the porosity of i-POPs synthesized via nucleophilic substitution reactions, a two-step method has been recently described to combine Friedel–Crafts alkylation with the nucleophilic substitution reaction. Wang et al. reported an imidazolinium-based i-POP (HCP-Cl-n) via such a two-step method using 2-phenylimidazoline (PhIm) and α,α′-dichloro-p-xylene (DCX) as building blocks.46 The self-condensation of DCX and the co-condensation of PhIm and DCX were conducted using the Friedel–Crafts alkylation, followed by quaternization resulting in an ionic hyper-crosslinking network with a remarkably increased porosity (1114 m2 g−1). Along this line, Yu et al. synthesized a series of imidazolinium-based i-POPs via a three-component polymerization with DCX and two imidazolinium-based monomers.47 The obtained POPs-B10 and POPs-B20 displayed a BET surface area of 823 and 662 m2 g−1, respectively. Later, Wang et al. conducted a tandem reaction involving Blanc chloromethylation, nucleophilic substitution and Friedel–Crafts alkylation to construct i-HCPs (iPT-n).48 After the condensation of 2,3-pyridinedicarboxylic anhydride (PDA) and biphenyl (DP) in the presence of chlorosulfonic acid, Blanc chloromethylation and quaternization produced the ionic modules in the networks. Then, Friedel–Crafts alkylation enabled the hyper-crosslinking of the building units. All of the obtained iPT-n possessed high BET surface areas in the range from 667 to 855 m2 g−1.
Other reactions such as the self-condensation of boronic acid and cyano cyclotrimerization have been explored to expand the diversity of i-POPs in structure. Zhu et al. synthesized boroxine-linked Cl-PAF-50 via the self-condensation of cyanuric chloride-containing 4-pyridinylboronic acid under solvothermal conditions at 120 °C for 36 h (Fig. 5a).49 Counterion exchange with other various halogen ions allowed the alteration of the pore sizes of the i-PAFs, of which F−-anchored PAF-50 possessed the optimal BET surface area of 580 m2 g−1. Coskun et al. reported the cyano cyclotrimerization of 1,1′-bis(4-cyanophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride in the presence of ZnCl2via an ionothermal method, producing the viologen-containing CTF (Fig. 5b).25 Such a positively charged network was featured with hierarchical pores including micropores and mesopores, of which the proportions could be adjusted via the reaction temperature. Accordingly, the BET surface areas were increased from 744 to 861 and to 1247 m2 g−1 at 400, 450 and 500 °C, respectively.
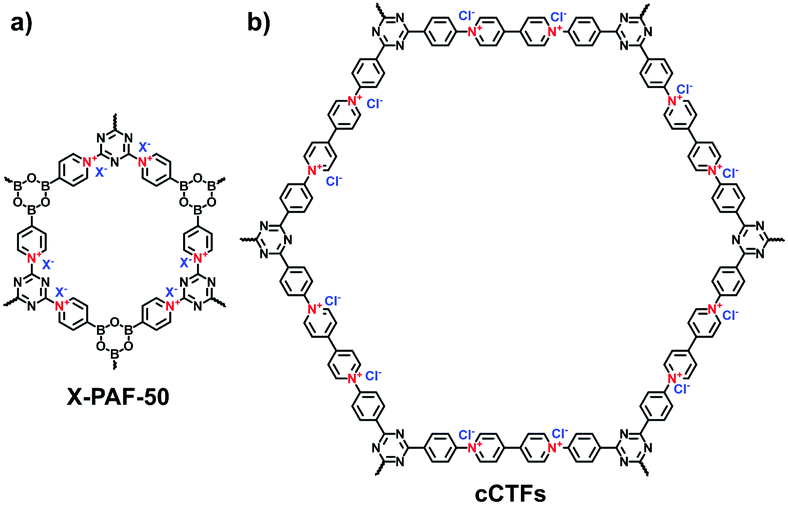 | ||
| Fig. 5 Structures of (a) X-PAF-50 and (b) cCTFs. | ||
 | ||
| Fig. 6 Reactions for ionizing amorphous POPs via post-modification strategies. | ||
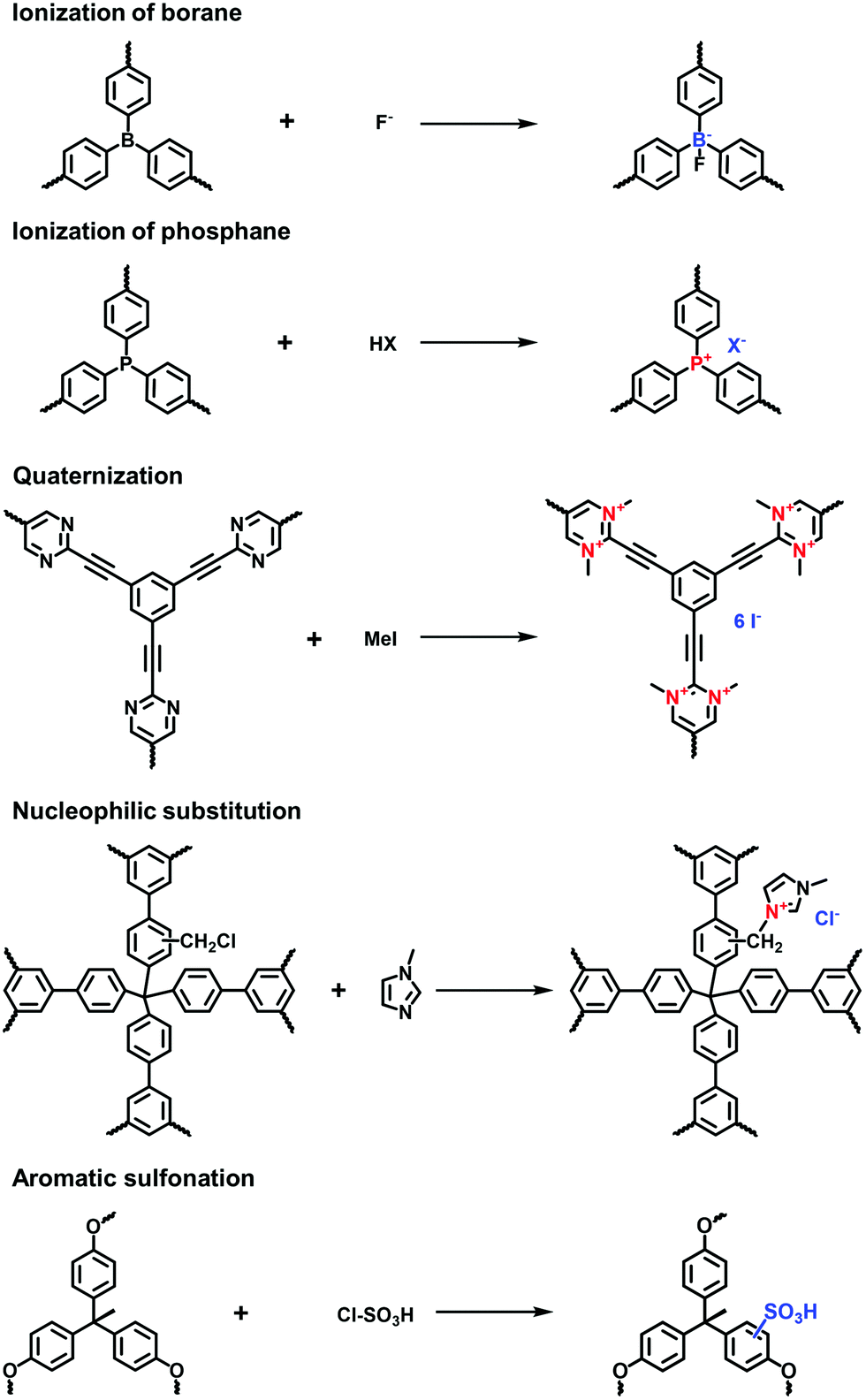
Building blocks containing the tetraphenylborate or tetraphenylphosphonium core have been widely exploited to construct neutral POPs through direct synthesis. Then, the post-modification of POPs could generate the ionic sites of P+ and B−, leading to a typical ionic framework. For example, Zhuang et al. accomplished the post-ionization of phosphorus-containing POPs (HC-TVP) prepared via the radical polymerization of tris(4-vinylphenyl)phosphane.50 Upon treatment with aqueous HI for 24 h, HC-TVP was transformed to i-POPs (HT-I) with P+ as the cations and I− as the counterions. Following this work, they continued to prepare the i-POPs with tetraaryl boron anions through the post-modification of neutral POPs (B-Ph-ae-n) with tridurylborane.32 The boron-containing B-Ph-ae-n was synthesized via the Sonogashira reaction of tris(bromoduryl)borane with different aryl alkynes. With the addition of tetrabutylammonium fluoride (Bu4NF), almost all of the boron atoms were coordinated with F− through the interaction of a Lewis acid and base. Thus, the neutral B-Ph-ae-n was converted to i-POPs with boron anions.
Post-quaternization is an effective strategy to incorporate quaternary ammonium moieties on the skeletons of POPs. Jiang et al. reported a zwitterionic porous network via the two-step post-modification of polyborane carbazole (PBC) using electrochemical synthesis. Tris(4-carbazolyl-1-phenyl-duryl)borane was electropolymerized to fabricate a borane-containing PBC film. Accompanied by the coordination of F− with B atoms, N-substituted carbazole units were quaternized via electro-oxidation, leading to a zwitterionic OFPBC film with radical cations and tetraaryl boron anions.51 Song et al. prepared a CMP (CMP-PM) through the Sonogashira reaction of 1,3,5-triethynylbenzene and 2,5-dibromopyrimidine. The pyrimidine moiety was methylated to N-methylpyrimidinium salts using CH3I, forming a cationic network with a moderate BET surface area of 241 m2 g−1.28
Imidazolium-derived ILs have been covalently attached to the skeletons of POPs using the post-modification method. Zhang et al. modified neutral HCPs with imidazolium cations via the Friedel–Crafts reaction.52 The remaining benzyl halides on the HCPs were applied to react with N-methylimidazole through nucleophilic substitution. The BET surface areas of the modified i-HCPs could reach as high as 926 m2 g−1. Zhu et al. proposed a two-step post-synthetic modification to immobilize imidazolium cations on the PAFs.27 The first step was to undergo the chloromethylation reaction with para-formaldehyde for grafting the chloromethyl groups on the PAF-111. Then N-methylimidazole was reacted with the attached chloromethyl groups via nucleophilic substitution, affording the imidazolium-modified PAF-111 with a BET surface area of 702 m2 g−1.
Aromatic sulfonation using chlorosulfonic acid has often been reported to endow the POPs with abundant anionic sites. Zhou et al. employed this reaction to build up a porous polymer network with the pendant sulfonic acid (PPN-6-SO3H).53 Although a number of sulphonic acid (–SO3H) groups were grafted on the skeletons of PPN-6, the BET surface area of PPN-6-SO3H still remained as high as 1254 m2 g−1. Compared with the direct synthesis of sulfonate i-POPs, the post-modification method is able to markedly increase the grafted amount of –SO3H groups in the porous networks.42
The alkali-metalation of POPs has attracted extensive attention as the metalated i-POPs exhibit an excellent performance in a wide field of applications such as H2 storage, catalysis and ion conduction. The naphthalene anion radical salt (Li+C10H8˙−) has been frequently used to accomplish the metalation of POPs through the interaction of Li+ cations with various active sites such as C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds and aromatic carbon atoms.54,55 For example, Cao et al. proposed a post-synthetic lithiated strategy, wherein the alkynyl groups of COP-1 were reacted with Li+C10H8˙− to form lithium carboxylate groups in the presence of dry ultrapure CO2.56 Zhu et al. adopted proton exchange with Li+C10H8˙− to anchor Li+ cations on the PAF-18-OH.57 Aside from Li+C10H8˙−, methyllithium (MeLi) has been proved effective to interact with the naphthyl groups of CMPs, preventing the clustering effect of Li+ during the lithiation process.58
C bonds and aromatic carbon atoms.54,55 For example, Cao et al. proposed a post-synthetic lithiated strategy, wherein the alkynyl groups of COP-1 were reacted with Li+C10H8˙− to form lithium carboxylate groups in the presence of dry ultrapure CO2.56 Zhu et al. adopted proton exchange with Li+C10H8˙− to anchor Li+ cations on the PAF-18-OH.57 Aside from Li+C10H8˙−, methyllithium (MeLi) has been proved effective to interact with the naphthyl groups of CMPs, preventing the clustering effect of Li+ during the lithiation process.58
2.2 Synthesis of crystalline i-POPs
In recent years, widespread attention has been gained on the reticular chemistry of ionic COFs including building units, linkages, reactions, synthetic methods and functionalized strategies. These studies have laid the foundation for the extension of emerging i-COFs. In this part, we summarize the milestones in the synthesis of i-COFs.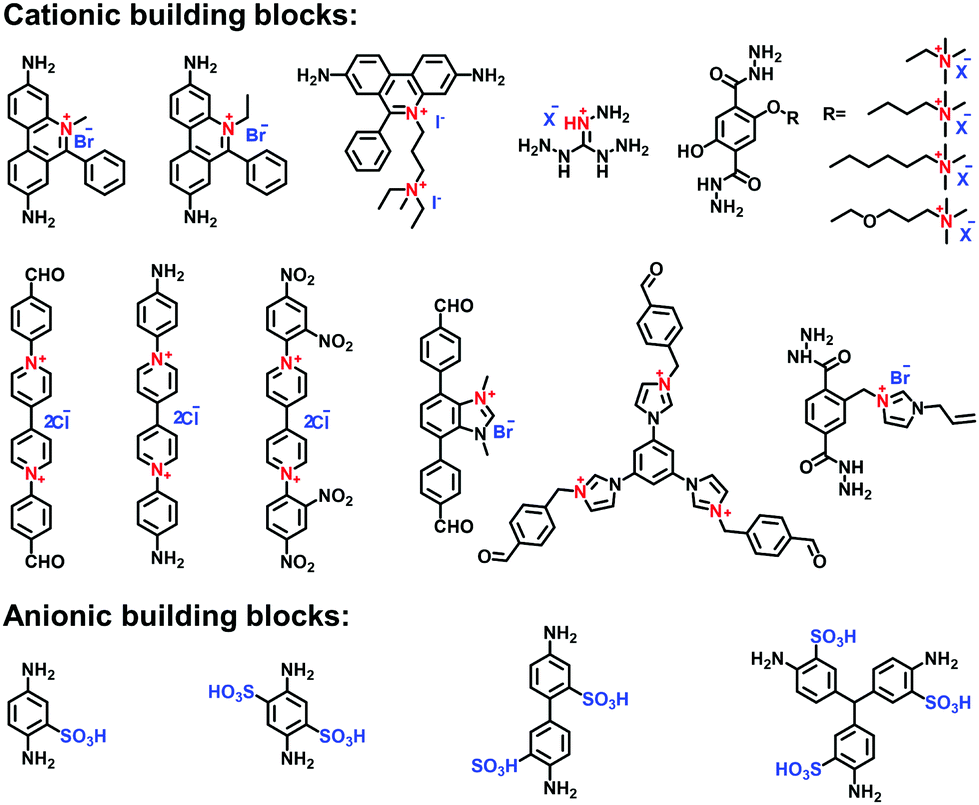 | ||
| Fig. 7 Building units for the construction of crystalline i-COFs. | ||
A series of ethidium bromide-based cationic COFs (EB-COF:X, X = F, Cl, Br, I) was reported by Zhu et al. in 2016, which is the first report of stable cationic crystalline frameworks (Fig. 8a).59 To construct the charged periodic frameworks, cationic ethidium bromide (EB) was designed as a C2 linker to couple with 1,3,5-triformylphloroglucinol (Tp), resulting in a β-ketoenamine-linked cationic COF. The ionized product retained the highly ordered arrangement by stacking the 2D hexagonal frameworks in an eclipsed mode. Furthermore, the porosity of EB-COF:X could be finely regulated via exchange of the counterions. Depending on the radius of the EB counterion, BET surface areas were achieved in the range from 616 to 1002 m2 g−1 and, correspondingly, the pore sizes were changed from 1.84 to 1.56 nm. This pioneering work elucidates the possibility of designing 2D i-COFs with adjustable porosity via a post-ion-exchange strategy.
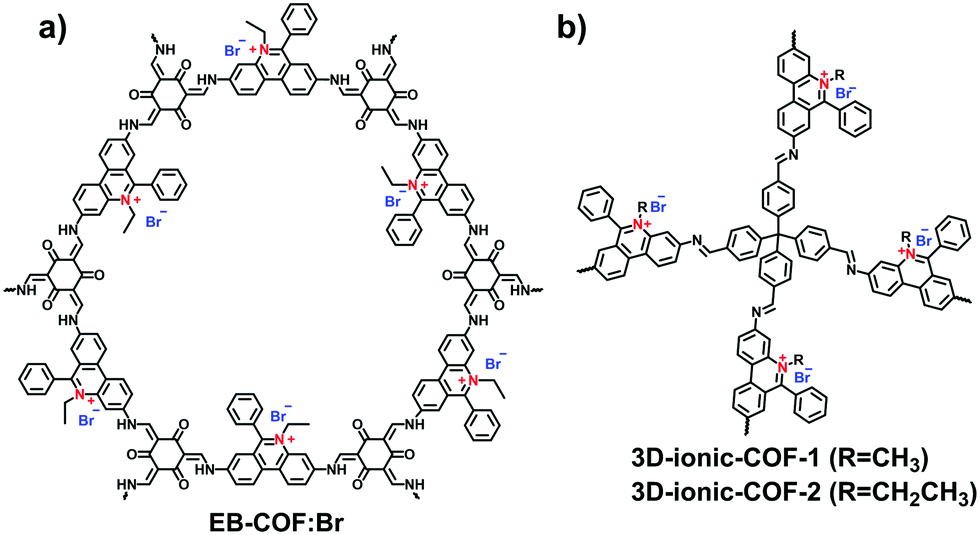 | ||
| Fig. 8 Structures of (a) EB-COF:Br and (b) 3D-ionic-COFs. | ||
Propidium iodide is a derivative of EB with C2 symmetry while linking a long dual-cation chain. Ajayaghosh et al. synthesized a propidium iodide-based COF (PI-TFP) via the aldimine reaction of 3,8-diamino-5-[3-(diethylammonio)propyl]-6-phenanthridinium diiodide (propidium iodide, PI) and 2,4,6-triformylphloroglucinol (TFP).60 Interestingly, such a cationic COF could be self-exfoliated in the aqueous solution owing to the strong electrostatic repulsion induced by the abundant quaternary nitrogen on the skeletons and the pendant diquats along the pore channels.
Compared with the layered stacking of 2D i-COFs, the interpenetrating structure of 3D i-COFs, to some degree, is conducive to alleviating the repulsion of adjacent charges within the framework, thereby enhancing the stability of the ordered structure. Qiu et al.61 fabricated two 3D i-COFs (3D-ICOF-1 and 3D-ICOF-2) utilizing tetrakis(4-formylphenyl)methane (TFPM) as a tetrahedral linker and diimidium bromide (DB) or EB as the building cations (Fig. 8b). Given the ionic properties of the skeletons, the two i-COFs had 3-fold penetration, which is lower than the 9-fold penetration of the neutral COF-320, while the BET surface areas were up to 966 m2 g−1 for 3D-ionic-COF-1 and 880 m2 g−1 for 3D-ionic-COF-2.
Triaminoguanidinium halide has been studied to ionize 2D COFs for post-exfoliation into nanosheets. In 2016, Banerjee et al. synthesized the guanidinium-based i-COFs (TpTGX, X = Cl, Br, I) through the aldimine reaction of Tp and triaminoguanidine hydrochloride in a mixture of dioxane and water under solvothermal conditions.62 The incorporated guanidinium cations in the skeletons could induce the self-exfoliation of TpTGXvia electrostatic repulsion. However, as the strong repulsion force existed between neighboring layers, the guanidinium-based COFs possessed limited crystallinity and porosity.63,64
Viologens based on cationic bipyridinium have a high C2 symmetry. Li et al. synthesized the viologen-based cationic COF (PC-COF) using 1,3,5-tris(4-aminophenyl)benzene (TAPB) as a knot and 1,1-bis(4-formylphenyl)-4,4′-bipyridinium dichloride (BFBP2+·2Cl−) as a strut.65 Although the viologen moiety was embedded in the cationic skeletons, the PC-COFs still adopted the eclipsed AA stacking as the chloride counterions were sandwiched between the adjacent layers. Ma et al. reported a soluble viologen-based COF (PyVg-COF) synthesized using 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetraaniline (PyTTA) and BFBP2+·2Cl− as monomers (Fig. 9a).66 Such a PyVg-COF dissolved readily in common solvents, showed good crystallinity and had a BET surface area of 348 m2 g−1. These characteristics were the result of strong π–π interactions provided by the pyrene knots in the structure. More interestingly, PyVg-COF could be processed into films in solution that afforded excellent conducting properties.
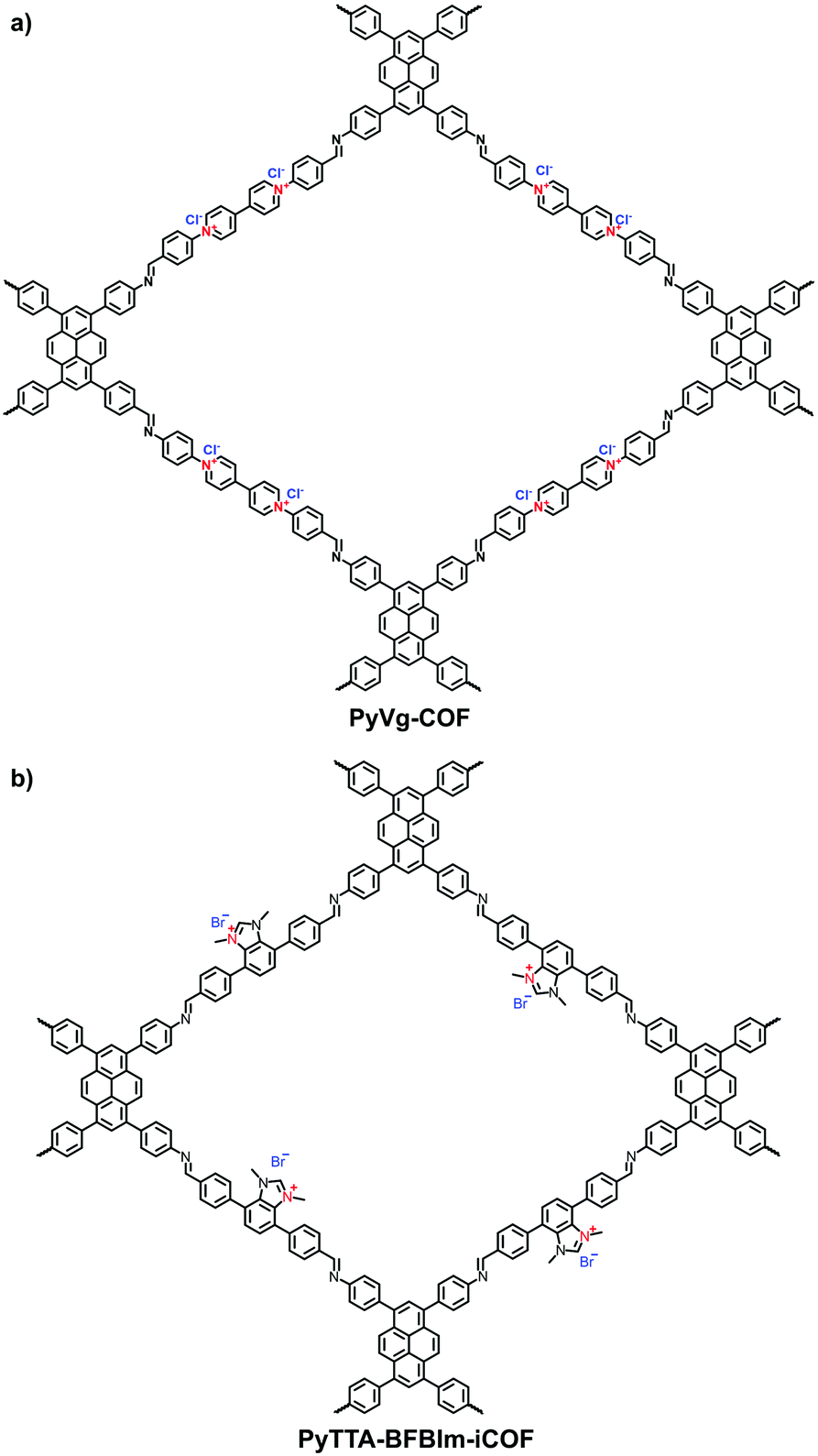 | ||
| Fig. 9 Structures of (a) PyVg-COF and (b) PyTTA-BFBIm-iCOF. | ||
Differing from viologens, imidazolium derivatives are less symmetric and often attached on the side of C2 building blocks. Jiang et al. reported a cationic COF (PyTTA-BFBIm-iCOF) with PyTTA as knots and 5,6-bis(4-formylbenzyl)-1,3-dimethyl-benzimidazolium bromide (BFBIm) as the linking cations (Fig. 9b).67 As the side cations at the strut of the frameworks could be coaxially aligned in the staggered way, PyTTA-BFBIm-iCOF was well crystallized and yielded a large BET surface area of 1532 m2 g−1. As such, benzimidazolium cations were sufficiently exposed at the pore–wall interfaces. In addition, imidazolium can be modified with allyl chains68 or immobilized on the branches of C3 building blocks. For example, Feng et al.69 synthesized the three-arm branched unit, 1,3,5-tris[3-(4-formylbenzyl)-1H-imidazol-1-yl]benzene bromide (TIBBr), as a central cation for the construction of hexagonal Im-COF-Br (Fig. 10).
