
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubphthalocyanine capsules: molecular reactors for photoredox transformations of fullerenes†
Marta
Moreno-Simoni
a,
Tomás
Torres
 *abc and
Gema
de la Torre
*ab
*abc and
Gema
de la Torre
*ab
aOrganic Chemistry Department, Universidad Autónoma de Madrid, Campus de Cantoblanco, 28049-Madrid, Spain
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Campus de Cantoblanco, 28049-Madrid, Spain. E-mail: gema.delatorre@uam.es
cIMDEA-Nanociencia, C/Faraday 9, 28049-Madrid, Spain
First published on 19th July 2022
Abstract
The internal cavity formed by a dimeric subphthalocyanine (SubPc) capsule (SubPc2Pd3, 2), ensembled by coordination of pyridyl substituents in the monomeric SubPc 1 to Pd centers, has proved an optimal space for the complexation of C60 fullerene. Taking advantage of the intense absorption of green light of the SubPc component at around 550 nm, we have tested different green-light induced photoredox addition reactions over the double bonds of guest C60. Both addition of amine radicals, generated by reductive quenching of the excited state of 2 by aromatic trimethylsilylamines, and addition of trifluoroethyl radicals, obtained from oxidative quenching of the photosensitizer, have successfully taken place with good yields in the 2:C60 host:guest complex. On the other hand, both the photoredox reactions result in much lower yields when the monomeric pyridyl-SubPc is used as a photocatalyst, demonstrating that encapsulation results in a strong acceleration of the reaction. Importantly, this is the first example of the use of a confined microenvironment to trigger photoredox chemical transformations of fullerenes.
Introduction
The use of cage-like host systems as catalysts to perform reactions in confined microenvironments has become the centre of attention since the central cavity of molecular capsules is reminiscent of an enzyme-active site.1 In particular, supramolecular coordination cages, easily prepared through modular approaches from metal ions and organic ligands, provide discrete microenvironments to bring the substrates together within the cavity, increasing the local concentration and triggering or accelerating chemical transformations.2On the other hand, the use of chromophores that absorb visible light and trigger reactions after their excitation is central for the search of “green” synthetic methods.3 The combination of the two fields, that is the formation of coordination cages by self-assembly of chromophores with photosensitizing abilities, offers powerful possibilities since it provides a local concentration of photoactive units surrounding a bound guest in a way that would not be possible in solution under normal conditions, affording an antenna effect in which excitation energy from several sources is directed to a single reaction site. Although there are examples in the literature of “antenna” cages performing as photocatalysts,4,5 most of them are focused on the production of hydrogen or the generation of reactive oxygen species, and the realization of more complex photoredox transformations remains largely unexplored.
The development of chemical reactions under photoredox catalysis is a central research field that has allowed us to overcome issues, such as decomposition or the lack of control over the reaction, associated with the direct excitation of organic substrates with high energy UV-light.6 Although Ir(III) and Ru(III) polypyridyl complexes are powerful visible-light catalysts for photoredox transformations, their low sustainability and potential toxicity have prompted the search for organic chromophores able to trigger electron transfer processes after photoexcitation with low-energy photon sources such as green or red light.7 Phthalocyanines (Pcs) and their lower homologs subphthalocyanines (SubPcs) have been addressed as optimal candidates for photoinduced electron transfer processes after excitation with visible light.8 In particular, the bowl-shaped SubPcs (Fig. 1)9 show intense transitions in the 550–630 nm region, and once photoexcited, they can act either as electron donors or acceptors as a function of the nature of the peripheral substituents. For that reason, they have been incorporated into multicomponent systems with other electroactive units to build photosynthetic models and advanced energy materials.10 Electron-acceptor fullerenes are by far the most widely used units merged with SubPcs, particularly in a supramolecular manner, due to the shape complementarity between the concave face of SubPcs and the spherical π-surface of the fullerenes.11 An outstanding illustration of this type of interaction is that exhibited by the SubPc-based coordination cage 2 reported by our group (Fig. 1), formed by Pd-directed dimerization of two C3-symmetry SubPcs 1 endowed with three peripheral 3-pyridyl units,12 which form stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complexes with C60 and C70.13,14
:1 host–guest complexes with C60 and C70.13,14
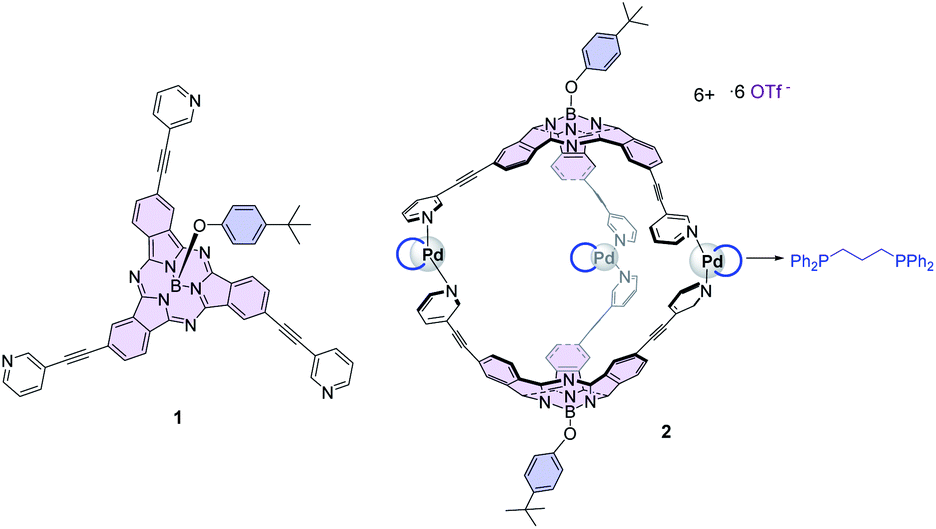 | ||
| Fig. 1 Structure of Pd3(SubPc)2 cage 2 and its precursor 1. | ||
The recognition of fullerenes using metallo-organic cages constructed with other aromatic panels has also been accomplished by several authors.15–25 The shape complementarity and space confinement in the host:guest complexes have a role in the reactivity of fullerenes, allowing reactions that do not proceed outside the cage,15 or the control of the formation of different types of addition compounds.19,26–29 However, the use of a confined microenvironment to trigger photoredox reactions over fullerenes has not yet been accomplished. In this regard, the use of SubPc cage 2 as an “antenna” pocket to induce photoredox transformations on fullerenes triggered by the surrounding SubPc excited states is a ground-breaking and conceptually innovative approach to obtain fullerene species that are not accessible by other means and can be of interest in areas such as solar energy conversion10 or biomedicine.30
Despite their well-recognized abilities as photosensitizers for different applications, there are only a few examples in the literature of Pcs31,32 and SubPcs33 working as photoredox catalysts for organic transformations; in particular, fluorinated SubPcs have been recently tested as photocatalysts for radical additions of alkenes.33 Our aim here is to take advantage of the strong interactions between the bowl-shaped SubPc units and the fullerene in the readily formed host:guest complexes in solution to trigger green-light induced photoredox addition reactions over the double bonds of C60. As we will show in the following, confinement of C60 in the void space of 2 is essential to effectively perform the radical addition. Importantly, the internal dimensions of the cavity (dPd–Pd = 18.4 Å; dB–B = 16.1 Å) may allow for co-binding of the reactive species.
Results and discussion
To prove that this SubPc cage can act as a molecular container for photoredox transformations, we have selected the addition of α-aminoalkyl radicals as a model reaction. Classically, reactions of C60 under high energy UV light irradiation with tertiary amines involve single electron transfer (SET) from the amines to the photoexcited C60, leading to radicals that enable addition reactions to form different fullerene derivatives.34–40 We have selected diphenyl(trimethylsilylmethyl)amine40 as a radical precursor to demonstrate the abilities of capsule 2 as a molecular photoreactor to accomplish addition reactions over fullerenes under benign green light illumination through SubPc-triggered SET processes. Importantly, to the best of our knowledge, this is the first time that a porphyrinoid cage works as a container for photocatalyzed fullerene derivatization. Then, with a protocol in hand, we have widened the applicability of our photocatalytic cage to other pioneering photocatalyzed radical additions over fullerenes.Initially, the reaction was performed with 1.5 equivalents of diphenyl(trimethylsilylmethyl)amine 3a, using toluene (a good solvent for C60) in the presence of 1% H2O as a source of H+ (necessary for the protonation of the intermediate anionic fulleroid, see the mechanism below) under continuous 530 nm irradiation, but in the absence of a photosensitizer. Under these conditions, as expected, no reaction took place due to the low energy of the light source (Table 1, entry 1). Before starting the photocatalyzed experiments, the stability of SubPc 1 and cage 214 under green light LED irradiation for periods of up to 12 h was confirmed. Then, the photocatalyzed reaction was set using 1 equivalent of SubPc cage 2 to ensure maximum complexation of C60 in the interior (Table 1, entry 2). After 9 h of reaction at rt, a fullerene derivative was isolated in 83% yield, with 1H-NMR and MS features that match with the addition of the α-diphenylaminomethylene radical (Fig. S1†). The 1,2-addition pattern of 4a was confirmed in the UV-vis spectrum, which shows a weak transition at 435 nm, characteristic for this type of adduct (Fig. S4†).
|
|
||||
|---|---|---|---|---|
| Entry | PS (equiv.) | Time (h) | Solvent | Yield |
| a PS = Photosensitizer. Conditions: C60 (0.003 mmol) 0.08 M, 3a (1.5 equiv.), at rt with irradiation by a 530 nm LED under Ar. Isolated yields of 4a. b 2 equiv. of 3a. | ||||
| 1 | — | 9 | Toluene | — |
| 2 | 2 (1.0) | 9 | Toluene | 83% |
| 3 | 1 (1.0) | 9 | Toluene | 20% |
| 4 | 2 (1.0) | 9 | DCM | 87% |
| 5b | 2 (1.0) | 9 | DCM | 75% |
| 6 | 2 (0.1) | 9 | DCM | 31% |
| 7 | 2 (1.0) | 6 | DCM | 86% |
| 8 | 2 (1.0) | 3 | DCM | 72% |
| 9 | 2 (1.0) | 1.5 | DCM | 43% |
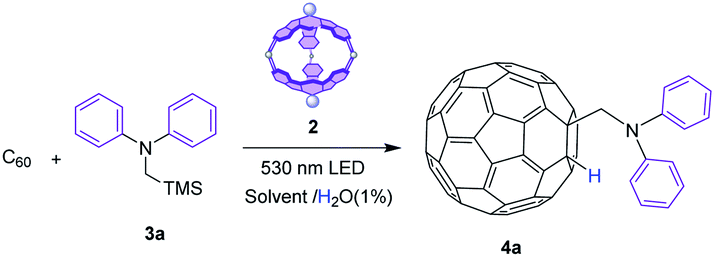
To determine if encapsulation is necessary for the reaction to proceed, SubPc 1 was tested as a photosensitizer (Table 1, entry 3). In this case, the addition product 4a was isolated in only 20% yield, which evidences the benefits of performing the reaction in a confined space. Another important conclusion from this experiment is that the SubPc chromophore is responsible for triggering the photoredox reaction, ruling out the possibility of Pd(II) behaving as catalytic centers. The only role of the metals is to bind the two SubPc units, although they also provoke a slight deactivation of the SubPc excited state (Fig. S2†), which does not otherwise reduce the photocatalytic activity of the cage.
To maximize the formation of the host–guest complex, we decided to change the reaction solvent to CH2Cl2, which is a poorer solvent for C60. In fact, binding constants have been derived from UV-vis titration experiments with values as high as 4.7 × 105 M−1 (Fig. S11†). It is also important to mention here that although SubPc 1 interacts with C60 also in CH2Cl2, binding isotherms (Fig. S12†) reveal poor fitting and lower binding constants (ca. 104). This finding points out again the importance of the confined microenvironment for the reaction to take place smoothly, which could probably be a consequence of the increased local concentration of the reactants inside the cavity and/or a larger antenna effect caused by the two SubPc units in cage 2.
Then, when the reaction was carried out in CH2Cl2 (entry 4), slightly higher yields of the isolated product (i.e., 87%) were achieved. However, when the amount of amine 3a was increased (entry 5), the yield of 4a decreased, while other products appeared in the reaction mixture, showing molecular peaks in the mass spectra that correspond with double additions of the amine radical to the fullerene (Fig. S3†). Finally, the amount of 2 was reduced to 10 mol% with respect to C60, but the yield of 4a dropped to 31% (entry 6) which may be probably caused by the competitive inhibition by the product, i.e., involvement of the resulting adduct 4a in the host–guest equilibrium with the SubPc cage, which hampers the complexation of pristine fullerene, as in previous examples of temperature-promoted reaction of fullerenes in metallocages.19,26–29 To demonstrate the inhibition by the product, we accomplished the assessment of the binding constants for the host:guest complex formation between 4a and 2. However, the insolubility of the adduct in CH2Cl2 (and also in toluene) prevented the realization of the experiments. Yet, it must be considered that this insolubility may be the driving force that pushes the adduct 4a to remain in the SubPc capsule (i.e., solvophobic effect). Importantly, although optimal results are achieved using 1 equiv. of the cage, 80% of the initial amount of 2 is recovered after treatment of the reactions, and it performed with the same efficiency in further photoredox cycles. Eventually, an assessment of the reaction kinetics was performed (entries 4 and 7–9), measuring the yields of 4a at different reaction times. As shown in Fig. 2, the reaction undergoes a strong acceleration in the first three hours, and then it slows down until a plateau is reached (i.e., 6 h) after which the yield remains stable.
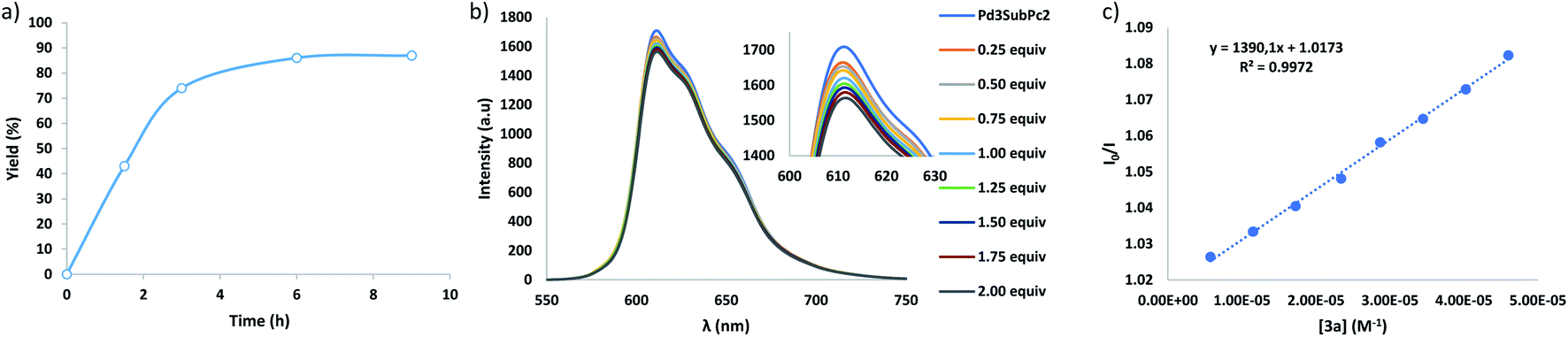 | ||
| Fig. 2 (a) Kinetic profile for the conversion of C60 in 4a; (b) fluorescence quenching of 2 (2.14 × 10−5M in CH2Cl2) upon addition of increasing amounts of 3a; (c) plot of the fluorescence quenching of 2versus [3a] and determination of the Stern–Volmer constant (I/I0 = 1 + KSV·x). | ||
With a general protocol in place (CH2Cl2, 1 equiv. of 2, 1.5 equiv. of amine vs. C60 and 6 h green-light irradiation at rt) we searched to broaden the scope of the reaction using different α-silylamines (Table 2). For the reactions with amines functionalized with strong or moderate electron donors (3b–d), the yields of the corresponding adducts 4b–e were moderately lower than those of the model reaction. Similar yields were achieved with other types of electron-rich aromatic amines (3f). Interestingly, the reaction with the NO2- functionalized silylamine 3e did not work out, which can be rationalized considering the proposed mechanism (see below). Finally, group substitution of one phenyl ring with an alkyl radical (amine 3g) led to yield reduction (i.e., 52%), while the use of dialkylamine 3h resulted in full inactivation of the process. Characterization of 4b–g is provided in the ESI (Fig. S7–S10)†.
| a Conditions: C60 (0.003 mmol) 0.08 M, 3b–h (1.5 equiv.), 2 (1 equiv.), at rt with irradiation by a 530 nm LED under Ar. |
|---|
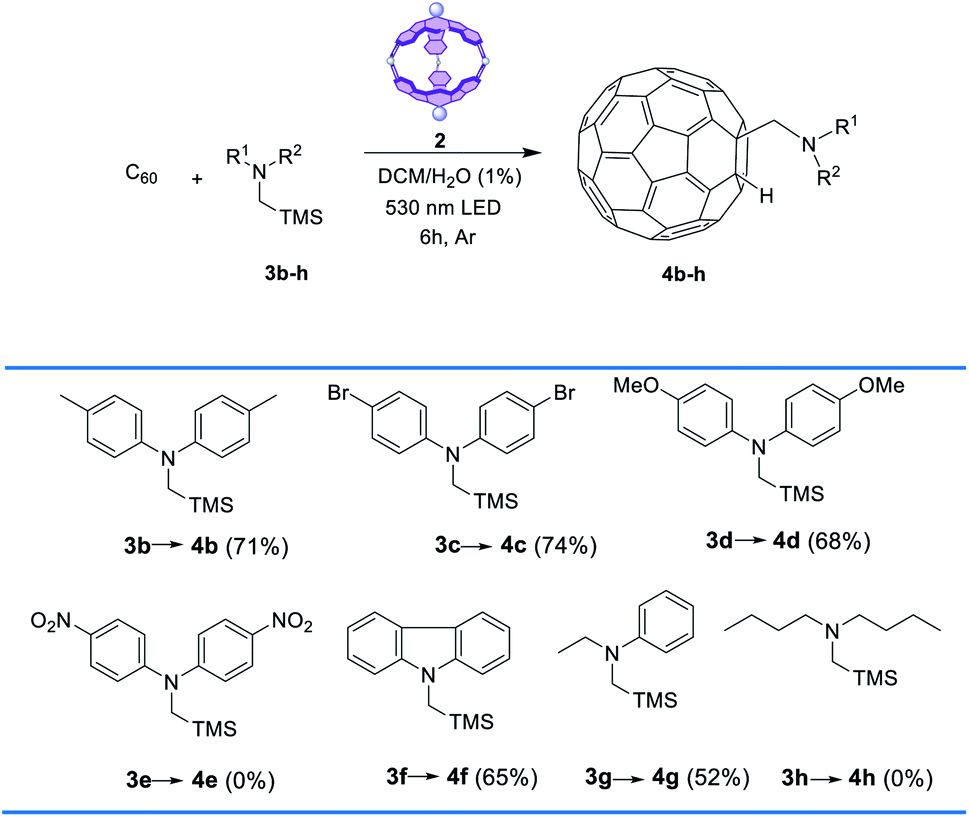
|
With a general protocol in place, the next step was the elucidation of the mechanism using the model reaction. Previous photophysical studies on C60 ⊂ 2 complexes concluded that after excitation of the SubPc, no SET to C60 was observed but energy transfer occurred,14 which excludes the oxidative quenching of 2 by C60. Nevertheless, the formation of a radical species in the herein described transformation is confirmed by the absence of reaction when it was carried out in the presence of 2 equiv. of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) radical scavenger, which yielded the diphenylmethylamine-TEMPO product by radical coupling (Fig. S13†).41 Therefore, the formation of the diphenylmethylamine radical species could be rationalized by initial SET from 3a to the photoexcited SubPc units in 2. This hypothesis was supported by Stern–Volmer fluorescence quenching experiments, which evidence that the amine 3a quenches the excited state of 2 (Fig. 2). Considering the high degree of occupancy of cage 2 with C60 under the reaction conditions, we have also performed Stern–Volmer experiments using a C60 ⊂ 2 complex synthesized by assembly of the cage in the presence of fullerene.14 As expected, the results derive a similar KSV (Fig. S15†).
To confirm the deactivation of the excited state of the cage by the amine, a reaction was set with a 1:1 mixture of 2 and 3a under green LED photoirradiation for 6 h, in the absence of C60, which led to the conversion of the trimethylsilylamine to Me3SiOH (Fig. S9†). This result is rationalized by the reductive quenching of the photoexcited SubPc units by 3a, leading to the radical cation form of the amine that suffers desilylation assisted by water to give an α-diphenylaminomethyl radical (Fig. 3). In an additional control reaction, a solution of 3a in DCM/H2O was irradiated, which provided the amine unaltered (Fig. S9†).
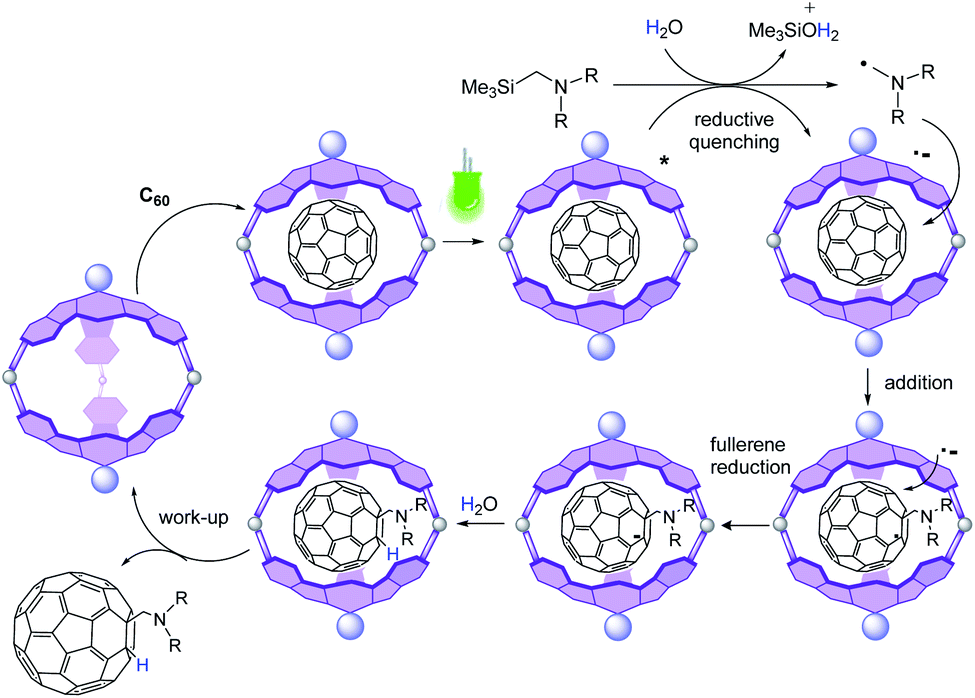 | ||
| Fig. 3 Postulated photoredox mechanism for the conversion of C60 to 4a. | ||
The reductive quenching of 2 by diphenyl(trimethylsilylmethyl) amine is plausible considering that the excited state reduction potential of 2 (0.771 eV vs. SCE, calculated from E*red = Ered(2/2−) + E00) exceeds the oxidation potential of 3a (0.471 eV vs. SCE) (Fig. S16 and 17†). Please note that the oxidation potential of amine 3e is 1.07 eV (Fig. S17†), which explains why the reaction with 3e failed to proceed. All these calculations are done considering the singlet as the involved excited state in the photoredox reaction, since the triplet energy (1.45 eV) is too low to be involved in the oxidation of the amine.
Then, the more plausible pathway is that after reductive quenching, addition of the α-aminomethyl radical to the fullerene gives a fulleroid radical, which can be further reduced by SET from the one-electron reduced form of 2 (2−) to give an anionic fullerene that is further protonated (Fig. 3). This last step of the catalytic cycle is supported by the fact that no reaction occurs in the absence of water. Moreover, the signal of the added H does not appear in the 1H-NMR spectrum when the reaction is carried out in DCM/D2O mixtures (Fig. S19†). Importantly, other pathways derived from the C60 excited state are excluded since the reaction between C60 and amine 3a under green LED irradiation does not take place in the absence of a photocatalyst (Table 1, entry 1).
After these outstanding results, we aimed to prove that SubPc cage 2 is a versatile molecular container for other types of photoredox reactions with fullerenes. Considering the increasing interest in fluoroalkyl groups for several applications, including medicinal chemistry,30 fluoroalkylation reactions over fullerenes are relevant targets of research.42 Radical addition of fluoroalkyl radicals to fullerenes has been achieved by thermal or photochemical decomposition of fluoroalkyl iodides. However, to the best of our knowledge, the photocatalyzed fluoroalkyl addition using low-energy light sources has not been undertaken yet.
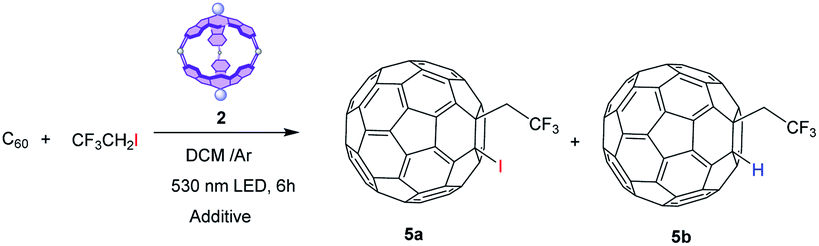
Among the different photocatalyzed fluoroalkylation reactions, we have selected the straightforward trifluoroethylation with CF3CH2I as a model reaction. The photocatalyzed reaction of fluorinated alkyl iodides with alkenes can yield atom transfer radical additions (ATRA), where the final alkene bears the two functionalities (i.e., CF3 and I),43 or hydrofluoralkylated compounds.44 Then, we have accomplished the photocatalytic activation of CF3CH2I by our cage 2 to generate radicals45 that subsequently react with fullerene to give 5a or 5b (Table 2).
As the high reduction potential of CF3CH2I (−1.4 eV vs. SCE) rules out the possibility of forming the trifluoromethylene radical by oxidative quenching of photoexcited 2 (excited state oxidation potential of SubPc cage 2 is −0.92 eV), the addition of sacrificial reductants seems necessary. After testing different additives (see Table 3), we found that the addition of 1 equiv. of PPh346 (entry 3) resulted in the formation of hydrotrifluoroethylated fullerene 5b (Fig. S13†) in 65% yield. The formation of this product requires hydrogen-atom transfer (HAT) from a hydrogen donor, most likely the solvent (CH2Cl2). Noteworthily, the formation of 5a was not detected under any of the tested conditions, probably because the incorporation of the iodide requires oxidation of the fullerene radical that results from the trifluoromethylene addition into a carbocation, and this step requires a strongly oxidative catalytic species (see the mechanism below). As in previous hydroaminations, the reaction performed more efficiently when using equimolar amounts of SubPc cage 2 rather than catalytic amounts (entry 4), but it is important to remark that 75% of 2 was recovered after treatment of the reaction, and it proved effective in further photoredox reactions. Again, the need of the molecular cavity for the reaction to proceed efficiently was proved, as the reaction yield dropped to 9% when SubPc 1 was used as the catalyst. Finally, the reaction performed in the absence of a SubPc derivative was ineffective, which demonstrates that the photocatalyst is necessary for the reaction to take place (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | PS (equiv.) | Additive (equiv.) | Yield (5a) | Yield (5b) |
| a PS = Photosensitizer. Conditions: C60 (0.003 mmol) 0.08 M, CF3CH2I (1.5 equiv.) at rt with irradiation by a 530 nm LED under an Ar atmosphere. Isolated yields of 5a and 5b. | ||||
| 1 | 2 (1.0) | Lutidine (1.0) | — | — |
| 2 | 2 (1.0) | Sodium ascorbate/NaOAc (1.0) | — | — |
| 3 | 2 (1.0) | PPh3 (1.0) | — | 65% |
| 4 | 2 (0.2) | PPh3 (1.0) | — | 27% |
| 5 | 1 (1.0) | PPh3 (1.0) | — | 9% |
| 6 | — | PPh3 (1.0) | — | — |
In subsequent mechanistic investigations, we found that the addition of TEMPO completely inhibited the reaction, confirming the radical character of the reaction (see Fig. S22† for the 19F-NMR spectrum of the CF3CH2-TEMPO coupling product). Stern–Volmer experiments revealed that neither CF3CH2I nor PPh3 alone quenched the excited state of 2 (Fig. 4). However, fluorescence titration with an equimolecular mixture of CF3CH2I and PPh3 yielded quenching of the emission of 2, with a Stern–Volmer constant of 2995 M−1. This finding implies that coordination of phosphine to CF3CH2I may lower the reduction potential of the haloalkane.‡ Halogen bonding between the iodine atom in CF3CH2I and the Lewis base PPh3 is plausible,47 because the “sigma-hole” of the iodine atom is enhanced by the three electron-withdrawing fluorines. From this result, it could be derived that after excitation of 2, an oxidative quenching is caused by the CF3CH2I–PPh3 complex (Fig. 5). The resulting radical anionic species could split into PPh3, iodide and the trifluoroalkyl radical, whose addition to fullerene results in the formation of the trifluoroethylated fullerene radical. The oxidized form 2 (2+) does not appear to oxidize this radical to its carbocationic form; instead, in the absence of an extra hydrogen source, CH2Cl2 solvent can act as a hydrogen donor for subsequent HAT to fullerene. To confirm this hypothesis, the reaction was performed in CD2Cl2, which provided the deuterated analog 5b (Fig. S24†). Reduction of the oxidized form of the SubPc photocatalytic cage 2 should be completed by PPh3 (E1/2ox = 0.98 vs. SCE), which also plays the role of a sacrificial reductant.
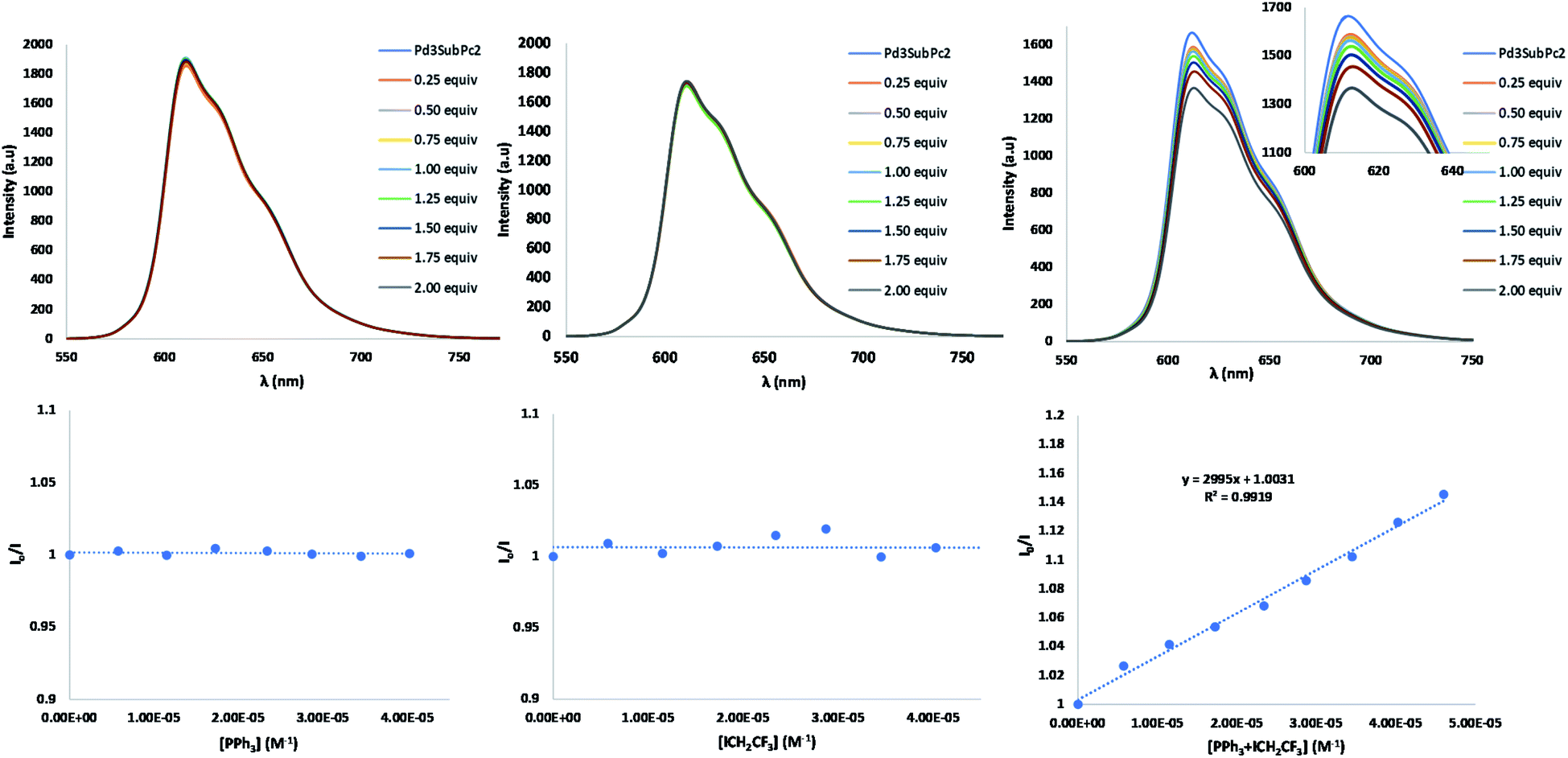 | ||
| Fig. 4 Fluorescence quenching and plot of the quenching of 2 (2.14 × 10−5M in CH2Cl2) versus: [PPh3], left column; [ICH2CF3], middle column; and [PPh3 + ICH2CF3], right column. Determination of the Stern–Volmer constant (I/I0 = 1 + KSV·x). | ||
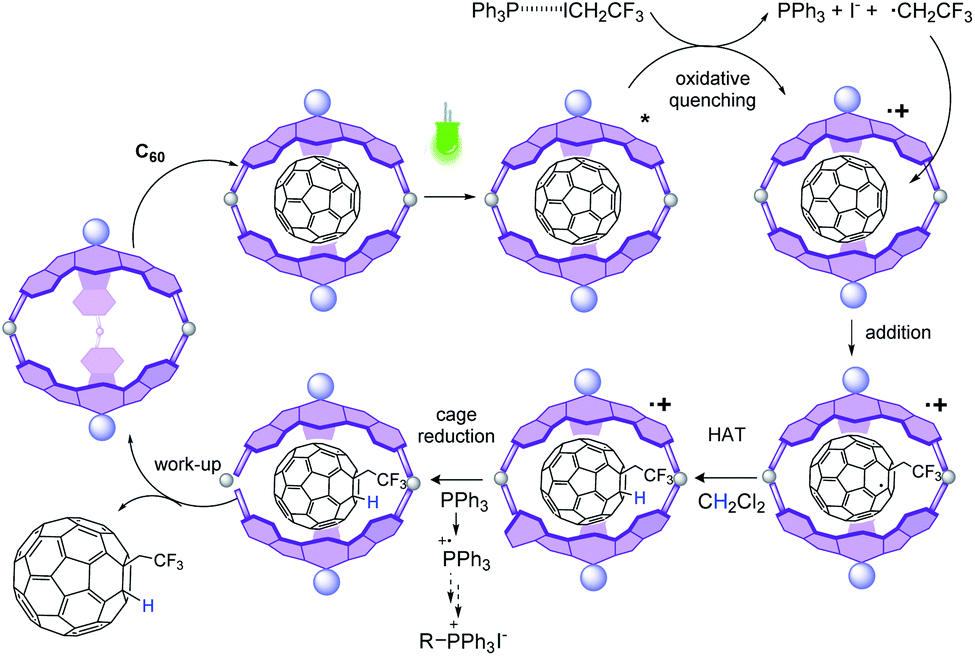 | ||
| Fig. 5 Postulated photoredox mechanism for the conversion of C60 to 5b. | ||
Conclusions
All the previous results indicate that SubPc capsule 2 is a powerful photosensitizer to trigger additions of (diaryl)methylamine and trifluoroethyl radicals to C60 fullerene. Taking advantage of the ready formation of host:guest complexes between 2 and C60 fullerene in CH2Cl2, the capsule behaves as a molecular photoreactor. Very remarkable is the versatility of the SubPc cage, which can initiate photoredox processes in the form of reductive or oxidative quenching. Probably due to the strong interaction between the C60 addition products and the SubPc cage, which do not allow a rapid exchange of the product by the pristine C60 substrate, equimolar amounts of 2 are necessary to maximize the yields. Despite that, the SubPc cage is recovered to a large extent after the reactions and can be used in further cycles. Undoubtedly, the reactions strongly benefit from being performed in the confined space, because when monomeric SubPc is used as a photocatalyst, the yields dropped considerably. This work is conceived as a proof-of-concept, demonstrating the capabilities of SubPc cages as molecular photoreactors, and setting the basis for the search for other “eco-friendly” green light-induced, photoredox transformations over fullerene species, which can provide interesting, functionalized fullerenes with applications in energy conversion schemes or biomedicine.Data availability
All data were given in ESI.†Author contributions
M. M.-S. performed all the the experimental work. T. T, coordinated the work. G. T. originated the central idea, coordinated the work, and analyzed the experimental data. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We want to dedicate this work to the memory of Christian G. Claessens, whose pioneering contribution to the synthesis of SubPc cages has allowed the development of the present research. Financial support from Spanish MINECO (PID2020-116490GB-I00 and PID2020-115801RB-C21) is acknowledged. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686).Notes and references
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- Y. Xue, X. Hang, J. Ding, B. Li, R. Zhu, H. Pang and Q. Xu, Coord. Chem. Rev., 2021, 430, 213656 CrossRef CAS.
- C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, Weinheim, Germany 2018 Search PubMed.
- T. Comerford, E. Zysman-Colman and M. D. Ward, in Reactivity in Confined Spaces, ed., R. S. Forgan and G. O Lloyd, The Royal Society of Chemistry, 2021, pp. 70–107 Search PubMed.
- H. Y. Lin, L. Y. Zhou and L. Xu, Chem.–Asian J., 2021, 16, 3805–3816 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Coord. Chem. Rev., 2021, 428, 213605 CrossRef CAS.
- C. G. Claessens, D. Gonzalez-Rodriguez, M. Salomé Rodriguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed.
- G. de la Torre, G. Bottari and T. Torres, Adv. Energy Mater., 2017, 7, 1–21 Search PubMed.
- G. Zango, M. Krug, S. Krishna, V. Mariñas, T. Clark, M. V. Martínez-Díaz, D. M. Guldi and T. Torres, Chem. Sci., 2020, 11, 3448–3459 RSC.
- C. G. Claessens and T. Torres, J. Am. Chem. Soc., 2002, 124, 14522–14523 CrossRef CAS PubMed.
- C. G. Claessens and T. Torres, Chem. Commun., 2004, 1298–1299 RSC.
- I. Sánchez-Molina, B. Grimm, R. M. Krick Calderon, C. G. Claessens, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2013, 135, 10503–10511 CrossRef PubMed.
- T. K. Ronson, B. S. Pilgrim and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 10417–10420 CrossRef CAS PubMed.
- W.-K. Han, H.-X. Zhang, Y. Wang, W. Liu, X. Yan, T. Li and Z.-G. Gu, Chem. Commun., 2018, 54, 12646–12649 RSC.
- E. Fazio, C. J. E. Haynes, G. de la Torre, J. R. Nitschke and T. Torres, Chem. Commun., 2018, 54, 2651–2654 RSC.
- C. García-Simón, A. Monferrer, M. Garcia-Borràs, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem. Commun., 2019, 55, 798–801 RSC.
- B. Chen, J. J. Holstein, S. Horiuchi, W. G. Hiller and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 8907–8913 CrossRef CAS PubMed.
- T. A. Barendt, W. K. Myers, S. P. Cornes, M. A. Lebedeva, K. Porfyrakis, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2020, 142, 349–364 CrossRef CAS PubMed.
- X. Chang, S. Lin, G. Wang, C. Shang, Z. Wang, K. Liu, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 15950–15960 CrossRef CAS PubMed.
- Y. Ni, F. Gordillo-Gámez, M. Peña Alvarez, Z. Nan, Z. Li, S. Wu, Y. Han, J. Casado and J. Wu, J. Am. Chem. Soc., 2020, 142, 12730–12742 CrossRef CAS PubMed.
- C. Fuertes-Espinosa, C. García-Simón, E. Castro, M. Costas, L. Echegoyen and X. Ribas, Chem.– Eur. J., 2017, 23, 3553–3557 CrossRef CAS PubMed.
- V. Martínez-Agramunt, T. Eder, H. Darmandeh, G. Guisado-Barrios and E. Peris, Angew. Chem., Int. Ed., 2019, 58, 5682–5686 CrossRef PubMed.
- D. M. Wood, W. Meng, A. K. Ronson, A. R. Stefankiewicz, J. K. M. Sanders and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 3988–3992 CrossRef CAS PubMed.
- V. Leonhardt, S. Fimmel, A.-M. Krause and F. Beuerle, Chem. Sci., 2020, 11, 8409–8415 RSC.
- C. Fuertes-Espinosa, C. García-Simón, M. Pujals, M. Garcia-Borràs, L. Gómez, T. Parella, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem, 2020, 6, 169–186 CAS.
- E. Ubasart, O. Borodin, C. Fuertes-Espinosa, Y. Xu, C. García-Simón, L. Gómez, J. Juanhuix, F. Gándara, I. Imaz, D. Maspoch, M. von Delius and X. Ribas, Nat. Chem., 2021, 13, 420–427 CrossRef CAS PubMed.
- N. Huang, K. Wang, H. Drake, P. Cai, J. Pang, J. Li, S. Che, L. Huang, Q. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 6383–6390 CrossRef CAS PubMed.
- E. Castro, A. Hernandez Garcia, G. Zavala and L. Echegoyen, J. Mater. Chem. B, 2017, 5, 6523–6535 RSC.
- Y. Katsurayama, Y. Ikabata, H. Maeda, M. Segi, H. Nakai and T. Furuyama, Chem.– Eur. J., 2022, 28, e202103223 CAS.
- Y. Ishikawa, T. Kameyama, T. Torimoto, H. Maeda, M. Segia and T. Furuyama, Chem. Commun., 2021, 57, 13594–13597 RSC.
- K. Matsuzaki, T. Hiromura, E. Tokunaga and N. Shibata, ChemistryOpen, 2017, 6, 226–230 CrossRef CAS PubMed.
- M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262–5321 CrossRef CAS PubMed.
- Y. Nakamura, M. Suzuki, K. Okawa, T. Konno and J. Nishimura, J. Org. Chem., 2005, 70, 8472–8477 CrossRef CAS PubMed.
- H. C. Jeong, S. H. Lim, D. W. Cho, S. H. Kim and P. S. Mariano, Org. Biomol. Chem., 2016, 14, 10502–10510 RSC.
- S. H. Lim, J. Yi, C. S. Ra, K. Nahm, D. W. Cho, G. Y. Lee, J. Kim, U. C. Yoon and P. S. Mariano, Tetrahedron Lett., 2015, 56, 3014–3018 CrossRef CAS.
- S. H. Lim, J. Yi, G. M. Moon, C. S. Ra, K. Nahm, D. W. Cho, K. Kim, T. G. Hyung, U. C. Yoon, G. Y. Lee, S. Kim, J. Kim and P. S. Mariano, J. Org. Chem., 2014, 79, 6946–6958 CrossRef CAS PubMed.
- H. C. Jeong, S. H. Lim, Y. Sohn, Y. Il Kim, H. Jang, D. W. Cho and P. S. Mariano, Tetrahedron Lett., 2017, 58, 949–954 CrossRef CAS.
- Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem.– Eur. J., 2014, 20, 6120–6125 CrossRef CAS PubMed.
- J. Y. Kim, Y. S. Lee, Y. Choi and D. H. Ryu, ACS Catal., 2020, 10, 10585–10591 CrossRef CAS.
- M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262–5321 CrossRef CAS PubMed.
- C. Rosso, J. D. Williams, G. Filippini, M. Prato and C. O. Kappe, Org. Lett., 2019, 21, 5341–5345 CrossRef CAS PubMed.
- S. Sumino, M. Uno, T. Fukuyama, I. Ryu, M. Matsuura, A. Yamamoto and Y. Kishikawa, J. Org. Chem., 2017, 82, 5469–5474 CrossRef CAS PubMed.
- J. R. Morton, F. Negri, K. F. Preston and G. Ruel, J. Phys. Chem., 1995, 99, 10114–10117 CrossRef CAS.
- V. S. Kostromitin, A. A. Zemtsov, V. A. Kokorekin, V. V. Levin and A. D. Dilman, Chem. Commun., 2021, 57, 5219–5222 RSC.
- Y. Xu, J. Huang, B. Gabidullin and D. L. Bryce, Chem. Commun., 2018, 54, 11041–11043 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental details, conditions for photoredox reactions, characterization of the new compounds and experiments for the elucidation of the mechanisms. See https://doi.org/10.1039/d2sc01931k |
| ‡ In a control reaction, it was checked that PPh3 and ICH2CF3 did not form the corresponding phosphonium derivative under these conditions, ruling out its participation in the reaction. |
| This journal is © The Royal Society of Chemistry 2022 |