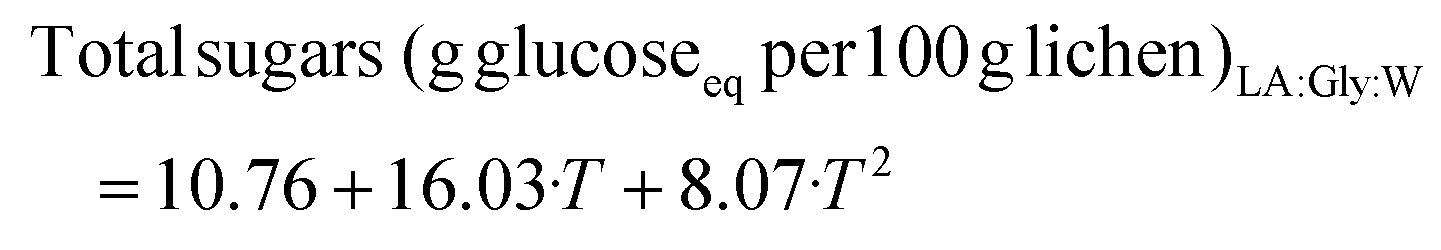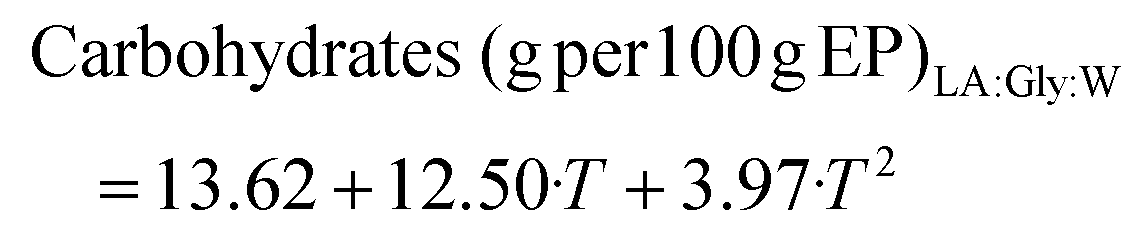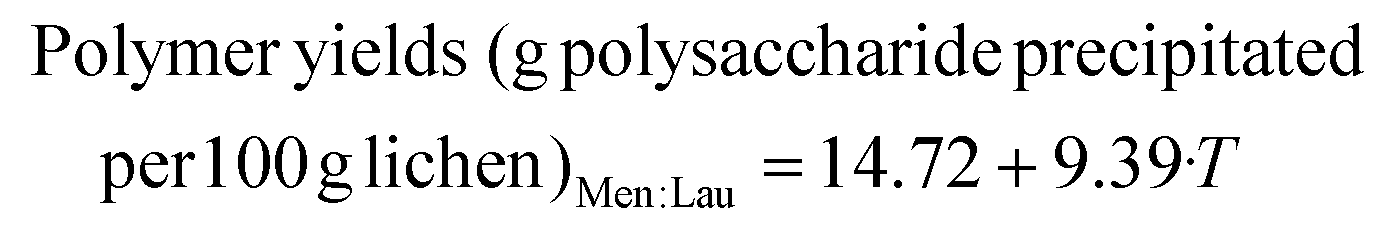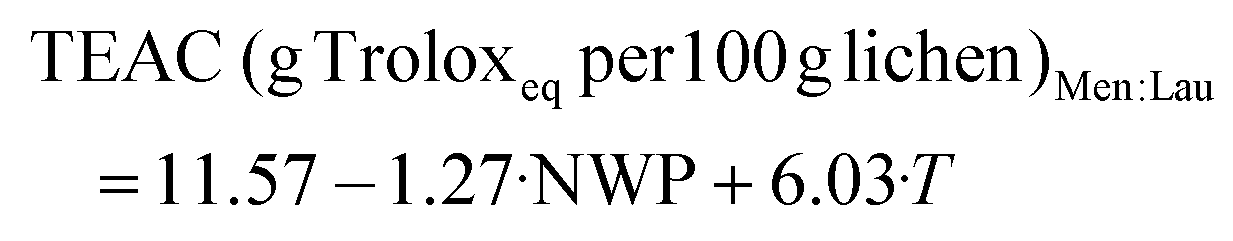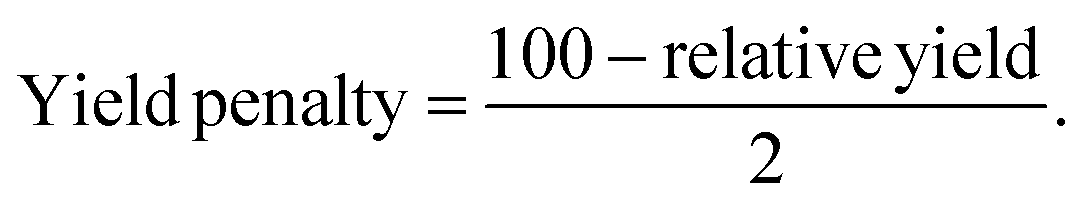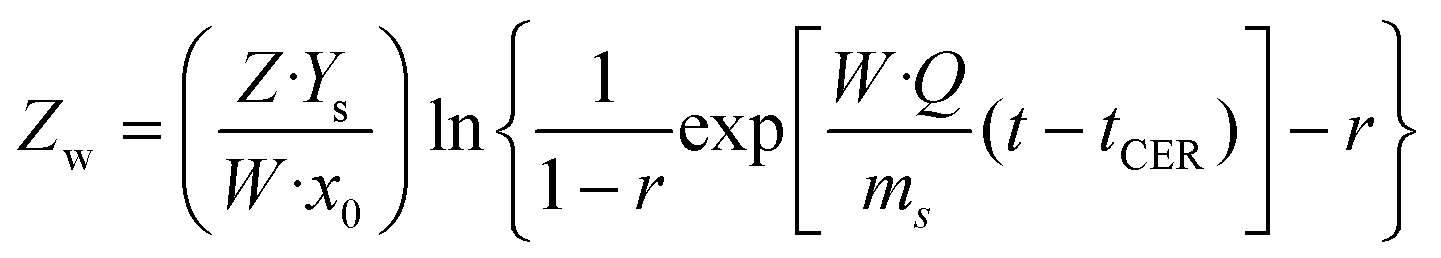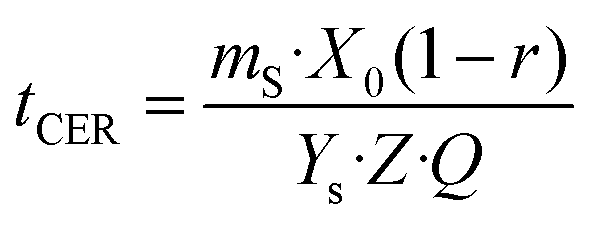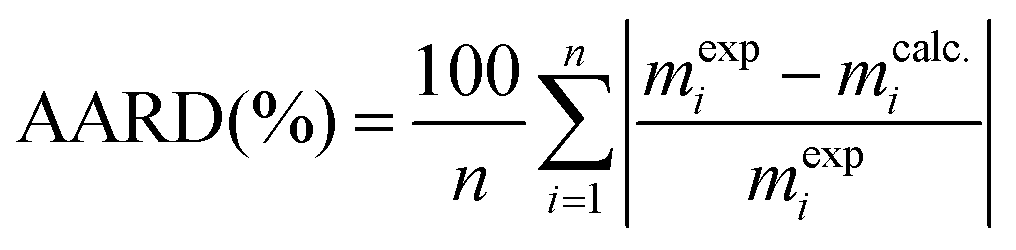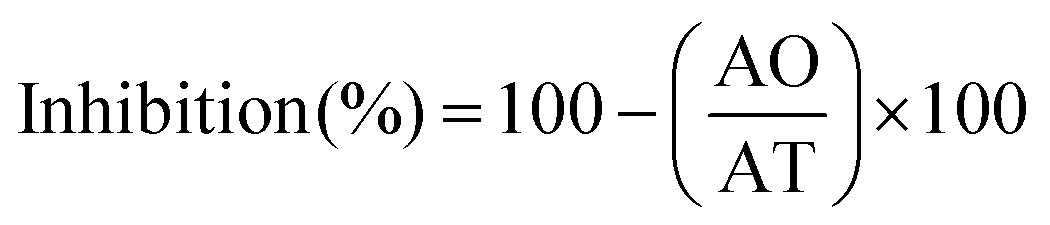DOI:
10.1039/D4GC02741H
(Paper)
Green Chem., 2024,
26, 10205-10224
Alternatives for the extraction of bioactives and biopolymers from Evernia prunastri for the formulation of antimicrobial bio-based films†
Received
6th June 2024
, Accepted 8th August 2024
First published on 13th August 2024
Abstract
The recent growing interest in the biological properties of lichen metabolites has evidenced different needs and challenges for further exploration, including the development of green processing with safer solvents and more efficient use of energy. Microwave assisted hydrothermal processing, applied after supercritical CO2 extraction, was proposed for the sequential extraction of bioactives and biopolymer fractions. Alternatively, it was combined with natural deep eutectics (NaDES) as cosolvents. Lichenic acids, antioxidants and oligosaccharides were simultaneously extracted using NaDES, and the recovered polysaccharides showed adequate mechanical properties for the formulation of films with antimicrobial action against Gram positive bacteria. An environmental assessment of the three different processes using the Eco-Scale suggested that the NaDES microwave extraction was, due to its low toxicity and good extraction yield of polysaccharides, the most sustainable of the three processes.
1. Introduction
Lichens have been used in traditional medicine, perfumery,1 and cosmetics2,3 and their unique metabolites provide new options for the development of functional foods and/or drugs.2–6 The major lichen components are polysaccharides, such as the linear 1,3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,4 β-D-glucan or lichenan, α-glucans or galactomannans that account for around 60% of the total.7 The hydrolysis of lichenan using lichenase has been proposed for bioconversion to bioethanol in a biorefinery approach,8 and more recently, attention is being paid to its anti-tumor, anti-inflammatory and immunostimulatory activities.8–10 Secondary metabolites include lichenic acids, terpenoids and sterols, and account for up to 5–20% of the dry weight.4,11 One extensively studied metabolite with a wide spectrum of activities is usnic acid12,13 although with controversial safety for oral or intravenous aplications.16 Lichens also produce simple phenolic compounds, playing an important role in the regulation of growth, development and protection. Their content has been correlated with the antioxidant and antimicrobial properties.5,12,17,18
:1,4 β-D-glucan or lichenan, α-glucans or galactomannans that account for around 60% of the total.7 The hydrolysis of lichenan using lichenase has been proposed for bioconversion to bioethanol in a biorefinery approach,8 and more recently, attention is being paid to its anti-tumor, anti-inflammatory and immunostimulatory activities.8–10 Secondary metabolites include lichenic acids, terpenoids and sterols, and account for up to 5–20% of the dry weight.4,11 One extensively studied metabolite with a wide spectrum of activities is usnic acid12,13 although with controversial safety for oral or intravenous aplications.16 Lichens also produce simple phenolic compounds, playing an important role in the regulation of growth, development and protection. Their content has been correlated with the antioxidant and antimicrobial properties.5,12,17,18
Secondary metabolites are mostly hydrophobic, soluble in organic solvents, such as acetone, trichloromethane, hexane, ethylacetate, methanol, chloroform11–14,17 and supercritical carbon dioxide.17 Conventional solvent extraction remains common,17–19 but greener alternatives are in increasing demand for safety and environmental issues.20 Representative examples of the use of green solvents can be mentioned. Komaty et al.21 selected nontoxic ionic liquids to selectively extract bioactives from Pseudevernia furfuracea, although degradation of certain compounds could occur. Kulinowska et al.22 proposed the extraction of usnic acid with thymol:camphor (1:0.3) in short times with higher yield than those attained with acetone, methanol and ethanol. Supercritical carbon dioxide at lab, pilot and industrial scales has been proposed for the extraction of bioactives from Usnea sp17,23–25 and Cladonia sp26 and microwave hydrothermal treatment has been proposed to extract bioactives and gelling polysaccharides from Evernia prunastri. The hydrogels separated showed antifreezing behaviour, were non-skin irritant and exhibited antioxidant, anti-tyrosinase, antiproliferative and anti-inflammatory properties.27
The extraction ability and selectivity of subcritical water extraction can be enhanced by the addition of eutectic solvents (DES) as co-solvents. This combination was used for the extraction of alginate and fucoidan from Saccharina japonica.28 Since then, it has been proposed for the extraction of xanthone and phenolics from mangosteen pericarp,29 anthocyanins from black beans hulls,30 pectin from Jaboticaba processing by-product,31 lipids from microalgae,32 phenolic compounds from winemaking by-products33 and sugar, protein, and phenolics from brewery spent grain.34 In addition, the assistance of DES can aid in improving the extraction efficiency of Lentinus edodes polysaccharides providing stronger antioxidant activity than subcritical water alone.35 There is no report on the hydrothermal extraction enhanced with DES for extracting polysaccharides from lichens.
The lichen slow-growth rate and the difficulties in cultivating in laboratories have limited the practical and commercial interest. In nature, lichen is present in a wide range of habitats and has ecological relevance in ecosystems with abundant lichen cover. For biodiversity conservation, sustainable forest management should consider that not all species are tolerant to intensification,36 and low-intensity harvesting is needed for their regeneration.37 However, lichens are present in waste forestal biomass and low-value tree fractions, often discarded or combusted. The implementation of an extraction stage as an initial stage of lignocellulosic biorefineries could lead to high value-added products, which favour the economics of the process.38,39 Some estimations can be illustrative of the number of resources that the lichen represents. In tropical dry forests the epiphyte crustose lichens account for 1.34–1.99 t ha−1, around 61% of the foliar biomass in the forest,40 whereas they are found as a small fraction in litter-fall.41 In old rockrose plants from Mediterranean areas, abundant amounts of lichens, especially Evernia prunastri, are found.42
The aim of this work is to evaluate different sequences based on green solvents for the extraction of the bioactive lichenic acids, antioxidant compounds and hydrocolloid fractions from Evernia prunastri biomass, including supercritical CO2 followed by microwave assisted hydrothermal extraction or a single stage of microwave assisted hydrothermal extraction with natural DES as cosolvent.
2. Results and discussion
2.1. Sequential supercritical extraction and microwave assisted hydrothermal processing
Extraction with apolar organic solvents can enhance further extraction and purification of solutes found in inner lichen cell structures and could be addressed before the aqueous extraction of oligosaccharides.50,51 The mechanical pretreatment of lichens is a key factor due to their unique and irregular distribution of metabolites onto the surface, intra- and intercellularly. The grinding technique markedly influences the morphology and length of the hyphae; milling generates a mixture of small pieces,52,53 whereas flaking avoids fine powders causing plugging and compaction.17,21,54 Microwave heating offers advantages regarding increased yields, lowered extraction time and solvent consumption compared to conventional methods, due to the enhanced heat and mass transfer. The structural matrix damage allows reduced severity of the mechanical pretreatment, which strongly influences the extraction rate, yield and quality of the extract.15 The influence of a short microwave pretreatment on supercritical fluid extraction was assessed for the extraction of usnic acid, as a representative metabolite.
Experimental data from the supercritical CO2 extraction of E. prunastri at 35 MPa and 40 °C using 5% ethanol as cosolvent are shown in Fig. 1, and also the predicted kinetics with the parameters from the model of Sovová, based on the adjustable parameters from Table 1. Pressure and temperature were fixed at values in the range of those leading to high yields and usnic acid purity from other species, avoiding higher values causing thermal degradation.17,26 The overall extraction curves (OECs) obtained show the typical behaviour of SFE kinetics. At the beginning of the extraction the curve exhibits a linear trend, the constant extraction rate (CER) stage, where the easily accessible solutes are extracted by convection. Once the most easily accessible solute was extracted, the extraction rate gradually decreased. In this falling extraction rate (FER) stage limitations to mass transfer by diffusion start to play a significant role. Finally, a diffusion-controlled (DC) process occurs, where the mass transfer is governed only by the diffusion mechanism.
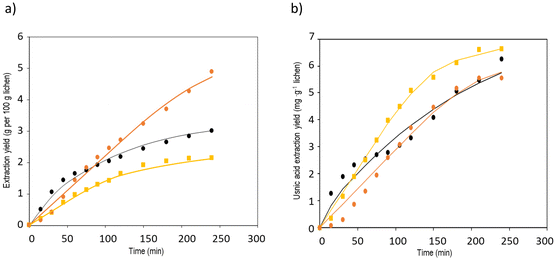 |
| | Fig. 1 Influence of the microwave ( , 1 min 480 W; , 1 min 480 W;  , 1 min 800 W) pretreatment on the supercritical carbon dioxide extraction from Evernia prunastri at 35 MPa and 40 °C and with 5% ethanol as cosolvent. (a) Overall extraction yield and (b) usnic acid extraction yield. Control experiment ( , 1 min 800 W) pretreatment on the supercritical carbon dioxide extraction from Evernia prunastri at 35 MPa and 40 °C and with 5% ethanol as cosolvent. (a) Overall extraction yield and (b) usnic acid extraction yield. Control experiment ( ). Lines represent Sovová's model fittings. ). Lines represent Sovová's model fittings. | |
Table 1 Kinetic parameters of the model of Sovová for the supercritical carbon dioxide extraction from E. prunastri
| Pretreatment |
k
fa (min−1) |
k
sa (min−1) |
r
|
Y
s (g kg−1) |
t
CER (min) |
t
FER (min) |
AARD (%) |
R
2
|
|
k
fa: fluid-phase mass transfer coefficient; ksa: solid-phase mass transfer coefficient; r: fraction of broken cells; Ys: pseudo-solubility of the extract in the solvent; tCER: extraction time at the end of the CER period; tFER: extraction time at the end of the FER period; and AARD: absolute average relative deviation. |
|
Control
|
| Overall extraction yield (g per 100 glichen) |
0.159 |
0.0024 |
0.286 |
0.364 |
12.9 |
47.4 |
4.71 |
0.988 |
| UA extraction yield (mg glichen−1) |
0.298 |
0.0015 |
0.120 |
0.053 |
4.4 |
29.3 |
12.45 |
0.957 |
|
MW 480 W
|
| Overall extraction yield (g per 100 glichen) |
0.107 |
0.0017 |
0.553 |
0.199 |
51.1 |
131.1 |
5.23 |
0.993 |
| UA extraction yield (mg glichen−1) |
0.090 |
0.0018 |
0.767 |
0.061 |
78.3 |
173.7 |
9.91 |
0.997 |
|
MW 800 W
|
| Overall extraction yield (g per 100 glichen) |
0.094 |
0.0011 |
0.782 |
0.316 |
126.0 |
277.4 |
19.65 |
0.993 |
| UA extraction yield (mg glichen−1) |
0.084 |
0.0015 |
0.902 |
0.043 |
120.0 |
246.5 |
63.94 |
0.981 |
The previous pretreatment with microwaves for 1 min at 480 W did not show any benefit in the overall extraction yield (Fig. 1a), but that at 800 W for 1 min showed an increase of the extraction yield by 67% after 4 h. The usnic acid yield was enhanced at 1–4 h in samples treated at 480 W (Fig. 1b). Comparable usnic acid yields have been reported from Usnea barbata at 30 MPa and 25 °C,17 from Cladonia sp. at 35 MPa at 40 °C and 40 min,26 from Usnea subflorifdiana at 75 °C, 20 MPa and 70 min,55 and from Usnea longissima at 42 °C and 7.5 h with 4.3% ethanol.25 The most marked effects of the pretreatment on the extraction rate and yield were observed for the second stage of SFE of falling extraction rates, when the amount of the solute available on the surface decreases and the solute inside the particles has to diffuse to the surface (Fig. 1a and b). Therefore, the extraction rate is limited by intraparticle diffusion of the solute, which controls the internal mass transfer, closely dependent on the particle size and on the disruption of the cell walls.15
The mass transfer coefficients in the fluid phase, kfa, were higher than the mass transfer coefficients in the solid phase, ksa (Table 1). The extraction resistance is higher when the solutes are located internally in the solid and the mass transport mechanism is controlled by diffusion, compared to the extraction resistance by convection of solutes from the surface of the solid. The parameter r represents the fraction of broken cells in the solid and is affected by the pretreatment of the raw material.56 The smallest value of r was obtained for the extraction without any pretreatment. Furthermore, a shorter CER period was also observed in this case; therefore the extraction was dominated by the mass transport in the solid phase from the very beginning of the process. The application of 1 min of microwave irradiation at 800 W increased the duration of the CER period up to 126 min, with an overall extraction yield at the end of this period of 2.8 g per 100 g lichen. In the case of the usnic acid extraction kinetics, the application of 480 W for 1 min increased the duration of the CER period from 4 to 78 min, with a yield at the end of this period of 3.4 mg usnic acid per 100 g lichen.
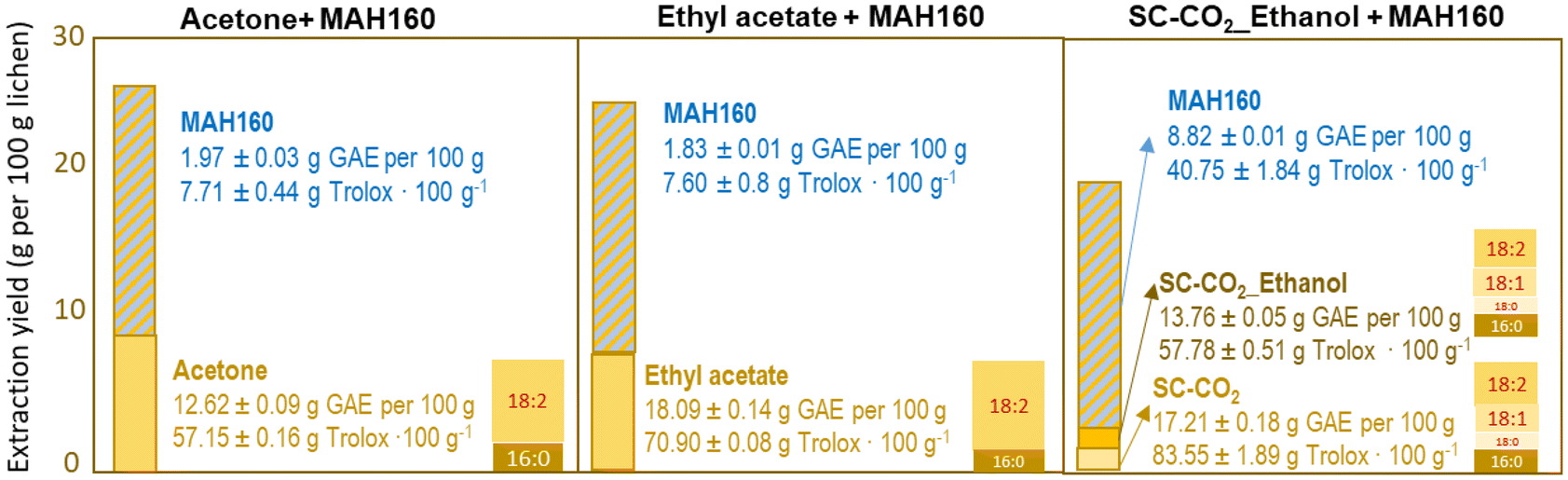
Acetone and ethyl acetate, selected based on the solubility of lichen bioactives,23,57 provided overall extraction yields of 8.09% and 7.65%, respectively (Fig. 2), in the range of those obtained by Popovici et al.57 but slightly lower compared to those of methanolic extracts.5 The highest total phenolic contents of 18.09 and 17.21 g GAE per 100 g extract, and antiradical capacities of 70.90 and 83.55 g Troloxeq. per 100 g extract were obtained, respectively, for the ethyl acetate and supercritical pure CO2 extracts. These contents were comparable to those of acetone and methanol extracts,5,58 but higher than those with hexane,59 methanol3,60,61 or acetone.17 Evernic acid59 and usnic acid are known to exhibit strong antioxidant activity, usually correlated with the content of total phenols, but may also depend on non-phenol components.62 The further application of a microwave assisted hydrothermal process allowed an additional extraction of active phenolics, but with lower selectivity since the polysaccharides were preferentially solubilized. Pure and ethanol modified CO2 extracts contained an analogous proportion of fatty acids, the most abundant being linoleic acid, followed by oleic, palmitic and stearic acids, whereas in Soxhlet extracts linoleic and palmitic acids were predominant (Fig. 2). Alfa-linolenic, stearic, linoleic and palmitic acids have been reported in this species.63
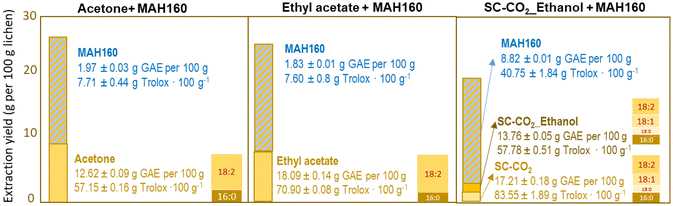 |
| | Fig. 2 Comparative performance of the three sequential processes: conventional Soxhlet acetone and ethyl acetate extraction and supercritical CO2 extraction followed by microwave assisted hydrolysis at 160 °C (MAH160) regarding the total phenolic content, Trolox equivalent antioxidant capacity and fatty acid content from Evernia prunastri. | |
2.2. Microwave assisted hydrothermal processing with NaDES as cosolvents
Microwave assisted hydrothermal treatment has been previously applied to extract polysaccharide fractions that could be further formulated as hydrogels with biological properties of interest for topical use. Optimal yields, mechanical properties and antiproliferative action were found in those obtained during operation at 160 °C.27 Natural deep eutectic solvents (NaDES) are usually non-toxic, easy to prepare, highly biodegradable and compatible with food and pharmaceutical products, provide a good response to microwaves64 and were tried as cosolvents during hydrothermal treatments.
NaDES characterization.
The two selected natural eutectic solvents have opposite physicochemical properties (Fig. S1†). The hydrophilic NaDES, synthesized with lactic acid, glycine, and water (LA:Gly:W) (3:1:3) presented a density of 1.21 ± 0.01 g cm−3 and viscosity around 82 mPa s at 100 s−1. The hydrophobic NaDES, made of menthol:lauric acid (Men:Lau) (2:1), was found to have a density of 0.87 ± 0.02 g cm−3 and to exhibit a pseudo-plastic fluid behaviour with a decrease in the apparent viscosity with an increased shear rate, to reach an apparent viscosity around 23 mPa s at 100 s−1. These results are coherent with those previously reported.65,66 Both solvents were stable after one month, maintaining similar viscosity at 10–100 s−1.
Whereas Nile Blue A dissolved in LA:Gly:W had a similar λmax to that in water, indicating that this solvent is polar, a blue shift to lower wavelengths was observed when dissolved in Men:Lau, as expected from a more apolar solvent. Similar results were found by Rebocho et al.65 and Vieira et al.67 using Nile Red dye. As microwave heating depends on the microwave absorption and heat transfer properties of the material, González-Rivera et al.64 proposed an indirect approach to estimate the microwave absorption properties of different DESs comparing their microwave heating profiles. Fig. S1b† shows the cumulative energy consumed (kW h) to heat LA:Gly:W and Men:Lau up to 170 °C as a function of time. Both NaDES showed a higher response to microwaves than water, achieving 170 °C in 70–78 s, whereas water required 83 s. Increasing NaDES concentration lowered the energy and time consumption. The cumulative energy consumed was also greater for water (3.54 × 10−3 kW h) than for Men:Lau (3.25 × 10−3 kW h at 50% and 3.10 × 10−3 at 80%) and for LA:Gly:W (2.96 × 10−3 kW h at 50% and 2.84 × 10−3 kW h at 80%).
Optimization of hydrothermal extraction with NaDES as cosolvents.
Hydrothermal treatment at high pressures and temperatures with water under subcritical conditions offers the possibility of modulating the water polarity since the dielectric constant decreases with increasing temperature. Additionally, the thermal and mechanical effects improve the mass transfer and extraction efficiency by destroying the cell wall. The autoionization, responsible for facilitating the degradation of polysaccharides, can be additionally enhanced by the incorporation of DESs, which promotes the production of catalytic ions. The effect of the NaDES concentration and temperature during microwave assisted hydrothermal extraction was studied with central composite experimental designs, defined for two different NaDES (LA:Gly:W and Men:Lau) and concentration ranges.
Range of high NaDES percentages during microwave assisted hydrothermal extraction.
Table 2 summarizes the independent and dependent variables assessed in the liquid phase for both NaDES. For the experiments with the hydrophobic NaDES, a too small aqueous phase was separated and the TEAC value was measured in the Men:Lau phase. Due to the incompatibility between Men:Lau and some spectrophotometric methods, the soluble protein and total sugar contents were only determined for the LA:Gly:W extracts.
Table 2 Experimental matrix for the study of the effect of the NaDES percentage (NP) in a range of high values and temperatures (T) on the extraction yield of carbohydrates and proteins, and on the Trolox Equivalent Antioxidant Capacity (TEAC)
| |
Uncoded variable |
Coded variable |
LA:Gly:W |
Men:Lau |
| Exp |
NP (%) |
T (°C) |
X
1
|
X
2
|
Extraction yield (g per 100 g lichen) |
Extraction yield (g per 100 g lichen) |
| Total sugarsa |
Carbohydrateb |
Protein |
TEAC |
TEACc |
|
Anthrone method.
HPLC method.
Men:Lau phase.
|
| 1 |
80 |
120 |
1 |
−1 |
9.60 |
12.27 |
1.24 |
14.81 |
12.59 |
| 2 |
86 |
145 |
1.41 |
0 |
28.05 |
30.89 |
2.91 |
25.13 |
14.58 |
| 3 |
65 |
145 |
0 |
0 |
15.60 |
19.91 |
1.21 |
16.58 |
16.35 |
| 4 |
65 |
110 |
0 |
−1.41 |
9.19 |
12.79 |
0.61 |
7.47 |
8.75 |
| 5 |
50 |
170 |
−1 |
1 |
29.56 |
30.87 |
1.64 |
17.89 |
36.50 |
| 6 |
50 |
120 |
−1 |
−1 |
12.21 |
18.31 |
0.62 |
9.66 |
11.90 |
| 7 |
80 |
170 |
1 |
1 |
34.81 |
25.73 |
4.88 |
28.22 |
22.90 |
| 8 |
65 |
180 |
0 |
1.41 |
28.04 |
23.69 |
3.97 |
25.07 |
31.21 |
| 9 |
44 |
145 |
−1.41 |
0 |
18.78 |
22.52 |
1.22 |
17.85 |
35.66 |
| 10 |
65 |
145 |
0 |
0 |
19.33 |
24.97 |
1.69 |
21.98 |
17.96 |
| 11 |
65 |
145 |
0 |
0 |
21.44 |
27.18 |
1.70 |
22.71 |
17.26 |
The increased protein and polysaccharide solubilization expected was synergistically enhanced when LA:Gly:W was used as cosolvent, with temperature being the most influencing variable. Higher temperatures increase the solvent power due to the decrease in viscosity and surface tension, leading to an enhanced diffusivity and dissolution of target components. Despite viscosity being important and that it could also be limiting at the tested concentration of LA:Gly:W, the maximum carbohydrate yield was obtained at the maximum tested NP. The enhanced protein and reducing sugar solubilization has been also reported for choline chloride:formic acid (1:2) at a lower content, 25%, and 20 min. Accelerating the disintegration of wet microalgal biomass with hydrothermal treatment at 160 °C and formic acid can also promote depolymerization.32
Under the conditions of experiment 7, with 80% NaDES at 170 °C, maximum carbohydrate and protein yields were obtained, with up to 35% polysaccharide yield, corresponding to half the content of this fraction. The addition of NaDES enhanced the polysaccharide extraction yield compared to the use of water, which yielded 25.5% carbohydrates for direct hydrothermal treatment at 180 °C and 15.2% at 160 °C.27 Usually, spectrophotometrically determined values are overestimated, but in this case the ranges were comparable (Table 2). Using NaDES, no polymer precipitated after freezing and thawing. Saravana et al.28 already found higher values for choline chloride:glycerol, with 60% water for alginate and fucoidan. The highest protein yield was almost 5%, half of the total amount present in the raw material, and was attained at 170 °C, close to the value reported for brewery spent grains,34 but during the treatment of microalgae, maximal protein solubilization in biomass occurred at lower temperatures.32
The maximum total phenolic extraction yield was attained at 65% NP/180 °C and 50% NP/170 °C, respectively, for each cosolvent, but these values are considerably higher than those attained with conventional solvents and in hydrothermal extraction at 160 and 180 °C. Therefore, due to the possibility of interferences this variable was not studied. The highest antiradical capacity, 36.50 g Troloxeq. per 100 g E. prunastri, was achieved with Men:Lau at 50%/170 °C, whereas the highest value with LA:Gly:W, 28.22 g Troloxeq. per 100 g of raw lichen, was obtained at 80%/170 °C. The model coefficients and test parameters for each objective function obtained from the multilinear regression, t-tests and ANOVA test are summarized in Tables S1–S4.† The objective functions of the reliable models are presented in eqs (1)–(4) for LA:Gly:W and in eqn (5) (Table S5†) for Men:Lau and the corresponding response surfaces are shown in Fig. 3a–d. Whereas this range of LA:Gly has no significant impact on the carbohydrate yield, it exhibits a linear positive effect on the TEAC value and the protein. The temperature shows both a strong positive linear effect and quadratic effect, which is negative for the TEAC value, probably due to the degradation of some bioactives at the highest values. A positive interaction between the NP and the temperature was observed for protein yield. Even if higher antioxidant activities were found for Men:Lau, its percentage has a negative linear effect on the TEAC value but a positive quadratic effect, whereas temperature has a positive linear impact on the response variables. A negative interaction between the temperature and NP was also observed.
| | 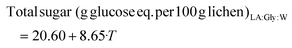 | (1) |
| | 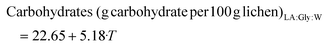 | (2) |
| | 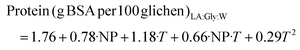 | (3) |
| | 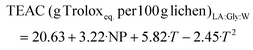 | (4) |
| | 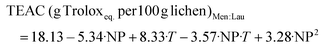 | (5) |
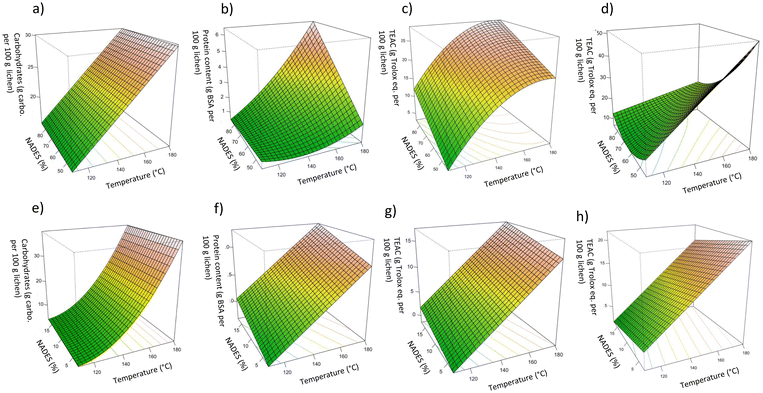 |
| | Fig. 3 Response surface for the effects of NaDES percentage and temperature in a range of high values with LA:Gly:W on (a) the extraction yield of carbohydrates, (b) the extraction yield of protein, (c) the ABTS antiradical capacity, and (d) for Men:Lau on the ABTS antiradical capacity, and in a range of low values with LA:Gly:W on (e) the extraction yield of carbohydrates, (f) the extraction yield of protein, and (g) TEAC, and (h) for Men:Lau on TEAC. | |
Range of low NaDES percentages during microwave assisted hydrothermal extraction.
The conditions defined in the previous experimental design proved suitable to maximize the extraction of compounds with antiradical properties, but such high NaDES content may be underoptimal for the recovery of the biopolymer by precipitation. Two effects could justify this. Viscosity could make precipitation difficult and the NaDES induced hydrolysis of the polysaccharides could result in low molecular weight fractions that do not precipitate by freezing and thawing. Therefore, another experimental design with lower NP was constructed according to the experimental matrix shown in Table 3.
Table 3 Experimental matrix for the study of the effect of the NaDES percentage and temperature in a range of low values on the extraction yield of carbohydrates and proteins, and the TEAC for LA:Gly:W as cosolvent, and total sugars, polymer yield and TEAC in the aqueous phase when Men:Lau was used as cosolvent
| |
Uncoded variable |
Coded variable |
|
LA:Gly:W |
Men:Lau |
| |
|
Extraction yield (g per 100 g lichen) |
Extraction yield (g per 100 g lichen) |
| Exp |
NP (w:w) |
T (°C) |
X
1
|
X
2
|
Total sugarsa |
Carbohydrates |
Protein |
TEAC |
Total sugarsb |
Polymer yield (%) |
TEACc |
|
Total sugars (anthrone method).
Total sugars in the aqueous phase (anthrone method).
TEAC in the aqueous phase after the polysaccharide precipitation.
|
| 1 |
15 |
120 |
1 |
−1 |
3.64 |
4.83 |
0.07 |
2.34 |
5.34 |
3.82 |
3.28 |
| 2 |
17 |
145 |
1.41 |
0 |
13.34 |
14.51 |
0.72 |
12.16 |
14.32 |
14.92 |
8.43 |
| 3 |
10 |
145 |
0 |
0 |
8.53 |
11.2 |
0.23 |
8.39 |
10.81 |
13.61 |
10.37 |
| 4 |
10 |
110 |
0 |
−1.41 |
3.08 |
3.53 |
0.07 |
1.11 |
3.33 |
3.39 |
5.24 |
| 5 |
5 |
170 |
−1 |
1 |
37.29 |
32.81 |
1.01 |
16.98 |
20.68 |
22.98 |
19.58 |
| 6 |
5 |
120 |
−1 |
−1 |
4.02 |
6.23 |
0.04 |
2.49 |
4.54 |
4.39 |
6.13 |
| 7 |
15 |
170 |
1 |
1 |
33.42 |
29.21 |
1.21 |
15.81 |
30.63 |
25.25 |
17.42 |
| 8 |
10 |
180 |
0 |
1.41 |
49.19 |
38.21 |
1.06 |
12.53 |
31.63 |
28.23 |
19.87 |
| 9 |
3 |
145 |
−1.41 |
0 |
7.63 |
11.37 |
0.09 |
4.80 |
8.84 |
15.38 |
12.07 |
| 10 |
10 |
145 |
0 |
0 |
10.67 |
13.98 |
0.43 |
5.02 |
10.48 |
14.78 |
12.81 |
| 11 |
10 |
145 |
0 |
0 |
12.10 |
15.68 |
0.32 |
5.39 |
10.09 |
15.15 |
12.06 |
Operating at 10% LAGly:W and 180 °C, the polysaccharide yield increased up to 49.2 g per 100 g lichen. Both eutectic solvents provided higher carbohydrate yields than hydrothermal extraction at the same temperature.27 Regardless of the NaDES, temperature favours the extraction of carbohydrates and proteins and the TEAC value. The objective functions defined considering only the statistically significant coefficients are shown in eqn (6)–(9), for the extraction yield of carbohydrates and proteins and TEAC activity for LA:Gly:W and in eqn (10)–(12), and for carbohydrates, polymer yield and TEAC with Men:Lau, based on the statistical information of Tables S6–S9 and S10–S12.†
With LA:Gly:W, the temperature was the only significant variable for carbohydrates and TEAC and the NP was significant for the protein content. For Men:Lau, both NP and T had a significant impact on the TEAC and sugar values. The corresponding response surfaces are plotted in Fig. 3e–h.
| | 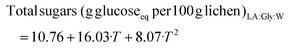 | (6) |
| | 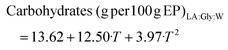 | (7) |
| | 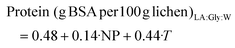 | (8) |
| | | TEAC (g Troloxeq per 100 g lichen)LA:Gly:W = 7.91 + 5.51·T | (9) |
| | 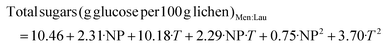 | (10) |
| | 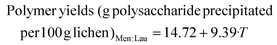 | (11) |
| | 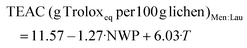 | (12) |
A certain water content is required for the NADES mixture to lower the viscosity, which could limit the mass transfer and solubilisation. When used as solvents in heating–stirring, Soxhlet, microwave- or ultrasound-assisted extraction, they can be added at 50–75%, but in semicontinuous pressurized liquid extraction, solvents have to be pumped through the system and the NADES cannot be employed as true solvents but as solvent modifiers at lower percentages.33 ChCl could be used as a solvent modifier and 30% solvent was suitable to maintain low viscosity. Optimal values of 40–50% were reported for the polysaccharide yield of Lentinus edodes at 147 °C for 17.6 min,35 and for subcritical water extraction at 122 °C modified with 8% DES to improve pectin yield from Jaboticaba processing by-products.31
Choline chloride and citric acid were used at 1% for the subcritical water extraction at 130 °C of phenolics and anthocyanins from black bean hulls, increasing the performance compared to that with the use of pure water.30 A higher content of ChCl:U at 30% and 100 °C was the best condition for the extraction of phenolic compounds from winemaking by-products.33 The beneficial effect of increasing the DES concentration from 10 to 30% on the phenolic and xanthone extraction from mangosteen pericarp was more marked at 120 °C than at higher temperatures, probably due to lignin depolymerization in the DES that may have acted as a catalyst.29 However, NaDES at 180 °C slightly improved the extraction efficiency of phenolics under subcritical hydrolysis conditions.34
When the eutectic solvent made of LA:Gly:W was used as cosolvent during hydrothermal treatment both at high and low concentrations, none or almost none of the polysaccharides precipitated after freezing and thawing. In general, polysaccharide solubility in water decreases with its molecular weight.68 The HPSEC chromatograms of the sample extracted with water exhibited two peaks under 786 kDa and one of higher molecular mass (Fig. 4). Almost no peak for structures with a value higher than that was observed in the sample with NaDES at high concentration. Note that the peak at 8–12 min is related to the NaDES itself and note also that this technique lacks accuracy to determine molecular mass, but it offers a first approach for determining the conformational properties of the oligosaccharides. The chromatograms of the sample with low NaDES concentrations exhibited a peak at high molecular weight coincident with that of the polysaccharides extracted with water, but its magnitude decreases with increasing temperature and NaDES ratio. The highest peak was observed at 5%–170 °C and the lowest at 17%–145 °C and 10%–180 °C. Thermodegradation is promoted by the NaDES33 and was confirmed on a previously water extracted, dried polysaccharide, which was dissolved in water or in 10 or 65% NaDES, stirred at 80 °C for 2 h.
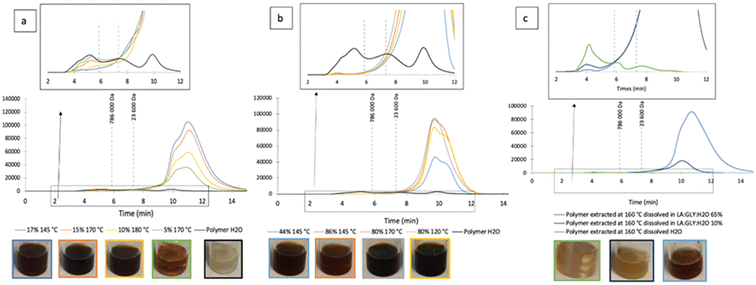 |
| | Fig. 4 HPSEC chromatograms of the extracts obtained during microwave assisted autohydrolysis with cosolvent (a) LA:Gly:W at high concentrations, (b) LA:Gly:W at low concentrations, and (c) the polymer extracted with water (MAH160), precipitated by freezing and thawing, dissolved in water and in LA:Gly:W at 10% and at 65%. | |
The samples in water and 10% LA:Gly:W exhibited a light-yellow color, but the sample at 65% NaDES exhibited a strong brown color, suggesting that the polysaccharide could react with the solvent. Moreover, after maintaining the samples at −18 °C for 48 h, the sample with 65% NaDES did not freeze and only the polysaccharides dissolved in water precipitated again. The HPSEC of the polysaccharides dissolved in water had mainly a high molecular weight (>786 kDa) (Fig. 4c). This peak at high molecular weight was also observed for the samples dissolved in 10 and 65% NaDES but with a height negatively correlated with the NaDES content. However, the promoted solubilization of biomacromolecules at 160 °C and 20 min with 25% ChCl:formic acid (1:2) was confirmed by the increased average molecular weight (from 1043 Da to 3203 Da) of solutes in the aqueous phase.32
Polymer recovery and film formation.
Polymer precipitation was observed after cooling the aqueous phase from Men:Lau experiments. The use of Men:Lau offered the possibility of extracting different components in each phase. This strategy has not been previously explored in lichens but has been reported in several cases, i.e. for the simultaneous extraction of flavonoids, terpene trilactones, procyanidine and polyprenyl acetates from Ginkgo biloba leaves in two-phase systems with two hydrophilic and one hydrophobic DESs.69 Also, biphasic systems with DESs and γ-valerolactone were used for glucan saccharification, furfural production, and lignin recovery.70 The yield of polymers extracted with MAH_ML5-170, which precipitated after freezing and thawing, is shown in Table 3. Rheological properties are summarized in Tables S13–S16.† The temperature had a positive significant impact on the polymer extraction yield and a negative one on the logarithm of the viscosity, and on the elastic and viscous moduli. However, models for the elastic modulus and viscosity presented a very low p-value for the lack of fit which means that these models are likely to be inaccurate. For none of the response variables evaluated, the NP has a statistically significant impact. The polymer's viscosity at 1 Hz varies from 114 to 82.8 × 104 mPa s. In comparison, the polymer extracted at 160 °C with water presented a viscosity around 5000 mPa s. Some of the precipitated polymers, before any other thermal treatment or formulation, already presented viscoelastic properties of a middle strength gel as their moduli are invariant with the frequency and the elastic modulus is almost 10 times higher than the viscous one. It is for example the case for 5%–120 °C and 15%–120 °C. The polymer extracted at 5%–170 °C was selected for further analysis and formulation as it presented a good balance between extraction yield and viscoelastic properties (a higher gap between the elastic and viscous moduli than those for samples from 17%–145 °C or 15%–170 °C).
2.3. Sequential solvent and hydrothermal extraction vs. hydrothermal extraction with NaDES as a modifier
The extraction technique has a great influence on the lichenic acid composition of the extracts (Table 4). Atranol and chloroatranol, which are degradation products of atranorin and chloroatranorin and well-known allergens, were identified in the hydrothermal extracts of the supercritical CO2 residual solid, but they were not detected in the hydrothermal extracts of the residual solids after the Soxhlet extraction. In terms of total peak area, the highest value was obtained with ethyl acetate in the Soxhlet system, followed by supercritical extraction with pure CO2, with ethanol modified CO2 and with acetone in the Soxhlet system. The results are coherent with the total phenolic content value obtained (Fig. 2).
Table 4 Comparative performance of different extraction technologies on the lichenic acids from E. prunastri, expressed as the area of the identified peaks by HPLC-MS and their relative percentage
| |
Extraction technology |
| Compounds (area × 105), % total |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
| 1: SA: Soxhlet (acetone); 2: SA-MAH160: Soxhlet (acetone)-microwave assisted hydrothermal extraction at 160 °C; 3: SEA: Soxhlet (ethyl acetate); 4: SEA-MAH160: Soxhlet (ethyl acetate)-microwave assisted hydrothermal extraction at 160 °C; 5: SFE: supercritical pure CO2 extraction; 6: SCF_E: supercritical ethanol modified CO2 extraction; 7: SFE_E-MAH160: supercritical ethanol modifed CO2 extraction-microwave assisted hydrothermal extraction at 160 °C; 8: LA:Gly:W_65%-145 °C and 80%-170 °C (same results): microwave assisted hydrothermal extraction at 145 or 170 °C with lactic acid:glycine:water at 65 or 80%; 9: Men:Lau_44%-145 °C: microwave assisted hydrothermal extraction at 145 °C with menthol:lauric acid at 44%; 10: Men:Lau_50%-170 °C: microwave assisted hydrothermal extraction at 170 °C with menthol:lauric acid at 50%. nd: not detected. Peak area × 100 per total area. |
| Total area of known compounds |
1277 |
X |
2053 |
X |
1442 |
1319 |
|
X |
6.27 |
1.08 |
| Orsellinic acid |
175.26, 14% |
Nd |
662.26, 30% |
nd |
5.95, 0.4% |
29.87, 2% |
X, 8% |
Nd |
|
|
| Methyl orsellinate |
|
|
|
|
|
|
|
|
2.94, 13% |
|
| Divaric acid |
40.45, 3% |
|
311.45, 14% |
|
|
|
|
|
|
|
| Everninic acid |
217.74, 17% |
|
481.58, 22% |
|
|
|
|
|
|
|
| Lecanoric acid |
60.95, 5% |
|
265.52, 12% |
|
|
15.47, 1% |
|
|
|
|
| Evernic acid |
520.42, 42% |
|
220.42, 10% |
|
253.06, 17% |
456.59, 37% |
|
|
|
|
| Usnic acid |
40.62, 2% |
|
39.30, 2% |
|
323.88, 22% |
57.96, 5% |
|
|
3.3, 14% |
1.08, 51% |
| Atranorine |
157.25, 10% |
|
33.41, 2% |
|
513.33, 34% |
488.73, 35% |
|
|
|
|
| Chloroatranorine |
54.19, 3% |
|
39.29, 2% |
|
345.53, 23% |
270.34, 17% |
|
|
|
|
| Atranol |
|
|
|
|
|
|
X, 7% |
|
|
|
| Chloroatranol |
|
|
|
|
|
|
X, 2% |
|
|
|
| Unidentified peaks, total % |
2% |
2 peaks, 100% |
7% |
1 peak, 100% |
4% |
3% |
83% |
2 peaks, 100% |
5 peaks, 73% |
4 peaks, 49% |
Evernic acid accounts for about 40% of the area in acetone extracts and in the ethanol modified supercritical solvent, whereas ethyl acetate extracts exhibited maximum values for orsellinic acid, divaric acid, everninic acid and lecanoric acid. Atranorin, chloroatranorin and usnic acid are more concentrated in supercritical extracts. However, no erverninic acid was found in these latter ones. As expected, supercritical extraction with pure CO2 enhanced the recovery of the more apolar compounds. The hydrothermal phase did not contain any of the major lichenic acids, but only one or two unidentified peaks. Usnic acid was more concentrated in the Men:Lau extract, but neither of the other major known peaks were found. The results were expected since evernic acid was the most abundant in the acetone extracts of E. prunastri, followed by usnic acid, chloroatranorin, atranorin, and lecanoric acid.19 Other metabolites identified in this lichen species, evernic and physodic acids, atranol and chlorobutanol, are the most studied.1,13,19,59 No common compounds were observed in the extracts with LA:Gly:W nor in the aqueous phase of Men:Lau. This extract contained more than 50% usnic acid, and could be obtained in a simple stage with higher selectivity than in conventional extraction. Therefore, three processes were further selected, and they involved sequential extraction with conventional and supercritical CO2 extraction before hydrothermal processing, as well as the use of a single stage of hydrothermal extraction using NaDES as cosolvents. The flow diagram is shown in Fig. 5.
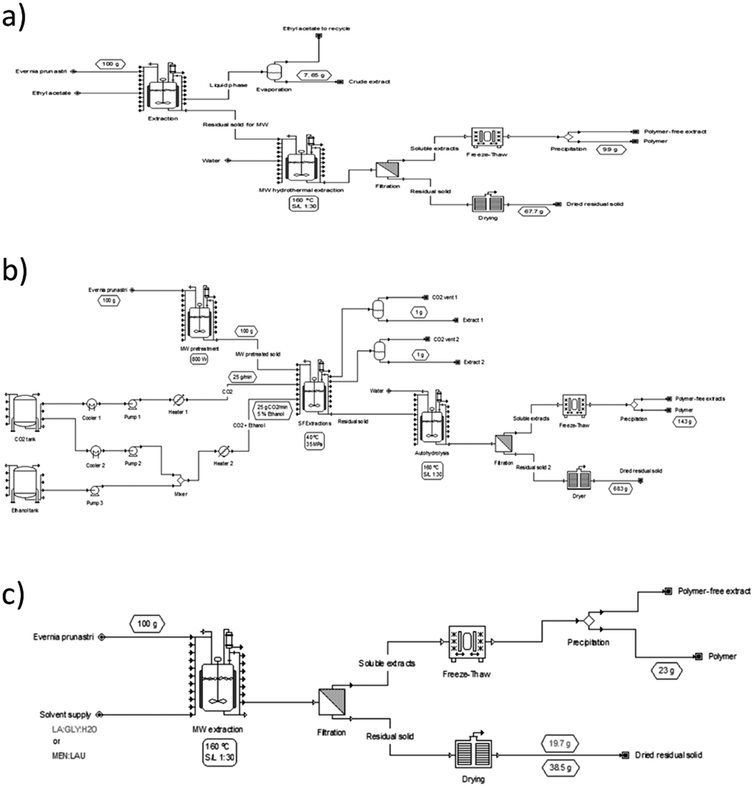 |
| | Fig. 5 Flow diagram of the different processes tested for the extraction of bioactives and biopolymers from E. prunastri: (a) SA-MAH160, (b) SFE_E-MAH160 and (c) NADES-modified hydrothermal extraction with LA:Gly:W 80% at 170 °C (bioactives) or with Men:Lau 5% at 170 °C (polysaccharides). | |
Residual solids.
The SEM images of the residual solids from the three different treatments, shown in Fig. 6, confirm that the smooth surface of untreated lichen changed to a rough, broken surface in treated solids. Furthermore, significant differences can be observed depending on the treatment; the residual solids from acetone and hydrothermal processing (SA-MAH160) present a grainy surface, whereas those from supercritical and hydrothermal processing (SCF-MAH160) present irregular folds. The residual solid after the hydrothermal processing with hydrophobic NaDES as cosolvent (MAH_ML5-170) appears to be the most damaged. Machmudah et al.29 also observed the disruption on the surface of biomass after subcritical water extraction with citric acid and alanine eutectic solvent as cosolvent, which may also act as a catalyst, facilitating the decomposition of the solid matrix components. Similarly, Roy et al.34 reported various degrees of damage in brewery spent grains; whereas subcritical water treatment showed a cracked and destroyed structure, the addition of DES led to a broken and internally damaged structure.
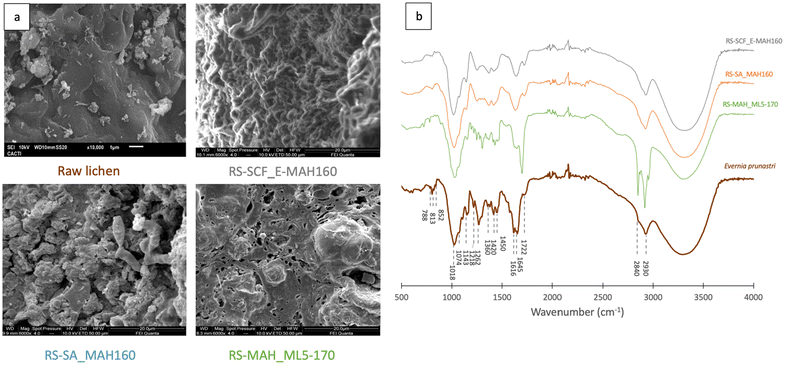 |
| | Fig. 6 (a) SEM photographs of the raw lichen and residual solids after SA-MAH160, SCF-MAH160 and MAH_ML5-170 and (b) their FT-IR spectra. | |
FTIR spectra of the residual solids, shown in Fig. 6b, confirm that the solvent extraction prior to the microwave assisted hydrothermal treatment did not affect the microstructure, since RS-SCF_MAH160 and RS-SA-MAH160 have similar profiles. The increase in the peak at 1713 cm−1 in RS-MAH_ML5-170 due to the stretching vibration of the carbonyl group of lauric acid shifted because of hydrogen bonding with menthol and an increase in the peaks at 2840–2930 cm−1, due to the presence of the eutectic solvent on the residual solid.66
In all spectra, the peaks at 813 cm−1 may be related to the β-glucosidic bonds of the β-D-mannopyranose units of galactomannans and/or β-glucopyranose of β-glucans and at 852 cm−1 due to the presence of α-D-galactopyranose. The presence of cellulose and hemicellulose can be identified by the peaks at 1110 cm−1, due to the stretching of C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and C–C–O in sugar rings and at 1150 cm−1 for the stretching vibration of C–O–C. Peaks related to lignin are seen at 1270 cm−1 (binding vibration of C–H bonds in lignin aromatic rings), at 1515 cm−1 (aromatic skeleton vibration) and 1580 cm−1 (aromatic ring vibration). The peak at 1645 cm−1 may be related to the stretching vibration of the carbonyl group in a conjugated ketone or aldehyde in lignin and hemicellulose, whereas the peak at 1722 cm−1 may be attributed to the stretching vibration in unconjugated CO ketone, aldehyde, carbonyl or aliphatic groups in hemicellulose. Finally, the wide band around 3300 cm−1 is related to intra- and intermolecular hydroxyl stretching of lignin, cellulose, hemicellulose and water.71,72 In the residual solids, the peaks from the primary aromatic blocks in lignin confirm that the basic lignin structures are maintained, but a reduction of peaks at 1270, 1515 and 1580 cm−1 related to lignin can be observed which might indicate that some lignin was extracted.73 Lignin solubilization with DES is well known, i.e. ChCl:LA treatments at 90–180 °C can selectively cleave ether linkages in wood lignin, and provide a high purity low molecular lignin.74 The extraction of hemicelluloses can be corroborated by the reduction of peaks at 813, 852, 1110, 1150, 1645, and 1712 cm−1.
C, and C–C–O in sugar rings and at 1150 cm−1 for the stretching vibration of C–O–C. Peaks related to lignin are seen at 1270 cm−1 (binding vibration of C–H bonds in lignin aromatic rings), at 1515 cm−1 (aromatic skeleton vibration) and 1580 cm−1 (aromatic ring vibration). The peak at 1645 cm−1 may be related to the stretching vibration of the carbonyl group in a conjugated ketone or aldehyde in lignin and hemicellulose, whereas the peak at 1722 cm−1 may be attributed to the stretching vibration in unconjugated CO ketone, aldehyde, carbonyl or aliphatic groups in hemicellulose. Finally, the wide band around 3300 cm−1 is related to intra- and intermolecular hydroxyl stretching of lignin, cellulose, hemicellulose and water.71,72 In the residual solids, the peaks from the primary aromatic blocks in lignin confirm that the basic lignin structures are maintained, but a reduction of peaks at 1270, 1515 and 1580 cm−1 related to lignin can be observed which might indicate that some lignin was extracted.73 Lignin solubilization with DES is well known, i.e. ChCl:LA treatments at 90–180 °C can selectively cleave ether linkages in wood lignin, and provide a high purity low molecular lignin.74 The extraction of hemicelluloses can be corroborated by the reduction of peaks at 813, 852, 1110, 1150, 1645, and 1712 cm−1.
2.4. Film formulation
Various techniques have been reported to efficiently recover DES after biomass pretreatment, including anti-solvent addition, crystallization, membrane filtration as well as solid–liquid, liquid–liquid or supercritical fluid extraction.32 However, the direct utilization of the final extract or its application for the formulation of novel products has operational advantages.27,75 The polymers recovered from the hydrothermal extracts from the different strategies tested were characterized for mechanical and biological properties. The surface morphologies of the films formulated with the recovered polymers, analysed by SEM (Fig. 7a–d), were quite similar, with irregularities and some polymer clumps. These granules were also observed by Ruggeri et al.75 and Coelho et al.76 in galactomannan films. Strong hydrogen bonds between the polysaccharide chains in the film forming solution or steric hindrance during the film formation might explain the presence of aggregates.77–79 However, the surface of MAH_ML5-170 seems slightly smoother and more homogeneous. All films presented similar FTIR spectra (Fig. 7e) with typical galactomannan bands at 852 and 813 cm−1, respectively, due to the presence of α-D-galactopyranose and β-D-mannopyranose units or β-glucopyranose units of β-glucans.80 The peaks at 1118 and 1143 cm−1 are characteristic of the stretching of C–O, CC, and C–C–O in sugar rings and of the stretching vibration of C–O–C in hemicellulosic materials. No peak deformation/addition due to the Men:Lau residue was observed in the spectra of F-MAH_ML-5-170.
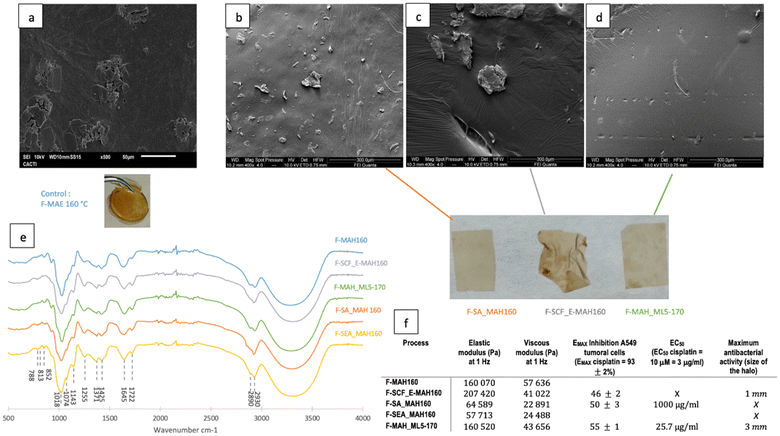 |
| | Fig. 7 SEM photographs of the films (F) formulated with direct processing of lichens by (a) microwave assisted extraction at 160 °C (F-MAH160), (b) microwave assisted extraction of the residual solid after Soxhlet acetone extraction (F-SA_MAH160), (c) microwave assisted extraction of the residual solids after ethanol modified supercritical CO2 extraction (F-SCF_E-MAH160), (d) and direct processing by microwave assisted hydrothermal extraction at 170 °C with 5% menthol-lauric acid as a modifier (MAH_ML5-170), (e) FT-IR spectra, and (f) rheological properties of these films and cytotoxicity on A549 tumoral cells. | |
The viscoelastic properties of the film are displayed in Fig. 7f. Although all the films have an elastic modulus higher than the viscous one and both are invariant with the frequency, a large difference can be observed between F-SA/SAE-MAH160, which has a lower elastic modulus value, and F-MAH160, F-SCF-MAH160, and MAH_ML5-170 for which the elastic component is more prevalent. This confirms for these latter that the application of a stress to the films results in a more reversible deformation. F-SA/SAE-MAH160 is a weaker film, and the values of tanδ = G/G′ of F-MAH160, F-SCF-MAH160, and MAH_ML5-170, of 0.36, 0.20, and 0.27, respectively, are higher but of the same order of magnitude as those obtained by Wang81 and Castro-Yobal et al.82 for carrageenan/galactomannan and alginate films (between 0.08 and 0.20).
Lichen metabolites exhibit promising antitumoral properties, but their low levels of cytotoxic compounds, the difficulties in their isolation and characterization, and the insufficient understanding of their mechanism of action limit the advances.62,83 None of the extracts were cytotoxic for normal MRC-5 cells. A low A549 tumoral cell growth inhibition (≤50%) at the maximum concentration evaluated (1000 μg mL−1) was found for the microwave assisted extracted polymer. However, the polymers obtained in a process modified with Men:Lau_5%_170 °C show a reasonable growth inhibition of 55% compared to 93% for cisplatin, and a good activity with a low EC50 of 25.7 μg mL−1. This value was similar to those reported for two acidic polysaccharides (IC50: 41.2 and 47.8 μg mL−1),84 and more favourable than those reported for synthetic chitosan selenate,85 for beta-glucans86,87 and for galactomannans.88
When the hydrophobic NaDES was used as cosolvent during hydrothermal extraction, usnic acid accounted for 50% of the identified peaks. The high usnic acid content in the Men:Lau extract could explain the potency of these extracts, whereas the hydrothermal treatment alone did not exhibit any action. The activity was in the range of those reported for pure usnic acid against HCT-116, HeLa and MCF-7, which showed IC50 values of 18, 24, and 76, respectively.89 However, conventional solvent extracts of E. prunastri showed higher IC50 values, i.e. 120.9 μg mL−1 for acetone extract in FemX and LS 174 cell lines lines,17 and 295.6 μg mL−1 for methanol extract in the colon cancer adenocarcinoma cell line HCT-116.3,61
Different compounds could be responsible for this action; evernic acid in the acetone and dichloromethane extracts from E. prunastri reduced cell migration and proliferation during the angiogenesis and inhibited the pro-inflammatory activation of leukocytes.90 Lecanoric acid and atranorin were responsible for the most effective anti-proliferative effects in various cancer cells.91 However, usnic acid exhibits different activities, including anti-inflammatory and antimicrobial activities, and supresses the proliferation of gastric,92 cervical93,94 and breast cancer cells.89,95 It might be responsible for the reduced tumor cell viability, apoptosis, and cell cycle inhibition.90 Its low water solubility could limit some potential applications and further studies are needed to develop different products.96 The hydrothermal process with Men:Lau as cosolvent allowed the production of films with more potent bioactivity than those elaborated with beta-glucan and galactomannan components, obtained without the cosolvent.
Antimicrobial activity.
The films developed with the polymer extracted after direct processing by microwave assisted hydrothermal extraction at 170 °C with 5% menthol-lauric acid as a modifier (MAH_ML-5-170) exhibited the best antimicrobial properties. They inhibited the growth of all tested Gram-positive bacterial strains, particularly Staphylococcus aureus and Bacillus cereus with a halo of 3 mm, Listeria monocytogenes and Staphylococcus epidermidis with a halo of 2 mm and Enterococcus faecalis with a halo of 1.5 mm. The polysaccharide-films prepared with extracts from ethanol modified supercritical CO2 (F-SCF_E-MAH160) exhibited less growth inhibition. In this case, only the proliferation of Enterococcus faecalis and Staphylococcus epidermidis was inhibited with a halo of 1 mm. Note here that those films formulated after Soxhlet acetone extraction (F-SA + MAH160) did not present an antimicrobial inhibition effect under the tested processing conditions. In all cases, the results with Escherichia coli, Pseudomonas aeruginosa and Salmonella spp. were negative. This suggests that the developed polysaccharide-films only exhibited a positive effect on gram positive bacterial strains, without evidence of any effect on Gram negative microorganisms. In the Men:Lau based process, the simultaneous extraction of this bioactive and biopolymers could be achieved, maintaining also biocompatibility and adequate mechanical properties.
Methanol followed by ethanol and acetone extracts from corticolous lichens were highly active against Gram-positive bacterial strains, whereas some aqueous infusions did not present antibacterial and antifungal activities.3 The antimicrobial impact of usnic acid was found against different bacteria, including E. faecalis, S. epidermidis, B. cereus and S. pyogenes.96 These authors also designed adhesive polymeric films to enhance usnic acid bioavailability to promote a prolonged release in the wounds and with antibacterial activity without affecting cell viability on human keratinocytes and fibroblasts.
2.5. Environmental analysis
The environmental impact of the three selected processes was estimated using the Eco-Scale method, slightly adapted from Sousa et al.97 and Van Aken et al.98 as it was initially invented for organic synthesis. In opposition to common green metrics like the E-factor or the atom economy factor, which only evaluate the sustainability of one aspect of the process, the Eco-Scale considers six criteria: the relative yield of products, the price/availability of the solvents, the technical setup needed, the operating conditions, and the downstream processes required (Table 5). However, it is important to note that the Eco-Scale is not as complete as the LCA as it does not consider the synthesis of the reagents and the use and end of life of the products. In the Eco-Scale, fixed penalty points are allocated to each process and substracted from 100 to give a final punctuation between 0 and 100, with 100 indicating an ideal process where a quantitative yield is reached, using inexpensive, non-toxic, non-flammable solvents, with a simple and not energy intensive technical setup and purification process. Usually, processes that have a final score of 75 or more are considered to have an excellent sustainability, those between 50 and 75 have an acceptable sustainability and those below 50 are not considered green. As two kinds of compounds were targeted, the lichenic acids/phenolic compounds and the polysaccharides, the relative final yield penalty was determined by calculating the average between, for the lichenic substances, the relative yield penalty of usnic acid and the antioxidant activity and for the biopolymer, the relative yield penalty of the recovered polysaccharides. The allocated penalty points are calculated from eqn (13)| | 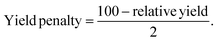 | (13) |
Table 5 Greenness assessment of the selected processes using the Eco-Scale
| |
Parameter |
Penalty pts |
SEA-MAH160 |
SCF_E-MAH160 |
MAH_ML-5-170 |
| Total point (100 – penalties) |
|
|
39 |
52 |
66 |
| (1) Penalty yield (mean of the two) |
|
(100-relative yield)/2 |
26.3 for bioactives |
19.3 for bioactives |
24.5 for bioactives |
|
|
|
|
28.5 for polymers |
19 for polymers |
0 for polymers |
| |
| (2) Price/availability (solvent) |
Inexpensive (<50 € per kg) |
0 |
|
X |
|
| Expensive (50–100 € per kg) |
3 |
X |
|
|
| Very expensive (>100 € per kg) |
5 |
|
|
X |
| |
| (3) Safety |
N (dangerous for the environment) |
5 |
X |
|
|
| T (toxic) |
5 |
X |
X |
|
| F (flammable) |
5 |
X |
X |
X |
| E (explosive) |
10 |
|
|
|
| F + (extremely flammable) |
10 |
|
|
|
| T + (extremely toxic) |
10 |
|
|
|
| |
| (4) Technical setup |
Common setup |
0 |
|
|
|
| Instruments for controlled addition of chemicals |
1 |
|
|
|
| Unconventional technique |
2 |
|
|
X |
| Pressure equipment, >1 atm |
3 |
X |
XX |
X |
| Any additional special glassware |
1 |
|
|
|
| (Inert) gas atmosphere |
1 |
|
|
|
| Glove box |
3 |
|
|
|
| |
| (5) Temperature/time |
Room temperature, <1 h |
0 |
|
|
|
| Room temperature, <24 h |
1 |
|
|
|
| Heating, <1 h |
2 |
X |
X |
X |
| Heating, >1 h |
3 |
X |
X |
|
| Cooling to 0 °C |
4 |
|
|
|
| Cooling, <0 °C |
5 |
X |
X |
X |
| |
| (6) Workup and purification |
None |
0 |
|
|
|
| Cooling to room temperature |
0 |
|
|
|
| Adding solvent |
0 |
|
|
|
| Simple filtration |
0 |
|
|
|
| Removal of solvent with bp <150 °C |
0 |
|
|
|
| Crystallization and filtration |
1 |
|
|
|
| Removal of solvent with bp >150 °C |
2 |
|
|
|
| Solid phase extraction |
2 |
|
|
(X) |
| Distillation/rotary evaporation |
3 |
X |
X |
|
| Sublimation |
3 |
|
|
|
| Liquid–liquid extraction |
3 |
|
|
|
| Classical chromatography |
10 |
|
|
|
The final penalty point for the yield was 27.4 for SEA-MAH160 (mean between 26.3 and 28.5) as it had the lowest polysaccharide yield and the second best usnic acid and antioxidant yield. SCF_E-MAH160 received 19.2 penalty points, having the second-best polysaccharide yield, the best usnic acid yield and the third antioxidant yield. Finally, MAH_ML5-170 received 12.3 penalty points, having the best yield of polysaccharides and antioxidant content and the third best usnic acid yield. Regarding the price, as ethanol and CO2 are inexpensive, SCF_E-MAH160 did not get any penalty points. As the price of ethyl acetate is around 52 € per L it was considered as expensive and received 3 penalty points, and the eutectic made with menthol and lauric acid was considered very expensive and had 5 penalty points allocated (menthol price for 1 kg is around 150 € and that of lauric acid is 60 €). For safety, all processes received 5 penalty points for being flammable, and ethyl acetate and ethanol were in addition penalised for certain toxicity (5 penalty points) and ethyl acetate for being dangerous for the environment (5 penalty points). As the microwave extraction is performed on pressurized vessels, all processes received 3 penalty points for “pressure equipment > 1 atm”. SCF_E-MAH160 received an extra 3 points as the supercritical CO2 equipment is also used under pressure. 2 penalty points were allocated to MAH_ML-5-170 for being an unconventional technique. All processes received 2 penalty points because of the short heating time during microwave extraction and 5 penalty points for cooling the samples below 0 °C to make the polysaccharides precipitate. During Soxhlet extraction and supercritical extraction, there was heating for >1 h which gave 3 additional penalty points. In the workup and purification category, ethyl acetate and ethanol need to be evaporated, coinciding with 3 penalty points. Regarding the lichenic acids in the NaDES phase, two options can be considered. One would be the direct use of the extracts in the eutectic and the second one would be a solid phase extraction. 2 penalty points would be allocated in the latter case.
To summarize, two of the three processes are considered to have an acceptable sustainability, SCF_E-MAH160 and MAH_ML-5-170, whereas SEA-MAH160 could not be considered a green process. The process using NaDES combined with microwaves received the best score mainly thanks to its low toxicity, its fast heating time and its good yield. However, its relatively high cost makes its recycling important to try, which would induce an additional solid phase extraction. In addition, despite NaDES being considered green solvents with low environmental impact compared to some organic solvents from non-renewable sources, the impact of the synthesis or bioconversion of their constituents could contribute to their life-cycle environmental impact.99 A summary of this comparative assessment is shown in Fig. 8.
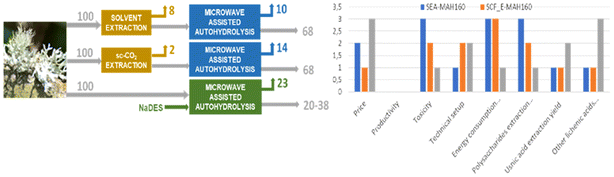 |
| | Fig. 8 Comparative summary of the tested processes. | |
3. Experimental
3.1. Materials
Samples from Evernia prunastri (L.) Ach. were collected in Nogueira de Ramuín (Ourense, Spain) (altitude: 608 m, 42° 24′ 47′′ North, 7° 43′ 34′′ West), air dried and ground in a lab knife miller before storage in airtight plastic bags in the dark. The specimen was identified by Prof. María Eugenia López de Silanes (University of Vigo, Spain) and a voucher specimen has been deposited at the Herbarium SANT (University of Santiago de Compostela, Spain, Ref. SANT-Lich 12461-A). The dried and ground (<0.5 cm) samples contained 11% moisture, and on dry basis contained 2.3 ± 0.3% ash, 9.9 ± 0.3% protein, 1.1 ± 0.1% lipids, 14.9 ± 0.1% acid insoluble residue, and 68.79 ± 2.36% carbohydrates, including 30.9% glucose, 13.7% galactose, 12.4% mannose, 3.79% arabitol, 1.29% mannitol, 3.7% glucuronic acid, 0.7% galacturonic acid, 1.7% acetic acid and minor amounts of arabitol and mannitol.27
Natural deep eutectic solvents were prepared with distilled water, L(+) lactic acid (Erbapharm 88–92%), glycine (Glentham Life Science >98%), DL-menthol (Thermo Scientific >98%), and lauric acid (Scharlau). Other reagents used in this study were pure liquid CO2 (Air Liquide); Folin-Ciocateau reagent, Na2HPO4, NaN3, K2S2O8, and KH2PO4 (Panreac Química); ABTS, BSA, gallic acid, Trolox, Bradford reagent, antrone, glucose, Nile Blue and Na2CO3 (Sigma-Aldrich); NaCl and NaOH (Thermo Scientific), KCl, ethyl acetate, ethanol 96%, acetone and sulfuric acid (Merck); and glycerol (Scharlau).
3.2. Conventional solvent extraction
Soxhlet extractions were performed for 8 h with acetone and ethyl acetate, at least in triplicate. The extracts were rotoevaporated and then suspended in ethanol for further analysis.
3.3. Supercritical fluid extraction
Pretreatment.
The control sample was compared with samples subjected to microwave pretreatment performed in a domestic microwave oven (BMO20SM-20, Bluesky, France) at 480 W or at 800 W for 1 min. After the treatments, the samples contained 9–10% moisture.
Extraction.
Dried lichens (10 g) were packed with glass beads into a 1 L cylinder extractor in a Thar Designs SFE-1000F-2-C10 (Pittsburgh, USA) equipment with two 500 mL separators. CO2 was precooled with a bath (PolyScience, USA, model 9506) and pumped with a P-200A piston pump (Thar Design Inc., Pittsburgh, PA, USA) at 25 g CO2 per min and the cosolvent was pumped with a HPLC pump (Scientific Systems, Inc., USA, model Series III). Extraction conditions were fixed at 35 MPa, 40 °C and 5% ethanol, based on previously reported data.26 The extract was collected at ambient temperature in the first separator, and was further washed with 96% ethanol. Ethanol was removed in a Rotavapor and the extracts were kept under N2 at −20 °C in the dark until analysis.
Kinetic assays were performed for 4 h by collecting extract samples at pre-established time intervals. The overall extraction curves (OECs) obtained were evaluated using the Broken and intact cell model,43 based on the assumption that a part of the extractable material is accessible to the solvent due to cell rupture caused by milling and pretreatment, whereas the rest of the solutes remain trapped inside the intact cells, and in order to dissolve them, the solvent has to penetrate by diffusion. According to the different mass transport phenomena occurring, three periods are observed: (i) a constant extraction rate (CER) period, associated with the extraction, mainly by convection, of easily accessible solutes; (ii) a falling extraction rate (FER) period, where mass transfer is controlled by both convection and diffusion in the solid phase and (iii) a diffusion controlled (DC) period, in which easily extractable solutes are removed and mass transfer is governed by diffusion. The extracted solute as a function of time is described by the following equations:
| | | mext = Q\cdot Ys[1 − exp(−Z)] for t ≤ tCER | (14) |
| | | mext = Q\cdot Ys[t − tCER exp(Zw − Z)] for tCER < t ≤ tFER | (15) |
| |  | (16) |
where
| | 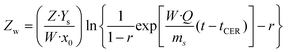 | (17) |
| | 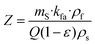 | (18) |
| | 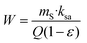 | (19) |
| | 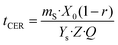 | (20) |
| | 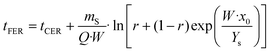 | (21) |
where
mext is the mass of the extracted solute (kg),
Q is the solvent flow rate (kg h
−1),
YS is the pseudo-solubility of the extract in the solvent (kg kg
−1),
x0 is the initial mass fraction of the extract in the inert material (kg kg
−1),
r is the fraction of broken cells,
mS is the mass of the inert solid material (kg),
ρf is the density of the solvent (kg m
−3),
ρs is the bed density (kg m
−1),
ε is the bed porosity,
kfa is the fluid-phase mass transfer coefficient (h
−1),
ksa is the solid-phase mass transfer coefficient (h
−1),
tCER is the extraction time at the end of the CER period (h) and
tFER is the extraction time at the beginning of the diffusional period (h).
The initial mass ratio (x0) was fixed as the asymptotic value at infinite time. The slope of the first linear part of the OEC curve was employed as an initial estimation of YS, and fitted to the experimental data along with the adjustable parameters kfa, ksa and r by minimizing the sum of least squares between the experimental and calculated values of mext.
The absolute average relative deviation (AARD) given by eqn (22) was used for error estimation:
| | 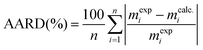 | (22) |
where
n is the experimental observation number, and
mexp and
mcalc. are the experimental and calculated extract mass, respectively.
3.4. Microwave assisted hydrothermal extraction
The aqueous extraction was performed in an Anton Paar Microwave reactor Monowave 450 (Austria). The residual solids after acetone and ethyl acetate extraction or after supercritical CO2 extraction were mixed with distilled water at a solid/liquid ratio of 1:30 (w/w) and after heating up to 160 °C (after around 1 min), isothermal operation was maintained for 5 min with an agitation speed of 800 rpm. After cooling down to 50 °C, the solid and liquid phases were separated by vacuum filtration.
3.5. Natural deep eutectic solvent (NaDES)
Selection and preparation of the solvent.
Two eutectic mixtures were selected based on their stability, non-toxicity, and different ranges of polarities to try to extract different kinds of bioactive compounds. A solvent with high polarity, prepared with lactic acid:glycine:water at a molar ratio (3:1:3), was selected as a more stable and transparent solvent than lactic acid:D-glucose monohydrate:water (5:1:1), proline:glutamic acid (2:1) and lactic acid:sodium citrate (4:1). The use of a hydrophobic solvent was also proposed to favor the extraction of lichen acids, forming a biphasic system with water in order to also extract polar compounds, using DL-menthol and lauric acid at a molar ratio (2:1). In both cases, all components were mixed for at least 40 min at 70 °C, until transparent monophasic liquids were obtained. They were then stored at room temperature in closed vials before being used within a week.
Microwave assisted hydrothermal extraction with NaDES as cosolvent.
The ground lichen biomass was processed by microwave assisted hydrothermal extraction, performed in an Anton Paar Microwave reactor Monowave 450 (Austria) using a solid/liquid ratio of 1:30 (w/w), for 5 min, with an agitation speed of 800 rpm. The vials were cooled down until 50 °C before vacuum filtration or centrifugation depending on the extracts. The soluble extracts were stored at −18 °C, whereas the solid ones were dried in an oven and stored at room temperature for further characterization.
Design of the experiment and data modelling.
In order to evaluate the effect of the NaDES percentage (NP) and the extraction temperature on different response variables (carbohydrates, protein, TEAC, and polymer mechanical properties), a Central Composite Design (CCD), consisting on 11 experimental runs including 4 axial or star points and a triplicated central point, was applied. The temperature in the range 110–180 °C and two NaDES percentages in the solvent, 5–17% and 44–86%, were evaluated. For each response variable, results were analyzed by multilinear regression using a full quadratic model from eqn (23):| | | Y = b0 + b1X1 + b2X2 + b1,2X1X2 + b1,1X12 + b2,2X22 | (23) |
where Y is the response variable, X1 and X2 are the independent factors (with X1 = NP and X2 = extraction temperature), b0 the intercept, b1 and b2 the coefficients of the linear effect of X1 and X2, b1,2 the coefficient for the interaction effect, and b1,1 and b2,2 the coefficients of the quadratic effects of X1 and X2.
A backward elimination was performed on all full quadratic models, eliminating all non-significant terms at a 10% level of acceptance through t-tests. ANOVA tests were also performed to evaluate the models’ accuracy. Only results where the null hypothesis “H0: The model fits the data adequately” for the lack of fit test was accepted at a 5% level of acceptance are displayed. All these steps have been performed on R Studio, using the “rsm” package.
3.6. Biopolymer recovery and formulation of film matrices
Freezing the liquid phases for at least 2 days and thawing were used to obtain biopolymers, after precipitation and recovery by centrifugation (5 °C, 10 min, 5000 rpm). Rheological analyses to compare their viscosity and viscoelastic properties were performed on the polymers before drying in an oven at 40 °C for 24 h. In order to prepare the film forming solutions, the polymers were dissolved in distilled water to obtain a 2% dw solution and glycerol (40% dw of polymer) was added as a plastifier. Two mL of the film forming solutions was placed in a circular cup (diameter = 2.4 cm) and oven dried at 40 °C for 24 h.
3.7. Analytical methods
Moisture and ash contents were gravimetrically determined at 105 °C for 24 h and at 575 °C for 6 h, respectively. The protein content was estimated using the universal factor 6.25 to convert the nitrogen content, determined by Kjeldahl. Hydrogen and carbon contents were measured on an elemental analyzer (Thermo Flash EA 1112, Germany). Carbohydrates were analyzed by high performance liquid chromatography (HPLC) after an acid hydrolysis of samples, with 72% sulfuric acid at 30 °C for 60 min and 4% sulfuric acid at 121 °C for 60 min and filtration to separate the acid-insoluble residue (AIR) and the filtrate was further filtered (0.45 μm). The monomers were quantified by HPLC (1100 series Agilent Technologies, California, USA) in an Aminex HPX87H column (300 × 7.8 mm, BioRad, USA) operated with 0.6 mL min−1 of 0.003 M sulfuric acid (Sigma-Aldrich, USA) as the mobile phase. 1 mg mL−1 solutions of extracts were analysed for secondary phenol metabolites.100 Secondary metabolites content of extracts was determined by HPLC/DAD/MS using a Prominence Shimadzu LC-20AD system and an ADVION expression CMS mass spectrometer. The chromatography was conducted on a Phenomenex Kinetex C18 column (2.6 μm, 100 × 4.6 mm), with a standard gradient program using 0.1% formic acid in HPLC-grade water and 0.1% formic acid in acetonitrile.101 An electrospray ionisation (ESI) source in negative mode was used and data were treated with MZmine 2v.53102 to identify lichen compounds by dereplication against our in-house databases.
3.8. NaDES characterization
Polarity.
Nile Blue A, exhibiting a maximum absorbance varying as a function of the polarity of the medium, with a blue shift in more apolar medium, was used to estimate the polarity of each eutectic mix used. A small amount of Nile Blue A dye was first diluted in ethanol and water, as LA:Gly:W is insoluble in ethanol and Men:Lau is insoluble in water, and this first solution was dissolved until an absorbance between 0.6 and 1.2 was reached. Then 50 μL of the dye dilution were added to 950 μL of the solvent and the absorbance was scanned from 400 to 700 nm.44,45
Viscosity.
Steady-state shear measurements were performed at 25 °C in an MCR 302 controlled stress-rheometer (Paar Physica, Austria) with a Peltier module (±0.01 °C) to determine the apparent viscosity. Solvents were placed in a sand blasted parallel plate of 25 mm diameter at a 1 mm gap. The apparent viscosity vs. the increasing shear rate of the prepared solutions was recorded from 1 to 100 s−1, as well as the corresponding time-dependence.
Density.
Density was manually determined weighing 3 times 1 mL of solvent.
Stability.
Solvents were placed at room temperature in vials. The apparent viscosity and visual appearance were assessed after 1 month.
3.9. Liquid phase characterization
Total phenolic content.
The phenolic content was estimated using the Folin–Ciocalteau method.46 In test tubes, 1.875 mL of distilled water and 0.125 mL of the Folin reactive diluted at a 1:1 ratio with distilled water and 0.25 mL of Na2CO3 at 20% were mixed with 0.25 mL of sample. The hydrolysate phase was diluted by 10–50 times as reported by other authors.32,34 After one hour in the dark, the absorbance was measured at λ = 765 nm. In the case of DL-menthol:lauric acid, 0.750 mL of NaOH at 4% was added to avoid NaDES precipitation, following the article in ref. 47 The standard curve was set up using gallic acid at concentrations between 0.1 and 0.00625 mg mL−1.
Carbohydrate content.
The total carbohydrate content was estimated with the anthrone method. 0.75 g of anthrone were dissolved in 500 mL of sulphuric acid at 70% (v/v) and 2.5 mL of this mixture was added to 0.1 mL of sample. The test tubes were then heated at 90 °C for 17 min before measuring their absorbance at 625 nm. The standard curve was set up using glucose from 0 to 0.5 g L−1.
Oligosaccharide content.
The liquid phases from hydrothermal treatment were subjected to hydrolysis with sulfuric acid (4%) at 121 °C for 20 min, and once cooled to room temperature were filtered (0.45 μm) before HPLC determination. In experiments with NaDES, their contribution during post-hydrolysis was considered in a blank sample.
Soluble protein content.
The soluble protein content was measured reading the absorbance at 595 nm after 16 min of 0.5 mL of Bradford reagent added to 0.5 mL of sample. Bovine serum albumin was used as a standard.48
The antiradical capacity of the samples was evaluated using the ABTS (2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid)) radical scavenging technique.49 The Trolox equivalent antioxidant capacity (TEAC) reagent was diluted in a PBS buffer to reach an absorbance at 734 nm of 0.7 ± 0.05 nm. One mL of the reagent was added to 10 μL of sample and incubated for 6 min at 30 °C. The blank was the PBS buffer, and a control with water was used for each 9 measurements. The standard curve was prepared using Trolox from 0.1 to 1 mM.
3.10. Structural characterization
The recovered biopolymers were analyzed by Fourier-transform infrared spectroscopy (FTIR) using a Nicolet 6700 spectrometer (Thermo Fisher Scientific, Massachusetts, USA). Samples were blended with KBr and the spectra were monitored from 500 to 4000 nm using a resolution of 4 cm−1 and 32 scans per min employing the OPUS-2.52 software (Opus Software Limited, UK).
The molecular weight distribution.
The molecular weight distribution was determined by High-performance size exclusion chromatography (HPSEC). The measurements were conducted on a HPLC (Agilent Technologies, California, USA) equipped with a TSKgel SuperMultiporePW-H column (6 mm × 15 cm) and a TSKgel SuperMP(PW)-H guard column (4.6 mm × 3.5 cm) (Tosoh, Tokio, Japan). Operating conditions were fixed at 40 °C, using Milli-Q water (0.4 mL min−1) as the mobile phase. Polyethylene oxide was employed as a standard, from 23.6 103 to 786 103 g mol−1.
SEM.
Scanning electron microscopy (SEM) was used to study the residual solid morphology (JEOL JSM6010LA, Japan). The different residual solids were previously coated with a 15 nm layer of gold.
3.11. Characterization of film matrices
Cytotoxicity on A549 human tumoral (Sigma Aldrich) cells was assessed for the recovered biopolymers, based on the fact that tetrazolium salts of MTT (3-[4,5-dimethylthiazol-2-yl]-2,-5 diphenyltretrazolium bromide) are turned into formazan when the cells are metabolically active. Briefly, 10,000 cells were seeded into each well of a 96-well sterile plate and incubated for 24 h in the growth medium (Eagle's minimum essential medium, EMEM, with 10% fetal bovine serum under an atmosphere of 95% air and 5% CO2 at 37 °C). The tested extracts were dissolved in water, added to the cells and incubated for 7 days. Then, 10 μL of 5 mg mL−1 MTT in PBS (0.136 M NaCl, 1.47 mM KH2PO4, 8 mM NaH2PO4, and 2.68 mM KCl) was added to the wells and the plate was incubated for 4 additional days. Finally, 100 μL of 10% SDS in 0.01 M HCl was added before incubating the plate for 12–14 h. The absorbance of the cell plate was measured at 595 nm in a Tecan M1000 infinite Pro microplate reader between a value for 10,000 cells in EMEM and in the absence of growth factors (to determine the stable cell concentration) and another value for those in the usual growth medium (to determine the maximum growth at 7 days). The control with water instead of extracts did not exhibit any growth inhibition. All experiments were carried out with triplicate. The growth inhibition was determined using the following formula:| | 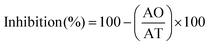 | (24) |
with AO = absorbance of the wells with the extracts and AT = absorbance of the control wells with H2O.
For the extracts showing a concentration-dependent inhibition, the inhibitory potency was evaluated by calculating the concentration − % inhibition curve adjusted to the equation:
| |  | (25) |
where
y is the observed effect at concentration
x,
Emax the maximum observed effect, IC
50 the concentration at which a 50% growth inhibition is reached and
n the slope of the curve. Cisplatin was used as a reference for the growth inhibition of A549 cells. The cytotoxicity of the polymers was also tested on MRC-5 non-cancerous cells.
Antimicrobial studies.
Antimicrobial studies were performed to assess the microbial inhibition capacity of the three selected extracts in the developed lichenan-based films. Gram positive and Gram negative bacterial strains from the Spanish Collection of Type Cultures (CECT), such as Bacillus cereus (CECT 148), Enterococcus faecalis (CECT 481), Listeria monocytogenes (CECT 935), Staphylococcus aureus (CECT 435), Staphylococcus epidermidis (CECT 231), Escherichia coli (CECT 516), Pseudomonas aeruginosa (CECT 108) and Salmonella spp. (CECT 4594), were used. They were seeded on Mueller–Hinton agar plates at a concentration of 104 ufc mL−1 in all cases. The inoculum was distributed homogeneously over the surface of the plates before a portion of each of the lichen films was aseptically applied in contact with the surface of the plate. The plates were incubated at 37 °C under the favourable conditions of each bacterium. The trials were performed at least in triplicate.
3.12. Statistical analysis
All the above measurements were performed at least in triplicate. Experimental data were assessed using one-factor analysis of variance, ANOVA, by means of a statistical software (PASW Statistics v.22, IBM SPSS Statistics, New York, USA). Whenever differences between means were distinguished, a post-hoc Scheffé test was carried out (95% confidence, p < 0.05).
4. Conclusions
The extraction of the major bioactives and biopolymers from lichens was proposed using a sequential process with conventional or with supercritical extraction followed by a hydrothermal stage and alternatively the extraction was carried out by a single hydrothermal extraction with a hydrophobic NaDES as cosolvent. When supercritical CO2 extraction was used for the selective separation of lichenic acids and phenolic compounds, the required time could be reduced compared to conventional extraction using acetone or ethyl acetate. In addition, the lichenic acids were more concentrated and the antiradical properties were enhanced. Further microwave assisted aqueous extraction under subcritical conditions solubilized up to 50% polysaccharides which could be used to formulate films. The simultaneous extraction of bioactives and biopolymers using menthol:lauric acid as cosolvent at 5% allowed the production of a material enriched in usnic acid, with antiproliferative action against lung cancer cells and antimicrobial activity against Gram positive bacteria. From a sustainability point of view, the conventional Soxhlet extraction followed by microwave assisted hydrothermal extraction cannot be considered green. This process uses more toxic and hazardous chemicals and has a lower polysaccharide extraction yield than the microwave extraction with menthol:lauric acid and a lower usnic acid extraction yield than that with supercritical CO2. The supercritical CO2 extraction followed by microwave hydrothermal extraction received an intermediate Eco-Scale score and can be considered to have an acceptable sustainability. The low toxicity and good yield (for polysaccharide and usnic acid) of the microwave extraction with NaDES make it the more sustainable of the three processes studied.
Author contributions
Conceptualization, H. D., M. D. T. and B. D-R.; methodology, J. Q., B. D-R., M. D. T., S. F., J. B., W. B. and I. R-G.; writing – review and editing, H. D., M. D. T., J. Q., B. D-R., S. F., J. B., W. B. and I. R-G.; supervision, H. D. and M. D. T.; funding acquisition, H. D. and M. D. T.; project administration, H. D. and M. D. T. All authors listed have made a substantial, direct, and intellectual contribution to the work and have agreed to the published version of the manuscript.
Data availability
The data supporting this work have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge Consellería de Cultura, Educación e Universidade da Xunta de Galicia (GRC-ED431C 2022/08), CINBIO (ED481D-2022/018) and measurements performed at the services of analysis of Universidade de Vigo (CACTI). M. D. T. acknowledges the Ministry of Science, Innovation and Universities of Spain for her postdoctoral grants (RYC2018-024454-I) and the Consellería de Cultura, Educación e Universidade da Xunta de Galicia (ED431F 2020/01). J. Q. thanks the Early Stage Researcher (CINBIO22/001). This article is based upon work from COST Action <CA 18224> and supported by COST (European Cooperation in Science and Technology). Funding for open access charge: Universidade de Vigo/CISUG.
References
- D. Joulain and R. Tabacchi, Flavour Fragrance J., 2009, 24(2), 49–61 CrossRef CAS.
- S. Huneck, Naturwissenschaften, 1999, 86(12), 559–570 CrossRef CAS PubMed.
- R. Sargsyan, A. Gasparyan, G. Tadevosyan and H. Panosyan, AMB Express, 2021, 11(1), 110 CrossRef CAS PubMed.
- V. Shukla, G. P. Joshi and M. S. M. Rawat, Phytochem. Rev., 2010, 9(2), 303–314 CrossRef CAS.
- C. Fernández-Moriano, P. K. Divakar, A. Crespo and M. P. Gómez-Serranillos, Food Chem. Toxicol., 2017, 105, 262–2771 CrossRef PubMed.
- M. Kello, M. Goga, K. Kotorova, D. Sebova, R. Frenak, L. Tkacikova and J. Mojzis, Plants, 2023, 12(3), 611 CrossRef CAS PubMed.
- G. Shrestha, J. Raphael, S. D. Leavitt and L. L. St Clair, Pharm. Biol., 2014, 52(10), 1262–1266 CrossRef PubMed.
- V. Menon, R. Divate and M. Rao, Fuel Process. Technol., 2011, 92(3), 401–406 CrossRef CAS.
- T. Majtan, E. M. Bublil, I. Park, E. Arning, T. Bottiglieri, F. Glavin and J. P. Kraus, Life Sci., 2018, 200, 15–25 CrossRef CAS PubMed.
- G. Shrestha, L. L. St Clair and K. L. O'Neill, Phytother. Res., 2015, 29(3), 3170–3322 CrossRef PubMed.
- A. P. Podterob, Pharm. Chem. J., 2008, 42(10), 582–588 CrossRef CAS.
- A. Zugic, I. Jeremic, A. Isakivic, I. Arsic and S. Savic, PLoS One, 2016, 11(1), 0146342 CrossRef PubMed.
- A. Galanty, P. Paśko, I. Podolak and P. Zagrodzki, Lichenologist, 2020, 52(5), 397–401 CrossRef.
- A. Galanty, P. Koczurkiewicz, D. Wnuk, M. Paw, E. Karnas, I. Podolak, M. Węgrzyn, M. Borusiewicz, Z. Madeja and J. Czyż, Toxicol. in Vitro, 2017, 40, 161–169 CrossRef CAS PubMed.
- J. Ivanovic, F. Meyer, M. Stamenic, P. Jaeger, I. Zizovic and R. Eggers, Chem. Eng. Technol., 2014, 37(9), 1606–1611 CrossRef CAS.
- S. P. Kwong and C. Wang, Environ. Toxicol. Pharmacol., 2020, 80, 103493 CrossRef CAS PubMed.
- J. Ivanovic, F. Meyer, D. Misic, J. Asanin, P. Jaeger, I. Zizivic and R. Eggers, J. Supercrit. Fluids, 2013, 76, 1–9 CrossRef CAS.
- M. Kosanić, N. Manojlović, S. Janković, T. Stanojković and B. Ranković, Food Chem. Toxicol., 2013, 53, 112–118 CrossRef PubMed.
- R. Staples, R. L. LaDuca, L. V. Roze, M. Laivenieks, J. E. Linz, R. Beaudry, A. Fryday, A. L. Schilmiller, A. V. Koptina, B. Smith and F. Trail, Chem. Biodiversity, 2020, 17, 1900465 CrossRef PubMed.
- B. Díaz-Reinoso, I. Rodríguez-González and H. Domínguez, Rev. Environ. Sci. Bio/Technol., 2021, 20, 917–942 CrossRef.
- S. Komaty, A. Sauvager, J. P. Bazureau, S. Tomasi and L. Paquin, Phytochem. Anal., 2020, 32(4), 592–600 CrossRef PubMed.
- M. Kulinowska, S. Dresler, A. Skalska-Kamińska, A. Hanaka and M. Strzemski, Molecules, 2023, 28, 5321 CrossRef CAS PubMed.
- I. Zizovic, J. Ivanovic, D. Misic, M. Stamenic, S. Djordjevic, J. Kukic-Markovic and S. D. Petrovic, J. Supercrit. Fluids, 2012, 72, 7–14 CrossRef CAS.
- T. A. Boitsova, O. S. Brovko, A. D. Ivakhnov and D. V. Zhil'tsov, Russ. J. Phys. Chem. B, 2020, 14(7), 1135–1114 CrossRef CAS.
- C. A. Dinçer, C. Gökalp, B. Getiren, A. Yildiz and N. Yildiz, Turk. J. Chem., 2021, 45(4), 1248–1256 CrossRef PubMed.
- O. S. Brovko, A. D. Ivakhnov, I. A. Palamarchuk and T. A. Boitsova, Russ. J. Phys. Chem. B, 2017, 11, 1306–1311 CrossRef CAS.
- J. Queffelec, N. Flórez-Fernández, M. D. Torres and H. Domínguez, Int. J. Biol. Macromol., 2024, 258, 128859 CrossRef CAS PubMed.
- P. S. Saravana, Y. N. Cho, H. C. Woo and B. S. Chun, J. Cleaner Prod., 2018, 198, 1474–1484 CrossRef CAS.
- S. Machmudah, S. D. Lestari, Widiyastuti, Wahyudiono, H. Kanda, S. Winardi and M. Goto, J. Supercrit. Fluids, 2018, 133, 615–624 CrossRef CAS.
- M. Kuasnei, J. P. Wojeicchowski, N. H. Santos, V. Z. Pinto, S. R. S. Ferreira and A. A. F. Zielinski, J. Supercrit. Fluids, 2022, 191, 105761 CrossRef CAS.
- L. Benvenutti, A. A. F. Zielinski and S. R. S. Ferreira, J. Supercrit. Fluids, 2022, 189, 105729 CrossRef CAS.
- R. Huang, Y. He, X. Yao, Y. Yu, W. Song, W. Yang and J. Cheng, Green Chem., 2022, 24(4), 1615–1626 RSC.
- L. Loarce, R. Oliver-Simancas, L. Marchante, M. C. Díaz-Maroto and M. E. Alañón, Food Res. Int., 2020, 137, 109728 CrossRef CAS PubMed.
- V. C. Roy, J. S. Park, A. R. Haque, M. S. Ali, H. J. Lee and B. S. Chun, J. Supercrit. Fluids, 2024, 204, 106108 CrossRef CAS.
- J. Zhang, Z. Ye, G. Liu, L. Liang, C. Wen, X. Liu, Y. Li, T. Ji, D. Liu, J. Ren and X. Xu, Molecules, 2022, 27(11), 3612 CrossRef CAS PubMed.
- P. Lohmus and A. Lohmus, Forests, 2019, 10(12), 1063 CrossRef.
- T. Tullus, R. Lutter, T. Randlane, A. Saag, A. Tullus, E. Roosaluste, P. Koresaar, M. Pärtel and H. Tullus, Can. J. For. Res., 2020, 50(12), 1268–1280 CrossRef.
- N. Bukhanko, T. Attard, M. Arshadi, D. Eriksson, V. Budarin, A. J. Hunt, P. Geladi, U. Bergsten and J. Clark, Ind. Crops Prod., 2020, 45, 112096 CrossRef.
- F. G. Fermoso, A. Serrano, B. Alonso-Fariñas, J. Fernández-Bolaños, R. Borja and G. Rodríguez-Gutiérrez, J. Agric. Food Chem., 2018, 66(32), 8451–8468 CrossRef CAS PubMed.
- R. Miranda-González and B. McCune, Biotropica, 2020, 52(6), 1298–1308 CrossRef.
- O. Krynytska, T. Bondarenko, J. Capuliak and M. Trenciansky, Cent. Eur. For. J., 2017, 63(1), 35–41 Search PubMed.
-
P. Martín and J. A. Oria, Guía técnica de gestión de matorrales ibéricos, Ministerio para la Transición Ecológica y el Reto Demográfico, Madrid, 2021, https://www.miteco.gob.es/content/dam/miteco/es/biodiversidad/temas/incendios-forestales/221275_937guiagestionmatorrales_web_tcm30-589399.pdf Search PubMed.
- H. Sovová, Chem. Eng. Sci., 1994, 49(3), 409–414 CrossRef.
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed.
- H. Tajalli, A. Ghanadzadeh Gilani, M. S. Zakerhamidi and P. Tajalli, Dyes Pigm., 2008, 78(1), 15–24 CrossRef CAS.
- V. L. Singleton and J. A. Rossi, Am. J. Enol. Vitic., 1965, 16, 144–158 CrossRef CAS.
- E. Boli, N. Prinos, V. Louli, G. Pappa, H. Stamatis, K. Magoulas and E. Voutsas, Separations, 2022, 9, 255 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 7(72), 248–254 CrossRef.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radicals Biol. Med., 1999, 26(9–10), 1231–1237 CrossRef CAS PubMed.
- E. S. Olafsdottir, S. Omarsdottir, B. S. Paulsen, K. Jurcic and H. Wagner, Phytomedicine, 1999, 6(4), 273–279 CrossRef CAS PubMed.
- R. Rashid, H. Ahmad, Z. Ahmed, F. Rashid and N. Khalid, Bioact. Carbohydr. Diet. Fibre, 2019, 19, 100194 CrossRef CAS.
- M. A. Fanovich, J. Ivanovic, D. Misic, M. V. Alvarez, P. Jaeger, I. Zizovic and R. Eggers, J. Supercrit. Fluids, 2013, 78, 42–53 CrossRef CAS.
- E. Calla-Quispe, J. Robles, C. Areche and B. Sepúlveda, Front Chem., 2020, 8, 450 CrossRef CAS PubMed.
- S. Komaty, M. Letertre, H. D. Dang, H. Jungnickel, P. Laux, A. Luch, D. Carrié, O. Merdrignac-Conanec, J. P. Bazureau, F. Gauffre, S. Tomasi and L. Paquin, Talanta, 2016, 150, 525–530 CrossRef CAS PubMed.
- T. A. Boitsova, O. S. Brovko, A. D. Ivakhnov and D. V. Zhil'tsov, Russ. J. Phys. Chem. B, 2020, 14, 1135–1141 CrossRef CAS.
- T. C. Confortin, I. Todero, L. Luft, G. A. Ugalde, M. A. Mazutti, Z. B. Oliveira, E. L. Bottega, A. E. Knies, G. L. Zabot and M. V. Tres, J. Environ. Chem. Eng., 2019, 7(2), 102972 CrossRef CAS.
- V. Popovici, L. Bucur, C. E. Gîrd, A. Popescu, E. Matei, G. C. Cozaru, V. Schröder, E. A. Ozon, A. C. Fiţa and D. Lupuliasa,
et al., Pharmaceuticals, 2022, 15, 829 CrossRef CAS PubMed.
- N. Aoussar, R. Manzali, I. Nattah, N. Rhallabi, P. Vasiljevic, M. Bouksaim, A. Douira, N. Manojlović and F. Mellouki, J. Mater. Environ. Sci., 2017, 8(6), 1968–1976 CAS.
- A. Shcherbakova, A. A. Strömstedt, U. Göransson, O. Gnezdilov, A. Turanov, D. Boldbaatar, D. Kochkin, G. Ulrich-Merzenich and A. Koptina, World J. Microbiol. Biotechnol., 2021, 37(8), 129 CrossRef CAS PubMed.
- A. Aslan, M. Güllüce, M. Sökmen, A. Adigüzel, F. Sahin and H. Özkan, Pharm. Biol., 2006, 44(4), 247–252 CrossRef.
- T. Mitrović, S. Stamenković, V. Cvetković, S. Tošić, M. Stanković, I. Radojević, O. Stefanović, L. Comić, D. Dačić, M. Curčić and S. Marković, Int. J. Mol. Sci., 2011, 12(8), 5428–5248 CrossRef PubMed.
- M. Kosanic, N. Manojlovic, S. Jankovic, T. Stanojkovic and B. Rankovic, Food Chem. Toxicol., 2023, 53, 112–118 CrossRef PubMed.
- V. M. Dembitsky, T. Rezanka and I. A. Bychek, Phytochemistry, 1992, 31(3), 851–853 CrossRef.
- J. González-Rivera, A. Mero, E. Husanu, A. Mezzetta, C. Ferrari, F. D'Andrea, E. Bramanti, C. S. Pomelli and L. Guazzelli, Green Chem., 2021, 23(24), 10101–10115 RSC.
- S. Rebocho, F. Mano, E. Cassel, B. Anacleto, M. d. R. Bronze, A. Paiva and A. R. C. Duarte, Curr. Res. Food Sci., 2022, 5, 571–580 CrossRef CAS PubMed.
- J. Panić, M. Rapaić, S. Gadžurić and M. Vraneš, Molecules, 2013, 28(15), 5723 CrossRef PubMed.
- C. Vieira, S. Rebocho, R. Craveiro, A. Paiva and A. R. C. Duarte, Front. Chem., 2022, 10, 954835 CrossRef CAS PubMed.
- X. Hu and H. D. Goff, Trends Food Sci. Technol., 2018, 81, 108–115 CrossRef CAS.
- J. Cao, L. Chen, M. Li, F. Cao, L. Zhao and E. Su Cao, Green Chem., 2018, 20(8), 1879–1886 RSC.
- S.-N. Chen, F.-H. Nan, M.-W. Liu, M.-F. Yang, Y.-C. Chang and S. Chen, Foods, 2023, 12, 659 CrossRef CAS PubMed.
- R. Javier-Astete, J. Jimenez-Davalos and G. Zolla, PLoS One, 2021, 16, 1–12 CrossRef PubMed.
- F. Xu, J. Yu, T. Tesso, F. Dowell and D. Wang, Appl. Energy, 2013, 104, 801–809 CrossRef CAS.
- F. Monteil-Rivera, G. H. Huang, L. Paquet, S. Deschamps, C. Beaulieu and J. Hawari, Bioresour. Technol., 2012, 104, 775–782 CrossRef CAS PubMed.
- C. Álvarez-Vasco, R. Ma, M. Quintero, M. Guo, S. Geleynse, K. K. Ramasamy, M. Wolcott and X. Zhang, Green Chem., 2016, 18, 5133 RSC.
- R. R. Ruggeri, F. F. Bressan, N. M. Siqueira, F. Meirelles, N. Frantz, Y. F. Watanabe, R. M. D. Soares and A. Bos-Mikich, Macromol. Res., 2014, 22(10), 1053–1058 CrossRef CAS.
- G. O. Coelho, M. J. A. Batista, A. F. Ávila, A. S. Franca and L. S. Oliveira, J. Food Eng., 2021, 289, 110083 CrossRef CAS.
- P. Guerrero, T. Garrido, I. Leceta and K. De La Caba, Eur. Polym. J., 2013, 49(11), 3713–3721 CrossRef CAS.
- F. Liu, W. Chang, M. Chen, F. Xu, J. Ma and F. Zhong, Food Hydrocolloids, 2020, 98, 105007 CrossRef CAS.
- V. R. F. Dos Santos, B. W. S. Souza, J. A. Teixeira, A. A. Vicente and M. A. Cerqueira, J. Food Sci. Technol., 2015, 52(12), 8292–8299 CrossRef CAS PubMed.
- J. A. D. A. Nascimento, L. K. S. Gomes, D. S. Duarte, M. A. C. D. Lima and D. D. Britto, Mater. Res., 2019, 22, e20190057 CrossRef.
- R. Wang, S. Zhang, S. Liu, Y. Sun and H. Xu, Polymers, 2023, 15(7), 1751 CrossRef CAS PubMed.
- M. A. Castro-Yobal, A. Contreras-Oliva, V. Saucedo-Rivalcoba, J. L. Rivera-Armenta, G. Hernández-Ramírez, J. Salinas-Ruiz and A. Herrera-Corredor, e-Polym., 2021, 21(1), 82–95 CrossRef CAS.
- M. Mohammadi, V. Zambare, L. Malek, C. Gottardo, Z. Suntres and L. Christopher, Expert Opin. Drug Discovery, 2020, 15(5), 575–601 CrossRef CAS PubMed.
- T. Xin, F. Zhang, Q. Jiang, C. Chen, D. Lv, Y. Huang, W. Shen and Y. Jin, Carbohydr. Polym., 2012, 90(4), 1671–1676 CrossRef CAS PubMed.
- J. Gao, Y. Zhao, C. Wang, H. Ji, J. Yu, C. Liu and A. Liu, Int. J. Biol. Macromol., 2020, 158, 689–697 CrossRef CAS PubMed.
- A. Choromanska, J. Kulbacka, J. Harasym, R. Oledzki, A. Szewczyk and J. Saczko, Pathol. Oncol. Res., 2018, 24(3), 583–592 CrossRef CAS PubMed.
- F. Peymaeei, F. Sadeghi, E. Safari, S. Khorrami, M. Falahati, S. R. Mohammadi and M. Roudbary, Asian Pac. J. Cancer Prev., 2020, 21(3), 837 CrossRef CAS PubMed.
- M. Zhou, L. Yang, S. Yang, F. Zhao, L. Xu and Q. Yong, Int. J. Biol. Macromol., 2018, 113, 1241–1247 CrossRef CAS PubMed.
- F. Brisdelli, M. Perilli, D. Sellitri, M. Piovano, J. A. Garbarino, M. Nicoletti, A. Bozzi, G. Amicosante and G. Celenza, Phytother. Res., 2013, 27(3), 431–437 CrossRef CAS PubMed.
- R. Ingelfinger, M. Henke, L. Roser, T. Ulshöfer, A. Calchera, G. Singh, M. J. Parnham, G. Geisslinger, R. Fürst, I. Schmitt and S. Schiffmann, Front. Pharmacol., 2020, 11, 1322 CrossRef CAS PubMed.
- L. A. Roser, P. Erkoc, R. Ingelfinger, M. Henke, T. Ulshöfer, A. K. Schneider, V. Laux, G. Geisslinger, I. Schmitt, R. Fürst and S. Schiffmann, Biomed. Pharmacother., 2022, 14, 112734 CrossRef PubMed.
- K. Kumar, J. P. N. Mishra and R. P. Singh, Chem.-Biol. Interact., 2020, 3155, 108898 CrossRef PubMed.
- T. X. Sun, M. Y. Li, Z. H. Zhang, J. Y. Wang, Y. Xing, M. Ri, C. H. Jin, G. H. Xu, L. X. Piao, H. L. Jin, H. X. Zuo, J. Ma and X. Jin, Phytother. Res., 2021, 35(7), 3916–3935 CrossRef CAS PubMed.
- B. Murugesan, A. Subramanian, S. Bakthavachalam, K. Rajendran, S. Raju and S. Gabriel, J. Biomol. Struct. Dyn., 2023, 11, 1–19 CrossRef PubMed.
- Ş.N Kalın, A. Altay and H. Budak, J. Appl. Toxicol., 2023, 43(8), 1148–1158 CrossRef PubMed.
- C. Pagano, M. R. Ceccarini, P. Calarco, S. Scuota, C. Conte, S. Primavilla, M. Ricci and L. Perioli, Colloids Surf., B, 2019, 178, 488–499 CrossRef CAS PubMed.
- S. Sousa, P. Paíga, D. Pestana, G. Faria, C. Delerue-Matos, M. J. Ramalhosa, C. Calhau and V. F. Domingues, Anal. Methods, 2023, 15, 1722–1733 RSC.
- K. Van Aken, L. Strekowski, L. Patiny and L. Strekowski, Beilstein J. Org. Chem., 2006, 2(3) DOI:10.1186/1860-5397-2-3.
- Q. Zaib, M. Eckelman, Y. Yang and D. Kyung, Green Chem., 2022, 24(20), 7924–793028 RSC.
- E. González-Burgos, P.M. Kirika, J. Boustie, S. Ferron, M. P. Gómez-Serranillos, H. T. Lumbsch and P. K. Divakar, J. Fungi, 2022, 8, 826 CrossRef PubMed.
- A. Gadea, A.-C. Le Lamer, S. Le Gall, C. Jonard, S. Ferron, D. Catheline, D. Ertz, P. Le Pogam, J. Boustie, F. Lohézic-Le Devehat and M. Charrier, J. Chem. Ecol., 2018, 44, 471–482 CrossRef PubMed.
- T. Pluskal, S. Castillo, A. Villar-Briones and M. Oresic, BMC Bioinformatics, 2010, 11, 395 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Herminia
Domínguez
a and
Herminia
Domínguez

 , 1 min 480 W;
, 1 min 480 W;  , 1 min 800 W) pretreatment on the supercritical carbon dioxide extraction from Evernia prunastri at 35 MPa and 40 °C and with 5% ethanol as cosolvent. (a) Overall extraction yield and (b) usnic acid extraction yield. Control experiment (
, 1 min 800 W) pretreatment on the supercritical carbon dioxide extraction from Evernia prunastri at 35 MPa and 40 °C and with 5% ethanol as cosolvent. (a) Overall extraction yield and (b) usnic acid extraction yield. Control experiment ( ). Lines represent Sovová's model fittings.
). Lines represent Sovová's model fittings.