
Iron oxide nanoparticle-stabilized Pickering emulsion-templated porous scaffolds loaded with polyunsaturated fatty acids (PUFAs) for bone tissue engineering†
W.
Aadinath
,
Teja
K. S. P. S.
,
Iniyan
Saravanakumar
and
Vignesh
Muthuvijayan
 *
*
Bhupat and Jyoti Mehta School of Biosciences, Indian Institute of Technology Madras, Chennai 600 036, Tamil Nadu, India. E-mail: vigneshm@iitm.ac.in
First published on 12th August 2024
Abstract
Dietary intake of ω-3-polyunsaturated fatty acids (PUFAs) can significantly improve the expression levels of alkaline phosphatase (ALP) and osteocalcin. However, PUFAs are hydrophobic and highly sensitive to temperature, oxygen concentration, pH, and ionic strength. Hence, it is challenging to use PUFAs as bioactive compounds for bone tissue engineering. Here, we encapsulated PUFAs in liposomes to improve their stability. The hydrodynamic size of the PUFA-loaded liposomes was found to be 121.3 ± 35 nm. GC-MS analysis showed that the encapsulation efficiency of the PUFAs was 19.9 ± 3.4%. These PUFA-loaded liposomes were loaded into porous scaffolds that were prepared by polymerizing glycidyl methacrylate and trimethylolpropane triacrylate monomers using the Pickering emulsion polymerization technique. Oleic acid-coated iron oxide nanoparticles were used as the stabilizing agent to prepare these acrylate-based scaffolds containing PUFA-loaded liposomes (P-Lipo-IO(GMA-TMPTA)). SEM micrographs confirmed the porous nature of the scaffolds and the presence of well-adhered liposomes. An in vitro cytotoxicity study conducted using MG63 cells confirmed that these scaffolds showed desirable cytocompatibility. Cell adhesion study showed a well-spread morphology, indicating firm adhesion of the cells. The alizarin red staining of P-Lipo-IO(GMA-TMPTA) scaffolds showed 3- and 2-fold higher calcium deposition compared to the control on days 7 and 14, respectively. ALP activity was also 2-fold higher than that of the control on day 14. RT-PCR analysis of cells exposed to P-Lipo-IO(GMA-TMPTA) scaffolds showed significantly higher expression of osteogenic markers compared to the control. An antibacterial study conducted on Staphylococcus aureus showed a higher percentage inhibition and reactive oxygen species generation in samples treated with P-Lipo-IO(GMA-TMPTA) scaffolds. These desirable biological properties indicate that the developed scaffolds are suitable for bone tissue engineering.
1. Introduction
The osteogenic potential of tissue-engineered scaffolds is one of the important prerequisites for successful clinical use.1 It is preferable that the polymers used for scaffold fabrication possess intrinsic osteogenic potential.1 If not, chemical moieties with osteogenic potential need to be incorporated into the scaffolds as bioactive compounds.1 Several polymers and chemical moieties have been used in tissue-engineered scaffolds to induce osteogenicity.2 ω-3-Polyunsaturated fatty acids (PUFAs) are well-known nutraceuticals that have diverse health benefits, including improving bone health.3 The consumption of oil rich in eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which are ω-3-polyunsaturated fatty acids, resulted in the increased intestinal uptake of calcium ions in both in vitro and in vivo studies.3,4 The Ca2+ ATPase enzyme is responsible for the intestinal uptake of calcium ions.3,4 Fish oil enriched with EPA and DHA helps increase the activity of the Ca2+ ATPase enzyme and thereby enhances calcium uptake.3,4 Increased osteoblast activity through PPAR-γ activation was also evidenced after treatment with PUFAs and their metabolites.3,4 Overall, PUFAs and their metabolites are known to facilitate bone health through multimodalities.However, PUFAs are highly sensitive to extreme conditions such as temperature, pH, light, and oxygen because of their high unsaturation.5,6 In addition, the hydrophobic nature of PUFAs further limits their usage as bioactive molecules.5,6 Various strategies, such as emulsions, liposomes, microcapsules, and nonlamellar liquid crystals, have been developed to mask the drawbacks associated with PUFAs and use them as bioactive or nutraceutical compounds.5 Among several encapsulation strategies, liposomes have drawn attention due to their biocompatibility and stability under diverse environmental conditions.5,6 Various studies have shown that liposomal encapsulation of PUFAs provides enhanced oxidative stability and bioavailability.2,5,6 In addition, the oxidative stability of PUFAs was shown to be better in nanoliposomes (d < 200 nm) than in larger liposomes.6 Hence, using PUFA-loaded liposomes as a bioactive agent in a scaffold for bone tissue regeneration is a feasible strategy.
Cancellous bone is highly porous and has high mechanical strength.7 It is noticed that the material with high porosity fails to have high mechanical strength.7 For the successful design of a scaffold for the cancellous bone tissue, it is a must to accommodate both the properties in the scaffold, which is a major challenge.7 Natural polymers fail to provide high mechanical strength, whereas it is easy to tune the mechanical strength of synthetic polymers.8 Among synthetic polymers, acrylate-based polymers yield very rigid material, whereas non-acrylate-based polymers yield elastomers.7 Additionally, the scaffold must interact with the cells and the bioactive molecules that mediate tissue regeneration.1,8 Bioactive molecules like PUFAs pose additional challenges because of their unsaturation and hydrophobicity. To counter the challenges mentioned above, careful selection of scaffold fabrication methods and choice of monomers is necessary. A suitable scaffold for bone tissue engineering would be highly porous with a high mechanical strength comparable to natural bone.7 In addition, desirable biological properties, such as biocompatibility, supporting cell adhesion, and pro-osteogenic activity, would greatly enhance the potential of the developed scaffold. Composite scaffolds have attracted a lot of attention because of their versatile nature.8 The use of such composite scaffolds for bone regeneration and their physicochemical properties are excellently summarized by Janmahommadi et al.8 Several commercially available bone scaffolds are composites in nature. Recently, glycidyl methacrylate-trimethylolpropane triacrylate (GMA-TMPTA) copolymer synthesized using the Pickering emulsion polymerization technique has shown promising physicochemical and biological properties for bone tissue engineering.7 Oleic acid-coated iron oxide nanoparticles (OA-IONPs) were used as a Pickering emulsifier. It was shown that micropatterns created on the porous scaffolds by OA-IONPs support firm cell adhesion.7 In addition, the antibacterial and pro-angiogenic functionality exhibited by the scaffolds proves that the scaffolds are better-suited candidates for bone regeneration.7
In the present work, PUFA-loaded liposomes were prepared and characterized. These PUFA-loaded liposomes were loaded onto the (GMA-TMPTA) scaffolds to prepare P-Lipo-IO(GMA-TMPTA) scaffolds. As PUFA-loaded liposomes also resemble the natural bilayer structure of cells, we expect firm adhesion of these liposomes onto the scaffold surface. Extensive physicochemical characterization of these scaffolds was performed to understand their properties. The cytocompatibility of these scaffolds was tested using osteosarcoma cell lines (MG63). Alkaline phosphatase (ALP) activity and alizarin red assays were carried out to test the osteogenic potential of the scaffolds. An RT-PCR study was conducted on these scaffolds to study the effect on the expression of osteogenic markers. Furthermore, the antibacterial potential of the scaffold was tested against a pathogenic Staphylococcus aureus. We hypothesized that the P-Lipo-IO(GMA-TMPTA) scaffolds would possess desirable physicochemical and biological properties, making them ideal for bone tissue engineering applications.
