
Progress in reaction mechanisms and catalyst development of ceria-based catalysts for low-temperature CO2 methanation
Yu
Xie
ad,
Junjie
Wen
ad,
Zonglin
Li
ad,
Jianjun
Chen
ad,
Qiulin
Zhang
 *ad,
Ping
Ning
ad,
Yaoqiang
Chen
c and
Jiming
Hao
*ab
*ad,
Ping
Ning
ad,
Yaoqiang
Chen
c and
Jiming
Hao
*ab
aFaculty of Environmental Science and Engineering, Kunming University of Science and Technology, Kunming 650500, PR China. E-mail: qiulinzhang_kmust@163.com
bState Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing 100084, PR China. E-mail: hjm-den@tsinghua.edu.cn
cCollege of Chemistry, Sichuan University, Chengdu 610064, PR China
dNational-Regional Engineering Center for Recovery of Waste Gases from Metallurgical and Chemical Industries, Kunming University of Science and Technology, Kunming 650500, PR China
First published on 14th December 2022
Abstract
With the development of carbon dioxide (CO2) capture, storage and utilization (CCSU) technologies, CO2 has gradually become a desired feedstock for the production of value-added chemicals like methane (CH4). Ceria (CeO2)-based catalysts have gained much attention because of their potential to efficiently hydrogenate CO2 to CH4 under mild conditions. Here we systematically outline the advances in CeO2-based catalysts for CO2 methanation mainly from the perspective of mechanism investigation and catalyst development. Various in situ/operando and ex situ technologies have verified that active metal and oxygen vacancies at the metal/metal oxide–CeO2 interface act as the prime active sites to promote the formation and hydrogenation of key intermediates during the CO2 methanation reaction. Kinetic analysis and in situ DRIFT characterization combined with theoretical calculations revealed that the reaction mechanism toward CO2 methanation is sensitive to active sites, and the formate route versus the carboxyl (CO*) route has been widely detected as the main methanation pathway over CeO2-based catalysts. Additionally, mainstream strategies to improve CeO2-based catalysts include optimizing reducibility, adjusting the distribution of basic sites, dispersing active metal supported on CeO2, and increasing the amount of oxygen vacancies or additional active sites for CO2 adsorption and selective hydrogenation into CH4. Finally, perspectives on the deeper understanding of active sites and intermediates’ evolution and the challenges of CeO2-based catalysts for CO2 methanation in future investigations are presented.
1. Introduction
The overutilization of fossil fuels has caused enormous amounts of CO2 to be emitted into the atmosphere over the past decades, contributing to a substantial waste of carbon feedstock and triggering a series of environmental concerns.1–4 Besides, the demand for natural gas in China is rising, driven by the coal-to-gas switching process.5 Confronting both of the above threats simultaneously has driven a lot of research on CO2 hydrogenation technologies for clean energy (i.e., methane and methanol) production.6–10 Concerning the hydrogenation of CO2 into methanol, the conversion of CO2 is about 20% even at 300 °C and 5 MPa,11–13 which does not contribute significantly to CO2 mitigation. For CO2 methanation, however, the CO2 conversion can reach 80% at 300 °C and the CH4 selectivity is up to 100%.14–16 Moreover, CH4 can be efficiently converted into methanol under a mild reaction condition (150 °C and 2 MPa).17 Therefore, the hydrogenation of CO2 to produce synthetic natural gas is a promising strategy to make a breakthrough on these above issues.Methane (CH4), as a major component of natural gas, is an important feedstock for fuel production.18 CO2 methanation, the so-called Sabatier reaction (CO2 + 4H2 → CH4 + 2H2O, ΔH298K = −165.0 kJ mol−1) is a highly exothermic process, which is favored at low temperatures.19–22 In addition, carrying out this reaction at a low temperature inhibits the endothermic reverse water–gas shift reaction (CO2 + H2 → CO + H2O, ΔH298K = 41.2 kJ mol−1),23 which effectively reduces the generation of by-product CO. The CO2 hydrogenation to CH4via heterogeneous catalysis has many advantages: (1) the CO2 methanation process can be integrated into industrial areas that produce substantial CO2 gas (e.g., coking and chemical plant, steel factory, cement factory, waste incineration plant, heavy crude refinery, etc.), due to the mature CO2 capture and storage (CCS) technologies; (2) the produced CH4 can be injected into the existing natural gas pipeline network for storage and transportation, which prevents CH4 from escaping into the atmosphere;24 (3) the required H2 can be generated by surplus renewable electricity (wind, solar, and hydro) via water electrolysis;25,26 (4) CO2 methanation can be performed at moderate temperatures and atmospheric pressure, which significantly reduces operating costs. In principle, the CO2 methanation technique is a promising power-to-gas concept for a sustainable future.
However, CO2 methanation is kinetically limited due to the chemical stability and highly C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond energy (806 kJ mol−1) of CO2 molecules.27 Hence, the current urgent demand is for the development of catalysts with superior activity for CO2 activation at low temperatures. Almost all the CO2 hydrogenation catalysts are developed as supported catalysts.28 Generally, active metal is the main active component, as it provides active sites for CO bond activation and dissociation of the H2 molecule. Existing studies have reported that noble metals (e.g., Rh, Pd, Ir, and Ru) or non-noble metals (e.g., Ni, Co, and Fe) loaded on various metal oxides (e.g., Al2O3, SiO2, ZrO2, TiO2, Y2O3, and CeO2, etc.) have been evaluated to be active for methanation.15,29–43 Among them, Ni-based catalysts have gained extensive attention because of their relatively high methane yields and cheaper prices than the noble ones.44 However, they are prone to deactivation due to coke formation and sintering at high temperatures.45 Ru-based catalysts are also a promising candidate for methanation applications, as they are more durable than Ni and less expensive than Rh, Pd, and Ir.46–48 Furthermore, it has been reported that the support may affect the reaction mechanism by adjusting surface properties and promoting metal dispersion, and thus govern catalytic performance.49 Hence, it is instructive to systematically understand the function of the support, specifically to account for the complex metal–support interaction (MSI).
O bond energy (806 kJ mol−1) of CO2 molecules.27 Hence, the current urgent demand is for the development of catalysts with superior activity for CO2 activation at low temperatures. Almost all the CO2 hydrogenation catalysts are developed as supported catalysts.28 Generally, active metal is the main active component, as it provides active sites for CO bond activation and dissociation of the H2 molecule. Existing studies have reported that noble metals (e.g., Rh, Pd, Ir, and Ru) or non-noble metals (e.g., Ni, Co, and Fe) loaded on various metal oxides (e.g., Al2O3, SiO2, ZrO2, TiO2, Y2O3, and CeO2, etc.) have been evaluated to be active for methanation.15,29–43 Among them, Ni-based catalysts have gained extensive attention because of their relatively high methane yields and cheaper prices than the noble ones.44 However, they are prone to deactivation due to coke formation and sintering at high temperatures.45 Ru-based catalysts are also a promising candidate for methanation applications, as they are more durable than Ni and less expensive than Rh, Pd, and Ir.46–48 Furthermore, it has been reported that the support may affect the reaction mechanism by adjusting surface properties and promoting metal dispersion, and thus govern catalytic performance.49 Hence, it is instructive to systematically understand the function of the support, specifically to account for the complex metal–support interaction (MSI).
As an easily accessible rare-earth material, ceria (CeO2) exhibits significant superiority as a support for methanation catalysts, because of its highly tunable nature involving MSI and oxygen vacancy distribution.50–58 In addition, CeO2 can modify the reducibility and surface properties of catalysts due to its ability to interact with other components.45,59–64 We used the keywords “CeO2” and “methanation” to search citation reports in Web of Science. As depicted in Fig. 1a, both the number of publications and citations have increased between 2016 and 2021, suggesting that CeO2-based catalysts are an ongoing research hotspot in the methanation field. Of note, Ru/CeO2 and Ni/CeO2 catalysts have been widely reported as promising candidates for industrial purposes due to their outstanding low-temperature methanation activity.19,32,65–70 However, the essential understanding of the reaction mechanism and structure–activity relationship between Ru/Ni and CeO2 remains limited, which impedes the development of new catalysts.
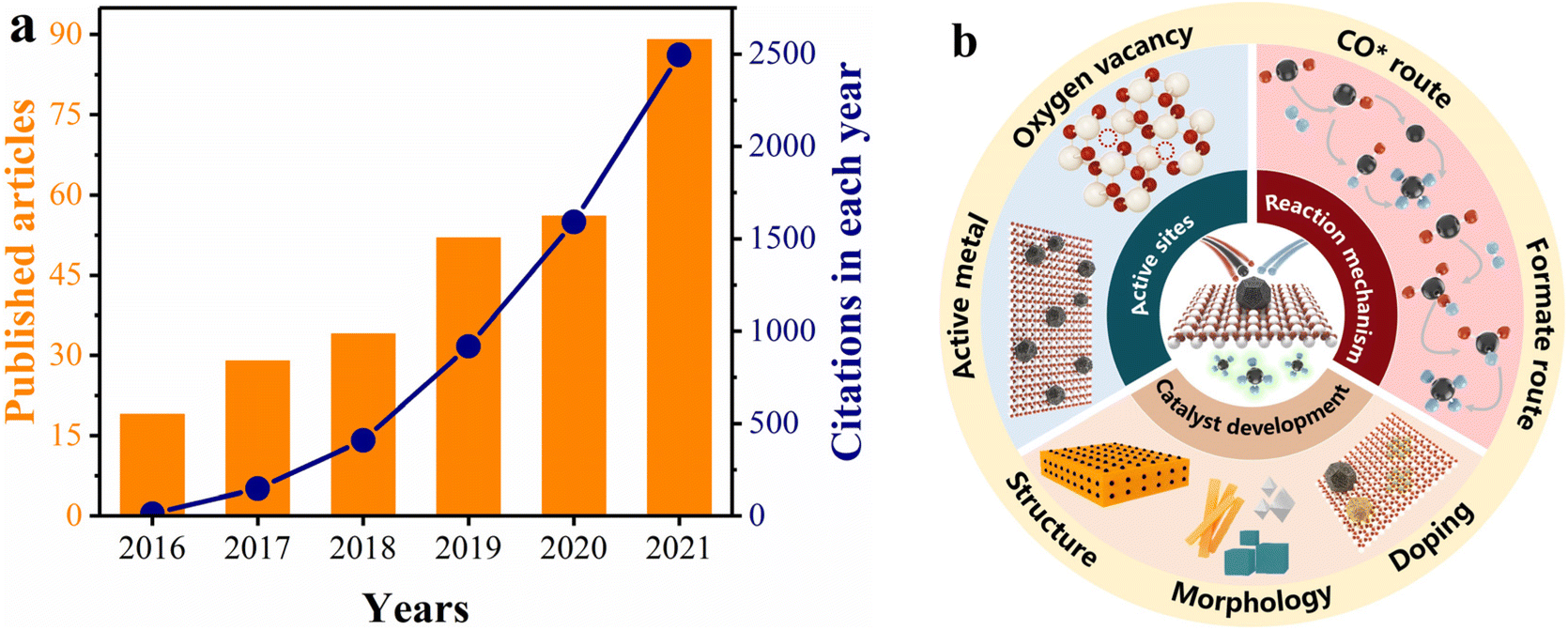 | ||
| Fig. 1 (a) The citation report from 2016 to 2021 in Web of Science for CeO2-based catalysts in methanation application. (b) Schematic illustration of active sites, reaction mechanism, and catalyst development for CO2 methanation. | ||
Several excellent reviews have been reported on CO2 methanation, covering methanation catalytic technology (e.g., thermocatalysis, photocatalysis, and plasma catalysis),71–73 catalyst development (including monometallic and bimetallic catalysts, alloys, and functionalized support),9,17,74,75 reaction mechanisms,76 product selectivity,77–79 and reactor technologies.73 Nevertheless, the role played by CeO2 in CO2 methanation catalysts has rarely been reviewed in detail. A comprehensive understanding is essential, considering that many valuable research articles on CeO2-based catalysts for CO2 methanation have been reported in the past few years.28,67,80
The specific objective of this review is to determine how the types of active sites, surface properties, and MSI of the Ru/CeO2 and Ni/CeO2 catalysts impact the reaction routes, and thus the catalytic performance. We discuss up-to-date findings about the promotion effects of CeO2 for low-temperature methanation and insight into the structure–activity relationship and reaction mechanism. This review is organized following the interaction between CeO2 and H2/CO2, the identification of active sites, the influence of MSI, methanation mechanisms involving adsorption and hydrogenation of the reactants, and the strategies for CeO2-based catalyst modification (Fig. 1b). Subsequently, the perspective and suggestions for further understanding the CO2 methanation mechanism and catalyst development of CeO2-based materials are discussed.
2. Understanding ceria-based catalysts for CO2 methanation
2.1. Interaction of CeO2 with H2 and CO2
The redox process with oxygen storage and release over CeO2 is one of the most interesting behaviors in the CO2 hydrogenation reaction. Schweke et al.81 performed a complementary temperature programmed reduction/desorption (TPR/TPD) measurement to identify the interaction of CeO2 with H2. They observed two peaks of H2 consumption assigned to the generation of surface and bulk oxygen vacancies at 350 °C and 700 °C, respectively, and found that the H2 consumption occurred concomitantly with the H2O desorption. Generally, the stoichiometric CeO2 with a face-centered cubic (FCC) structure (Fig. 2Ia) is unstable in an H2-rich environment. The dislodgment of an O atom under an H2 atmosphere would leave two electrons to reduce the adjacent two Ce4+ to Ce3+ (Fig. 2Ib).82 This process triggers the generation of oxygen vacancies due to the electrostatic balance mechanism.83 It is generally accepted that the oxygen vacancy plays a critical role in the adsorption and activation of the oxygen-containing molecule, i.e., CO2.84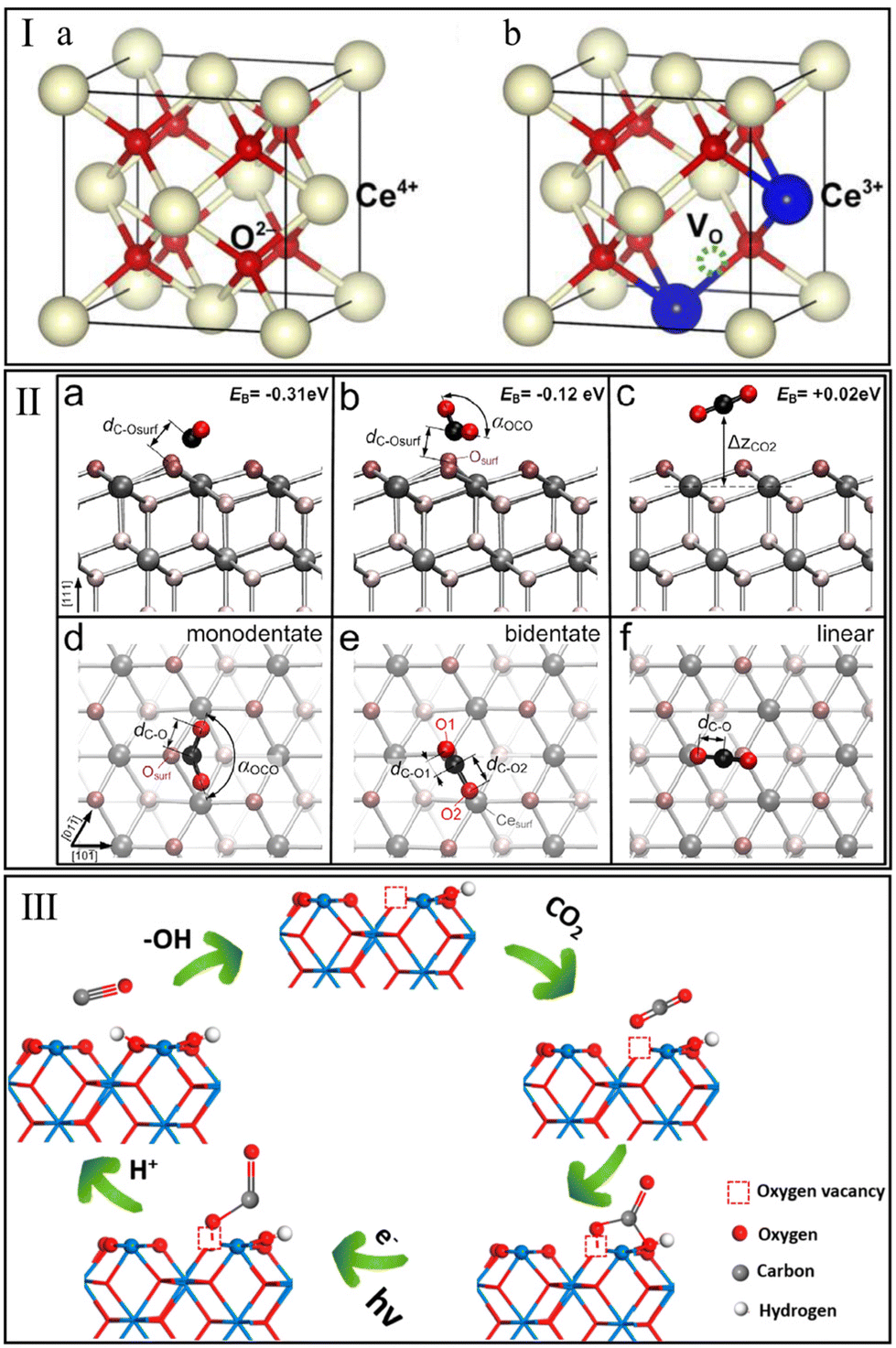 | ||
| Fig. 2 (I) Ideal face-centered cubic structure of CeO2 (a) and crystal structure of CeO2 with one oxygen vacancy (b). Reproduced from ref. 82 with permission from Elsevier, copyright 2018. (II) Stable configurations of CO2 molecule adsorbed on a CeO2(111) facet (a–c cross-sectional and d–f top view). Reproduced from ref. 86 with permission from American Chemical Society, copyright 2013. (III) Adsorption and photoreduction of CO2 at oxygen vacancy and OH site. Reproduced from ref. 89 with permission from American Chemical Society, copyright 2020. | ||
Understanding the interaction of CO2 with CeO2 is of high importance for CO2 hydrogenation. In 2014, Yoshikawa et al.85 performed CO2 pulse injection measurements and found that the CeO2-based adsorbents showed the strongest CO2 adsorption capacity among various single metal oxides (Al2O3, SiO2, and ZrO2). In situ FT-IR spectroscopy observed the bonds assigned to monodentate carbonate, bidentate carbonate, polydentate carbonate, and hydrogen carbonate on the tested CeO2 surface. Detailed adsorption mechanisms of CO2 on CeO2 were studied by Hahn et al.86 using hybrid functional (PBE0) and density functional theory (DFT)+U calculations. This indicated that the isolated CO2 molecules interact with CeO2(111) more favorably to form two stable carbonate configurations, monodentate and bidentate carbonate, rather than the linear adsorption configuration (Fig. 2IIa–f). For these two carbonate species, the anti-bonding orbitals of the C atom could interact with the O atom on the CeO2 surface and subsequently form a stable chemical bond. However, the role of oxygen vacancy was not discussed in these studies.