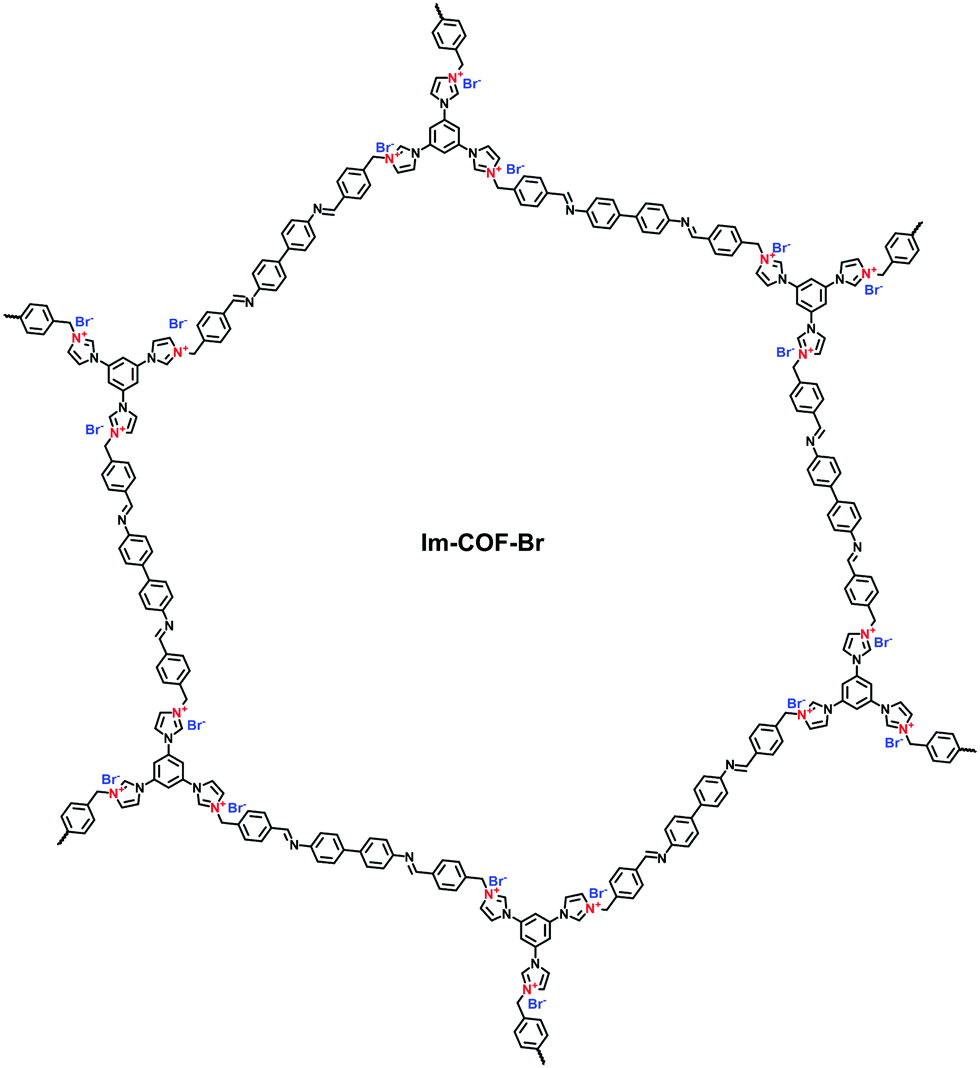 | ||
| Fig. 10 Structure of Im-COF-Br. | ||
So far, there are just a few anionic building blocks available for 2D COFs as displayed in Fig. 7. Zhang et al. adopted the transesterification reaction between trimethyl borate and diol-functionalized macrocyclic molecules to synthesize the spiroborate anionic COFs, ICOF-1 and ICOF-2, containing Me2NH2+ or Li+ as counter ions.70 The alkali catalyst, dimethylamine or lithium hydroxide, was necessary for the crystallization of i-COFs (Fig. 11a). In 2017, Feng et al.71 first used γ-cyclodextrin to build a spiroborate-linked 3D COF, of which a variety of counter cations such as Li+, HDMA+, H2PPZ2+, etc., could be entrapped.
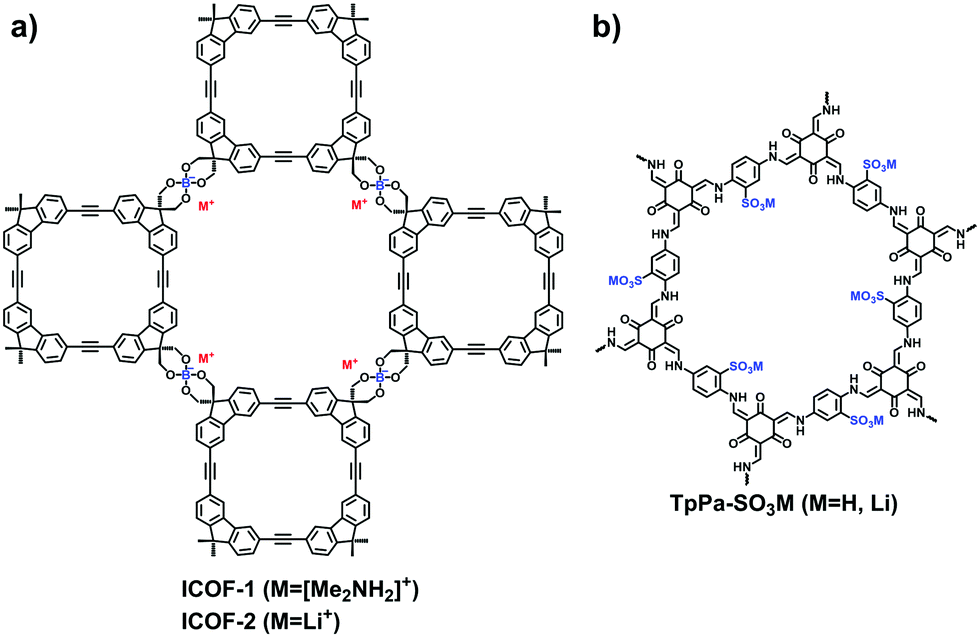 | ||
| Fig. 11 Structures of (a) ICOF-1 and ICOF-2, and (b) TpPa-SO3M. | ||
Given the applicability of sulfonates, the side groups of building blocks have been functionalized for the exploration of anionic COFs. In 2019, Lee et al. utilized the sulfonic-acid-modified 1,4-phenylenediamine (Pa-SO3H) to synthesize a TpPa-SO3 COF for creating numerous lithiated sites.72 As the pendant SO3Li groups were densely aligned in the pore channels, the resulting COF exhibited excellent single-lithium-ion conductivity. In 2021, Jiang et al.73 fabricated a self-supporting TpPa-SO3H COF membrane through the interface polymerization method (Fig. 11b). The membrane possessed parallel 1D pore channels and a high charge density in favor of the high salinity gradient energy conversion. To increase the density of acid sites in the COFs, Gu et al.74 designed a 2,2′-benzidine disulfonic acid as a strut of the framework. Qiu et al. modified a C3-symetric amine monomer with sulfuric acid on the three aromatic rings around the central carbon, 5,5′,5′′-methanetriyl tris(2-aminobenzenesulfonic acid).75 However, the ordering arrangement of atoms in the frameworks has been compromised due to the interference of strong acid groups.
The reported zwitterionic COFs have either zwitterionic linkages or zwitterions hanging at the side groups of the backbones. In 2013, Jiang et al. synthesized a squaraine-linked CuP-SQ COF via the condensation of squaric acid (SQ) and copper(II) 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAP-CuP) under solvothermal conditions (Fig. 12).76 The zwitterions were positioned at the strut of 2D tetragonal frameworks while they were coaxially overlapped for superior crystallinity and a high specific surface area (539 m2 g−1).
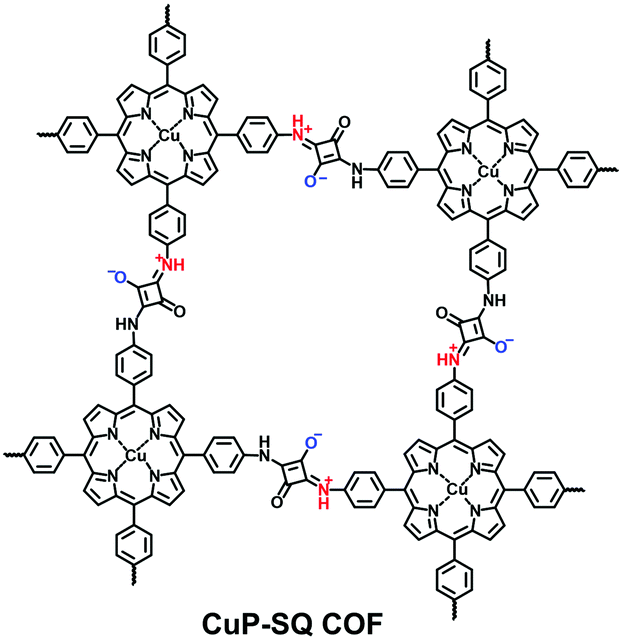 | ||
| Fig. 12 Structure of CuP-SQ-COF. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and hydrazone, and charged linkages including squaraine, spiroborate and viologen (Fig. 13).
C bonds and hydrazone, and charged linkages including squaraine, spiroborate and viologen (Fig. 13).
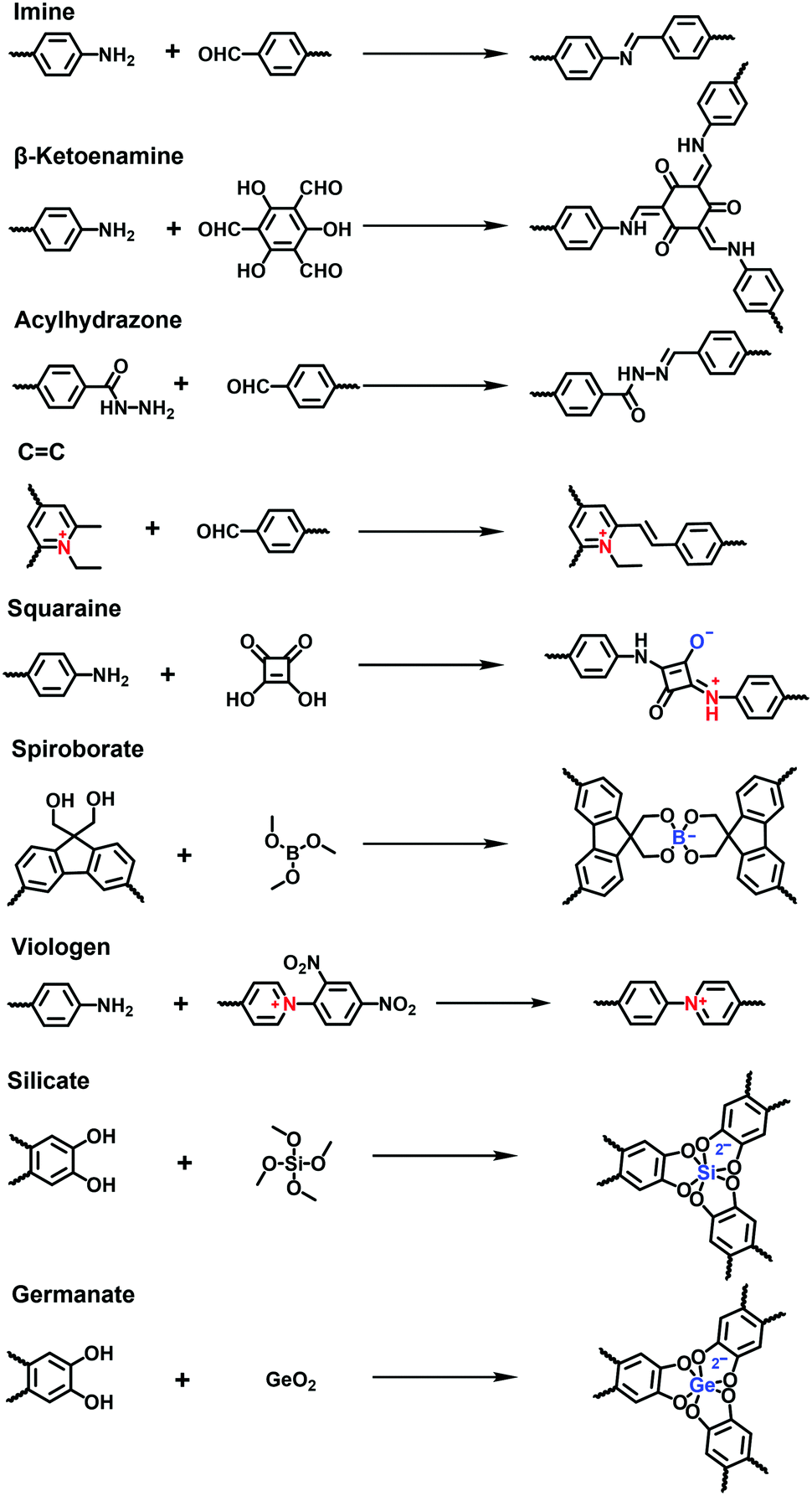 | ||
| Fig. 13 Synthetic reactions to produce i-COFs with different linkages. | ||
Most i-COFs reported so far are linked by imine bonds, which are formed via the Schiff-base condensation reaction of aromatic aldehydes and amines in the presence of an acid catalyst. Usually, the solvothermal method is superior to other methods to obtain highly crystalline imine-linked i-COFs. Even so, it is always challenging to incorporate ionic building blocks due to their solubility and reactivity, which are unfavourable of the growth of i-COFs. Thus, solvents, temperatures, reaction times and catalysts should be rigorously optimized to improve the crystallinity and porosity of i-COFs.
It has been reported that imine-linked COFs are susceptible to hydrolysis, especially in acidic solutions. To improve their chemical stability, imine bonds can be converted to β-ketoenamine bonds via irreversible enol-to-keto tautomerization in the frameworks formed by Tp and amines. Thus, the i-COFs maintain their chemical stability, expanding the scope of their applications under relatively harsh conditions. In addition to the commonly used solvothermal method, mechanochemical methods and interfacial polymerization are also applied to prepare i-COFs. For example, Ma et al. reported stable β-ketoenamine-linked EB-COF:Br nanosheets via the interfacial crystallization approach.78 Zhao et al. adopted a liquid-assisted grinding method to synthesize sulfonate COFs using Tp as a knot and 2,5-diaminobenzenesulfonic acid (DABA) or 2,5-diaminobenzene-1,4-disulfonic acid (DABDA) as a linker. The method could realize the scaled-up synthesis of NUS-9 and NUS-10 (Fig. 14).79 It is worth noting that NUS-10 could not be obtained using the solvothermal method owing to the extremely low solubility of DABDA in organic solvents. Besides, based on the aldimine reaction, the hydrazide building blocks modified with imidazolium68 or quaternary ammonium80 were available to react with aldehydes for producing a series of i-COFs with ionic side groups.
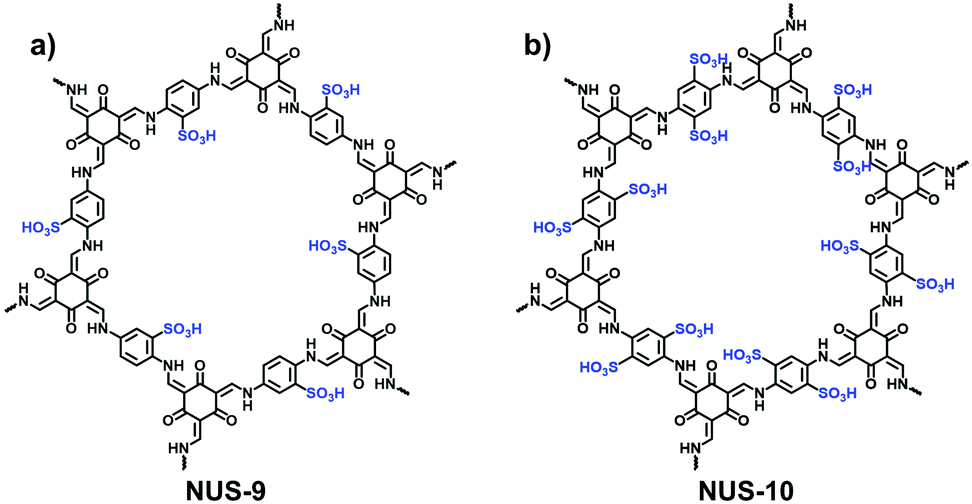 | ||
| Fig. 14 Structures of (a) NUS-9 and (b) NUS-10. | ||
The sp2-carbon COFs bearing stable CC linkages allow a fully conjugated and periodic framework, although their synthesis often suffers from the inferior reversibility of the Knoevenagel condensation reaction. Thus, there have only been a few sp2-carbon COFs reported so far and, consequently, their ionization has rarely been explored. Recently, Zhang et al. addressed a quaternization-promoted reversibility method to synthesize a high-quality sp2-carbon i-COFs via the Knoevenagel reaction of N-ethyl-2,4,6-trimethylpyridinium halide with 1,3,5-tris-(4′-formyl-biphenyl-4-yl)triazine.81 The resultant i-COFs were positively charged by the pyridine quaternary ammonium salts at the knots of frameworks and exhibited a high surface area of 1343 m2 g−1.
Except for neutral linkages, the reactions forming charged bonds have been studied to crystallize COFs. The condensation of squaric acid with aromatic amines leads to a squaraine linkage, which is a zwitterion consisting of oxygen anions and protonated imine cations. Jiang et al.76 first reported a squaraine-linked CuP-SQ COF through the reaction of squaric acid (SQ) and copper(II) 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAP-CuP) via SQ self-catalytic condensation under solvothermal conditions.
Spiroborate is structured with a tetrahedral tetrakis(spiroborate) containing an anionic boron center. It is formed via a transesterification reaction between polyols and trimethyl borate (B(OMe)3) in the presence of an alkali catalyst. The early reported boron-based linkages with strong Lewis acidic properties were prone to hydrolytic decomposition even in the presence of atmospheric moisture at ambient temperature. By contrast, spiroborate-linked i-COFs with anionic boron centers showed remarkable hydrolytic stability. This was ascribed to the additional Lewis-base coordination with boron in anionic spirocyclic esters, which suppressed the hydrolysis reaction. As an alternative method to solvothermal synthesis, the microwave-assisted solvothermal method has been developed to synthesize i-COFs with spiroborate linkages.71 Feng et al. obtained a highly crystalline 3D anionic COF through the condensation of γ-cyclodextrin (γ-CD) and B(OMe)3 under microwave-assisted solvothermal conditions, leading to the BET surface area of 760 m2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g−1.
g−1.
A π-conjugated viologen-based cationic COF (COGF) has been synthesized via the Zincke reaction between 1,1′-bis(2,4-dinitrophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride (BDB) and TAPB in a mixture of ethanol and water (1/1, v/v) under the microwave conditions (Fig. 15).82 Specifically, the nucleophilic N-(2,4-dinitrophenyl)pyridinium was attacked by the aromatic aldehyde amine. Subsequently, electrocyclic ring opening formed the König salt. Finally, the ionic pyridinium was obtained by ring closure after the elimination of 2,4-dinitroaniline.
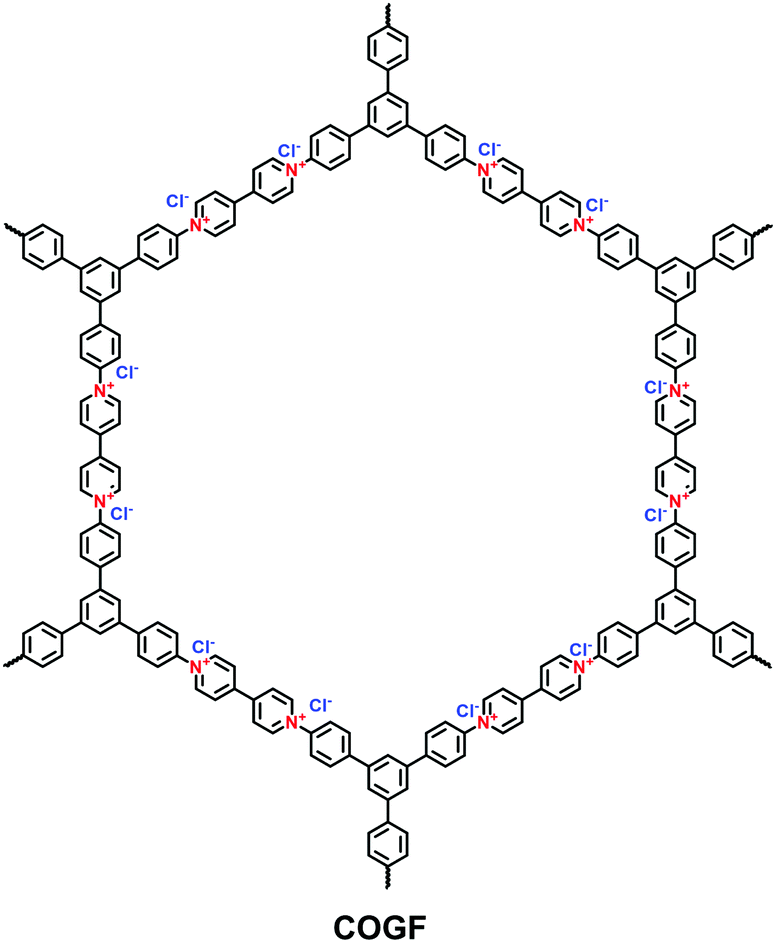 | ||
| Fig. 15 Structure of COGF. | ||
Besides the aforementioned reactions, Thomas et al. developed a hyper-coordinated silicon anion to constitute a silicate-based i-COF (Fig. 16a).83 The reversible Si–O bonds ensured the crystallization of the frameworks in the condensation of hexacoordinate silicon with 9,10-dimethyl-2,3,6,7-tetrahydroxyanthracene (DMTHA) under basic conditions. Also, it was available to build 3D SiCOF-5 with dianionic hexacoordinate [SiO6]2− as a node and 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) as a linker.84 Analogously, the hexa-coordinated germanate (Ge) center could be incorporated into the crystalline frameworks through the microwave heating of germanium dioxide and 9,10-dimethyl-2,3,6,7-tetrahydroxy-anthracene (DMTHA) in the presence of 1 M CH3OLi in methanol at 180 °C for 6 h (Fig. 16b).85,86 The obtained Ge-COF-1 maintained a well-defined primary- and higher-ordered structure as well as a high BET surface area of 808 m2 g−1.
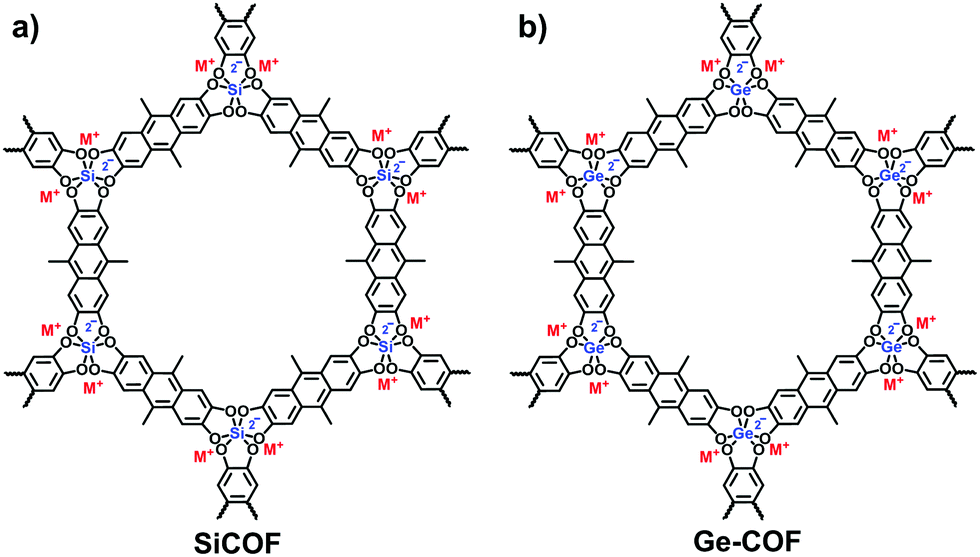 | ||
| Fig. 16 Structures of (a) SiCOF and (b) Ge-COF. | ||
Viologen-based i-COFs usually suffer from the weak stacking interactions between the layers, causing limited crystallinity and porosity. As a result, the advantages of ordered and accessible 1D pore channels are hard to be exhibited. To tackle this issue, our group proposed a novel strategy to convert 2,2′-bipyridine-based COFs from neutral to positively charged viologen-based frameworks and subsequently to cationic radical frameworks via a two-step post-synthetic approach.87 The pristine neutral COF (Py-BPy-COF) was synthesized via the Schiff-base condensation of 4,4,′4,′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetraaniline (Py-TA) and (2,2′-bipyridine)-5,5′-dicarbaldehyde (2,2′-BPy-DCA) in the presence of 6 M HOAc (Fig. 17). The acid catalyst facilitated the interconversion of the trans- form of 2,2′-BPy-DCA to the more stable cis conformer. Also, the interlayered stacking of 2D COFs via π-interaction inhibited the configuration transformation of cis-2,2′-bipyridine units, providing the essential prerequisite for the formation of cyclic alkylated diquats via the post-quaternization of Py-BPy-COF with 1,2-dibromoethane. The maximum conversion of quaternization was up to 72.7% and the ordered layer-stacking could be maintained in this case. Subsequently, the redox reaction on Py-BPy2+-COF was conducted using aqueous sodium dithionite (Na2S2O4). The cyclic diquat [BPy]2+ was reduced to cationic radical [BPy]+˙ moieties, giving the radical-containing COF (Py-BPy+˙-COF). Py-BPy2+-COF and Py-BPy+˙-COF both showed high crystallinity and the BET surface areas were 461 and 398 m2 g−1, respectively. The maintained overlapping arrangement of the COF layers enabled the ordering alignment of charged radicals, resulting in strengthened radical properties and an excellent photothermal conversion efficiency. This post-synthetic strategy was demonstrated to be applicable to different 2,2′-bipyridine-based COFs,88 elucidating its generality toward the development of i-COFs.
 | ||
| Fig. 17 Schematic representation of the reactions employed for the post-modification of skeletons. | ||
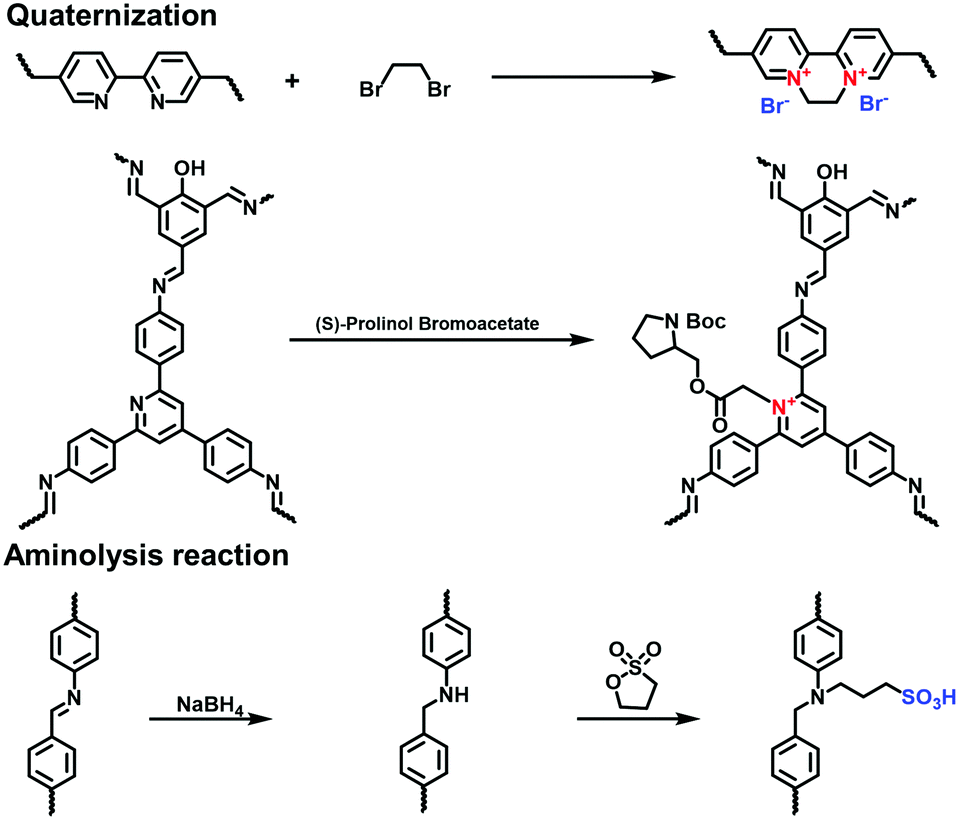
To incorporate the ionic chiral units, Zhang et al. prepared the pyridine-based COFs (TPP-HTD COF) via the condensation of 2-hydroxyl tribenzaldehyde (HTD) and 2,4,6-(4′-triaminophenyl)pyridine (TPP).89 (S)-Prolinol bromoacetate was linked to the N atom of the pyridine unit in the TPP-HTD COF via post-modification in acetonitrile at room temperature. The formed pyridinium salts positively charged the skeletons and allowed the immobilization of the chiral auxiliary on the pore walls. The resulting chiral i-COF maintained its highly crystalline structure.
The post-synthetic functionalization of linkages with ionic groups is an alternative method to ionize COFs. Gao et al. reported a strategy to immobilize ionic units onto the linkages via the post-modification method.90 Using NaBH4, the imine linkages of TFPPy-PyTTA-COF were quantitatively reduced to amine bonds without he loss of crystallinity and porosity. The amine linkages subsequently underwent the aminolysis reaction with 1,3-propane sulfone, giving a sulfonate anionic COF, SO3H@TFPPy-PyTTA-COF.
Han et al. reported a post-modification strategy to anchor ionic modules on the structural defects of 2D COFs (Fig. 18).91 To introduce a number of reactive sites, monoaldehyde DHA was employed as the end-capping units for condensation with TAPB and DHTA. Thus, there were a certain number of unreacted amine groups left as anchoring sites for ionic units in dCOF-NH2-Xs. The cationic aldehyde monomer, 3-benzyl-2-formyl-1-methyl-1H-imidazol-3-ium bromide (ImBr) was linked with the remaining amines via the Schiff-base reaction. The work offers a defect-controlled methodology to precisely implant ionic units in the frameworks via post-modification.
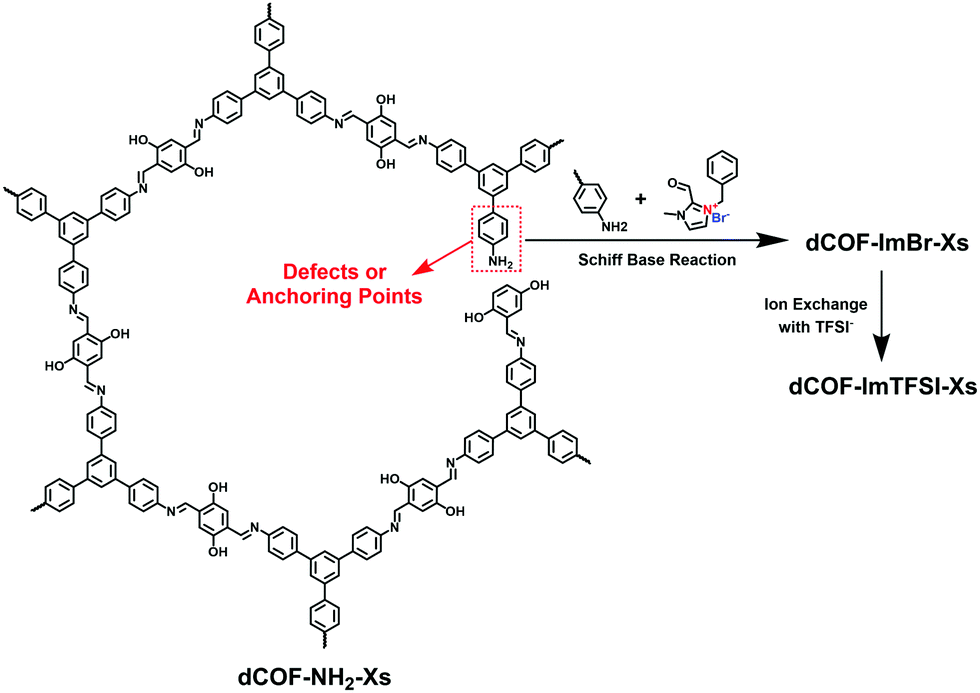 | ||
| Fig. 18 Schematic of the synthesis of dCOFs via the post-modification strategy to anchor ionic modules at the defects of 2D COFs. | ||
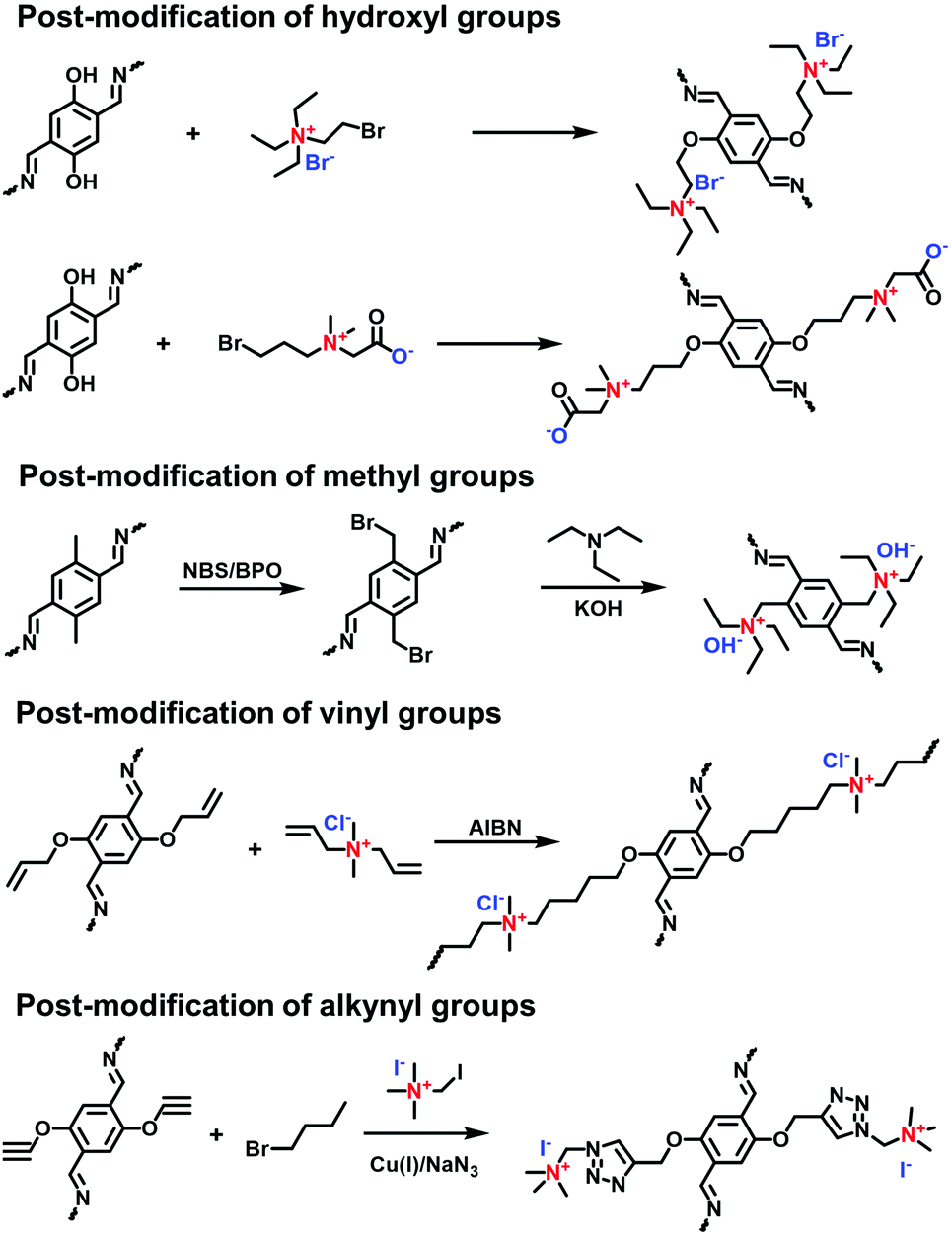
The post-synthetic approach is an important strategy to convert neutral pores to ionic channels. The reactive sites attached on the pore walls are given by the side groups of COF skeletons such as hydroxyl, methyl, vinyl and alkynyl groups for the functionalization of neutral COFs (Fig. 19).
 | ||
| Fig. 19 Schematic representation of the reactions employed for the post-modification of pores. | ||
Incorporating ILs into the pores of neutral COFs by post-modification via the Williamson ether reaction of hydroxyl and bromo groups has been widely reported.92,93 In 2016, Gao et al. reported a multivariate i-COF furnished with ILs.94 The neutral pristine COFs were obtained via the co-condensation of PyTTA as a node and 2,5-dihydroxyterephthalaldehyde (DHPA) and 1,4-phthalaldehyde (PA) as dual linkers. The molar ratios of DHPA and PA were varied to prepare [HO]X%-Py-COFs containing different phenol contents, where X% represents the molar percentage of DHPA relative to the dialdehyde monomers. (2-Bromoethyl)triethylammonium bromide was immobilized on the COFs via reaction with phenol groups in [HO]X%-Py-COFs, giving [Et4NBr]X%-Py-COFs. Therefore, the ionization degree was controlled by the content of the phenol groups in the pristine neutral COFs. Along this line, the zwitterionic 2-((3-bromopropyl)-dimethylammonio) acetate was anchored on the skeletons of [OH]X%-TD-COFs through the same strategy by Han et al.95
Yan et al. reported the post-modification of methyl groups in neutral COFs to synthesize cationic COFs. The pristine COF (TpBD-Me) was formed via the Schiff-base condensation of Tp and BD-Me.96 The Wohl–Ziegler reaction was applied to brominate TpBD-Me using NBS and BPO and, in turn, TpBD-MeBr was quaternized by trimethylamine (TMA) at room temperature, obtaining a cationic COF (TpBD-MeQA+Br−) with quaternary ammonium (QA) ions in the pores.
Unsaturated vinyl and alkynyl groups have been employed as the side groups of building blocks for the post-modification of COFs. A vinyl-containing COF (DhaTab-V) prepared using vinyl-modified 2,5-dihydroxyterephthalaldehyde (Da-V) and 3,5-tris(4-aminophenyl)benzene (Tab) had modifiable pores.97 A cationic surfactant diallyldimethylammonium chloride (DMDAAC) with vinyl groups was covalently anchored onto DhaTab-V via radical polymerization, resulting in cationic-surfactant-modified pores. Likewise, Zhang et al. reported the copolymerization of 4-vinylbenzyl chloride with a vinyl-decorated COF, followed by quaternization with tertiary amines for the modification of COFs with linear poly(ionic liquid)s in the pores.98 In addition, the efficient click reaction was applied to functionalize alkynyl-containing COFs, enabling a high load of quaternary ammonium salts within the pore channels.99
3. Applications
3.1 Adsorption and separation
Porous materials have been intensively explored for the adsorption and separation of gases and chemical species. The predominant factors determining the adsorption and separation performances of porous materials include the specific surface area, pore volume, pore size and chemical environments inside the pores. i-POPs have the characteristic of charged skeletons with pre-designable configurations, thereby providing strong ionic interactions with targeted molecules. Also, precise control over the pore properties will strengthen the size-sieving effect for exceptional selectivity. Therefore, i-POPs have great potential in gas storage and separation and in ionic pollution removal. Technically, the separation of gases and chemical species with i-POPs is correlated with their specific forms, including powders and membranes. Although they confer the same pore sizes and environments, i-POP powders enable the separation of single-component products in the mixtures, while i-POP membranes allow the separation of each component in the mixtures. This promotion arises from the different diffusion rates and the size-exclusion effect of the membranes, of which the ordered vertical channels act as nano-funnels to play a key role.| i-POP | S BET (m2 g−1) | T (K) | P (bar) | CO2 (wt%) | Ref. |
|---|---|---|---|---|---|
| a CO2 uptake (cm3 g−1). | |||||
| PCP-Cl | 755 | 273 | 1 | 10.2 | 24 |
| PCP-BF4 | 586 | 273 | 1 | 9.7 | 24 |
| PCP-PF6 | 433 | 273 | 1 | 7.8 | 24 |
| cCTF | 1247 | 273 | 1 | 12.6 | 25 |
| CPN-1-Br | 1455 | 273 | 1 | 11.0 | 22 |
| CPN-1-Cl | 1504 | 273 | 1 | 12.5 | 22 |
| CPN-2-Br | 540 | 273 | 1 | 6.8 | 22 |
| HIP-Cl-1 | 1114 | 273 | 1 | 16.7 | 46 |
| HIP-Br-1 | 743 | 273 | 1 | 14.5 | 46 |
| POPs-B10 | 824 | 273 | 1 | 71.7a | 47 |
| POPs-B20 | 662 | 273 | 1 | 73.6a | 47 |
| F-PAF-50 | 580 | 273 | 1 | 14.7 | 49 |
| Cl-PAF-50 | 337 | 273 | 1 | 8.7 | 49 |
| Br-PAF-50 | 158 | 273 | 1 | 5.0 | 49 |
| 2I-PAF-50 | 96 | 273 | 1 | 1.3 | 49 |
| POM1-IM | 926 | 273 | 1 | 13.9 | 52 |
| POM2-IM | 653 | 273 | 1 | 14.5 | 52 |
| POM3-IM | 575 | 273 | 1 | 14.2 | 52 |
| PPN-6-SO3Li | 1186 | 295 | 1 | 13.5 | 53 |
| Li@COP-1 | 573 | 298 | 18 | 22 | 56 |
| PAF-18-OLi | 981 | 273 | 1 | 14.4 | 57 |
| 3D-ionic-COF-1 | 966 | 273 | 1 | 16.1 | 61 |
| 3D-ionic-COF-2 | 880 | 273 | 1 | 13.3 | 61 |
| PyTTA-BFBIm-iCOF | 1532 | 273 | 1 | 17.7 | 67 |
| COF-IL | 291 | 273 | 1 | 106a | 68 |
| 5%Li@PAF-1 | 479 | 273 | 1.22 | 39.6 | 103 |
| TAPOP-1 | 940 | 273 | 1 | 15.4 | 104 |
| TAPOP-2 | 930 | 273 | 1 | 13.6 | 104 |
| CB-PCP-1 | 419 | 273 | 1 | 9.0 | 105 |
3.1.1.1 Hydrogen adsorption. H2 is an environmentally friendly fuel with a high energy density. Over the decades, tremendous effort has been devoted to the capture and storage of H2. The goal for the H2 storage capacity proposed by the U.S. Department of Energy is 4.5 wt% at −40 to 85 °C and 5–12 bar.100 In order to achieve the goal, strategies to enhance the interaction between H2 and i-POPs are in urgent need of exploration. From a theoretical point of view, hybridizing POPs with alkali-metal ions can improve the H2 uptake capability.56 This is attributed to the electrostatic charge-quadrupole and charge-induced dipole interactions between the H2 molecules and the atomically dispersed alkali-metal ions (e.g., Li+ and Na+).101,102 To prove this, the alkali-metalation strategy for H2 storage was experimentally demonstrated by Deng et al. on a CMP platform.54 The pristine CMP (HCMP-1; Fig. 20a) was synthesized using 1,3,5-triethynylbenzene via the oxidative homocoupling reaction of terminal alkynes and showed good chemical and thermal stability and high surface areas that arise from the micropores. As the CMPs were immersed in a THF solution of the naphthalene anion radical salt (Li+C10H8˙−), Li+ ions were coordinated with the electron-rich C
C bonds, resulting in a Li-doped CMP (Li-CMP). Theoretical calculations demonstrated that the adsorbed H2 molecules were mostly clustered around the Li atoms. The H2 uptake value of the pristine CMP was only 1.6 wt% at 77 K and 1 bar, while the doped CMP with 0.5 wt% Li conferred an adsorbed H2 capacity of up to 6.1 wt%. The noticeable difference was ascribed to the strong interaction between the Li+ and H2 molecules as well as the high surface area (795 m2 g−1) and large pore volume (1.61 cm3 g−1).
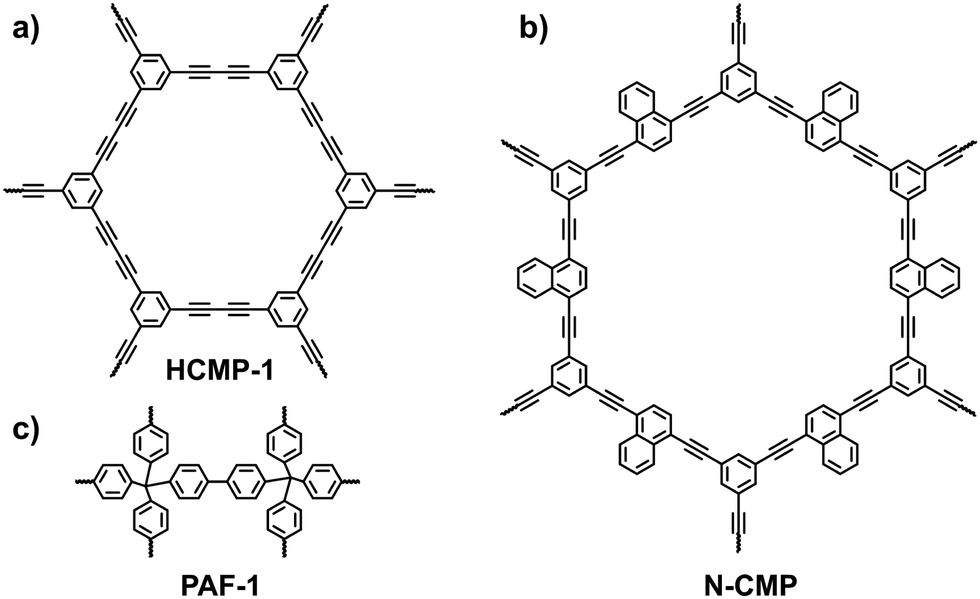 | ||
| Fig. 20 Structures of (a) HCMP-1, (b) N-CMP and (c) PAF-1 that are employed for alkali-metalation to improve the H2 uptake capability. | ||
Although Li atoms can stay on alkyne bonds, they tend to aggregate as a cluster upon an increase in the feed contents. Compared with the pristine POPs, the specific surface areas and total pore volumes of the metalated i-POPs are declined severely and impair H2 uptake. To deal with the issue, other lithiation dopants were alternatively anchored at the active sites in i-POPs. Deng et al. found that methyllithium (MeLi) was an effective lithiation dopant that significantly enhanced the H2 storage performance of the Li-POPs.58 With the assistance of theoretical simulation, it was demonstrated that MeLi provided a more readily accessible binding environment for H2. When naphthyl-containing CMP (N-CMP) was synthesized as the host (Fig. 20b), the clustering of lithium with an increase in its loading amount was suppressed because MeLi could be dispersed on both sides of the naphthyl plane. H2 uptake capacities of 6.5 wt%, 4.7 wt% and 2.6 wt% were achieved at 77 K and 80 bar for MeLi-doped N-CMP (3 wt%-Li), Li+C10H8˙−-doped N-CMP (9 wt%-Li) and pristine N-CMP, respectively. A promising H2 uptake under both cryogenic conditions and near room temperature is challenging but crucial for practical applications. MeLi-doped N-CMP (3 wt% Li) showed an H2 uptake of 1.0 wt% at 273 K and 80 bar. This study provides an accessible route for improving the H2 storage performance of metalated i-POPs.
