
Active tumor targeting by core–shell PDMS–HA nanoparticles with sequential delivery of doxorubicin and quercetin to overcome P-glycoprotein efflux pump†
Madhu
Verma
ab,
Krishna
Yadav
c,
Rashmi
Parihar
c,
Debjani
Dutta
 *b,
Surabhi
Chaudhuri
*b and
Sri
Sivakumar
*ade
*b,
Surabhi
Chaudhuri
*b and
Sri
Sivakumar
*ade
aDepartment of Chemical Engineering, Indian Institute of Technology Kanpur, Kanpur, Uttar Pradesh, India. E-mail: srisiva@iitk.ac.in
bDepartment of Biotechnology, National Institute of Technology Durgapur, Durgapur, West Bengal, India. E-mail: ddutta.bt@nitdgp.ac.in; schaudhuri.bt@nitdgp.ac.in
cCentral Experimental Animal Facility, Indian Institute of Technology Kanpur, Kanpur, Uttar Pradesh, India
dMaterial Science Programme, Indian Institute of Technology Kanpur, Kanpur, Uttar Pradesh, India
eCentre for Environmental Science and Engineering, Center for Nanosciences, Mehta Family Centre for Engineering in Medicine, Gangwal School of Medical Sciences and Technology Indian Institute of Technology Kanpur, Kanpur, Uttar Pradesh, India
First published on 14th January 2025
Abstract
The therapeutic efficacy of chemotherapy in various malignancies and solid tumors is significantly limited when used as monotherapy. This study explored a combined treatment approach for breast cancer cells involving sequential delivery of doxorubicin followed by quercetin, both delivered via polydimethylsiloxane nanoparticles decorated with hyaluronic acid. Quercetin inhibits P-glycoprotein efflux action to enhance doxorubicin activity by increasing its intracellular accumulation; hence, both synergistically suppress cancer cell growth by promoting cytotoxicity and apoptosis. Quercetin reverses multidrug resistance, induces arrest in the cell cycle, and alters the mitochondrial membrane potential. The successful delivery and internalization of these drugs into breast cancer cells were confirmed through CD44 ligand recognition, inhibiting cell viability via apoptosis (caspase-induced) and cell arrest in the G2/M phase of the cell cycle. Furthermore, in an MCF-7 (breast cancer) cell-derived xenograft tumor model using NOD/SCID mice, the core–shell PDMS–HA nanoparticle system carrying quercetin and doxorubicin resulted in approximately 65% tumor volume reduction, outperforming the loaded single drug and free drug combination. These results were supported by the TUNEL assay and proliferation index by Ki-67 immunohistochemistry staining, which show substantial cell death and tissue necrosis in the tumor sections. Histological studies of tumor tissues confirm enhanced anticancer efficacy with negligible systemic toxicity to normal organs. Overall, the PDMS–HA delivery system efficiently transports quercetin and doxorubicin to tumor cells, enhancing the antitumor effects against the MCF-7 tumor xenograft model in mice without adverse effects. This study suggests that the targeted co-delivery of phytochemicals and anti-cancer agents can synergistically overcome many barriers associated with tumor treatment.
Introduction
Combination therapy is the foremost approach for combating tumor resistance and reoccurrence by optimizing therapeutic efficacy through targeting multiple pathways.1–3 However, the use of chemotherapy combinations has been associated with adverse consequences, including compromised immunity, impaired liver function, and cardiovascular issues.4,5 Consequently, attention has shifted to anti-cancer phytochemicals due to their potential to mitigate the adverse effects of chemotherapy.6–8 Phytochemicals inhibit the progression of cancer by exhibiting a variety of cell death mechanisms, involving autophagy, apoptosis, necrosis, and ferroptosis.9 Several studies have demonstrated synergistic outcomes, reversal of MDR,2 and a reduction in drug dose and toxic effects (cardiotoxicity, nephrotoxicity, and hepatotoxicity) linked with chemotherapy.10–14 Therefore, synergistic interactions between these agents may amplify anti-tumor effects, reverse chemoresistance, and reduce side effects.15–17Therefore, in the present study, the synergistic outcome of quercetin (Que) and doxorubicin (Dox) has been considered as both have been reported to have broad anti-cancer effects against many cancers.18–20 Que is a naturally occurring flavonoid (3,3′,4′,5,7-pentahydroxyflavone (C15H10O7)) that is reported to modulate many cellular activities (apoptosis, inflammation, autophagy, inhibition of angiogenesis, and TME re-modulation in different cancers).21,22 Apart from this, it is reported that Que lowers the MDR effects by inhibiting P-glycoprotein (P-gp), MDR1, BCRP, and MRP1, which are accountable for the efflux of chemotherapeutic agents.23–25 Hence, Que could increase the Dox intracellular accumulation in cancer cells. Moreover, Dox has anti-tumor effects by inhibiting anti-apoptotic proteins, particularly the Bcl-2-associated X protein (BAX),26,27 and it can induce caspase-mediated apoptosis. Hence, the co-delivery of Que and Dox demonstrates the capability for synergistic cancer treatment. However, the solubility and bioavailability of these drugs and phytochemical molecules are not adequate, resulting in an unexpectedly diminished efficacy when used in in vivo models compared with in vitro cell-based studies. In order to overcome these challenges, the utilization of nanoparticles offers distinct advantages and promising potential for drug delivery applications.28,29
There are several advantages of NPs compared with free drugs: enhanced accumulation, targeted delivery, and the delivery of multiple drugs.30–32 Several NPs have been engineered33,34 to deliver a combination of drugs and phytochemicals (Table S1†). For instance, Ferguson et al. formulated curcumin-loaded nanoparticles with 5-fluorouracil to elicit synergistic effects on breast carcinoma cells.35 Mengyao Liu et al.36 explored the synergistic effect of paclitaxel with quercetin delivered via functionalized mesoporous silica NPs, yielding noteworthy outcomes. The study suggests that the combination successfully reduced tumor volume, with the least adverse effects, and the combination effectively overcame the multi-drug resistance in the breast cancer model. Furthermore, these studies have indicated limitations, such as non-specific release, potentially causing damage to healthy tissues, and reduced tumor accumulation due to the burst release.37,38 Therefore, it is imperative to develop more advanced nanocarriers that will enhance efficacy while decreasing drug dosage. This can be achieved by utilizing stimulus-responsive sequential delivery of therapeutic agents.39 Additionally, this approach enables a precise and stimulus-responsive controlled release of pharmaceutical molecules specific to tumor cells, thereby minimizing adverse effects.40–43
In this regard, polydimethylsiloxane (PDMS) is a promising polymer that can be employed for drug delivery applications. Intriguingly, a few reports suggested the virgin PDMS NPs44,45 for drug delivery. It emerges as a promising drug delivery vehicle attributed to its exceptional biocompatibility, physiochemical stability, and significant drug loading capability and thus it can enhance the bioavailability of hydrophobic drugs, all while minimizing systemic side effects. Recently, our group also developed PDMS nanoparticles with a unique capability to reach the nucleus, making them well-suited for delivering anticancer drugs.46 Their distribution within the cell, encompassing the cytoplasm and nucleus, renders them ideal for co-delivering drugs targeting multiple pathways in cancer progression. Exploiting these properties, PDMS NPs hold significant potential to serve as versatile platforms for NP-based combination chemotherapy, offering a potent strategy to address limitations associated with conventional combination approaches.47,48
This study endeavors to address existing challenges and introduce innovative strategies to develop stable and long-circulating NPs, notably suitable for combination therapy with targeted and stimulus-responsive drug delivery having a sequential release of loaded cargo. Therefore, we propose the synthesis of hyaluronic acid (HA) functionalized PDMS–HA core–shell NPs that can co-encapsulate Que and Dox while targeting CD44 receptors on breast cancer cells.49 The loading/encapsulation of Que and Dox in PDMS–HA NPs aimed to overcome challenges associated with their free-form obstacles. Through HA-functionalisation, we expect to achieve more efficient drug loading and stabilization, thereby maintaining optimal active drug dosages and actively targeting CD44 receptors to enhance cellular uptake potentially.50,51 Targeting the key HA receptor involved in HA-related cell adhesion and endocytosis can facilitate enhanced tumor penetration and uptake, thereby enhancing treatment efficacy while minimizing off-target side effects and systemic toxicity. The synergistic anti-tumor efficacy was systematically investigated in vitro and in vivo using the MCF-7 tumor model. Taken together our findings indicate that the formulated hybrid NPs synergistically inhibited cancer cell growth compared with their free drug equivalents, demonstrating promising potential for combating cancer.
Results and discussion
Fabrication and characterization of silica, PDMS, HA–PDMS, and Dox–Que loaded HA–PDMS nanoparticles
The fabrication of SCMS (solid core mesoporous shell) silica NPs and PDMS NPs was based on our previous46 study with slight modifications. The schematic illustration of silica NPs and PDMS NPs is represented in Fig. S1a.† Initially, silica NPs were made using the sol–gel technique; PDMS NPs were fabricated using prepared silica NPs as sacrificial templates. Subsequently, HA-modified PDMS NPs were fabricated by sequential physical adsorption of PEI and HA polymer onto the surface of bare PDMS NPs. The result was a core–shell structured nanoparticle, which gives us the advantage of loading dual drugs. One drug can be loaded to the core nanoparticle, and the second drug on the shell structure. Since Que has low solubility and structural instability at physiological temperature and pH, it was selected to load in the core of the PDMS NPs. Dox is relatively water-soluble due to its hydrophilic sugar moiety, but also its solubility is limited in physiological conditions and thus it was loaded in the outer shell of the PEI–HA polymer. Quercetin is loaded into the core of nanoparticles for chemotherapy because the core can protect quercetin from premature degradation, ensuring its stability and bioavailability. Additionally, encapsulation allows for controlled release, enabling a precise timing of quercetin delivery. The development of multidrug resistance (MDR) is one of the major challenges in the success of chemotherapy. P-Glycoprotein (P-gp) is an ATP-dependent efflux pump that plays a pivotal role in the development of MDR in cancer. These transporters exhibit wide drug specificity, enabling them to transport a wide range of structurally diverse compounds. This reduces drug accumulation within cells, thereby diminishing drug efficacy. Quercetin is a P-gp inhibitor that can improve the efficacy of chemotherapy drugs like doxorubicin by reducing drug efflux and enhancing intracellular retention and accumulation. The later release of quercetin relative to doxorubicin was designed to allow the initial cytotoxic activity of doxorubicin and furthermore quercetin blocks P-gp to limit its efflux (Scheme S1a†). This timing ensures that the chemotherapeutic agent can act first, followed by the inhibitor boosting its effectiveness by overcoming resistance mechanisms. This sequential strategy optimizes the therapeutic outcome while minimizing potential drug–drug interactions. Hence, this approach maximizes quercetin's effectiveness as a P-gp inhibitor, helping to overcome drug resistance in cancer cells and enhancing the efficacy of the combined treatment (Scheme S1b†). Scheme 1 represents PEI and HA modification of PDMS NPs and sequential drug loading, further illustrating the uptake of Que–Dox–PDMS–HA NPs exhibiting the process of sequential release of Dox and Que and demonstrating the mode of action and synergistic impact.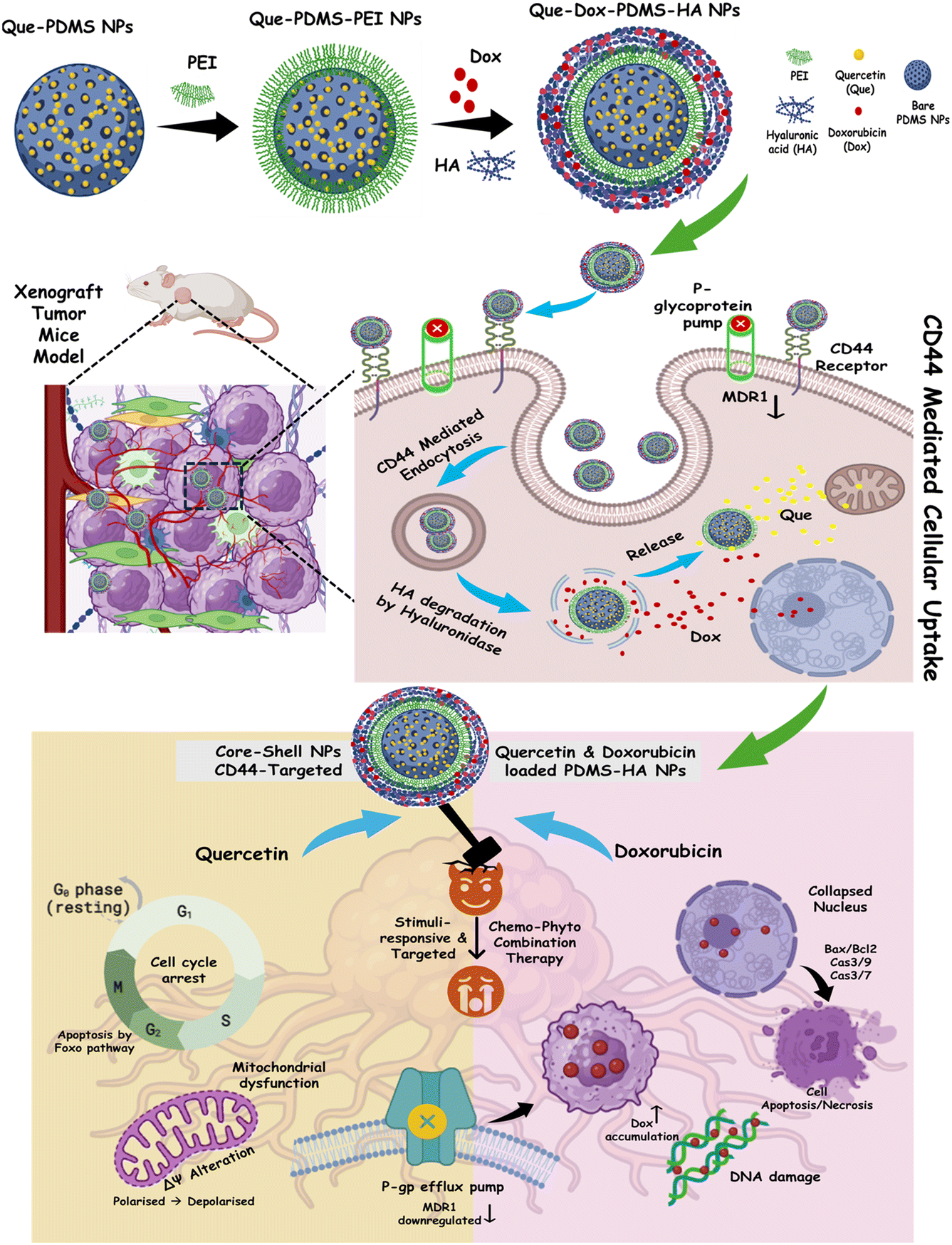 | ||
| Scheme 1 The schematic illustration of the fabrication process of PDMS–HA core–shell NPs loaded with Que and Dox, their cellular uptake, and Que and Dox mechanism of action. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to investigate the morphology of the fabricated nanoparticles. The SEM images of silica NPs, PDMS NPs, and PDMS–HA NPs exhibit uniformly distributed spherical-shaped nanoparticles with sizes of 270–280 nm, 145–165 nm, and 190–210 nm, respectively (Fig. S1b and e,†Fig. 1a). The TEM images show that silica NPs were well dispersed and spherical, and had a solid core and mesoporous shells (Fig. S1c†). Furthermore, the TEM image (Fig. S1f†) of PDMS NPs validates the formation of monodisperse porous PDMS NPs. There was a decrease in the size of fabricated PDMS NPs relative to silica NPs, representing the dissolution of template silica NPs and the subsequent formation of crosslinked PDMS NPs after the HF treatment. Fig. 1b displays a TEM image of drug-loaded core–shell PDMS–HA NPs. The SEM and TEM images of PDMS–HA NPs demonstrate that they were not uniformly spherical due to surface modification by PEI and HA polymers. The darker contrast in TEM images was due to the loaded drug in the core as well as in the shell. The only difference we observed was a polymeric shell on the surface, which was attributed to an increase in size compared with the bare PDMS NPs. The size of PDMS–HA NPs was increased by nearly 30–40 nm compared with PDMS NPs, indicating the effective coating of HA polymer on the PDMS NPs. Hence, the polymer coating gives PDMS NPs a rough structure, and the drug loading saturating its pores gives a darker contrast to the core and the shell polymers, illustrating the successful preparation of Que–PDMS NPs, Que–PDMS–PEI NPs (Fig. S2†) and Dox–Que loaded PDMS–HA NPs (Fig. 1b).
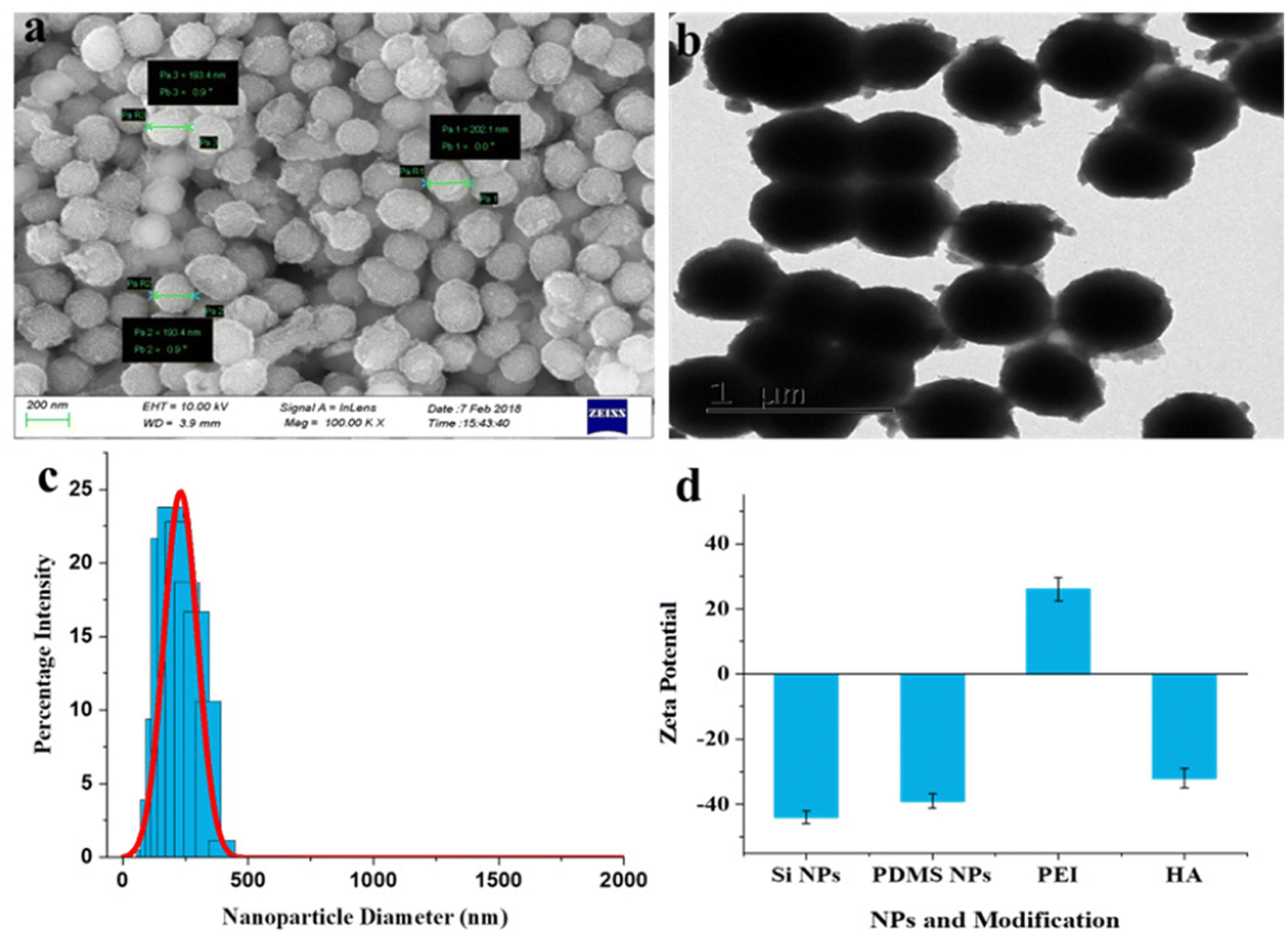 | ||
| Fig. 1 Morphology and surface characterization of PDMS–HA NPs. (a) SEM image of PDMS–HA NPs, (b) TEM image of drug (Que and Dox) loaded PDMS–HA NPs, (c) dynamic light scattering (DLS) data of PDMS–HA NPs, and (d) zeta potential of silica NPs, PDMS NPs, PDMS–PEI NPs and PDMS–PEI–HA NPs. | ||
Moreover, the particle, hydrodynamic diameter, zeta potential, and polydispersity index (PDI) were evaluated by a dynamic light scattering (DLS) technique. Silica NPs (template) formed a uniform dispersion and had a narrow peak with 0.11 PDI, which indicates a monodisperse population (Fig. S1d†) with an average hydrodynamic diameter ∼ 283 nm. Similarly, PDMS NPs (Fig. S1g†) and HA–PDMS NPs (Fig. 1c) had narrow particle size distributions, PDI was 0.18 and 0.29, and there was an average hydrodynamic diameter of ∼ 165 nm and ∼ 206 nm, respectively (Fig. S3a and c†). Furthermore, a change in the zeta potential was observed for PEI-modified PDMS NPs compared with bare PDMS NPs (−41.7 mV), followed by HA modification, which imparts a negative zeta potential to the surface of HA–PDMS NPs (Fig. S3b† and Fig. 1d). The zeta potential value for PDMS NPs–PEI was elevated to 31.5 mV upon PEI coating, attributed to the considerable abundance of amine (–NH2) groups. Followed by the coupling of HA polymer, the corresponding zeta potential values of PDMS–PEI–HA NPs changed to −37.6 mV, indicating an abundance of carboxyl (–COOH) groups. Accordingly, the changed surface zeta potential corroborated the synthesis of PDMS–HA NPs. It was noted that the negative charges on the NPs will facilitate enhanced retention and long-term circulation in the blood by avoiding untargeted binding. The DLS and zeta potential values of the Que- and Dox-loaded nanoformulation is reported in Fig. S3a and b.† HA surface modification will bind to the CD44-positive cells specifically. The diameter of the PDMS–HA NPs (206 ± 18 nm) and their softness was believed to enhance their potential to accumulate preferentially at the tumor area via an enhanced permeability and retention (EPR) effect.52
Next, Fourier transform infrared (FTIR) spectra were employed to examine the functional groups and validate the successful preparation of nanoparticles and their surface modification. The PDMS NPs exhibit Si–O peaks at 807 and 1078 cm−1, at 874 cm−1 which is attributed to bending vibrations of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, and at 1626 cm−1 which corresponds to vinyl groups CC (Sylgard 184). In addition, the effective coupling of HA with PDMS NPs–PEI was validated by the presence of a peak of CO at 1617 and 1409 cm−1 (Fig. S4, Table S3†). Moreover, Dox had peaks at 3432, 2925, 1725, 1623, 1407 and 1008 cm−1 that corresponds to the quinone and ketone carbonyl groups. In contrast, the spectrum peaks at 1650–1700 cm−1 correspond to CO stretching from the carbonyl group in the ketone moiety. Furthermore, the peaks at 1500–1600 cm−1 represent aromatic CC stretching vibrations, and finally peaks near 1200–1300 cm−1 are related to C–O stretching and bending of hydroxyl groups belonging to Que. Thus, the FTIR spectra of Que–Dox–PDMS–HA NPs showed the most absorption bands, confirming both the drugs were incorporated in the PDMS–HA core–shell NPs (Fig. S5, Table S4†). Based on the outcomes, it was confirmed that a successful modification and drug loading had been achieved.
CH, and at 1626 cm−1 which corresponds to vinyl groups CC (Sylgard 184). In addition, the effective coupling of HA with PDMS NPs–PEI was validated by the presence of a peak of CO at 1617 and 1409 cm−1 (Fig. S4, Table S3†). Moreover, Dox had peaks at 3432, 2925, 1725, 1623, 1407 and 1008 cm−1 that corresponds to the quinone and ketone carbonyl groups. In contrast, the spectrum peaks at 1650–1700 cm−1 correspond to CO stretching from the carbonyl group in the ketone moiety. Furthermore, the peaks at 1500–1600 cm−1 represent aromatic CC stretching vibrations, and finally peaks near 1200–1300 cm−1 are related to C–O stretching and bending of hydroxyl groups belonging to Que. Thus, the FTIR spectra of Que–Dox–PDMS–HA NPs showed the most absorption bands, confirming both the drugs were incorporated in the PDMS–HA core–shell NPs (Fig. S5, Table S4†). Based on the outcomes, it was confirmed that a successful modification and drug loading had been achieved.
In vitro cytocompatibility and uptake studies for PDMS–HA NPs
Biocompatibility is the utmost fundamental property of any nanoformulation before their utilization in any biomedical application. Hence, to examine the in vitro cytotoxicity, an MTT assay was employed for the PDMS–HA NPs with different concentrations and time points. Therefore, the assay was performed using cancerous cells (MCF-7, MDA-MB-231, HeLa, and HepG2) and noncancerous cells (HaCaT and HEK) for 24 h and 48 h at varying concentrations from 0–500 μg ml−1 to demonstrate the cytotoxicity (Fig. S6†). It was found that the relative cell viability was higher than 90% at all concentrations to which cells were exposed, even when incubated with a high concentration (200 μg mL−1) for 48 h. This indicates that PDMS–HA NPs had no evident cytotoxicity to cells and had good biocompatibility. Hence, the PDMS–HA NPs were safe for further in vitro and in vivo investigations. In addition, HA coating can promote cell proliferation within a certain concentration range, possibly because HA is a part of the cellular ECM and may provide compatibility for cell growth and proliferation.35,36The appropriate distribution and localization of nanoformulations within cells is crucial for achieving therapeutic effects on cells via cancer-specific target molecules. The evaluation of cellular uptake of PDMS–HA NPs was performed using a flow cytometer using cells treated with dye-loaded nanoparticles and bare nanoparticles. In this regard, cell uptake was performed on the MCF-7, MDA-MB-231, HepG2, HeLa, HaCaT, and HEK-293 cells to assess the uptake of PDMS–HA NPs loaded with RITC dye. The results of the flow cytometry analysis in Fig. S7† demonstrate that the uptake of PDMS–HA NPs was facilitated by the presence of the CD44 marker on the cells. Since the cancer cells have more CD44 markers on the cell surface, there will be more affinity towards the HA. This indicates that red fluorescence from PDMS–HA NPs–RITC can be seen only in cancer cells in contrast to non-cancerous cells. Hence, according to the results, we can summarise that the uptake was higher in the cancer cells (MCF-7 > MDA-MB-231 > HepG2 > HeLa) than in non-cancerous HaCaT and HEK-293 cells. These results suggest that the PDMS–HA NPs can enter specifically into the tumor cells mediated by HA polymer on the shell of the nanoformulation and can be used in targeted therapy.
Reactive oxygen species (ROS) generation detection for PDMS–HA NPs
ROS are the byproducts of cellular oxidative metabolism, mostly arising in the cell mitochondria.53 ROS play a vital physiological role in several cellular activities and cell signaling. Upon exposure to NPs, cells can experience an increase in ROS that can lead to a significant rise in intracellular ROS levels.54 This is due to the unique physio-chemical properties of different NPs, leading to cellular oxidative stress in response to the imbalanced redox equilibrium.55 We calculated the quantity of ROS present in cells using DCFDA, which emits green fluorescence when encountering ROS. This study investigates the intracellular effects connected to oxidative stress caused by ROS formation within the cells by PDMS–HA NPs. After being oxidized by ROS, the DCFDA dye breaks down into a de-esterified molecule within the cells and produces green fluorescence. As demonstrated by the results, no sudden surge in green fluorescence was detected when cells were exposed to PDMS–HA NPs up to 200 μg ml−1 for 24 and 48 h (Fig. S8a and b† respectively). From the confocal images, the mean fluorescence intensity was evaluated and the results indicated that the cells subjected to the PDMS–HA NPs exhibited a similar MFI value as untreated (negative control) cells (Fig. S8c†). This indicates no increase in the ROS levels inside mitochondria, and the PDMS–HA NPs caused no oxidative damage to the cells. However, there was a huge increase in the green fluorescence intensity for the positive control (80 μM hydrogen peroxide treated) cells. This result was also supported by the flow cytometry analysis at the same concentration of PDMS–HA NPs and the cells were stained with CellROX Green to check the ROS generation after PDMA–HA NP treatment (Fig. S8†). We found no ROS generation in the presence of the PDMS–HA NPs. This suggests that the PDMS–HA NPs induce no oxidative stress and can be used for further in vivo studies to validate their capabilities as a drug delivery system.Hemolysis assay for PDMS–HA NPs
Before any in vivo investigations, the nanoformulations must be checked for their interactions with the blood cells. Any nanoformulations injected into the body are directly exposed to the blood circulation and interact with the blood cells.56,57 These studies can be useful in deciding the NPs’ feasibility with RBCs for further biomedical application since the interactions may result in potentially fatal blood disorders including affecting erythrocyte membrane integrity and impacting the factors involved in blood clotting.58,59To assess the toxicity and safety profile of PDMS–HA NPs, the degree of hemolysis upon exposure to these NPs was systematically evaluated. The PDMS–HA NPs demonstrated a high level of blood compatibility by exhibiting no hemolysis (0.5%) at different concentrations (0–200 μg ml−1) (Fig. S9†). This hemolysis assay confirmed the exceptional hemocompatibility of PDMS–HA NPs in biological systems. The findings from the hemolysis assay provide strong evidence supporting the biocompatibility of PDMS–HA NPs. Therefore, the PDMS–HA NPs are safe for in vivo administration which thus highlights their promise for biomedical use.
Drug loading, release studies and in vitro cytotoxicity studies of Que–Dox–PDMS–HA NPs
The assessment of drug loading capability and entrapment efficiency was done by using the Dox and Que standard curves (Fig. S10a and b†). The Que and Dox loading was confirmed by UV-Vis scanning, represented in Fig. S11.† The release study was performed in the presence of the hyaluronidase enzyme and PBS (pH 7.4). Hyaluronidase enzyme will facilitate the breaking down of HA and the release of drugs in a controlled sequence. As indicated in Fig. 2a, the cumulative non-linear release profile of Que and Dox from PDMS–HA NPs strongly depended on the presence of an enzyme in the buffer. The initial release of the Dox was followed by the release of Que, which was contingent upon the delivery vehicle's arrival at the target site, i.e., TME, where hyaluronidase enzymes were more prevalent. Attaining the regulated release of the two payloads in a spatiotemporal manner is a critical aspect of our approach.60 Dox must reach the tumor and accumulate in the cell, to employ a cytotoxic effect. Subsequently, Que must be released onto the tumor site to act on reversing MDR, leading to a reduction in Dox efflux.24 According to the release profile, approximately 65% of Que and 88% of Dox were released from the nanoformulation in the initial 12 hours, while the remaining quantity was gradually released thereafter, observed up to 24 hours. The observed pattern of drug release in response to stimuli may be ascribed to the deliberate and prolonged release of the drug. Sequential drug release has many benefits compared with, i.e., TME, burst release. By gradually releasing medications, sequential release allows for enhanced penetration of the pharmaceuticals into tumor cells. This, in turn, facilitates the synergistic death of the tumor cells. This release profile can be ascribed to Dox release by disruption of the outer HA polymer matrix followed by the Que release by diffusion from the porous core PDMS NPs.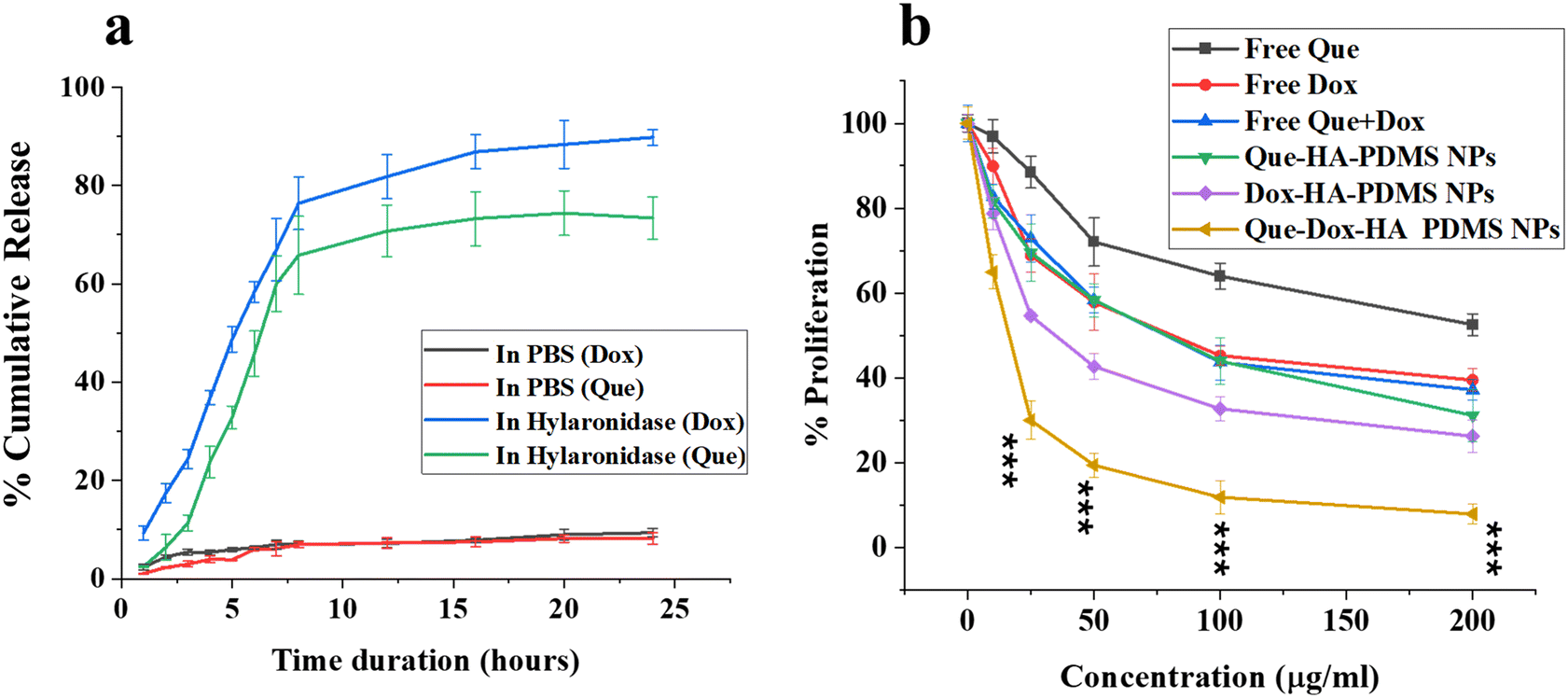 | ||
| Fig. 2 (a) Cumulative release of Que and Dox from PDMS–HA NPs in PBS and the presence of hyaluronidase enzyme. (b) In vitro cytotoxicity profile of Que–Dox–PDMS–HA NPs, Que–PDMS–HA NPs, and Dox–PDMS–HA NPs for concentrations (0–200 μg ml−1) and free drug (Que and Dox) equivalents incubated for 24 h against MCF-7 cells, evaluated by MTT assay. The measure of cytotoxicity is typically expressed as a percentage of proliferating cells remaining. Each of the data points signifies the average ± standard error (n = 3) (***p < 0.001). | ||
To assess the therapeutic efficacy of single and dual drug-loaded PDMS–HA NPs, we conducted an MTT cytotoxicity assay on MCF-7 cells for 24 h. The quantity of loaded drug for concentrations (0–200 μg mL−1) of PDMS–HA NPs was calculated (Table S5†), and consequently, an equivalent of free Dox and Que was administered. A reduction in the values of IC50 for Que and Dox carried through PDMS–HA NPs against MCF-7 was observed as represented in Fig. 2b. The evaluated values were Que (67.6 μM) and Dox (13.6 μM) when delivered separately through PDMS–HA NPs compared with their free equivalents of Que (153.3 μM), and Dox (7.5 μM). Furthermore, we compared the dual-loaded formulation with free equivalents of dual delivery, and the results show that Que (14.4 μM) and Dox (1.5 μM) were sufficient to kill the cells when delivered with PDMS–HA NPs. The IC50 fitting curves (Fig. S12†) represent the dose-dependent non-linear fitting. The findings indicate that the drugs administered through PDMS–HA NPs exhibit increased cytotoxicity compared with their free drug equivalents and an approximately 10-fold reduction in the IC50 values. Thus, HA-modified PDMS nanoparticles were believed to possess the potential to function as an efficient drug delivery system for anticancer agents.
Que anticancer mechanism studies
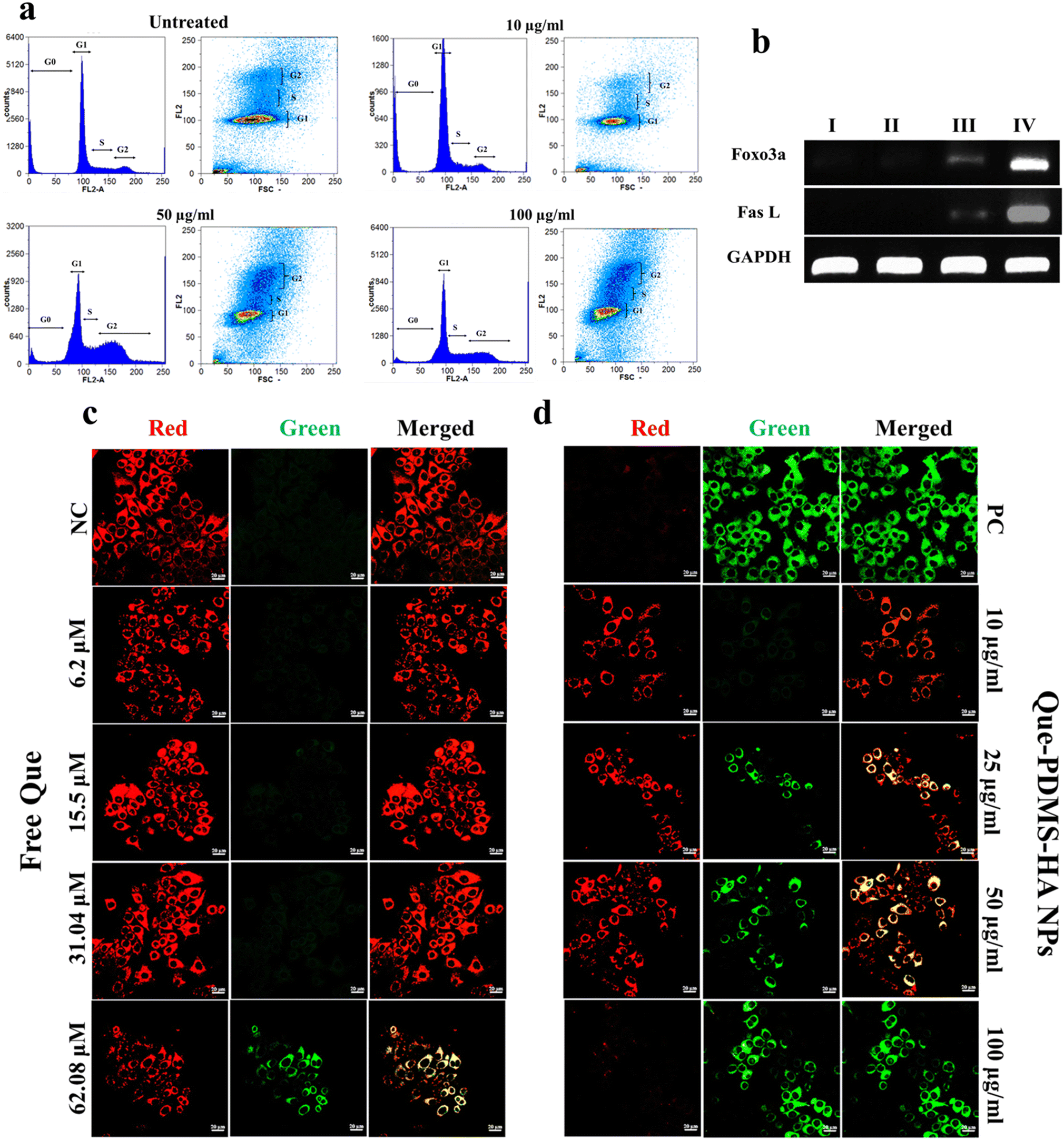 | ||
| Fig. 3 (a) Cell cycle arrest assay using PI-based flow cytometry study with bare PDMS–HA NPs (untreated) and Que–PDMS–HA NPs (10, 50, 100 μg ml−1) for 24 h with MCF-7 cells. (b) Cell cycle (FOXO pathway) gene expression studies in MCF-7 cells treated with Que–PDMS–HA NPs and bare PDMS–HA NPs (I. bare-PDMS–HA NPs, II. 10 μg ml−1, III. 50 μg ml−1, IV. 100 μg ml−1) and GAPDH used as a housekeeping gene. (c) and (d) CLSM images to detect Δψ with JC-1 dye with MCF-7 cells treated with Que–PDMS–HA NPs and free Que equivalents. Red fluorescence (JC-1 aggregates); green fluorescence (JC-monomers). NC: negative control (untreated cells), PC: positive control (CCCP treated cells). Scale bar 20 μm. | ||
The ability of quercetin to suppress P-gp expression in MCF-7 cells was assessed using flow cytometry with a primary P-gp monoclonal antibody and a secondary antibody conjugated to Alexa Fluor 488. Nonspecific binding of the P-gp antibody was evaluated using a blank or negative control, where the primary antibody was omitted, and no significant fluorescence was observed. As shown in Fig. S13a,† cells treated with Que–PDMS–HA NPs (25, 50, and 100 μg ml−1) for 24 hours demonstrated a dose-dependent decrease in fluorescence intensity, indicating reduced P-gp expression with increasing concentrations of quercetin-loaded NPs. Cells were categorized into green-positive and green-negative populations, where green-positive cells indicate P-gp expression, and green-negative cells reflect P-gp suppression (Fig. S13b†). To evaluate the efficacy of Que-loaded PDMS–HA nanoparticles, MCF-7 cells were also treated with free quercetin equivalents. The results showed that free quercetin led to less suppression of P-gp expression compared with Que–PDMS–HA nanoparticles, highlighting the enhanced efficacy of the nanoformulation.
Further to corroborate the above finding, the change in membrane potential was analyzed by rhodamine 123 (Rh 123) dye. It is a lipophilic cationic dye with a specific affinity for mitochondria and is known to selectively accumulate within the mitochondrial membrane, making it a reliable marker for assessing mitochondrial health. The healthy mitochondrial membrane (polarised state), which allows the dye to localize efficiently, results in bright green fluorescence. Conversely, dysfunctional mitochondria (depolarized state) demonstrate reduced fluorescence due to a diminished capacity to retain Rh 123. In this assay, the untreated cells exhibit a scattered pattern of bright green fluorescence within mitochondria when stained with Rh 123 (Fig. S15a and b†). In contrast, the mitochondria subjected to the Que-loaded PDMS–HA NPs displayed a dose dose-dependent reduction in the green fluorescence intensity. Hence, the findings indicate that the administration of quercetin induced alterations in the Δψmwith a more pronounced depolarisation effect on treated cells. This alteration in membrane potential reflects a shift in mitochondrial health, potentially contributing to the therapeutic effects of Que in a dose-dependent manner observed in MCF-7 cells.
Dox localization and its anticancer activity
Time-dependent intracellular distribution of Dox-loaded PDMS–HA NPs was monitored with confocal microscopy for HA-mediated intracellular drug release. The nuclear accumulation and localization of Dox–PDMS–HA NPs were validated by a DAPI-stained nucleus. According to Fig. 4a, the MCF-7 cell nucleus was stained with DAPI (blue), and Dox (red) appears to be co-localized. The observation of complete overlap between the extremely strong red fluorescence (Dox–PDMS–HA) and the deep blue fluorescence originating from the nuclei over a duration (12 and 24 h) provides evidence supporting the effectiveness of NPs in carrying the Dox inside the nucleus. As shown in the images, a decrease in red fluorescence intensity was observed at 24 h, which indicates the commencement of the apoptosis pathway and cell death. Hence, cell number was drastically reduced, and it reveals the effective transport of Dox into the cell nuclei. This observation demonstrated that PDMS–HA NPs enhanced the delivery of Dox to MCF-7 cells. | ||
| Fig. 4 (a) Time-dependent Dox internalization by MCF-7 cells at the concentrations (25, 50, and 100 μg ml−1) incubated for 12 h and 24 h. (b) Apoptotsis gene expression profile of MCF-7 cells treated with (I) bare PDMS–HA NPs, and Dox-PDMS-HA NPs ((II) 25 μg ml−1, (III) 50 μg ml−1, (IV) 100 μg ml−1) GAPDH used as housekeeping gene. (c) Caspase 3/7 activity assay in MCF-7 cells treated with free Dox and Dox–PDMS–HA NPs. The caspase activity was depicted as the fold rise relative to the untreated cells. Scale bar 20 μm (n = 3, *p < 0.05, **p < 0.01 ***p < 0.001). | ||
Quantitative PCR (Q-PCR) was conducted to evaluate the transcriptional expression levels of specific genes that play crucial roles in regulating of apoptosis. The analysis focused on Bcl-2, a gene known for its anti-apoptotic properties, and Bax, a pro-apoptotic gene that promotes cell death. Additionally, the expression levels of caspase-3 and caspase-9, key mediators in the apoptotic signaling cascade, were also assessed for cell apoptosis. By examining the transcriptional dynamics of these genes, Q-PCR provided insights into the molecular mechanisms underlying apoptotic regulation. Hence, the mRNA expression levels of these genes were evaluated followed by treatment with Dox–PDMS–HA NPs (25, 50, and 100 μg ml−1) in MCF-7 cells for 24 hours. The apoptosis mediated by these genes happens indirectly via the upregulation of apoptotic pathways.67 The Bcl-2 protein promotes the development of tumors and inhibits cellular death.68GAPDH (used as a housekeeping gene) was selected to serve as a control in qPCR. In the present investigation, the delivery of Dox–PDMS–HA NPs to MCF-7 cells resulted in a substantial increase in the mRNA levels of cas-3 and cas-9 when evaluated against the control groups (Fig. 4b). Furthermore, the use of Dox–PDMS–HA NPs has shown a notable ability to suppress the mRNA levels of anti-apoptotic genes, Bcl-2, and enhance the expression of Bax, in MCF-7 cells. The observed differences in mRNA expression levels in cells treated with blank NPs were found to be statistically negligible when compared with cells that were not subjected to any treatment. The mRNA expression levels of several genes linked with apoptosis demonstrate a considerable enhancement in the efficacy of the Dox delivered through PDMS–HA NPs.
Certain anti-cancer medications have been shown to provoke apoptosis, a form of programmed cell death, by activating caspase 3/7. This activation can occur through either the intrinsic or extrinsic pathway, ultimately leading to the destruction of cancerous cells. Some anti-cancer drugs can induce apoptosis by activating caspase 3/7, involving either pathway (intrinsic or extrinsic) of cell death.69 Therefore, evaluating caspase 3/7 levels can offer insights into cell death, regardless of the specific pathway involved. To assess whether cell death in MCF-7 cells was mediated by apoptosis, the activity of caspase 3/7 was evaluated for all treatments. The cells were treated with bare PDMS–HA NPs and Dox–PDMS–HA NPs (25, 50, and 100 μg ml−1). From the cytotoxicity profile, 100 μg ml−1 concentration was chosen as the maximal concentration, as it was approximately 10-fold above the IC50 value. The data for caspase 3/7 activity in MCF-7 cells are demonstrated in Fig. 4c. The results indicate that the fold increase in caspase 3/7 activity for MCF-7 cells treated with 25, 50, and 100 μg ml−1 was greater than that of free Dox equivalents cells. Consequently, Dox was efficaciously delivered to the cells via PDMS–HA NPs, resulting in a significant enhanced efficacy.
Dox induces oxidative stress and DNA damage, which can lead to mitochondrial dysfunction and apoptosis. Hence, it is noted that the Dox also produces ROS which hampers the mitochondrial function of the cells and thus we report the change in the emission wavelength of JC-1 dye from the red to green region. The JC-1 assay is used to evaluate mitochondrial membrane potential (Δψm), a key indicator of apoptosis. In this context the bare PDMS–HA NPs shows minimal effect on Δψm, indicating no intrinsic toxic effects by nanoparticles. Furthermore, Dox disrupts mitochondrial function, leading to depolarization of mitochondrial membrane potential and results in a shift in flourescence of JC-1 dye from red (JC-1 aggregates) to green (JC-1 monomer). This has been represented in the images (Fig. S16†) when the cells were treated by the Dox-loaded PDMS–HA NPs, when compared with the Dox free equivalents. This enhanced toxic effect is due to the nanoparticle formulations which enhance the targeted delivery of Dox, potentially increasing its efficacy in inducing mitochondrial damage while minimizing off-target effects.
Synergistic activity studies of Que and Dox delivered through PDMS–HA NPs
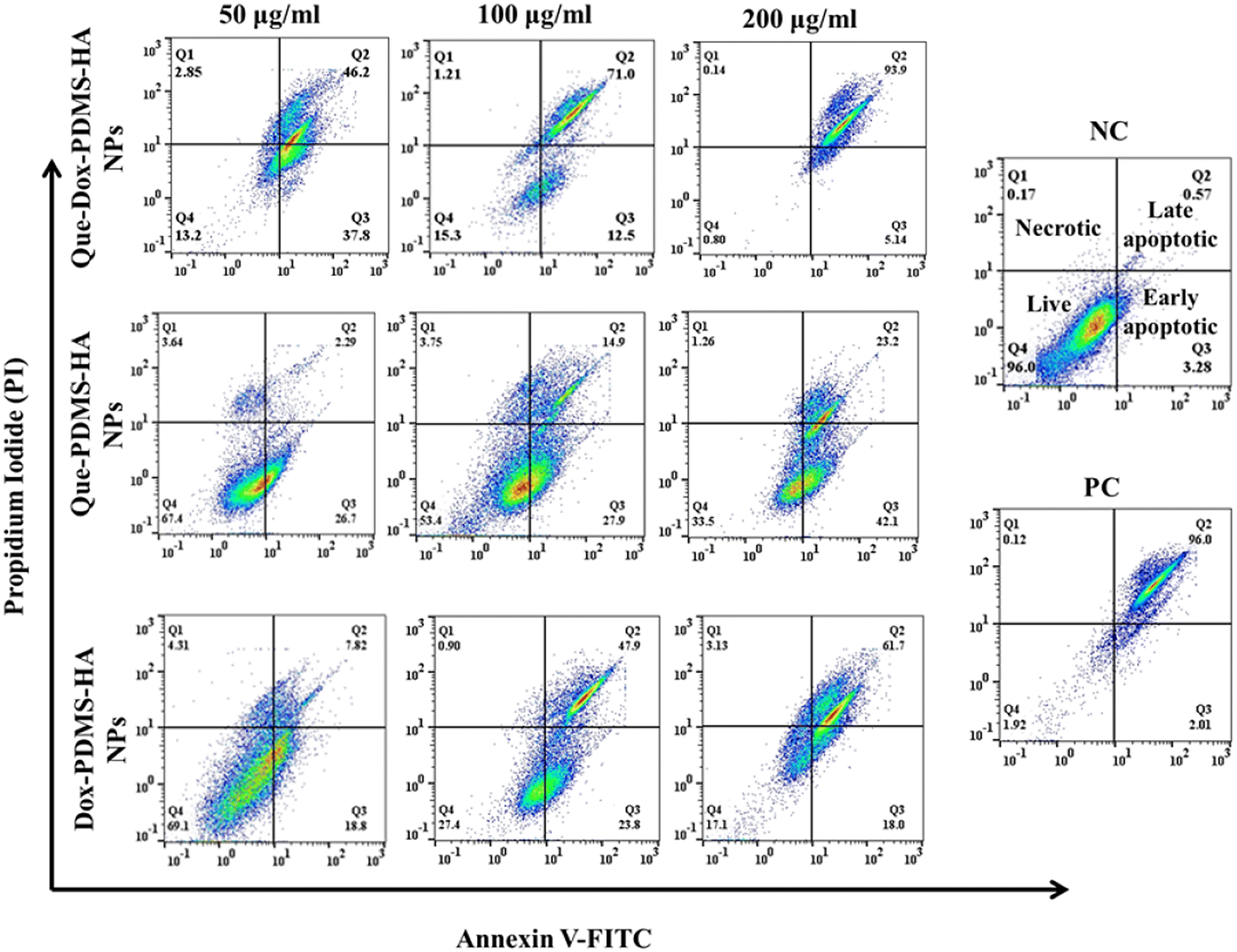 | ||
| Fig. 5 Apoptosis detection in MCF-7 cells using Annexin V–FITC and PI assay analyzed by flow cytometry. The cells were exposed to Dox–PDMS–HA NPs, Que–PDMS–HA NPs, and Que–Dox–PDMS–HA NPs at different concentrations (50, 100, and 200 μg ml−1). NC: negative control (PBS) and PC: positive control (H2O2). | ||
Apoptotic cell death is a cellular process that serves as a mechanism for suppressing cell proliferation. DAPI staining is a fluorescent dye with a high affinity for DNA, specifically targeting the nuclei. DAPI labeling/staining has been employed to characterize the impact of drug-induced apoptosis in cancer cells. A fluorescent dye (DAPI) was used to selectively visualize the alterations in the nucleus that occur during the process of apoptosis, enabling the quantification of the proportion of cells undergoing apoptosis based on the presence of condensed and fragmented chromatin. Fig. S17b† illustrates the magnitude of nuclear alterations occurring during apoptosis, as assessed using DAPI staining in both the control and treatment groups. The findings indicate that apoptosis was seen in MCF-7 cells, as shown by increased permeability to DAPI, nuclear apoptotic bodies, and chromatin condensation. This outcome was consistent with the finding of Annexin V–FITC/PI staining. Altogether, we conclude that PDMS–HA NPs possibly will be exceptional candidates for drug delivery, and the utilization of Que and Dox with PDMS–HA NPs potentially improves antitumor efficacy by activating apoptosis pathways.
Synergistic mechanistic studies through gene expression profile
Apoptosis is a potent mechanism of halting the progression of cancer cells and most anti-cancer agents target apoptotic pathways via targeting transcription factors and proteins. This can induce the pro-apoptotic and/or suppress the antiapoptotic proteins and in either way has proved effective in curbing the growth of cancer cells. To explore the Que and Dox synergistic effect at the molecular level on different apoptosis and cell cycle signaling pathways of cells after treatment with Que–Dox–PDMS–HA NPs, the mRNA expression levels were evaluated after incubation with bare PDMS–HA NPs, Que–Dox–PDMS–HA NPs (50 μg ml−1), and their free equivalents in MCF-7 cells for 24 hours. Q-PCR was used as to ascertain the transcriptional expression levels of several genes implicated in the apoptosis pathway and cell cycle arrest mechanism including for cas-3, cas-9, Bax, Bcl-2, FasL, and Foxo3a. Dox will initiate the apoptosis pathway by DNA damage and upregulating apoptotic genes. Primarily Bax is recognized for its ability to initiate cellular apoptotic pathways by meddling with the mitochondrial membrane integrity and enabling cytochrome c to be released from mitochondria to the cytoplasm; this results in the activation of cas-3 and cas-9.70 Then, the cas-9 acts as an intermediate and can induce alterations in mitochondrial morphology and facilitate the production of ROS. Cas-3 is triggered by cas-9 during the early phases of apoptosis, leading to DNA damage events inside the apoptotic cells.71,72 The Bcl-2 protein promotes the development of tumors and inhibits cellular death. From the gene expression profile, it was summarised that the tumor-supporting genes (bcl-2) were downregulated, and apoptosis-inducing genes (bax, cas3, and cas9) were upregulated to kill the tumor cells (Fig. 6a). Simultaneously, due to the effect of Que, Fas L and Foxo 3a genes were upregulated to participate in the arrest of the cell cycle of tumor cells. Hence, together these two pathways will inhibit the cancer cell progression and proliferation. Moreover, the Que administration with Dox also downregulates the MDR effect by regulating the P-glycoprotein efflux pump since the Mdr1 gene codes to produce a protein known as P-gp. Hence, the Mdr1 gene, mainly responsible for therapeutic agent efflux, was examined, and we found that it was downregulated due to the action of Que (Fig. 6b). We performed a gene expression experiment to confirm the expression of P-gp in the MCF-7 cells with and without quercetin treatment and GAPDH was the housekeeping gene. The groups were as follows: MCF-7 cells treated with PBS and PDMS–HA NPs groups served as a control, 100 μg ml−1 of Que-loaded PDMS–HA NPs and its free equivalent. The result suggests that Que successfully acts on the MDR genes to overcome the Dox efflux and enhance its accumulation, and this led to enhancement of the cell apoptosis signalling. GAPDH (used as a housekeeping gene) was selected to serve as a control in qPCR.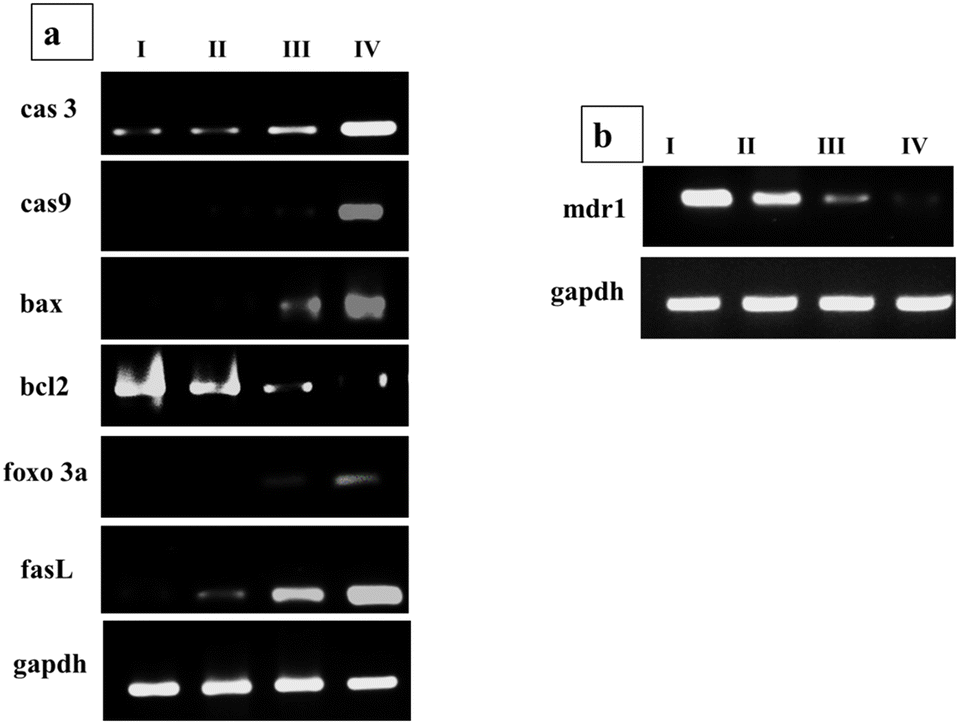 | ||
| Fig. 6 (a) Apoptosis pathway and (b) MDR reversal gene expression profile of MCF-7 cells incubated with (I) PBS, (II) PDMS–HA NPs, (III) free Que + Dox, and (IV) Que–Dox–PDMS–HA NPs (50 μg ml−1) for 24 h. GAPDH was used as a housekeeping gene. | ||
Cell viability studies by live/dead analysis (FDA/PI)
To determine the quantity of live and dead cells after treating Que–Dox-loaded PDMS–HA NPs. The MCF-7 cells were exposed to 50 μg ml−1 Que–Dox–PDMS–HA NPs for 24 hours. Then, the cells were subjected to dual staining with fluorescein diacetate (FDA) and propidium iodide (PI) to selectively mark the live and dead cells, respectively. The fluorescence microscopy images were acquired on the fluorescence microscope (Fig. S19†). The FDA (green fluorescence) specifically marks viable cells, and in contrast, PI (red fluorescence) is exclusively for necrotic/dead cells. The drug loaded in NPs shows more toxicity, in contrast to free drug equivalents. With both drugs, Que and Dox delivered by PDMS–HA NPs, an exponential increase in dead cells can be observed due to the synergistic effect compared with their free equivalents.Cytotoxicity studies on multicellular spheroid (3D model)
The antitumor effectiveness of anticancer medications has already been extensively studied using the traditional 2D cell culture paradigm. However, the reliability of simulating the 3D tumor environment in vivo in a 2D setting is under scrutiny. Alternatives include numerous 3D cancer models replicating a real solid tumor natural form and microenvironment.60 Due to their ease of production and repeatability, 3D multicellular spheroid models have gained significant popularity. Therefore, using a modified hanging drop approach, we assessed the anticancer effectiveness of Dox- and Que-loaded PDMS–HA-NPs (100 μg ml−1) in spheroids (3D tumor model) of MCF-7/NIH3T3 cells. The viability of cells in spheroids was assessed using a live/dead staining assay that incorporated FDA (viable cells) stain and PI (dead cells) stain. The observation of the green fluorescence over the spheroid, unaccompanied by a substantial red signal, suggests that most cells inside the spheroid maintained their viability during a 7 day culture period. To assess the cytotoxicity after the administration of drug-NPs, the spheroids were subjected to a 5 day incubation period, after which they were examined using confocal microscopy at various time points (1, 3, and 5 days).In order to assess the effectiveness of Que–Dox loaded PDMS–HA NPs in inhibiting growth, MCF-7 spheroids were subjected to a 5 day exposure. The results are shown in Fig. 7, spheroids that were exposed to bare PDMS–HA NPs served as a control group and thus did not exhibit any notable suppression of spheroid growth over a period of five days. In contrast, singly loaded Dox–PDMS–HA and Que–PDMS–HA NPs administration resulted in some extent of suppression of spheroid formation. In these groups, after three days, an increased presence of red-stained cells was noticed, indicating cell death (PI-stained cells), but a high number of green-stained cells was also visible indicating that the cells in the spheroids were viable and were proliferating. In contrast, the Que–Dox–PDMS–HA NPs group demonstrates effectiveness of the treatment on the spheroids as at the third day most of the cells were seen to be PI stained and a small number of green-labeled living cells were observable for the dual delivery group. Hence, the collapse of the spheroids was noted after five days of treatment. This finding also indicates that co-delivery of Dox and Que with PDMS–HA NPs has a greater capacity to suppress the proliferation of solid tumors compared with free drug and single-loaded drug, which aligns well with the previously observed cytotoxicity data.
 | ||
| Fig. 7 In vitro antitumor efficacy test with a multicellular (MCF-7 and NIH3T3) spheroid model. (I) Bare-PDMS–HA NPs, (II) free Que, (III) free Dox, (IV) free Que + Dox, (V) Que–PDMS–HA NPs, (VI) Dox–PDMS–HA NPs and (VII) Que–Dox–PDMS–HA NPs. Scale bar 200 μm. | ||
In vivo tumor regression studies in the MCF-7 tumor xenograft model
To investigate the therapeutic effectiveness of the nanoformulations, Que–Dox–PDMS–HA NPs were compared with single-loaded (Que–PDMS–HA NPs, Dox–PDMS–HA NPs), free drug equivalents, and control groups (PBS, bare PDMS–HA NPs) in the MCF-7 tumor xenograft mice model. Fig. 8a illustrates the scheme which includes information on the in vivo experiments on the timeline and the subsequent treatments. Initially, MCF-7 cells were subcutaneously injected to induce the tumor in the NOD/SCID mice. Once the volume of the grown tumor reached approximately 100 mm3, the mice were randomly split into eight groups (four in each group). The nanoformulations were intravenously administered at the dose of 100 mg per kg of mouse body weight every alternate day for 15 days. As shown in Fig. 8b and c, tumor inhibition was observed in the Que–PDMS–HA NPs and Dox–PDMS–HA NPs exposed groups, as compared with the control groups, but unfortunately, the efficacy was limited due to tumor drug resistance and the hostile TME. In contrast, the Que–Dox–PDMS–HA NPs group achieved significantly improved therapeutic efficacy due to the synergistic effect compared with the other groups. As proposed, Que–Dox–PDMS–HA NPs showed better anticancer effects than the free combination of Que and Dox, primarily due to their CD44-targeted delivery and the EPR effect. This leads to increased accumulation of the NPs at tumor locations through the EPR effect. Furthermore, it was confirmed that the superiority of this combination treatment is the result of remodeling TME and reversing MDR by Que–Dox–PDMS–HA NPs, which evidently suppresses tumor growth.73 Moreover, the body weight graph pattern of all the treated and control groups was consistent and stable (Fig. S20†), indicating the drug dose fell within the acceptable range of tolerability. Considering the overall findings, PDMS–HA NPs seem to be a potential nanocarrier for intracellular drug delivery with enhanced therapeutic efficiency and least systemic toxicity. Co-delivery gives the advantage of a synergistic effect as well, which can lessen the systemic toxicity imparted by Dox.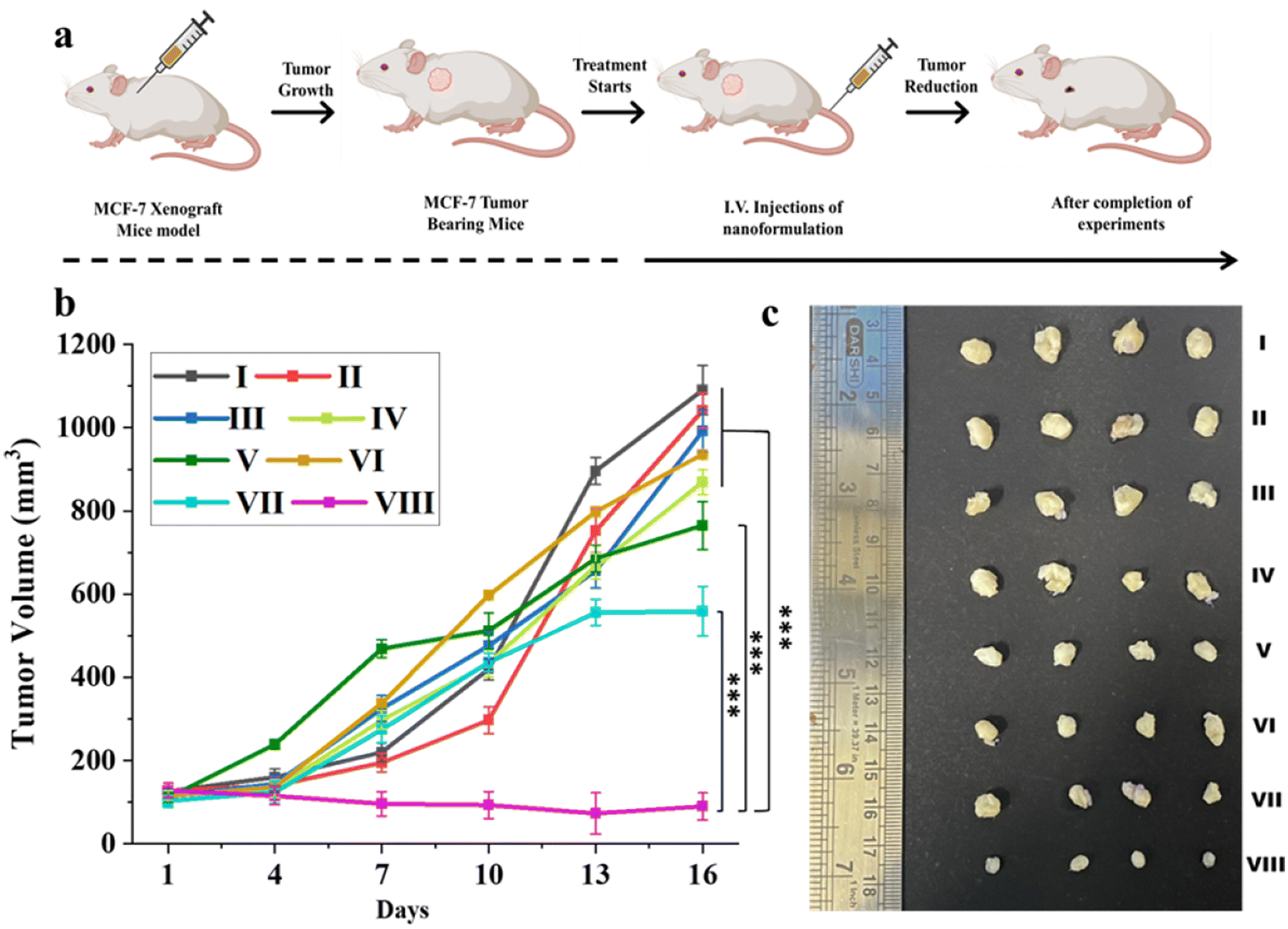 | ||
| Fig. 8 In vivo antitumor efficacy studies (a) Schematic diagram of the timeline followed for tumor growth and treatment schedules. (b) Tumor volume measurements at different time points after the initiation of the treatments. (c) Representative images of tumors extracted after the completion of the treatment which corresponds to the different treatment groups. Different groups for the experiment: (I) PBS, (II) bare-PDMS–HA NPs, (III) free Que, (IV) free Dox, (V) free Que + Dox, (VI) Que–PDMS–HA NPs, (VII) Dox–PDMS–HA NPs, and (VIII) Que–Dox–PDMS–HA NPs (n = 4, ***p < 0.001). | ||
The antitumor performance of fabricated nanoformulations was further estimated by histological staining (H&E, TUNEL, and Ki-67 staining). In the H&E-stained tumor tissues from the control group, we observed a substantial number of tightly packed tumor cells with healthy nuclei, and very few apoptotic or necrotic tumor cells were visible (Fig. 9a). In contrast, the Que–Dox–PDMS–HA NPs-treated tumor tissue experienced significant damage, and the majority of tumor cells were necrotic with a shrunk nucleus and DNA fragmentation, resulting in lysis of tumor cells. However, some tumor cells showed apoptotic features when treated with Dox–PDMS–HA NPs when compared with Que–PDMS–HA NPs, free Que, and free Dox. In these groups, tightly packed proliferating tumor cells were present with normal morphology and varying amounts of the extracellular matrix. Hence, compared with single drug-loaded formulations and free drug equivalents, most tumor cells treated with Que–Dox–PDMS–HA NPs exhibited necrotic tissue and showed apoptotic cell death in tumor sections.
 | ||
| Fig. 9 In vivo studies to check antitumor efficacy in MCF-7 tumor-bearing xenograft mice models. (a) Characteristic histological images of H&E staining of tumor sections. (b) CLSM images of TUNEL assay of tumor sections: blue (DAPI) and green (TUNEL). (c) CLSM images for Ki-67 expression by IHC for tumor sections. Blue (DAPI) and red (anti-Ki-67). Different groups for the experiment: (I) PBS, (II) free Que, (III) free Dox, (IV) free Que + Dox, (V) bare-PDMS–HA NPs, (VI) Que–PDMS–HA NPs, (VII) Dox–PDMS–HA NPs and (VIII) Que–Dox–PDMS–HA NPs. Scale bar 50 μm. | ||
To provide more conclusive evidence of the synergistic anticancer effectiveness of the fabricated nanoformulation, TUNEL assay and Ki-67 immunohistochemical (IHC) staining were conducted. These assays were performed with tumor sections of the control and experimental groups. The TUNEL assay, which is mediated by terminal deoxynucleotidyl transferase enzyme and utilizes dUTP nick end labeling, was employed to evaluate the levels of proliferation and apoptosis in the tumor sections. As depicted in Fig. 9b, in healthy cells, the nuclei were stained blue with DAPI, while the apoptotic cells appeared bright green. No visible cell apoptosis was detected in the tumor cells that received treatment with the free drug, including free Dox, Que free, and their combination. The group with single-drug-loaded Dox–PDMS–HA NPs also demonstrated increased apoptotic activity when compared with Que–PDMS–HA NPs. However, apoptotic cell percentages remarkably increased due to the synergistic impact of Dox–Que–PDMS–HA NPs (Fig. S21†). Moreover, Ki-67, the proliferation marker, was used further to analyze tumor cell viability and proliferation. As shown in Fig. 9c, the Ki-67 nuclear antigen appears as the red stain in the nuclei of epithelial cells. The results revealed that Que–Dox–PDMS–HA NPs could significantly sensitize MCF-7 tumors specifically, and the expression of Ki-67 was inhibited, which can be attributed to the reduced tumor size. In contrast, Ki-67 expression was much more evident in Dox–PDMS–HA NPs, Que–PDMS–HA NPs, free drugs, and control groups. Fig. S22† illustrates that the MFI of the Ki-67 marker expression corresponds to the proliferation index of the tumor sections. The Ki-67 proliferation index was significantly decreased in tumors treated with Que–Dox–PDMS–HA NPs compared with other groups. As demonstrated by the results, these smart nanoformulations could reduce tumor cell proliferation and inhibit cancer progression in cancer models. This conclusion validates the synergistic impact of using two therapeutic agents in combination, which were delivered through PDMS–HA NPs.
In vivo tumor targeting and biodistribution
To demonstrate the CD44 targeting mechanism of HA-decorated nanoparticles, immunohistochemistry (IHC) was conducted to confirm the overexpression of CD44 on tumor cells. Anti-CD44 antibodies were used to specifically bind to and highlight the presence of the CD44 receptor in tumor tissue sections. Upon visualization, green fluorescence was observed on the tumor cell walls (Fig. S23†), indicating the localization of CD44 receptors. This fluorescence pattern confirms the surface expression of CD44, which serves as the binding site for PDMS–HA NPs. The tumor cell morphology was further validated by DAPI staining, which marks the cell nuclei with a distinct blue fluorescence, ensuring the structural integrity and proper identification of tumor cells. This dual-staining approach provides robust evidence of CD44 overexpression in tumor cells, supporting the hypothesis that HA-coated NPs target these regions specifically due to the affinity between HA and CD44.The biodistribution of NPs plays a critical role in determining their therapeutic efficacy and safety profile, as it governs the fate of NPs under physiological conditions. Specifically, assessing the NPs localization at a target site is essential for evaluating the success of both passive and active targeting strategies. In this study, biodistribution analysis was performed using MCF-7 xenograft models to investigate the localization of PDMS–HA NPs at tumor sites over time. The tissues were obtained at 12, 24, and 48 hour intervals and were digested and the Si content was measured using inductively coupled plasma mass spectrometry (ICP-MS).
ICP-MS was utilized to measure the concentration of silicon (Si) in each organ, enabling a quantitative assessment of the distribution of PDMS–HA NPs. As shown in Fig. S24,† 12 h post injection (PDMS–HA NPs), the Si amounts in tumor appears ≈23% of ID, while that in other organs ranged from 5 to 24% of ID. For 24 h, the concentration of the NPs in the tumor increased to approximately 45% of the injected dose (ID), while the concentration in the lungs elevated to about 24% of the ID, likely due to the blood supply, and it remains unchanged in other major tissues (liver, heart and kidneys). At 48 h postinjection, the concentration of Si in most organs had further decreased, while the NPs concentration in the tumor remained elevated at 75% of the injected dose. In comparison with nonspecific NPs, the HA-modified PDMS NPs demonstrated additional substantial accumulation in the tumor sites in contrast to other major organs (Fig. S24†). The amounts of Si in the tumors were approximately 22%, 40%, and 78% of ID at different time intervals of 12, 24, and 48 h, respectively, with the maximum detected at 48 h. In comparison, low Si amounts (3–18%) were detected in other organs (kidneys, spleen, heart, lung, and liver) up to 48 h. These findings clearly indicated that PDMS–HA NPs preferentially accumulated in tumor xenografts in mice, likely due to the enhanced permeability and retention (EPR) effect. Furthermore, conjugation with HA–CD44 receptors significantly enhanced this tumor-specific accumulation of these NPs. Results indicated that the presence of a tumor-targeting moiety on the nanoparticles significantly influenced their overall biodistribution. The functionalization enhanced their specificity and resulted in increased accumulation within tumor tissues compared with non-targeted controls. This enhanced tumor localization can be attributed to active targeting mechanisms mediated by interactions between the HA on the nanoparticle surface and CD44 receptors overexpressed in tumor cells. The prolonged retention and progressive increase in accumulation observed over the 48 hour period further underscore the efficacy of the targeting approach.
Demonstrating CD44 expression in tumor sections is a pivotal step in validating the targeting mechanism of HA-coated nanoparticles. The combination of IHC and nanoparticle biodistribution studies establishes a direct link between HA binding and nanoparticle accumulation, providing strong evidence for the efficacy of the active targeting strategy. From the biodistribution analysis it was certain that the HA-decorated PDMS NPs was targeted towards the CD44-overexpressed tumor cells and hence the accumulation of nanoparticles at the tumor sites. To illustrate the CD44 targeting mechanism, IHC was performed using anti-CD44 antibodies to visualize CD44 overexpression on tumor cells, with the green fluorescence (CD44) on the tumor cell walls, and blue (DAPI)-stained nuclei. To further verify the colocalization of PDMS–HA NPs with CD44-positive cells, nanoparticles were loaded with rhodamine B isothiocyanate (RITC), a red fluorescent dye, and administered intravenously into mice. Biodistribution was monitored at 12, 24, and 48 hours post-injection. Tumor tissues were harvested, sectioned, and stained with anti-CD44 antibodies. Over time, increasing red fluorescence from RITC-loaded PDMS–HA NPs was observed within tumor sections (Fig. S25†), indicating progressive nanoparticle accumulation in the tumor. The co-localization of red fluorescence from PDMS–HA NPs with green fluorescence from CD44-positive regions confirmed that the nanoparticles actively targeted CD44-expressing tumor cells. The observed time-dependent increase in red fluorescence intensity suggests enhanced nanoparticle accumulation, driven by the high affinity of HA for CD44. By 48 hours, the accumulation reached a maximum, demonstrating the effectiveness of the HA coating in improving tumor targeting of the PDMS nanoformulation. To this end, these results underscore the critical role of HA decoration in enabling precise and efficient targeting of CD44-overexpressing tumor cells, validating PDMS–HA NPs as a promising platform for tumor-targeted drug delivery or imaging applications.
Histological studies of tumor-bearing mice models
To examine the potential toxic effects on normal tissues by the nanoformulations and drug treatments of the tumor-bearing mice models, the organs (lungs, liver, kidneys, heart, and spleen) were obtained and subsequently fixed with formaldehyde, followed by H&E staining. As depicted in Fig. S26,† there was no evidence of necrosis or toxicity in the tissues in the control groups and experimental groups, indicating no systemic toxicity. The H&E-stained liver sections have healthy and normal hepatocyte morphology with intact nuclei and well-defined sinusoidal structure. In addition, the spleen sections show normal morphology of spleen cells and lymphoid follicles. Similarly, lung tissue sections also indicate normal and unaffected cells with intact alveolar architecture. Moreover, the H&E-stained kidney tissue sections show no abnormalities in renal structure, with unaltered glomeruli morphology. However, free Dox has been implicated in causing cardiac toxicity, which was characterized by acute inflammatory cell infiltration and necrosis compared with the Dox–PDMS–HA NPs. This could potentially be attributed to free Dox, which exhibits non-specific targets and results in toxicity to vital organs, in contrast to stimulus-responsive targeted delivery. In the presence of Que, the inflammation can be reduced, and the co-delivery of Que and Dox can also be the reason for reduced toxicity when compared with the free Que and Dox groups. Meanwhile, no visible differences were observed in the organs of mice between the control group and PDMS–HA NPs and drug-loaded PDMS–HA NPs, which indicates no pathological damage to the vital tissues.Conclusion
The utilization of Dox in combination with Que proves to be an efficacious synergistic approach for improving cytotoxicity by the enhanced accumulation of Dox within breast cancer cells. This was potentially achieved by suppressing the expression of efflux transporters, such as MDR1, thereby effectively eradicating cancerous cells. This combination allows Dox to be more efficacious at a lower dosage, resulting in the desired anti-tumor impact while minimizing the usual harmful side effects associated with high-dose Dox therapy. Que boosts the chemotherapeutic efficacy of Dox against cancer cells while minimizing the adverse effects. Additionally, the doses of Que have a lethal effect on tumor cells by causing mitochondrial dysfunction and arrest in the cell cycle. Loading these drugs in a core–shell polymeric nanoparticle, i.e., PDMS–HA NPs, designed for combination therapy can actively target cancer cells, expressing the CD44 marker, and employs stimulus-responsive sequential delivery. This approach has increased the potency of the drugs relative to their free equivalents. Higher Dox accumulation induces the apoptosis by DNA damage, activating apoptotic genes and suppressing bcl2. The in vitro results suggest an approximately 10-fold reduction in the drug doses when compared with single drug-loaded formulations and a much greater increase in activity when compared with free drugs. Moreover, these outcomes were corroborated and validated by in vivo experiments conducted on xenograft tumor mice models. The result demonstrates an evident reduction in tumor size and an enhanced number of necrotic cells within the tumor tissue when treated with Que–Dox–PDMS–HA NPs. Hence, we summarise that the stimulus-responsive tumor-targeted PDMS–HA core–shell NPs loaded with Que and Dox defeats tumor barriers and synergistically combats drug resistance by inhibiting efflux pumps and enhancing efficacy. By efficiently co-delivering hydrophobic drugs, PDMS–HA nanoparticles offer a promising strategy to improve treatment outcomes by reducing doses, and potentially decreasing chemotherapy-related systemic toxicity.Materials and methods
Chemicals
N-Octadecyltrimethoxysilane (TMS), tetraethyl orthosilicate (TEOS), dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide (MTT), polyethyleneimine (PEI), trypsin–EDTA, ethidium bromide (EtBr), hyaluronic acid (HA), rhodamine 123 (Rh 123), Dulbecco's modified eagle's medium (DMEM), fluorescein diacetate (FDA), dichlorodihydrofluorescein diacetate (DCFDA), rhodamine B isothiocyanate (RITC), propidium iodide (PI), hydrofluoric acid (HF), penicillin–streptomycin antibiotic, Annexin V–Alexa Fluor 488 reagent, and anti-Ki-67 antibody were acquired from Sigma-Aldrich. PDMS elastomer kit (SYLGARD® 184) was procured from Dow Corning. Fetal bovine serum (FBS) was acquired from Invitrogen. Sulfuric acid, Triton X-100, dimethyl sulfoxide (DMSO), ethanol, tetrahydrofuran (THF), and formaldehyde were obtained from Merck. Ammonium fluoride and ethylenediaminetetraacetic acid (EDTA) were acquired from Loba Chemie. Acetone, methanol, sodium hydroxide (NaOH), aqueous ammonia, and chloroform came from Fisher Scientific. DAPI was purchased from Thermo Fisher Scientific. JC-1 kit, TUNNEL assay kit, and Cas3/7 detection kit were acquired from Elabscience, and iScript cDNA synthesis kit and SYBR green PCR master mix from Bio-Rad. The cell lines used for this work were procured from the NCCS Pune, India.Fabrication process of Si, PDMS NPs and PDMS–HA NPs
The synthesis of silica NPs and PDMS NPs was referred to in our previous study, with slight modifications. The PDMS NPs were synthesized using SCMS (solid core mesoporous shell) silica (Si) NPs for a template.74 The first step was SCMS silica NPs synthesis, which were prepared using the sol–gel method. Briefly, 74 ml of absolute ethanol was added to 10 ml DI water in a round bottom (RB) flask, and then 12.7 ml of aq. ammonia was added and mixed at 30 °C. Then, 5.9 ml of TEOS was rapidly added and stirred. After one hour, a blend of TEOS and TMS was added drop by drop to the solution for 20 minutes. The solution was then maintained idle for 1 h, and then the solvent was removed. Silica NPs obtained were maintained in an oven at 100 °C overnight and were then calcined at 550 °C for 5 hours to eliminate the porogen and generate the desired mesoporous silica NPs. Furthermore, for crosslinking, the base of the PDMS elastomer was blended with its linker in a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. It was thoroughly mixed for 5 minutes and degassed to drive out air bubbles. By adding THF, 100 mg ml−1 PDMS stock solution was prepared, and this solution was stirred for 90 minutes. The silica NPs (10 mg) used as a template were placed in a tube, and 2 ml of 10 mg ml−1 PDMS solution prepared from the stock solution was used. The mixture was sonicated for the next 10 minutes, and the mixture was maintained for 18 hours to adsorb and crosslink the PDMS polymer onto the silica NP template. The mixture was settled at 6500 rpm and washed with THF. The fabricated PDMS–Si NPs were treated at 100 °C. The silica NPs template was exterminated by the etching process; for this, a buffer was used with ammonium fluoride and HF (1:4) and then obtained NPs were DI-washed to eliminate any remnant of the etching buffer and allowed to dry at RT.
:1. It was thoroughly mixed for 5 minutes and degassed to drive out air bubbles. By adding THF, 100 mg ml−1 PDMS stock solution was prepared, and this solution was stirred for 90 minutes. The silica NPs (10 mg) used as a template were placed in a tube, and 2 ml of 10 mg ml−1 PDMS solution prepared from the stock solution was used. The mixture was sonicated for the next 10 minutes, and the mixture was maintained for 18 hours to adsorb and crosslink the PDMS polymer onto the silica NP template. The mixture was settled at 6500 rpm and washed with THF. The fabricated PDMS–Si NPs were treated at 100 °C. The silica NPs template was exterminated by the etching process; for this, a buffer was used with ammonium fluoride and HF (1:4) and then obtained NPs were DI-washed to eliminate any remnant of the etching buffer and allowed to dry at RT.
Drug loading
Furthermore, for sequential drug loading and modification (Scheme 1), quercetin (50 mg ml−1) was dissolved in DMSO:PBS (1:4), and the PDMS NPs (10 mg ml−1) were dispersed together and sonicated for 5 min. The solution was maintained on a shaker for the next 24 hours. After the pores were saturated with the loaded quercetin, the PDMS NPs were washed to remove the unbound quercetin. Then, the Que-loaded PDMS NPs were exposed to PEI, and finally, for the Dox loading, the PEI-modified NPs were maintained with the Dox–HA solution, hence facilitating the simultaneous binding of HA and entrapment of the Dox. They were then rinsed with PBS to remove unbound Dox and HA and then the prepared nanoformulations were lyophilized and stored at 4 °C till further use.
Characterization of PDMS–HA NPs
SEM and TEM analyzed the morphology of PDMS NPs. For the SEM characterization, both the NPs (1 mg) were mixed in 1 ml DI water and then maintained in a sonicator for 5 min to get dispersed NPs, and then ethanol-cleaned aluminium foil was used to drop-cast the sample (10 μl drop). Samples were dried in a vacuum desiccator for the next 1 h. For TEM examination, the sample (0.1 mg) was mixed in 1 ml DI water and maintained in a sonicator for 5 min for homogenized dispersion, and then the NP dispersion (15 μl) was drop-cast on a TEM grid. Then, it was allowed to dry for 24 hours under vacuum in a desiccator. The PDMS–HA NP size (hydrodynamic size) measurements and surface charges (zeta potential) were analyzed by a Malvern Zetasizer. Briefly, approx. 1 mg of NPs was dispersed in 2 ml of DI water through 1 min sonication, and then readings were obtained immediately. All the measurements were taken in triplicate. For FTIR analysis, different samples (PDMS–HA NPs and drug-loaded PDMS–HA NPs) were mixed with KBr.Encapsulation efficiency (% EE) and loading Dox and Que on PDMS–HA NPs
The Que and Dox % EE and % Loading capacity of PDMS–HA NPs were estimated using a UV spectrometer. This was evaluated by quantifying the OD values of the unentrapped Que and Dox at 415 nm and 480 nm, respectively. The Que (core) and Dox (shell) were separately loaded on the PDMS–HA NPs core–shell structure. Then this was centrifuged at 6000 rpm for 15 min to separate un-entrapped Dox and Que from the PDMS–HA NPs. The supernatant obtained through centrifugation comprises the unencapsulated Que and Dox, which were subsequently utilized to determine the % EE and % Loading.75The calculation of the % EE and % Loading was derived from the formulae provided below:
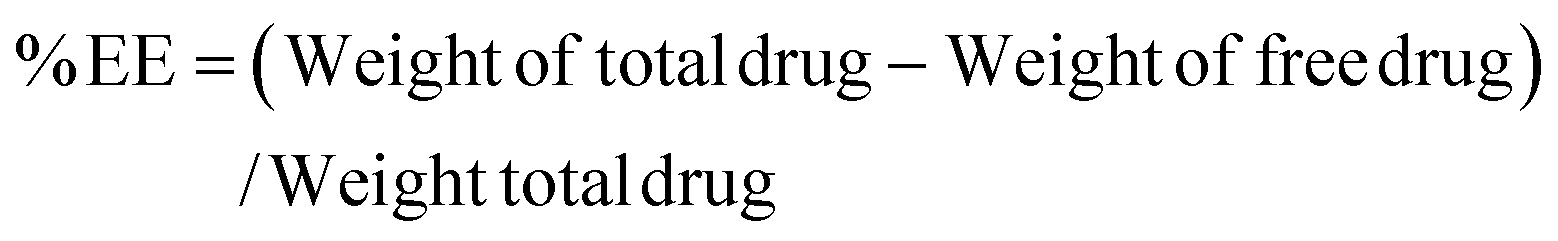
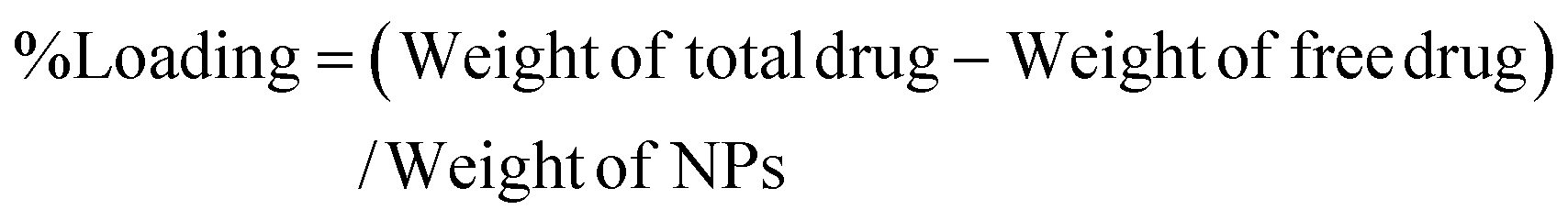
Drug release studies
To assess the drug (Que and Dox) release kinetics from PDMS–HA NPs, the dialysis method was employed. Drug release was checked in two conditions: first, only PBS, and second, PBS supplemented with hyaluronidase enzyme. Que–Dox–PDMS–HA NPs (10 mg) were dispersed in 100 ml PBS maintained at 37 °C for 24 h with mild stirring. At every few hours, 1 ml buffer from the solution was withdrawn, and an equal amount of buffer was supplemented back to the solution to maintain the volume. The amount of released Que and Dox was evaluated by UV-Vis spectrometry at 415 nm and 580 nm, respectively.In vitro cytotoxicity and cellular uptake
The influence of the PDMS–HA NPs on cell growth and viability was investigated by an MTT assay with MCF-7, HeLa, MDA-MB-231, HepG2, HaCaT, and NIH3T3 cells. Briefly, 5000 cells per well were seeded and incubated for cell attachment and morphology. The cells were exposed to PDMS–HA NPs (00–200 μg ml−1) for 24 h and 48 h. After incubation, MTT (0.5 mg ml−1) in basal media was added and this was maintained for the next 4–5 h; then DMSO was added and absorbance was recorded at 570.76For drug toxicity and IC50 evaluation, Que and Dox-loaded PDMS–HA NPs (0–200 μg ml−1) were exposed to MCF-7 for 24 h. Untreated cells, free Que, and Dox equivalents served as experimental controls. After incubation, MTT was added and this was maintained for 4–5 h, followed by DMSO treatment, and absorbance was recorded at 570. For each experimental group, dose-dependent cell proliferation was calculated, and then the IC50 values were determined by using proliferation curve GraphPad Prism software.
Different cell lines (MCF-7, MDA-MB-231, HeLa, and HaCaT) were utilized to study the HA-mediated uptake of PDMS–HA NPs. Cells were cultured in a 24-well plate with culture medium at 37 °C and 5% CO2. After cells had adhered and maintained their morphology, they were treated for 24 h with RITC-loaded PDMS NPs and RITC-loaded PDMS–HA NPs. Cells were harvested and directly taken to flow cytometry for further analysis.
Hemolytic study
The blood compatibility of PDMS–HA NPs was assessed through a hemolysis assay. Whole blood was collected and serum was separated from the red blood cells (RBC). Separated RBCs were diluted with PBS, and a time-dependent analysis was executed for PDMS–HA NPs (50, 100, and 200 μg ml−1). Each sample was dispersed in PBS and exposed to prediluted blood at 37 °C for 2, 4, and 6 h. Triton-X-100 served as positive control and PBS as negative control. This was further centrifuged at 1500 for 5 minutes and the supernatant with released hemoglobin was analyzed by UV–Vis spectrophotometry (541 nm).77 Furthermore, to determine the rate of hemolysis the following formula was used.S = sample, NC = negative control, PC = positive control.
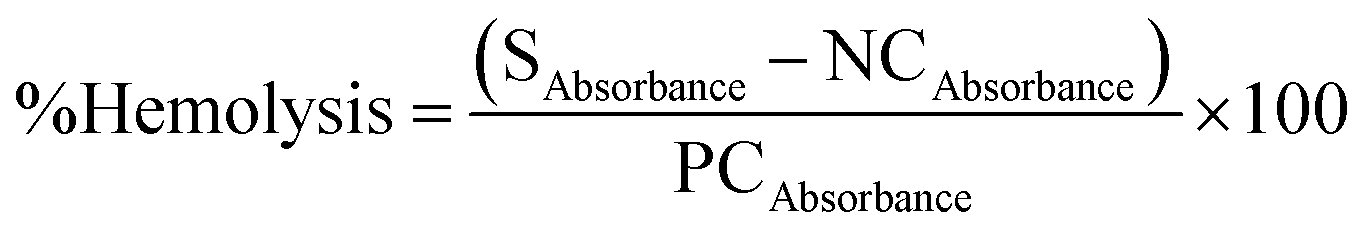
ROS detection
The intracellular ROS generation was evaluated by DCFDA assay with MCF-7 cells. Briefly, 5000 cells per well were seeded and incubated for cell attachment and morphology. The cells were exposed to PDMS–HA NPs (00–200 μg ml−1) for two different time points (24 and 48 h). After nanoparticle incubation, cells were exposed to 20 μM DCFH–DA for 45 min in the dark at 37 °C. ROS generation was examined by imaging the cells via a confocal microscope (Ex: 480 nm and Em: 525 nm).Immunostaining studies of P-gp expression levels in MCF-7 cells
The MCF-7 cells were seeded and incubated for cell attachment and morphology. Following a 24 hour treatment with PDMS–HA loaded quercetin, the cells were washed with PBS and treated with primary antibody (anti-P-gp) and incubated for 1 h; next the cells were washed and treated with the secondary antibody tagged with Alexa Fluor 488 and further incubated for 2 h. The samples were collected, and signal was measured in the green channel by flow cytometry.Cell cycle analysis
105 MCF-7 cells per well were taken and incubated for cell attachment and morphology. Following a 24 hour treatment with PDMS–HA loaded quercetin, the cells underwent two washes with PBS. Subsequently, they were ethanol (70%) fixed for one hour. The cells were then resuspended in PBS consisting of PI (20 mg ml−1) and RNase A (50 mg ml−1), followed by incubation at RT for 45 min. Subsequently, measured by flow cytometry, the fluorescence was represented in histograms.62Study of mitochondrial membrane potential (Δψm)
JC-1 stain was used to evaluate Δψm, and the methodology was carried out per the manufacturer's instructions. 105 MCF-7 cells were seeded, followed by incubation for 24 h with the Que–PDMS–HA NPs (10, 25, 50, 100 μg ml−1) and their free equivalents. The experimental control groups include a negative control (untreated cells) and positive control CCCP (50 μM, treated cells). Then, JC-1 dye was added and the cells were incubated in the dark for 30 minutes. In a polarized mitochondrial membrane, JC-1 forms an aggregate and hence emits red fluorescence (Ex/Em: 525/595 nm), whereas in an unpolarised mitochondrial membrane, JC-1 monomers were scattered due to damaged mitochondria and seen as green fluorescence (Ex/Em: 488/530 nm).105 MCF-7 cells were seeded and followed by incubation with Que–PDMS–HA NPs (10, 25, 50, 100 μg ml−1) and their free equivalents, followed by rhodamine 123 (10 μg ml−1) staining for 30 minutes. Following the incubation time, the cells underwent a PBS wash and were then subjected to analysis using a confocal microscope.
Apoptosis assay
The levels of apoptosis were assessed by annexin V-FITC and PI staining methods. The methodology was followed as described in the apoptosis detection kit. Briefly, MCF-7 cells were incubated for 24 h with drug-loaded PDMS–HA NPs (50, 100, 200 μg ml−1). Following the treatment, the cells were harvested and suspended in binding buffer (1×), and then untreated (control) and treated cells were exposed to Annexin V–FITC and PI stain for 30 min in the dark. Then, the cell suspension was taken for analysis by flow cytometry.77Apoptosis determination by DAPI staining
MCF-7 cells were seeded on a gelatin-coated glass coverslip and incubated for 24 h with drug-loaded PDMS–HA NPs (50, 100, 200 μg ml−1). This was followed by formaldehyde (4%) fixation, PBS washing, and DAPI staining for 20 min at 37 °C. Finally, the cells were fixed with 4% formaldehyde and examined using a confocal microscope.(AO/EtBr) double staining
The MCF-7 cells were used for the study, and 50000 cells per well cells were seeded. Then the cells were incubated for 24 h with drug-loaded PDMS–HA NPs (50 μg ml−1). The double-staining solution mixture was prepared (100 μg mL−1 AO and EtBr) and 5 μL was added in 50 μL cell suspension and the cells were further maintained for 5 min in the dark. The cells were studied using a confocal microscope.
Gene expression studies
MCF-7 cells were seeded and incubated with Que–Dox–PDMS–HA NPs for 24 h. After the incubation process, the cells were harvested, and their RNA was isolated using the TRIzol method. After checking the purity and concentration by a Nanodrop, cDNA was synthesized by RNA. Quantitative PCR was performed with the prepared cDNA. GAPDH was used as a housekeeping gene.3D spheroid model
The hanging droplet method prepared multicellular spheroids with MCF-7 and NIH3T3 cells. The cells were counted and grown in a flask, followed by trypsinization. The cells were harvested, and each cell type was diluted appropriately to maintain particular cell density in the spheroids. Furthermore, both cell suspensions were mixed seeded and cultured for 4 days. After carefully transferring the spheroids to untreated Petri plates, they were provided with fresh growth media to support their development. This was followed by the treatment of Que–Dox–PDMS–HA NPs, Que–PDMS–HA NPs, Dox–PDMS–HA NPs, and their free equivalents. For control groups, the spheroids were treated with bare PDMS–HA NPs. The treatment was maintained for different durations (1, 3, and 5). After incubation, at each time point the spheroids were washed and stained with FDA/PI and images were obtained by confocal microscopy to determine the live and dead cells.In vivo animal tumor model studies
The in vivo studies were conducted under the guidelines that were approved by the Institute Animal Ethical Committee (IAEC) of the Indian Institute of Technology Kanpur (Project Approval number IITK/IAEC/2020/1113) in compliance with the established regulations of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA).In vivo tumor regression study
The study used 7–8 weeks-old NOD SCID mice and were housed carefully in a pathogen-free animal facility. In vivo antitumor studies with Que–Dox–PDMS–HA NPs were conducted on MCF-7 xenografts in NOD SCID mice. The MCF-7 cells were added to Matrigel and a suspension of 107 cells was further injected subcutaneously into the mice. Once the tumor was developed and measured, as its size reached approximately 100 mm3, these animals were allotted to eight groups (4 animals per group) and were intravenously injected with PBS, bare PDMS–HA NPs, free Que, free Dox, free Que–Dox, Que–PDMS–HA NPs, PDMS–HA NPs, and Que–Dox–PDMS–HA NPs (100 mg per kg mice body weight) every three days. The volume of tumors was assessed, and the mice's weight was obtained every third day throughout the experimental period, to assess the efficacy and safety of the treatment. After completing the studies, the mice were euthanized, and the tumor was excised to measure and determine the regression. Additionally, the major tissues were collected to assess tissue-specific toxicity. The tumors that were taken out were measured, and sliced sections (10 μm) for further experiments.In vivo biodistribution and tumor targeting studies
To study the tissue retention and biodistribution of PDMS–HA NPs, the mice with MCF-7 xenograft tumors were allocated into three groups and kept separately in different cages. The mice were treated with intravenous injections of PDMS–HA NPs in 100 μl of PB and 100 mg kg−1. To study the effect of the dosage, after incubations of 12, 24 and 48 h from the injection, the animals were sacrificed by asphyxiation, liver, kidney, spleen, heart, lung, and muscle and tumor tissue were harvested, and blood was collected instantly after sacrificing the animal. To study the effect at the time points, the animals were divided into three groups (4 mice in each group) at each time point (12, 24 and 48 h). This biodistribution was done with ICPMS study. Briefly, tissues were dried at 80 °C in a hot air oven for 24 hours. The samples were treated with 6 ml of HNO3, 2 ml of HCl, and 2 ml of HF (3:1:1) at 120 °C for 24 h, followed by dilution with 2% HNO3. Si (silicon) quantification was done using a standard curve plotted at a range of 50 PPB to 500 PPB in 2% HNO3 with a R2 value of 0.999 using an ICPMS instrument.
For tumor-targeting studies, RITC-loaded PDMS–HA NPs were used which were injected intravenously into the mice in the MCF-7 xenograft tumor model. After different time points (12, 24 and 48 h) the tumor was harvested and processed and sliced sections (10 μm) were used for further experiments (IHC for CD44 marker) and imaging.
H&E staining and immunohistochemistry studies
The tumor was obtained and processed with 4% formaldehyde and a gradient of ethanol, to embed the tissues in paraffin and cut them into sections measuring 10 μm in thickness for H&E staining, IHC for Ki-67, CD44 and TUNEL assay. TUNEL was performed according to the instructions provided by the manufacturer, to evaluate cell proliferation and apoptosis in tumor tissues. Furthermore, the organs (heart, lungs, liver, kidneys, and spleen) were harvested and paraffin-embedded and then sectioned for H&E staining.Statistical analysis
Statistical analysis was done by GraphPad Prism 5.0 software using one-way analysis of variance (ANOVA). Data were statistically significant if *p < 0.05.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted at the Indian Institute of Technology, Kanpur (India). We acknowledge DST Nanomission for funding. We acknowledge the Department of Biotechnology, India, for funding the project (BT/PR21693/NNT/28/1180/20 and DBT-Sahaj BT/INF/22/SP54119/2024).References
- Y. Xiang, Q. Chen, Y. Nan, M. Liu, Z. Xiao, Y. Yang, J. Zhang, X. Ying, X. Long and S. Wang, Adv. Funct. Mater., 2024, 34, 2312092 CrossRef CAS.
- A. Sajid, H. Rahman and S. V. Ambudkar, Nat. Rev. Cancer, 2023, 23, 762–779 CrossRef CAS PubMed.
- A. K. Ghosh, A. Ghosh and P. K. Das, ACS Appl. Nano Mater., 2024, 7(5), 4946–4959 CrossRef CAS.
- Y. Loriot, G. Perlemuter, D. Malka, F. Penault-Lorca, V. Boige, E. Deutsch, C. Massard, J. P. Armand and J.-C. Soria, Nat. Clin. Pract. Oncol., 2008, 5, 268–278 CrossRef CAS.
- J. Herrmann, Nat. Rev. Cardiol., 2020, 17, 474–502 CrossRef CAS PubMed.
- J. Jeyabalan, F. Aqil, R. Munagala, L. Annamalai, M. V. Vadhanam and R. C. Gupta, J. Agric. Food Chem., 2014, 62, 3963–3971 CrossRef CAS PubMed.
- B. Rizeq, I. Gupta, J. Ilesanmi, M. AlSafran, M. D. M. Rahman and A. Ouhtit, J. Cancer, 2020, 11, 4521 CrossRef CAS PubMed.
- Y.-Q. Liu, X.-L. Wang, D.-H. He and Y.-X. Cheng, Phytomedicine, 2021, 80, 153402 CrossRef CAS PubMed.
- R. Jain, S. Shukla, N. Nema, R. Panday and H. S. Gour, J. Agric. Sci. Food Res., 2020, 11, 274 Search PubMed.
- N. Drinković, M. Beus, R. Barbir, Ž. Debeljak, B. Tariba Lovaković, N. Kalčec, M. Ćurlin, A. Bekavac, D. Gorup, I. Mamić, D. Mandić, V. Micek, P. Turčić, N. Günday-Türeli, E. Türeli and I. Vinković Vrček, Nanoscale, 2024, 16, 9412–9425 RSC.
- J. M. Estrela, S. Mena, E. Obrador, M. Benlloch, G. Castellano, R. Salvador and R. W. Dellinger, J. Med. Chem., 2017, 60, 9413–9436 CrossRef CAS.
- W. Li, Y. Guo, C. Zhang, R. Wu, A. Y. Yang, J. Gaspar and A.-N. T. Kong, Chem. Res. Toxicol., 2016, 29, 2071–2095 Search PubMed.
- R. P. Singh, S. Dhanalakshmi and R. Agarwal, Cell Cycle, 2002, 1, 155–160 CrossRef.
- A. P. Subramanian, S. K. Jaganathan, A. Manikandan, K. N. Pandiaraj, N. Gomathi and E. Supriyanto, RSC Adv., 2016, 6, 48294–48314 RSC.
- F.-L. Mi, L.-F. Wang, P.-Y. Chu, S.-L. Peng, C.-L. Feng, Y.-J. Lai, J.-N. Li and Y.-H. Lin, ACS Biomater. Sci. Eng., 2018, 4, 2847–2859 CrossRef CAS.
- H. Hu, Z. Liao, M. Xu, S. Wan, Y. Wu, W. Zou, J. Wu and Q. Fan, ACS Omega, 2023, 8, 976–986 CrossRef CAS PubMed.
- A. K. Jain, K. Thanki and S. Jain, Mol. Pharm., 2013, 10, 3459–3474 CrossRef CAS.
- S. Liu, R. Li, J. Qian, J. Sun, G. Li, J. Shen and Y. Xie, Mol. Pharm., 2020, 17, 1415–1427 CrossRef CAS.
- J. Fang, S. Zhang, X. Xue, X. Zhu, S. Song, B. Wang, L. Jiang, M. Qin, H. Liang and L. Gao, Int. J. Nanomed., 2018, 13, 5113–5126 CrossRef CAS.
- H. Zhou, Y. Yuan, Z. Wang, Z. Ren, M. Hu, J. Lu, H. Gao, C. Pan, W. Zhao and B. Zhu, Colloids Surf., A, 2023, 658, 130654 CrossRef CAS.
- S.-M. Tang, X.-T. Deng, J. Zhou, Q.-P. Li, X.-X. Ge and L. Miao, Biomed. Pharmacother., 2020, 121, 109604 CrossRef CAS PubMed.
- J. Pan, T. Wu, L. Chen, X. Chen, C. Zhang, Y. Wang, H. Li, J. Guo and W. Jiang, Nanoscale, 2024, 16, 2955–2965 RSC.
- T. Nabekura, T. Yamaki, T. Hiroi, K. Ueno and S. Kitagawa, Pharmacol. Res., 2010, 61, 259–263 CrossRef CAS.
- P. Limtrakul, O. Khantamat and K. Pintha, J. Chemother., 2005, 17, 86–95 CrossRef CAS PubMed.
- G. Scambia, F. O. Ranelletti, P. B. Panici, R. De Vincenzo, G. Bonanno, G. Ferrandina, M. Piantelli, S. Bussa, C. Rumi, M. Cianfriglia and S. Mancuso, Cancer Chemother. Pharmacol., 1994, 34, 459–464 CrossRef CAS.
- M. B. Wolf and J. W. Baynes, Biochim. Biophys. Acta, 2006, 1760, 267–271 CrossRef CAS.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- N. Murali, S. K. Rainu, A. Sharma, S. Siddhanta, N. Singh and S. Betal, ACS Omega, 2024, 9(26), 28937–28950 CrossRef CAS.
- K. Bhattacharjee and B. L. V. Prasad, Chem. Soc. Rev., 2023, 52, 4121 RSC.
- R. Lagoa, J. Silva, J. R. Rodrigues and A. Bishayee, Biotechnol. Adv., 2020, 38, 107382 CrossRef CAS.
- Y.-L. Liu, T.-H. Wang, N.-T. Yeh, W.-J. Huang, B.-S. Tzang, I.-T. Wu, H.-Y. Chin, S.-H. Hu, T.-C. Hsu and W.-H. Chiang, Nanoscale, 2024, 16, 1415–1427 RSC.
- M. Ahmadi, C. A. Ritter, T. von Woedtke, S. Bekeschus and K. Wende, Chem. Sci., 2024, 15, 1966–2006 RSC.
- F. Dai, F. Chen, J. Zhang, X. Chen, H. Liang, Z. Liang, S. Zhang, H. Tan and L. Zhao, ACS Appl. Nano Mater., 2024, 7, 7289–7299 CrossRef CAS.
- B. Xiao, X. Si, M. K. Han, E. Viennois, M. Zhang and D. Merlin, J. Mater. Chem. B, 2015, 3, 7724–7733 RSC.
- J. E. Ferguson and R. A. Orlando, J. Med. Food, 2015, 18, 497–502 CrossRef CAS.
- M. Liu, M. Fu, X. Yang, G. Jia, X. Shi, J. Ji, X. Liu and G. Zhai, Colloids Surf., B, 2020, 196, 111284 Search PubMed.
- B. A. Baldo and N. H. Pham, Cancer Metastasis Rev., 2013, 32, 723–761 CrossRef CAS PubMed.
- Q. Gao, J. Feng, W. Liu, C. Wen, Y. Wu, Q. Liao, L. Zou, X. Sui, T. Xie, J. Zhang and Y. Hu, Adv. Drug Delivery Rev., 2022, 188, 114445 CrossRef CAS.
- C.-M. J. Hu and L. Zhang, Biochem. Pharmacol., 2012, 83, 1104–1111 CrossRef CAS PubMed.
- J. Lee, J. Kim, M. Jeong, H. Lee, U. Goh, H. Kim, B. Kim and J.-H. Park, Nano Lett., 2015, 15, 2938–2944 CrossRef CAS PubMed.
- D. Hwang, J. D. Ramsey and A. V. Kabanov, Adv. Drug Delivery Rev., 2020, 156, 80–118 CrossRef CAS PubMed.
- D. Pooja, T. S. Reddy, H. Kulhari, A. Kadari, D. J. Adams, V. Bansal and R. Sistla, Eur. J. Pharm. Biopharm., 2020, 154, 377–386 CrossRef CAS.
- H. Y. Huang, A. Skripka, L. Zaroubi, B. L. Findlay, F. Vetrone, C. Skinner, J. K. Oh and L. A. Cuccia, ACS Appl. Bio Mater., 2020, 3, 7219–7227 CrossRef CAS.
- A. K. Maparu, P. Singh, B. Rai, A. Sharma and S. Sivakumar, Nanotechnology, 2022, 33, 495102 CrossRef CAS.
- A. K. Maparu, P. Singh, B. Rai, A. Sharma and S. Sivakumar, Mater. Sci. Eng., C, 2021, 119, 111577 CrossRef CAS PubMed.
- G. Mishra, S. Bhattacharyya, V. Bhatia, B. Ateeq, A. Sharma and S. Sivakumar, ACS Appl. Mater. Interfaces, 2019, 11, 39395 CrossRef CAS.
- T. H. Tran, J. Y. Choi, T. Ramasamy, D. H. Truong, C. N. Nguyen, H.-G. Choi, C. S. Yong and J. O. Kim, Carbohydr. Polym., 2014, 114, 407–415 CrossRef CAS PubMed.
- Y. Zhang, K. Wu, H. Sun, J. Zhang, J. Yuan and Z. Zhong, ACS Appl. Mater. Interfaces, 2018, 10, 1597–1604 CrossRef CAS PubMed.
- T. L. Nascimento, H. Hillaireau, M. Noiray, C. Bourgaux, S. Arpicco, G. Pehau-Arnaudet, M. Taverna, D. Cosco, N. Tsapis and E. Fattal, Langmuir, 2015, 31, 11186–11194 CrossRef CAS PubMed.
- S. Agrawal, M. Dwivedi, H. Ahmad, S. B. Chadchan, A. Arya, R. Sikandar, S. Kaushik, K. Mitra, R. K. Jha and A. K. Dwivedi, Nanomedicine, 2018, 14, 327–337 CrossRef CAS PubMed.
- B. Xiao, M. K. Han, E. Viennois, L. Wang, M. Zhang, X. Si and D. Merlin, Nanoscale, 2015, 7, 17745–17755 RSC.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS.
- Y. Zhang, M. Dai and Z. Yuan, Anal. Methods, 2018, 10, 4625–4638 RSC.
- T. Popescu, C. O. Matei, I. D. Vlaicu, I. Tivig, A. C. Kuncser, M. Stefan, D. Ghica, L. C. Miclea, T. Savopol and D. C. Culita, Sci. Rep., 2020, 10, 18062 CrossRef CAS.
- H. Yin, P. S. Casey, M. J. McCall and M. Fenech, Langmuir, 2010, 26, 15399–15408 CrossRef CAS PubMed.
- S. Mukhopadhyay, H. Veroniaina, T. Chimombe, L. Han, W. Zhenghong and Q. Xiaole, RSC Adv., 2019, 9, 35566–35578 RSC.
- L. Chen, J. J. Glass, R. De Rose, C. Sperling, S. J. Kent, Z. H. Houston, N. L. Fletcher, B. E. Rolfe and K. J. Thurecht, ACS Appl. Bio Mater., 2018, 1, 756–767 CrossRef CAS PubMed.
- Y. Han, X. Wang, H. Dai and S. Li, ACS Appl. Mater. Interfaces, 2012, 4, 4616–4622 CrossRef CAS.
- M. A. Agotegaray, A. E. Campelo, R. D. Zysler, F. Gumilar, C. Bras, A. Gandini, A. Minetti, V. L. Massheimer and V. L. Lassalle, Biomater. Sci., 2017, 5, 772–783 RSC.
- T. Lang, Y. Liu, Z. Zheng, W. Ran, Y. Zhai, Q. Yin, P. Zhang and Y. Li, Adv. Mater., 2019, 31, 1806202 CrossRef PubMed.
- T. Waldman, Y. Zhang, L. Dillehay, J. Yu, K. Kinzler, B. Vogelstein and J. Williams, Nat. Med., 1997, 3, 1034–1036 CrossRef CAS PubMed.
- L. Ren, W. Li, X. Zheng, J. Liu, Y. Yang, S. Li, S. Zhang, W. Fu, B. Xiao and J. Wang, Acta Pharmacol. Sin., 2022, 43, 194–208 CrossRef CAS.
- L. Xiong, N. Deng, B. Zheng, T. Li and R. H. Liu, Food Funct., 2021, 12, 6513–6525 RSC.
- Y. Zhao, K. Wang, Y. Zheng, X. Zeng, Y. C. Lim and T. Liu, Front. Chem., 2021, 8, 601649 CrossRef.
- X. Zhang, N. Tang, T. J. Hadden and A. K. Rishi, Biochim. Biophys. Acta, 2011, 1813, 1978–1986 CrossRef CAS PubMed.
- Y. Furukawa-Hibi, Y. Kobayashi, C. Chen and N. Motoyama, Antioxid. Redox Signaling, 2005, 7, 752–760 CrossRef CAS.
- X. Shi, Y. Cao, H. Wang, Q. Zhao, C. Yan, S. Li and L. Jing, J. Cardiovasc. Transl. Res., 2024, 1–17 Search PubMed.
- L. Xu, Y. Cao, Y. Xu, R. Li and X. Xu, Macromol. Biosci, 2024, 24, 2300238 CrossRef CAS PubMed.
- D. A. Ang, J.-M. Carter, K. Deka, J. H. L. Tan, J. Zhou, Q. Chen, W. J. Chng, N. Harmston and Y. Li, Nat. Commun., 2024, 15, 2513 CrossRef CAS.
- Q. He, S. Huang, S. Xu and L. Wang, RSC Adv., 2015, 5, 43148–43154 RSC.
- Y. Hailing, L. Xiufang, W. Lili, L. Baoqiang, H. Kaichen, H. Yongquan, Z. Qianqian, M. Chaoming, R. Xiaoshuai, Z. Rui, L. Hui, P. Pengfei and S. Hong, Nanoscale, 2020, 12, 17222–17237 RSC.
- P. Soltantabar, E. L. Calubaquib, E. Mostafavi, M. C. Biewer and M. C. Stefan, Biomacromolecules, 2020, 21, 1427–1436 CrossRef CAS PubMed.
- K. Hu, L. Miao, T. J. Goodwin, J. Li, Q. Liu and L. Huang, ACS Nano, 2017, 11, 4916–4925 CrossRef CAS PubMed.
- B. Mahaling, M. Verma, G. Mishra, S. Chaudhuri, D. Dutta and S. Sivakumar, Nanotoxicology, 2020, 4(5), 577–594 CrossRef.
- S. M. Motevalli, A. S. Eltahan, L. Liu, A. Magrini, N. Rosato, W. Guo and M. Bottini, Biophys. Rep., 2019, 5, 19–30 CrossRef CAS.
- H. Shin, M. Kwak and T. G. Lee, Nanoscale, 2020, 2020(12), 15743–15751 RSC.
- A. Bhattacharjee, A. Gupta, M. Verma, P. A. Murugan, P. Sengupta, S. Matheshwaran, I. Manna and K. Balani, Ceram. Int., 2019, 45, 12225–12233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03040k |
| This journal is © The Royal Society of Chemistry 2025 |