Recently, Ruhaimi et al.87 confirmed that oxygen vacancies on the CeO2 surface could serve as CO2 adsorption sites. Typically, the electronegative O atom of CO2 could provide electrons to oxygen vacancies on CeO2, lead to the polarization of adsorbed CO2 molecules and reduce the activation energy of CO2. Additional evidence has been provided by Zhu et al.88 Compared with the ideal CeO2(110) facet, the CO2 molecule on CeO2(110) with one oxygen vacancy exhibited more pronounced geometrical changes (more tortuous O–C–O angle and stretched CO bond lengths), and resulted in a lower CO2 adsorption energy (−1.93 eV vs. −1.44 eV). Besides oxygen vacancy, Zhu et al.89 demonstrated that the hydroxyl (OH) group on the CeO2 surface could act as an adsorption site for CO2. Generally, the O atom of OH groups on CeO2 tends to act as Lewis basicity to interact with the C atom of CO2 due to its electronegativity, thus promoting the adsorption and reduction of CO2 (Fig. 2III). In summary, the above-mentioned experimental and theoretical studies indicate that the interactions between CeO2 and methanation reactants (CO2 and H2) may play a positive role in CO2 methanation.
2.2. Identification of active sites over ceria-based catalysts for methanation of CeO2 with H2 and CO2
The interaction of CeO2 with CO2 methanation reactants is a prerequisite for the application of CeO2 in this reaction. Unfortunately, the single CeO2 catalyst is extremely inefficient for CO2 activation. Varvoutis et al.90 recently evaluated the CO2 methanation performance over hydrothermally synthesized CeO2-nanorods and commercial CeO2 (Sigma Aldrich, >99.5%) samples and found that only little CO was formed via reverse water–gas shift (RWGS) reaction, indicating that the CH4 yield over bare CeO2 is negligible. Similarly, Li et al.32 found no apparent CO2 conversion over the physical mixture of Ni particles and CeO2 compared with a high-performance Ni/CeO2 catalyst prepared by a wet-impregnation method. Therefore, it is essential to systematically identify the catalytic sites for CO2 methanation over CeO2-based catalysts.Zhang et al.91 investigated the CO2 methanation reaction over Ni supported on an ideal CeO2(111) surface by first principles calculations based on DFT. They concluded that the CeO2 support promoted the dispersion of Ni particles and the electronic interaction of charge transfer between Ni and CeO2. As shown in Fig. 3I, the top site of the Ni particle exhibited a lower CO2 adsorption energy (−158.89 kJ mol−1) than that of Ni particle side (−85.58 kJ mol−1) and metal–support interface (−136.53 kJ mol−1), indicating that the top of Ni is the predominant catalytic site for CO2 methanation. In contrast, Li et al.32 argued that CO2 prefers to adsorb and activate at the interface of the Ni/CeO2 catalyst, while the Ni particle is the site of H2 dissociation to afford active H atoms for the subsequent hydrogenation reactions. This view was approved by Cárdenas-Arenas et al.,92 who suggested that the reduced NiO–CeO2 interface is the chemisorption region of CO2, and the adsorbed CO2 species could react with H atoms to produce CH4 (Fig. 3II).
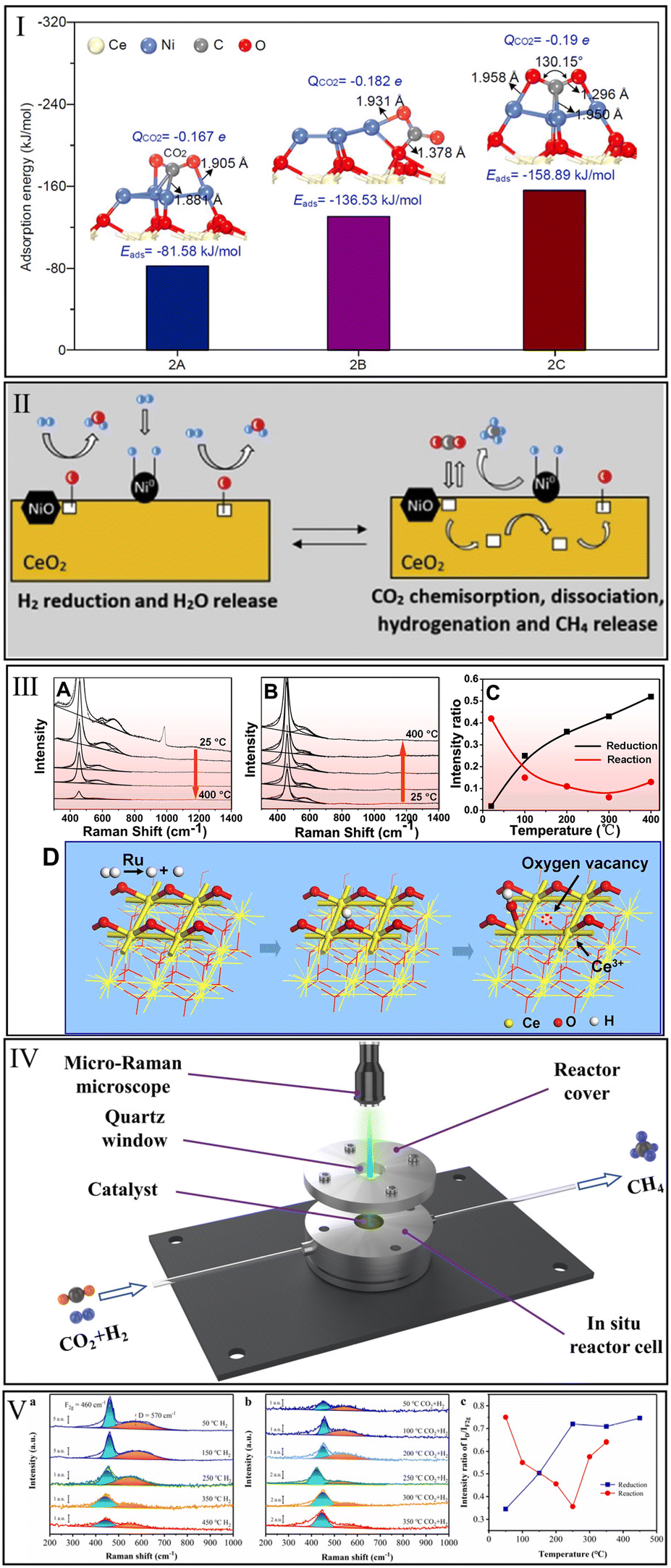 | ||
| Fig. 3 (I) Adsorption energies of CO2 adsorbed at top, side sites of Ni particle and Ni–CeO2 interface. Reproduced from ref. 91 with permission from Elsevier, copyright 2021. (II) Reduction of NiO–CeO2 interface and hydrogenation of CO2. Reproduced from ref. 92 with permission from Elsevier, copyright 2020. (III) Operando visible Raman spectra of Ru/CeO2 in the pre-reduction process (A), the CO2 methanation process (B), and the corresponding ID/IF2g value (C). Generation of oxygen vacancy, OH group, and Ce3+ in the reduction process of Ru/CeO2 catalyst (D). Reproduced from ref. 84 with permission from American Chemical Society, copyright 2016. (IV) Schematic diagram of reactor cell for in situ Raman. (V) In situ Raman spectra in the reduction process (a), methanation reaction process (b), and the corresponding intensity ratio of ID/IF2g over Ni/CeO2 (c). Reproduced from ref. 99 with permission from American Chemical Society, copyright 2022. | ||
Recent findings further identified the catalytic position of Ni/CeO2 catalysts. Lin et al.24 performed a series of reduction–oxidation treatments to tune the interaction of Ni particles and CeO2 support. They found that strong Ni–CeO2 interaction was detected after a high-temperature reduction of Ni/CeO2, which resulted in the partial encapsulation of Ni particles as well as subsequent decreased ability to activate H2 molecules. This further limited the hydrogenation of CO2. However, Varvoutis et al.90 claimed both Ni sites and the Ni–CeO2 interface cannot be regarded as methanation performance descriptors of Ni/CeO2 catalysts. They synthesized a series of CeO2 nanorods loaded with Ni of different particle sizes, and found that the optimum methanation activity could be obtained only by coordinating the competitive relationship between Ni–CeO2 perimeter and large Ni particles. This boosted activity could be mostly correlated to the under-coordinated step/edge and kink/corner sites on the Ni surface. It can be seen that detailed characterizations and meticulous catalyst synthesis technologies combined with theoretical models are required to determine the exact location of the active sites over Ni/CeO2 catalysts.
In addition to discerning where the active sites are, it is important to understand what the active sites are. It was reported that the basic and oxygen vacancies sites on CeO2 make it promising support for CO2 adsorption and conversion at low temperatures.88 Muroyama et al.15 prepared a series of Ni loaded on various metal oxides (Y2O3, Sm2O3, ZrO2, CeO2, Al2O3, and La2O3) catalysts and combined CO2-TPD techniques to explore the distribution of basic sites over these samples. For Ni/CeO2, the main CO2 desorption was detected below 180 °C, denoting the existence of a high concentration of weak basic sites on the Ni/CeO2 surface. Subsequently, Yan et al.14 claimed that the adsorbed species of CO2 at OH sites with weak basicity can react with H atoms to produce CH4. It should be noted that the CO2 methanation activity is particularly relevant to the number of weak (OH groups) and medium (metal–oxygen pairs) basic sites.49,93
Furthermore, Hao et al.94 attributed the high performance of Ni/CeO2 mainly to the oxygen vacancies with the interaction of Ni–CeO2. They observed that Ni/CeO2 exhibited hundreds of times higher CO2 conversion rates than that of Ni/SiO2 without oxygen vacancy in a temperature range from 200 to 300 °C. Subsequently, their work confirmed that the turnover frequency (TOF) of CO2 increased with the decrease of Ni sizes (8–21 nm) over Ni/CeO2 catalysts. A similar experiment has also been performed by Marconi et al.;95 however, a positive correlation was observed between the TOF for CO2 conversion and Ni particle size, and CH4 prefers to be produced on large Ni particles (8 nm). Moreover, Lin et al.96 suggested that the larger Ni particles (8 nm) on the CeO2 surface accelerated the hydrogenation of key intermediates for CO2 methanation. In this light, Varvoutis et al.90 believed that the differences in the extent of H2 spillover and the dispersion of Ni particles led to these contradicting reports.
López-Rodríguez et al.97 monitored the active sites on Ni/CeO2 catalyst for CO2 methanation by in situ near-ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS). The Ni0 sites and oxygen vacancies located at the Ni–CeO2 interface were confirmed to be the active sites for H2 dissociation and CO2 dissociation, respectively, while a small amount of NiO and Ni-carbonates hydroxyls acted as spectators in methanation. Besides, they found that the reduction rates of Ni2+ and Ce4+ gradually became faster than their CO2 oxidation rates as the reaction temperature increased. Therefore, an appropriate CO2 methanation temperature may be beneficial for maintaining the chemical state of the active sites on Ni/CeO2 catalysts.
In terms of Ru/CeO2 catalysts, Guo et al.28 reported that the Ce3+–OH and Ru sites on Ru/CeO2 nanowires were recognized as the catalytic sites for CO2 dissociation and carbonyl hydrogenation, respectively. The TOF over as-prepared Ru/CeO2 catalysts followed the order of Ru nanoclusters/CeO2 > Ru single atoms/CeO2 > Ru nanoparticles/CeO2. It should be noted that the CeO2-supported Ru nanoclusters catalyst with the lowest oxygen vacancy concentration exhibited the highest CO2 methanation performance, suggesting that the rate-determining step occurred on Ru species. However, the oxygen vacancy content on the reacted Ru/CeO2 catalysts showed a great decrease after the methanation process, indicating that the oxygen vacancies indeed contributed to CO2 methanation. Subsequently, these authors examined the impact of Ru particle size on H2 dissociation capacity by H2-TPR and H2-TPD experiments. The H2 spillover intensity detected on CeO2-supported Ru nanoparticles (ca. 4 nm) was much higher than for Ru nanoclusters (ca. 1.2 nm) and Ru single-atom samples. This may be due to the low coverage of H2 on these small metal particles and single-atom sites. Moreover, the in situ DRIFTS results confirmed that the Ce3+–OH structure greatly facilitated the transformation of CO2 to formate species. Overall, it is well established that active Ru sites, oxygen vacancies, and OH groups play important roles in the hydrogenation of CO2 into CH4 over metal nanoparticles-supported CeO2 catalysts. Also, the structural sensitivity induced by metal size effects is prevalent in CO2 methanation.
Nevertheless, based on the results of kinetic analysis, Wang et al.98 presented the opposite view. They deposited Ru particles on CeO2 supports with different shapes. Although these Ru/CeO2 catalysts displayed different CO2 conversion, the similar apparent activation energies (75 kJ mol−1) indicated the same active site on as-prepared Ru/CeO2 catalysts. It is worth mentioning that the TOF values with respect to the surface oxygen vacancy were similar (2.7 × 10−4 s−1), while the values with respect to exposed Ru atom were markedly different for all Ru/CeO2 catalysts (6.61 × 10−5–6.06 × 10−4 s−1). Therefore, the oxygen vacancy, not the Ru site, was the major active site for the rate-determining step in CO2 methanation over Ru/CeO2 catalysts.
Oxygen vacancy, one of the vital defects on CeO2-based catalysts, plays an important role in CO2 adsorption and hydrogenation. Detecting the evolution of oxygen vacancy during the process of pre-reduction and CO2 methanation reaction by in situ/operando Raman characterization is beneficial for revealing the role of oxygen vacancy in methanation. Generally, the ratio of peak intensities at ∼570 cm−1 (defect-induced (D) mode) and ∼460 cm−1 (first-order F2g mode) is a quantitative measurement of oxygen vacancy concentration.84 Wang et al.84 found that the oxygen vacancy concentration increased as the reduction temperature was raised from room temperature to 400 °C (Fig. 3IIIA and C). In the methanation process, the oxygen vacancy concentration decreased sharply from room temperature to 100 °C and remained at a low level in a temperature range of 200–400 °C (Fig. 3IIIB and C). These results indicated that oxygen vacancies were gradually generated on the Ru/CeO2 surface in the reduction process, and the oxygen vacancies were indeed actively involved in the methanation reaction. Combining operando X-ray absorption near edge structure (XANES) and operando infrared (IR) results, they deemed that oxygen vacancies, OH groups, and Ce3+ sites were generated almost synchronously during the reduction process of Ru species. As shown in Fig. 3IIID, metallic Ru with the ability to split H2 molecules was first produced in the reduction process. Afterward, the Ce–O bond on the CeO2 surface was attacked by active H atoms due to the hydrogen-spillover effect, which accelerated the generation of oxygen vacancies, OH groups, and Ce3+. The detailed evolution of these active species in CO2 methanation will be presented later in this review.
Recently, our group performed in situ Raman spectra (Fig. 3IV) to systematically monitor the changes in oxygen vacancy density over Ni/CeO2 catalyst during the reduction and subsequent methanation reaction processes.99 In the reduction process (Fig. 3Va and c), the oxygen vacancy concentration over Ni/CeO2 surged with the increment of temperature from 50 to 250 °C, and stayed relatively stable from 250 to 450 °C. This confirmed the progressive generation of oxygen vacancies during the reduction treatment over Ni/CeO2. Subsequently, when the inlet gas was switched from 25% H2/75% N2 to 5% CO2/20% H2/75% N2 (Fig. 3Vb and c), the evolution of oxygen vacancy concentration exhibited a concave-like shape, which decreased from 50 to 250 °C and then increased from 250 to 350 °C. This suggested that CO2 could dramatically occupy the oxygen vacancies and participate in the subsequent hydrogenation process.
2.3. Effect of metal–support interaction for catalytic performance
In the field of heterogeneous catalysis, the MSI effect plays an extremely important role in dispersing metal particles, tuning redox properties, and resisting catalyst sintering and coke.100This significant effect may alter the microstructure of the catalyst due to geometric and/or electronic effects, and thus the catalytic performance.24 MSI is a well-recognized concept that mainly relies on strong metal–support interaction (SMSI) and electronic metal–support interactions (EMSIs), in which the SMSI effect was studied more than 40 years ago by Tauster et al.101 Generally, the SMSI effect acts solely on the catalysts supported on a reducible support (such as CeO2, TiO2, Fe3O4, and ZnO),102–105 and is highly dependent on the reaction conditions. The major characteristic of the typical SMSI effect is the migration and encapsulation of partially reduced oxides to metal particles during the reaction process.106
Matte et al.107 discovered a CeO2−x encapsulation overlayer on Ni nanoparticles after an H2 reduction treatment at 500 °C. A similar phenomenon on a Ni/CeO2 sample was observed by Li et al.32 They deemed that the coverage of Ni particles by reduced CeO2 decreased the exposed surface of active Ni, which accordingly impaired the capacity for CO2 and/or H2 activation. Specifically, the CO2 consumption rate over Ni/CeO2 catalysts at 523 K significantly decreased (from ∼32 to ∼7 h−1) due to SMSI induced by the increase of pre-reduction temperature (from 723 K to 973 K) (Fig. 4I). Ho et al.108 prepared a series of CeO2 supported by Ni or Ru with different loadings by a direct coating process. They found the low metal content enhanced the MSI effect, but these samples exhibited a poor catalytic performance (CO2 conversion below 10% and CH4 selectivity below 70%) at 350 °C, while the catalyst with high Ni loading (45%) achieved ∼65% CO2 conversion and ∼92% CH4 selectivity. This could be attributed to SMSI and unbalanced sites for H2 dissociation and CO2 activation. In this respect, Lin et al.24 found that the oxidative treatment alleviated the encapsulation of the CeO2−x layer on the Ni surface, but this did not fully recover the CO2 methanation activity over Ni/CeO2. Recently, Pu et al.109 reported that SMSI on Ni/CeO2 catalysts was greatly influenced by CeO2 shapes. They found that the extent of encapsulation of Ni particles induced by SMSI followed the sequence of Ni/CeO2-nanooctahedra > Ni/CeO2-nanocube > Ni/CeO2-nanorod (Fig. 4II). Therefore, SMSI between Ni and CeO2 is not only affected by Ni content, but is also affected by CeO2 properties. The geometric surface restructuring process induced by the SMSI effect may significantly affect the CO2 methanation reaction, due to the metal nanoparticles undertaking the role of CO2 and H2 activation.
 | ||
| Fig. 4 (I) HRTEM images of Ni/CeO2 after a reduction treatment at (A) 773 K, (B) 973 K, and (C) effect of strong metal–support interaction induced by reduction temperature on CO2 consumption rate over Ni/CeO2 catalysts. Methanation condition: GHSV = 6.6 × 104 h−1; Treact = 523 K. Reproduced from ref. 32 with permission from Elsevier, copyright 2018. (II) HR-TEM images and schematic illustration of SMSI influence on Ni particles supported on CeO2 with different shapes. Reproduced from ref. 109 with permission from Elsevier, copyright 2022. (III) HRTEM images for reduced Ni/CeO2 samples with different Ni content (A) 1%, (B) 2.5%, and (C) 5%. Reproduced from ref. 94 with permission from Elsevier, copyright 2021. | ||
In addition, the modification of methanation catalysts by the appropriate degree of MSI effect has been extensively established. On the one hand, Hao et al.94 observed that spherical Ni particles were moderately embedded in the nanocrystalline CeO2 due to the MSI effect, which is beneficial for maintaining the stability of Ni particles at low Ni content (Fig. 4IIIA–C). On the other hand, the redox properties of Ni/CeO2 and Ni/γ-Al2O3 catalysts were compared by Cárdenas-Arenas et al.92 They found that the reduction of NiO on CeO2 occurred at a much lower temperature than that on γ-Al2O3 support. This implied that the moderate interaction between NiO and CeO2 support improved the reduction of NiO species. Thus, Ni/CeO2 with more metallic Ni sites achieved ∼60% CO2 conversion at 300 °C, much higher than that of Ni/γ-Al2O3 (∼2%).
Rui et al.66 investigated the MSI effect toward Ni/CeO2 catalyst prepared by plasma decomposition (Ni/CeO2-P) and conventional thermal calcination (Ni/CeO2-C) strategies. They suggested that the stronger MSI induced by plasma decomposition treatment prompted the generation of Ni–O–Ce coordination. This not only inhibited the aggregation of NiO particles by an anchoring effect, but also enhanced the activity of O atoms at Ni–CeO2 interfacial regions. As expected, Ni/CeO2-P with better reducibility and reactivity exhibited a high CO2 conversion of 84.2% at 275 °C, while it is only 34.7% over Ni/CeO2-C. In this regard, Ruiz Puigdollers et al.110 believed that the O atoms in the periphery between metal and support are more reactive than those in other regions, and accordingly, generation of oxygen vacancies is easier at the metal–support interface sites.
In the case of Ru/CeO2 catalysts, a recent report explored the effect of Ru content (1–5 wt%) on Ru/CeO2 catalysts for CO2 methanation. López-Rodríguez et al.111 found that the optimum catalyst with 2.5 wt% Ru detected the highest proportion of Run+ species with strong interaction with the CeO2 support. This MSI effect improved the reducibility of Ru cations at low temperatures, and thus achieved a high CO2 conversion of ∼80% with 100% CH4 selectivity at 290 °C.
In the CO2 activation situation, the EMSI effect can induce partial charge transfer from the support to raise the electron density at metal sites, which facilitated the active intermediate conversion.73 Tada et al.112 reported that the activation from carbonyl species into CH4 on metal can be promoted due to the EMSI effect. This is attributed to the ability of the electron-rich metal to donate electrons to the antibonding π-orbital of the adsorbed carbonyl species, and thus the C–O bond is easily activated. However, Wang et al.98 found a different phenomenon about the EMSI effect. They employed different CeO2 shapes (nanorods, nanopolyhedrons, and nanocubes, denoted as NRs, NPs, and NCs) as the support for Ru particles. Subsequently, Run+ species at the interface region attributed to the electron transfer from Ru to CeO2 support were observed in Ru/CeO2-NRs and Ru/CeO2-NPs samples. This electronic interaction impacted the concentration of oxygen vacancies on Ru/CeO2 catalysts. According to Raman spectra analysis, the oxygen vacancy concentration on the three bare CeO2 supports is decreased in the following order: CeO2-NRs > CeO2-NCs > CeO2-NPs. As for Ru/CeO2 samples, it should be noted that the above order was changed to Ru/CeO2-NCs > Ru/CeO2-NRs > Ru/CeO2-NPs. This may be due to the electronic interaction in Ru/CeO2-NRs and Ru/CeO2-NPs leading to the decrease of Ru0 proportion, thus weakening the H2 activation ability. Finally, the Ru/CeO2-NCs exhibited an outstanding catalytic rate of 4.85 × 10−8 mol gcat−1 s−1 even at a low temperature of 150 °C.
Likewise, Guo et al.28 prepared CeO2-supported Ru single atoms (SA), nanoclusters (NC), and nanoparticles (NP) by adjusting the deposition amount of RuCl3 to investigate the effect of electronic interaction strength in Ru/CeO2 catalysts for CO2 methanation. The X-ray photoelectron spectroscopy (XPS) results indicated that the strength of electronic interaction is decreased in the sequence Ru(SA)/CeO2 > Ru(NC)/CeO2 > Ru(NP)/CeO2. However, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) combined with the DFT calculations further demonstrated that the strong charge transfer induced by the EMSI effect is disadvantageous for the activation of carbonyl species. The TOFCO2 of as-prepared Ru/CeO2 samples at 190 °C ranked in the following order: Ru(NC)/CeO2 (7.41 × 10−3 s−1) > Ru(SA)/CeO2 (4.59 × 10−3 s−1) > Ru(NP)/CeO2 (5.30 × 10−4 s−1). It is important to note that the methanation activity is not only affected by electronic interaction but also by the H-spillover effect, hence it is difficult to obtain a direct relationship between EMSI strength and CO2 methanation performance.
Collectively, the SMSI effect is widely recognized to be unfavorable for CO2 methanation as it may lead to the encapsulation of active metal sites. However, based on the existing literature, the impacts of the EMSI effect on CO2 methanation are still unclear, and more in-depth experimental and theoretical studies are required to fully understand this interaction.
3. CO2 methanation mechanism over ceria-based catalysts
Fundamentally understanding the mechanism of heterogeneous catalytic reaction is a pivotal premise for designing efficient catalysts.113,114 Although much effort has been made to reveal the reaction mechanism of CO2 methanation by experimental or/and theoretical methods, a consensus has not yet been reached. As shown in Fig. 5, the C–O bond cleavage mechanism and the formate mechanism are widely recognized. The former involves the direct dissociation or hydrogen-assisted dissociation of CO2 on the active metal surface to CO* (* represents an adsorbed species). Subsequently, the CO* species react with H*, leading to CH4 production. The latter involves the CO2 adsorbed on the support or metal–support interface hydrogenation to formate (HCOO*) intermediates, which are subsequently hydrogenated to CH4. | ||
| Fig. 5 Possible CO2 methanation pathways. | ||
3.1. CO2 methanation mechanism over Ni/CeO2 catalysts
CO2 methanation pathways over Ni/CeO2 catalysts have been examined by many researchers because of the potential practical application of inexpensive Ni-based catalysts.91,115–117 On the basis of DFT calculations, in 2021, Zhang et al.91 reported that the RWGS + CO-hydro route over the Ni/CeO2(111) surface is more favorable than the direct CO2 dissociation route and formate route. Ni nanocluster acted as the major activation site and facilitated the following pathway: CO2* → HOCO* → CO* → HCO* → H2CO* → CH2* → CH3* → CH4*. Among them, the C–O bond cleavage of the H2CO* intermediate is the rate-limiting step of this route. Interestingly, in 2015, Ren et al.118 suggested that the direct dissociation of CO2 is the optimal methanation pathway over the Ni(111) surface, and that CO* → C* + O* is the rate-limiting step. Hence, Zhang et al.91 argued that the CeO2 support played a key role in changing the optimal route and the corresponding rate-determining step of CO2 methanation over Ni-based catalysts.Instead, Hongmanorom et al.119 prepared an ordered mesoporous CeO2 support (mpCeO2) to experimentally explore the difference in methanation pathway between Ni/mpCeO2 and conventional Ni/CeO2. As shown in Fig. 6I, CH4 was produced over conventional Ni/CeO2 mainly via an associative mechanism (*CO2 → *CO3/*HCO3 → *HCOO → CH4). In situ DRIFTS results indicated that the carbonates and bicarbonates were converted into formate species that would further hydrogenate to CH4 by dissociated H atoms, and no bands for *CO species were detected on the conventional Ni/CeO2 surface. However, Ni/mpCeO2 with rich oxygen vacancies, abundant Ni–O–Ce interfacial sites, and highly dispersed Ni particles exhibited a simultaneous dissociative (*CO2 → *CO → *HCO → CH4) and associative mechanism.
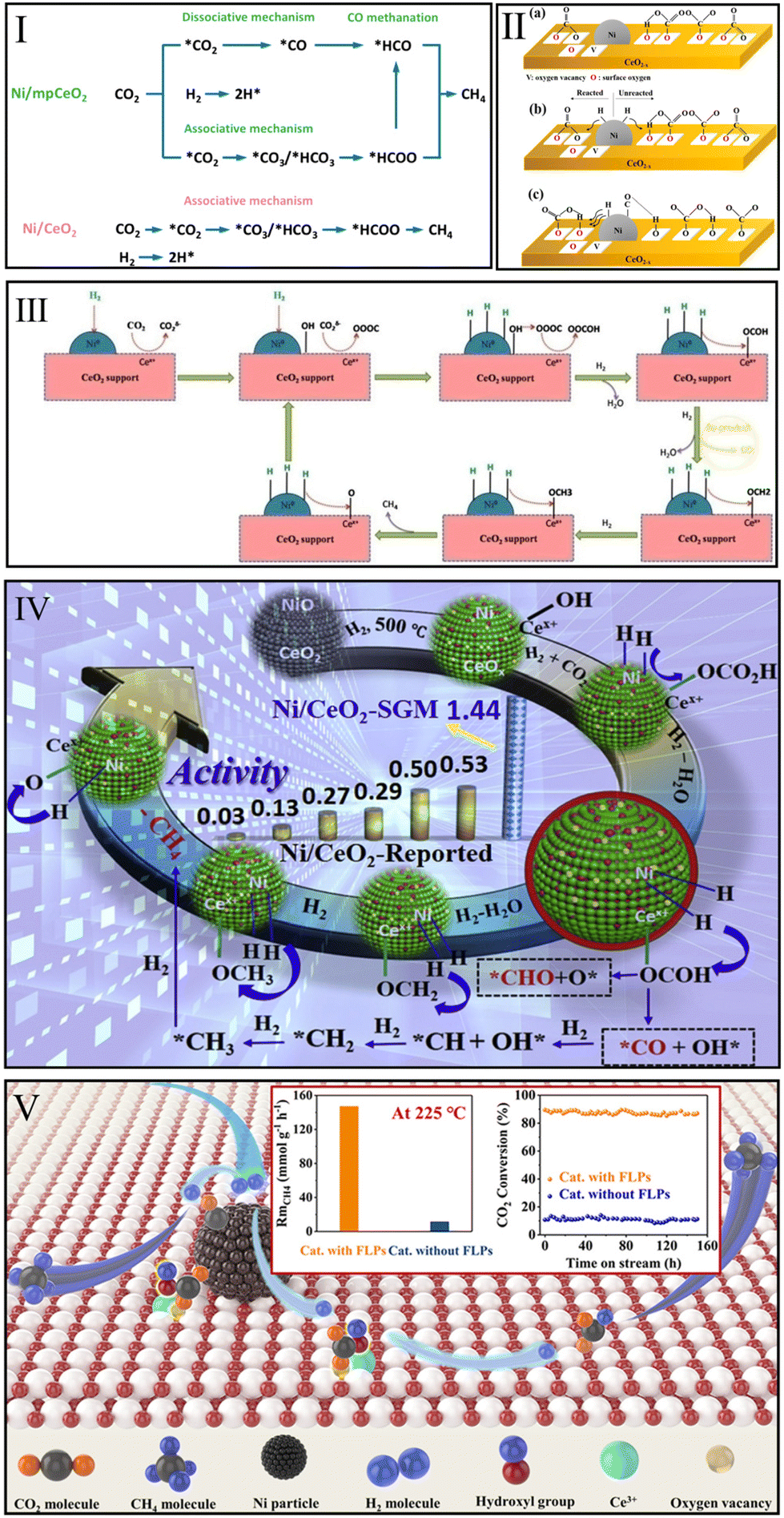 | ||
| Fig. 6 (I) Proposed CO2 methanation mechanism over Ni/mpCeO2 and Ni/CeO2 catalysts. Reproduced from ref. 119 with permission from Elsevier, copyright 2021. (II) Proposed reaction pathway over Ni/CeO2−x: CO2 adsorption at 180 °C (a), CO2 methanation at 180 °C (b), and CO2 methanation over 260 °C (c). Reproduced from ref. 120 with permission from American Chemical Society, copyright 2019. (III) Proposed CO2 methanation mechanism over Ni–CeO2. Reproduced from ref. 121 with permission from Elsevier, copyright 2018. (IV) CO2 possible methanation route over Ni/CeO2-SGM. Reproduced from ref. 80 with permission from Elsevier, copyright 2020. (V) Proposed CO2 methanation mechanism and catalytic performance over Ni/CeO2. Reproduced from ref. 99 with permission from American Chemical Society, copyright 2022. | ||
Lee et al.120 suggested that different reaction pathways dominated at different temperatures. In the low-temperature range of 180–240 °C (Fig. 6IIa and b), Ce3+ sites with oxygen vacancies over Ni/CeO2 catalyst activated CO2 to form carbonate species and then hydrogenated to formate and finally CH4. When the temperature exceeded 260 °C (Fig. 6IIc), the CO2 could be dissociated into CO* on the Ni surface due to the high activation energy. Then, the CO* species were hydrogenated until the CH4 generation. However, Yu et al.121 argued CO* species over Ni–CeO2 can be attributed to the formate decomposition at the Ce3+ site. Moreover, they found that the CO* species was not an active intermediate and existed as a by-product (Fig. 6III).
In this light, Ye et al.80 believed that the hydrogenation of CO* species was a highly dissociated H demands process. They prepared a Ni/CeO2 catalyst with 16.75% Ni loading to provide enough sites for H2 dissociation, thus the DRIFTS results indicated that some CO* presented at Ni sites can react with H* to form formyl groups that can produce CH4 by CO* hydrogenation. In addition, they traced the evolution of formate species by injecting formic acid into the reaction system. Stable formate bands on the Ni/CeO2 surface were observed at 300 °C. Subsequently the band associated with CH4 appeared when H2 was introduced into the system. Therefore, the formate route played an important role in CO2 methanation over Ni/CeO2 catalyst (Fig. 6IV). Recently, Onrubia-Calvo et al.122 performed 124 kinetic experiments to establish a kinetic model that reflects the intrinsic CO2 methanation activity over Ni/CeO2 catalyst. They found the formate Langmuir–Hinshelwood–Hougen–Watson mechanistic model, which considered the hydrogenation of bicarbonate to formate as rate-determining step, in good line with the kinetic data and the results of in situ DRIFTS.
Of note, although the formate route has been considered to be the dominant CO2 methanation pathway over Ni/CeO2 catalysts, the contribution of the CO* route cannot be ignored. It is a pity that the exact origin of CO* is still controversial because of the structure-sensitivity of the CO2 methanation reaction. Typically, formate species can be converted to CO* species or directly hydrogenated to CH4.123,124 However, Pan et al.125 argued that CO* can be obtained by direct dissociation of CO2. The dynamic evolutions of CO* species on Ni–CeO2–Y2O3 catalyst have been explored by Zhu et al.88 They observed that the consumption rate of CO* was faster than that of formate species after the reaction gas was switched from CO2/H2 to H2. This confirmed that the CO* was generated by the direct CO2 dissociation, not by formate. However, no CO* was detected on the Ni–CeO2 surface due to the lower oxygen vacancy concentration than Ni–CeO2–Y2O3. A similar finding was reported by Zhang et al.126 They found that the high population of oxygen vacancies and medium-strength basic sites may be promoted by both the formate and CO* pathways.
Recently, our group found that the frustrated Lewis pairs constructed by oxygen vacancies and OH groups could promote CO2 dissociation at a low temperature of 225 °C, and the CO* route as well as formate pathway together enhanced the low-temperature methanation activity over Ni/CeO2 catalyst (Fig. 6V).99 Unfortunately, it remains unclear what role oxygen vacancies play in the dissociation of carbon dioxide.
3.2. CO2 methanation mechanism over Ru/CeO2 catalysts
Ni-based catalysts may be deactivated because of the loss of active metal induced by the formation of mobile nickel subcarbonyls at low temperatures.2 Although Ru is more expensive than Ni, the CO2 methanation mechanism over Ru/CeO2 catalysts still commands great attention due to the considerable methanation activity and stability of Ru-based catalysts.Ma et al.127 investigated the methanation mechanism and site requirements on the ideal CeO2(110)-supported Run (n represents the number of atoms) surface by a first-principles approach (Fig. 7Ia). The reaction pathway was observed to be sensitive to the number of Ru atoms. On the Ru1/CeO2 surface, the single Ru atom and nearby lattice O atoms were recognized as active sites, and CH4 was generated by a carboxylate (COOH*) route. By comparison, the Ru nanocluster was identified as the major catalytic site on the Ru4/CeO2 and Ru8/CeO2 surface, and the CO* became the dominant route. Compared with the CO mechanism, which exhibited a significant ensemble effect and required 3–4 Ru atoms, the COOH mechanism was less structure sensitive and only required 1–2 Ru atoms. Of note, the elementary step of the accepted formate route (CO2* + H* → HCOO*) on all Ru/CeO2 surfaces needs to overcome a higher barrier than that of CO* and COOH* routes. Moreover, Ru4/CeO2 showed a lower apparent activation energy of 0.91 eV than that of Ru1/CeO2 (1.65 eV) and Ru8/CeO2 (1.41 eV), indicating that the CO2 molecule could be easily activated over Ru4/CeO2 (Fig. 7Ib). The first four influential steps are depicted in Fig. 7Ic. It appeared that the rate-determining step switched from CO* + H* → HCO* over Ru1/CeO2 and Ru4/CeO2 to CH2* + H* → CH3* over Ru8/CeO2 with the increasing of Ru atom numbers. Meanwhile, the second most influential step switched from CO2* + H* → COOH* (Ru1/CeO2) to CO2* + * → CO* + O* (Ru4/CeO2 and Ru8/CeO2), indicating that CO2 dissociation required more Ru atoms.
 | ||
| Fig. 7 (I) CO2 methanation over CeO2(110)-supported Run (n represents the number of atoms) (a), Arrhenius plot (b), and the influence of the elementary steps on the TOF of CH4 (c). Reproduced from ref. 127 with permission from American Chemical Society, copyright 2021. (II) In situ DRIFTS spectra recorded at 220 °C on reduced (a and d) Ru(SA)/CeO2, (b and e) Ru(NC)/CeO2, and (c and f) Ru(NP)/CeO2. The results were recorded under atmosphere switched from 1% CO2/4% H2/He to 5% H2/He after stabilization for 60 min for Ru(SA)/CeO2 and Ru(NP)/CeO2, and 30 min for Ru(NC)/CeO2. (g) The CO2 methanation reaction pathways over Ru/CeO2 catalysts based on the results of in situ DRIFTS. Reproduced from ref. 28 with permission from American Chemical Society, copyright 2018. (III) (A) Formate route for CO2 methanation over Ru/CeO2, and (B) CO2 conversion over Ru/CeO2 (a) and Ru/α-Al2O3 (b). Reproduced from ref. 84 with permission from American Chemical Society, copyright 2016. | ||
A finding raised questions about this interpretation. Gou et al.28 argued that the rate-determining step of methanation occurred at Ru sites on CeO2-supported Ru singles atom (SA), nanoclusters (NC), and nanoparticles (NP) catalysts. According to the results of in situ DRIFTS (Fig. 7IIa–f), the CO route was confirmed as the dominant CO2 methanation pathway over all Ru/CeO2 samples, and the bicarbonate (HCO3* at 3623, 1396, and 1048 cm−1), carboxylate (O2C at 1559, 1508, and 1305 cm−1), and bridged formate (HCOO* at 3017, 2836, 1580, and 1339 cm−1) were the active intermediates. Additionally, they found that different reaction pathways took place on CeO2 supported by different Ru structures. The reaction pathway is summarized in Fig. 7IIg. Specifically, the CO2-derived HCO3* was converted to HCOO* (path 1), bidentate CO3* (path 2) or bridged CO3* (path 3) species and then to form Ru–CO*. Subsequently, the CO* species react with the active H* atoms to produce CH4 or direct desorption. In parallel, the adsorbed CO2 could convert to O2C* (path 4) and then form Ru–CO*.
Sharma et al.128 reported a different methanation pathway involving methoxy species over a Ru-substituted CeO2 sample (Ce0.95Ru0.05O2). Based on TPR, DRIFTS and DFT results, the CO* species was first hydrogenated by dissociated H and underwent CO2* → CO* → OCH2* → OCH3* → CH4. In sharp contrast, for the conventional supported metal/oxide sample, the CO* was evolved to formate followed by hydrogenation to form CH4. According to their earlier study,129 Ru was substituted in a +4 oxidation state in the CeO2 lattice and thus formed Ce0.95Ru0.05O2. These works indicated that the CO2 methanation pathway over Ru/CeO2 catalysts was significantly impacted by the chemical environment of the Ru species.
Recently, more details of methanation behavior and mechanism over Ru/CeO2 have been provided by López-Rodríguez et al.130 The (NAP-XPS) results indicated the reduced Ru/CeO2 can be slightly oxidized below 300 °C under reaction conditions of CO2 methanation (CO2 + H2). Combined with the in situ DRIFTS results, it can be seen that the chemisorbed CO2 on Ru sites was dissociated into Ru–CO* and O atoms, which could potentially oxidize the surface of Ru0 and CeO2. Then, DFT calculations demonstrated that the hydrogenation of Ru–CO* is the rate-determining step in this reaction system. Besides, CO2 can be adsorbed at the surface oxygen site and oxygen vacancy site of CeO2 to form carbonate and carboxylate species, respectively, while carboxylates can evolve into formate species. These formate species which followed decomposed to CO on CeO2 above 200 °C and served as an additional reservoir of Ru–CO*.
To systematically understand the catalytic behavior and the relationship between the reaction pathway and active site over Ru/CeO2, three kinds of operando characterizations (XANES, Raman, and IR) were used by Wang et al.84 They found that the concentrations of Ce3+, OH groups, and oxygen vacancies decreased at different temperatures, which indicated these active sites function at different steps of this reaction system. A complete formate route is displayed in Fig. 7IIIA on the basis of the steady-state isotope transient kinetic analysis (SSITKA) type operando DRIFTS results. First, Ce3+ was responsible for the activation of CO2 into carboxylate at 25 °C, then the carboxylate species was hydrogenated to formate by the surface OH group at the same temperature. As the temperature increased to 150 °C, the oxygen vacancy was activated and thus promoted the conversion of formate to methanol (detected as the rate-determining step). Finally, the methanol was easily hydrogenated to form CH4. In contrast, the CO* route activated by metallic Ru was not observed over the Ru/α-Al2O3 sample until 250 °C, resulting in a low CO2 conversion (Fig. 7IIIB).
In summary, the CO2 methanation mechanism over CeO2-based catalysts has been disputed and no consensus has yet been reached (Table 1). Based on the above findings, it can be concluded that mechanistic studies are largely limited by the characterization techniques employed by researchers. For example, Zhang et al.91 and Ma et al.127 used DFT calculations to explore the methanation pathway over CeO2-based catalysts. However, the theoretical results were inconsistent with the experimental results, because the influence of the abundant OH groups and oxygen vacancies over the CeO2 surface for the reaction pathway was not taken into account. Additionally, although in situ DRIFTS is a reliable characterization technique for the detection of surface-adsorbed species on the tested catalyst, it is difficult to specifically probe the evolution of active species during the reaction. Perhaps pulse technology combined with spectrokinetics studies could make breakthroughs on this issue.
| Sample | Method | Dominant reaction pathway | Ref. |
|---|---|---|---|
| Ni/CeO2 | DFT calculations | CO* route | 91 |
| Ni/CeO2 | In situ DRIFTS | Formate route | 119 |
| Ni/mpCeO2 | Formate route | ||
| Ni/CeO2−x | In situ DRIFTS | Formate route (180–240 °C) CO* route (>260 °C) | 120 |
| Ni/CeO2-SGM | In situ DRIFTS | Formate route | 80 |
| Ni/CeO2 | In situ DRIFTS | Formate route | 99 |
| Ru1/CeO2 | DFT calculations | Carboxylate route | 127 |
| Ru4/CeO2 | CO* route | ||
| Ru8/CeO2 | CO* route | ||
| Ru(SA)/CeO2 | In situ DRIFTS + DFT calculations | CO* route | 28 |
| Ru(NC)/CeO2 | CO* route | ||
| Ru(NP)/CeO2 | CO* route | ||
| Ce0.95Ru0.05O2 | In situ DRIFTS + DFT calculations | CO* route | 128 |
| Ru/CeO2 | Formate route | ||
| Ru/CeO2 | In situ DRIFTS + DFT calculations | CO* route | 130 |
| Ru/CeO2 | SSITKA type Operando DRIFTS | Formate route | 84 |
4. Strategies to raise the catalytic performance over ceria-based catalysts
From the above discussion, it is clear that the CO2 methanation undergoes CO2 adsorption, generation of intermediates via interaction between the active site and CO2 molecule, H2 dissociation, hydrogenation of active intermediates, and CH4 production. So, the enhanced catalytic performance could be established on CeO2-based catalysts by increasing the number of sites for CO2 adsorption/activation and accelerating the dissociation and spillover of the H2 molecule. Several strategies for modifying the existing CeO2-catalysts and corresponding CO2 methanation activity are summarized in Table 2.| Strategy | Sample | Reaction gases | GHSV (mL gcat−1 h−1) | Temperature (°C) | CO2 conv. (%) | CH4 select. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a The unit is h−1. — not shown in the literature. | |||||||
| Constructing efficient structure | Ni/CeO2-SGM | 20% CO2/80% H2 | 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
250 | 82.5 | 94.8 | 80 |
| NiCe(35)_HT | 11% CO2/48% H2/41% N2 | 72000 |
300 | 76 | 99 | 131 | |
| NiCe(15)_IWI | 66 | 98 | |||||
| Ni/CeO2-600 °C | 10% CO2/40% H2/50% N2 | 45000 |
300 | ∼56 | ∼99 | 132 | |
| 10Ni/CeO2-M-1 | 20% CO2/80% H2 | 45000 |
325 | ∼79 | ∼100 | 133 | |
| 10Ni/CeO2-M | ∼69 | ∼100 | |||||
| 10Ni/CeO2-C | ∼30 | ∼100 | |||||
| Pellet Ni3Ce1 | 17% CO2/67% H2/17% N2 | 38200a |
300 | ∼62 | ∼96 | 108 | |
| Foam Ni3Ce1 | ∼22 | ∼95 | |||||
| Ni/CeO2–NCT | 10% CO2/40% H/50% Ar | 45000 |
340 | 91.1 | 100 | 134 | |
| NiO–CeO2 (np) | 16% CO2/64% H2/20% N2 | 60000 |
300 | ∼55 | ∼95 | 135 | |
| NiO–CeO2 (3DOM) | ∼3 | ∼50 | |||||
| Ni–CeO2-CSC | 10% CO2/40% H/50% Ar | 120000 |
300 | 70 | ∼100 | 136 | |
| Porous NC3 | 10% CO2/40% H/50% Ar | 60000 |
300 | 85.6 | 100 | 137 | |
| NiNPs@CeO2NF | 20% CO2/80% H2 | 36000 |
300 | 82.3 | ∼99 | 138 | |
| NiNPs/CeO2 | ∼68 | ∼98 | |||||
| NiNPs/CeO2NF | ∼70 | ∼98 | |||||
| Synthesizing special morphology | Ni/CeO2-NR | 10% CO2/40% H2/50% He | 24000 |
250 | ∼23 | ∼98 | 139 |
| Ni/CeO2-NC | ∼10 | ∼96 | |||||
| Ni/CeO2-PH | 10% CO2/40% H2/50% He | 21000 |
250 | 41 | ∼99 | 140 | |
| Ni/CeO2-NR | 21.7 | ∼97 | |||||
| Ni/CeO2-NP | 15.7 | ∼98 | |||||
| Ni/CeO2-NC | 11 | ∼95 | |||||
| Ni/CeO2-P | 19% CO2/76% H2/5% N2 | 6000 | 260 | 73 | 100 | 141 | |
| Ru/CeO2/r | 10% CO2/40% H2/50% N2 | 72000 |
300 | ∼72 | ∼99 | 67 | |
| Ru/CeO2/o | ∼56 | ∼99 | |||||
| Ru/CeO2/c | ∼14 | ∼99 | |||||
| Metal/oxide loading | Na/Ni/CeO2 | 1% CO2/50% H2/49% He | 60000 |
250 | ∼97 | ∼ | 142 |
| Ni/Ca0.1Ce0.9Ox | 20% CO2/80% H2 | 36000 |
290 | 75 | 99 | 143 | |
| Ni/Sr0.1Ce0.9Ox | ∼66 | ∼99 | |||||
| Ni/Mg0.1Ce0.9Ox | ∼58 | ∼99 | |||||
| Ni/Ba0.1Ce0.9Ox | ∼53 | ∼99 | |||||
| 12Ni3Co/M–Ce80Zr20 | 20% CO2/80% H2 | 12000 |
300 | ∼72 | ∼99 | 144 | |
| 12Ni3Fe/M–Ce80Zr20 | ∼68 | ∼98 | |||||
| 12Ni3Mn/M–Ce80Zr20 | ∼62 | ∼98 | |||||
| 12Ni3Cu/M–Ce80Zr20 | ∼8 | — | |||||
| 1Co15Ni/CeO2 | 15% CO2/60% H/25% Ar | 52000a |
250 | 74 | ∼100 | 145 | |
| RuNi/CZ | 20% CO2/80% H2 | 24000 |
350 | 53 | 93 | 146 | |
| Ni/Pr–Ce | 10% CO2/40% H/50% Ar | 25000 |
350 | 54.5 | 100 | 147 | |
| Ni/Sm–Ce | 44.9 | 100 | |||||
| Ni/Mg–Ce | 43.2 | 100 | |||||
| Ni/Pr10Ce | 20% CO2/80% H2 | 25000 |
300 | 46 | 98 | 148 | |
| 5Ni/CeO2–Y2.0% | 15% CO2/60% H/25% Ar | 60000 |
300 | ∼74 | ∼100 | 149 | |
| NiCeO2–La-600 | 18% CO2/72% H/10% N2 | 30000 |
300 | ∼88 | ∼100 | 126 | |
| Ru/Ce0.9Cr0.1Ox | 20% CO2/80% H2 | 36000 |
250 | ∼70 | ∼100 | 19 | |
| Ru/Ce3PrOx | 10% CO2/40% H2/50% He | 9000a | 270 | ∼55 | 100 | 150 | |
| Activation treatment | Ni/CeO2–H2 | 20% CO2/80% H2 | 36000 |
300 | 70.9 | 99.7 | 24 |
| Ni/CeO2–N2 | 67.7 | 99.8 | |||||
| Ni/CeO2–air | 64.4 | 99.8 | |||||
| Ni/CeO2-250 | 3.5% CO2/10% H2/87.5% N2 | 20000 |
300 | 0 | 0 | 151 | |
| Ni/CeO2-350 | 74.7 | 99.1 | |||||
| Ni/CeO2-450 | 53.1 | 99 | |||||
| Ni/CeO2-650 | 16.7 | 91.1 | |||||
4.1. Constructing efficient structured CeO2-based catalysts
The structure of the support not only impacts the dispersion of active metal, but also greatly changes the reducibility and basicity of the catalyst.49,152 Ye et al.80 synthesized a nanostructured Ni/CeO2-SGM that has a higher specific surface area than a conventional Ni/CeO2-IM catalyst by a sol–gel method (Fig. 8I). The finding showed that the modified structure offered an array of advantages in Ni dispersion, reducibility, and CO2 adsorption. Accordingly, even at a low temperature of 250 °C, the Ni/CeO2-SGM presented a high CO2 conversion of 80.5% and maintained it for more than 100 h. Therefore, modification of the CeO2-based catalyst structure is a direct means to improve the catalytic activity.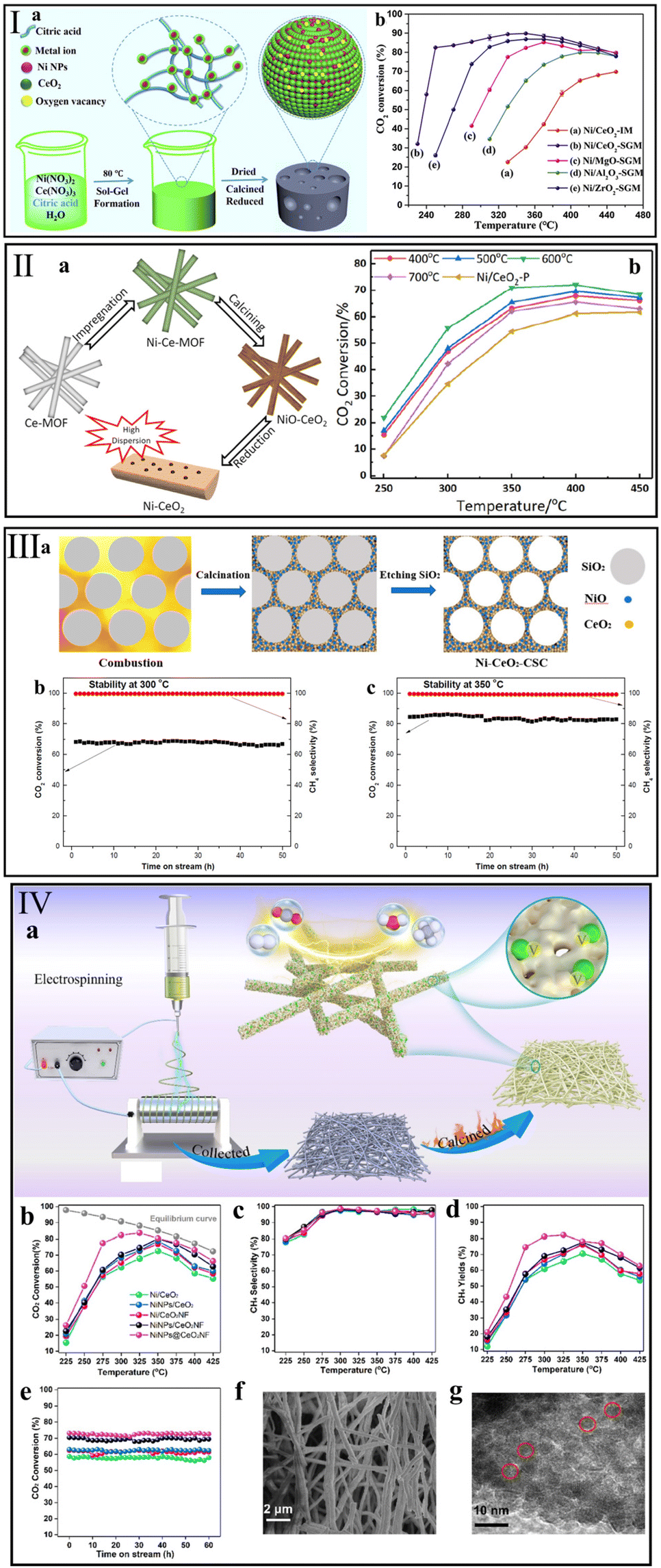 | ||
| Fig. 8 Synthesis route and corresponding catalytic performance of (I) Ni/CeO2-SGM (reaction conditions: GHSV = 10000 mL g−1 h−1, n(H2):n(CO2) = 4:1). Reproduced from ref. 80 with permission from Elsevier, copyright 2020, (II) Ni/CeO2-MOF (reaction conditions: GHSV = 45000 mL g−1 h−1, n(H2):n(CO2):n(N2) = 4:1:5). Reproduced from ref. 132 with permission from Elsevier, copyright 2021, (III) Ni–CeO2-CSC (reaction conditions: GHSV = 120000 mL g−1 h−1, n(H2):n(CO2):n(Ar) = 4:1:5). Reproduced from ref. 136 with permission from MDPI, copyright 2020, and (IV) NiNPs@CeO2NF (reaction conditions: GHSV = 36000 mL g−1 h−1, n(H2):n(CO2) = 4:1). Reproduced from ref. 138 with permission from Elsevier, copyright 2022. | ||
To avoid the sintering of Ni particles on conventional metal/support structure during the preparation and reaction process, Atzori et al.131 prepared a series of NiO–CeO2 mixed oxides (NiCe(x)_HT) with different Ni loadings (5–35 wt%) using SBA-15 as the template. A counterpart pure CeO2 sample was prepared by the same hard template method, and then Ni particles were deposited by incipient wetness impregnation (NiCe(x)_IWI). For the NiCe(x)_HT catalysts, the MSI effectively prevents the sintering and aggregation of active Ni particles even at a high Ni content (35 wt%). Accordingly, CO2 conversion reached 76% at 300 °C over NiCe(35)_HT, which is higher than that over NiCe(35)_IWI (57%). On the contrary, NiCe(15)_IWI exhibited the highest CO2 conversion of 66% among the as-prepared NiCe(x)_IWI catalysts. Based on the TEM results of NiCe(x)_IWI samples, an obvious Ni aggregation was observed with the increase in Ni content, leading to a decrease in methanation activity.
For the same purpose, Feng et al.132 designed a Ni/CeO2 catalyst with high Ni dispersion and good durability using a Ce-based metal–organic framework (MOF) (Fig. 8II). At 350 °C, the Ni/CeO2 sample with MOF structure (Ni/CeO2-600 °C) yielded a higher CO2 conversion of 71.0% than the conventional catalyst (∼55%) prepared by co-precipitation method (Ni/CeO2-p). Also, benefiting from the confinement effect of the MOF structure, only slight sintering of Ni nanoparticles on the Ni/CeO2 sample was observed (from 2.3 to ∼3 nm) after a 30 h durability test at 350 °C. Tang et al.133 reported the difference in methanation performance between Ni deposited on Ce-MOF (Ni/CeO2-M) and Ni/CeO2-MOF (Ni/CeO2-M-1) prepared by a one-pot method to further explore the effect of catalyst structure on CO2 methanation. Although both the Ni/CeO2 catalysts contained the same component and content, Ni/CeO2-M-1 possessed a higher methanation activity of ∼79% at 325 °C compared with Ni/CeO2-M (∼69%). This can be attributed to the stronger MSI over Ni/CeO2-M-1, and further induced higher NiO dispersion and oxygen vacancy concentration.
The synthesis method largely determines the structural characteristics of the Ni/CeO2 catalysts. Generally, CeO2-based materials are less porous than conventional Al2O3, which may inhibit the dispersion of active metals. Fortunately, the utilization of templates in the synthesis process can optimize the pore structure of the CeO2-based catalyst, and thus promote catalytic activity. Zhou et al.134 prepared a series of CeO2, prepared by hard-template (CT), soft-template (CS), and precipitation (CP) methods, that served as support of Ni particles. They found the Ni crystal particle size, specific surface area, and pore structure were governed by the CeO2 support with different structures. Among the Ni/CeO2 samples, the NCT catalyst with the largest specific surface area and the highest Ni dispersion had the strongest capacity to hydrogenate CO2, and thus achieved a high CO2 conversion of 91.1% at 340 °C. Similarly, Cárdenas-Arenas et al.135 synthesized NiO–CeO2 nanoparticles (NiO–CeO2 (np)) prepared by a reversed microemulsion method, and the resulting catalyst exhibited CO2 conversion of ∼55% at 300 °C. However, the NiO–CeO2 sample (NiO–CeO2 (3DOM)) with a three-dimensionally ordered macroporous (3DOM) structure, by hard-template method, showed a poor CO2 conversion of ∼5%. The difference in specific surface area and reducibility was the primary reason for the substantial difference in methanation.
Wang et al.136 synthesized a three-dimensional mesoporous Ni–CeO2 sample (Ni–CeO2-CSC) containing highly dispersed Ni particles (Fig. 8III). This special structure supplied an efficient Ni–CeO2 interface for CO2 activation, and thus enhanced the methanation activity over Ni–CeO2-CSC. Also, they observed that Ni particles smaller than 5 nm can be embedded in the pore walls, which prevented the Ni sintering during the methanation reaction. Consequently, the CO2 conversion of ∼70% can be maintained at 300 °C under a high GHSV of 120000 mL g−1 h−1 for 50 h over Ni–CeO2-CSC. To further optimize the pore structure of the Ni/CeO2 catalyst, Zhou et al.137 prepared a series of NiCe composite samples with different nNi/nCe ratios by a soft template method. The physicochemical properties of as-prepared NiCe catalysts were highly correlated with the nNi/nCe ratio. They found that the NC3 (nNi/nCe ratio = 3) with porous structure and high specific surface area showed high performance for CO2 hydrogenation to CH4 at 300 °C, with CO2 conversion and CH4 selectivity of 85.6% and 100%, respectively. Meanwhile, this nano-porous structure of NC3 was beneficial for the maintenance of methanation activity.
Recently, Hu et al.138 reported Ni nanoparticles encapsulated in highly mesoporous CeO2 nanofiber (NiNPs@CeO2NF) fabricated by the co-electrospinning method (Fig. 8IV). They found that the structure of CeO2 nanofibers could promote the production of oxygen vacancies, and those confined NiNPs could further enhance the methanation activity. Compared with the samples (NiNPs/CeO2 and NiNPs/CeO2NF) prepared by traditional impregnation, NiNPs@CeO2NF possessed the highest oxygen vacancy concentration and thus exhibited a superior CO2 conversion of 82.3% at 300 °C, which was higher than that of NiNPs/CeO2 (∼68%) and NiNPs/CeO2NF (∼70%). In addition, attributed to the confined effect of NiNPs@CeO2NF, no significant deactivation was observed on NiNPs@CeO2NF during a 60 h durability testing at 400 °C.
In summary, these studies confirmed that the catalytic activity over Ni/CeO2 catalysts in CO2 methanation can be enhanced by optimization of the structure. An efficient structured CeO2-based catalyst not only decelerated the sintering of active metal, but also accelerated the generation of oxygen vacancy, and consequently, the CO2 hydrogenation processes.
4.2. Synthesizing specific morphology
In addition to optimizing the structure of CeO2-based catalysts, synthesizing the specific morphology is another approach to improve the catalyst's nature and activity. The effect of different morphology of CeO2 support on the activity of CeO2-based catalysts for CO2 hydrogenation has been studied extensively due to the highly tunable crystal texture.153–156 For CO2 methanation, the activity over the CeO2-based catalysts is strongly affected by the morphology of CeO2. Compared with a commercial nano-CeO2 (Aladdin, 20 nm, 99.5% metals basis)-supported Ni catalyst (∼60%), Ni/CeO2-nanoplates (Ni/CeO2-P) exhibited a higher CO2 conversion of 73% at 260 °C.141 Besides, the TOF of Ni/CeO2-P was up to 1.7 fold higher than Ni/CeO2 sample at 200 °C. It should be noted that the H2-TPD results indicated a similar Ni particle size on these Ni/CeO2 catalysts. Thus, Du et al.141 suggested that the difference in distinct catalytic activity over Ni/CeO2 and Ni/CeO2-P was attributed to the CeO2 supports with different shapes.Generally, CeO2 with different shapes have been synthesized by hydrothermal process under different hydrothermal conditions.157,158 Bian et al.139 deposited Ni particles on two CeO2 supports with different shapes (nanorod and nanocube, denoted as NR and NC), and evaluated the low-temperature methanation activity over these samples. At a temperature range of 200–250 °C, the CO2 conversion over Ni/CeO2-NR was consistently higher than that over Ni/CeO2-NC. They proposed that the high activity was related to a large number of oxygen vacancies resulting from the CeO2-NR structure. This apparent discrepancy in oxygen vacancy concentration has a direct influence on the generation of a key intermediate (formate).
Subsequently, Jomjaree et al.140 prepared additional CeO2 supports including nanopolyhedron (PH) and nanoparticle (NP) to further complement this study. The results of a series of characterizations indicated the significant difference in textural property, reducibility, and oxygen vacancy content, which further impacted the catalytic activity over these Ni/CeO2 catalysts. As a result, the CO2 conversion ranked in the following order: Ni/CeO2-PH > Ni/CeO2-NR > Ni/CeO2-NP > Ni/CeO2-NC. Interestingly, a lower CO2 conversion of 21.7% than that over Ni/CeO2-PH (41%) was observed over the Ni/CeO2-NR at 250 °C, even though it contained the highest surface area and reducibility. The authors suggested that the negative SMSI effect between Ni and CeO2-NR was responsible for the worse low-temperature CO2 methanation performance over the Ni/CeO2-NR.
Furthermore, the preferentially exposed facet of CeO2 is highly correlated with its morphology.153,159–161 This characteristic offered an appropriate model for exploring the impact of the CeO2 crystal facet on CO2 methanation. Generally, the behaviors of oxygen vacancy generation and lattice oxygen migration on the CeO2 surface were highly correlated with its crystal structure.27,162 The DFT calculations indicated that the stabilities of CeO2 crystal facets decreased in the order of (111) > (110) > (100), while the oxygen vacancy formation energy decreased in the following order: (111) > (100) > (110).163 Hence, the CeO2(110) facet has the potential to produce oxygen vacancies and maintain the stability of these vacancy sites, which is favorable for the hydrogenation of CO2 into CH4.
Wang et al.98 linked the oxygen vacancy content to CeO2 exposed facets, and further revealed the structure–activity relationship over Ru/CeO2-nanorods (NRs), Ru/CeO2-nanopolyhedrons (NPs), and Ru/CeO2-nanocubes (NCs) catalysts. The HR-TEM images displayed the primary CeO2 lattice fringes of (110), (100), and (111) facets observed on Ru/CeO2-NRs, Ru/CeO2-NCs, and Ru/CeO2-NPs, respectively. The subsequent catalytic activity experiments indicated that the reaction rates at 150 °C decreased in the order: Ru/CeO2-NCs > Ru/CeO2-NPs > Ru/CeO2-NRs. The Raman spectra certified that Ru/CeO2-NCs contained the highest oxygen vacancy concentration among these Ru/CeO2 samples, which greatly benefited the activation of CO2. However, the difference in Ru dispersion on these catalysts may impact the CO2 conversion. Hence, the exclusion of the influence of particle size is warranted for further exploration of the crystal facet effect.
Sakpal et al.67 prepared three CeO2 supports with different shapes by a hydrothermal method, and supported Ru particles with similar size (∼2 nm). SEM and TEM images (Fig. 9a–l) showed that CeO2(111) and (100) lattice fringes were observed on CeO2 rods (CeO2/r) and CeO2 cubes (CeO2/c), respectively, while CeO2 octahedra (CeO2/o) exposed (110) and (111) facets. Raman spectra combined with the results of TPR and XPS demonstrated that the addition of Ru particles enhanced the reducibility of CeO2, and further increased oxygen vacancy concentration. Ru/CeO2/r exhibited a higher CO2 conversion of ∼72% than Ru/CeO2/o (∼56%) and Ru/CeO2/c (∼14%) at 300 °C (Fig. 9m), and no significant deactivation was observed over these catalysts within 24 h testing (Fig. 9n). This trend was consistent with the trend in oxygen vacancy content on as-prepared catalysts.
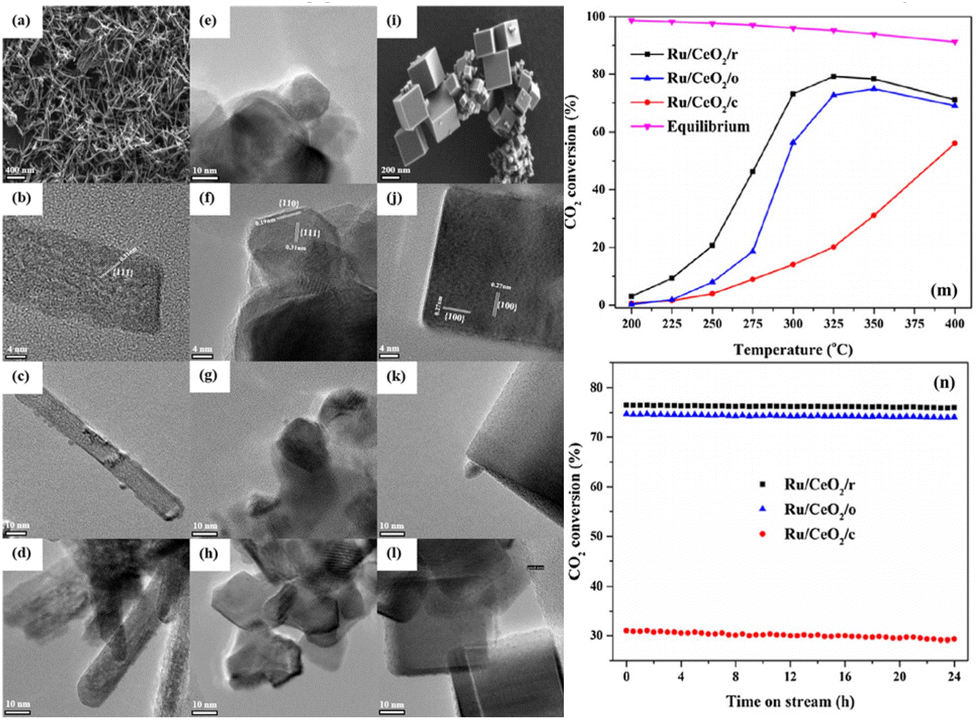 | ||
| Fig. 9 SEM and TEM images of CeO2/r (a and b), CeO2/o (e and f), and CeO2/c (i and j). TEM images of Ru/CeO2/r (fresh, c and spent, d), Ru/CeO2/o (fresh, g and spent, h), and Ru/CeO2/c (fresh, k and spent, l). CO2 conversion over Ru/CeO2 catalysts (m), and CO2 conversion at 350 °C against time on stream (n) (reaction conditions: GHSV = 72000 mL g−1 h−1, n(H2):n(CO2):n(N2) = 4:1:5). Reproduced from ref. 67 with permission from Elsevier, copyright 2018. | ||
Overall, the CeO2 morphology indeed impacted the CO2 methanation reaction over the CeO2-based catalyst. Generally, there were substantial differences in specific surface area and pore structure for CeO2-based catalysts with different shapes. Therefore, because of these significant differences in textural properties of CeO2-based catalysts, the influence of the CeO2 crystal facet involved in CO2 methanation has not been clearly understood. Unfortunately, the calcination temperature and Ce precursors can alter the crystal facets on the CeO2 sample,163 which further increases the difficulty in linking methanation activity with the exposed crystal facets on CeO2.
4.3. Loading metal or oxide
The incorporation of metal or oxide is a simple, effective, and economical strategy to raise the methanation activity over CeO2-based catalysts.164–167 In general, CO2 is an acidic gas, which can be adsorbed at alkaline sites on the catalyst surface. Thus, the addition of alkalis on the CeO2-based catalyst would benefit the CO2 methanation. Sodium (Na), a readily available alkali metal, has been considered for the modification of the Ni/CeO2 catalyst. Le et al.142 prepared Ni/SiO2 and Ni/CeO2 samples with 0.1 and 1 wt% Na to understand the effects of Na content for methanation. Based on the results of H2 chemisorption, the Ni dispersion on Ni/SiO2 and Ni/CeO2 obviously decreased with the increasing of Na content, and further impaired the ability to activate H2. In respect of CO2 adsorption, the adsorption sites on the Ni/SiO2 surface increased with the Na ratio. The loss of H2 dissociation sites and the gain of CO2 adsorption sites reached a balance on 0.1% Na/Ni/SiO2 catalyst, and thus a higher CO2 conversion than Ni/SiO2 and 1% Na/Ni/SiO2 samples. On the contrary, a negative influence both on CO2 adsorption and conversion was observed over Ni/CeO2 catalysts even with a Na content as low as 0.1 wt%. According to the XPS analysis, Na tended to interact with CeO2, which may lead to coverage of active sites on the CeO2 surface.To explore the influence of adding alkaline earth metal oxides on CO2 methanation over Ni/CeO2 catalyst, Liu et al.143 prepared a series of NiM0.1Ce0.9Ox (M = Ca, Sr, Mg, and Ba) samples. They found that the distributions of these alkaline additives were different. CaO and MgO tended to dissolve into the CeO2 lattice to form a solid solution, while BaO and SrO tended to exist on the surface of CeO2. Overall, the addition of alkaline earth metal oxides increased Ni dispersion, the number of CO2 adsorption sites and oxygen vacancies, and further enhanced the intrinsic catalytic activity over as-prepared catalysts. These positive effects were most pronounced on Ni/Ca0.1Ce0.9Ox, thus it showed the highest CO2 conversion of 75% at 290 °C among all the catalysts. It should be noted that a Ni/Ca0.1Ce0.9Ox simply synthesized by a traditional impregnation method possessed a lower methanation activity, which indicated that the Ca species dissolved into the CeO2 lattice were more effective than that on the CeO2 surface.
Moreover, it was reported that bimetallic CeO2-based catalysts can provide additional active sites for the activation of CO2 and H2.168–172 Xu et al.144 found that the loading of transition metal on Ni monometallic catalyst to build bimetallic catalyst could improve methanation activity over a mesoporous Ce0.8Zr0.2O2-supported Ni particles catalyst (12Ni/M–Ce80Zr20) (Fig. 10I). The influences of adding different 3 wt% transition metals on CO2 methanation over 12Ni/M–Ce80Zr20 samples were different. Specifically, Co and Fe enhanced the methanation activity, and the contribution of Mn could be neglected, while the negative effect of Cu on CO2 conversion was observed. Among the as-prepared bimetallic catalysts, 12Ni3Co/M–Ce80Zr20 achieved the highest CO2 conversion of ∼72% and CH4 selectivity of ∼99% at 300 °C. In situ DRIFTS and temperature-programmed surface reaction (TPSR) results indicated that the incorporation of Co accelerated the conversion of intermediates and decreased the activation temperature of CO2, due to the synergistic effect of Ni and Co on the 12Ni3Co/M–Ce80Zr20 catalyst. Recently, Hasrack et al.145 found that CO2 methanation performance could be improved over 15Ni/CeO2 catalysts via adding 1 and 5 wt% of Co. On the basis of activity evaluation and CO2-TPD results, the relationship between catalytic activity and the number of medium basic sites could be well established. Although the addition of Co decreased the total basicity, the amount of medium basic sites greatly increased, which was widely considered a key factor in the methanation reaction.
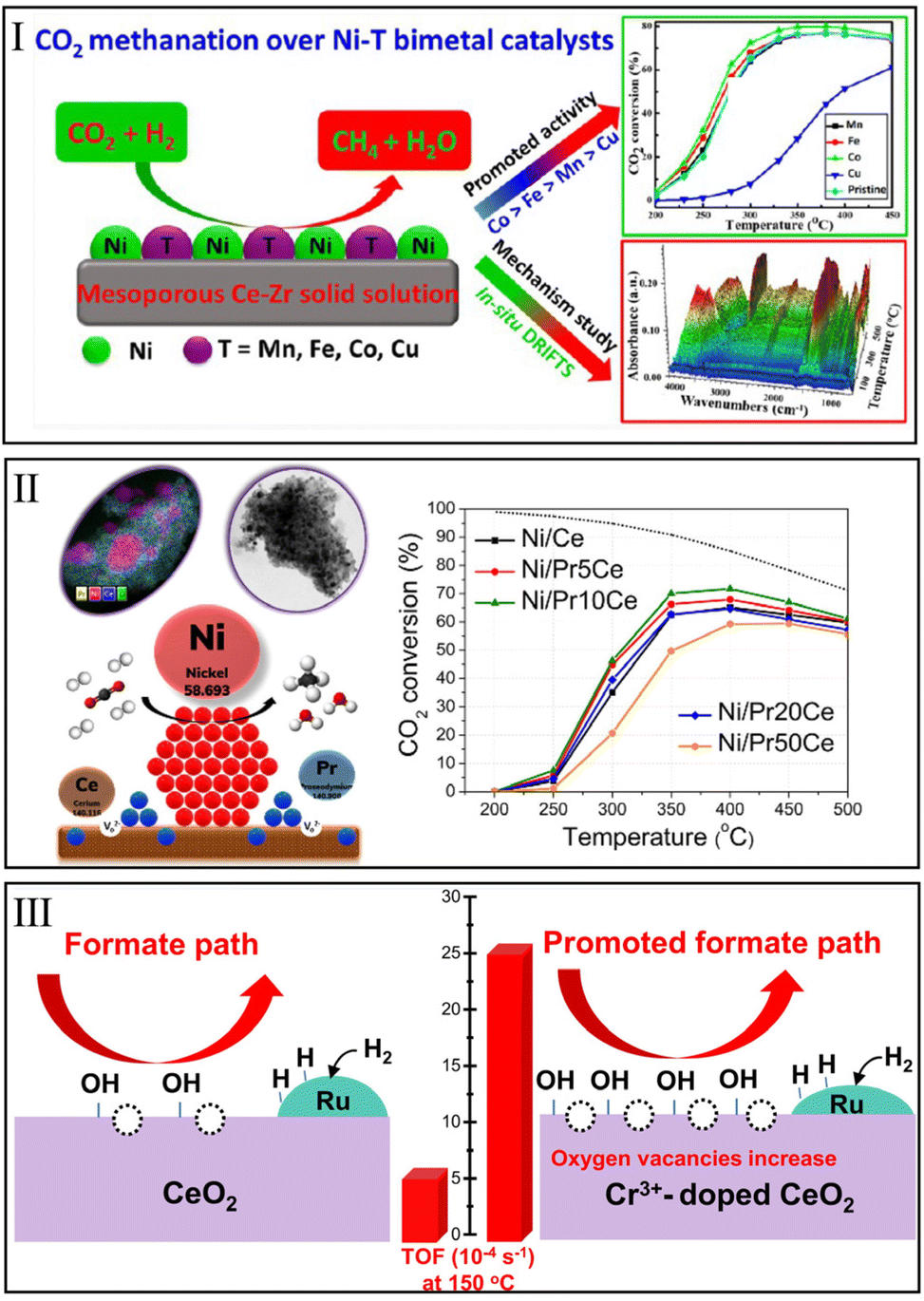 | ||
| Fig. 10 (I) CO2 methanation over Ni–T bimetal catalysts. Reproduced from ref. 144 with permission from American Chemical Society, copyright 2021. (II) Influence of Pr3+ on CO2 conversion over Ni/CeO2 catalyst. Reproduced from ref. 148 with permission from Elsevier, copyright 2022. (III) Promotion of Cr3+ on Ru/CeO2 sample for CO2 methanation. Reproduced from ref. 19 with permission from American Chemical Society, copyright 2021. | ||
Saché et al.146 prepared a series of supported metal catalysts on CeZr (ZC) solid solution, including Ni/CZ, FeNi/CZ, RuNi/CZ, and RuFeNi/CZ. They found that the incorporation of small amounts of Ru (1 wt%) could greatly enhance CO2 methanation performance, and achieved a high CO2 conversion of 53% at 350 °C. However, the addition of Fe inhibited CO2 conversion over FeNi/CZ catalyst. In the terms of the trimetallic RuFeNi/CZ, the improvement effect of the promoter was not significant. The benefits of adding Ru were mainly reflected in Ni dispersion and electronic properties. Besides, Ru provided additional active sites on the RuNi/CZ catalyst, which accelerated the H2 dissociation and CO2 hydrogenation. Recently, Elia et al.171 found a similar trend in their study of monometallic and bimetallic CeO2-based methanation catalysts. They suggested that the addition of Ru does indeed enhance the methanation activity by improving the dispersion of the Ni phase. It is noteworthy that no deactivation was observed on the Ru–Ni/CeO2 catalyst during 75 h-on-stream at 350 °C.
The incorporation of oxides is a commonly used method to improve the surface properties of catalysts. Siakavelas et al.147 reported the CO2 methanation over these Ni/CeO2 (Ni/Ce) catalysts modified by Sm2O3 (Sm), Pr2O3 (Pr), and MgO (Mg). They suggested that the additional Pr3+ and Sm3+ cations could not only dissolve into the CeO2 lattice to promote the generation of oxygen vacancies, but also inhibited the agglomeration of Ni particles. It was noted that the Pr3+ tended to form Ce4+–OV–Pr3+ species to produce more oxygen vacancies than the Ni/Sm–Ce sample. As for Ni/Mg–Ce, the MSI effect induced by the addition of Mg2+ effectively improved the stability at the CO2 methanation reaction. Moreover, these additional cations optimized the distribution of basic sites and increased the number of moderate adsorption sites, thus enhancing the catalytic activity over the as-prepared Ni/Ce catalysts. As a result, Ni/Pr–Ce exhibited higher CO2 conversion of 54.5% than Ni/Sm–Ce (44.9%), Ni/Mg–Ce (43.2%), and Ni/Ce (39.4%) at 350 °C.
Recently, Tsiotsias et al.148 found that an appropriate amount of Pr3+ cations tended to substitute Ce4+ ions in the CeO2 lattice and further induced the generation of oxygen vacancies. On the contrary, a high Pr loading (>20%) decreased the concentration of oxygen vacancies. Thus, Ni/Pr–CeO2 catalysts with low Pr content (Ni/Pr10Ce) showed a higher CO2 conversion of 46% than Ni/Ce (35%) and Ni/Pr50Ce (21%) at 300 °C (Fig. 10II). In addition, López-Rodríguez et al.150 found the dual effect of Pr on CO2 methanation over Ru/CeO2 catalysts. Specifically, a Ru/CeO2 sample with low Pr content (∼3 wt%) exhibited a relatively high CO2 conversion of ∼55% at 270 °C due to a positive effect in oxygen mobility induced by Pr. However, a high Pr addition (∼25 wt%) hindered the dissociation of CO2 at interface sites, and thus showed a low CO2 conversion of ∼20%.
Sun et al.149 found that Y played an important role in modifying Ni/CeO2 catalyst for CO2 methanation. To systematically understand the promotion effect of Y, they prepared 5Ni/CeO2, 5Ni/Y2O3, and 5Ni/CeO2–Y samples with different Y content. Subsequently, it was found that the 5Ni/CeO2–Y catalyst with appropriate Y content possessed a higher CO2 methanation activity than 5Ni/CeO2 and 5Ni/Y2O3. The results of CO2-TPD and H2-TPR indicated that the basic distribution and reducibility of 5Ni/CeO2–Y catalysts were largely influenced by Y content. The activity over these 5Ni/CeO2–Y samples followed the order: 5Ni/CeO2–Y2.0% > 5Ni/CeO2–Y1.0% > 5Ni/CeO2–Y0.5% > 5Ni/CeO2–Y5.0%. This agreed well with the trend of the number of weak and medium basic sites, oxygen vacancy content, and reduction temperature of NiO.
In terms of Ru/CeO2 catalysts, Xu et al.19 reported a high-performance CeO2 support doped with Cr cation (Cr/Ce = 1:9) using a precipitation method (Fig. 10III). Then, 3 wt% Ru was loaded on the Ce0.9Cr0.1Ox support to hydrogenate CO2 to CH4. They found that the Cr3+ could incorporate into the CeO2 lattice, and thus promoted the formation of surface oxygen vacancies on Ru/Ce0.9Cr0.1Ox. Meanwhile, XPS results indicated that the Ru/Ce0.9Cr0.1Ox catalyst contained more OH groups than Ru/CeO2 after a reduction treatment. Similar apparent activation energies were calculated on Ru/Ce0.9Cr0.1Ox (74.9 kJ mol−1) and Ru/CeO2 (79.2 kJ mol−1), indicating that the same methanation mechanism proceeds on these two samples. However, Ru/Ce0.9Cr0.1Ox showed a considerable TOFCO2 (25.1 × 10−4 s−1) at a low temperature of 150 °C, which was 5.3 times higher than that of Ru/CeO2. These illustrated that the addition of Cr could indeed improve the intrinsic activity of the Ru/CeO2 catalyst. In situ FTIR spectroscopy demonstrated the CO* and formate pathways that occurred on Ru/Ce0.9Cr0.1Ox and Ru/CeO2. Moreover, the number of bicarbonates and formates increased markedly as the oxygen vacancies and OH group increased, which greatly enhanced the catalytic performance of Ru/Ce0.9Cr0.1Ox for low-temperature CO2 methanation.
In summary, for CO2 methanation over CeO2-based catalysts, the addition of metal or oxide often causes certain positive or negative effects. This depends not only on the nature of the additive, but also on the interaction between CeO2 and the additive. Generally, increasing the amounts of oxygen vacancies and basic sites, and improving the metal dispersion and reducibility on CeO2-based catalysts by the addition of promotors can effectively improve the activity for CO2 methanation.
4.4. Exposing more active sites by pre-treatment
The purpose of pre-treatment is to expose more active sites before the methanation reaction is started. Generally, a reduction treatment is needed to generate active metal sites on the catalyst surface for CO2 and H2 activation. Lin et al.24 suggested that the activation treatment could determine the strength of MSI, and further impact the methanation activity over Ni/CeO2 catalysts (Fig. 11Ia). They found that the thermal treatment of Ni/CeO2 catalysts under N2 or air atmosphere decreased Ni dispersion and the number of oxygen vacancies, while annealed Ni/CeO2 under H2 atmosphere was distinctly beneficial for CO2 methanation due to an appropriate MSI effect. Subsequently, a series of Ni/CeO2 catalysts were pre-treated under an H2 atmosphere with different temperatures (400–550 °C) to verify the interaction between H2 and the catalyst. The conversion rate of CO2 over these catalysts followed the order: Ni/CeO2-450 °C > Ni/CeO2-400 °C > Ni/CeO2-500 °C > Ni/CeO2-550 °C (Fig. 11Ib). The results of H2 chemisorption indicated the exposed metallic Ni decreased with the increase in reduction temperature. This is attributed to the encapsulation of the Ni surface induced by the SMSI effect. A similar finding was reported by Li et al.;32 the appropriate reduction temperature for Ni/CeO2 catalyst prepared by wet-impregnation was 400–450 °C.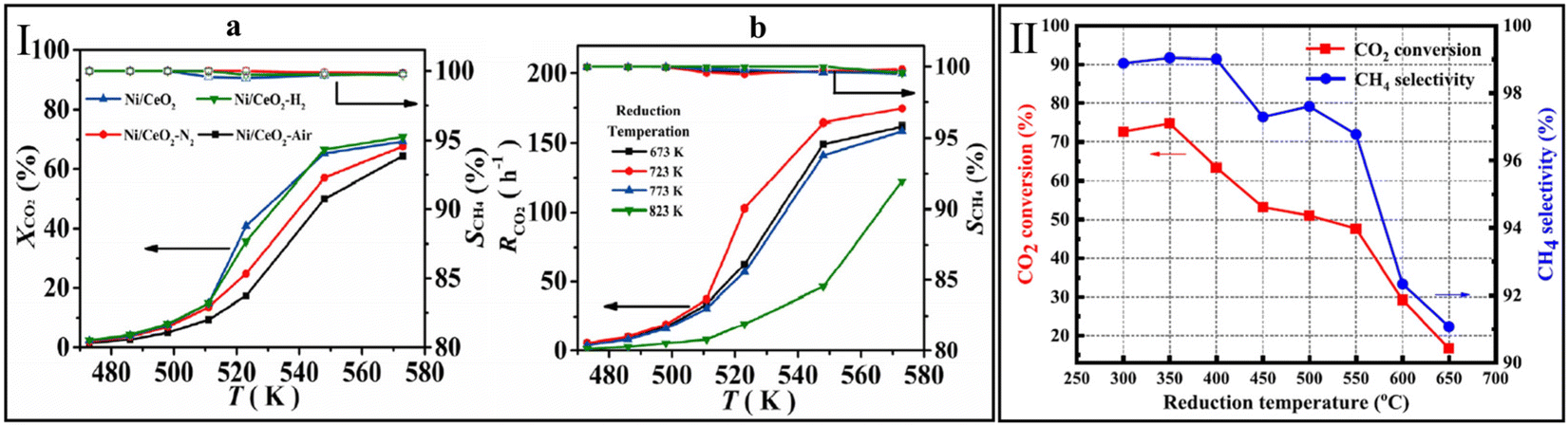 | ||
| Fig. 11 (I) Effect of heat treatment atmosphere (a) and reduction temperature (b) on CO2 conversion and CH4 selectivity over Ni/CeO2. Reproduced from ref. 24 with permission from Elsevier, copyright 2021. (II) CO2 methanation performance over the Ni/CeO2 samples reduced at different temperatures. Reproduced from ref. 151 with permission from American Chemical Society, copyright 2021. | ||
Huang et al.151 further investigated the effect of reduction temperature for CO2 methanation over Ni/CeO2 catalysts synthesized by a wet mixing method. Before the activity evaluation experiment, Ni/CeO2 was reduced under H2 flow at a set temperature (300–650 °C). The experimental results indicated the reduction temperature substantially affected CO2 conversion and CH4 selectivity over Ni/CeO2 catalysts. As shown in Fig. 11II, the highest methanation catalytic activity was observed over Ni/CeO2 sample reduced at 350 °C, and then declined rapidly as the reduction temperature raised to 650 °C. Unexpectedly, the high reduction temperature had a marginal impact on the Ni dispersion and Ni surface area of the Ni/CeO2 samples. However, many surface oxygen vacancies may be consumed at a high reduction temperature (>450 °C), causing poor catalytic performance.
In summary, an appropriate reduction temperature can indeed enhance the activity toward CO2 methanation. Unfortunately, little attention has been focused on the effect of pre-treatment conditions for CO2 methanation. Moreover, the previous views on the poor activity over Ni/CeO2 catalyst reduced at high temperatures remain controversial. This may be attributed to the different synthetic methods and the content of the metal. The reduction pre-treatment is almost necessary to expose more active sites for CO2 methanation catalysts. Thus, it is worthwhile to further investigate the interaction between CeO2-based catalysts and H2.
4.5. Tolerance of CeO2-based catalysts
Benefiting from the above-mentioned strategies, the low-temperature CO2 methanation activity over CeO2-based catalysts under laboratory-scale experiments has essentially met the requirements. Additionally, carrying out a methanation reaction at low temperature is effective for avoiding catalyst deactivation induced by sintering and carbon deposition.49,99 However, extra poisoning components such as chlorine, CO, and especially sulfur normally exist in industrial conditions.173–176 Generally, the poisoning effects are mainly caused by the occupation of active sites and the strong chemisorption of species.76Konishcheva et al.173 deemed that chlorine could interact with Ce3+ to form CeOCl-like species, which occupied active sites on the surface of CeO2 and inhibited CO2 hydrogenation. Afterward, they found that the existence of CO also inhibited the CO2 methanation reaction due to the active metal sites being clogged by strongly adsorbed CO species.174 Ahn et al.175 investigated the impact of H2S on Ni–Ce–Zr catalyst for CO2 methanation. They observed that the CO2 conversion decreased dramatically from ∼70% to ∼10% after the injection of 20–100 ppm H2S at 220 °C. H2S preferred to interact with CeO2 to form Ce2O2S species rather than Ni.177 This may be due to the fact that the formation of Ce2O2S is thermodynamically more favorable than that of NiS. In contrast to Ni/CeO2 and Ru/CeO2 catalysts, Mo-based methanation catalysts may be promising candidates due to their good sulfur-resistant properties.178–180 However, the current CO2 conversion and CH4 selectivity over Mo-based catalysts cannot meet the demand for efficient CO2 mitigation and fuel production.
Although the poisoning phenomenon and mechanism of methanation catalysts have been widely reported,181–184 few studies have focused on the strategies to improve the tolerance of CeO2-based methanation catalysts. Fortunately, many reports focusing on the preparation of anti-poisoning catalysts can provide constructive information for the perspectives of CeO2-based catalysts in CO2 methanation.60,185–191 Jeong et al.192 proposed that the hydrothermal treatment of Pd/CeO2 catalyst promoted the redispersion of Pd species and the formation of surface OH groups. This further increased the CO oxidation activity over Pd/CeO2 and the resistance of the sample to surface poisoning compounds (Fig. 12I). Using the citric acid-assisted hydrothermal method, Zheng et al.193 synthesized a porous Fe-doped CeO2 catalyst with a flower-like shape to achieve efficient H2S-selective oxidation activity. Meanwhile, the addition of Fe cations could alleviate the deactivation of CeO2 induced by sulfation (Fig. 12II). In addition to the modification of the CeO2 support, Xie et al.194 reported that the well-dispersed electron-deficient Rh particles may be beneficial for the enhancement of sulfur-tolerant performance.
 | ||
| Fig. 12 (I) Promoting effects of hydrothermal treatment on sulfur resistance of Pd/CeO2 catalysts. Reproduced from ref. 192 with permission from American Chemical Society, copyright 2017. (II) H2S oxidation over the Fe-doped CeO2 nanoflowers. Reproduced from ref. 193 with permission from American Chemical Society, copyright 2020. (III) Enhanced sulfur resistance of octahedral nano-ceria. Reproduced from ref. 195 with permission from American Chemical Society, copyright 2021. | ||
Kim et al.195 prepared a Pt catalyst supported on CeO2–Al2O3 with the octahedral morphology of CeO2 for CO oxidation in the presence of SO2. They found that this support with few Ce3+ defects exhibited better sulfur-resistant properties than that of the defective one (Fig. 12III). Recently, Hu et al.196 studied the effects of the Ce3+/Ce4+ ratio of CeO2/MoO3 catalyst on the selective catalytic reduction (SCR) of NOx with NH3. Contrary to the report of Kim et al.,195 Hu et al.196 suggested that a high Ce3+/Ce4+ ratio (≥0.5) could induce the appearance of the non-bulk electronic states for the CeO2 species, which led to the enhanced sulfur resistance of CeO2-based catalyst. Therefore, the applicability of these strategies for enhancing catalyst resistance to CeO2-based methanation catalysts requires further experimental verification.
5. Summaries and future prospects
This paper thoroughly reviewed the application of CeO2-based catalysts for CO2 hydrogenation to CH4. The existing literature has been summarized to understand the interaction between CO2/H2 and CeO2, to identify active sites, and to reveal the role of metal–support interaction for CO2 methanation. It was demonstrated that the reaction mechanism was highly correlated with the properties of the active site. Oxygen vacancy, as a key active site on CeO2, can be created by interacting with H2, and further promoting CO2 activation and conversion, while the metallic sites dissociated H2 to accelerate the hydrogenation of key intermediates. The formate route (CO2* → CO3*/HCO3* → HCOO* → CH4) was observed to be dominant on most of the CeO2-based catalysts with abundant oxygen vacancies, although the CO* route (CO2* → CO* → OCH2* → OCH3* → CH4) was possible.Based on the above discussion, CeO2-based catalysts could be optimized through the following ideas: (1) weakening the Ce–O bond to improve the reducibility, (2) adjusting the distribution of basic sites to boost the CO2 adsorption, (3) dispersing active metal supported on CeO2 to assist the dissociation of H2 and CO2, and (4) increasing the number of oxygen vacancies and additional active sites (such as active metal and OH groups) for CO2 hydrogenation. Popular strategies such as optimizing the structure of the catalysts, synthesizing specific morphology, and doping additional metal or oxide could be used to effectively achieve these purposes. Also, the pre-treatment was found to impact metal–support interaction, and thus the catalytic activity. The highly tunable nature of CeO2 makes the CeO2-based catalyst a promising material for application in CO2 methanation.
Despite the significant advances that have been made in the understanding and development of CeO2-based catalysts for CO2 methanation, several issues remain to be elucidated and solved. First, a current urgent demand is to clearly identify the evolution of active sites during the CO2 methanation reaction process by multiple in situ/operando characterizations combined with temperature-programmed surface reaction experiments, and further precisely establish the structure–performance relationship. Second, the evolution of active sites under reaction conditions should be linked to the transformation of active intermediates. Toward this goal, spectrokinetics studies and a more realistic model for DFT calculations is needed. Third, exploring a new way to precisely tailor the extent of MSI between active metal and CeO2 is necessary to understand the roles of MSI in CO2 methanation. Such a fundamental understanding of the methanation mechanism and catalytic sites at the atomic level is a pivotal premise for designing a better CeO2-based catalyst.
The CO2 methanation activity over CeO2-based catalysts under ideal reaction conditions has shown relatively satisfactory methanation performance. However, from a practical industrial point of view, challenges remain in developing a durable and low-cost CeO2-based methanation catalyst with considerable low-temperature activity. From a long-term perspective, one major challenge to ensuring a large-scale application of CO2 methanation is to obtain H2 economically and sustainably. Water electrolysis driven by renewable energy is an economic and environment-friendly technology for H2 generation. Besides, other methods of H2 production such as electrocatalysis, photocatalysis, thermal splitting, and high-temperature chemical cycles have been developed considerably in the last decades. Hence, the recycling of atmospheric CO2 into fuels or chemicals using cost-effective hydrogen will contribute to a sustainable society. To this end, an in-depth understanding of the reaction mechanism and catalyst development is the direction of future efforts.
Author contributions
Yu Xie: conceptualization, data curation, investigation, and writing – original draft; Junjie Wen: data curation and investigation; Zonglin Li: data curation and investigation; Jianjun Chen: writing – review & editing; Qiulin Zhang: conceptualization, funding acquisition, writing – review & editing; Ping Ning: supervision; Yaoqiang Chen: supervision; Jiming Hao: conceptualization and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (no. 2019YFC0214403) and Yunnan Fundamental Research Projects (no. 202201BE070001-025).Notes and references
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- W. Liu, L. Li, S. Lin, Y. Luo, Z. Bao, Y. Mao, K. Li, D. Wu and H. Peng, J. Energy Chem., 2022, 65, 34–47 CrossRef CAS.
- N. Altaf, S. Liang, L. Huang and Q. Wang, J. Energy Chem., 2020, 48, 169–180 CrossRef.
- Y. Gan, H. M. El-Houjeiri, A. Badahdah, Z. Lu, H. Cai, S. Przesmitzki and M. Wang, Nat. Commun., 2020, 11, 824 CrossRef CAS PubMed.
- M. Fan, J. D. Jimenez, S. N. Shirodkar, J. Wu, S. Chen, L. Song, M. M. Royko, J. Zhang, H. Guo, J. Cui, K. Zuo, W. Wang, C. Zhang, F. Yuan, R. Vajtai, J. Qian, J. Yang, B. I. Yakobson, J. M. Tour, J. Lauterbach, D. Sun and P. M. Ajayan, ACS Catal., 2019, 9, 10077–10086 CrossRef CAS.
- B. Rivas-Murias, J. M. Asensio, N. Mille, B. Rodriguez-Gonzalez, P. F. Fazzini, J. Carrey, B. Chaudret and V. Salgueirino, Angew. Chem., Int. Ed., 2020, 59, 15537–15542 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- E. Furimsky, Ind. Eng. Chem. Res., 2020, 59, 15393–15423 CrossRef CAS.
- Q. Song, N. Altaf, M. Zhu, J. Li, X. Ren, J. Dan, B. Dai, B. Louis, Q. Wang and F. Yu, Sustainable Energy Fuels, 2019, 3, 965–974 RSC.
- X. Jia, K. Sun, J. Wang, C. Shen and C.-j. Liu, J. Energy Chem., 2020, 50, 409–415 CrossRef.
- N. Rui, Z. Wang, K. Sun, J. Ye, Q. Ge and C.-j. Liu, Appl. Catal., B, 2017, 218, 488–497 CrossRef CAS.
- J. L. Snider, V. Streibel, M. A. Hubert, T. S. Choksi, E. Valle, D. C. Upham, J. Schumann, M. S. Duyar, A. Gallo, F. Abild-Pedersen and T. F. Jaramillo, ACS Catal., 2019, 9, 3399–3412 CrossRef CAS.
- Y. Yan, Y. Dai, Y. Yang and A. A. Lapkin, Appl. Catal., B, 2018, 237, 504–512 CrossRef CAS.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef CAS.
- S. Tada, S. Ikeda, N. Shimoda, T. Honma, M. Takahashi, A. Nariyuki and S. Satokawa, Int. J. Hydrogen Energy, 2017, 42, 30126–30134 CrossRef CAS.
- F. Hu, R. Ye, Z.-H. Lu, R. Zhang and G. Feng, Energy Fuels, 2021, 36, 156–169 CrossRef.
- J. Wang, Y. Mao, L. Zhang, Y. Li, W. Liu, Q. Ma, D. Wu and H. Peng, Fuel, 2022, 315, 123167 CrossRef CAS.
- X. Xu, L. Liu, Y. Tong, X. Fang, J. Xu, D.-e. Jiang and X. Wang, ACS Catal., 2021, 11, 5762–5775 CrossRef CAS.
- M. Mikhail, P. Da Costa, J. Amouroux, S. Cavadias, M. Tatoulian, M. E. Gálvez and S. Ognier, Appl. Catal., B, 2021, 294, 120233 CrossRef CAS.
- A. Vita, C. Italiano, L. Pino, P. Frontera, M. Ferraro and V. Antonucci, Appl. Catal., B, 2018, 226, 384–395 CrossRef CAS.
- N. Ullah, R. Tang and Z. Li, J. Taiwan Inst. Chem. Eng., 2022, 134, 104317 CrossRef CAS.
- J. Wang, G. Zhang, J. Zhu, X. Zhang, F. Ding, A. Zhang, X. Guo and C. Song, ACS Catal., 2021, 11, 1406–1423 CrossRef CAS.
- S. Lin, Z. Hao, J. Shen, X. Chang, S. Huang, M. Li and X. Ma, J. Energy Chem., 2021, 59, 334–342 CrossRef CAS.
- H. L. Huynh, J. Zhu, G. Zhang, Y. Shen, W. M. Tucho, Y. Ding and Z. Yu, J. Catal., 2020, 392, 266–277 CrossRef CAS.
- L. Wei, H. Grénman, W. Haije, N. Kumar, A. Aho, K. Eränen, L. Wei and W. de Jong, Appl. Catal., A, 2021, 612, 118012 CrossRef CAS.
- S. Zhang, Z. Xia, Y. Zou, F. Cao, Y. Liu, Y. Ma and Y. Qu, J. Am. Chem. Soc., 2019, 141, 11353–11357 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- A. Karelovic and P. Ruiz, ACS Catal., 2013, 3, 2799–2812 CrossRef CAS.
- S. Chen, A. M. Abdel-Mageed, M. Dyballa, M. Parlinska-Wojtan, J. Bansmann, S. Pollastri, L. Olivi, G. Aquilanti and R. J. Behm, Angew. Chem., Int. Ed., 2020, 59, 22763–22770 CrossRef CAS PubMed.
- M. Schubert, S. Pokhrel, A. Thomé, V. Zielasek, T. M. Gesing, F. Roessner, L. Mädler and M. Bäumer, Catal. Sci. Technol., 2016, 6, 7449–7460 RSC.
- M. Li, H. Amari and A. C. van Veen, Appl. Catal., B, 2018, 239, 27–35 CrossRef CAS.
- L. Torrente-Murciano, R. S. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Phys. Chem. Chem. Phys., 2016, 18, 15496–15500 RSC.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- X. Zou, Z. Shen, X. Li, Y. Cao, Q. Xia, S. Zhang, Y. Liu, L. Jiang, L. Li, L. Cui and Y. Wang, J. Colloid Interface Sci., 2022, 620, 77–85 CrossRef CAS PubMed.
- Z. Zhang, K. Feng and B. Yan, Catal. Sci. Technol., 2022, 12, 4698–4708 RSC.
- Y. Zhang, Z. Zhang, X. Yang, R. Wang, H. Duan, Z. Shen, L. Li, Y. Su, R. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Green Chem., 2020, 22, 6855–6861 RSC.
- T. Siudyga, M. Kapkowski, P. Bartczak, M. Zubko, J. Szade, K. Balin, S. Antoniotti and J. Polanski, Green Chem., 2020, 22, 5143–5150 RSC.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Mater. Today, 2020, 19, 100591 CrossRef.
- N. M. Martin, F. Hemmingsson, A. Schaefer, M. Ek, L. R. Merte, U. Hejral, J. Gustafson, M. Skoglundh, A.-C. Dippel, O. Gutowski, M. Bauer and P.-A. Carlsson, Catal. Sci. Technol., 2019, 9, 1644–1653 RSC.
- D. Jampaiah, D. Damma, A. Chalkidis, P. Venkataswamy, S. K. Bhargava and B. M. Reddy, Catal. Today, 2020, 356, 519–526 CrossRef CAS.
- G. Botzolaki, G. Goula, A. Rontogianni, E. Nikolaraki, N. Chalmpes, P. Zygouri, M. Karakassides, D. Gournis, N. Charisiou, M. Goula, S. Papadopoulos and I. Yentekakis, Catalysts, 2020, 10, 944 CrossRef.
- T. H. Nguyen, H. B. Kim and E. D. Park, Catalysts, 2022, 12, 212 CrossRef CAS.
- M. Safdar, M. González-Castaño, A. Penkova, M. A. Centeno, J. A. Odriozola and H. Arellano-García, Appl. Mater. Today, 2022, 29, 101577 CrossRef.
- F. Hu, S. Tong, K. Lu, C.-M. Chen, F.-Y. Su, J. Zhou, Z.-H. Lu, X. Wang, G. Feng and R. Zhang, J. CO2 Util., 2019, 34, 676–687 CrossRef CAS.
- Y. Zhang, X. Su, L. Li, H. Qi, C. Yang, W. Liu, X. Pan, X. Liu, X. Yang, Y. Huang and T. Zhang, ACS Catal., 2020, 10, 12967–12975 CrossRef CAS.
- Z. Zhao, Q. Jiang, Q. Wang, M. Wang, J. Zuo, H. Chen, Q. Kuang and Z. Xie, ACS Sustainable Chem. Eng., 2021, 9, 14288–14296 CrossRef CAS.
- E. Moioli and A. Züttel, Sustainable Energy Fuels, 2020, 4, 1396–1408 RSC.
- L. Shen, J. Xu, M. Zhu and Y.-F. Han, ACS Catal., 2020, 10, 14581–14591 CrossRef CAS.
- A. Parastaev, V. Muravev, E. Huertas Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CrossRef CAS.
- N. Ta, J. J. Liu, S. Chenna, P. A. Crozier, Y. Li, A. Chen and W. Shen, J. Am. Chem. Soc., 2012, 134, 20585–20588 CrossRef CAS PubMed.
- B. Chen, Y. Ma, L. Ding, L. Xu, Z. Wu, Q. Yuan and W. Huang, J. Phys. Chem. C, 2013, 117, 5800–5810 CrossRef CAS.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- F. Wang, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem. A, 2016, 4, 5773–5783 RSC.
- S. Lin, J. Wang, Y. Mi, S. Yang, Z. Wang, W. Liu, D. Wu and H. Peng, Chin. J. Catal., 2021, 42, 1808–1820 CrossRef CAS.
- M. Boaro, S. Colussi and A. Trovarelli, Front. Chem., 2019, 7, 28 CrossRef CAS.
- S. Jantarang, E. C. Lovell, T. H. Tan, B. Xie, J. Scott and R. Amal, Catal. Sci. Technol., 2021, 11, 5297–5309 RSC.
- S. Lin, Z. Li and M. Li, Fuel, 2023, 333, 126369 CrossRef CAS.
- C. Sun, K. Świrk, Y. Wang, K. S. Scheidl, D. W. Breiby, M. Rønning, C. Hu and P. Da Costa, Catal. Today, 2021, 382, 104–119 CrossRef CAS.
- C. Tang, H. Zhang and L. Dong, Catal. Sci. Technol., 2016, 6, 1248–1264 RSC.
- K. Chang, H. Zhang, M.-j. Cheng and Q. Lu, ACS Catal., 2019, 10, 613–631 CrossRef.
- X. Yuan, T. Pu, M. Gu, M. Zhu and J. Xu, ACS Catal., 2021, 11, 11966–11972 CrossRef CAS.
- X. Guo, H. He, A. Traitangwong, M. Gong, V. Meeyoo, P. Li, C. Li, Z. Peng and S. Zhang, Catal. Sci. Technol., 2019, 9, 5636–5650 RSC.
- T. T. V. Nguyen, N. Phung Anh, T. G. Ho, T. T. P. Pham, P. H. D. Nguyen, B. L. Do, H. K. P. Huynh and T. Nguyen, ACS Omega, 2022, 7, 36623–36633 CrossRef CAS PubMed.
- C. Italiano, J. Llorca, L. Pino, M. Ferraro, V. Antonucci and A. Vita, Appl. Catal., B, 2020, 264, 118494 CrossRef CAS.
- N. Rui, X. Zhang, F. Zhang, Z. Liu, X. Cao, Z. Xie, R. Zou, S. D. Senanayake, Y. Yang, J. A. Rodriguez and C.-J. Liu, Appl. Catal., B, 2021, 282, 119581 CrossRef CAS.
- T. Sakpal and L. Lefferts, J. Catal., 2018, 367, 171–180 CrossRef CAS.
- V. Alcalde-Santiago, A. Davó-Quiñonero, D. Lozano-Castelló, A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, ChemCatChem, 2018, 11, 810–819 Search PubMed.
- W. Gac, W. Zawadzki, M. Rotko, M. Greluk, G. Slowik, H. Pennemann, S. Neuberg, R. Zapf and G. Kolb, ChemPlusChem, 2021, 86, 889–903 CrossRef CAS PubMed.
- C. Han, Z. Cao, J. Yang, X. Lu, H. Liu, Z. Jin, Y. Zhang, S. Yang, X. Zheng and L. Wang, Crystals, 2022, 12, 702 CrossRef CAS.
- S. Singh, A. Modak, K. K. Pant, A. Sinhamahapatra and P. Biswas, ACS Appl. Nano Mater., 2021, 4, 8644–8667 CrossRef CAS.
- P. Da Costa, G. Hasrack, J. Bonnety and C. Henriques, Curr. Opin. Green Sustainable Chem., 2021, 32, 100540 CrossRef CAS.
- W. J. Lee, C. Li, H. Prajitno, J. Yoo, J. Patel, Y. Yang and S. Lim, Catal. Today, 2021, 368, 2–19 CrossRef CAS.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Catalysts, 2017, 7, 59 CrossRef.
- W. Gac, W. Zawadzki, M. Rotko, M. Greluk, G. Słowik and G. Kolb, Catal. Today, 2020, 357, 468–482 CrossRef CAS.
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- L. X. Wang, L. Wang and F. S. Xiao, Chem. Sci., 2021, 12, 14660–14673 RSC.
- Y. Wang, L. R. Winter, J. G. Chen and B. Yan, Green Chem., 2021, 23, 249–267 RSC.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- R.-P. Ye, Q. Li, W. Gong, T. Wang, J. J. Razink, L. Lin, Y.-Y. Qin, Z. Zhou, H. Adidharma, J. Tang, A. G. Russell, M. Fan and Y.-G. Yao, Appl. Catal., B, 2020, 268, 118474 CrossRef CAS.
- D. Schweke, L. Shelly, R. Ben David, A. Danon, N. Kostirya and S. Hayun, J. Phys. Chem. C, 2020, 124, 6180–6187 CrossRef CAS.
- Y. Ma, W. Gao, Z. Zhang, S. Zhang, Z. Tian, Y. Liu, J. C. Ho and Y. Qu, Surf. Sci. Rep., 2018, 73, 1–36 CrossRef.
- H. Li, Y. Cui, Q. Liu and W.-L. Dai, ChemCatChem, 2018, 10, 619–624 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- K. Yoshikawa, H. Sato, M. Kaneeda and J. N. Kondo, J. CO2 Util., 2014, 8, 34–38 CrossRef CAS.
- K. R. Hahn, M. Iannuzzi, A. P. Seitsonen and J. Hutter, J. Phys. Chem. C, 2013, 117, 1701–1711 CrossRef CAS.
- A. H. Ruhaimi and M. A. Ab Aziz, Chem. Phys. Lett., 2021, 779, 138842 CrossRef CAS.
- M. Zhu, P. Tian, X. Cao, J. Chen, T. Pu, B. Shi, J. Xu, J. Moon, Z. Wu and Y.-F. Han, Appl. Catal., B, 2021, 282, 119561 CrossRef CAS.
- C. Zhu, X. Wei, W. Li, Y. Pu, J. Sun, K. Tang, H. Wan, C. Ge, W. Zou and L. Dong, ACS Sustainable Chem. Eng., 2020, 8, 14397–14406 CrossRef CAS.
- G. Varvoutis, M. Lykaki, S. Stefa, V. Binas, G. E. Marnellos and M. Konsolakis, Appl. Catal., B, 2021, 297, 120401 CrossRef CAS.
- J. Zhang, Y. Yang, J. Liu and B. Xiong, Appl. Surf. Sci., 2021, 558, 149866 CrossRef CAS.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Catal., B, 2020, 265, 118538 CrossRef.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS.
- Z. Hao, J. Shen, S. Lin, X. Han, X. Chang, J. Liu, M. Li and X. Ma, Appl. Catal., B, 2021, 286, 119922 CrossRef CAS.
- E. Marconi, S. Tuti and I. Luisetto, Catalysts, 2019, 9, 375 CrossRef CAS.
- L. Lin, C. A. Gerlak, C. Liu, J. Llorca, S. Yao, N. Rui, F. Zhang, Z. Liu, S. Zhang, K. Deng, C. B. Murray, J. A. Rodriguez and S. D. Senanayake, J. Energy Chem., 2021, 61, 602–611 CrossRef CAS.
- S. López-Rodríguez, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, I. J. Villar-Garcia, V. P. Dieste, J. A. O. Calvo, J. R. G. Velasco and A. Bueno-López, J. CO2 Util., 2022, 60, 101980 CrossRef.
- F. Wang, C. Li, X. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177–186 CrossRef CAS.
- Y. Xie, J. Chen, X. Wu, J. Wen, R. Zhao, Z. Li, G. Tian, Q. Zhang, P. Ning and J. Hao, ACS Catal., 2022, 12, 10587–10602 CrossRef CAS.
- R. K. Singha, A. Shukla, A. Yadav, L. N. Sivakumar Konathala and R. Bal, Appl. Catal., B, 2017, 202, 473–488 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- M. G. Willinger, W. Zhang, O. Bondarchuk, S. Shaikhutdinov, H. J. Freund and R. Schlogl, Angew. Chem., Int. Ed., 2014, 53, 5998–6001 CrossRef CAS PubMed.
- X. Liu, M. H. Liu, Y. C. Luo, C. Y. Mou, S. D. Lin, H. Cheng, J. M. Chen, J. F. Lee and T. S. Lin, J. Am. Chem. Soc., 2012, 134, 10251–10258 CrossRef CAS PubMed.
- J. Dong, Q. Fu, H. Li, J. Xiao, B. Yang, B. Zhang, Y. Bai, T. Song, R. Zhang, L. Gao, J. Cai, H. Zhang, Z. Liu and X. Bao, J. Am. Chem. Soc., 2020, 142, 17167–17174 CrossRef CAS PubMed.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang and M. Haruta, Sci. Adv., 2017, 3, e1700231 CrossRef PubMed.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS PubMed.
- L. P. Matte, A. S. Kilian, L. Luza, M. C. M. Alves, J. Morais, D. L. Baptista, J. Dupont and F. Bernardi, J. Phys. Chem. C, 2015, 119, 26459–26470 CrossRef CAS.
- P. H. Ho, G. Sanghez de Luna, A. Poggi, M. Nota, E. Rodríguez-Castellón, G. Fornasari, A. Vaccari and P. Benito, Ind. Eng. Chem. Res., 2021, 60, 6730–6741 CrossRef CAS.
- T. Pu, J. Chen, W. Tu, J. Xu, Y.-F. Han, I. E. Wachs and M. Zhu, J. Catal., 2022, 413, 821–828 CrossRef CAS.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493–6513 CrossRef CAS.
- S. López-Rodríguez, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló and A. Bueno-López, Mol. Catal., 2021, 515, 111911 CrossRef.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- X. Li, J. Lin, L. Li, Y. Huang, X. Pan, S. E. Collins, Y. Ren, Y. Su, L. Kang, X. Liu, Y. Zhou, H. Wang, A. Wang, B. Qiao, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19983–19989 CrossRef CAS PubMed.
- L. P. L. Gonçalves, J. Mielby, O. S. G. P. Soares, J. P. S. Sousa, D. Y. Petrovykh, O. I. Lebedev, M. F. R. Pereira, S. Kegnæs and Y. V. Kolen'ko, Appl. Catal., B, 2022, 312, 121376 CrossRef.
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef CAS.
- I. Luisetto, S. Stendardo, S. M. Senthil Kumar, K. Selvakumar, J. K. Kesavan, G. Iucci, U. Pasqual Laverdura and S. Tuti, Processes, 2021, 9, 1899 CrossRef CAS.
- M. V. Konishcheva, D. I. Potemkin, S. D. Badmaev, P. V. Snytnikov, E. A. Paukshtis, V. A. Sobyanin and V. N. Parmon, Top. Catal., 2016, 59, 1424–1430 CrossRef CAS.
- J. Ren, H. Guo, J. Yang, Z. Qin, J. Lin and Z. Li, Appl. Surf. Sci., 2015, 351, 504–516 CrossRef CAS.
- P. Hongmanorom, J. Ashok, P. Chirawatkul and S. Kawi, Appl. Catal., B, 2021, 297, 120454 CrossRef CAS.
- S. M. Lee, Y. H. Lee, D. H. Moon, J. Y. Ahn, D. D. Nguyen, S. W. Chang and S. S. Kim, Ind. Eng. Chem. Res., 2019, 58, 8656–8662 CrossRef CAS.
- Y. Yu, Y. M. Chan, Z. Bian, F. Song, J. Wang, Q. Zhong and S. Kawi, Int. J. Hydrogen Energy, 2018, 43, 15191–15204 CrossRef CAS.
- J. A. Onrubia-Calvo, A. Quindimil, A. Davó-Quiñonero, A. Bermejo-López, E. Bailón-García, B. Pereda-Ayo, D. Lozano-Castelló, J. A. González-Marcos, A. Bueno-López and J. R. González-Velasco, Ind. Eng. Chem. Res., 2022, 61, 10419–10435 CrossRef CAS.
- X. Jia, X. Zhang, N. Rui, X. Hu and C.-j. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- A. Solis-Garcia, J. F. Louvier-Hernandez, A. Almendarez-Camarillo and J. C. Fierro-Gonzalez, Appl. Catal., B, 2017, 218, 611–620 CrossRef CAS.
- Y.-x. Pan, C.-j. Liu and Q. Ge, J. Catal., 2010, 272, 227–234 CrossRef CAS.
- T. Zhang, W. Wang, F. Gu, W. Xu, J. Zhang, Z. Li, T. Zhu, G. Xu, Z. Zhong and F. Su, Appl. Catal., B, 2022, 312, 121385 CrossRef CAS.
- H.-Y. Ma and G.-C. Wang, J. Phys. Chem. C, 2021, 125, 18161–18169 CrossRef CAS.
- S. Sharma, K. B. Sravan Kumar, Y. M. Chandnani, V. S. Phani Kumar, B. P. Gangwar, A. Singhal and P. A. Deshpande, J. Phys. Chem. C, 2016, 120, 14101–14112 CrossRef CAS.
- S. Sharma, Z. Hu, P. Zhang, E. W. McFarland and H. Metiu, J. Catal., 2011, 278, 297–309 CrossRef CAS.
- S. López-Rodríguez, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, F. C. Herrera, E. Pellegrin, C. Escudero, M. García-Melchor and A. Bueno-López, J. Phys. Chem. C, 2021, 125, 25533–25544 CrossRef.
- L. Atzori, M. G. Cutrufello, D. Meloni, R. Monaci, C. Cannas, D. Gazzoli, M. F. Sini, P. Deiana and E. Rombi, Int. J. Hydrogen Energy, 2017, 42, 20689–20702 CrossRef CAS.
- X. Feng, K. Wang, M. Zhou, F. Li, J. Liu, M. Zhao, L. Zhao, X. Song, P. Zhang and L. Gao, Ceram. Int., 2021, 47, 12366–12374 CrossRef CAS.
- R. Tang, N. Ullah, Y. Hui, X. Li and Z. Li, Mol. Catal., 2021, 508, 111602 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, H. Xie, Z. Jiao, G. Zhang, K. Xiong and X. Zheng, Int. J. Hydrogen Energy, 2017, 42, 16108–16117 CrossRef CAS.
- A. Cárdenas-Arenas, H. S. Cortés, E. Bailón-García, A. Davó-Quiñonero, D. Lozano-Castelló and A. Bueno-López, Fuel Process. Technol., 2021, 212, 106637 CrossRef.
- L. Wang, J. Hu, H. Liu, Q. Wei, D. Gong, L. Mo, H. Tao and C. Zhang, Catalysts, 2020, 10, 523 CrossRef CAS.
- G. Zhou, S. Xu, Y. Li, Q. Jiang, B. Xu, H. Xie and G. Zhang, ChemNanoMat, 2022, 8, e202100533 CAS.
- F. Hu, R. Ye, C. Jin, D. Liu, X. Chen, C. Li, K. H. Lim, G. Song, T. Wang, G. Feng, R. Zhang and S. Kawi, Appl. Catal., B, 2022, 317, 121715 CrossRef CAS.
- Z. Bian, Y. M. Chan, Y. Yu and S. Kawi, Catal. Today, 2020, 347, 31–38 CrossRef CAS.
- T. Jomjaree, P. Sintuya, A. Srifa, W. Koo-amornpattana, S. Kiatphuengporn, S. Assabumrungrat, M. Sudoh, R. Watanabe, C. Fukuhara and S. Ratchahat, Catal. Today, 2021, 375, 234–244 CrossRef CAS.
- Y. Du, C. Qin, Y. Xu, D. Xu, J. Bai, G. Ma and M. Ding, Chem. Eng. J., 2021, 418, 129402 CrossRef CAS.
- T. A. Le, T. W. Kim, S. H. Lee and E. D. Park, Catal. Today, 2018, 303, 159–167 CrossRef CAS.
- K. Liu, X. Xu, J. Xu, X. Fang, L. Liu and X. Wang, J. CO2 Util., 2020, 38, 113–124 CrossRef CAS.
- L. Xu, Y. Cui, M. Chen, X. Wen, C. Lv, X. Wu, C.-e. Wu, Z. Miao and X. Hu, Ind. Eng. Chem. Res., 2021, 60, 8056–8072 CrossRef CAS.
- G. Hasrack, M. C. Bacariza, C. Henriques and P. Da Costa, Catalysts, 2021, 12, 36 CrossRef.
- E. le Saché, L. Pastor-Pérez, B. J. Haycock, J. J. Villora-Picó, A. Sepúlveda-Escribano and T. R. Reina, ACS Sustainable Chem. Eng., 2020, 8, 4614–4622 CrossRef.
- G. I. Siakavelas, N. D. Charisiou, S. AlKhoori, A. A. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, Appl. Catal., B, 2021, 282, 119562 CrossRef CAS.
- A. I. Tsiotsias, N. D. Charisiou, A. AlKhoori, S. Gaber, V. Stolojan, V. Sebastian, B. van der Linden, A. Bansode, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, J. Energy Chem., 2022, 71, 547–561 CrossRef CAS.
- C. Sun, P. Beaunier, V. La Parola, L. F. Liotta and P. Da Costa, ACS Appl. Nano Mater., 2020, 3, 12355–12368 CrossRef CAS.
- S. L. Rodríguez, A. Davo-Quinonero, J. Juan-Juan, E. Bailon-Garcia, D. Lozano-Castello and A. Bueno-Lopez, J. Phys. Chem. C, 2021, 125, 12038–12049 CrossRef.
- P. Huang, Y. Guo, G. Wang, J. Yu, C. Zhao, X. Wang and T. Wang, Energy Fuels, 2021, 35, 20185–20196 CrossRef CAS.
- W. L. Vrijburg, J. W. A. van Helden, A. Parastaev, E. Groeneveld, E. A. Pidko and E. J. M. Hensen, Catal. Sci. Technol., 2019, 9, 5001–5010 RSC.
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS.
- L. Fan, J. Zhang, K. Ma, Y. Zhang, Y.-M. Hu, L. Kong, A.-p. Jia, Z. Zhang, W. Huang and J.-Q. Lu, J. Catal., 2021, 397, 116–127 CrossRef CAS.
- B. Yang, Y. Wang, L. Li, B. Gao, L. Zhang and L. Guo, Catal. Sci. Technol., 2022, 12, 1159–1172 RSC.
- R. Khobragade, M. Roškarič, G. Žerjav, M. Košiček, J. Zavašnik, N. Van de Velde, I. Jerman, N. N. Tušar and A. Pintar, Appl. Catal., A, 2021, 627, 118394 CrossRef CAS.
- Y. Bian, C. Xu, X. Wen, L. Xu, Y. Cui, S. Wang, C.-e. Wu, J. Qiu, G. Cheng and M. Chen, Fuel, 2023, 331, 125755 CrossRef CAS.
- M. Konsolakis, M. Lykaki, S. Stefa, S. A. C. Carabineiro, G. Varvoutis, E. Papista and G. E. Marnellos, Nanomaterials, 2019, 9, 1739 CrossRef CAS PubMed.
- X. Zhang, R. You, D. Li, T. Cao and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 35897–35907 CrossRef CAS.
- X. Gao, S. Zhu, M. Dong, J. Wang and W. Fan, J. Catal., 2020, 389, 60–70 CrossRef CAS.
- Y. Zhang, W. Feng, F. Yang and X. Bao, Chin. J. Catal., 2019, 40, 204–213 CrossRef CAS.
- S. Zhang, Z. Q. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C. R. Chang and Y. Qu, Nat. Commun., 2017, 8, 15266 CrossRef CAS.
- W. Huang and Y. Gao, Catal. Sci. Technol., 2014, 4, 3772–3784 RSC.
- Z. Zhang, Z. Yu, K. Feng and B. Yan, Appl. Catal., B, 2022, 317, 121800 CrossRef CAS.
- I. Iglesias, A. Quindimil, F. Mariño, U. De-La-Torre and J. R. González-Velasco, Int. J. Hydrogen Energy, 2019, 44, 1710–1719 CrossRef CAS.
- C. Mebrahtu, S. Abate, S. Perathoner, S. Chen and G. Centi, Catal. Today, 2018, 304, 181–189 CrossRef CAS.
- Z. Zhang, Y. Tong, X. Fang, J. Xu, X. Xu and X. Wang, Fuel, 2022, 316, 123191 CrossRef CAS.
- P. Frontera, A. Malara, V. Modafferi, V. Antonucci, P. Antonucci and A. Macario, Can. J. Chem. Eng., 2020, 98, 1924–1934 CrossRef CAS.
- P. Frontera, A. Macario, A. Malara, V. Antonucci, V. Modafferi and P. L. Antonucci, Catal. Today, 2020, 357, 565–572 CrossRef CAS.
- L. Pastor-Pérez, E. L. Saché, C. Jones, S. Gu, H. Arellano-Garcia and T. R. Reina, Catal. Today, 2018, 317, 108–113 CrossRef.
- N. Elia, J. Estephane, C. Poupin, B. El Khoury, L. Pirault-Roy, S. Aouad and E. A. Aad, ChemCatChem, 2021, 13, 1559–1567 CrossRef CAS.
- P. Frontera, A. Macario, G. Monforte, G. Bonura, M. Ferraro, G. Dispenza, V. Antonucci, A. S. Aricò and P. L. Antonucci, Int. J. Hydrogen Energy, 2017, 42, 26828–26842 CrossRef CAS.
- M. V. Konishcheva, D. I. Potemkin, P. V. Snytnikov, O. A. Stonkus, V. D. Belyaev and V. A. Sobyanin, Appl. Catal., B, 2018, 221, 413–421 CrossRef CAS.
- M. V. Konishcheva, D. I. Potemkin, P. V. Snytnikov and V. A. Sobyanin, Int. J. Hydrogen Energy, 2019, 44, 9978–9986 CrossRef CAS.
- J. Ahn, W. Chung and S. Chang, Catalysts, 2021, 11, 1292 CrossRef CAS.
- B. Wang, J. Zhao, D. Meng, Z. Li, Y. Xu and X. Ma, Appl. Organomet. Chem., 2018, 32, e4515 CrossRef.
- A. Alarcón, J. Guilera, R. Soto and T. Andreu, Appl. Catal., B, 2020, 263, 118346 CrossRef.
- Q. Wang, Z. Yin, B. Wang, Y. Xu, X. Ma and Z. Li, Mol. Catal., 2022, 517, 112057 CrossRef CAS.
- M. Jiang, B. Wang, Y. Yao, H. Wang, Z. Li, X. Ma, S. Qin and Q. Sun, Catal. Commun., 2013, 35, 32–35 CrossRef CAS.
- B. Wang, W. Yu, W. Wang, Z. Li, Y. Xu and X. Ma, Chin. J. Chem. Eng., 2018, 26, 509–513 CrossRef CAS.
- B. Legras, V. V. Ordomsky, C. Dujardin, M. Virginie and A. Y. Khodakov, ACS Catal., 2014, 4, 2785–2791 CrossRef CAS.
- K. Lee, C. Song and M. J. Janik, Appl. Catal., A, 2010, 389, 122–130 CrossRef CAS.
- I. Czekaj, R. Struis, J. Wambach and S. Biollaz, Catal. Today, 2011, 176, 429–432 CrossRef CAS.
- M. Wolf, C. Schüler and O. Hinrichsen, J. CO2 Util., 2019, 32, 80–91 CrossRef CAS.
- J. Zhou, R.-t. Guo, X.-f. Zhang, Y.-z. Liu, C.-p. Duan, G.-l. Wu and W.-g. Pan, Energy Fuels, 2021, 35, 2981–2998 CrossRef CAS.
- W. Yang, J. Gong, X. Wang, Z. Bao, Y. Guo and Z. Wu, ACS Catal., 2021, 11, 12446–12468 CrossRef CAS.
- X. Qi, L. Han, J. Deng, T. Lan, F. Wang, L. Shi and D. Zhang, Environ. Sci. Technol., 2022, 56, 5840–5848 CrossRef CAS PubMed.
- C. Lv, J. Zhang, L. Yan, H. Chen and M. Hu, J. Environ. Chem. Eng., 2022, 10, 108372 CrossRef CAS.
- Z. Lu, J. Kullgren, Z. Yang and K. Hermansson, J. Phys. Chem. C, 2012, 116, 8417–8425 CrossRef CAS.
- L. Zhang, H. Qu, T. Du, W. Ma and Q. Zhong, Chem. Eng. J., 2016, 296, 122–131 CrossRef CAS.
- C. Liu, J.-W. Shi, C. Gao and C. Niu, Appl. Catal., A, 2016, 522, 54–69 CrossRef CAS.
- H. Jeong, J. Bae, J. W. Han and H. Lee, ACS Catal., 2017, 7, 7097–7105 CrossRef CAS.
- X. Zheng, Y. Li, Y. Zheng, L. Shen, Y. Xiao, Y. Cao, Y. Zhang, C. Au and L. Jiang, ACS Catal., 2020, 10, 3968–3983 CrossRef CAS.
- C. Xie, Y. Chen, M. H. Engelhard and C. Song, ACS Catal., 2012, 2, 1127–1137 CrossRef CAS.
- B.-S. Kim, J. Bae, H. Jeong, C. Choe and H. Lee, ACS Catal., 2021, 11, 7154–7159 CrossRef CAS.
- X. Hu, J. Chen, W. Qu, R. Liu, D. Xu, Z. Ma and X. Tang, Environ. Sci. Technol., 2021, 55, 5435–5441 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |