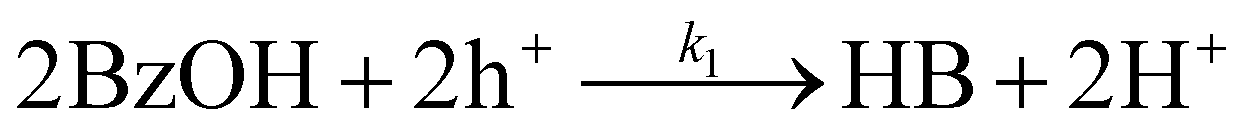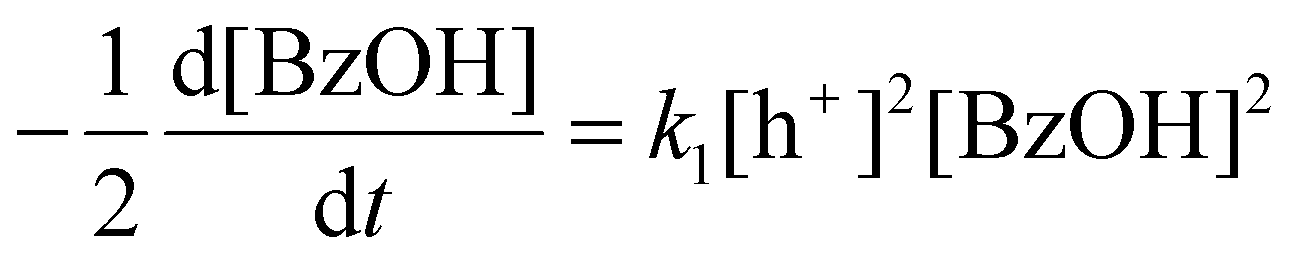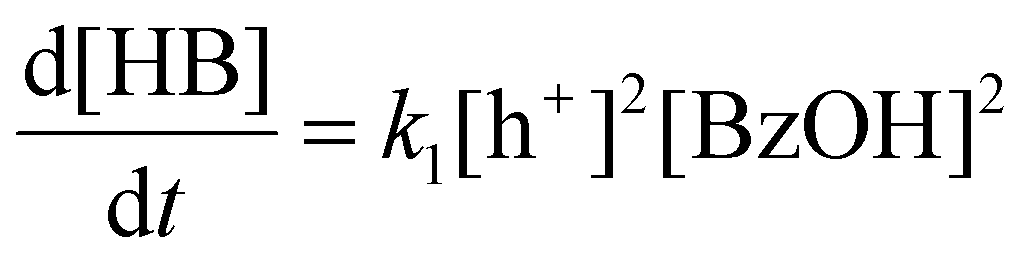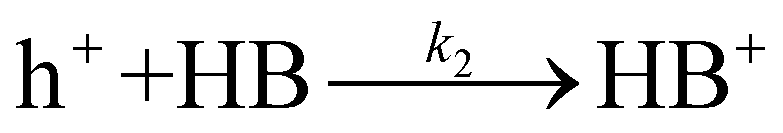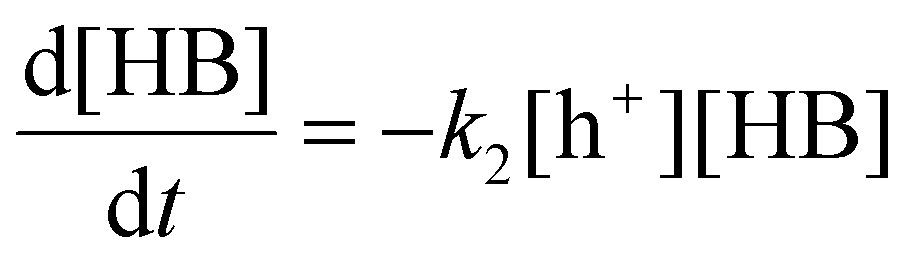DOI:
10.1039/D4NR03148B
(Paper)
Nanoscale, 2024,
16, 20977-20985
Selective photocatalytic C–C coupling of benzyl alcohol into hydrobenzoin using Pt-deposited CdS nanosheets passivated with cysteamine†
Received
31st July 2024
, Accepted 20th October 2024
First published on 21st October 2024
Abstract
Achieving high selectivity towards hydrobenzoin (HB) from photocatalytic carbon–carbon (C–C) coupling reaction of benzyl alcohol (BzOH) remains a challenge due to side competing reactions and subsequent conversions of HB into its derivatives. In this study, we have developed a high-performance CdS-based photocatalyst for synthesizing HB with precisely controlled surface properties and structure, achieving high selectivity for HB synthesis. We employed strategies such as cysteamine passivation and Pt deposition to address issues related to photogenerated charge trapping and recombination, thereby enhancing the photocatalytic capability of CdS. With optimized Pt/CdS NSs as the photocatalyst, we investigated the impact of the Pt/CdS heterostructure on intermediate reactions, which in turn altered product selectivity. Specifically, excessive Pt suppresses the electron-induced benzaldehyde-to-intermediate reaction by consuming electrons for the competing hydrogen evolution reaction (HER), leading to high selectivity toward benzaldehyde. In contrast, bare CdS without Pt suffers from insufficient charge supply for BzOH conversion due to the charge recombination issue, which promotes the subsequent conversion of HB to its derivatives. Notably, when Pt is precisely loaded to avoid dominant HER competition, the overall reaction rate increases, maintaining high selectivity towards HB and ensuring faster conversion of BzOH to HB rather than subsequent conversions of HB into its derivatives, thereby maximizing the HB yield. Subsequently, we have developed a photocatalyst that achieves a 93.4% conversion of 0.24 mmol BzOH with 85.3% selectivity toward HB under solar simulator irradiation (AM 1.5G). This work is expected to offer instructive guidance on rationally designing the photocatalyst for efficient C–C coupling reactions.
Introduction
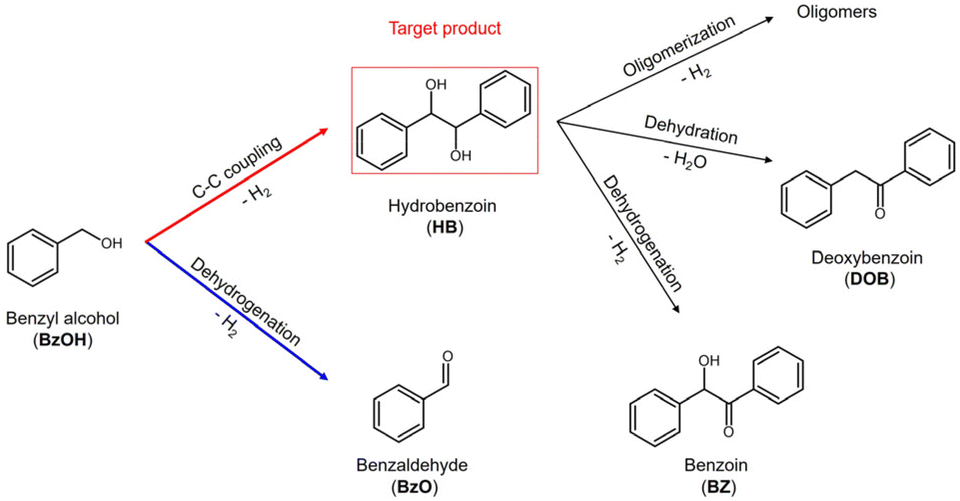
Photocatalytic carbon–carbon (C–C) coupling reactions, driven by light and facilitated by photocatalysts, enable the formation of new C–C bonds between organic molecules.1,2 These reactions provide a method for synthesizing complex organic structures from simpler precursors, eliminating the need for harsh reaction conditions. A notable application of this technology is the photocatalytic C–C coupling of benzyl alcohol (BzOH) to synthesize hydrobenzoin (HB), an essential intermediate in the synthesis of cyclopentitols3 and in facilitating the asymmetric addition of diethylzinc to aldehydes.4 Derivatives of HB, including benzoin (BZ) and deoxybenzoin (DOB), are versatile building blocks in chemical and pharmaceutical industries.5 However, conventional syntheses of HB mostly rely on the dihydroxylation of alkenes and the pinacol coupling of benzaldehyde (BzO), which often involve the use of oxidants and reductants, respectively.6,7 In this context, photocatalytic conversion of BzOH to HB has emerged as an attractive approach, due to the absence of scavenging reagents and the use of solar light as a renewable energy input.
The photocatalytic conversion of BzOH involves a complex reaction pathway, as depicted in Scheme 1, which presents a significant challenge in achieving efficient synthesis of HB with high selectivity. A crucial step in this process is the Cα–H bond cleavage of BzOH to achieve the desired C–C coupling reaction, yielding the target product HB. However, an additional O–H bond cleavage can trigger a dehydrogenation reaction, leading to the production of BzO as a by-product.8,9 Moreover, the HB produced from the C–C coupling reaction of BzOH can subsequently transform into derivatives such as BZ, DOB, and oligomers, resulting in a mixture of products with low selectivity.10–14 Xu et al. demonstrated a significant selectivity (99.1%) towards HB by shortening the reaction time, which suppressed the subsequent production of derivatives. However, 14.6% of BzOH remained unreacted, leading to a low overall conversion rate.15 The low reaction rates of reported semiconductor-based photocatalysts are due to severe charge recombination. Although metal–semiconductor heterostructures (MSH) have emerged as an effective strategy to mitigate charge recombination and boost photocatalytic efficiency,16 reported MSH photocatalysts often lead to low selectivity toward HB.14,17 This selectivity issue can be addressed by introducing a hole scavenger,18 but the use of sacrificial reagents limits practical applications. These findings highlight the urgent need to develop photocatalysts that efficiently catalyze the C–C coupling reaction of BzOH for HB production without relying on sacrificial agents, moving towards more sustainable and feasible photocatalytic practices. Achieving this objective requires not only a high-performance photocatalyst but also a good understanding of the effect of photocatalyst structure on reaction mechanism and kinetics.
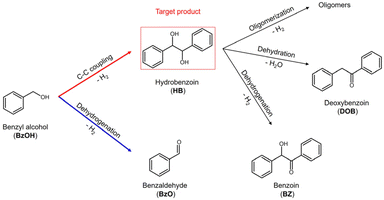 |
| | Scheme 1 Reaction pathways of conversion of BzOH. | |
In this study, we have achieved high-efficiency C–C coupling of BzOH for HB production using platinum-deposited CdS nanosheets (Pt/CdS NSs) as the photocatalyst. CdS was chosen as the photocatalyst base due to its well-documented capability in facilitating the C–C coupling reaction of BzOH.14,15 The two-dimensional morphology of CdS NSs with Pt deposition extends charge-separated state lifetime, enhancing the photocatalytic efficiency.19 We selected cysteamine as a surface ligand to suppress the hole trapping process in CdS, efficiently catalyzing the C–C coupling reaction of BzOH. By studying the MSH configuration, we revealed that photocatalyst structure affects reaction intermediate in the BzOH conversion, impacting the reaction mechanism and kinetics, and consequently altering the selectivity toward HB. Through judicious material design, we developed a photocatalyst that substantially improves HB production over previous ones, as summarized in Table 1. This research not only offers a promising avenue for efficient C–C coupling reactions but also sets the stage for further investigations into heterostructure models for adeptly manipulating semiconductor nanocrystals in solar energy-driven organic transformations.
Table 1 Overview of the recent literature for photocatalytic BzOH conversion
| Photocatalyst |
Photocatalyst mass (mg) |
Light source |
BzOH (mmol) |
Conversiona |
Selectivityb |
HB yield;c H2 formation rated |
Ref. |
|
Conversion = (n0(BzOH) − n(BzOH))/n0(BzOH) × 100%.
For HB, BZ, or DOB, selectivity = 2 × n(product)/(n0(BzOH) − n(BzOH)) × 100%; for BzO, selectivity = n(product)/(n0(BzOH) − n(BzOH)) × 100%.
Yield = conversion × selectivity.
For references where the H2 formation rate was not directly reported, the values were calculated based on the experimental data provided in the respective references.
|
| Zn0.2In2S3.2 |
10 |
86 W blue LED |
0.2 |
>99% (12 h) |
9% BzO |
HB: 0%; H2: 1.82 mmol g−1 h−1 |
11
|
| 61% BZ |
| 30% DOB |
| ZnIn2S4 |
10 |
LED lamp, λ = 450 nm |
0.1 |
>99% (10 h) |
58% BZ |
HB: 0%; H2: 0.98 mmol g−1 h−1 |
12
|
| 40% DOB |
| CdS |
15 |
LED lamp, λ = 440 nm |
0.1 |
45% (24 h) |
10% HB
|
HB: 4.5%; H2: 0.075 mmol g−1 h−1 |
13
|
| 5% BzO |
| 40% BZ |
| CdS |
6 |
300 W Xe-lamp |
0.2 |
50% (20 h) |
38% HB
|
HB: 19%; H2: 0.78 mmol gcds−1 h−1 |
14
|
| 9% BzO |
| 37% BZ |
| CdS/SiO2 |
20 |
300 W Xe-lamp |
0.24 |
85.4% (12 h) |
99.1% HB
|
HB: 84.6%; H2: 17.9 mmol gcds−1 h−1 |
15
|
| Pt/CdS |
1 |
100 W Xe-lamp |
0.24 |
93.4% (3 h) |
85.3% HB
|
HB: 79.7%; H2: 41.1 mmol g−1 h−1 |
This work |
| 4.7% BzO |
Results and discussion
Material characterizations and photocatalysis
We prepared CdS NSs following a previously reported synthesis protocol,19 with detailed experimental procedures provided in the ESI.† The absorption spectra of the CdS NSs in hexane, shown in Fig. S1(a),† reveal distinct electron-heavy hole and electron-light hole transitions at 414 nm and 407 nm, respectively. This pattern indicates that CdS NSs are 6.5 monolayers thick.20,21 The scanning transmission electron microscopy (STEM) image in Fig. S1(b)† shows that the synthesized CdS NSs tend to fold into scroll-like structures. To measure their thickness, an upright nanosheet was identified in the TEM image, as shown in Fig. S1(c).† The measured thickness is approximately 1.8 nm, which is consistent with previous reports.21 Additionally, X-ray diffraction (XRD) analysis confirmed the zinc blende (ZB) crystalline structure of the CdS NSs,21 as shown in Fig. S1(d).† Subsequently, we deposited Pt on CdS NSs to form an MSH configuration. Herein, the Pt is to not only suppress the charge recombination in CdS but also provide a means to control electron behavior. This approach aimed to understand the influence of MSH configuration on the reduction mechanism in the conversion of BzOH. Therefore, we deposited Pt on the CdS NSs utilizing a previously reported thermal reduction method,19 with successful deposition evidenced by STEM images, as shown in Fig. S1(e).† The absorption spectrum changed slightly after Pt deposition, primarily due to inherent light absorption of Pt, as shown in Fig. S1(f).† The XPS spectrum of the Pt 4f band in the Pt/CdS NSs is presented in Fig. S1(g).† The peaks at binding energies of 70.9 eV and 74.2 eV correspond to the Pt 4f7/2 and Pt 4f5/2 states, respectively, which are characteristic of Pt in its metallic state (Pt0).22 This confirms the successful growth of metallic Pt particles on the CdS NSs.
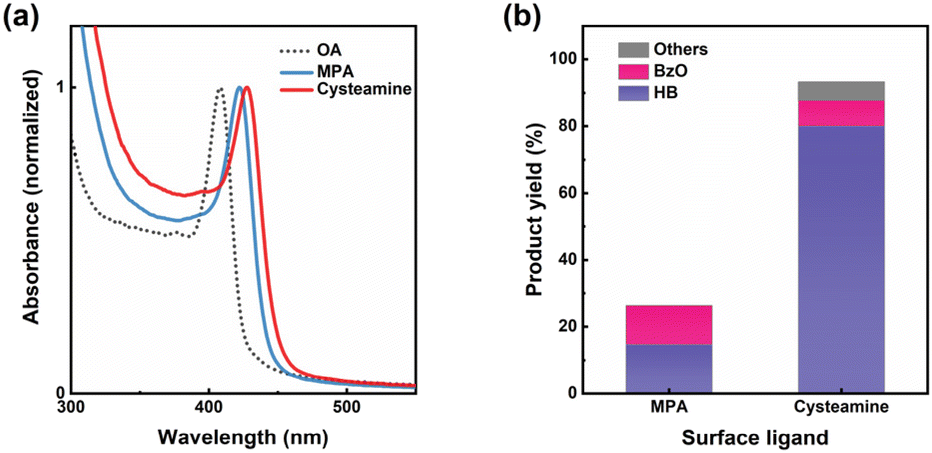
The as-synthesized Pt/CdS NSs cannot be used directly as photocatalyst because the surface was hydrophobic and insulating due to the passivation of oleate (OA) ligand, which is to facilitate synthesis and prevent aggregation of CdS NSs in organic solvents.23,24 The OA-passivated surface hinders the dispersion and charge extraction of CdS NSs for photocatalysis in polar solvent, necessitating the substitution of these ligands with charged short-chain molecules. Although 3-mercaptopropionic acid (MPA) is commonly used for passivating Cd-based nanocrystals in photocatalytic applications,25 research has shown that amine-based ligands are superior in enhancing oxidation reaction, owing to their efficiency in passivating hole traps on CdS-based nanocrystals.26–30 Considering this, we selected cysteamine as the passivation ligand for Pt/CdS NSs to control the carrier dynamics of holes by altering hole traps. For comparative purposes, we also prepared samples passivated with MPA. The ligand exchange process was carried out as previously described,31 with the successful exchange indicated by the phase transfer of colloid from a nonpolar to a polar solvent, as shown in Fig. S2.† Following the ligand exchange process, the absorption peaks of Pt/CdS NSs passivated with MPA and cysteamine were shifted to 422 nm and 424 nm, respectively, as shown in Fig. 1(a). This shift was due to the alteration in the surrounding medium (from hexane to dimethyl sulfoxide (DMSO)) and the different delocalization states of exciton on the ligand shell (transitioning from carboxylate to thiolate head).32 Fig. S3† further revealed that the initially curled CdS nanocrystals expanded after the ligand exchange process.19,33 The Pt to Cd mass ratio was determined to be 0.017, quantified by inductively coupled plasma optical emission spectrometry (ICP/OES).
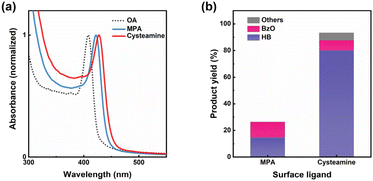 |
| | Fig. 1 (a) Normalized absorption spectra of colloidal Pt/CdS NSs capped with OA in hexane, MPA in DMSO, and cysteamine in DMSO. (b) Products distribution of photocatalytic BzOH conversion by Pt/CdS NSs with different surface ligands. Yield (HB) = 2 × n(HB)/n0(BzOH) × 100%; yield (BzO) = n(BzO)/n0(BzOH) × 100%; yield (others) = 100% – yield (HB) – yield (BzO). | |
Effect of surface modification on photocatalysis
We conducted the photocatalytic BzOH conversion using Pt/CdS NSs, passivated with either MPA or cysteamine, in a 10 mL acetonitrile (ACN) solution containing 0.24 mmol of BzOH and 1.0 mg of the chosen photocatalyst, under an inert atmosphere. Light exposure was provided for 3 hours using AM 1.5 illumination at an intensity of 100 mW cm−1.2 The quantification of products was carried out using gas chromatography (GC). Control experiments, shown in Fig. S4,† indicated that no products were detected in the absence of light or photocatalyst, confirming that both are necessary for the photocatalytic reaction to proceed. The identified products included HB and BzO, with the corresponding reaction equations shown as follows:
Fig. 1(b) demonstrates that Pt/CdS NSs, when passivated with cysteamine, achieved a significantly higher BzOH conversion rate of 93.3%, surpassing those passivated with MPA, which exhibited only 26.3% conversion. In a supplementary experiment, CdS NSs passivated with cysteamine also displayed a higher BzOH conversion rate of 52.0% compared to the 13.3% conversion of those passivated with MPA, as shown in Fig. S5(a).† Moreover, we conducted hydrogen evolution reaction (HER) experiments in water using the same Pt/CdS NSs catalysts, and found that the cysteamine-passivated photocatalyst also showed high reaction rate than MPA-passivated one, as shown in Fig. S5(b).† This consistency suggests that the differences in photocatalytic activity due to the surface ligand are likely a result of the distinct photogenerated charge dynamics induced by surface passivation. The on–off switching behavior of photocurrent (Fig. S5(c)†) of the CdS NSs showed that both ligand passivation types resulted in transient photocurrent responses. The photocurrent density of cysteamine capped CdS NSs was significantly higher than that of MPA-capped sample, indicating that more photogenerated carriers were available for the photoelectric signal in CdS NSs passivated with cysteamine, similar to the result of photocatalytic activity. To further investigate the effect of surface ligand on the charge separation dynamics in the hybrid nanoparticles, transient absorption (TA) spectroscopy measurements were performed as described next.
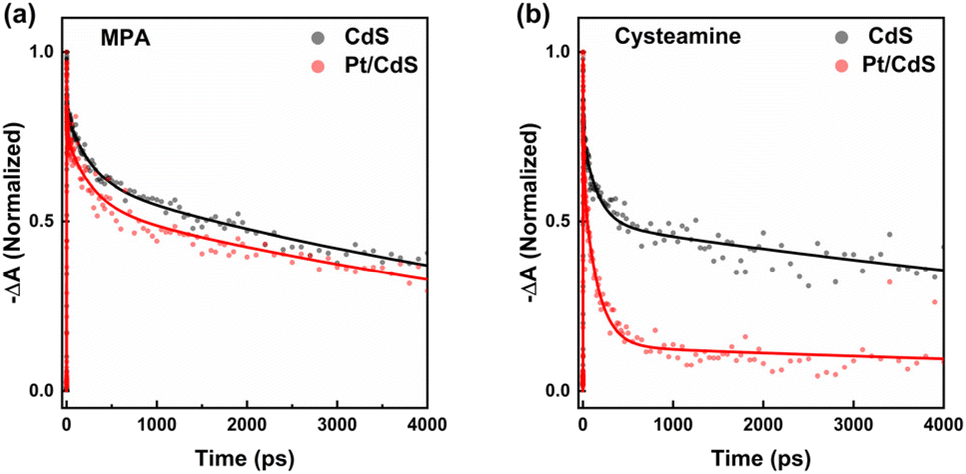
We monitored the population of the band-edge (1S) exciton state of CdS NSs dissolved in DMSO via TA spectroscopy. Fig. S6(a and b)† shows the TA spectra at indicated delay time for CdS NSs passivated with MPA and cysteamine, recorded with an excitation photon energy of 3.88 eV, generating above-band-edge carriers. Fig. 2 shows the corresponding dynamics of the band-edge (1S) absorption changes. The decay curves of the absorption at 1S absorption wavelength of the CdS NSs were fit to tri-exponential functions characterized by distinct time constants (τ1, τ2, τ3), as summarized in Table S1.† Detailed analysis of TA data is documented in ESI Note 1.† In the band-edge bleach recovery kinetics, we observed rapid decays on the order of several picoseconds (τ1) and several hundred picoseconds (τ2), followed by a slower decay on the order of nanoseconds (τ3). Considering that the charge recombination rate in nanosheets or nanoplatelets is on the order of nanoseconds,34–36 the two fast decays are attributed to carrier trapping. For Cd-chalcogenide quantum dots (QDs), TA spectra are dominated by state-filling-induced bleach, the magnitude of which is defined by the occupancy of the band-edge 1S electron level because of high degeneracy of the valence band.34 Therefore, the two fast decays are ascribed to electron trapping. The amplitudes of these two fast decays are 52.2% and 58.6% for MPA-passivated and cysteamine-passivated CdS NSs, respectively, indicating that the extent of electron trapping is comparable.
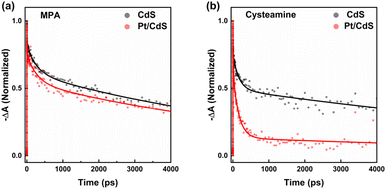 |
| | Fig. 2 Normalized TA bleaching recovery kinetics of CdS NSs (grey) and Pt/CdS NSs (red): (a) at 422 nm with MPA as the capping ligand, and (b) at 424 nm with cysteamine as the capping ligand. The experimental data are represented by dots, with corresponding model fits from ESI Note 1† shown as solid lines. | |
We subsequently examined the band-edge bleach recovery of the two samples following the introduction of Pt, as shown in Fig. S6(c and d).† Exponential fitting (Table S1†) revealed that the amplitude of the fast components increased for both samples upon Pt attachment. This increase is attributed to electron transfer from CdS NSs to Pt. Notably, the distinction in the normalized TA bleaching recovery kinetics between cysteamine-passivated Pt/CdS and CdS is more pronounced than that between MPA-capped Pt/CdS and CdS, as shown in Fig. 2. The amplitude of the fast decay component increased by 5.2% and 31.5% for MPA-passivated and cysteamine-passivated CdS NSs, respectively. Given that the comparable extent of electron trapping in both samples, we attribute the significant difference in electron transfer dynamics to Pt between the two samples to hole-trapping-induced exciton localization. This occurs because when holes are trapped, exciton localization arises due to the Coulomb interaction between the trapped hole and the electron, inhibiting electron transfer to Pt.25,37,38 Thus, the substantial increase in the fast component amplitude suggests that cysteamine passivates more hole traps than MPA. Consequently, we propose that the cysteamine-passivated surface harbors fewer hole traps, facilitating more efficient electron transfer from CdS to Pt and providing a larger number of photogenerated holes available for photocatalytic chemical reactions.
Effect of Pt deposition on photocatalysis
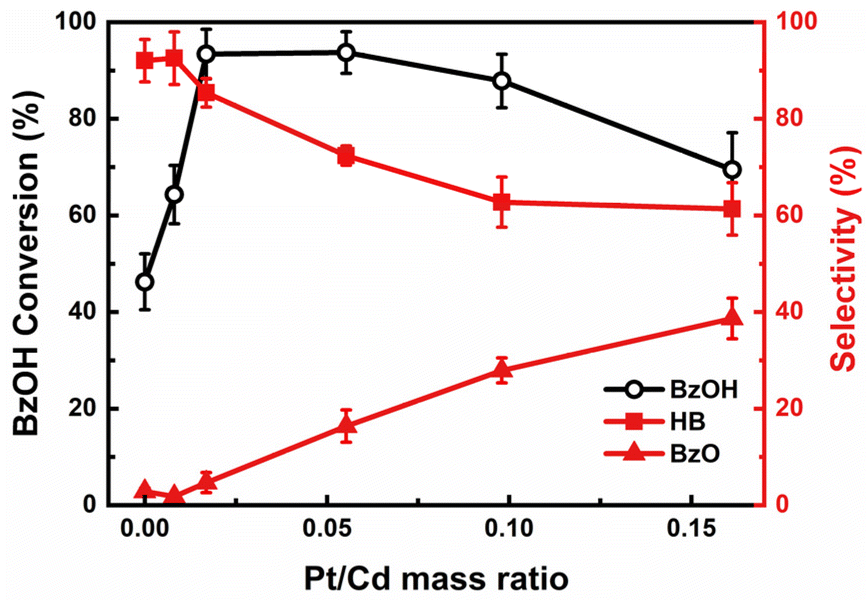
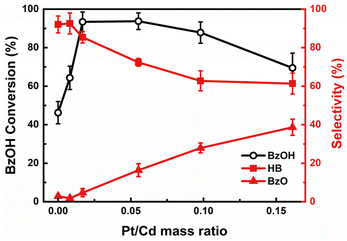
To assess the influence of the Pt on photocatalytic performance, we synthesized a series of Pt/CdS NSs catalysts with Pt/Cd mass ratio varying from 0 to 0.16. Pt/CdS NSs with a higher Pt loading have a higher density of Pt nanoparticles (NPs) on the CdS NSs, as shown in Fig. S7(a–f).† The absorption spectra reflecting these variations are illustrated in Fig. S7(g).† The photocatalytic performance of these varied Pt/CdS samples, as shown in Fig. 3, revealed a significant improvement in the conversion rates of BzOH compared to the bare CdS. As previously mentioned, the Pt/CdS heterojunction makes electron transfer from CdS to Pt. This electron transfer process can suppress charge recombination, thus prolonging the lifetime of the charge-separated state, which is vital to achieve high-efficiency photocatalysis.19,39 Despite the enhanced reaction activity due to Pt deposition, excessive amounts of Pt proved counterproductive, leading to decreased BzOH conversion rates, as shown in Fig. 3. Considering Pt NPs themselves are capable of absorbing light, as shown in Fig. S8(a),† the decreased activity with an excess of Pt was linked to diminished light absorption by the CdS, which was overshadowed by the Pt NPs. To further explore the influence of Pt on light absorption, a comparative photocatalytic experiment was performed using a mixture of separately prepared CdS and Pt as the photocatalyst. The outcomes demonstrated that the CdS and Pt mixture had a lower conversion rate compared to CdS alone, as shown in Fig. S8(b),† thereby confirming the detrimental effects of excessive Pt on photocatalytic activity. Moreover, the selectivity of the photocatalytic conversion of BzOH to HB showed significant variation with changes in the Pt loading on the CdS. In the absence of Pt, CdS demonstrated high selectivity towards the production of HB. However, as the Pt loading increased, there was a noticeable shift towards the formation of BzO, leading to a reduction in HB selectivity, as shown in Fig. 3. These results demonstrate the critical role of the Pt on the photocatalytic performance.
 |
| | Fig. 3 Photocatalytic BzOH conversion and product selectivity by Pt/CdS with varying mass fractions of Pt. Conversion (BzOH) = (n0(BzOH) − n(BzOH))/n0(BzOH) × 100%; selectivity (HB) = 2 × n(HB)/(n0(BzOH) − n(BzOH)) × 100%; selectivity (BzO) = n(BzO)/(n0(BzOH) − n(BzOH)) × 100%. | |
Reaction mechanism analysis
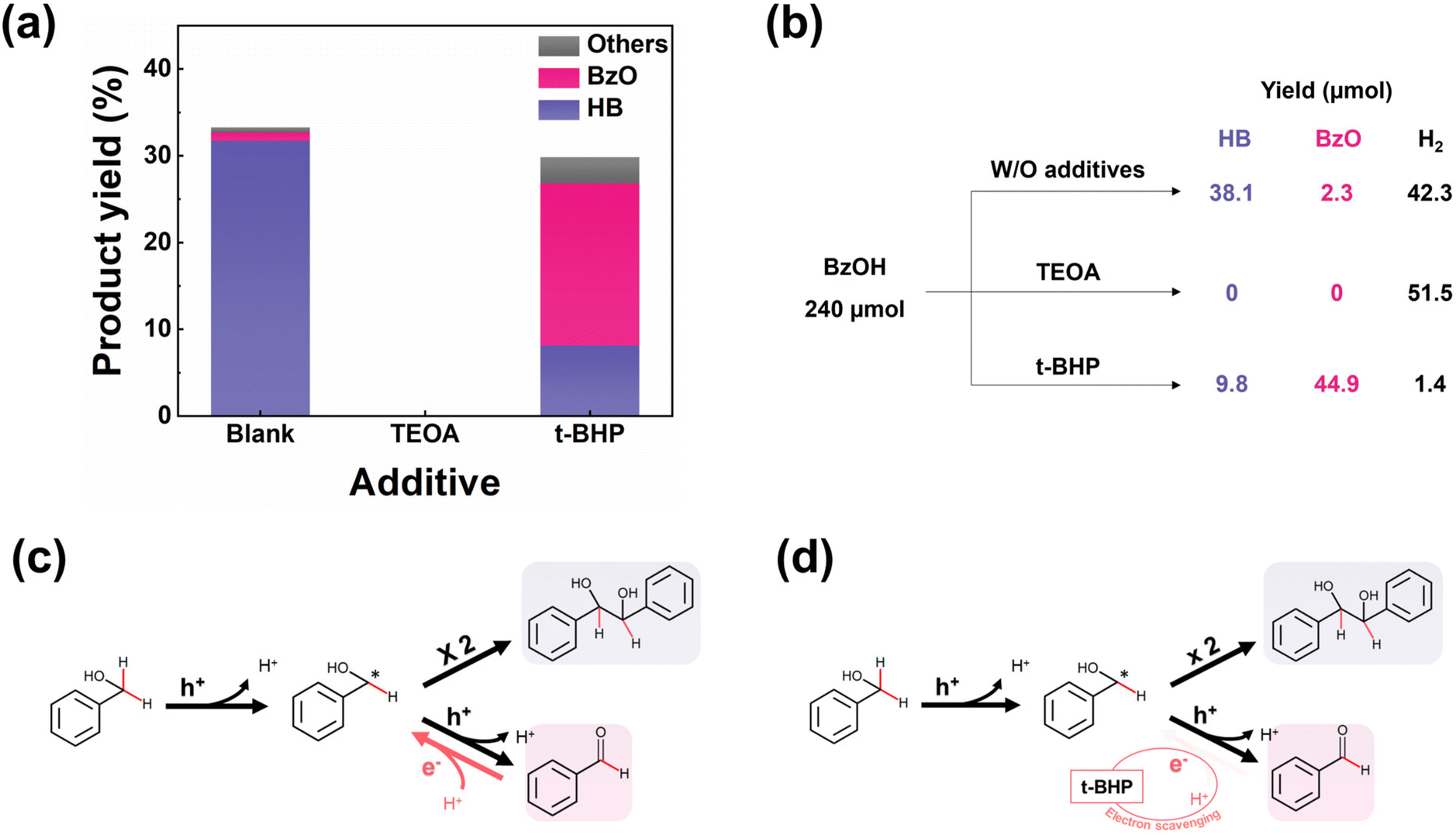
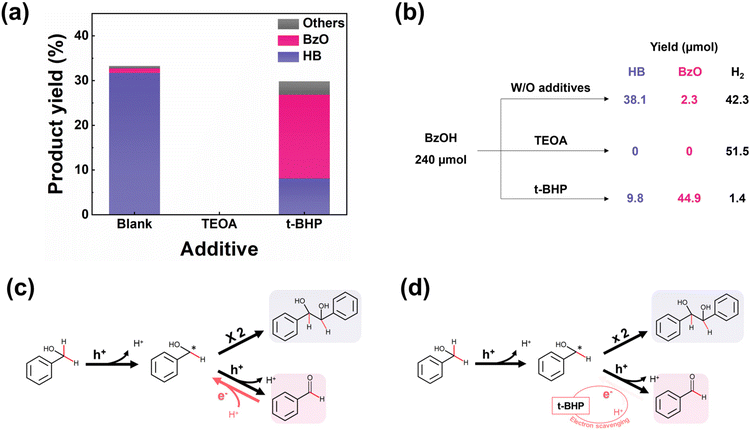
We next consider the possible mechanisms underlying the selectivity concerns in the photocatalytic conversion of BzOH. It is well-known that the photocatalytic reaction is motivated by the interaction between the electrons/holes and the reactants. To clarify the roles of the electrons and holes in reaction mechanism, we introduced triethanolamine (TEOA) as a hole scavenger and tert-butyl hydroperoxide (t-BHP) as an electron scavenger into the reaction system, respectively. To sufficiently scavenge photogenerated charges, we added excessive charge scavengers into the system. It is noteworthy that high concentration of t-BHP degrades photocatalyst. We found that 48 mM is the maximum concentration to maintain the stability of photocatalyst under light irradiation. Fig. 4(a) shows that the presence of TEOA completely halted the conversion of BzOH, highlighting the important role of holes in initiating the reaction. Conversely, the addition of t-BHP led to a noticeable decrease in the selectivity towards HB and hydrogen production, as shown in Fig. 4(a and b). Additionally, the use of 1,1-diphenyl ethylene (DPE) as a radical capture confirmed the generation of the ketyl radical (BzOH*),15 as shown in Fig. S9.† Based on these observations, we propose a reaction mechanism for the photocatalytic conversion of BzOH, as presented in Fig. 4(c). The process begins with the oxidation of BzOH by a photogenerated hole, yielding a ketyl radical (BzOH*) and a proton (eqn (3)). The presence of a hole scavenger interrupts this initial oxidation step, thereby stopping the BzOH conversion process. The subsequent steps involve the combination of two ketyl radicals to form HB (eqn (4)), or the further oxidation of a ketyl radical by another hole to produce BzO and a proton (eqn (5)).12,18 This oxidation process is reversible, with BzO potentially being reduced back to a BzOH* in the presence of a proton and an electron (eqn (6)).14,15 The HER (eqn (7)) acts as a counter-reaction to these hole-induced oxidation reactions.| | | BzOH + h+ → BzOH* + H+ | (3) |
 |
| | Fig. 4 (a) Photocatalytic BzOH conversions and (b) product distributions by Pt/CdS with different charge scavengers in 1 h. Additive: 10%v/v TEOA or 48 mM t-BHP. Proposed reaction mechanism of the photocatalytic BzOH conversion (c) with and (d) without t-BHP as the electron scavenger. The BzO reduction reaction (eqn (6)) is marked as red arrow. When electron scavenger exists, the electrons and protons are consumed, subsequently suppressing the BzO reduction reaction (eqn (6)). | |
Given that electron scavengers primarily impede reduction reactions (eqn. (6) and (7)), we deduced that inhibiting the reduction of BzO back to BzOH* results in a decrease in HB selectivity. In this context, we outline a mechanism stimulated by the electron scavengers, as depicted in Fig. 4(d). Electron scavengers interfere with the dynamic redox balance between BzO and BzOH* by reducing the availability of electrons necessary for the reduction of BzO. This disruption leads to an accumulation of BzO and a corresponding reduction in HB production, as the electron scavengers prevent the reverse reaction from taking place.
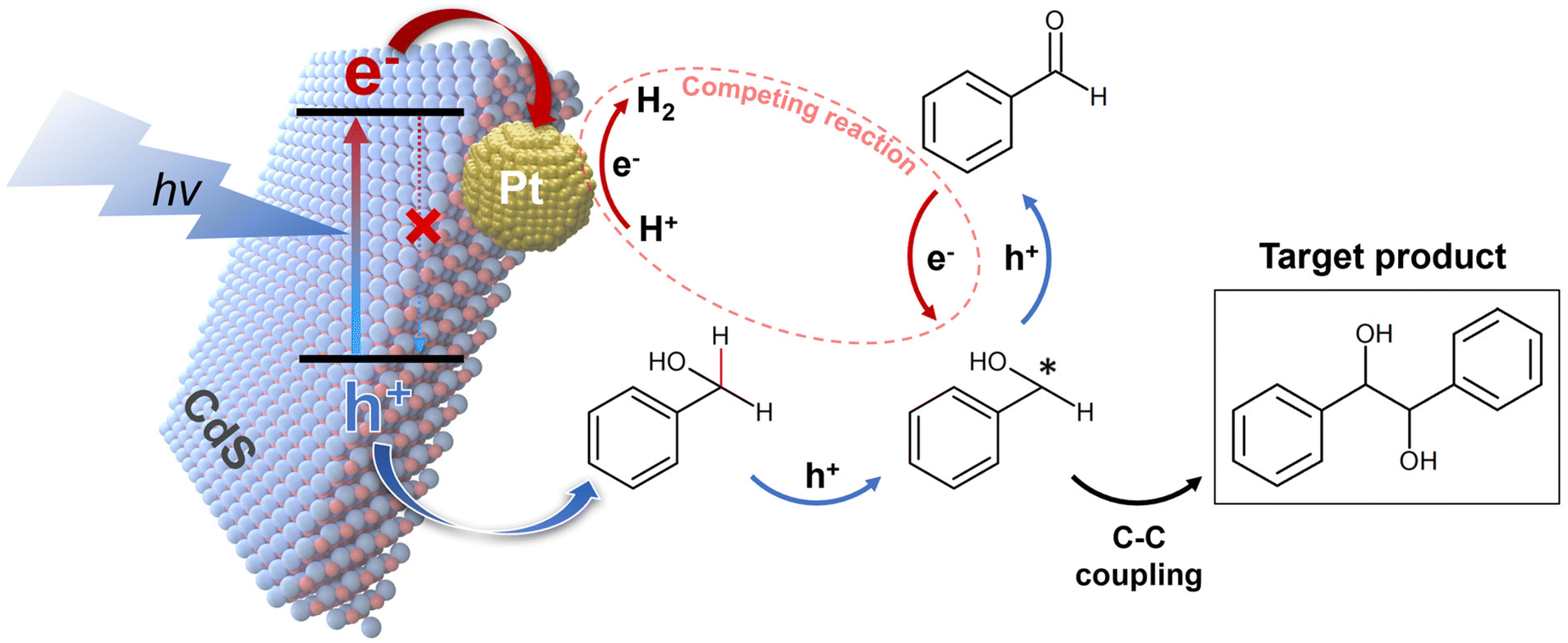
The discussions above provide insights into how selectivity towards HB changes with the deposition of Pt on CdS. The primary difference between the effects of electron scavenger and Pt lies in their interaction with electrons: electron scavenger consumes electrons, whereas Pt acts as an electron acceptor, facilitating electron transfer and promoting HER.40,41 Consequently, while electron scavengers significantly reduce hydrogen production as shown in Fig. 4(b), the introduction of Pt still permits observable hydrogen production, as depicted in Fig. S10.† Moreover, given exceptional catalytic activity of Pt in HER (eqn (7)),42,43 it is reasonable to conclude that Pt, similar to the electron scavenger, restricts electron availability for the reduction of BzO back to BzOH* (eqn (6)), thereby influencing HB selectivity. Fig. 5 summarizes the reaction mechanisms and role of Pt in the BzOH conversion process.
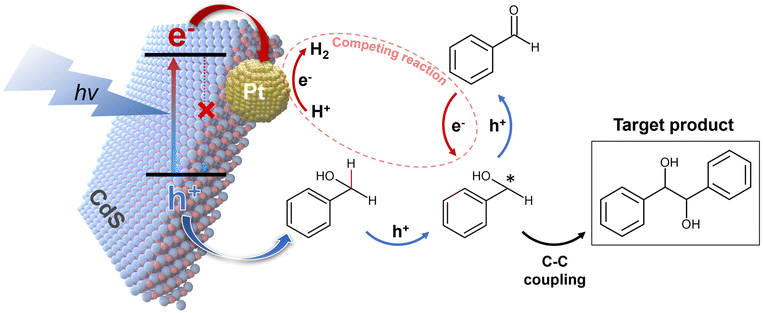 |
| | Fig. 5 Postulated reaction mechanism of photocatalytic BzOH conversion by Pt/CdS. The CdS acting as the light absorber, generates electrons (e−) and holes (h+) upon exposure to light. The introduction of deposited Pt suppresses charge recombination within CdS by facilitating electron transfer from CdS to Pt, resulting in increased reaction activity. The hole and electron, extracted from CdS, move to participate in BzOH oxidation and HER, respectively. The BzOH is oxidized by the holes, producing the BzOH*, which is essential for HB production through C–C coupling. Additionally, there is a dynamic redox equilibrium between BzO and the BzOH*. In this context, the Pt intensifies HER, the competing reaction to BzO reduction reaction, leading to the accumulation of BzO and subsequently decreased HB selectivity. | |
Our observations reveal that a moderate amount of Pt deposition not only accelerates the overall reaction rate but also maintains high selectivity towards HB. This preservation of selectivity can be understood by examining the hydrogen production rates relative to the quantity of Pt in the photocatalytic reaction, as depicted in Fig. S10.† We noticed that between a Pt-to-Cd ratio of 0.01 to 0.05, the rate of BzOH conversion saw a notable increase, while the HER experienced only a slight enhancement. This pattern indicates that moderate levels of Pt deposition effectively maintain a preferential pathway towards the BzO reduction reaction over HER. Such a dynamic ensures that the redox equilibrium between BzO and BzOH* remains relatively undisturbed, thereby preventing excessive accumulation of BzO and favoring its conversion to HB, as illustrated in Fig. S11.† Therefore, the precise control of Pt deposition amount suggests the possibility of enhancing the overall reaction rate through effective charge separation while preserving high selectivity towards HB.
Reaction kinetic modeling
Suppressing the competitive dehydrogenation reaction that converts BzOH to BzO is indeed essential for attaining high selectivity towards HB. However, it's also critical to consider that the subsequent transformation of HB into its derivatives, such as BZ, DOB, and various oligomers, contributes to decreasing the selectivity towards HB,11–13 as highlighted in Scheme 1. This aspect introduces a complexity that results in a volcano-like yield pattern of HB over time, as shown in Fig. 6(a). This pattern indicates that there's a point of maximized yield of HB, situated at the peak of the curve. Thus, a photocatalyst that can realize a “volcano” curve with a high peak is particularly desirable, as it signifies the potential for maximized HB production before the onset of significant transformation into its derivatives.
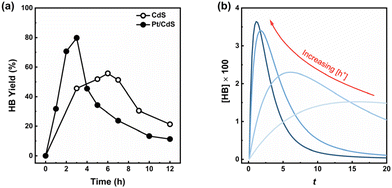 |
| | Fig. 6 (a) HB yield of CdS and Pt/CdS NSs with Pt/Cd mass ratio of 0.017 during 12 h reaction. HB yield = 2 × n(HB)/n0(BzOH) × 100%; (b) Relationship between [HB] and t in the eqn (11) at different appointed values of [h+]. To enable the calculation by Python, we appointed the values of 0.24, 5, and 1 for [BzOH]0, k1, and k2, respectively. The initial value of [HB] is 0. | |
Although CdS shows a high selectivity towards HB during a 3 hour photocatalysis period, as indicated in Fig. 3, an optimized Pt/CdS catalyst, with a Pt/Cd mass ratio of 0.017, outperforms CdS by achieving an HB peak yield of 79.8% compared to the 55.8% yield observed with CdS, as shown in Fig. 6(a). To understand this phenomenon, we investigated the reaction kinetics. We hypothesize that the hole-induced oxidative reaction (eqn (8)) could act as the rate-determining step (RDS) in the C–C coupling reaction of BzOH, and establish the corresponding reaction rate equations (eqn (9) and (10)):
| | 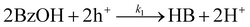 | (8) |
| | 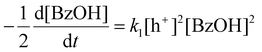 | (9) |
| | 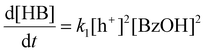 | (10) |
Here k1 denotes the reaction rate constant for eqn (8), with [BzOH], [HB], and [h+] representing the concentrations of BzOH, HB, and photogenerated holes, respectively. t is the reaction time.
The transformation of HB into its derivatives involves several pathways, including dehydrogenation to DOB, dehydration to BZ, and oligomerization to various oligomers.11 Analysis of the reaction products, as shown in Fig. S12,† reveals DOB and BZ were not the dominant products of HB's consecutive conversion. Given the larger molecular size and lower volatility of oligomers, which precludes their detection by GC, it is likely that the main products resulting from HB conversion are indeed oligomers. This suggests that the majority of HB's conversions are primarily oxidative reactions, requiring the involvement of photogenerated holes for their activation. Herein, we hypothesize that the activation of HB by photogenerated holes (h+) represents the RDS in its conversion to these products.
| | 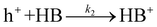 | (11) |
Where k2 is the reaction rate constant of the reaction eqn (11). HB+ represents the reaction intermediate produced by HB oxidation with a hole (h+). Herein, we are assuming that the derivatives of HB are produced from subsequent HB+ conversion. The corresponding reaction rate equation is:
| | 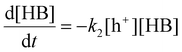 | (12) |
With [HB] denoting the concentration of HB. Integrating eqn (9)–(11), we derive:
| | 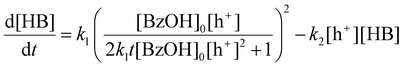 | (13) |
Here, [BzOH]0 denotes the initial concentration of BzOH. Given the complexity of eqn (13), a direct analytical solution is impractical. To overcome this challenge, we employed Python for computational analysis, with the code provided in the ESI Note 2.† The results of this analysis, reveal that an increase in the concentration of photogenerated holes ([h+]) not only advances the peak of HB occurrence but also amplifies its magnitude, as shown in Fig. 6(b). These computational findings suggest that attaining a high peak yield of HB requires a substantial presence of photogenerated holes, which corroborates with the experimental data shown in Fig. 6(a). Based on this result, we attribute the higher maximum HB yield with Pt/CdS compared to CdS to the higher availability of photogenerated charges for the reaction.
Conclusions
In conclusion, our research has demonstrated the effective photocatalytic C–C coupling reaction of BzOH for synthesizing HB with a 93.4% conversion and 85.3% selectivity, utilizing 1 mg Pt/CdS NSs and 0.24 mmol BzOH in ACN over a period of 3 hours. We identified cysteamine as a highly effective passivation ligand that boosts the reaction rate by improving surface properties. The MSH configuration of Pt/CdS is pivotal in promoting charge transfer, which substantially minimizes charge recombination in CdS, thus elevating reaction efficiency. However, the function of Pt as an electron reservoir, similar in effect to an electron scavenger, emerges as a double-edged sword. While it enhances charge separation, it simultaneously reduces selectivity towards the targeted C–C coupling product by inhibiting the formation of essential reaction intermediates, BzOH*. By precise control of Pt loading, we managed to balance high catalytic activity with sustained selectivity. Moreover, kinetic modeling simulations highlight the superiority of Pt/CdS over CdS alone, demonstrating a significantly greater maximum HB yield, despite slight BzO production. This study not only proposes an effective photocatalyst model for C–C coupling reactions but also provides valuable insights into the intricate relationship between photogenerated charge dynamics and reaction mechanisms, offering profound insights for future advancements in photocatalysis.
Author contributions
Conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, software, supervision, visualization, writing – original draft, writing – review & editing. P. L.: conceptualization, data curation, formal analysis, investigation, methodology, software, visualization, writing – original draft; G. K.: data curation, formal analysis; N. W.: data curation, formal analysis, writing – review & editing; J. P.: writing – review & editing; W. D. K.: formal Analysis, writing – original draft, writing – review & editing, D. C. L.: conceptualization, writing – review & editing, funding acquisition, project administration, resources, supervision.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† Raw data that support the findings of this study are available from the corresponding authors upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is funded by National Research Foundation of Korea (2021M3H4A3A01062960, 2022R1A5A1033719 and RS-2024-00350615), Saudi Aramco-KAIST CO2 Management Center, and the Korea Institute of Industrial Technology as “Development and commercialization for clean hydrogen production/storage and CO2 monitoring system in the field of industrial complex (KITECH EH-24-0007)”.
References
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed.
- M.-Y. Qi, M. Conte, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed.
- K. S. Kim, J. Park II and P. Ding, Tetrahedron Lett., 1998, 39, 6471–6474 CrossRef CAS.
- M. A. Dean and S. R. Hitchcock, Tetrahedron: Asymmetry, 2008, 19, 2563–2567 CrossRef CAS.
- K. Okano, Tetrahedron, 2011, 67, 2483–2512 CrossRef CAS.
- A. Bensari, J.-L. Renaud and O. Riant, Org. Lett., 2001, 3, 3863–3865 CrossRef CAS PubMed.
- Z.-M. Wang and K. B. Sharpless, J. Org. Chem., 1994, 59, 8302–8303 CrossRef CAS.
- Z.-G. Liu, Y. Wei, L. Xie, H.-Q. Chen, J. Wang, K. Yang, L.-X. Zou, T. Deng and K.-Q. Lu, Mol. Catal., 2024, 553, 113738 CrossRef CAS.
- R. Paul, I. R. Warkad, S. Arulkumar, S. Parthiban, H. R. Darji, M. Naushad, R. G. Kadam and M. B. Gawande, Mol. Catal., 2022, 530, 112566 CrossRef CAS.
- X. Sun, D. Jiang, L. Zhang and W. Wang, Appl. Catal., B, 2018, 220, 553–560 CrossRef CAS.
- N. Luo, T. Hou, S. Liu, B. Zeng, J. Lu, J. Zhang, H. Li and F. Wang, ACS Catal., 2019, 10, 762–769 CrossRef.
- G. Han, X. Liu, Z. Cao and Y. Sun, ACS Catal., 2020, 10, 9346–9355 CrossRef CAS.
- T. Mitkina, C. Stanglmair, W. Setzer, M. Gruber, H. Kisch and B. König, Org. Biomol. Chem., 2012, 10, 3556–3561 RSC.
- Z. Chai, T.-T. Zeng, Q. Li, L.-Q. Lu, W.-J. Xiao and D. Xu, J. Am. Chem. Soc., 2016, 138, 10128–10131 CrossRef CAS PubMed.
- M.-Y. Qi, Y.-H. Li, M. Anpo, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2020, 10, 14327–14335 CrossRef CAS.
- M. Passi and B. Pal, Korean J. Chem. Eng., 2022, 39, 942–953 CrossRef CAS.
- D. Gunawan, L. Y. Lau, J. A. Yuwono, P. V. Kumar, L. Oppong-Antwi, I. Kuschnerus, S. L. Chang, R. K. Hocking, R. Amal and J. Scott, Chem. Eng. J., 2024, 486, 150215 CrossRef CAS.
- K. P. McClelland and E. A. Weiss, ACS Appl. Energy Mater., 2018, 2, 92–96 CrossRef.
- Q. Li, F. Zhao, C. Qu, Q. Shang, Z. Xu, L. Yu, J. R. McBride and T. Lian, J. Am. Chem. Soc., 2018, 140, 11726–11734 CrossRef CAS PubMed.
- S. Ithurria, M. Tessier, B. Mahler, R. Lobo, B. Dubertret and A. L. Efros, Nat. Mater., 2011, 10, 936–941 CrossRef CAS PubMed.
- Z. Li, H. Qin, D. Guzun, M. Benamara, G. Salamo and X. Peng, Nano Res., 2012, 5, 337–351 CrossRef CAS.
- D. You, B. Pan, Y. He, X. Wang and W. Su, Res. Chem. Intermed., 2017, 43, 5103–5112 CrossRef CAS.
- D.-E. Yoon, W. D. Kim, D. Kim, D. Lee, S. Koh, W. K. Bae and D. C. Lee, J. Phys. Chem. C, 2017, 121, 24837–24844 CrossRef CAS.
- H. Lee, D.-E. Yoon, S. Koh, M. S. Kang, J. Lim and D. C. Lee, Chem. Sci., 2020, 11, 2318–2329 RSC.
- Y. Ben-Shahar, F. Scotognella, N. Waiskopf, I. Kriegel, S. Dal Conte, G. Cerullo and U. Banin, Small, 2015, 11, 462–471 CrossRef CAS PubMed.
- D. P. Morgan and D. F. Kelley, J. Phys. Chem. C, 2018, 122, 25661–25667 CrossRef CAS.
- M. Micheel, B. Liu and M. Wächtler, Catalysts, 2020, 10, 1143 CrossRef CAS.
- C. Pu and X. Peng, J. Am. Chem. Soc., 2016, 138, 8134–8142 CrossRef CAS PubMed.
- Y. Gao and X. Peng, J. Am. Chem. Soc., 2015, 137, 4230–4235 CrossRef CAS PubMed.
- J. Jasieniak and P. Mulvaney, J. Am. Chem. Soc., 2007, 129, 2841–2848 CrossRef CAS PubMed.
- A. Nag, M. V. Kovalenko, J.-S. Lee, W. Liu, B. Spokoyny and D. V. Talapin, J. Am. Chem. Soc., 2011, 133, 10612–10620 CrossRef CAS PubMed.
- M. T. Frederick and E. A. Weiss, ACS Nano, 2010, 4, 3195–3200 CrossRef CAS PubMed.
- N. Wang, S. Cheong, D.-E. Yoon, P. Lu, H. Lee, Y. K. Lee, Y.-S. Park and D. C. Lee, J. Am. Chem. Soc., 2022, 144, 16974–16983 CrossRef CAS PubMed.
- K. Wu, Q. Li, Y. Du, Z. Chen and T. Lian, Chem. Sci., 2015, 6, 1049–1054 RSC.
- A. Dutta, A. Medda, R. Bera, K. Sarkar, S. Sain, P. Kumar and A. Patra, ACS Appl. Nano Mater., 2020, 3, 4717–4727 CrossRef CAS.
- A. Medda, A. Dutta, D. Bain, M. K. Mohanta, A. De Sarkar and A. Patra, J. Phys. Chem. C, 2020, 124, 19793–19801 CrossRef CAS.
- K. Wu, L. J. Hill, J. Chen, J. R. McBride, N. G. Pavlopolous, N. E. Richey, J. Pyun and T. Lian, ACS Nano, 2015, 9, 4591–4599 CrossRef CAS PubMed.
- M. J. Berr, A. Vaneski, C. Mauser, S. Fischbach, A. S. Susha, A. L. Rogach, F. Jäckel and J. Feldmann, Small, 2012, 8, 291–297 CrossRef CAS PubMed.
- M. Zhukovskyi, P. Tongying, H. Yashan, Y. Wang and M. Kuno, ACS Catal., 2015, 5, 6615–6623 CrossRef CAS.
- S. Park, D. Choi, D. W. Lee, B. B. Choi and S. J. Yoo, Korean J. Chem. Eng., 2023, 40, 1549–1562 CrossRef CAS.
- D. Yoo, B. Hwang, S. Oh and K. Park, Korean J. Chem. Eng., 2023, 40, 2004–2009 CrossRef CAS.
- J. Zhang, P. Cai and J. Lin, J. Phys. Chem. C, 2022, 126, 7896–7902 CrossRef CAS.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. M. Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Whi Dong
Kim
*c and
Doh C.
Lee
b,
Whi Dong
Kim
*c and
Doh C.
Lee