Likewise, the lithiation strategy was applied to the frameworks of PAFs and COFs for optimizing the H2 and CH4 adsorption capacities. Hill et al. reported a lithiated PAF (Li@PAF-1) by conjugating Li+C10H8˙− with PAF-1 (Fig. 20c).103 Compared with the parent material, the pore size of 5%_Li@PAF-1 shrank to 1.1 nm, which was more appropriate for H2 storage at 77 K. Accordingly, 5%_Li@PAF-1 gave a high H2 uptake capacity of 2.2 wt% at 77 K and 1.22 bar. Also, the exposed Li+ sites allowed the materials to adsorb 2.1 wt% CH4 at 273 K and 1.22 bar. Zhang et al. reported a spiroborate-linked i-COF (ICOF-2) with sp3-hybridized boron anions and Li+ counterions.70 ICOF-2 exhibited a high BET surface area of 1259 m2 g−1 and a narrowed pore-size distribution of around 1.1 nm. As the ordering frameworks promoted the availability of the pore channels, the binding energy of H2 and CH4 to ICOF-2 increased remarkably in the presence of Li counterions, achieving 3.11 wt% of H2 (77 K, 1 bar) and 4.62 wt% of CH4 (273 K, 1 bar), respectively.
3.1.1.2 Carbon dioxide adsorption. CO2 is one of the main greenhouse gases that induces global warming. Capturing CO2 using porous materials via physical adsorption is economical and technically feasible among various techniques. Different strategies to ionize the POPs for high CO2 storage have been developed, which are mainly related to the post-metallation and bottom-up synthesis of charged frameworks in order to form acid–base or quadrupolar interactions with CO2.
The elaborate design of i-POPs with electric dipole structures has been demonstrated to boost CO2 uptake. Thomas et al. comprehensively investigated the CO2 adsorption performance of a cationic POP.22 Positively charged tetrakis(4-bromophenyl)phosphonium bromide was coupled with 1,3,5-triethynylbenzene or tetrakis(4-bromophenyl)phosphonium bromide via the Suzuki reaction, producing CPN-1-Br (Fig. 21a) and CPN-2-Br (Fig. 21b), respectively. The cationic CPN-1-Br had a much higher BET surface area (1455 m2 g−1) than CPN-2-Br (540 m2 g−1). Correspondingly, the CO2 uptake of CPN-1-Br (11.0 wt%) was nearly double that of CPN-2-Br (6.8 wt%) at 273 K and 1 bar. Using an anion-exchange route, CPN-1-Cl was prepared without any decrease in the surface area (1504 m2 g−1). The CO2 uptake of CPN-1-Cl was further elevated to 12.5 wt%, which increased by around 40% comparison with the neutral PAF-1 that has an extremely high surface area (5600 m2 g−1). Besides, other cationic building units have been designed to constitute a series of i-POPs used as CO2 adsorbents. Han et al. reported a variety of triazatriangulenium (TATA)-based i-POPs prepared employing the thiophene-/carbazole-functionalized TATA derivatives via FeCl3-catalyzed oxidative polymerization.104 The abundant nitrogen sites and positive charges in the porous structure endowed TAPOP-1 with a CO2 uptake capacity of up to 15.4 wt% at 273 K and 1.0 bar (Fig. 21c).
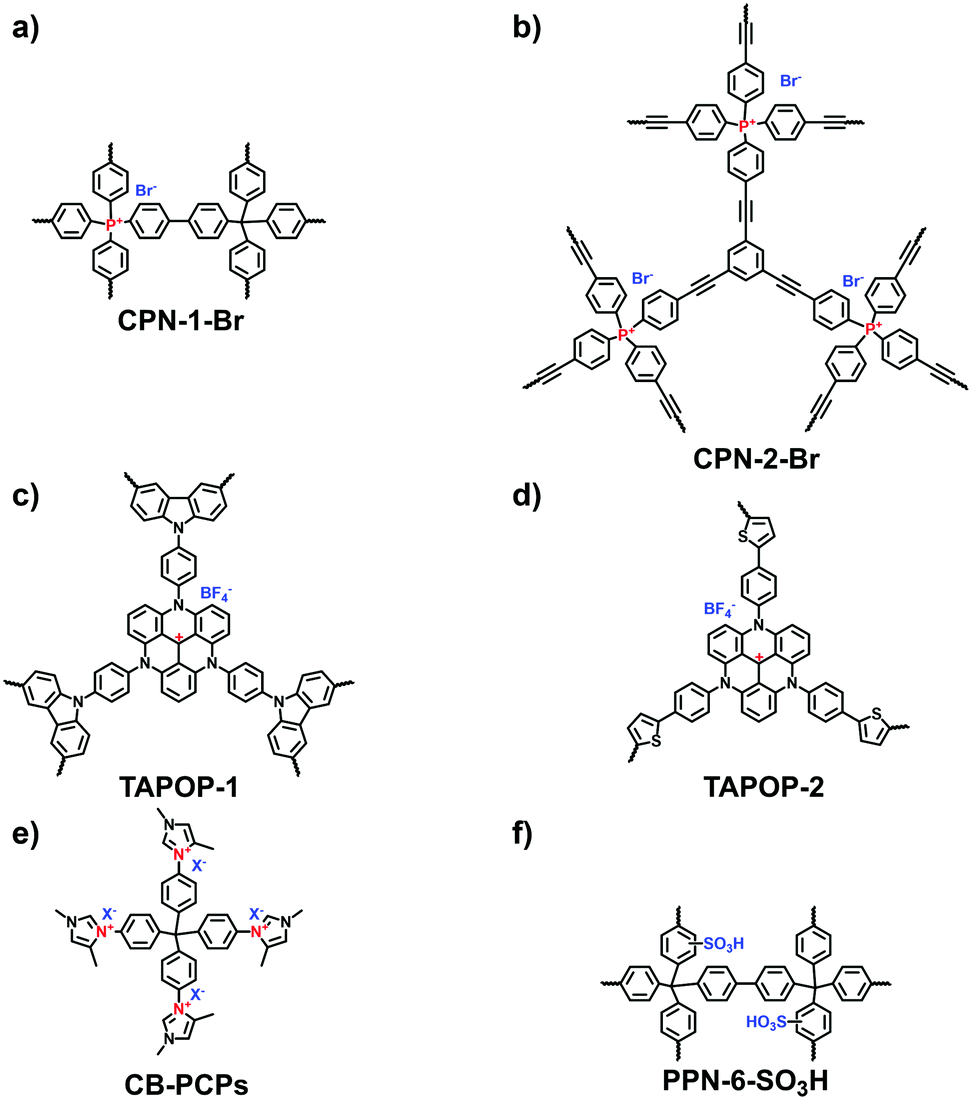 | ||
| Fig. 21 Structures of (a) CPN-1-Br, (b) CPN-2-Br, (c) TAPOP-1, (d) TAPOP-2, (e) CB-PCP and (f) PPn-6-SO3H for carbon dioxide adsorption. | ||
As ILs and CO2 have strong van der Waals and electrostatic interactions, constructing porous poly-IL frameworks is an effective strategy for CO2 adsorption and transformation.94 The concept was verified by Zhang et al. through developing a series of imidazolinium-based i-HCPs via the post-synthetic modification of the N-methylimidazole moiety. The optimal CO2 uptake capacity was as high as 14.5 wt% at 273 K and 1.0 bar.52 Inspired by these pioneering studies, a variety of imidazolinium-containing i-POPs have been successively reported using different approaches. Wang et al. constructed imidazolinium-based i-HCPs (HIP-X, X = Cl, Br, I) via the Friedel–Crafts alkylation reaction of 2-phenylimidazoline and benzyl halides.46 Of those species, HIP-Cl-1 exhibited the maximum CO2 uptake capacity of 16.7 wt% at 273 K and 1.0 bar. Analogously, Bordiga et al. synthesized the imidazolium-derived i-POPs (CB-PCPs) via the click reaction (Fig. 21e).105 As the different anions, including PF6−, Tf2N−, TfO−, BF4− and AcO−, were coordinated with the cationic skeletons of CB-PCPs via the ion-exchange process, CO2 adsorption was affected by interaction with the anions; the quantity of adsorbed CO2 with respect to the imidazolium moiety followed the trend of Tf2N− > PF6− ≈ TfO− > BF4− > AcO−. Additionally, as N-heterocyclic carbenes (NHCs) were incorporated in the CB-PCPs, the formation of the adduct between the NHC and CO2 could improve the adsorption ability of i-POPs as well.
Besides the ionization of backbones over the POPs, the organic cations dangled on the side groups of the skeletons have been investigated for CO2 adsorption. Yu et al. synthesized the IL-POPs with p-dichloroxylene (p-DCX) and N-methylimidazole (N-MI) for grafting the imidazole cations.47 By tuning the molar ratios of N-MI to p-DCX, the isosteric heat of adsorption was increased from 7.84 to 17.35 kJ mol−1. The optimal CO2 uptake of the IL-POPs eventually reached 73.6 cm3 g−1 at 273 K and 1 bar. Jiang et al. synthesized a PyTTA-BFBIm i-COF via the aldimine reaction of 5,6-bis(4-formylbenzyl)-1,3-dimethyl-benzimidazolium bromide (BFBIm) with PyTTA.67 As the benzimidazolium cations were uniformly distributed on the frameworks, the charged pores allowed the strong affinity for CO2 molecules. Thus, the CO2 uptake capacities of PyTTA-BFBIm-i-COF were as high as 9.3 wt% at 298 K and 17.7 wt% at 273 K and 1 bar. With a similar design, Dong et al. reported an acyl-hydrazone-linked COF (COF-IL) using allyl-imidazolium building blocks.68 The CO2 uptake capacity of COF-IL was 106.04 cm3 g−1 at 273 K and 59.37 cm3 g−1 at 298 K and 1 bar.
In addition to molecular engineering, the design of topological structures is another feasible strategy to adjust the CO2 adsorption properties. 3D COFs are featured with higher surface areas and smaller pore sizes than those of 2D COFs, holding promise for CO2 capture. Qiu et al. investigated the CO2 uptake capacity of 3D-ionic-COF-1, which was as high as 16.1 wt% at 273 K and 9.3 wt% at 298 K.61
The high selective adsorption of CO2/N2 under low pressure and near room temperature is essential for porous materials to meet industrially applicable challenges. To improve the CO2/N2 adsorption selectivity, endowing i-POPs with polar functionalities is responsible for an increase in the isosteric heat of CO2 adsorption. Zhou et al. adopted chlorosulfonic acid to modify PPN-6 (PPN-6-SO3H; Fig. 21f), followed by lithiation to produce PPN-6-SO3Li.53 PPN-6-SO3H and PPN-6-SO3Li both showed an exceptionally high adsorption selectivity for CO2 over N2 in flue-gas streams (15:85 v/v) at 295 K and 1 bar. The selectivity values calculated using ideal adsorption solution theory (IAST) were 155 and 414 for PPN-6-SO3H and PPN-6-SO3Li, respectively. Correspondingly, the isosteric heat of CO2 adsorption reached 35.7 kJ mol−1 for PPN-6-SO3Li, which was slightly higher than that of PPN-6-SO3H (30.4 kJ mol−1). This pioneering work confirms that the metalation of POPs is an effective method for improving the selective CO2 adsorption, which is also confirmed by alkali metal-conjugated PAF materials, PAF-18-OLi57 and PAF-26-COOMg.29 Following this strategy, crystalline COFs with ordering and uniform pores are more beneficial for implementing highly selective adsorption over CO2/N2. Wang et al. employed safraninet-/guanidine hydrochloride-/dimidium bromide (DB)-derived ILs as ionic building blocks to react with neutral building blocks for the construction of multivariate COFs.106 Through modulation via the multicomponent aldimine reaction, DB10%-Pa-Tp generated the optimal crystallinity and porosity and, in turn, the CO2 uptake arrived at a maximum of 21.3 wt% at 273 K and 1 bar. The adsorption selectivity for CO2 over N2 was calculated via IAST to be 1 × 1014. Based on the superior theoretical result, the column breakthrough experiment of DB10%-Pa-Tp with a CO2–N2 mixture (15:85 v/v) was conducted at 308 K. Since N2 penetrated rapidly while CO2 was retained, the distinctive CO2/N2 separation was accomplished under practical dynamic conditions.
3.1.1.3 Adsorption of other gases. Apart from the significant potential of i-POPs for the capture of H2 and CO2, the versatile design of building blocks, topological structures and functionality enables the adsorption to be extended to other gases or vapours such as acid/base gases and hydrocarbon gases. This is highly desirable in industrial production and is of critical importance for environmental protection.
The capture and storage of iodine is conducive to facilitating the development of nuclear energy. To explore iodine-capturing materials with a high affinity and loading capacity, Zhu et al. designed anionic PAFs based on tetrahedral lithium tetrakis(4-iodophenyl)borate, which simultaneously carries three adsorption sites including ions, phenyl rings and alkynyl groups for I2 capture.38 Thus, PAF-23, PAF-24, and PAF-25 all exhibited a high affinity and capacity for iodine. Among them, PAF-24 (Fig. 22a) showed the best adsorption ability at 75 °C (2760 mg g−1), which could be ranked among the top reported porous materials.
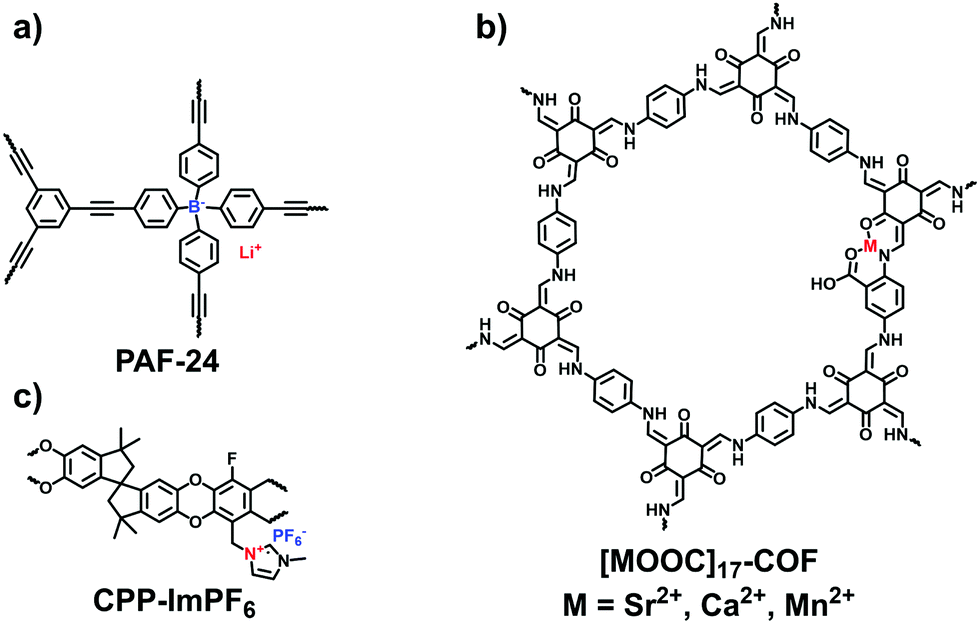 | ||
| Fig. 22 Structures of (a) PAF-24, (b) [MOOC]17-COF and (c) CPP-ImPF6 for the adsorption of other gases. | ||
Dai et al. reported a cationic porous polymer (CPP-ImPF6, Fig. 22c) for SO2 adsorption, which was synthesized by using 5,5,6,6-tetrahydroxy-3,3,3,3-tetramethyl-1,1′-spirobisindane (TTSBI) and imidazolium salts via the ball-grinding method.41 The obtained CPP-ImPF6 was subjected to an ion exchange with proline anions, leading to a rising SO2 adsorption capacity (24.6 wt%) at 298 K and 1 bar. Such superior performance is attributed to the strong basicity of proline anions giving the chemisorption interaction with SO2.
Combining the physisorption and chemisorption effects, Zhu et al. reported a series of multivariate COFs ([HOOC]X-COFs) with various binding groups for NH3 enrichment.107 The –HOOC sites in the COFs were flexibly regulated by varying the molar ratios of 2,5-diaminobenzoic acid (DAA) and p-phenylenediamine (PA-1) in the aldimine reaction with triformylphloroglucinol (TFP). Metal ions including Ca2+, Mn2+, and Sr2+ were coordinated with [HOOC]17-COF, respectively, for achieving the maximum uptake of NH3. Of the metalated COFs, the largest adsorption capacity was given by [SrOOC]17-COF (Fig. 22b), reaching 19.8 mmol g−1 at 283 K and 1 bar and possessing the high isosteric heat of 91.2 kJ mol−1. In addition to the metal sites from [MOOC]17-COFs, polar groups including –N–H, –CO and –COOH acted as the active sites for NH3 capture, although their binding capability was far lower than the coordinated metal ions.
Depending on the surface engineering strategy, Luo et al. explored a sulfonic acid-containing COF-ECUT-1 that consisted of 2,5-diaminobenzenesulfonic acid as a linker and 2,4,6-triformylphloroglucinol as a knot for the selective adsorption over a C2H2/C2H4 binary mixture (1/99, v/v).108 Calculated using IAST, the adsorption selectivity of COF-ECUT-1 was 6.33 at 298 K and 1 bar. After ion exchange with a low content of Na+ ions (4.0 wt%), the selectivity could be increased to 9.41 for the doped COF-ECUT-1, which was desirable for C2H2/C2H4 separation in practice.
Separating gases and liquids via membranes is an alternative energy-efficient technique. Preparing ionic porous membranes is of great significance since the pore size and thickness of the membrane can be easily controlled via the building blocks and synthetic methods. Interfacial polymerization and the spin-coating technique are the prevailing strategies for preparing i-POP-based membranes. Zhao et al.109 proposed a layer-by-layer (LbL) assembly strategy to obtain ultrathin 2D membranes using cationic TpEBr covalent organic nanosheets (CONs) and anionic TpPa-SO3Na CONs (Fig. 23). The strong electrostatic interactions between the two i-CONs formed the staggered packing of the nanosheets, thereby making the pore sizes smaller and approaching the kinetic diameters of the gases. When a 41-nm-thick TpEBr@TpPa-SO3Na membrane was applied as a polymeric sieve for H2/CO2 separation, the H2 permeance reached 2566 gas permeation units (GPUs) and the H2/CO2 separation factor was 22.6 at 423 K. Such a separation efficiency was much higher than those of the single-phase TpEBr or TpPa-SO3Na membranes, surpassing the Robeson upper bound as well. This strategy shows promise for developing i-COFs-based sieve membranes.
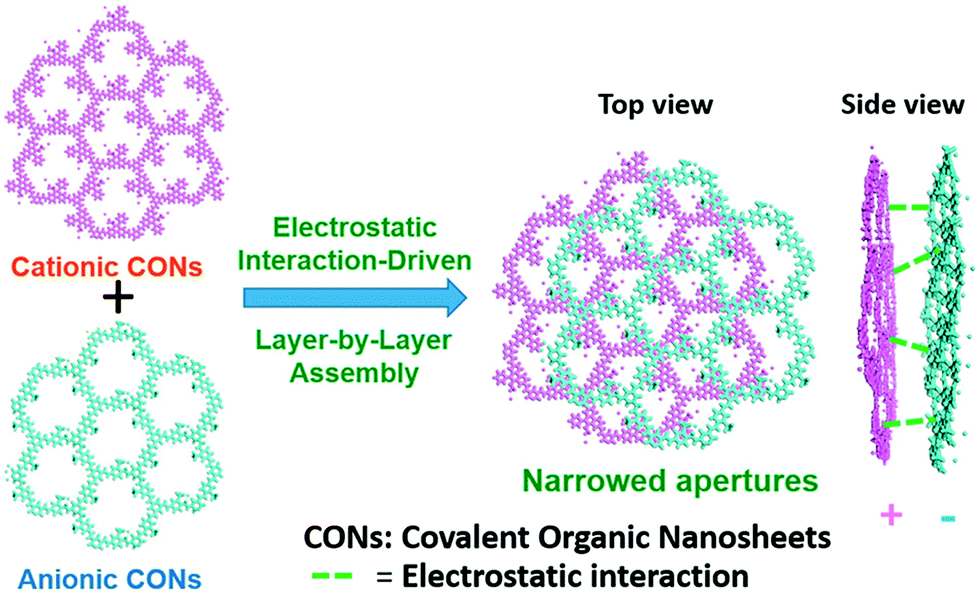 | ||
| Fig. 23 Schematic representation of 2D membranes fabricated via layer-by-layer assembly with cationic TpEBr CONs and anionic TpPa-SO3Na CONs. Reprinted with permission from ref. 109. Copyright (2020) American Chemical Society. | ||
Li et al. reported a cationic bipyridinium-based COF (PC-COF) using 1,3,5-tris(4-aminophenyl)benzene (TAPB) and 1,1′-bis(4-formylphenyl)-4,4′-bipyridinium dichloride (BFBP2+·2Cl−) as building blocks for the removal of anionic dyes.65 Combined with the accessible 1D pore channels, the adsorbing sensitivity of the anionic dyes was quantified at a very low concentration of 3.2 × 10−5 M, validating the possibility of eliminating traces of dye pollutants, such as methyl orange (MO), acid green 25 (AG-25), Direct Fast Brown M (DFBM), indigo carmine (IC) and acid red 27 (AR-27), in waste water (to >97%). These prominent performances originated not only from the electrostatic interactions between the cationic skeletons of PC-COF and the anionic dyes but also from the ion exchange of dye anions with the Cl− counterions in PC-COF, which could form the two more stable soft base–soft acid (BIPY) and hard base–hard acid (Na+) ion pairs.
Along this line, anionic COFs were designed for the selective adsorption of cationic dyes. Qiu et al. demonstrated that sulfonate-based TpPa-SO3Na was effective for adsorbing cationic molecules including methylene blue (MB; ∼96%) and various antibiotics (norfloxacin, 99%) within 1 min.111 Furthermore, the selective capture of dyes and antibiotics with different charges and sizes was accomplished. The increase in selectivity and capacity of TpPa-SO3Na was ascribed to a combination of electrostatic interactions and H-bonding between the sulfonate groups grafted on the pores and the amine groups in the organic pollutants.
Adsorption based on ion exchange between ionic pollutants and the counterions of i-POPs has been extensively employed to leverage the strengths of i-POPs at their best. Wang et al. designed the imidazolium-based i-POPs (POP-Ims; Fig. 24a) through the Yamamoto reaction of 1,3-bis(4-bromophenyl)imidazolium bromide and tetrakis(4-bromophenyl)ethylene.112 Given the high density of imidazolium units, POP-Im1 could be highly dispersed in aqueous solution to facilitate the mass distribution in the porous structure. The adsorbed capacity of POP-Im1 for Cr2O72− was 171.99 mg g−1 and the removal efficiency was 87.9% in 5 min. Nevertheless, the neutral POP with a similar surface area could not capture Cr2O72−; this confirmed that the adsorption mechanism was derived from the ion exchange between Cr2O72− and Br− of the POP-Ims.
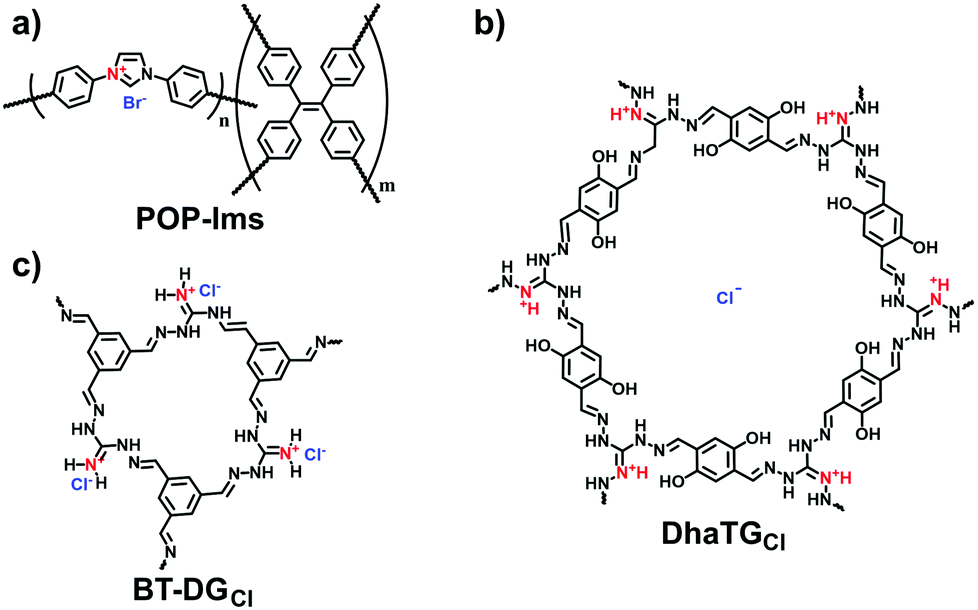 | ||
| Fig. 24 Structures of (a) POP-Ims, (b) DhaTGCl and (c) BT-DGCl. | ||
As guanidinium groups are prone to binding oxoanions,113 guanidinium-containing COFs (BT-DGCl) were developed for removing CrO42− in aqueous solution by Moyer et al. (Fig. 24c).64 The adsorption capacity of BT-DGCl for CrO42− reached 200 mg g−1 at pH = 2. More importantly, BT-DGCl was capable of diminishing the CrO42−concentration from 1 ppm to 10 ppb within minutes, which was much lower than that specified by the U.S. Environmental Protection Agency (100 ppb). Also, such guanidinium-based COFs have been validated to enable the removal of the ionic organic pollutant 2,4-dichlorophenol from water.114
Yan et al. adopted a post-polymerization method to graft the cationic surfactant diallyldimethylammonium chloride onto a vinyl-containing COF (DhaTab-V).97 Using the ion exchange route, the cationic surfactant-modified COF (DhaTab-S) was confirmed to be a promising adsorbent for NO3−. The adsorption capacity was up to 108.8 mg g−1, which is approximately 15 times that of the un-modified COF. Trabolsi et al. presented a porphyrin-based COF (PV-COF; Fig. 25) containing bipyridinium as a linker for the removal of toxic bromate in water.115 The designed PV-COF was unique in that the bipyridinium and porphyrin moieties could confer the ion-exchange sites and H-bonding interactions, respectively. As a result, PV-COF displayed an extraordinary BrO3− removal efficiency of 203.8 mg g−1 along with a high uptake rate of 191.45 g mg−1 min−1.
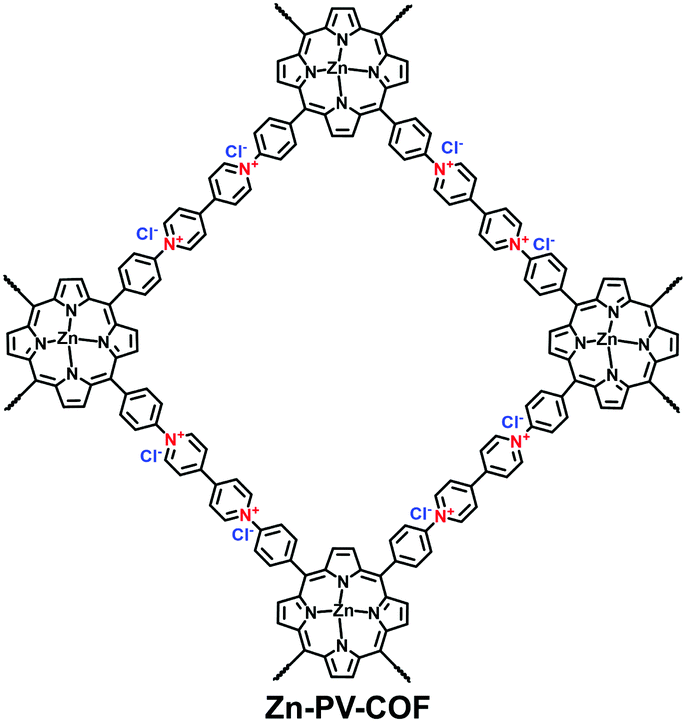 | ||
| Fig. 25 Structure of Zn-PV-COF. | ||
With an ultrahigh energy density, nuclear energy is one of the important energy resources all around the world. However, with the development of the nuclear industry, radioactive 99Tc and the fission products of 235U and 239Pu present a severe environmental issue. It is therefore urgent to explore specific absorbents for radioactive 99TcO4− in nuclear waste. Qiu et al. investigated the potential of 3D-ionic-COFs for the removal of 99TcO4− in nuclear waste.61 MnO4− was selected as a model ion of 99TcO4− since these are both group VII oxoanions. A certain quantity of 3D-ionic-COF (20 mg) could remove almost 100% of MnO4− in an aqueous solution of KMnO4 (0.5 mg mL−1, 20.0 mL) at room temperature within 30 min. Furthermore, 3D-ionic-COFs could be recovered in NaBr aqueous solution via the reversible sorption process. To precisely estimate the ability of i-COFs to capture radioactive 99TcO4−, the following studies adopted ReO4− as a non-radioactive surrogate of 99TcO4− due to their identical charge density and with similar anion-exchange properties. Wang et al. reported a viologen-based i-COF (SCU-COF-1; Fig. 26) to treat nuclear waste streams.116 The selective adsorption of ReO4− over NO3− and SO42− occurred even under strong acidity and radiation fields. The uptake capacity reached 702.4 mg g−1 within 1 min, which was more competitive than commercial resins. Yan et al. reported a guanidinium-based cationic CON (DhaTGCl i-CON; Fig. 24b) for the efficient uptake of ReO4−.63 Since a number of cationic sites were exposed on the DhaTGCl i-CON, the ultrafast exchange kinetics facilitated the efficient adsorption of ReO4− (437 mg g−1) and remained stable in the pH range of 3–12. Even in the presence of interfering ions such as NO3−, CO32−, PO43− and SO42−, DhaTGCl i-CON still could selectively capture ReO4−.
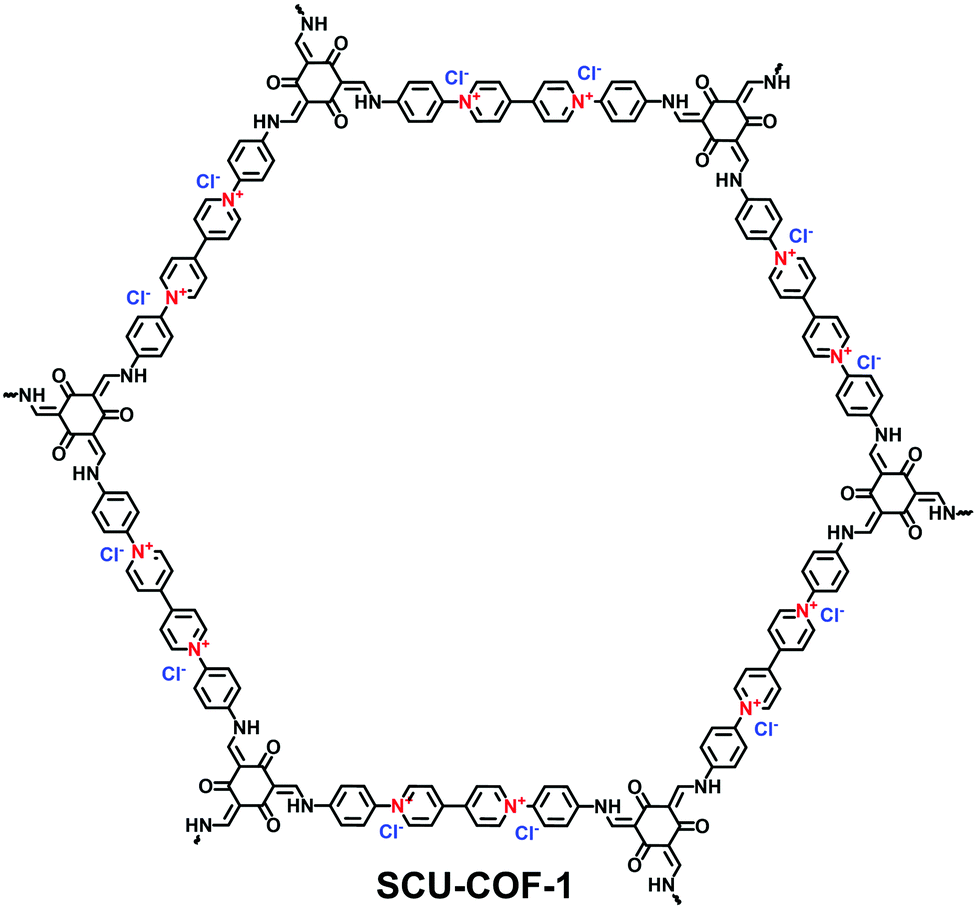 | ||
| Fig. 26 Structure of SCU-COF-1. | ||
Extracting uranium selectively from sea water is challenging as the uranium concentration is extremely low and there is a huge amount of other metal ions such as Na+, K+, and Sr+. Luo et al. synthesized a sulfonate-modified anionic COF (COF-SO3H), followed by ammoniation, resulting in the [NH4]+[COF-SO3−] adsorbent for uranium.117 Depending on the ion exchange between NH4+ and UO22+ as well as the strong coordination interaction derived from the oxygen atoms of –SO3−, the UO22+ uptake capacity of [NH4]+[COF-SO3−] was as high as 851 mg g−1, which could be ranked among the top reported adsorbents for UO22+. Furthermore, the sustainable chemical stability of [NH4]+[COF-SO3−] allowed its utilization under harsh conditions (pH = 1, and 8). Also, [NH4]+[COF-SO3−] exhibited a high adsorption selectivity for UO22+ over various metal ions, e.g., SU/Cs = 821, SU/Na = 277 and SU/Sr = 124. In tests using real seawater, a considerable capacity of 17.8 mg g−1 was achieved to prove the feasibility of uranium extraction.
Membrane-type absorbents are ideal in molecular sieving for removing organic pollutants. Inspired by the striking progress of POPs in synthesis, recent studies have made a significant contribution to the related applications with i-POP-based membranes. Ma et al. fabricated cationic COF (EB-COF:Br) nanosheets with EB as a linker via an interfacial polymerization and crystallization approach.78 A continuous and dense membrane was obtained using a layer-by-layer restacking process through the handy vacuum filtration of EB-COF:Br nanosheets on a substrate of nylon 66 (Fig. 27a). The thickness of the EB-COF:Br membrane could be easily tuned using different amounts of the nanosheet dispersion. The membrane exhibited a 90% rejection of PEG at 882 Da via the size-sieving effect. Due to the abundant positive charge sites along the pore channels, the EB-COF:Br membrane effectively intercepted anionic dyes while maintaining a high solvent permeability, resulting in a rejection efficiency as high as 98%.
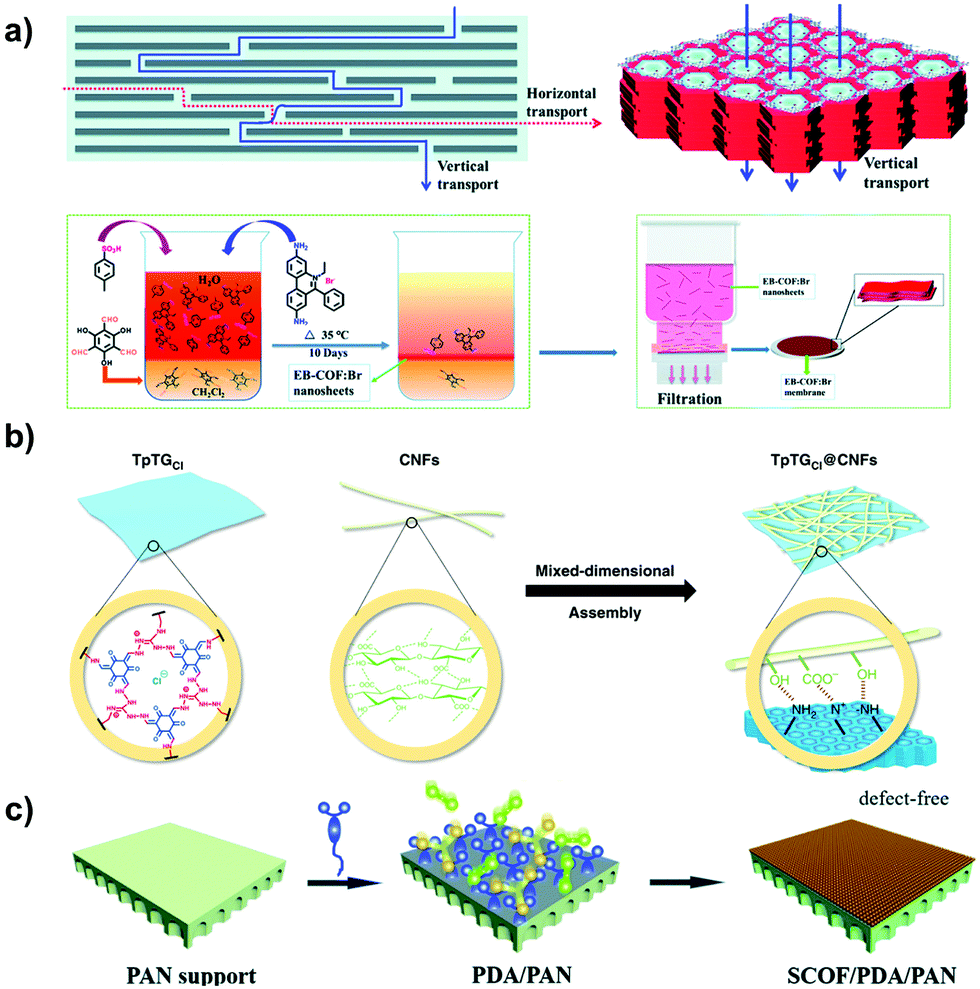 | ||
| Fig. 27 (a) Schematic representation of fabrication of the EB-COF:Br membrane using the nanosheet layer-by-layer restacking process. Reprinted with permission from ref. 78. Copyright (2018) Royal Society of Chemistry. (b) Illustration of the synthesis of guanidinium-based COF composite membranes with different pore sizes by virtue of the sheltering effect of 1D cellulose nanofibers (CNFs). Reprinted with permission from ref. 118. Copyright (2019) Springer Nature. (c) Schematic of PDA-modulated fabrication of the SCOF/PDA/PAN membrane using the bottom-up method. Reprinted with permission from ref. 119. Copyright (2019) Royal Society of Chemistry. | ||
To make the pore sizes of the COFs applicable to separating targeted molecules, Jiang et al. fabricated a series of guanidinium-based COF composite membranes by virtue of the sheltering effect of cellulose nanofibers (Fig. 27b).118 As the pore sizes were limited in the range of 0.45–1.0 nm, the molecular sieving ability of the membranes was largely promoted. They afforded a separation factor of 3876 for n-butanol dehydration at a flux of 8.53 kg m−2 h−1 and the high-permeance removal of Na2SO4 with a rejection of 96.8%. To reduce the solvent-transport resistance, it is desirable for the i-POP membranes to produce ordered pore channels and few defects in the structures. Jiang et al. prepared a sulfonate-COF (SCOF) composite membrane supported on a polydopamine-modified porous polyacrylonitrile (PAN) substrate (Fig. 27c).119 The role of polydopamine (PDA) was exerted on the controlled growth of SCOF on PAN, resulting in a high ordering of the SCOF composite. The resultant SCOF/PDA/PAN membrane showed an outstanding water permeance of up to 1346 L m−2 h−1 MPa−1 with a remarkable dye rejection, e.g., Eriochrome black T and Congo red, of >99.0%.
3.2 Ion conduction
Thanks to the collective merits of i-POPs, a variety of artificial ion-conduction systems have been constituted on platforms of such materials, which hold great promise for sensors, supercapacitors, batteries and fuel cells. Compared with natural biological ion channels, shuttling ions through the artificial channels is more challenging as cations and anions are strongly paired, especially in the solid state. To surmount these barriers, solvating the conduction channels has been addressed to benefit ion-pair dissociation and charge transport. 2D i-COFs are featured with highly periodic atomic arrangements and 1D available pore channels, both of which are conducive to ion conduction. Also, these are energetically desirable because the uniform distribution of ions in the periodic structures of i-COFs allows for a decrease in the energy barriers of ion transport. Meanwhile, the coaxially aligned pore channels provide a shortcut pathway for continuous ion hopping. Therefore, the progress of i-COFs achieved for Li-ion conduction, proton conduction, and anion conduction have attracted mounting interest recently.| i-COF | E a (eV) | T Li + | Conductivity (S cm−1) | Conditions | Ref. |
|---|---|---|---|---|---|
| ICOF-2 | 0.24 | 0.80 | 3.05 × 10−5 | r.t., 55 wt% PC/15 wt% PVDF | 70 |
| CD-COF-Li | 0.26 | 2.7 × 10−3 | 30 °C, 20 wt% LiPF6/EC/DMC | 71 | |
| TpPa-SO3-Li | 0.18 | 0.90 | 2.7 × 10−5 | r.t. | 72 |
| CH3-Li-ImCOF | 5 × 10−7 | r.t. | 120 | ||
| CH3-Li-ImCOF | 0.27 | 0.93 | 8.0 × 10−5 | r.t., 20 wt% PC | 120 |
| H-Li-ImCOF | 0.12 | 0.88 | 5.3 × 10−3 | r.t., 20 wt% PC | 120 |
| CF3-Li-ImCOF | 0.10 | 0.81 | 7.2 × 10−3 | r.t., 20 wt% PC | 120 |
| Li-CON-TFSI | 0.34 | 0.61 | 5.74 × 10−5 | 30 °C, LiTFSI | 121 |
| PEG-Li+@CD-COF-ClO4 | 0.17 | 0.20 | 2.6 × 10−5 | 30 °C | 123 |
| PEG-Li+@EB-COF-ClO4 | 0.21 | 0.60 | 1.93 × 10−5 | 30 °C | 123 |
| LiCON-1 | 0.25 | 0.86 | 2.13 × 10−7 | 20 °C | 124 |
| LiCON-2 | 0.22 | 0.83 | 4.36 × 10−6 | 20 °C | 124 |
| LiCON-3 | 0.13 | 0.92 | 3.21 × 10−5 | 20 °C | 124 |
| Ge-COF-1 | 0.26 | 1.1 × 10−6 | 30 °C | 85 | |
| Ge-COF-1 | 0.24 | 1.6 × 10−5 | 30 °C, 20 wt% EC/DEC | 85 | |
| Ge-COF-1 | 0.29 | 0.67 | 2.5 × 10−4 | 30 °C, 20 wt% LiPF6 | 85 |
Zhang et al. reported an anionic spiroborate-linked ICOF-2 that was applied for Li+ conduction.70 A hybrid membrane was prepared by mixing ICOF-2 solids with polyvinylidene difluoride (PVDF). The Li+ conductivity of the resulting membrane was 3.05 × 10−5 S cm−1 at room temperature using propylene carbonate (PC) as an electrolyte, and the activation energy (Ea) of Li+ diffusion was calculated to be 0.24 eV.
When the electronic properties of the skeletons were modulated using different electron donating/withdrawing substituents, the interactions between the COF anions and Li+ ions were varied accordingly to alter the ion conductivity. Zhang et al. synthesized a series of anionic COFs based on imidazole units, which were further functionalized with different substituents, including –H, –CH3 and –CF3, on the pyrrolyl N sites (Fig. 28). When introducing Li+ ions to the imidazole moieties, the –CF3-modified i-COF exhibited the highest Li+ conductivity of up to 7.2 × 10−3 S cm−1 at room temperature using PC as an electrolyte.120 The electron-withdrawing –CF3 groups delocalized the negative charge of the imidazolate anions, thus weakening the electrostatic interaction between the Li+ ions and the anionic skeletons for increasing the Li+ mobility. As a result, the Ea value of CF3-Li-ImCOF was as low as 0.10 eV. Besides the 2D i-COFs, 3D anionic COF-containing cyclodextrin as a building block was confirmed to be conducive to ion transport, giving the conductivity of 2.7 × 10−3 S cm−1 at 30 °C using LiPF6 as an ionic carrier and ethylene carbonate (EC) and dimethyl carbonate (DMC) as the electrolytes.71
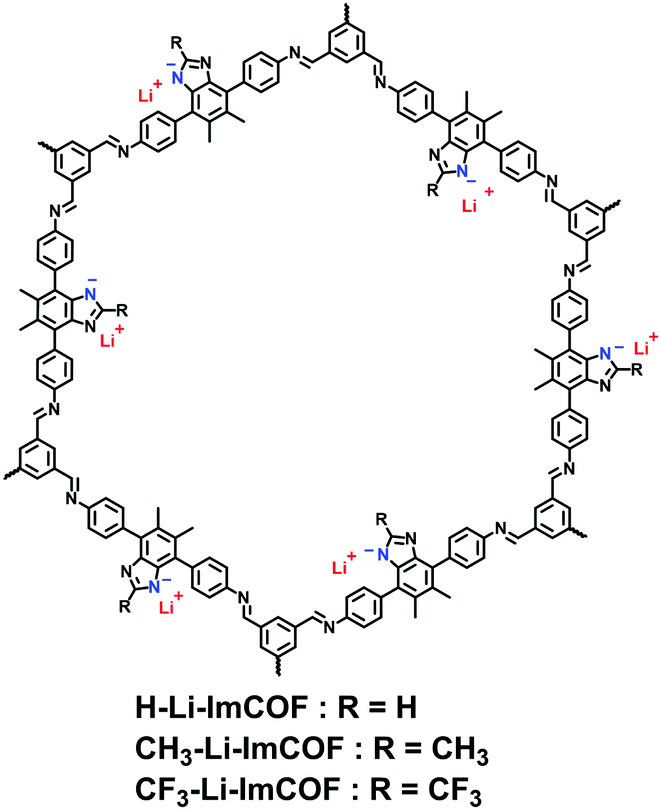 | ||
| Fig. 28 Structure of the Li-ImCOFs. | ||
As commonly used organic electrolytes such as PC, EC and DMC can contribute to the mobility of the lithium counterions, studies have been devoted to the exploration of solid-state i-COFs for Li+ conduction. As reported by Chen et al., without employing any solvent or quasi-solid-state plasticizers, lithium bis(trifluoromethane) sulfonimide (LiTFSI) was incorporated onto the positively charged skeletons of COFs via an ion-exchange process and, in turn, the guanidinium-derived cationic linkers and TFSI− were mutually adsorbed to allow the unbound Li+ ions to hop along the pore channels (Fig. 29a). The Li+ conductivity reached as high as 2.09 × 10−4 S cm−1 at 70 °C.121
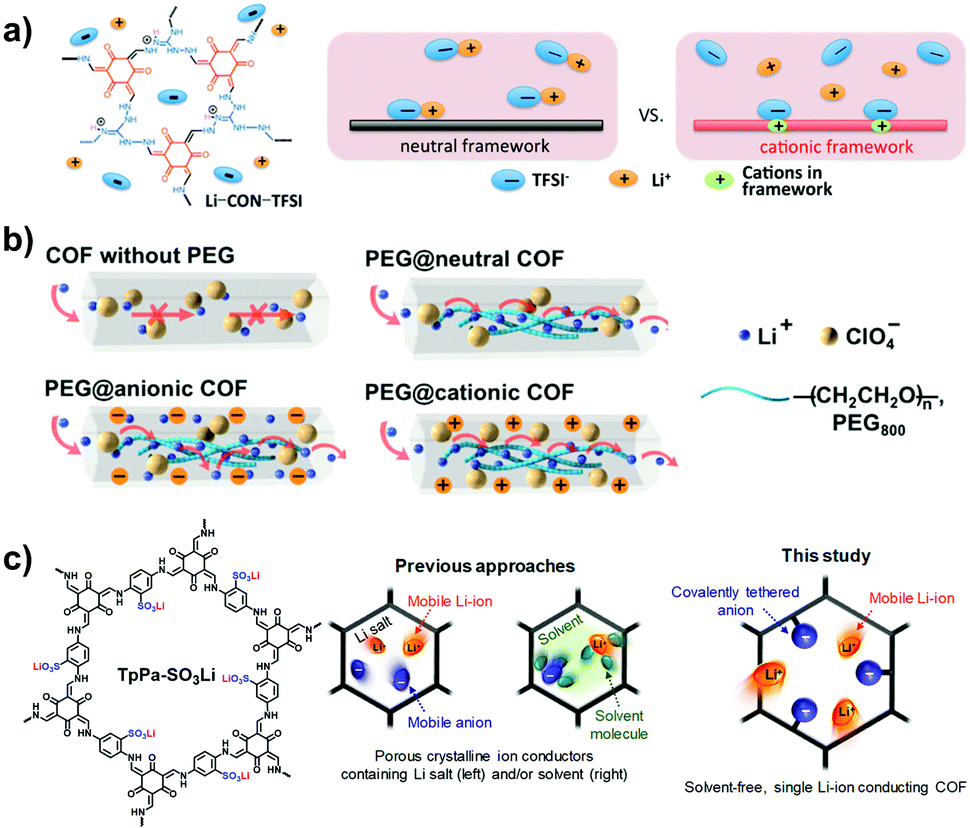 | ||
| Fig. 29 (a) Illustration of ion association in neutral and cationic Li-CON-TFSI. Reprinted with permission from ref. 121. Copyright (2018) American Chemical Society. (b) Schematic of Li+-transport behaviour in PEG@COFs. Reprinted with permission from ref. 123. Copyright (2019) American Chemical Society. (c) Schematic of Li+-transport mechanism of TpPa-SO3Li. Reprinted with permission from ref. 72. Copyright (2019) American Chemical Society. | ||
To simulate the solvated conduction, short-chain polyethers such as polyethylene glycol (PEG) were non-covalently assembled in pores or covalently grafted to the skeletons of i-COFs, providing a polyelectrolyte interface for quick Li+ conduction.122 The encapsulation of short PEG chains (Mw = 800) into the COFs with anionic, neutral, or cationic skeletons could boost the Li+ conduction significantly (Fig. 29b).123 A further investigation demonstrated that the highest conductivity (1.78 × 10−3 S cm−1 at 120 °C) was obtained on the PEG-Li+@EB-COF-ClO4, which contained cationic skeletons derived from the attached EB groups. The superior performance originated from the fact that the cationic skeletons trapped anions to promote the dissociation of ion pairs, thus causing fast Li+ transport in the 1D pore channels containing liquid PEG at elevated temperatures.