2. Experimental
2.1 Materials
Glycidyl methacrylate (C7H10O3; Mol. Wt.: 142.1546 g mol−1) and trimethylolpropane triacrylate (C15H20O6; Mol. Wt.: 296.32 g mol−1) were purchased from Tokyo Chemical Industry (TCI), India. Oleic acid (C18H34O2; Mol. Wt.: 282.46 g mol−1), 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), and heptadecanoic acid (C17H34O2: Mol. Wt.: 270.45 g mol−1) were purchased from Sigma-Aldrich, India. Fish oil capsules enriched with PUFAs were purchased from WOW Skin Science, India. Ferric chloride (FeCl3; Mol. Wt.: 162.20 g mol−1), ferrous sulfate heptahydrate (FeSO4·7H2O; Mol. Wt.: 278.01 g mol−1), azobisisobutyronitrile (AIBN) (C8H12N4; Mol. Wt.: 164.21 g mol−1), glutaraldehyde, sodium lauryl sulfate (SDS) (C12H25NaO4S; Mol. Wt.: 288.38 g mol−1), ethanol, methanol, sodium hydroxide (NaOH; Mol. Wt.: 40 g mol−1), chloroform, fluorescein diacetate (FDA), lecithin, propidium iodide (PI), cholesterol (C27H46O; Mol. Wt.: 386.65 g mol−1), Nile red, p-nitrophenyl phosphate (p-NPP), and Alamar blue were purchased from Himedia, India. Hydrochloric acid (HCl) and hexane were purchased from RANKEM, India. Fetal bovine serum (FBS), antibiotic–antimycotic solution, bicinchoninic acid assay kit, and Dulbecco's modified Eagle's medium (DMEM) were purchased from Thermo Fisher Scientific, India. All reagents used in this study were of analytical grade.2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :02 s on:off cycle; 60% amplitude). Control liposomes were also prepared in a similar fashion without the addition of PUFA-enriched fish oil. Microscopy images of the liposomes were captured at 40× magnification. Dynamic light scattering and zeta potential (Malvern Zetasizer nano ZS90) of the liposomes were measured by sufficiently diluting the liposomes in water. ImageJ software (ImageJ v1.54g) was used to calculate the size distribution of the liposomes. The SEM images of the liposomes-adhered scaffolds were fed to the ImageJ software to calculate the size distribution.
:02 s on:off cycle; 60% amplitude). Control liposomes were also prepared in a similar fashion without the addition of PUFA-enriched fish oil. Microscopy images of the liposomes were captured at 40× magnification. Dynamic light scattering and zeta potential (Malvern Zetasizer nano ZS90) of the liposomes were measured by sufficiently diluting the liposomes in water. ImageJ software (ImageJ v1.54g) was used to calculate the size distribution of the liposomes. The SEM images of the liposomes-adhered scaffolds were fed to the ImageJ software to calculate the size distribution.
The encapsulation efficiency of PUFA into the liposomes was estimated by GC-MS analysis, and heptadecanoic acid (C17:0) was used as an internal standard.10,11 PUFA-loaded and control liposomes were centrifuged at 25000 rpm for an hour at 4 °C to settle the liposomes. The liposome pellet was air-dried. To the dried pellet, 200 μL of chloroform was added and mixed well until all the lipids dissolved in the solvent. Later, lipids dissolved in the solvent were transferred to another container, and then the solvent was evaporated under a nitrogen stream. To the lipid extract, 0.5 mL of methanol/NaOH and 1 mL of hexane were added and incubated at 50 °C for 30 minutes. After 30 minutes of incubation, 0.5 mL of methanolic HCl was added and incubated again at 50 °C for 15 minutes. The upper hexane phase containing fatty acid methyl esters (FAMEs) was carefully collected and evaporated under a nitrogen stream. Dried FAMEs were resuspended in 300 μL of hexane and analyzed using GC-MS.
After fabricating the scaffolds, PUFA-loaded and control liposomes were loaded by incubating with the scaffolds for 24 h under stirring conditions at 200 rpm. After that, loosely adhered liposomes were removed by washing multiple times with water. The liposome-loaded scaffolds were lyophilized and stored at 4 °C until further use.
Scanning electron microscopy (SEM) (Apreo S) images of the scaffold without any liposomes (IO(GMA-TMPTA)), scaffold with control liposome-loaded scaffold (Lipo-IO(GMA-TMPTA)), and scaffold with PUFA-loaded liposomes (P-Lipo-IO(GMA-TMPTA)) were taken after sputter coating. Liposome adhesion onto the scaffolds was further verified by loading Nile red-stained liposomes onto the scaffolds.12 Briefly, 50 μL of Nile red (stock concentration 5 mg mL−1 in chloroform) was mixed with the lipid solution that was used for the liposome preparation. Nile red-stained liposomes were prepared and then loaded onto the scaffolds in a similar fashion as explained above. Fluorescence images of the Nile red-stained liposomes adhered to the scaffolds were captured.
Thermogravimetric analysis (TGA) (NETZSCH DSC 204F1 Phoenix) was carried out to understand the thermal degradation behavior of the samples. Approximately 10 mg of the scaffold samples were weighed and heated at a rate of 10 °C min−1 from room temperature to 1000 °C under an inert environment.
A vibrating sample magnetometer (VSM) (Lakeshore 7410S) study was carried out to understand the magnetic susceptibility of the samples. Approximately 10 mg of the scaffold samples were weighed and subjected to a magnetic field of ±15000 G at room temperature.
The percentage porosity of the scaffold samples was calculated using the ethanol displacement method.7 Briefly, scaffold samples were cut into defined dimensions and weighed (Wi). The scaffold samples were subsequently immersed in ethanol for 24 h to allow the solvent to penetrate the micropores of the scaffold samples. After 24 h, scaffold samples were taken out of the solvent, tapped gently onto the tissue paper to dislodge surface-adsorbed ethanol, and weighed again (Wf). The percentage porosities of the scaffold samples were calculated using the formula below,
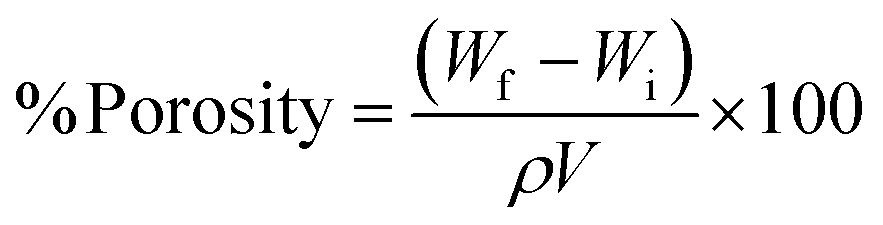
2.2.4.1 Cytotoxicity study. MG63 cells were seeded at a density of 104/well in a 96-well plate. Once the cells reached 70% confluency, sterilized scaffold samples were placed along with the cells. After a certain interval of time (24 and 48 h), the media was removed from the wells, and the scaffolds were washed several times with phosphate-buffered saline (PBS). To each well, 200 μL of Alamar blue dye (0.1 mg mL−1) was added, and the plate was transferred back to the CO2 incubator and left for 2 h. Wells without any scaffold were considered as the control. The cell viability in the wells treated with the scaffold was normalized against the control.7
2.2.4.2 Live/dead assay. Fluorescein diacetate/propidium iodide (FDA/PI) staining was carried out to qualitatively image the live and dead cells. Scaffolds were treated with the cells for 24 h using the protocol described above. Then, the media was removed from the wells, and the scaffolds were washed multiple times with PBS. To each well, 200 μL of the working staining solution was added (8 μg of FDA and 20 μg of PI per mL of PBS) and incubated under dark conditions for 5 min. The excess dye solution was removed by gently washing the wells with PBS twice. To each well, 100 μL of PBS was added to prevent dehydration of cells. The cells were then imaged under an inverted fluorescence microscope (Olympus IX83).7
2.2.4.3 Cell adhesion. MG63 cells were used to study cell adhesion onto the scaffolds.7 Scaffolds were placed into a 96-well plate and soaked in DMEM supplemented with 10% FBS and 1% antibiotic–antimycotic solution for 24 h. Then, cells were seeded onto the scaffolds at a density of 1 × 104 cells per well and incubated for 24 h. In the next step, the media was removed, and the cells were washed multiple times with PBS. Cells adhered to the scaffolds were fixed by adding 200 μL of 2.5% (v/v) glutaraldehyde and incubating for 2 h. Excess glutaraldehyde was removed from the wells, and the cells were washed multiple times with PBS. Scaffold samples were dehydrated with graded ethanol series and then air dried. Scaffolds were sputter coated and then imaged under SEM.
2.2.7.1 Alizarin red assay. Scaffolds were placed in a 96-well plate and incubated with DMEM supplemented with 10% FBS and 1% antibiotic–antimycotic solution for 24 h. Then, the media was removed, and osteosarcoma cells (MG63) were seeded at a density of 104 cells per well and incubated in a CO2 incubator at 37 °C temperature for a specific period of time (7 and 14 days). Every 2–3 days, the media was changed. After incubation for a specific period of time, the media was removed. Cells were fixed with 2.5% glutaraldehyde by incubating them with the solution for 2 h at room temperature. Excess glutaraldehyde was removed by washing multiple times with water. Subsequently, 200 μL of alizarin red dye (pH: 4.1–4.3) was added to each well and kept under mild shaking for an hour. After that, the excess dye was removed by multiple washes and stored at −20 °C until further processing. Scaffolds were chopped into small pieces. Bound dye was dissolved by adding 200 μL of 10% acetic acid and kept under mild shaking. This process was repeated until all the bound dye was eluted from the scaffold. Eluted dye from the scaffolds was pooled in the centrifuge tube and vortexed for 15 s. 500 μL of mineral oil was added to the centrifuge tube, and the mixture was heated at 85 °C for 10 min. After cooling for 5 min at room temperature, the upper mineral oil was removed and centrifuged at 15
000×g for 15 min. Then, 500 μL of the supernatant was collected and transferred to a fresh Eppendorf tube. 200 μL of ammonium hydroxide solution (10% v/v) was added to neutralize the acid. Aliquots of 150 μL were transferred to a 96-well plate and read at 405 nm.15,16 The results were expressed in terms of μmoles of Ca2+ by using the standard curve of known quantities of alizarin red stain.
2.2.7.2 Alkaline phosphatase activity. Cells were cultured on the scaffolds for 7 and 14 days, similar to the alizarin red assay. Every 2–3 days, the media was changed, and the spent media was collected. After a specific period, the scaffolds were removed and washed multiple times with PBS. In the next step, 200 μL of Triton-X-100 was added to each well and left for 20 min. All the contents were transferred to fresh Eppendorf tubes and sonicated twice for 30 s. Then, the tubes were vortexed for 15 s and centrifuged at 2500×g for 10 min at 4 °C. 50 μL of the supernatant was transferred to a 96-well plate, to which 50 μL of spent media collected was added. The reactant p-nitrophenyl phosphate (pNPP) (50 μL of 5 mM) was added and stored in an incubator at 37 °C for an hour. After that, the reaction was stopped by adding 2 M NaOH, and the absorbance was read at 405 nm. The results are expressed in terms of μg of p-nitrophenol (pNP) produced/hour by using a standard curve of pNP.15,16
2.2.7.3 RT-PCR study. MG63 cells were seeded in 6-well plates at a cell density of 1 × 106 cells per well. The cells were cultured in a CO2 incubator at 37 °C temperature, and the media was changed every 24 h. After reaching 70% confluency, scaffold samples were placed in these wells. The cells were cultured for 7 and 14 days. Cells without any scaffold treatment were considered as the control. After culturing for a specific period of time, the scaffolds and media were discarded. Total RNA was extracted from the cells as per the manufacturer's protocol. Following RNA isolation, 500 μg of the extracted RNA sample was used to synthesize cDNA using a reverse transcription kit. Next, RT-PCR (Applied Biosystems) was performed using the SYBR Green master mix. Gene expression was expressed as the fold change by normalizing the CT value with the housekeeping gene (β-actin). Details of the primer sequences are provided in the ESI† (Table S1).
A single colony was picked from the overnight culture and inoculated in 5 mL of Luria–Bertani (LB) broth. Once the optical density (OD) of the culture reached the mid-log phase, it was diluted 102 times, and 100 μL of the diluted culture was added to a 96-well plate containing the scaffolds. To each well, 100 μL of LB medium was added. Bacterial cultures without scaffolds were considered as the control. The well plates were transferred to an incubator maintained at 37 °C for 24 h. After 24 h of incubation, scaffold samples were removed, and the OD was taken at 600 nm. The percentage bacterial inhibition was calculated by comparing the OD values of the wells containing the scaffolds with those of the control wells.7
The generation of reactive oxygen species (ROS) in the different scaffold-treated bacteria was studied using the H2DCFDA assay.7 Briefly, a single colony was picked from the overnight culture and inoculated in 5 mL of LB medium. Once the OD reached the mid-log phase (0.5–0.6 OD), the bacterial culture was centrifuged at 5000 rpm for 5 min to settle the bacterial cells. The supernatant was discarded. 5 mL of fresh LB was added to the pellet containing cells and mixed well. Then, 200 μL of the culture was added to wells in the 96-well plate. Later, cultures were treated with different scaffolds, and the culture without any scaffold treatment was used as a control. To each well containing bacterial culture, 20 μL of H2DCFDA (1 mM) was added and incubated for 24 h at 37 °C temperature. After incubation, the conversion of non-fluorescent H2DCFDA to fluorescent 2′,7′-dichlorofluorescein (DCF) was measured using a fluorescence spectrophotometer at excitation and emission wavelengths of 485 and 515 nm, respectively.
3. Results and discussion
3.1 Preparation and characterization of PUFA-loaded and control liposomes
Optical microscopy images, size distribution (DLS method and ImageJ-based), and zeta potential of the PUFA-loaded and control liposomes (without PUFA) are presented in Fig. 1. The size distribution measured by DLS (Fig. 1c and d) showed that the hydrodynamic diameters of the PUFA-loaded liposomes were 121.3 ± 35 nm, and those of the control liposomes were 146.3 ± 68 nm. The hydrodynamic sizes close to 100 nm indicate that the prepared liposomes were nanoliposomes.6 As mentioned earlier, the oxidative stability of the PUFA was better in nanoliposomes (d < 200 nm) compared to the larger-sized liposomes.6 The polydispersity index (PDI) of the control and PUFA-loaded liposomes were 0.3 ± 0.03 and 0.5 ± 0.08, respectively. The zeta potentials of the control and PUFA-loaded liposomes were −4.4 ± 1.2 and −5.0 ± 2.6 mV, respectively. Despite the smaller zeta potentials of the liposomes, optical microscopy images showed no agglomeration of the particles. This indicates that the liposomes were stable in the aqueous medium. The size distribution of the liposomes (with and without PUFA), measured by the ImageJ software, showed smaller particle sizes than that shown by the DLS study. This is because the DLS study gives the hydrodynamic diameter of the particles, whereas ImageJ software measures the actual sizes of the particles.17 The amount of PUFA that was encapsulated within the liposomes was calculated using GC-MS analysis (Fig. S1, ESI†). The PUFA-enriched fish oil chromatogram showed two major peaks at retention times of 41.3 and 45.5 min, which are representative of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), respectively. The chromatograph of PUFA-loaded liposomes also showed peaks at the same retention times, indicating successful encapsulation of PUFA-enriched fish oil. Heptadecanoic acid (C17:0) was used as an internal standard to calculate the encapsulation efficiency of PUFAs. A known mass of heptadecanoic acid was used while preparing the fatty acid methyl esters and eluted through GC-MS along with other contents of PUFA-loaded liposomes. Based on the area under the curve of the internal standard, the encapsulation efficiency of PUFA-enriched fish oil was estimated to be 19.9 ± 3.4%. | ||
| Fig. 1 Optical microscopy images (a) and (b), DLS size distribution (c) and (d), zeta potential, and ImageJ-based size distribution (g) and (f) of liposomes and PUFA-loaded liposomes (scale: 10 μM). | ||
3.2 Scaffold fabrication and characterization
SEM micrographs of the scaffolds prepared by the Pickering emulsion polymerization technique are shown in Fig. 2. As seen from the figure, the scaffolds were highly porous in nature, with pore sizes in the range of 5–20 μm. In the case of scaffolds containing control liposomes (Lipo-IO(GMA-TMPTA)) and PUFA-loaded liposomes (P-Lipo-IO(GMA-TMPTA)), we could see densely adhered liposomes. In addition, we did not notice any liposomal membrane distortion, indicating that liposomes were stable after loading onto the scaffolds. Thin sections of scaffolds were loaded with Nile red-stained liposomes. Fluorescence images of Lipo-IO(GMA-TMPTA) and P-Lipo-IO(GMA-TMPTA) are shown in Fig. S2 (ESI†). As seen from the figure, liposomes were densely loaded onto the scaffolds, which agrees with the SEM micrographs. | ||
| Fig. 2 SEM micrographs of control scaffolds (IO(GMA-TMPTA)), scaffolds with control liposomes (Lipo-IO(GMA-TMPTA)), and scaffolds with PUFA-loaded liposomes (P-Lipo-IO(GMA-TMPTA)) (scale: upper panel: 20 μM; lower panel: 10 μM). | ||
The thermal degradation behavior of the scaffolds is presented in Fig. 3a. Scaffold IO(GMA-TMPTA) started to degrade at 295 °C. After the incorporation of control and PUFA-loaded liposomes into the scaffolds, there was a slight decrease in the melting temperature to 260 °C. Significant incorporation of lipids in these scaffolds led to a decrease in the melting temperature.
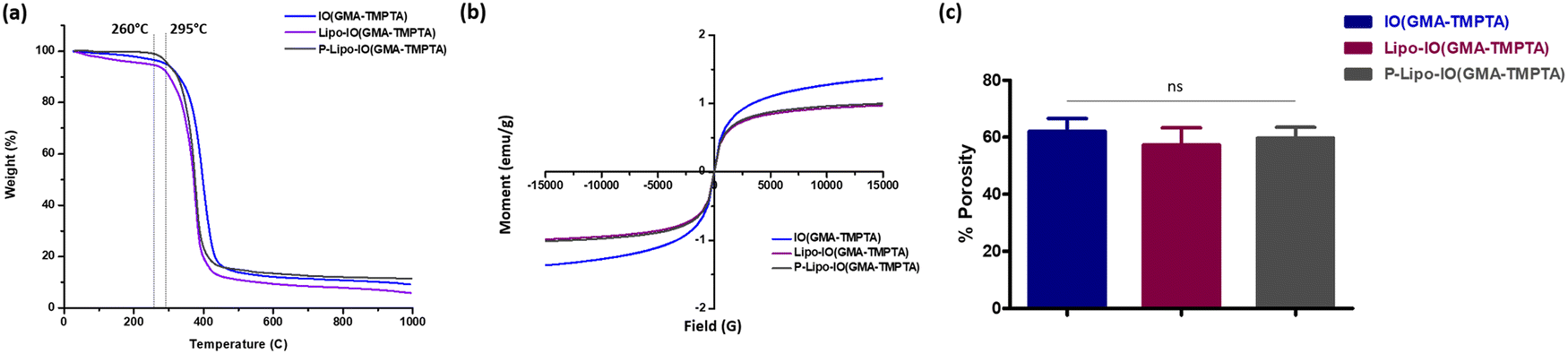 | ||
| Fig. 3 Physicochemical characterization of scaffolds, (a) TGA thermographs, (b) VSM magnetic susceptibilities, and (c) percentage porosity of the different scaffold preparations. | ||
The magnetic susceptibility of the scaffolds was tested using VSM by applying a magnetic field of ±15000 G at room temperature (Fig. 3b). The magnetic susceptibility of the control scaffold samples was higher than that of Lipo-IO(GMA-TMPTA) and P-Lipo-IO(GMA-TMPTA) scaffolds. This was possibly due to the higher nonmagnetic organic content. However, none of the scaffolds showed any hysteresis loop, indicating a superparamagnetic nature. Various reports have shown that magnetic stimulation of osteoblasts induces osteogenesis.18–21 Both static and alternating magnetic fields of different strengths have been used to stimulate osteoblasts both in vitro and in vivo.18–21 The saturation magnetization (Ms) values obtained for Lipo-IO(GMA-TMPTA) and P-Lipo-IO(GMA-TMPTA) scaffolds are in the range sufficient to stimulate bone cells for osteogenesis.21–23 The composite scaffold reported in the literature showed a magnetic moment in a similar range as our scaffolds with no hysteresis loop and exhibited excellent magnetic responsiveness both in vivo and in vitro conditions.22,24 Hence, the magnetic properties of these scaffolds can potentially be used to stimulate the adhered osteoblasts to improve bone formation and enhance the healing process.
The percentage porosity of the scaffold samples was calculated using the ethanol displacement method. The results are represented in Fig. 3c. The percentage porosities of the scaffold samples were in the range of 57–62%. The porosities of the cancellous bone are in the range of 50–90%. Hence, all the scaffold samples showed comparable porosities.1 This result confirms that Pickering emulsion polymerization produced highly porous scaffold materials, which would facilitate fluid transportation.1,7
3.3 Protein adsorption
Protein adsorption onto the scaffold material facilitates the cell adhesion process. Hence, it is necessary to understand the protein adsorption ability of the scaffolds. Protein adsorption onto the different scaffolds was estimated using a BCA assay, and the results are shown in Fig. 4a. Results show that 47.1 ± 5.3 μg of the protein from FBS adsorbed to 1 mg of the IO(GMA-TMPTA) scaffolds. However, in the case of Lipo-IO(GMA-TMPTA) and P-Lipo-IO(GMA-TMPTA) scaffolds, the protein adsorption was found to be 99.4 ± 9.2 and 91.7 ± 10.9 μg mg−1 of the scaffold, respectively. Protein–lipid interactions are a well-known phenomenon.25,26 The interaction between the lipids of liposomes and the proteins in FBS resulted in significantly higher protein adsorption in Lipo-IO(GMA-TMPTA) and P-Lipo-IO(GMA-TMPTA) scaffolds.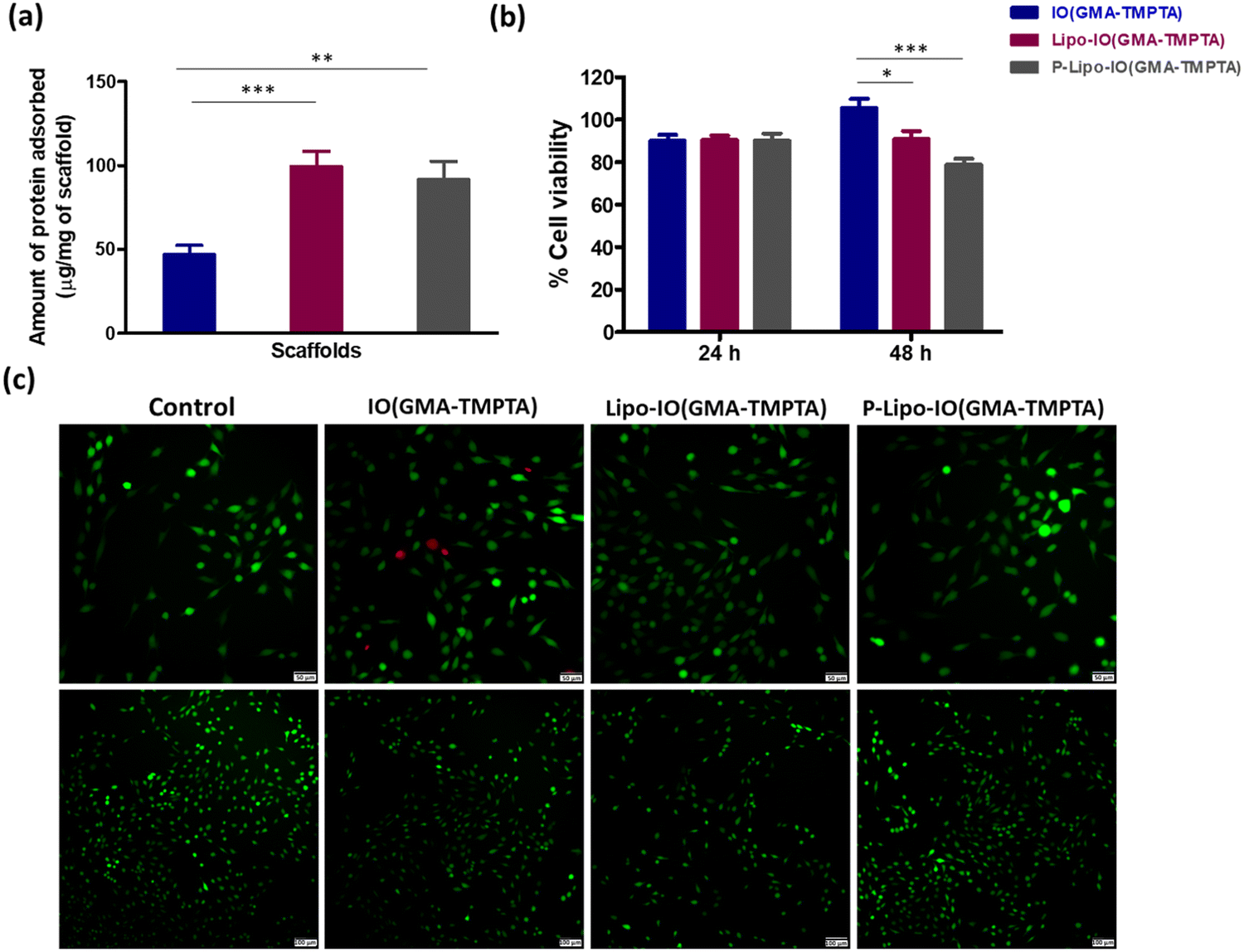 | ||
| Fig. 4 (a) Protein adsorption onto the different scaffold preparations quantified using BCA assay, (b) cell viability of the different scaffold preparations conducted on MG63 cell lines for 24 and 48 h using Alamar blue assay, and (c) live/dead assay conducted on the scaffolds using FDA/PI dyes (scale: upper panel: 50 μM; lower panel: 100 μM). * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 (n = 3). | ||
3.4 In vitro cell culture study
 | ||
| Fig. 5 SEM micrographs of different scaffold preparations showing adhesion of MG63 cells (scale: 30 μM). | ||
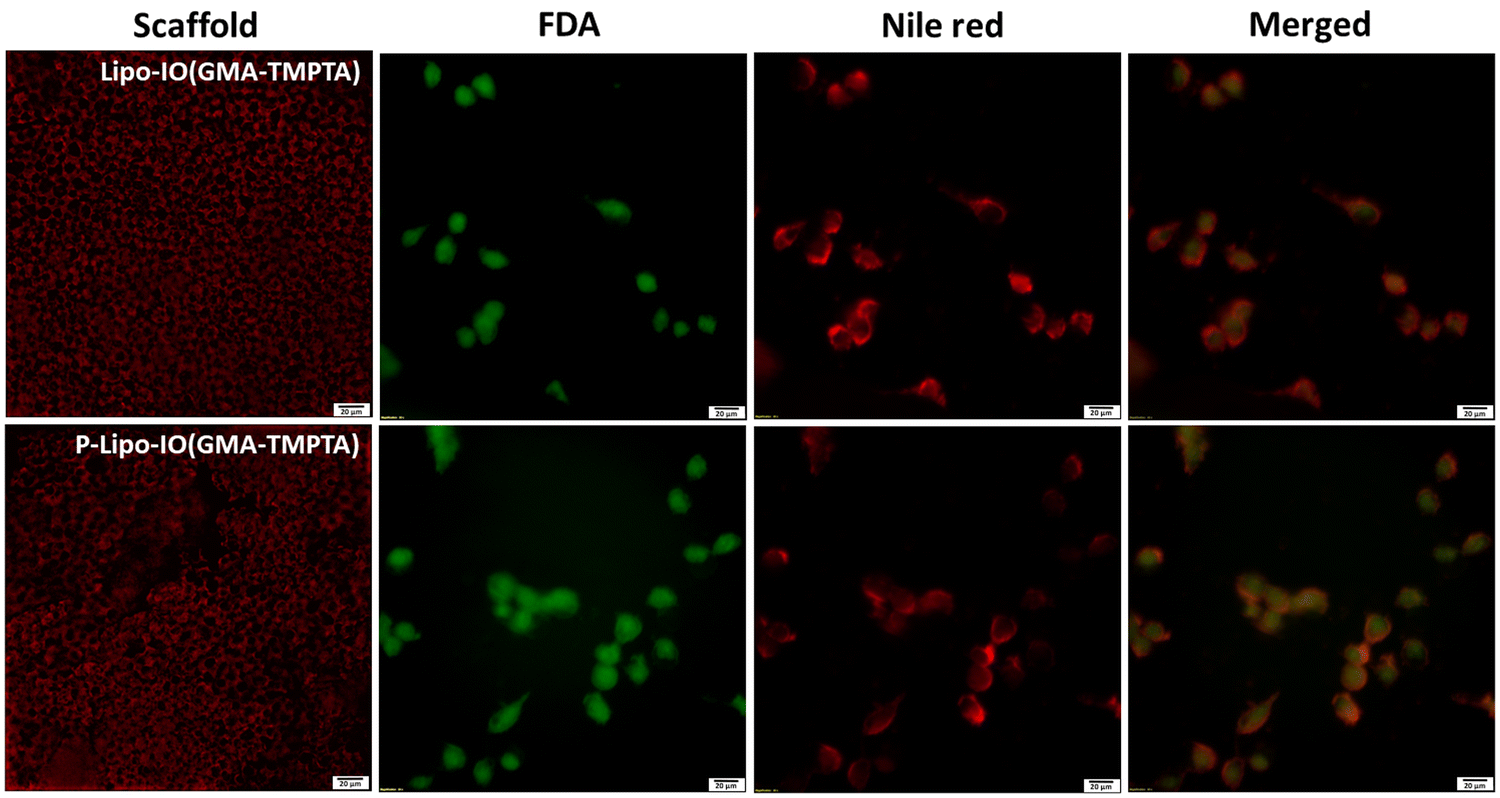 | ||
| Fig. 6 Release and uptake of the Nile red-stained liposomes by the MG63 cell lines that were loaded onto the scaffolds (scale: 20 μM). | ||
3.5 Simulated body fluid (SBF) mineralization assay
The mineralization of the scaffolds was evaluated by incubating the scaffold samples in the SBF media for 14 days. The composition of the SBF media closely resembles the ionic concentration of human plasma. Hence, it acts as a suitable biomimetic in vitro medium.34 The SEM images of the hydroxyapatite crystal formation on the scaffold surface are presented in Fig. 7. As seen from the SEM images, all the scaffold samples showed a mineral layer deposition on the scaffold surface. The EDAX analysis of those regions on the scaffold surfaces indicates the presence of calcium and phosphorous ions. There are several reports of a positive correlation between PUFA consumption and bone mineral density.35 It was observed that consumption of EPA and DHA mediates increased expression of markers of mineralization and thereby promotes differentiation of mesenchymal stem cells to osteoblasts.36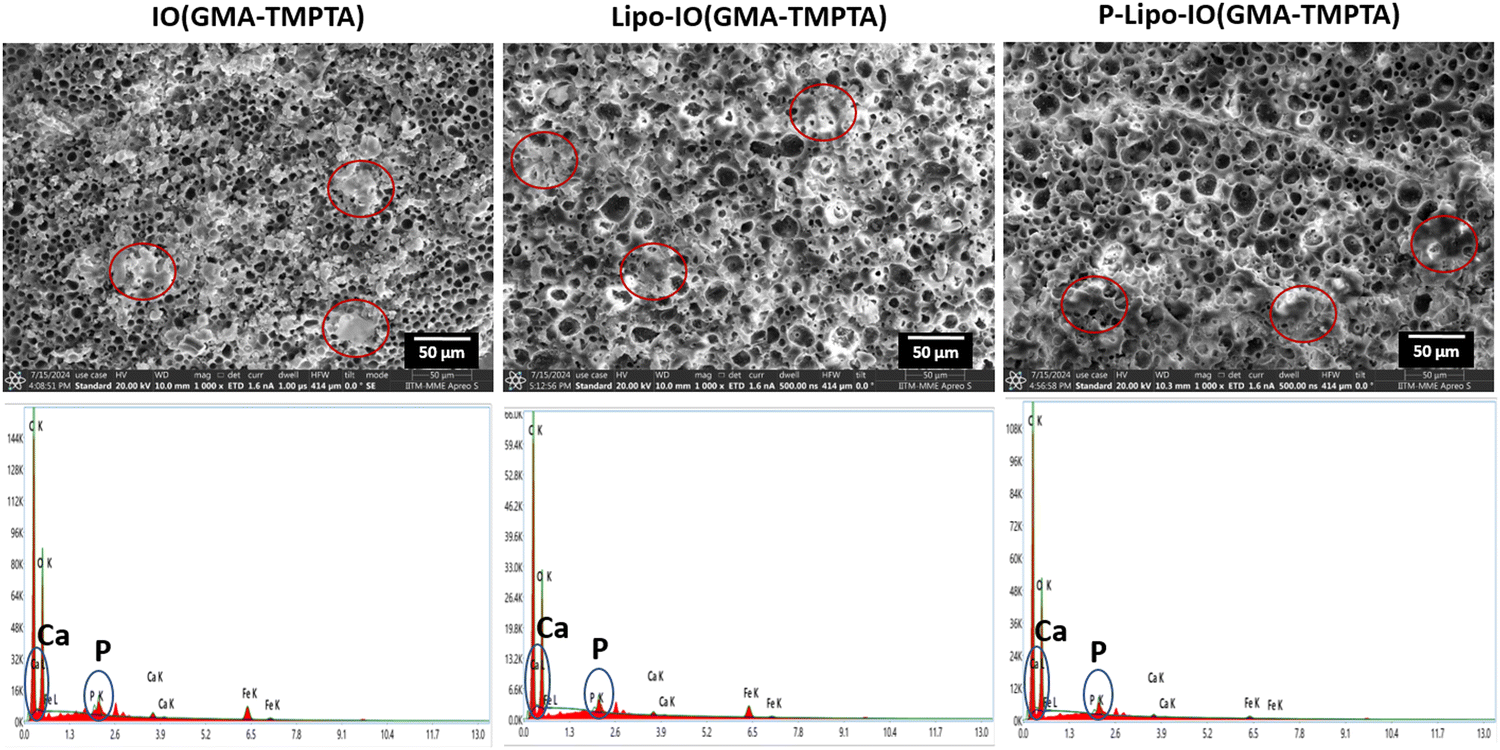 | ||
| Fig. 7 SEM micrographs of different scaffold preparations showing mineralization after incubating the scaffolds in the SBF media for 14 days (scale: 50 μM). EDAX study shows the presence of calcium and phosphorous onto the scaffold surface post incubation. | ||
3.6 Osteogenic potential
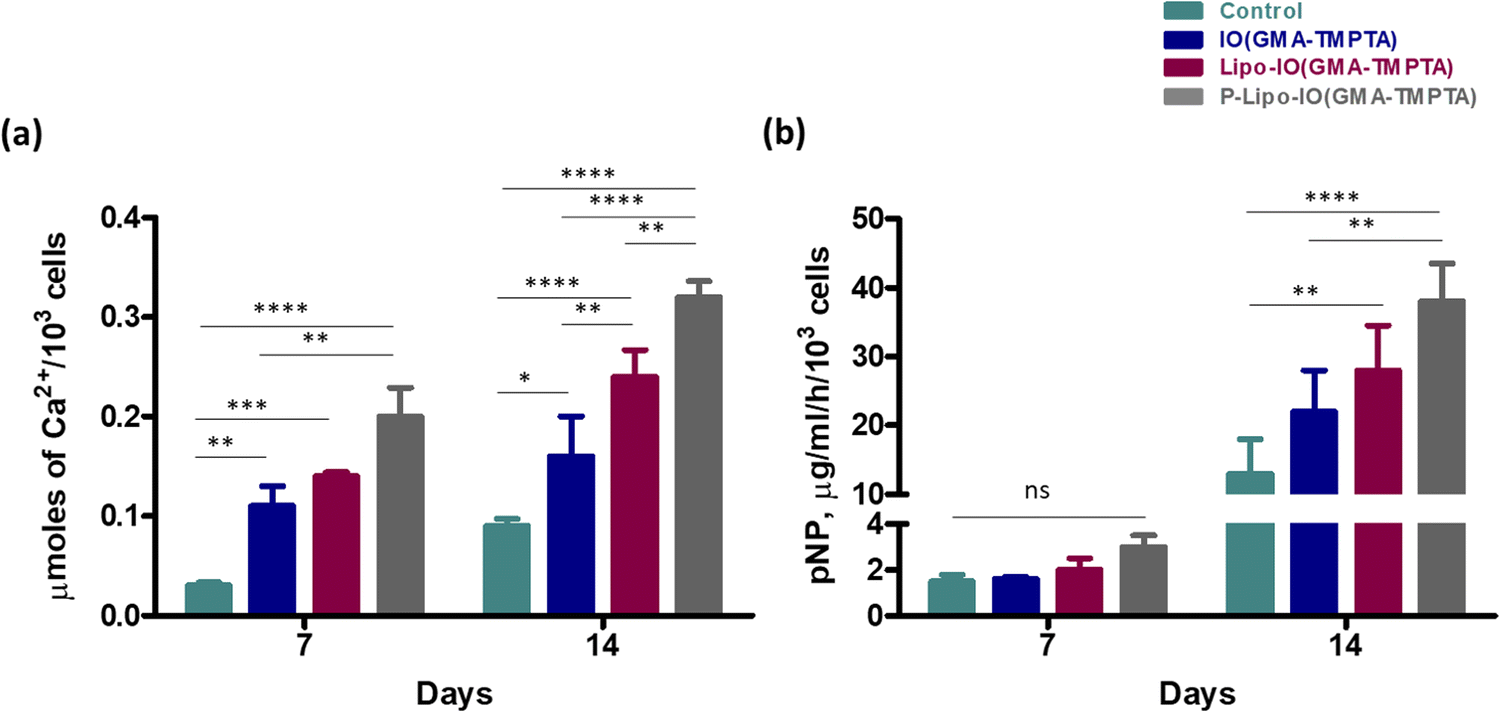 | ||
| Fig. 8 (a) Calcium deposition by alizarin red assay and (b) ALP activity after incubation of MG63 cells with the scaffolds for 7 and 14 days. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 (n = 3). | ||
 | ||
| Fig. 9 mRNA expression levels of osteogenic markers (a) BMP-2, (b) RUNX-2, (c) ALP, and (d) osteocalcin quantified using RT-PCR study. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 (n = 3). | ||
3.7 Antibacterial activity
The antibacterial activity of the scaffolds was tested against Staphylococcus aureus MTCC 1144, which is a well-known clinical strain. Staphylococcus species are known to cause 75% of osteomyelitis cases.7 The antibacterial potential of the different scaffolds is represented in Fig. 10a. The IO(GMA-TMPTA) scaffold showed 21.7 ± 9.2% inhibition after 24 h incubation, which was similar to our previous study.7 IONPs are well-known antibacterial agents and leaching of IONPs from the scaffold surface causes bacterial death. In addition, IONPs can interact with the bacteria that adhere to the scaffold surface, thereby killing it. Fenton-mediated ROS generation and membrane damage by physical rupture are two main known phenomena by which IONPs exert antibacterial activity.7 P-Lipo-IO(GMA-TMPTA) scaffolds showed 31.9 ± 6.8% antibacterial activity after a 24 h period. PUFAs are known to exhibit antibacterial activity against various pathogenic strains.39 High unsaturation of PUFA leads to an increase in oxidative stress by peroxidation and thereby imparts antibacterial activity.39 The presence of IONPs in the scaffold further accelerated the process of peroxidation in our study.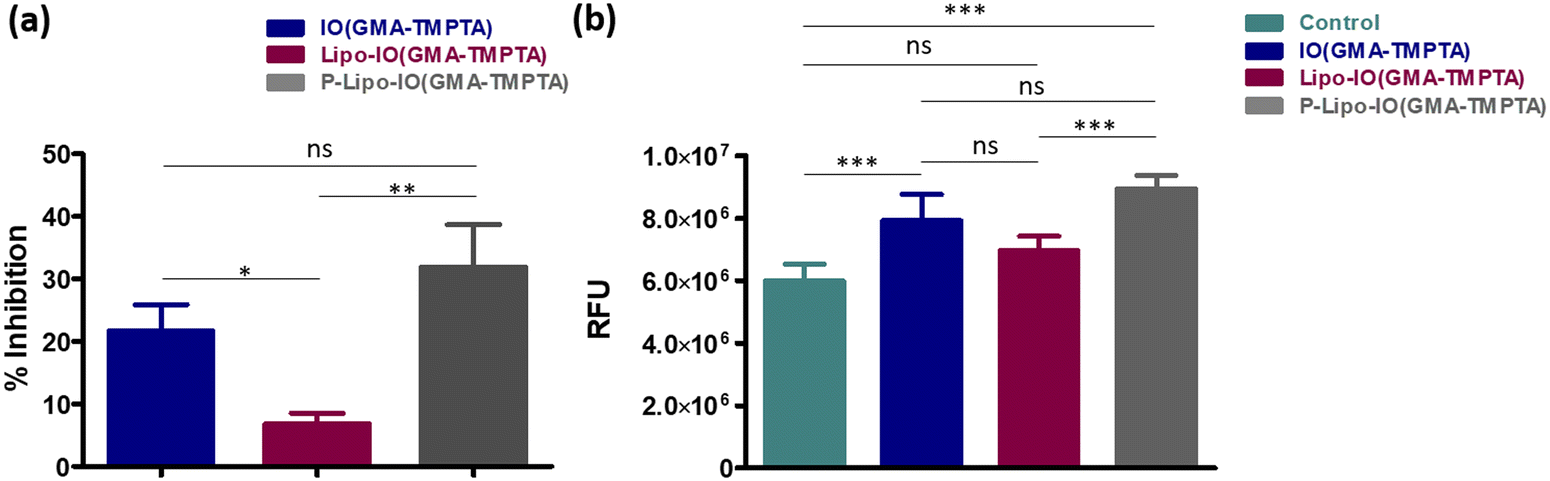 | ||
| Fig. 10 (a) Percentage inhibition of the bacteria S. aureus MTCC 1144 and (b) ROS generated in the bacteria measured by H2DCFDA assay after being incubated with the different scaffolds for 24 h. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 (n = 3). | ||
ROS generation by different scaffold-treated bacteria was studied by the H2DCFDA assay, and the results are presented in Fig. 10b. The non-fluorescent dye H2DCFDA converts into the fluorescent dye DCF in the presence of free radicals. The amount of DCF produced gives the quantitative value of free radicals present in the system.7 A schematic representation of the probable mechanism of antibacterial activity mediated by IONPs is presented in Fig. 11. As seen from Fig. 10b, ROS generation by different scaffold treatments followed a similar trend as that of % inhibition. The presence of IONPs in the scaffolds causes Fenton reaction-mediated ROS generation, which was evident in the IO(GMA-TMPTA) scaffolds. In the case of P-Lipo-IO(GMA-TMPTA) scaffolds, a significantly larger ROS generation was observed. As explained earlier, peroxidation of PUFA might have contributed to the significantly higher ROS generation, and the presence of IONPs might have accelerated the process.7,39
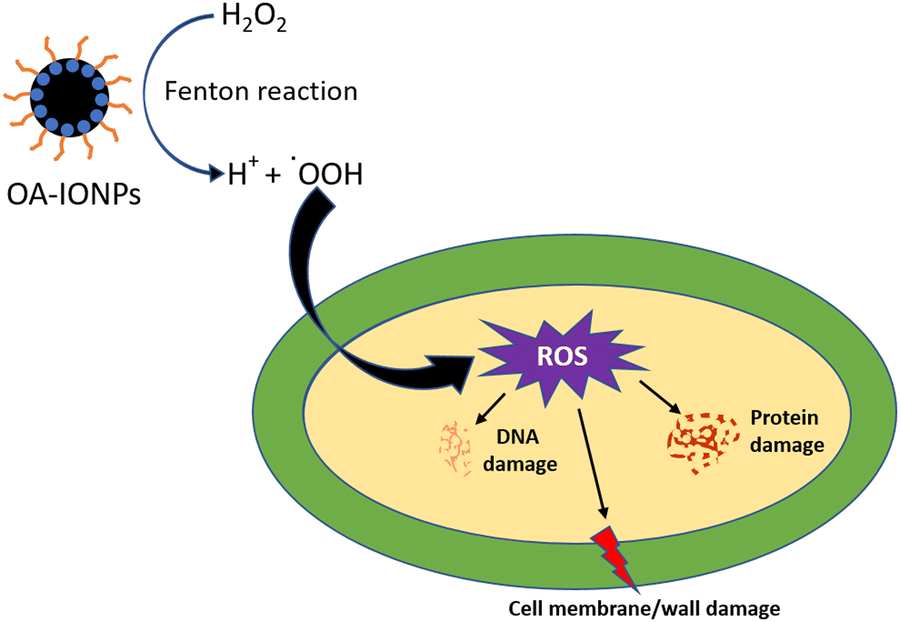 | ||
| Fig. 11 Schematic diagram representing the antibacterial activity mediated by IONPs present in the scaffolds. | ||
4. Conclusion
In the present work, oleic acid-coated iron oxide nanoparticle-templated Pickering emulsions were prepared. Glycidyl methacrylate and trimethylolpropane triacrylate monomers were dissolved in the oil phase of the Pickering emulsion and subsequently polymerized to yield a porous scaffold. To induce osteogenesis, PUFAs were encapsulated in liposomes and loaded onto these porous scaffolds. SEM and fluorescence microscopy images showed dense loading of liposomes on the scaffolds. In vitro cytotoxicity study on osteosarcoma cells showed a slight loss in viability after 48 h, which might be due to the peroxidation of highly sensitive PUFAs. The SEM images showed a well-spread morphology onto the scaffold surface, indicating firm adhesion of the cells. The SBF mineralization assay showed the deposition of a hydroxyapatite layer on the surface of all the scaffolds. Alizarin red and ALP activity studies showed that the P-Lipo-IO(GMA-TMPTA) scaffolds exhibited significantly higher calcium deposition and ALP activity. The RT-PCR study showed that the P-Lipo-IO(GMA-TMPTA) scaffolds exhibited significantly higher expression of osteogenic marker genes on day 14. Cell adhesion, together with alizarin red, ALP activity, and RT-PCR studies, indicate that the P-Lipo-IO(GMA-TMPTA) scaffolds have the potential to induce osteogenesis. The percentage bacterial inhibition of the P-Lipo-IO(GMA-TMPTA) scaffold was also significantly higher than that of the other two scaffold treatments. Although P-Lipo-IO(GMA-TMPTA) scaffolds showed better osteogenic potential and antibacterial activity, further optimization is needed to enhance the oxidative stability of PUFAs and improve the viability of the cells that are present on the scaffold.Author contributions
Aadinath W.: conceptualization, methodology, software, validation, formal analysis, investigation, visualization, writing – original draft; K. S. P. S. Teja: methodology, formal analysis, investigation; Iniyan Saravanakumar: methodology; Vignesh Muthuvijayan: conceptualization, formal analysis, fund acquisition, project administration, writing – review & editing.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
Aadinath W would like to acknowledge the Ministry of Education (MoE), Government of India, for awarding the fellowship. The authors would like to thank Mr Vimal Kumar Dewangan and Mr Sachin Latiyan for helping with the RT-PCR studies.References
- D. Jeyachandran and M. Cerruti, Adv. Eng. Mater., 2023, 25, 2201743 CrossRef CAS.
- A. Szwed-Georgiou, P. Płociński, B. Kupikowska-Stobba, M. M. Urbaniak, P. Rusek-Wala, K. Szustakiewicz, P. Piszko, A. Krupa, M. Biernat, M. Gazińska, M. Kasprzak, K. Nawrotek, N. P. Mira and K. Rudnicka, ACS Biomater. Sci. Eng., 2023, 9, 5222–5254 CrossRef CAS PubMed.
- M. J. Zhang and M. Spite, Annu. Rev. Nutr., 2012, 32, 203–227 CrossRef CAS PubMed.
- M. Bao, K. Zhang, Y. Wei, W. Hua, Y. Gao, X. Li and L. Ye, Cell Proliferation, 2020, 53, e12735 CrossRef PubMed.
- Q. Du, L. Zhou, M. Li, F. Lyu, J. Liu and Y. Ding, Food Front., 2022, 3, 239–255 CrossRef CAS.
- V. K. Venugopalan, L. R. Gopakumar, A. K. Kumaran, N. S. Chatterjee, V. Soman, S. Peeralil, S. Mathew, D. J. McClements and R. C. Nagarajarao, Foods, 2021, 10, 1566 CrossRef CAS PubMed.
- W. Aadinath and V. Muthuvijayan, Colloids Surf., B, 2023, 231, 113572 CrossRef CAS.
- M. Janmohammadi, Z. Nazemi, A. O. M. Salehi, A. Seyfoori, J. V. John, M. S. Nourbakhsh and M. Akbari, Bioact. Mater., 2023, 20, 137–163 CAS.
- R. A. Schwendener and H. Schott, Methods in Molecular Biology, Humana Press Inc., 2017, vol. 1522, pp. 73–82 Search PubMed.
- S. M. Alinafiah, A. Azlan, A. Ismail and N. K. M. A. Rashid, Molecules, 2021, 26, 6592 CrossRef PubMed.
- H. H. Chiu and C. H. Kuo, J. Food Drug Anal., 2020, 28, 60–73 CrossRef CAS PubMed.
- M. Zaremba-Czogalla, A. Jaromin, K. Sidoryk, A. Zagórska, M. Cybulski and J. Gubernator, Materials, 2020, 13, 1–18 CrossRef.
- K. M. Woo, J. Seo, R. Zhang and P. X. Ma, Biomaterials, 2007, 28, 2622–2630 CrossRef CAS PubMed.
- A. Dridi, K. Z. Riahi and S. Somrani, J. Phys. Chem. Solids, 2021, 156, 110122 CrossRef CAS.
- I. R. Orriss, S. E. B. Taylor and T. R. Arnett, Methods Mol. Biol., 2012, 816, 31–41 CrossRef CAS PubMed.
- M. Brennan, A. Renaud, A. L. Gamblin, C. D’Arros, S. Nedellec, V. Trichet and P. Layrolle, Biomed. Mater., 2015, 10, 045019 CrossRef PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS.
- C. Shuai, W. Yang, S. Peng, C. Gao, W. Guo, Y. Lai and P. Feng, Int. J. Bioprint., 2018, 4, 138 CrossRef CAS.
- C. Shuai, W. Yang, C. He, S. Peng, C. Gao, Y. Yang, F. Qi and P. Feng, Mater. Des., 2020, 185, 108275 CrossRef CAS.
- M. M. Fernandes, D. M. Correia, C. Ribeiro, N. Castro, V. Correia and S. Lanceros-Mendez, ACS Appl. Mater. Interfaces, 2019, 11, 45265–45275 CrossRef CAS PubMed.
- N. Bock, A. Riminucci, C. Dionigi, A. Russo, A. Tampieri, E. Landi, V. A. Goranov, M. Marcacci and V. Dediu, Acta Biomater., 2010, 6, 786–796 CrossRef CAS PubMed.
- P. Li, S. Zhang, K. Li, J. Wang, M. Liu, X. Gu and Y. Fan, J. Mater. Chem. B, 2018, 6, 4952–4962 RSC.
- J. Meng, Y. Zhang, X. Qi, H. Kong, C. Wang, Z. Xu, S. Xie, N. Gu and H. Xu, Nanoscale, 2010, 2, 2565–2569 RSC.
- M. Parmaksiz, Ö. Lalegül-Ülker, M. T. Vurat, A. E. Elçin and Y. M. Elçin, Mater. Sci. Eng., C, 2021, 124, 112065 CrossRef CAS.
- J. Guo, Y. Zhang, H. Li, H. Chu, Q. Wang, S. Jiang, Y. Li, H. Shen, G. Li, J. Chen and C. Xu, PLoS Biol., 2018, 16, e2006525 CrossRef.
- T. Izard and D. T. Brown, J. Biol. Chem., 2016, 291, 2548–2555 CrossRef CAS.
- P. Ji, X. Wang, J. Yin, Y. Yao and W. Du, Biomater. Sci., 2022, 10, 1544–1553 RSC.
- Y. Yang, S. Zuo, L. Li, X. Kuang, J. Li, B. Sun, S. Wang, Z. He and J. Sun, Asian J. Pharm. Sci., 2021, 16, 784–793 CrossRef PubMed.
- R. Guo, C. L. Ward, J. M. Davidson, C. L. Duvall, J. C. Wenke and S. A. Guelcher, Biomaterials, 2015, 54, 21–33 CrossRef CAS PubMed.
- C. Gögele, S. Wiltzsch, A. Lenhart, A. Civilleri, T. M. Weiger, K. Schäfer-Eckart, B. Minnich, L. Forchheimer, M. Hornfeck and G. Schulze-Tanzil, Mater. Sci. Eng., C, 2021, 130, 112421 CrossRef PubMed.
- N. Bar-Shai, O. Sharabani-Yosef, M. Zollmann, A. Lesman and A. Golberg, Sci. Rep., 2021, 11, 11843 CrossRef CAS PubMed.
- L. González-Rodríguez, S. Pérez-Davila, R. Lama, M. López-Álvarez, J. Serra, B. Novoa, A. Figueras and P. González, RSC Adv., 2023, 13, 15947–15959 RSC.
- L. Pan, X. Pei, R. He, Q. Wan and J. Wang, Colloids Surf., B, 2012, 93, 226–234 CrossRef CAS PubMed.
- X. Wu, K. Walsh, B. L. Hoff and G. Camci-Unal, Bioengineering, 2020, 7, 1–24 CrossRef PubMed.
- H. Wang, X. Li, M. Xuan, R. Yang, J. Zhang and J. Chang, Giant, 2024, 19, 100298 CrossRef CAS.
- B. Y. Y. Lau, D. J. A. Cohen, W. E. Ward and D. W. L. Ma, Molecules, 2013, 18, 14203–14227 CrossRef PubMed.
- R. Rajesh and Y. D. Ravichandran, RSC Adv., 2015, 5, 41135–41143 RSC.
- H. Kanniyappan, P. Thangavel, S. Chakraborty, V. Arige and V. Muthuvijayan, Int. J. Biol. Macromol., 2020, 143, 30–40 CrossRef CAS PubMed.
- W. N. Beavers, A. J. Monteith, V. Amarnath, R. L. Mernaugh, L. J. Roberts, W. J. Chazin, S. S. Davies and E. P. Skaar, mBio, 2019, 10, e01333-19 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00286e |
| This journal is © The Royal Society of Chemistry 2024 |