Single lithium-ion conducting polyelectrolytes (TLi+ = 1) have become fascinating materials since they can effectively alleviate the concentration gradients during the anion motion and suppress lithium dendrite proliferation, thereby improving the stability and safety of lithium-ion batteries. The lithium-ion transference number (TLi+) is an important factor for evaluating the performance of electrolytes, although it is challenging as the opening pores of frameworks are accessible to all the ions and solvents. In this context, Lee et al. synthesized a sulfonated β-ketoenamine-linked COF using –SO3−-functionalized aromatic rings as building blocks.72 Theoretical calculations demonstrated that the Li+ ions preferred to hop along the O atoms of keto groups aligned in the axial direction. Thanks to such a molecular design, TpPa-SO3-Li, with well-ordered ionic channels and a high density of Li+ ions, resulted in excellent Li+-conduction characteristics, i.e., σ = 2.7 × 10−5 S cm−1 at room temperature and Ea = 0.18 eV. Without the freely mobile anions and solvents, the TLi+ value of TpPa-SO3-Li was measured to be 0.9, showing potential as a solid single lithium-ion conductor (Fig. 29c). Another example reported was SD_COF-2-3, which was fabricated via the thiol–ene click reaction of 3-mercaptopropanoic acid and sodium 2-mercaptoethane sulfonate, respectively, with a neutral hydrazine-COF. Lithiation of SD_COF-2-3 yielded LiCON-2-3. The anionic side chains were anchored in the skeletons via the post-modification route for Li+ hopping. The optimized Li conductivity was as high as 0.9 × 10−5 S cm−1 at −40 °C with a TLi+ value of 0.92, demonstrating single-Li-ion conduction under harsh conditions.124
Based on the strong lithiophilicity and ion conductivity of i-COFs, they have further been assembled into lithium-ion batteries to serve as solid electrolytes. Han et al. incorporated imidazolium groups into the structural defects of COFs using post-functionalization treatment.91 Then the anion exchange was applied to load numerous TFSI− anions within the cationic Im-COFs. The hydrogen bonding given by the amine N–H groups and the F atom of TFSI− facilitated the dissociation of Li+ ion pairs and, in turn, improved the Li+ conductivity in the solid state. Depending on the excellent properties of the COF-based solid electrolytes, an initial discharge capacity of the all-solid Li-ion battery was up to 143.7 mAh g−1 at 0.1C at 353 K.
Chen et al. built a sulfonated-COF layer on an inorganic solid electrolyte garnet Li6.4La3Zr1.4Ta0.6O12 (LLZTO; Fig. 30a).125 The in situ electrochemical lithiation transformed the i-COF layer into a solid electrolyte, leading to a significant decrease in the interfacial resistance. The resulting lithium-ion battery showed an excellent rate performance with a discharge capacity of 97 mAh g−1 at 2C. This was attributed to the quick Li+ mobility through the long-range ordering array within the sulfonated-COF. Chen et al. proposed a facile approach to prepare a composite membrane using the layered Kevlar polymer and the cationic guanidinium-based COF.126 The Li conductivity of the composite membrane was 1.62 × 10−4 S cm−1 at room temperature. When it was assembled for an all-solid LiFePO4/Li battery, the cycling performance was rather stable with a decay ratio of 0.052% per cycle over 300 cycles.
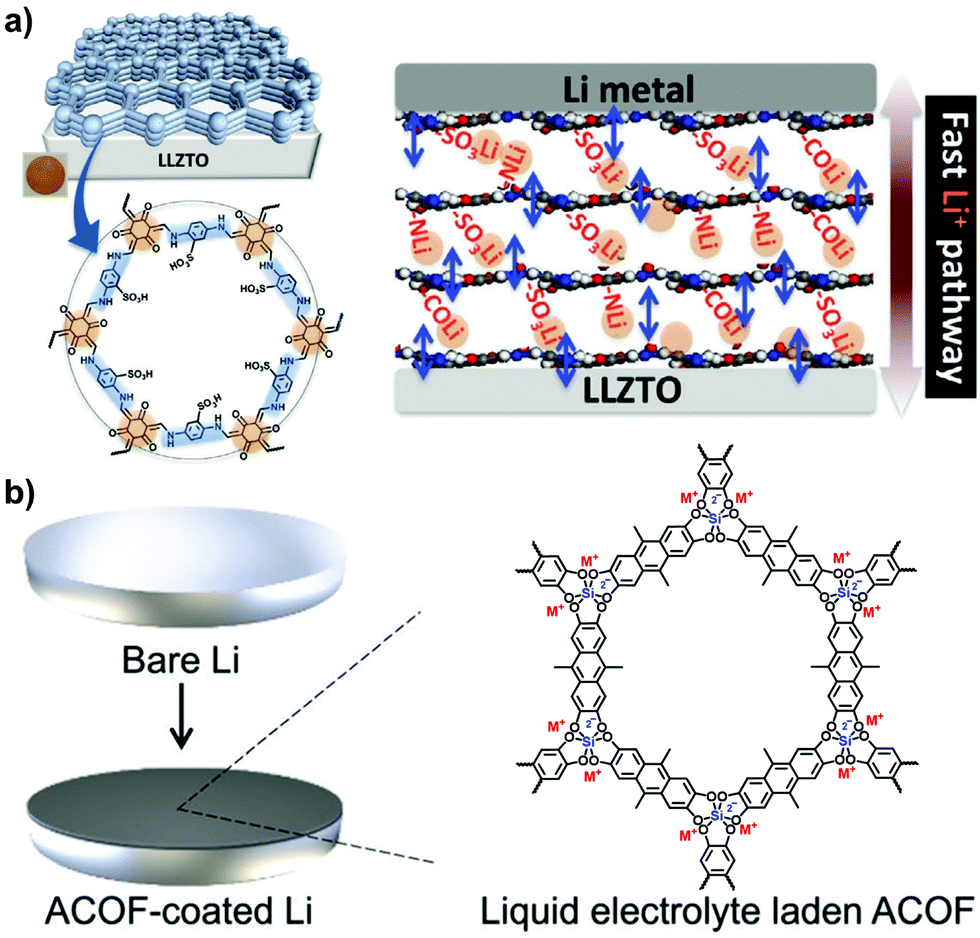 | ||
| Fig. 30 (a) Schematic diagram of Li+ transport in the S-COF membrane. Reprinted with permission from ref. 125. Copyright (2020) Wiley-VCH. (b) Illustration of ACOF for protecting the Li metal electrode. Reprinted with permission from ref. 127. Copyright (2021) Wiley-VCH. | ||
Individual i-COF-based membranes are promising for use in the solid electrolyte interphase (SEI) to suppress Li dendrites. Lu et al. reported an anionic COF (ACOF) that consists of hexacoordinate silicon centres (SiO62−) at the knots and anthracene at the struts for protecting the Li metal electrode (Fig. 30b).127 As the i-COF was lithophilic and offered straight and opening pathways for ion mobility, the resulting electric properties showed a high TLi+ value of 0.82 and an excellent conductivity of 3.7 × 10−3 S cm−1. Also, the dendrite growth in the LiCoO2-based battery was largely suppressed, affording an exceptional cycling stability at 4.5 V and a high capacity of 89 mAh g−1 at 10C.
| i-COF | Conductivity (S cm−1) | Temperature (°C) | RH | Description | Ref. |
|---|---|---|---|---|---|
| EB-COF:Br | 6.06 × 10−9 | r.t. | 11% | 59 | |
| EB-COF:Br | 2.82 × 10−6 | r.t | 97% | 59 | |
| EB-COF:PW12 | 3.32 × 10−3 | r.t | 97% | 59 | |
| NUS-9(R) | 1.5 × 10−4 | r.t | 33% | 79 | |
| NUS-9(R) | 1.24 × 10−2 | r.t | 97% | 79 | |
| NUS-10(R) | 2.8 × 10−4 | r.t | 33% | 79 | |
| NUS-10(R) | 3.96 × 10−2 | r.t | 97% | 79 | |
| NUS-9(R) | 2.06 × 10−3 | r.t | Water | 50 wt% PVDF | 79 |
| NUS-10(R) | 5.19 × 10−3 | r.t | Water | 50 wt% PVDF | 79 |
| COF-1-Na | 2.5 × 10−2 | 40 | 98% | 128 | |
| COF-1-Li | 2.7 × 10−2 | 40 | 98% | 128 | |
| IPC-COF | 3.8 × 10−1 | 80 | 35% | 129 | |
| TaPa-SO3H | 1.2 × 10−5 | r.t | Water | 130 | |
| TaPa-SO3H | 1.7 × 10−5 | 120 | Anhydrous | 130 | |
| TaPa-SO3H | 7.5 × 10−5 | 120 | Anhydrous | With phytic | 130 |
| TpPa-Py | 3.0 × 10−4 | 120 | Anhydrous | With phytic | 130 |
| TpPa-(SO3H-Py) | 5.0 × 10−4 | 120 | Anhydrous | With phytic | 130 |
The commercially available Nafion polymer shows an excellent proton conductivity at high humidity and moderate temperatures (<80 °C), where the sulfonic acid groups grafted on the backbone of Nafion plays a key role in proton conduction. As such, introducing sulfonic acid into the COF framework offers a promising route for designing COF-based proton conductors. Ghosh et al. reported a sulfonic acid-modified i-POP (PCF-1-SO3H) via post-synthetic modification.42 The proton conductivity at 30 °C and 95% RH was about 2.6 × 10−2 S cm−1, increasing by almost 130-fold that of the pristine PCF-1. As it was difficult to achieve a uniform distribution of ions within the framework via heterogeneous post-modification, Jiang et al. adopted a bottom-up approach to synthesize sulfonic acid-functionalized COF nanosheets (NUS-9).129 The NUS-9 nanosheets could be further assembled into a self-standing membrane (IPC-COF membrane) that has an intrinsic proton-conducting ability (Fig. 31). The proton conductivity was calculated to be 3.8 × 10−1 S cm−1 at 80 °C and 98% RH, which could be maintained at around 10−1 S cm−1 at 30% RH. Under anhydrous conditions, proton carriers such as organic heterocyclic compounds and phosphoric acid are often applied. Banerjee et al. reported a sulfonic acid-modified COF (TpPa-SO3H) for use in anhydrous proton conduction.130 Based on the loaded sulfonic acid units, the proton conductivity was 1.7 × 10−5 S cm−1 at 120 °C under anhydrous conditions. Next, TpPa-(SO3H-Py) was synthesized using the multicomponent aldimine reaction of Tp, 2,5-diaminobenzenesulfonic acid and 2,5-diaminopyridine in which sulfonic acid acted as the acidic site and pyridine as the basic site. After phytic acid molecules were introduced inside the framework, the anhydrous proton conductivity was increases up to 5 × 10−4 S cm−1 at 120 °C.
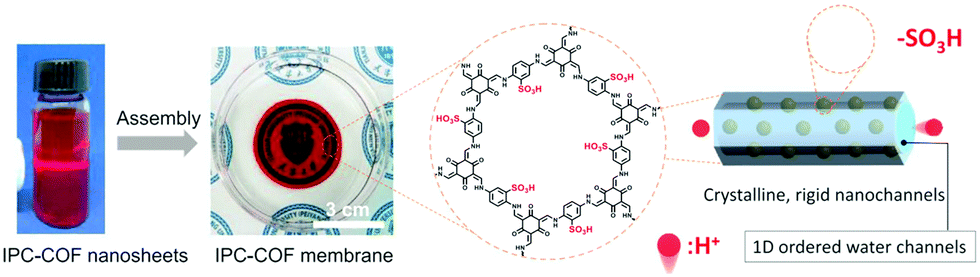 | ||
| Fig. 31 Synthesis of IPC-COF membrane for proton conduction. Reprinted with permission from ref. 129. Copyright (2020) Wiley-VCH. | ||
3.3 Catalysis
The immobilization of metal ions such as Ir(I), Ru(II) and Co(II) on the skeletons of POPs via the chelating units is a typical strategy for improving the catalytic performance of i-POPs via organic transformation. Tahir et al. synthesized an Ir(I)-anchored CTF through post-coordination with the bipyridine moiety.131 In the presence of B2Pin2 as a boron source, the resulting Ir(I)@bipyCTF efficiently catalyzed the C–H borylation of various aromatic compounds (Fig. 32a). With a loading of 1.5 mol% Ir, a 95% yield was achieved in the C–H borylation of 1,2-dichlorobenzene under mild conditions. The robust CTF ensured the catalytic stability over at least 5 cycles without any decrease in the yield. Feng et al.32 prepared a series of boron-containing CMPs with hierarchical porous structures, which were further converted into anionic porous polymers via Lewis acid–base interaction with fluoride ions (Fig. 32b). Using subsequent ion-exchange treatment, various heavy metal cations including Co2+, Fe2+ and Ni2+ were interacted with the anionic skeletons of the i-CMPs. Of these species, the Co(II)-loaded i-CMPs exhibited an outstanding catalytic activity for the homocoupling of aryl Grignard reagents with a high yield of 91%. Huang et al. incorporated Re(CO)3Cl in an imine-linked 2D COF (Fig. 32e).132 Photogenerated electron transfer occurred between the COF framework and the Re complex, giving rise to an enhanced light absorbability and efficient charge separation. Using triethanolamine as an electron sacrificial donor, the Re-COF showed an excellent photocatalytic performance for the reduction of CO2 to CO, with a high selectivity of 95%. Thomas et al. presented a variety of microporous anionic borate networks (ABNs) with a weakly coordinating anionic tetraphenylborate.31 Through exchange with lithium ions, the resulting [Mn(bpy)2]2+-ABN exhibited a distinctive character in the catalytic oxidation of alkenes towards epoxides, with a high selectivity of 81% (Fig. 32c). Gao et al. synthesized an anionic COF through the ring-opening reaction of 1,3-propane sultone with phenolic hydroxyl on the skeletons of DhaTab.133 Mn2+ and Mn(II) bipyridine complexes ([Mn(bpy)2]2+) were trapped by the anionic sulfonates attached on the side backbones of the COF via an ion-exchange process, respectively. The conversion of styrene reached as high as 99% and the yield of styrene oxide was 74% for the [SO3Mn]-DhaTab (Fig. 32d). With [SO3Mn(bpy)2]-DhaTab as the catalyst, the epoxidation of trans-stilbene was converted completely into 2,3-diphenylethylene oxide.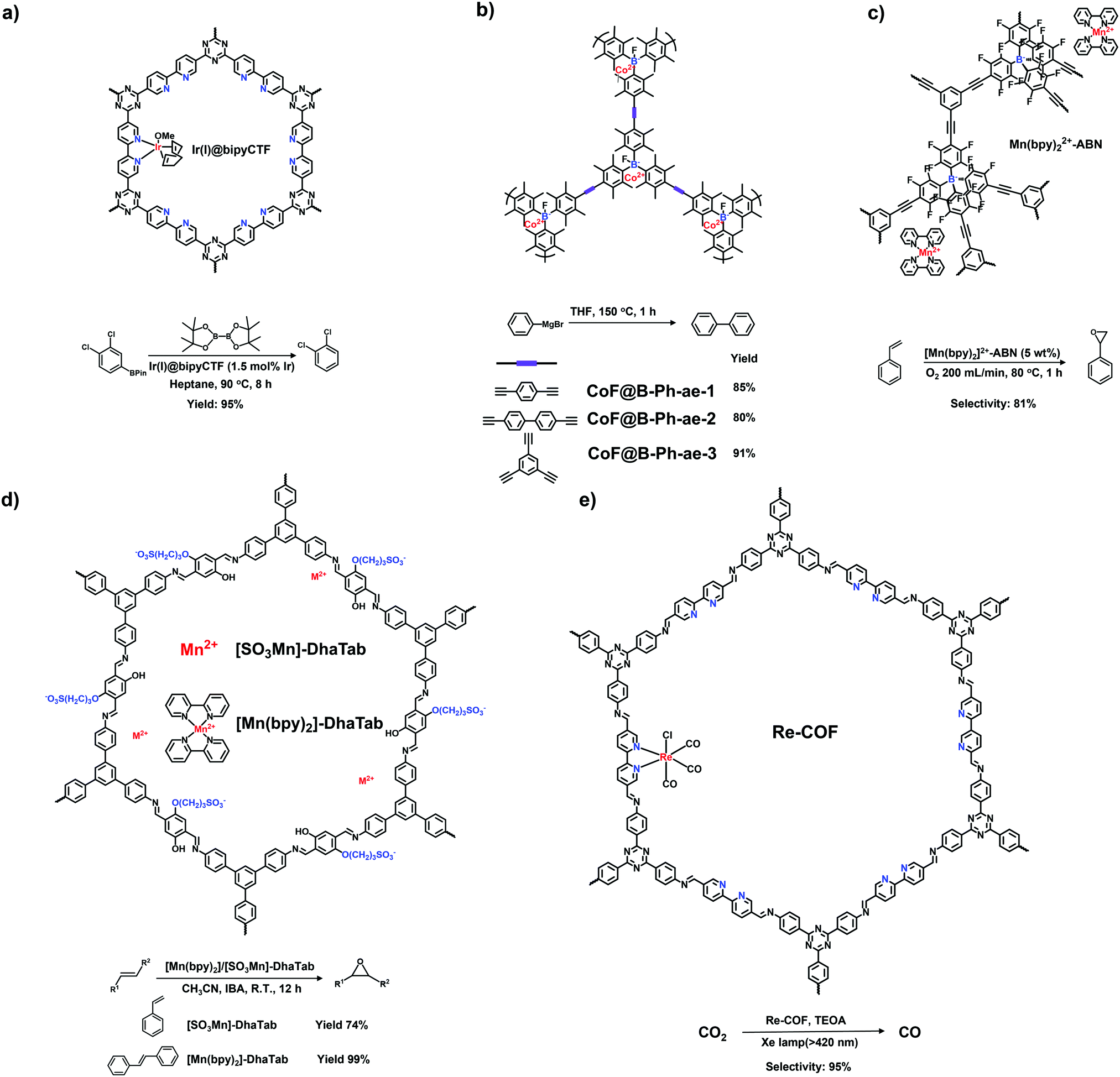 | ||
| Fig. 32 Structures of (a) Ir(I)@bipyCTF, (b) CoF@B-Ph-ae, (c) Mn(bpy)2+-ABN, (d) Mn-DhaTab, and (e) Re-COF and their catalytic reaction schemes. | ||
As an alternative, metal-free i-POPs have been intensively explored for the sake of green chemistry developments. Coskun et al.25 adopted the ionothermal trimerization of cyanophenyl-substituted viologen cations into the i-CTFs for CO2 capture and conversion. Compared with the neutral CTF, the viologen-containing i-CTF showed a greater CO2 uptake ability and was applied as a metal-free catalyst for the cycloaddition of CO2 with epoxides into cyclic carbonates, giving a high yield of 99% and 95% for propylene oxide and epichlorohydrin, respectively.
IL-functionalized COFs can serve as ionic catalysts and provide better stability than other known organic catalysts. Zhang et al.134 reported a series of Brønsted-acid IL (BIL)-containing imine-linked COFs with high crystallinity and porosity. Taking BIL-COF-30 as an example for the catalytic dehydration of sorbitol into isosorbide, yields of up to 97% at 160 °C and could be recycled over at least 5 runs without losing activity (Fig. 33a). Along this line, they continued and explored a poly-IL-decorated COF through the quaternization reaction of tertiary amines.98 The COF particles with covalently grafted poly-ILs could reach a yield of up to 83% in the transformation of sorbitol into isosorbide and remained the catalytic activity over 10 cycles. Dong et al. decorated COFs with allyl imidazolium-based ILs,68 demonstrating the highly selective adsorption for CO2 and catalytic activity towards the cycloaddition of epoxides with CO2 without any co-catalysts (Fig. 33b). To scale up the reaction, they fabricated a COF-IL@chitosan aerogel using a thiol–ene click reaction of COF-ILs and SH-modified chitosan binders, which they then processed into a cup-like reactor. Likewise, a quinoline-linked COF-IL was designed using a multicomponent Povarov reaction and post-modification with 1,3-propane sultone.135 The obtained COF-IM-SO3H served as an efficient Brønsted-acid heterogeneous catalyst for the Biginelli reaction under solvent-free conditions (Fig. 33c).
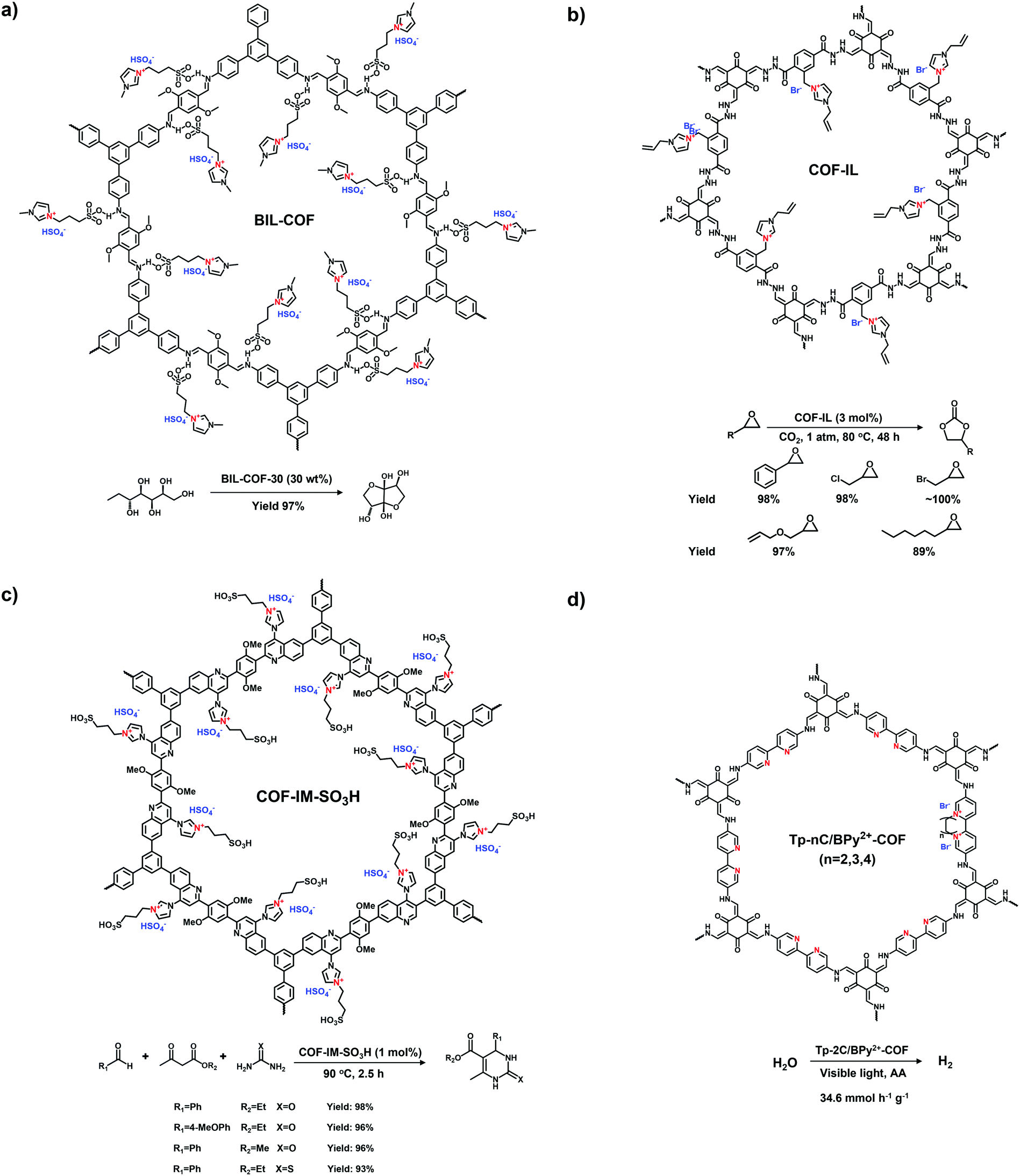 | ||
| Fig. 33 (a) Catalytic dehydration of sorbitol with BIL-COF. (b) Cycloaddition of CO2 with epoxides using COF-IL. (c) COF-IM-SO3H catalyzed Biginelli reaction. (d) Photocatalytic hydrogen evolution with Tp-nC/BPy2+-COF. | ||
Viologen-derived electron-transfer mediators (ETMs) can relay electrons to greatly improve the reaction efficiency. To boost the photocatalytic H2 evolution in water splitting, our group88 designed a COF-based multivariate system to fuse ETM modules for promoting the electron transfer from photosensitizers to catalytically active sites in photocatalytic reactions (Fig. 33d). A series of 2,2′-bipyridine-based Tp-nC/BPy2+-COF (n = 2–4) materials were synthesized by varying the content of the cationic cyclic diquat with different alkyl chains via a post-quaternization route. Given the dual effect of electrostatic repulsion and steric hinderance, the distribution of cationic cyclic diquat (BPy2+) moiety was discrete and site-isolated in the frameworks, thus effectively inhibiting the generation of electron-trapped BPy2+ dimers and, in turn, playing the role of an ETM in the photogenerated exciton dissociation and free-electron transfer along the skeletons. In comparative studies, Tp-2C/BPy2+-COF that contained 19.10 mol% of 2C/BPy2+ exhibited the highest photocatalytic hydrogen production activity of 34600 μmol h−1 g−1 with a high apparent quantum efficiency of 6.93% at 420 nm; this performance allowed such materials to be ranked among the top organic photocatalysts for H2 evolution.
3.4 Sensing
As the optically or electrically active molecules are functionalized as ionic building blocks, the built i-POPs are enabled to combine the porosity with the specific functions for sensing applications. In this respect, Coskun et al. presented a cationic porous polymer (pDAP, Fig. 34a) by incorporating diazapyrenium units via a nucleophilic substitution reaction. Exposing the resulting i-POP that contains aliphatic amines resulted in a broad absorption band around 595 nm and an obvious change in colour from orange to dark green.136 Such a visual change was largely due to the charge transfer between the diazapyrenium moieties and aliphatic amines within the porous networks.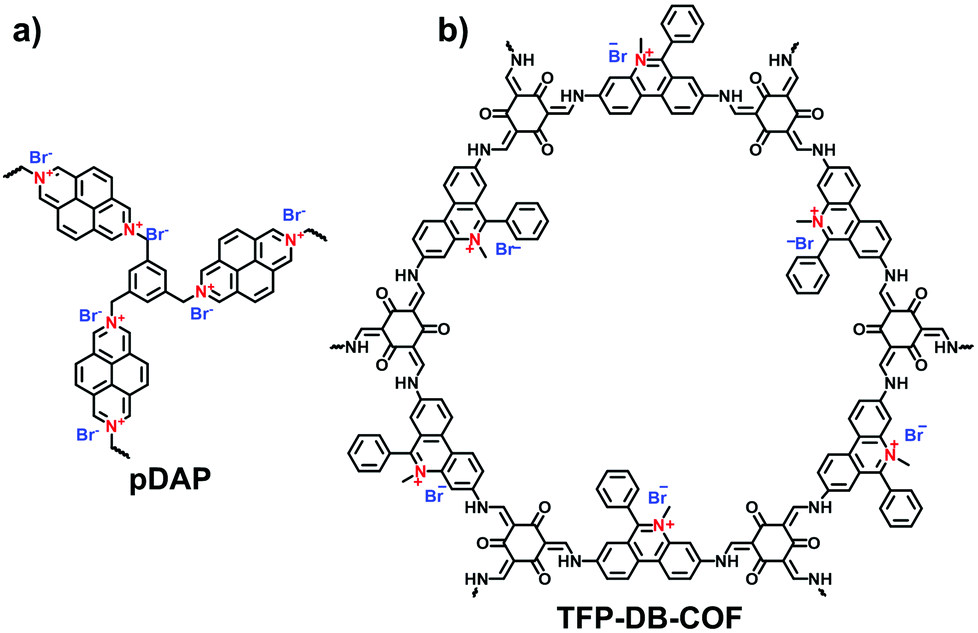 | ||
| Fig. 34 Schematics of (a) pDAP and (b) TFP-DB-COF. | ||
Lanthanide complexes feature narrow emissions and long lifetimes, which are desirable fluorescence properties for sensing and imaging. Anchoring lanthanide complexes onto POPs is a potent strategy to suppress their aggregation, which is often detrimental to the stability and photoactivity of complexes. To this end, Liao et al. reported a lanthanide-containing COF (DhaTab-COF-EuIL) obtained via a post-synthetic method including the Williamson ether reaction with the IL and subsequent ion displacement with a europium (Eu) IL complex.137 The post-modified i-COFs showed dual luminescent emission originating from COF and Eu3+. By virtue of the photoinduced energy transfer, the fluorescent emission was quenched by specific chemicals. Further studies demonstrated that the materials exhibited high sensitivity and selectivity toward acetone at the lowest detected concentration of 1% (Fig. 35a). Likewise, Eu/Tb and Dy acetylacetone complexes were successfully grafted onto TpBPy-COF via a similar route. As reported by Kaczmarek et al., the obtained i-COFs were used as a luminescent ratiometric thermometer that could be operated in the ranges of 10–360 K and 280–440 K.138
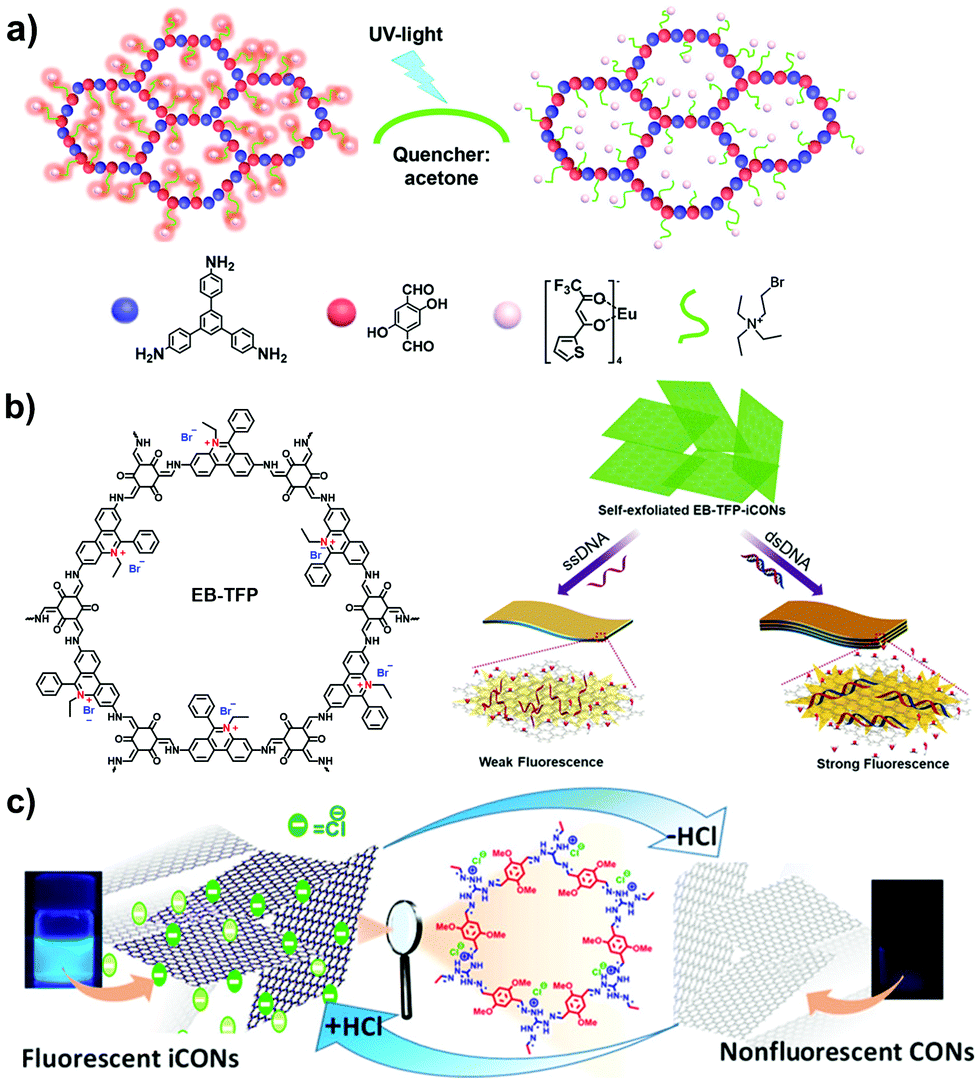 | ||
| Fig. 35 Schematics of detecting (a) acetone with DhaTab-COF-EuIL, (b) DNA with EB-TFP, and (c) fluoride anions with DATGCl i-CONs. (a) Reprinted with permission from ref. 138. Copyright (2019) American Chemical Society. (b) Reprinted with permission from ref. 143. Copyright (2018) Wiley-VCH. (c) Reprinted with permission from ref. 141. Copyright (2020) American Chemical Society. | ||
Apart from the post-grafting of metal complexes onto POPs, ionized organic modules have also been employed to directly prepare an i-POP sensor. Mi et al. synthesized TFP-DB-COF, using dimidium bromide as the cationic linker, which demonstrated an ability as a triboelectric nanogenerator (TENG).139 Compared with the neutral counterpart, the TFP-DB-COF TENG exhibited the better triboelectric output performance owing to the highly efficient charge transport within the framework (Fig. 34b). Such an i-COF-based TENG even could light up 1388 low-power green LEDs connected in series. Zhang et al. investigated the TpPa-SO3Na i-COF for humidity sensing applications.140 The pendant –SO3Na groups on the linkers endowed the i-COF with a water adsorption capacity (411 cm3 g−1). Furthermore, the conductivity showed a linear dependence on the relative humidity, a fast response over a broad detection range and superior long-term stability, all of which were attributed to the fast ion mobility in the i-COF.
Exfoliation of 2D i-COFs into i-CONs is conducive to fully exposing the active sites and enhancing the sensing ability. Pal et al. designed fluorescent DATGCl i-CONs via the condensation of triaminoguanidinium chloride (TGCl) and 2,5-dimethoxyterephthalaldehyde (DA).141 The fluorescence intensity of the i-CONs could be drastically reduced by 67.3-fold with the addition of fluoride ions. This was due to the proton abstraction of F− ions causing deprotonation of the i-CONs (Fig. 35c). The sensitivity and selectivity of DATGCl towards the F− ions could be down to the ppb level, making it more competitive among other reported F− sensors.
For the borate ester-linked COFs, the COFs often fail to delaminate in solution due to the instability of the linkages. However, ion pairs fixed in the frameworks are available to trigger the self-exfoliation process. With BF3·OEt2 as the catalyst, the i-COFs-A frameworks were formed through the condensation of boronic acid and pinacol-capped catechol, accompanied by the formation of stable ion pairs of BF3·OEt2 with N atoms on the linkers.142 With the aid of electrostatic repulsion, the i-COFs could be easily exfoliated in acetonitrile into nanosheets with an average thickness of 3.5 ± 0.5 nm. The i-CONs-A could be further castg on SiO2 wafers to obtain micro-gap electrodes with good electroconductive and photoconductive properties.
Ajayaghosh et al. synthesized EB-TFP-iCOFs through imine condensation (Fig. 35b), using EB as an ionic linker and 2,4,6-triformylphloroglucinol (TFP) as a neutral knot.143 The obtained 2D i-COFs were self-exfoliated via electrostatic repulsion between the charged layers, thereby producing aqueous dispersions of i-CONs with the ability for optically detecting DNA. As demonstrated in the study, DNA chains were electrostatically assembled with the i-CONs to enhance the hydrophobicity. Accordingly, excited-state proton transfer was affected to result in the emission change from green to orange under 365 nm UV light.
3.5 Biomedical applications
Guanidinium-based materials are usually soluble, limiting their application as recyclable antibacterial agents. Introducing guanidinium units into the COF skeletons will provide an opportunity for improving the bio-applicability. Banerjee et al.62 fabricated three guanidinium-based i-CONs (TpTGX, X = Cl−, Br−, I−) via Schiff base condensation between Tp and amines TGX (Fig. 36a). Thanks to the intrinsic charges in the frameworks and the counterions sandwiched between the layers, TpTGX could self-exfoliate into few-layered i-CONs, which were produced with difficulty from the neutral COFs. The i-CONs remained stable in colloidal suspensions for 20 days and reduced the colony forming units (CFU) towards Gram-positive (S. aureus) and Gram-negative (E. coli) bacteria. This may arise from the electrostatic interaction between the cationic guanidinium moiety and the anionic phospholipid bilayer of the bacterial cell (Fig. 37a). Also, the good dispersibility of i-CONs allowed the mixture of TpTGX and polysulfone (PSF) to produce an antibacterial coating (TpTGCl@PSF). However, the bacterial-inhibition dynamics of TpTGX were irreversible and uncontrollable. To meet this challenge, Ajayaghosh et al.60 reported a bacterial-growth strategy based on a propidium iodide-linked i-COF (PI-TFP) (Fig. 36b). The PI-TFP featured a high charge density, stable β-ketoenamine linkages and self-exfoliation behavior in water. After being exfoliated into few-layered i-CONs, the restacking process was driven by the complexation of the quaternary ammonium salts with cucurbit[7]uril (CB[7]), leading to a decrease in the surface charges and antibacterial effectiveness. The bacterial CFU counts increased from 65% for E. coli and 58% for S. aureus to 90% and 95%, respectively. By introducing a competitive guest molecule 1-adamantylamine hydrochloride (AD), the decomplexation of CB[7] and re-exfoliation of i-COF took place, resulting in the regeneration of the PI-TFP i-CONs and inhibition of bacterial growth. The CFU recovered to 51% for E. coli and 57% for S. aureus. With such a synergistic effect of the host–guest interaction and electrostatic interaction, the bacterial growth could be finely tuned as required (Fig. 37b).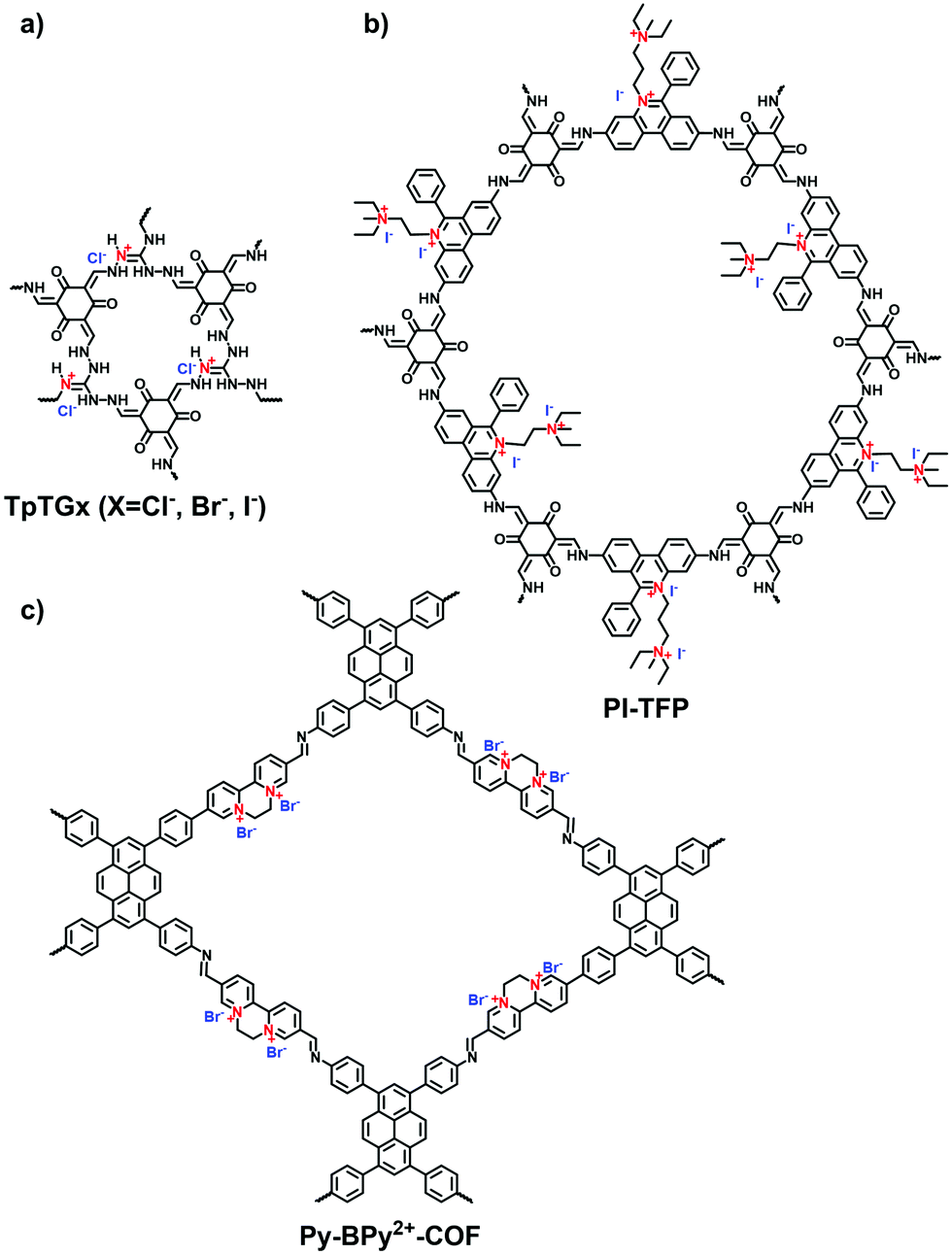 | ||
| Fig. 36 Schematics of (a) TpTGX (X = Cl−, Br−, I−), (b) PI-TFP and (c) Py-BPy2+-COF. | ||
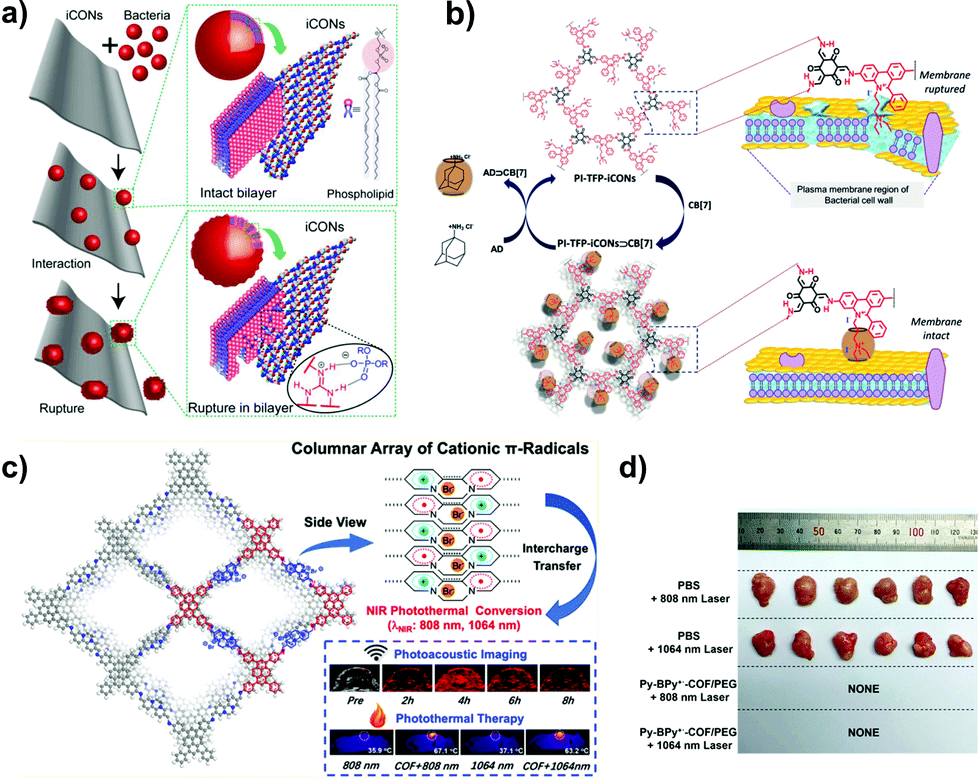 | ||
| Fig. 37 Antibacterial mechanism of (a) TpTGX (X = Cl−, Br−, I−) and (b) PI-TFP i-CONs, respectively. (c) Photoacoustic and photothermal performance of Py-BPy+˙-COF. (d) In vivo treatment of mouse tumors using Py-BPy+˙-COF as a NIR photothermal agent. (a) Reprinted with permission from ref. 62. Copyright (2016) American Chemical Society. (b) Reprinted with permission from ref. 60. Copyright (2019) Wiley-VCH. (c) Reprinted with permission from ref. 87. Copyright (2019) American Chemical Society. | ||
Photothermal therapy (PTT) and photoacoustic (PA) imaging are considered as non-invasive approaches for cancer treatment and diagnosis. They require prominent photophysical properties such as a large near-infrared (NIR) absorption and intense non-radiative relaxation under excitation. Our group reported a multistep post-synthetic strategy that converted a 2,2′-bipyridine-containing neutral COF into the cationic i-COF (Py-BPy2+-COF) (Fig. 36c) and the cationic radical i-COF (Py-BPy+•-COF) for photothermal therapy (PTT) and photoacoustic imaging (PAI) (Fig. 37c).87 The cationic radical structure of Py-BPy+˙-COF originated from the in situ cyclic quaternization at the 2,2′-bipyridine unit, followed by a one-electron reduction to achieve radical cations. Compared with neutral and cationic frameworks, Py-BPy+˙-COF/PEG exhibited an enhanced absorption in the NIR region as the uniaxial stacking of cationic radicals enhanced the electron delocalization. Under irradiation of 808 nm and 1064 nm lasers (1 W cm−2), the aqueous dispersion of Py-BPy+˙-COF allowed an increase in temperature by 50 °C and 40 °C, respectively, yielding photothermal conversion efficiencies of as high as 63.8% and 55.2%, respectively. On the basis of such remarkable performances, Py-BPy+˙-COF/PEG served as a good PTT and PA imaging agent for treating the mouse tumor model (Fig. 37d).
4. Conclusions and future outlook
In this review, the synthesis and applications of polyelectrolyte porous frameworks are summarized to survey the recent progress of emerging porous materials. Originating from the classifications of POPs, two different bottom-up synthesis approaches for polyelectrolyte-based POPs have been detailed, i.e., irreversible coupling reactions for amorphous i-POPs, and reversible dynamic reactions for crystalline i-POPs. Some allow for the generation of charged backbones via the direct incorporation of ionic building blocks in the frameworks, and the others are enabled to produce charged linkages with neutral building blocks. Post-modification is an alternative method for the preparation of polyelectrolyte-based frameworks that often fail to afford crystalline or amorphous high-surface-area structures in a direct way. A variety of ionic functional units can be immobilized at the pendant groups of skeletons via post-synthetic reactions. With these synthesis methods, porous polyelectrolyte frameworks show a close synergy between charges and pores that defines new structure-to-activity connections. Accordingly, appealing properties can be generated to serve a wide range of fields, including gas storage and separation, ionic pollution removal, heterogenous catalysis, ion conduction, sensing, antibiotics and phototherapy. Compared with neutral POPs, the advantages of i-POPs in applications are mainly attributed to the controlled ionic sites, which not only have their own functions in catalysis and sensing but also offer Coulombic interactions with pore confinement to play a crucial role in adsorption, separation and conduction.In contrast to amorphous ionic frameworks, the synthesis of highly crystalline i-COFs remains challenging. The typical solvothermal routes available for the crystallization of neutral COFs have often failed in the formation of the periodic ionic arrangement and ordering alignment of charged skeletons in 2D or 3D motifs. The key point is due to the presence of numerous ionic units or charged linkages that impart strong electrostatic interactions to the solvated backbones during the reaction. In most cases, the ionic units or charged linkages preclude the π-interaction of aromatic building blocks and, in turn, disorder the planar configurations of frameworks for access to the expected stacking or interpenetrating structures. Therefore, among the few ionic building blocks reported so far, charge sites are often located at the pendant groups of repeating units rather than on the supporting backbones. To address these challenges, fused aromatic rings integrated into the skeletons can provide extended π-conjugation for balancing the electrostatic repulsion. Meanwhile, reactions for new linkages containing anions, cations or zwitterions would open up an avenue for greatly expanding i-COFs as a vast number of neutral building blocks are applicable. To accompany the development of synthesis, theoretical modelling is needed to reasonably understand the stacking array of ionic frameworks as charges can offset the alignment of adjacent layers, especially for 2D COFs, causing a weakened performances in, for example, interlayered charge mobility. By contrast, post-modification is a more flexible strategy to achieve synergy between charges, pores and skeletons for bolstering specific functions. Furthermore, the post-modification technique is accessible to fabricate i-POPs with technologically more relevant forms such as membranes, thin films, fibers and particles, since all have been well documented for neutral POPs.
Most research regarding the applications of i-POPs is still at an early stage. In terms of ion conduction, i-POPs show distinctive performances in transferring lithium ions, protons and anions, while research on the use of i-POPs for solid electrolytes and as ion-exchange membranes is just beginning. Challenges regarding battery performance and assembly techniques need to be addressed to highlight values. For example, miniaturizing i-POPs to microspheres, nanofibers and nanosheets has been much less explored and represents vast opportunities for energy applications. Polyelectrolytes have been investigated for a long time in the biomedical field, of which the Coulombic interaction plays a pivotal role in specific utilities. Taking advantage of the accessible pores and functional skeletons, polyelectrolyte-derived i-POPs show great potential for the pursuit of drug delivery, gene therapy, protein enrichment, imaging, and so on. In terms of catalysis, metalated i-POPs are advanced owing to the abundant choice of metal elements. The anchored metal moieties can co-operate with pore environments, regulating the confinement effect, ionic interactions with substrates and the functions of skeletons for individual reactions. The rapid development of i-POPs for gas storage and separation and ionic-pollution removal have been achieved, while in the presence of counterions, control over the surface area and pore-size distributions of i-POPs is not yet satisfactory and is far lower than for the corresponding neutral POPs. These challenges and hurdles will greatly advance research into the practical application of porous polymers and maintain the original motivation in the future development.
This overview of emerging porous polyelectrolyte frameworks emphasizes the enormous potential of i-POPs for a broad range of scientific research and advanced applications. Much work on the emerging synthetic chemistry, characterization technologies and material engineering of i-POPs is expected in the coming years to fulfil the ever-increasing demands for functional materials. We hope that the current review will arouse the interest of scientists for boosting this emerging field and leading to industrial applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was supported financially from the National Natural Science Foundation of China (Grants No. 21774023, 51973039, and 52173197).References
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- R. Liu, K. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- M. Liu, L. Guo, S. Jin and B. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- J. Meng, J. Li, J. Liu, X. Zhang, G. Jiang, L. Ma, Z. Hu, S. Xi, Y. Zhao, M. Yan, P. Wang, X. Liu, Q Li, J. Liu, T. Wu and L. Mai, ACS Cent. Sci, 2020, 6, 1431–1440 CrossRef CAS PubMed.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- J. Wu, F. Xu, P. Ma, X. Zhang, Q. Liu, R. Fu and D. Wu, Adv. Mater., 2019, 31, 1802922 CrossRef PubMed.
- Y. Xie, J. Yin, J. Zheng, L. Wang, J. Wu, M. Dresselhaus and X. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 32244–32250 CrossRef CAS PubMed.
- B. Zheng, X. Lin, X. Zhang, D. Wu and K. Matyjaszewski, Adv. Funct. Mater., 2020, 30, 1907006 CrossRef CAS.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- X. Zhao, Y. Chen, Z. Wang and Z. Zhang, Polym. Chem., 2021, 12, 4874–4894 RSC.
- J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- J. Yang, X. Zhang, C. Liu, Z. Wang, L. Deng, C. Feng, W. Tao, X. Xu and W. Cui, Prog. Mater. Sci., 2021, 118, 100768 CrossRef CAS.
- W. Chen, P. Chen, G. Zhang, G. Xing, Y. Feng, Y. W. Yang and L. Chen, Chem. Soc. Rev., 2021, 50, 11684–11714 RSC.
- W. Zhang, Q. Zhao and J. Yuan, Angew. Chem., Int. Ed., 2018, 57, 6754–6773 CrossRef CAS PubMed.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- S. Fischer, A. Schimanowitz, R. Dawson, I. Senkovska, S. Kaskel and A. Thomas, J. Mater. Chem. A, 2014, 2, 11825–11829 RSC.
- J. Wang, J. G. Wei Yang, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 15708–15711 RSC.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- W. Zhang, F. Ma, L. Ma, Y. Zhou and J. Wang, ChemSusChem, 2020, 13, 341–350 CrossRef CAS PubMed.
- P. Chen, L. Zhang, J. S. Sun, E. K. Xiao, X. T. Wu and G. Zhu, ChemPlusChem, 2020, 85, 943–947 CrossRef CAS PubMed.
- Y. Liu, Y. Cui, C. Zhang, J. Du, S. Wang, Y. Bai, Z. Liang and X. Song, Chem. – Eur. J., 2018, 24, 7480–7488 CrossRef CAS PubMed.
- H. Ma, H. Ren, X. Zou, S. Meng, F. Sun and G. Zhu, Polym. Chem., 2014, 5, 144–152 RSC.
- Y. Pan, X. Zhai, J. Yin, T. Zhang, L. Ma, Y. Zhou, Y. Zhang and J. Meng, ChemSusChem, 2019, 12, 2231–2239 CrossRef CAS PubMed.
- S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174–12178 CrossRef CAS PubMed.
- W. Zhao, F. Zhang, L. Yang, S. Bi, D. Wu, Y. Yao, M. Wagner, R. Graf, M. R. Hansen, X. Zhuang and X. Feng, J. Mater. Chem. A, 2016, 4, 15162–15168 RSC.
- C. Gu, N. Huang, Y. Chen, H. Zhang, S. Zhang, F. Li, Y. Ma and D. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3049–3053 CrossRef CAS PubMed.
- Q. Sun, S. Ma, Z. Dai, X. Meng and F. Xiao, J. Mater. Chem. A, 2015, 3, 23871–23875 RSC.
- C. Li, W. Wang, L. Yan, Y. Wang, M. Jiang and Y. Ding, J. Mater. Chem. A, 2016, 4, 16017–16027 RSC.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS.
- H. C. Cho, H. S. Lee, J. Chun, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 917–919 RSC.
- Z. Yan, Y. Yuan, Y. Tian, D. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12733–12737 CrossRef CAS PubMed.
- H. Zhao, Y. Wang and R. Wang, Chem. Commun., 2014, 50, 10871–10874 RSC.
- D. Wei, F. Wang, H. Sun, Z. Zhu, W. Liang and A. Li, Eur. Polym. J., 2021, 147, 110281 CrossRef CAS.
- P. Zhang, X. Jiang, S. Wan and S. Dai, Chem. – Eur. J., 2015, 21, 12866–12870 CrossRef CAS PubMed.
- P. Samanta, A. V. Desai, B. Anothumakkool, M. M. Shirolkar, A. Karmakar, S. Kurungot and S. K. Ghosh, J. Mater. Chem. A, 2017, 5, 13659–13664 RSC.
- P. Zhang, Z. A. Qiao, X. Jiang, G. M. Veith and S. Dai, Nano Lett., 2015, 15, 823–828 CrossRef CAS PubMed.
- A. A. Raja and C. T. Yavuz, RSC Adv., 2014, 4, 59779–59784 RSC.
- K. Kim, O. Buyukcakir and A. Coskun, RSC Adv., 2016, 6, 77406–77409 RSC.
- J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- S. Hao, Y. Liu, C. Shang, Z. Liang and J. Yu, Polym. Chem., 2017, 8, 1833–1839 RSC.
- K. Wang, W. Cui, Z. Bian, Y. Liu, S. Jiang, Y. Zhou and J. Wang, Appl. Catal., B, 2021, 281, 119425 CrossRef CAS.
- Y. Yuan, F. Sun, L. Li, P. Cui and G. Zhu, Nat. Commun., 2014, 5, 4260 CrossRef CAS PubMed.
- S. Han, Y. Feng, F. Zhang, C. Yang, Z. Yao, W. Zhao, F. Qiu, L. Yang, Y. Yao, X. Zhuang and X. Feng, Adv. Funct. Mater., 2015, 25, 3899–3906 CrossRef CAS.
- C. Gu, N. Huang, Y. Chen, H. Zhang, S. Zhang, F. Li, Y. Ma and D. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3049–3053 CrossRef CAS PubMed.
- J. Wang, W. Sng, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 12076–12079 RSC.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- A. Li, R. F. Lu, Y. Wang, X. Wang, K.-L. Han and W.-Q. Deng, Angew. Chem., Int. Ed., 2010, 49, 3330–3333 CrossRef CAS PubMed.
- K. Konstas, J. W. Taylor, A. W. Thornton, C. M. Doherty, W. X. Lim, T. J. Bastow, D. F. Kennedy, C. D. Wood, B. J. Cox, J. M. Hill, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2012, 51, 6639–6642 CrossRef CAS PubMed.
- Z. Xiang, D. Cao, W. Wang, W. Yang, B. Han and J. Lu, J. Phys. Chem. C, 2012, 116, 5974–5980 CrossRef CAS.
- H. Ma, H. Ren, X. Zou, F. Sun, Z. Yan, K. Cai, D. Wang and G. Zhu, J. Mater. Chem. A, 2013, 1, 752–758 RSC.
- D. Xu, L. Sun, G. Li, J. Shang, R. X. Yang and W. Q. Deng, Chem. – Eur. J., 2016, 22, 7944–7949 CrossRef CAS PubMed.
- H. Ma, B. Liu, B. Li, L. Zhang, Y. G. Li, H. Q. Tan, H. Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS PubMed.
- A. Mal, S. Vijayakumar, R. K. Mishra, J. Jacob, R. S. Pillai, B. S. Dileep Kumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2020, 59, 8713–8719 CrossRef CAS PubMed.
- Z. Li, H. Li, X. Guan, J. Tang, Y. Yusran, Z. Li, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2017, 139, 17771–17774 CrossRef CAS PubMed.
- S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS PubMed.
- H. J. Da, C. X. Yang and X. P. Yan, Environ. Sci. Technol., 2019, 53, 5212–5220 CrossRef CAS PubMed.
- S. Jansone Popova, A. Moinel, J. A. Schott, S. M. Mahurin, I. Popovs, G. M. Veith and B. A. Moyer, Environ. Sci. Technol., 2019, 53, 878–883 CrossRef CAS PubMed.
- S. Yu, H. Lyu, J. Tian, H. Wang, D. Zhang, Y. Liu and Z. Li, Polym. Chem., 2016, 7, 3392–3397 RSC.
- L. Wang, C. Zeng, H. Xu, P. Yin, D. Chen, J. Deng, M. Li, N. Zheng, C. Gu and Y. Ma, Chem. Sci., 2019, 10, 1023–1028 RSC.
- N. Huang, P. Wang, M. A. Addicoat, T. Heine and D. Jiang, Angew. Chem., Int. Ed., 2017, 56, 4982–4986 CrossRef CAS PubMed.
- L. G. Ding, B. J. Yao, F. Li, S. C. Shi, N. Huang, H. B. Yin, Q. Guan and Y. B. Dong, J. Mater. Chem. A, 2019, 7, 4689–4698 RSC.
- Z. Li, Z. W. Liu, Z. J. Mu, C. Cao, Z. Li, T. X. Wang, Y. Li, X. Ding, B. H. Han and W. Feng, Mater. Chem. Front., 2020, 4, 1164–1173 RSC.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS PubMed.
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed.
- K. Jeong, S. Park, G. Y. Jung, S. H. Kim, Y. H. Lee, S. K. Kwak and S. Y. Lee, J. Am. Chem. Soc., 2019, 141, 5880–5885 CrossRef CAS PubMed.
- S. Hou, W. Ji, J. Chen, Y. Teng, L. Wen and L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 9925–9930 CrossRef CAS PubMed.
- Y. Yan, S. Wu, Y. Yan, X. Tang, S. Cai, S. Zheng, W. Zhang and F. Gu, Chem. J. Chin. Univ., 2021, 42, 956–964 Search PubMed.
- W. Jiang, W. R. Cui, R. P. Liang and J. D. Qiu, Chem. Eng. J., 2021, 417, 128034 CrossRef CAS.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- W. Zhang, L. Zhang, H. Zhao, B. Li and H. Ma, J. Mater. Chem. A, 2018, 6, 13331–13339 RSC.
- Y. Peng, G. Xu, Z. Hu, Y. Cheng, C. Chi, D. Yuan, H. Cheng and D. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 18505–18512 CrossRef CAS PubMed.
- X. He, Y. Yang, H. Wu, G. He, Z. Xu, Y. Kong, L. Cao, B. Shi, Z. Zhang, C. Tongsh, K. Jiao, K. Zhu and Z. Jiang, Adv. Mater., 2020, 32, 2001284 CrossRef CAS PubMed.
- F. Meng, S. Bi, Z. Sun, B. Jiang, D. Wu, J. Chen and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 13614–13620 CrossRef CAS PubMed.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gandara, M. Lusi, D. S. S. Rao, S. Vimala, S. K. Prasad, J. Raya, D. S. Han, R. Jagannathan, J. Olsen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- J. Roeser, D. Prill, M. J. Bojdys, P. Fayon, A. Trewin, A. N. Fitch, M. U. Schmidt and A. Thomas, Nat. Chem., 2017, 9, 977–982 CrossRef CAS PubMed.
- O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Fröba, A. Thomas and J. Roeser, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS PubMed.
- S. Ashraf, Y. Zuo, S. Li, C. Liu, H. Wang, X. Feng, P. Li and B. Wang, Chem. – Eur. J., 2019, 25, 13479–13483 CrossRef CAS PubMed.
- S. Ashraf, C. Liu, S. Li, I.-U. Haq, M. Mehmood, P. Li and B. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 40372–40380 CrossRef CAS PubMed.
- Z. Mi, P. Yang, R. Wang, J. Unruangsri, W. Yang, C. Wang and J. Guo, J. Am. Chem. Soc., 2019, 141, 14433–14442 CrossRef CAS PubMed.
- Z. Mi, T. Zhou, W. Weng, J. Unruangsri, K. Hu, W. Yang, C. Wang, K. A. I. Zhang and J. Guo, Angew. Chem., Int. Ed., 2021, 60, 9642–9649 CrossRef CAS PubMed.
- M. Chen, J. Zhang, C. Liu, H. Li, H. Yang, Y. Feng and B. Zhang, Org. Lett., 2021, 23, 1748–1752 CrossRef CAS PubMed.
- Q. Yan, H. Xu, X. Jing, H. Hu, S. Wang, C. Zeng and Y. Gao, RSC Adv., 2020, 10, 17396–17403 RSC.
- Z. Li, Z. W. Liu, Z. Li, T. X. Wang, F. Zhao, X. Ding, W. Feng and B. H. Han, Adv. Funct. Mater., 2020, 30, 1909267 CrossRef CAS.
- J. Qiu, Y. Zhao, Z. Li, H. Wang, Y. Shi and J. Wang, ChemSusChem, 2019, 12, 2421–2427 CAS.
- Y. Zhang, H. Hu, J. Ju, Q. Yan, V. Arumugam, X. Jing, H. Cai and Y. Gao, Chin. J. Catal., 2020, 41, 485–493 CrossRef CAS.
- B. Dong, L. Y. Wang, S. Zhao, R. L. Ge, X. D. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- Z. J. Mu, X. S. Ding, Z. Y. Chen and B. H. Han, ACS Appl. Mater. Interfaces, 2018, 10, 41350–41358 CrossRef CAS PubMed.
- H. Guo, J. Wang, Q. Fang, Y. Zhao, S. Gu, J. Zheng and Y. Yan, CrystEngComm, 2017, 19, 4905–4910 RSC.
- S. L. Ji, H. L. Qian, C. X. Yang, X. Zhao and X. P. Yan, ChemPlusChem, 2020, 85, 828–831 CrossRef CAS PubMed.
- Y. R. Du, B. H. Xu, S. P. Xia, G. R. Ding and S. J. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 552–562 CrossRef CAS PubMed.
- S. Wang, Y. Liang, T. Dai, Y. Liu, Z. Sui, X. Tian and Q. Chen, J. Colloid Interface Sci., 2021, 591, 264–272 CrossRef CAS PubMed.
- Office of Energy Efficiency & Renewable Energy DOE, Technical Targets for Onboard Hydrogen Storage for Light-Duty Vehicles, https://www.energy.gov/eere/fuelcells/doe-technicaltargets-onboard-hydrogen-storage-lightdutyvehicles#main-content.
- B. K. Rao and P. Jena, Europhys. Lett., 1992, 20, 307–312 CrossRef CAS.
- W. Deng, X. Xu and W. A. Goddard, Phys. Rev. Lett., 2004, 92, 166103 CrossRef PubMed.
- K. Konstas, J. W. Taylor, A. W. Thornton, C. M. Doherty, W. X. Lim, T. J. Bastow, D. F. Kennedy, C. D. Wood, B. J. Cox, J. M. Hill, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2012, 51, 6639–6642 CrossRef CAS PubMed.
- X. M. Hu, Q. Chen, Z. Y. Sui, Z. Q. Zhao, N. Bovet, B. W. Laursen and B. H. Han, RSC Adv., 2015, 5, 90135–90143 RSC.
- A. Dani, V. Crocellà, C. Magistris, V. Santoro, J. Yuan and S. Bordiga, J. Mater. Chem. A, 2017, 5, 372–383 RSC.
- J. Cao, W. Shan, Q. Wang, X. Ling, G. Li, Y. Lyu, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6031–6041 CrossRef CAS PubMed.
- Y. Yang, M. Faheem, L. Wang, Q. Meng, H. Sha, N. Yang, Y. Yuan and G. Zhu, ACS Cent. Sci., 2018, 4, 748–754 CrossRef CAS PubMed.
- Y. Tao, R. Krishna, L. X. Yang, Y. L. Fan, L. Wang, Z. Gao, J. B. Xiong, L. J. Sun and F. Luo, Inorg. Chem. Front., 2019, 6, 2921–2926 RSC.
- Y. Ying, M. Tong, S. Ning, S. K. Ravi, S. B. Peh, S. C. Tan, S. J. Pennycook and D. Zhao, J. Am. Chem. Soc., 2020, 142, 4472–4480 CrossRef CAS PubMed.
- H. Jiang, F. Xue, J. Sun, J. Lin, C. Zhang, X. Wang and R. Zhao, Microchim. Acta, 2021, 188, 47 CrossRef CAS PubMed.
- W. Jiang, D. Peng, W. Cui, R. Liang and J. D. Qiu, ACS Omega, 2020, 5, 32002–32010 CrossRef CAS PubMed.
- Y. Su, Y. Wang, X. Li, X. Li and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18904–18911 CrossRef CAS PubMed.
- P. Blondeau, M. Segura, R. Pérez-Fernández and J. de Mendoza, Chem. Soc. Rev., 2007, 36, 198–210 RSC.
- H. Da, C. Yang, H. Qian and X. Yan, J. Mater. Chem. A, 2020, 8, 12657–12664 RSC.
- T. Skorjanc, D. Shetty, F. Gándara, L. Ali, J. Raya, G. Das, M. A. Olson and A. Trabolsi, Chem. Sci., 2020, 11, 845–850 RSC.
- L. He, S. Liu, L. Chen, X. Dai, J. Li, M. Zhang, F. Ma, C. Zhang, Z. Yang, R. Zhou, Z. Chai and S. Wang, Chem. Sci., 2019, 10, 4293–4305 RSC.
- X. H. Xiong, Z. W. Yu, L. L. Gong, Y. Tao, Z. Gao, L. Wang, W. H. Yin, L. X. Yang and F. Luo, Adv. Sci., 2019, 6, 1900547 CrossRef PubMed.
- H. Yang, L. Yang, H. Wang, Z. Xu, Y. Zhao, Y. Luo, N. Nasir, Y. Song, H. Wu, F. Pan and Z. Jiang, Nat. Commun., 2019, 10, 2101 CrossRef PubMed.
- J. Shen, R. Zhang, Y. Su, B. Shi, X. You, W. Guo, Y. Ma, J. Yuan, F. Wang and Z. Jiang, J. Mater. Chem. A, 2019, 7, 18063–18071 RSC.
- Y. Hu, N. Dunlap, S. Wan, S. Lu, S. Huang, I. Sellinger, M. Ortiz, Y. Jin, S.-H. Lee and W. Zhang, J. Am. Chem. Soc., 2019, 141, 7518–7525 CrossRef CAS PubMed.
- H. Chen, H. Tu, C. Hu, Y. Liu, D. Dong, Y. Sun, Y. Dai, S. Wang, H. Qian, Z. Lin and L. Chen, J. Am. Chem. Soc., 2018, 140, 896–899 CrossRef CAS PubMed.
- Q. Xu, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 4557–4563 CrossRef CAS PubMed.
- Z. Guo, Y. Zhang, Y. Dong, J. Li, S. Li, P. Shao, X. Feng and B. Wang, J. Am. Chem. Soc., 2019, 141, 1923–1927 CrossRef CAS PubMed.
- X. Li, Q. Hou, W. Huang, H.-S. Xu, X. Wang, W. Yu, R. Li, K. Zhang, L. Wang, Z. Chen, K. Xie and K. P. Loh, ACS Energy Lett., 2020, 5, 3498–3506 CrossRef CAS.
- Z. Cheng, M. Xie, Y. Mao, J. Ou, S. Zhang, Z. Zhao, J. Li, F. Fu, J. Wu, Y. Shen, D. Lu and H. Chen, Adv. Energy Mater., 2020, 10, 1904230 CrossRef CAS.
- W. Sun, J. Zhang, M. Xie, D. Lu, Z. Zhao, Y. Li, Z. Cheng, S. Zhang and H. Chen, Nano Lett., 2020, 20, 8120–8126 CrossRef CAS PubMed.
- X. Li, Y. Tian, L. Shen, Z. Qu, T. Ma, F. Sun, X. Liu, C. Zhang, J. Shen, X. Li, L. Gao, S. Xiao, T. Liu, Y. Liu and Y. Lu, Adv. Funct. Mater., 2021, 31, 2009718 CrossRef CAS.
- B. Zhou, J. Le, Z. Cheng, X. Zhao, M. Shen, M. Xie, B. Hu, X. Yang, L. Chen and H. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 8198–8205 CrossRef CAS PubMed.
- L. Cao, H. Wu, Y. Cao, C. Fan, R. Zhao, X. He, P. Yang, B. Shi, X. You and Z. Jiang, Adv. Mater., 2020, 32, 2005565 CrossRef PubMed.
- S. Chandra, T. Kundu, K. Dey, M. Addicoat, T. Heine and R. Banerjee, Chem. Mater., 2016, 28, 1489–1494 CrossRef CAS.
- N. Tahir, F. Muniz-Miranda, J. Everaert, P. Tack, T. Heugebaert, K. Leus, L. Vincze, C. V. Stevens, V. Van Speybroeck and P. Van Der Voort, J. Catal., 2019, 371, 135–143 CrossRef CAS.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- H. Hu, Q. Yan, M. Wang, L. Yu, W. Pan, B. Wang and Y. Gao, Chin. J. Catal., 2018, 39, 1437–1444 CrossRef CAS.
- Y. Du, B. Xu, J. Pan, Y. Wu, X. Peng, Y. Wang and S. Zhang, Green Chem., 2019, 21, 4792–4799 RSC.
- B. J. Yao, W. X. Wu, L. G. Ding and Y. B. Dong, J. Org. Chem., 2021, 86, 3024–3032 CrossRef CAS PubMed.
- K. Kim, O. Buyukcakir and A. Coskun, RSC Adv., 2016, 6, 77406–77409 RSC.
- H. Y. Zuo, Y. Li and Y. Z. Liao, ACS Appl. Mater. Interfaces, 2019, 11, 39201–39208 CrossRef CAS PubMed.
- A. M. Kaczmarek, Y. Y. Liu, M. K. Kaczmarek, H. S. Liu, F. Artizzu, L. D. Carlos and P. Van Der Voort, Angew. Chem., Int. Ed., 2020, 59, 1932–1940 CrossRef CAS PubMed.
- L. P. Zhai, W. T. Wei, B. W. Ma, W. Y. Ye, J. Wang, W. H. Chen, X. B. Yang, S. W. Cui, Z. J. Wu, C. Soutis, G. S. Zhu and L. W. Mi, ACS Mater. Lett., 2020, 2, 1691–1697 CrossRef CAS.
- Y. Zhang, W. X. Zhang, Q. Z. Li, C. Chen and Z. C. Zhang, Sens. Actuators, B, 2020, 324, 128733 CrossRef CAS.
- H. Singh, M. Devi, N. Jena, M. M. Iqbal, Y. Nailwal, A. De Sarkar and S. K. Pal, ACS Appl. Mater. Interfaces, 2020, 12, 13248–13255 CrossRef CAS PubMed.
- N. Zhang, T. S. Wang, X. Wu, C. Jiang, F. Chen, W. Bai and R. K. Bai, RSC Adv., 2018, 8, 3803–3808 RSC.
- A. Mal, R. K. Mishra, V. K. Praveen, M. A. Khayum, R. Banerjee and A. Ajayaghosh, Angew. Chem., Int. Ed., 2018, 57, 8443–8447 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |