
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing DFT and machine learning for accurate optical gap prediction in conjugated polymers†
Bin
Liu‡
ab,
Yunrui
Yan‡
ab and
Mingjie
Liu
 *ab
*ab
aDepartment of Chemistry, University of Florida, Gainesville, FL 32611, USA. E-mail: mingjieliu@ufl.edu
bQuantum Theory Project, University of Florida, Gainesville, FL 32611, USA
First published on 18th February 2025
Abstract
Conjugated polymers (CPs), characterized by alternating σ and π bonds, have attracted significant attention for their diverse structures and adjustable electronic properties. However, predicting the optical band gap (Eexpgap) of CPs remains challenging. This study presents a rational model that integrates density functional theory (DFT) calculation with a data-driven machine learning (ML) approach to predict the experimentally measured Eexpgap of CPs, using 1096 data points. Through alkyl side chain truncation and conjugated backbone extension, the modified oligomers effectively capture the electronic properties of CPs, significantly improving the correlation between the DFT-calculated HOMO–LUMO gap (Eoligomergap) and Eexpgap (R2 = 0.51) compared to the unmodified side-chain-containing monomers (R2 = 0.15). Moreover, we trained six ML models with two categories of features as input: Eoligomergap to represent the extended backbone and molecular features of unmodified monomers to capture the alkyl-side-chain effect. The best model, XGBoost-2, achieved an R2 of 0.77 and an MAE of 0.065 eV for predicting Eexpgap, falling within the experimental error margin of ∼0.1 eV. We further validated XGBoost-2 on a dataset of 227 newly synthesized CPs collected from literature without further retraining. Notably, XGBoost-2 exhibits both excellent interpolation for BT-, BTA-, QA-, DPP-, and TPD-based CPs, and exceptional extrapolation for PDI-, NDI-, DTBT-, BBX-, and Y6-based CPs, which are attributed to the integration of DFT methods with rationally designed oligomer structures. For the first time, we demonstrated a novel and effective strategy combining quantum chemistry calculations with ML modeling for accurate and efficient prediction of experimentally measured fundamental properties of CPs. Our study paves the way for the accelerated design and development of high-performance CPs in photoelectronic applications.
 Mingjie Liu | Dr. Mingjie Liu received her Ph.D. in Materials Science and Nanoengineering from Rice University. After doing a postdoc at Brookhaven National Laboratory and Massachusetts Institute of Technology, she joined University of Florida in 2022, as an Assistant Professor in Chemistry. Dr. Liu's research interests center on leveraging materials informatics, data-driven methodologies and generative AI to advance high-impact research in the realms of materials design and discovery. |
1 Introduction
Conjugated polymers (CPs) are organic macromolecules composed of electron donor and electron acceptor units linked by carbon–carbon bonds. The alternation of σ and π bonds along the backbone chain of CPs enables the delocalization of π-electrons, forming a semiconductor band structure and thus endowing CPs with exceptional optical and electronic properties.1–3 These characteristics can be effectively tuned through a variety of material engineering strategies, such as the combination of various electron donator and electron acceptor aromatic units,4,5 halogenation,6 the introduction of non-covalent intra- and inter-molecular interactions,7,8 and modifications to alkyl side chains.9 Owing to their structural diversity, facile synthesis, ease of chemical modification and functionalization, excellent photo-physical properties, and relatively low cost, CPs have been extensively explored for a wide range of applications in optoelectronic devices, electrochemical sensors and transistors, drug delivery systems and bio-medical applications.10–13 So far, hundreds of thousands of CPs are available, but the scientific community predominantly relies on a laborious trial-and-error approach for the discovery, design, and optimization of CP materials, resulting in a substantial number of unexplored structures. The relationships between the structures of CP materials and their electronic properties are complex and not well understood.The optical band gap is one of the most essential electronic properties of CP materials for their use in photonic and electronic devices, such as organic solar cells (OSCs), organic light-emitting diodes, and organic field-effect transistors.14 Quantum chemistry simulations, particularly Density Functional Theory (DFT) and Time-Dependent DFT (TDDFT), are indispensable in polymer science for predicting and rationalizing the properties of polymeric materials.15,16 These methods offer insights into molecular properties across both ground and excited states and facilitate the prediction of optical gaps. While these simulations can handle sizeable molecular systems efficiently, the correlation between experimental measurements and DFT/TDDFT calculated values often remains weak for several reasons.17,18 The optical band gap is the energy required for a photon to excite an electron from the ground state to the first excited state, typically measured using UV-Vis absorption or photoluminescence spectroscopy. These measurements incorporate all physical effects presented in the system, such as solvent effects, vibronic coupling, and other fine details of the electronic structure. In contrast, the DFT-calculated HOMO–LUMO gap measures the energy required to move an electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), ignoring the coulombic interactions between the excited electron and the hole, which are involved in the optical band gap. Also, the HOMO–LUMO gap represents a vertical electronic transition and misses the relaxation transition due to interactions of the excited states with the surrounding environment. On the other hand, the TDDFT method calculates excited state energies based on the time-dependent response of the electronic system to an external perturbation. The lowest excited state in TDDFT corresponds to the energy required to promote an electron from the ground state to the first excited state, including excitonic effects and accounting for the dynamic response of the electrons. This provides a more accurate description of excitation energies compared to static DFT. The accuracy of both DFT and TDDFT depends on the choice of exchange–correlation (xc) functional, with hybrid functionals generally yielding better results. However, discrepancies can still arise due to functional approximations and the lack of consideration for experimental conditions. Moreover, higher-level quantum mechanical theories, such as GW method coupled with Bethe-Salpeter equation, which might offer improved accuracy, are impractical for large systems such as CPs due to their computational demands.19
Data-driven machine learning (ML) approaches are powerful tools for the rapid property predictions and virtual structure screening of organic molecules and CP materials, offering substantial time and cost advantages over traditional experimental and computational methods.17,20–22 The effectiveness of ML models depends crucially on the availability of adequate and reliable training data, as well as the selection of appropriate descriptors that capture the structural and physicochemical properties of CPs. These descriptors include topological, electronic, geometrical, and molecular fragment attributes.23,24 Previous studies indicate that different ML algorithms trained with identical descriptors often yield similar accuracy. However, descriptor selection significantly impacts model performance, underscoring its dominant role in determining prediction accuracy.25 So far, ML methods are increasingly used to predict the electronic properties of CPs and their derivatives, with most studies focusing on small molecules. The unique complexities of CP systems remain less explored, resulting in a scarcity of robust models that accurately reflect the behavior of these polymers. Additionally, many studies utilize DFT-calculated HOMO–LUMO gaps (EDFTgap) as reference data, which are generally less correlated with experimentally measured optical gaps (Eexpgap) of CPs. Some studies incorporate Eexpgap data from small CP datasets,26 but these models generally show low performance and debatable robustness and transferability. Furthermore, due to the absence of underlying physical principles and the use of obscure descriptors, even well-trained ML models for interpolation struggle with robust performance when extrapolating to new CP design spaces.20 To the best of our knowledge, there remains a significant gap in the development of well-established ML models that can predict the experimentally measured optical gap of CPs with high accuracy and transferability.
In this study, we developed a sophisticated approach that combines DFT calculations and data-driven ML models to accurately predict the Eexpgap values of CPs. We demonstrated that by modifying oligomer structures—specifically, removing alkyl side chains and extending conjugated backbones—we can effectively capture the electronic properties of CPs. This modification significantly improved the correlation between EDFTgap and Eexpgap, achieving an R2 value of 0.51, while also considerably reducing computational time consumption. In contrast, unmodified monomer structures yielded a notably low R2 value of 0.15. To further enhance the prediction accuracy of Eexpgap, we trained a variety of ML models using both EDFTgap and conventional molecular representations as inputs. Compared to the baseline model trained with only molecular representations, incorporating EDFTgap of modified oligomers not only improves prediction accuracy—reflected by an R2 of 0.77 and a mean absolute error (MAE) of 0.065 eV achieved by XGBoost—but also enhances the models’ transferability in predicting the optical gaps of new polymers outside the design space of the training dataset. Our work outlines a rational strategy for predicting fundamental properties of polymers by segmenting them into different substructures. These substructures are then characterized using different levels of theoretical or methodological approaches based on how well they correlate with the target properties. This methodological framework provides a robust basis for enhancing the predictive capabilities of computational models in polymer science.
2 Computational details
2.1 Dataset
The original dataset with 1203 data points was adopted from Saeki et al.'s work,27 in which experimentally measured data of synthesized polymers for OSC applications were manually collected from 503 literatures. For each polymer, the simplified molecular input line entry system (SMILES) string of its repeating unit was provided, together with a list of experimental parameters, including HOMO, LUMO, and Eexpgap. We removed 88 duplicate entries based on the SMILES strings and 18 non-conjugated polymer structures containing sp3-hybridized N atom along backbone chain (see Fig. S1†). An extra polymer containing Tellurium atoms was also excluded due to being out of the applicable range for the 6-31G* basis set in DFT calculations. Therefore, the final dataset comprised 1096 unique CPs. The distribution plots and statistical analysis of HOMO, LUMO, and Eexpgap values are presented in Fig. S2 and Table S1.†2.2 DFT calculations
All the DFT and TDDFT calculations were performed with Gaussian 16 package.28 The B3LYP hybrid functional29–31 together with D3 dispersion correction32 and 6-31G* basis set were employed for both geometry optimization and electronic property calculations. The maximum force tolerance is 0.02 eV Å−1. The initial xyz coordinates of polymers were generated from the SMILES strings with OpenBabel package.33 Before geometry optimization with DFT, we manually adjusted the oligomer backbone to be coplanar using the Avogadro package34 to more closely resemble a realistic configuration, as high planarity is favored in experiments to promote the performance of polymer-based electronic devices.352.3 Molecular features
The chemical structures of CPs were represented with SMILES strings. RDKit library36 was used to convert SMILES strings into three types of molecular features (MFs), including RDKit Descriptor,37 molecular access system (MACCS),38 and extended connectivity fingerprints (ECPF6).39 RDKit Descriptor consists of the 209 molecular properties calculated by RDKit package, covering structural connectivity, geometry, electronic properties, and chemical composition. MACCS is a pre-defined fragment library with a subset of 166 keys which counts the presence of 166 various chemical fragments, such as S–N and alkaline metal, whereas one extra key with zero value is added as a consequence of Python's array-indexing-by-zero convention, resulting in a 167-bit vector. ECFP6, a flavor of Morgan fingerprints, considers the neighboring connectivity of atoms with 1024 keys, which was generated by selecting the maximum diameter of the circular atom neighborhood to be six. We performed feature selection on 209 RDKit descriptors to eliminate irrelevant or redundant features, with the details presented in Note S1.†2.4 Machine learning models
We employed six conventional ML algorithms: Hist Gradient Boosting regression (HGBR),40 Gradient Boosting Regression (GBR),41 LightGBM regression (LGBM),42 Extreme Gradient Boosting regression (XGBoost),43 AdaBoost regression (AdaBoost),44 random forest (RF).45 These models are widely used in materials science and chemistry to uncover structure–property relationships.46,47 ML model training was performed with Scikit-learn library.48 The details of the training process can be found in Note S2.† Performance metrics, including coefficient of determination (R2), Pearson correlation coefficient (r), root mean square error (RMSE), and mean absolute error (MAE), were defined in Note S3.† The comparison of six machine learning models can be found in Note S8.†3 Results and discussion
3.1 Rational design of oligomer model
CPs adopt a one-dimensional periodic structure, consisting of a conjugated backbone and various lengthy alkyl side chains. This periodicity and the lengthy side chains present significant challenges in modeling CP systems accurately and efficiently. CPs feature an extended backbone, but their poor crystallization results in an ill-defined lattice, complicating the construction of a periodic model for simulation. Furthermore, using a monomer—a single repeating unit—to represent a CP, fails to effectively capture the characteristics of π-electron delocalization in CP structures. Here, we employed monomer structures to calculate the EDFTgap values at the B3LYP level, denoted as Emonomergap. As shown in Fig. 1b, Emonomergap show no linear relationship with Eexpgap of the corresponding CPs, as evidenced by a markedly low R2 value of 0.15. This weak correlation is attributed to both the intrinsic limitations of the DFT method for predicting optical gaps16,49 and the inadequacy of monomer models in accurately representing the properties of CPs.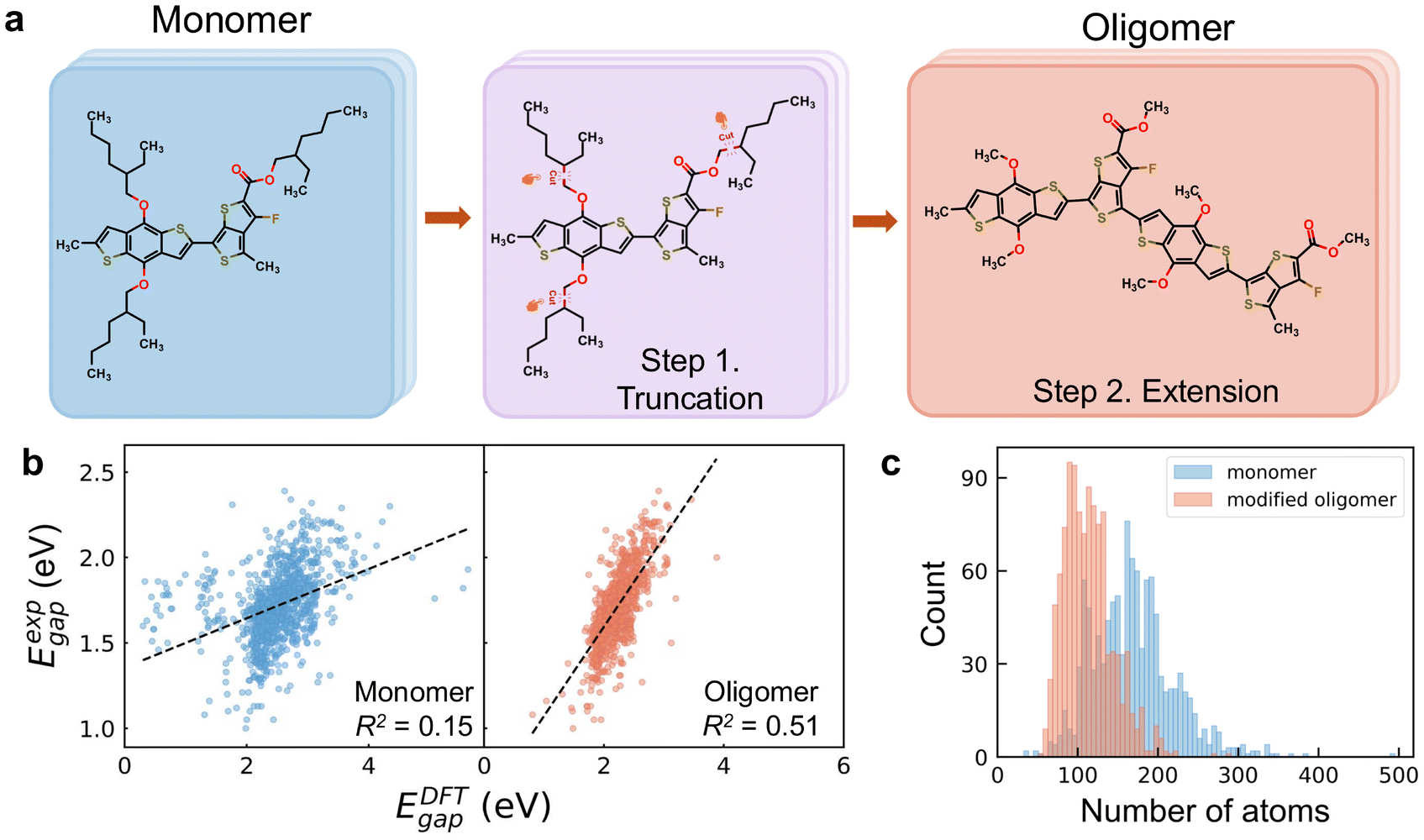 | ||
| Fig. 1 (a) Scheme of converting the monomer structure of PTB7 into a modified oligomer with a two-step procedure, namely, alkyl side chain truncation and conjugated backbone extension. (b) The parity plots of DFT calculated HOMO–LUMO gaps (EDFTgap) based on monomer and modified oligomer structures versus experimentally measured optical gaps (Eexpgap). The black dashed lines correspond to linear fitting. (c) The distributions of atom counts in the monomers and modified oligomers. | ||
In this study, we rationally designed an oligomer model to represent CP materials based on their fundamental characteristics. The two-step procedure to construct the oligomer structure is depicted in Fig. 1a with polymer PTB7 as an example. In the first step, we replaced the long alkyl side chains with methyl groups. While alkyl side chains are critical for influencing aggregation and morphology in thin films, which in turn would impact the electronic properties of the polymer, their primary role in single-molecule calculations is to affect solubility and steric effects. This substitution simplifies the computational model while retaining the electronic properties of the polymer backbone.50,51 As the conjugated backbone contributes most to the electronic properties of CPs, following side chain truncation, we replicated the monomer to form an oligomer structure, such as dimer or trimer. Previous studies suggested that using oligomer structures—such as a dimer, trimer, or tetramer, which extends the conjugated backbone chains to capture the characteristic of π-electron delocalization—enhances the predictive accuracy of DFT methods for experimental gaps, compared to employing monomer.52 To determine the optimal number of monomers repeating units for constructing the oligomer, we applied two key principles: first, preserving the inherent charge-transfer characteristics, such as donor–acceptor alternation; and second, ensuring a sufficiently long conjugated backbone to achieve convergence of the electronic properties. Given the limited understanding of the correlation between the conjugation length of the backbone chain and the electronic properties of CPs, we performed systematic convergence tests using four conjugated polymers. Based on the results, we propose the following two guidelines(see Note S4† for details): (1) the oligomer should contain at least four aromatic blocks linked by C–C single bonds along the backbone chain; (2) the oligomer should consist of at least six aromatic rings. Additionally, after side chain truncation, the obtained monomer can be regarded as an oligomer if it simultaneously contains more than four aromatic blocks and more than eight aromatic rings. It should be noted that all oligomers discussed in this study refer to these modified oligomer structures.
To validate the effectiveness of the simplified oligomer in capturing the electronic properties of CPs in comparison with the monomer, we also calculated the EDFTgap values with oligomers at the B3LYP level, denoted as Eoligomergap. Other xc functionals, including PBE,53 ωB97XD,54 and CAM-B3LYP,55 exhibit high linear correlations with B3LYP for HOMO–LUMO gap calculations, achieving Pearson correlation coefficients above 0.96 among each other (see Note S5†). As shown in Fig. 1b, the modified oligomers exhibit a significant improvement in correlating DFT-calculated and experimental gap values compared to the monomers, with the R2 value increasing from 0.15 to 0.51. While the moderate correlation (R2 = 0.51) indicates potential deviations between experimental and computational values, the substantial enhancement underscores the importance of selecting appropriate configurations to accurately represent the fundamental characteristics of CP materials. Moreover, the moderate correlation suggests that incorporating additional features beyond DFT-calculated HOMO–LUMO gaps is crucial to addressing these deviations and enhancing predictive accuracy. On the other hand, our results suggest that ML models trained with DFT-calculated HOMO–LUMO gaps of monomers as reference data may not effectively predict experimental optical gaps of CPs due to the poor correlation observed.
Besides accuracy improvement, our two-step simplification procedure also significantly reduces computational demands by decreasing the number of atoms in CPs. Fig. 1c illustrates the histogram distributions of atom counts for monomer and oligomer structures. Around 80% of the monomers, originally with atom counts ranging from 107 to 232, were reduced to between 78 and 156 atoms. Particularly, the largest monomer, which contains 494 atoms, is reduced to 200 atoms in its oligomer form, while the smallest with 33 atoms increases to 68 in the oligomer. This reduction is primarily achieved through the truncation of long alkyl side chains, while the increase in the number of atoms is due to the extension of the backbone chains. These two modifications synergistically lead to an overall decrease in system size for the majority of CPs.
CPs are composed of electron donor and acceptor units as building blocks linked by C–C single bonds. These donor and acceptor units are crucial for tuning the electronic properties, particularly the optical band gap.56–58 Donor units donate electron density to the polymer backbone, raising the HOMO level, while acceptor units withdraw electron density, lowering the LUMO level. Thus, combining donor and acceptor units creates a push–pull effect, significantly narrowing the band gap and enhancing charge transport. By strategically incorporating donor and acceptor units into the polymer backbone, a variety of CPs can be designed with tailored electronic properties for specific applications. Given the importance of donor and acceptor units in determining the experimental optical gap, we aimed to investigate the effectiveness of our oligomer model in capturing the electronic properties of various donor and acceptor units. We categorized the 1096 CPs in our dataset based on donor or acceptor unit types.
Fig. 2c and f show the chemical structures of four commonly used donor and acceptor units. The categorization follows a specific order from D1 to D4, with polymers containing multiple donor types assigned to the latter type in the search order (e.g., D4 if containing both D1 and D4). D1 represents benzodithiophene and its derivatives with S atoms replaced by O and Se. D2, D3, and D4 represent carbazole, dithieno[3,2-b:2′,3′-d]pyrrole, and pyrroloindacenodithiophene, respectively, along with their derivatives where N is substituted by C, Si, O, S, and Se.56 Polymers lacking these donor units are grouped as “others”. The same approach was applied for acceptor units. A1 represents the benzazole series, encompassing benzothiadiazole (BT), benzotriazole (BTA), benzoxazole, and related derivatives.57 A2, A3, and A4 denote diketopyrrolo[3,4-c]-pyrrole-1,4-dione (DPP), quinoxaline (QA), and thieno[3,4-c]pyrrole-4,6-dione (TPD), respectively.56 Based on the percentage distributions (see Table S8†), D1 and A1 are the predominant donor and acceptor units, with ratios of 46.7% and 32.8%, respectively. As shown in Fig. 2b and e, oligomers in each CP group exhibited significantly improved R2 values compared to monomers (Fig. 2a and d), reinforcing the widespread efficacy of our two-step approach in capturing the electronic properties of copolymers via modified oligomer structures, regardless of the specific donor and acceptor types.
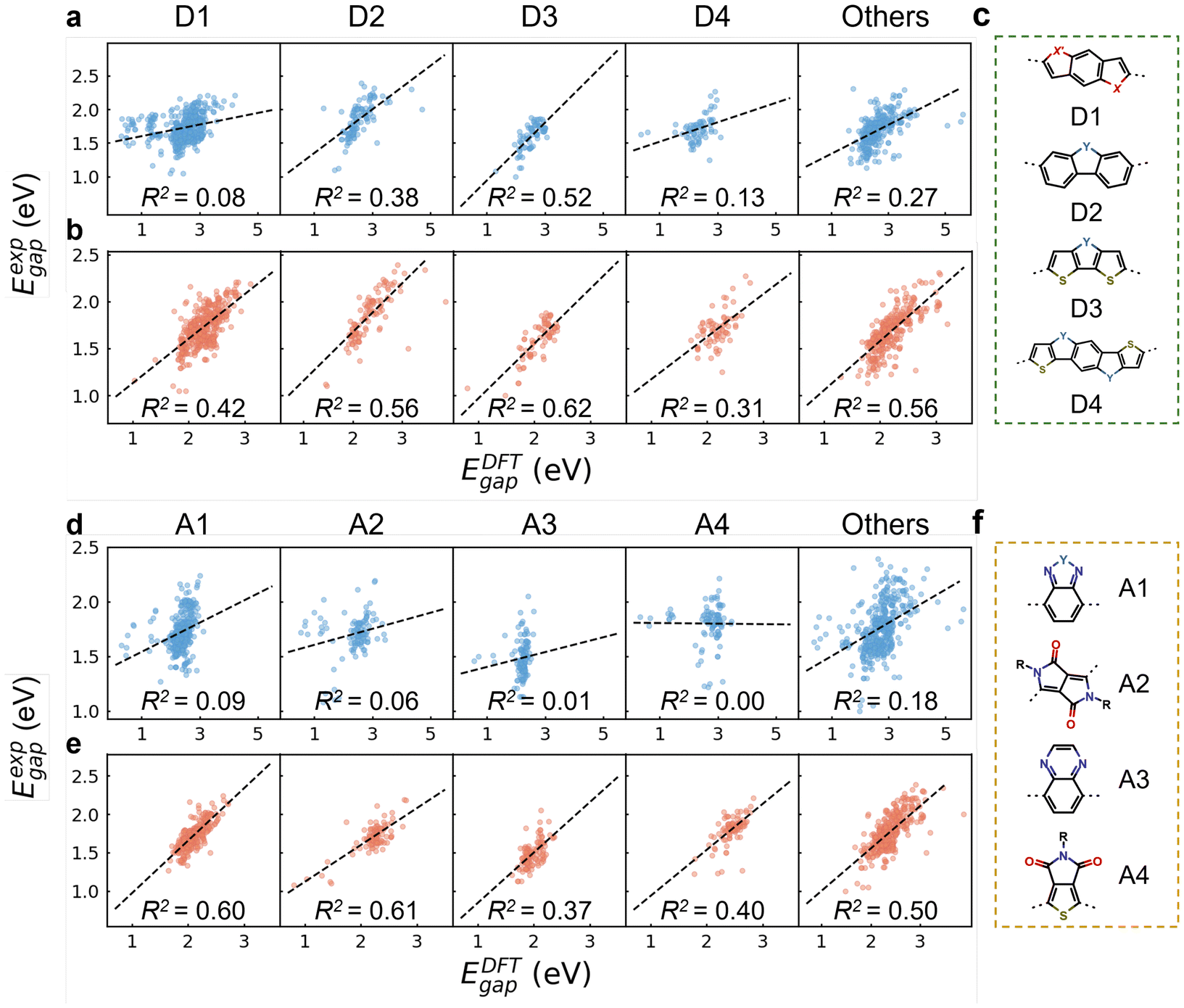 | ||
| Fig. 2 The linear correlation between DFT-calculated HOMO–LUMO gaps (EDFTgap) and experimental optical gaps (Eexpgap) for different groups of conjugated polymers categorized based on donor and acceptor units, respectively. The EDFTgap is calculated from (a and d) monomers with alkyl side chains and (b and e) modified oligomers after two-step procedure shown in Fig. 1a. The black dashed lines correspond to linear fitting. (c and f) The chemical structures of four donor and acceptor units. X or X′ denotes O, S, or Se atom, and Y represents C, N, O, Si, S, or Se atom. | ||
3.2 Effect of alkyl side chains
As demonstrated above, our two-step procedure for oligomer model construction through side chain truncation and backbone extension significantly improves the linear correlation between EDFTgap and Eexpgap, increasing the R2 value from 0.15 to 0.51 (see Fig. 1b). Particularly, side chain truncation largely reduces computational cost, which is beneficial for high-throughput screening. Although less impactful on electronic properties than the conjugated backbone, alkyl side chains still require consideration in order to further improve the prediction accuracy of experimentally measured optical gaps. Consequently, we applied two categories of descriptors for ML modeling: DFT calculated HOMO–LUMO gaps of modified oligomers to represent the extended backbone, and molecular features from SMILES strings of monomers to capture the effect of alkyl side chains.In this study, we evaluated the effectiveness of three types of MFs for capturing the impact of alkyl side chains on the optical gaps of CPs: RDKit Descriptors, MACCS, and ECFP6 fingerprints, which were calculated from the SMILES strings of monomer structures containing alkyl side chains as detailed in the Methods section. Previous studies have shown these MFs are effective for training ML models to predict the photo-electronic properties of CP-based OSCs.27,59 The workflow for database preparation, feature engineering, model training, and transferability test is summarized in Fig. 3. We trained six ML algorithms that were commonly used in materials sciences, including HGBR, LGBM, GBR, XGBoost, AdaBoost, and RF, using EDFTgap combined with different types of MFs as input parameters to predict Eexpgap. Performance metrics, including R2, r, RMSE, and MAE, were detailed in Tables S9–S11.† Notably, the prediction accuracy of optical gap values is significantly enhanced, with the R2 value increasing from 0.51 to as high as 0.77, by incorporating information from both the conjugated backbone (captured by Eoligomergap) and the side chains (captured by MFs). Particularly, ECFP6 combined with Eoligomergap consistently achieved the highest prediction accuracy across all ML models, demonstrating its superiority in capturing the side chain information of CPs compared to RDKit and MACCS.
 | ||
| Fig. 3 The workflow for the machine learning model training procedure to predict the experimentally measured optical gaps of conjugated polymers (CPs). | ||
To further investigate the impact of Eoligomergap on optical gap prediction, we summarized the R2 and MAE values for the six ML models trained using ECFP6 alone and in combination with Eoligomergap in Fig. 4. From SHAP (SHapley Additive exPlanations) analysis, the DFT-calculated HOMO–LUMO gap consistently dominates as the most important feature across all models, followed by two RDKit descriptors (details are in Note S7 and Fig. S7†). In addition, these features are independent, as demonstrated by the low correlations shown in Fig. S8,† which illustrates the relationships among the key features in each model. All models achieved higher accuracy using Eoligomergap and ECFP6, with R2 values over 0.62, compared to 0.51 for a simple linear regression model (Fig. 1c). Notably, XGBoost emerged as the top performer with an R2 of 0.77 and MAE of 0.065 eV. This level of accuracy falls within the experimental error margin of approximately 0.1 eV. For instance, polymer P3HT has an Eexpgap between 1.9 and 2.14 eV,60–63 while PDB7 ranges from 1.6 to 1.7 eV,64–66 influenced by molecular weight, regioregularity, and processing conditions.67 In addition, when retrained with only ECFP6, all models had lower accuracy; for example, the XBGoost model achieved an R2 of 0.7 and MAE of 0.075 eV.
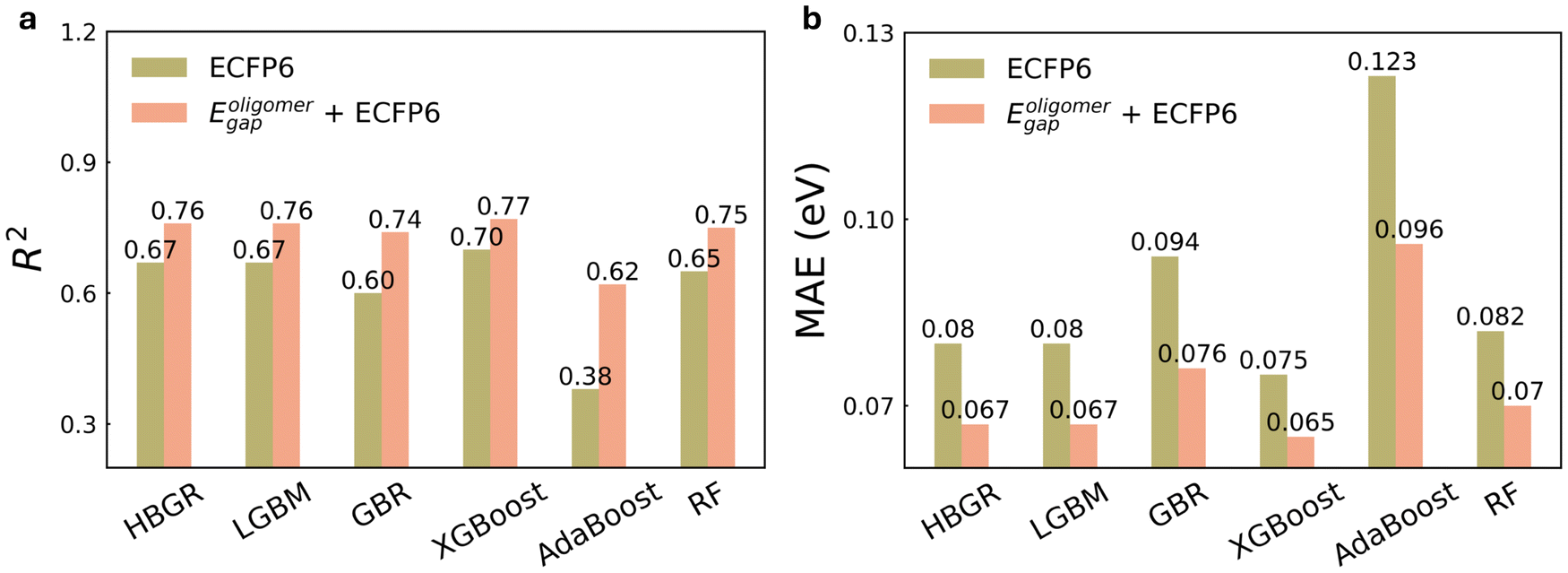 | ||
| Fig. 4 (a) R2 and (b) mean absolute error (MAE) (eV) of six machine learning models for predicting experimental optical gaps of conjugated polymers with different descriptors as input. Eoligomergap is HOMO–LUMO gap calculated from modified oligomer structures. | ||
In summary, descriptors are essential for ML models to capture critical information influencing targeted properties and learn structure–property relationships effectively. DFT-calculated HOMO–LUMO gaps of modified oligomers and ECFP6 MF derived from unmodified monomers can effectively capture fundamental characteristics of both the extended backbone and alkyl side chains, enabling accurate and efficient prediction of Eexpgap values.
3.3 Model transferability
We have demonstrated that ML models can leverage rationally designed Eoligomergap and MFs to improve the prediction accuracy of experimental optical gaps. Beyond accuracy, it is essential to validate the model's robustness and transferability with new datasets which have not been used in the ML model training process. In this study, we manually collected 227 newly synthesized CP structures from the literature, categorizing them into two groups based on their electron acceptor units. As shown in Fig. 5a, CP structures from group 1 contain at least one of the five acceptor units which were included in the training set; for example, BT and BTA units belong to A1 type (see Fig. 2f). This subset of CP structures is applied to evaluate the interpolation performance of the trained ML models considering the close similarity of this subset of CP structures as compared to the training set. In contrast, group 2 contains five acceptor units which have not been seen in the training set (see Fig. 5b), including Perylene Diimide (PDI),68 naphthalenediimide (NDI),69 dithieno[3′,2′:3,4;2′′,3′′:5,6]benzo[1,2-c][1,2,5]thiadiazole (DTBT),70 benzobisoxazole (BBX),71 and Y6.72 These acceptor units are important components of electron-accepting semiconductors for organic photovoltaic applications. For example, the Y6-based small molecule, first announced in 2019, achieved a record power conversion efficiency of 15.7% as an acceptor in OSCs.73 In 2020, the first Y6-series-based polymer acceptor was reported, and since then, these acceptors have been recognized as the best n-type materials.74 Additionally, Liu et al. introduced a novel family of polymer donors named D18 based on DTBT and fluorinated BDTT in 2020, achieving the first single-junction OSC with an efficiency of over 18% when blended with Y6 small molecule.75 The CP structures from group 2 containing one of these five acceptor units are used to assess the extrapolation performance of the trained ML models.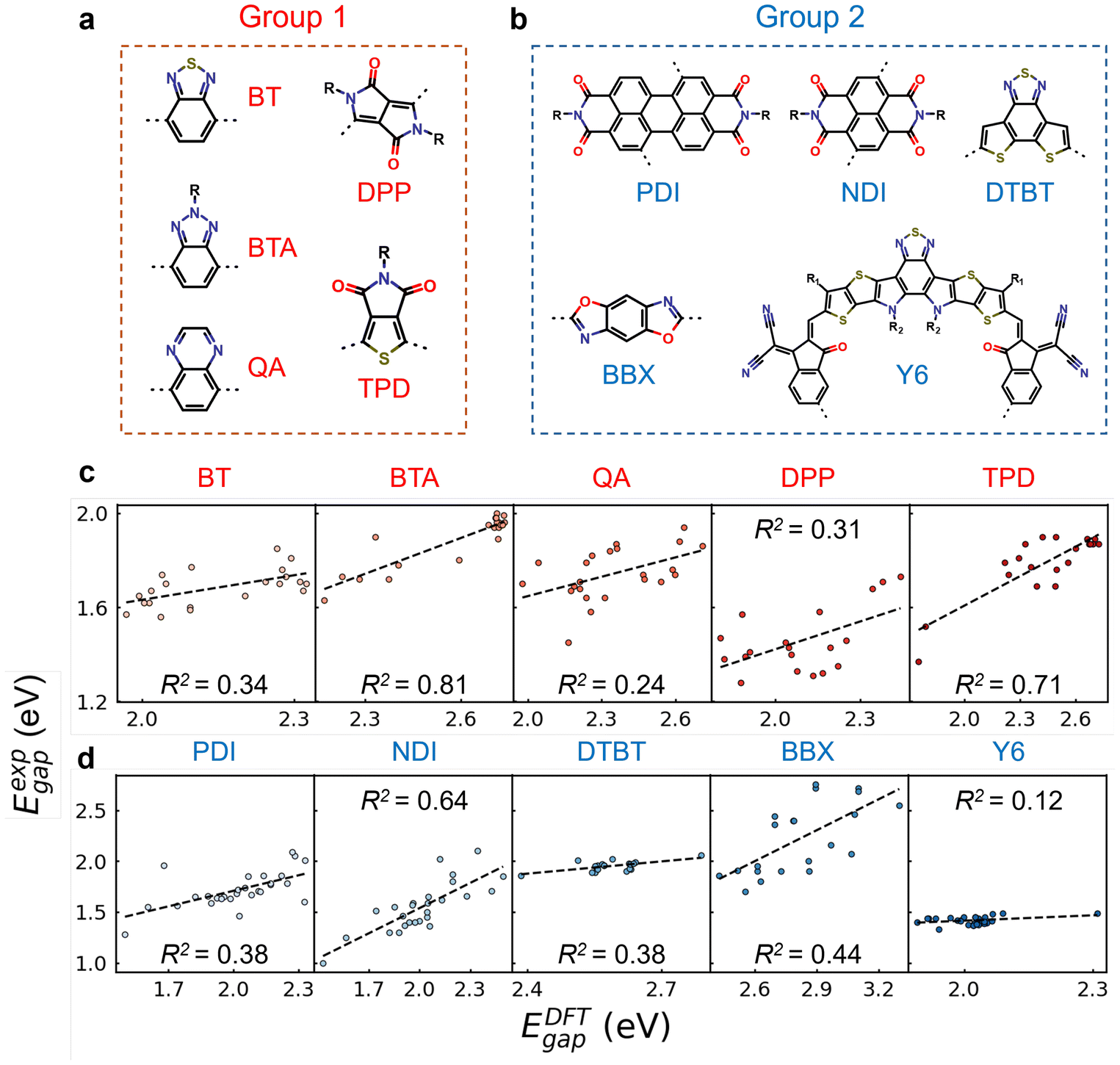 | ||
| Fig. 5 The chemical structures of (a) five acceptor units exiting in the training set and (b) five acceptor units not existing in the training set. (c and d) The linear correlation between DFT-calculated HOMO–LUMO gaps (EDFTgap) with modified oligomer structures and experimental optical gaps (Eexpgap) for (c) group 1 and (d) group 2 conjugated polymers. The black dashed lines correspond to linear fitting. | ||
For both group 1 and group 2 CP structures, we constructed the modified oligomers using the two-step procedure (see Fig. 1a) to obtain the Eoligomergap values and converted the SMILES strings of the alkyl-side-chain-containing monomers into ECFP6 features. Then, we applied the XGBoost models previously trained with the 1096 dataset to predict the Eexpgap of both groups without further retraining. The performance metrics are presented in Table 1. XGBoost-2, trained with Eoligomergap and ECFP6, accurately predicted the optical gaps of group 1 CPs with most MAEs below 0.1 eV, demonstrating excellent interpolation performance. Interestingly, XGBoost-1, trained with only ECFP6, also showed superior interpolation performance, resulting in lower RMSE and MAE than XGBoost-2 across all CP types in group 1. These results suggest that ECFP6 effectively captures the electronic properties of similar CPs within the same chemical design space. In fact, ECFP6 has been widely used to measure the similarities of various organic molecules in previous studies.76
| Acceptor unit | #Data points | XGBoost-1 | XGBoost-2 | ||
|---|---|---|---|---|---|
| RMSE | MAE | RMSE | MAE | ||
| Group 1 acceptor units are included in the training set, whereas group 2 units are not. Chemical structures are illustrated in Fig. 5. XGBoost-1 is trained using ECFP6 alone, while XGBoost-2 is trained with both Eoligomergap and ECFP6. Both root mean square error (RMSE) and mean absolute error (MAE) are measured in eV. | |||||
| Group 1 | |||||
| BT | 21 | 0.073 | 0.052 | 0.085 | 0.069 |
| BTA | 20 | 0.106 | 0.085 | 0.112 | 0.099 |
| QA | 23 | 0.075 | 0.063 | 0.114 | 0.092 |
| DPP | 19 | 0.068 | 0.054 | 0.133 | 0.102 |
| TPD | 20 | 0.077 | 0.063 | 0.09 | 0.068 |
| Group 2 | |||||
| PDI | 28 | 0.181 | 0.158 | 0.189 | 0.147 |
| NDI | 26 | 0.262 | 0.211 | 0.126 | 0.094 |
| DTBT | 20 | 0.157 | 0.135 | 0.059 | 0.041 |
| BBX | 21 | 0.583 | 0.49 | 0.338 | 0.253 |
| Y6 | 29 | 0.427 | 0.418 | 0.217 | 0.211 |
We then assessed the extrapolation performance of both models with group 2 CPs. As shown in Table 1, XGBoost-2 significantly outperformed XGBoost-1, yielding substantially lower RMSE and MAE values across all CP types. For example, the MAE for Y6 based CPs decreases from 0.418 eV to 0.211 eV with XGBoost-2. Previous studies have also shown that conventional ML models trained with molecular descriptors may perform well in the chemical structure space similar to the training set, whereas the extrapolation to the new structure space is challenging due to the lack of physical/chemical insight from the input descriptors.77,78 Our results demonstrate that the XGBoost-2 model, trained with both Eoligomergap and ECFP6, excels in both high interpolation and extrapolation performance. This superior transferability is originated from the excellent robustness of DFT methods and rationally designed oligomer structures, effectively capturing the electronic properties of the CPs. Indeed, as shown in Fig. 5c and d, Eoligomergap are highly correlated with Eexpgap for both group 1 and group 2 CPs.
3.4 Further discussion
As detailed above, the XGBoost-2 model, trained with 1096 CPs, demonstrated the superior transferability on a new dataset of 227 CPs. It is well acknowledged that conventional ML models such as XGBoost can benefit from larger datasets to further improve prediction accuracy.79,80 Therefore, we selected one structure with the highest prediction error from each category in group 1 and group 2, forming a new test set of 10 CP structures (see Fig. S9†). The remaining 217 data points were combined with the original 1096, creating a new training set of 1313 structures, thus augmenting the training set by around 20%. We retrained a new XBGoost model (labeled as “XGBoost-2-plus”) with 10-fold cross-validation on 1313 data points and calculated the average RMSE and MAE for predicting the Eexpgap of 10 CPs in the new test set. As shown in Table S12,† XGBoost-2-plus achieved enhanced prediction accuracy, with a lower RMSE of 0.241 eV and MAE of 0.213 eV compared to XGBoost-2 (0.333 eV and 0.3 eV, respectively). Particularly, the prediction errors of the XGBoost-2-plus were significantly reduced for each polymer in the test set, demonstrating the effectiveness of data augmentation in improving model performance. It is important to note that experimentally measured optical gap values for CPs can vary across different labs and experiments, introducing potential inconsistencies and errors. Factors such as processing conditions, solvents, additives, and film morphology can influence these measurements.81 Utilizing larger and more accurate experimental datasets can enhance the predictive accuracy of ML models for optical gaps. It is important to note that the torsion angle in C–C single bonds and the quinoidal resonance can significantly influence the band gap. Specifically, larger torsion angles lead to an increase in the band gap,82,83 while an enhanced quinoidal character tends to reduce it.8 However, to ensure consistency and comparability across our extensive and diverse dataset, these factors were not explicitly incorporated into our model. Accurately capturing the effects of torsion and quinoidal resonance would require a more specialized and carefully curated dataset that explicitly accounts for these structural variations.In addition to predicting optical gaps, our training strategy, which combines quantum chemistry calculations and MFs, extends to predicting other fundamental properties of CPs such as HOMO and LUMO levels, which are also vital for their applications in electronics and solar cells.84,85 In our dataset of 1096 structures, HOMO levels were measured via cyclic voltammetry, while LUMO levels were derived from substituting optical gap values with HOMO values. Following the methodology detailed in the Methods section, we retrained the XGBoost model and presented the performance metrics in Table S13.† Notably, XGBoost-2-H trained with DFT-calculated HOMO values of modified oligomers and ECFP6 exhibits higher accuracy in predicting experimentally measured HOMO values, achieving an R2 of 0.5 and an MAE of 0.109 eV. Similarly, incorporating DFT-calculated LUMO levels enhances the accuracy of XGBoost-2 in predicting LUMO levels, with an R2 of 0.6 and an MAE of 0.112 eV.
4. Conclusions
In this study, we introduced a model that combines DFT calculations with a ML approach to accurately predict the experimentally measured optical band gaps of CPs, utilizing a dataset of 1096 data points. We first proposed a two-step modification procedure for constructing oligomers to effectively capture π-electron delocalization in CPs: alkyl side chain truncation and conjugated backbone extension. This approach significantly improves the correlation between the DFT-calculated HOMO–LUMO gaps and experimental gaps (R2 = 0.51) compared to the unmodified side-chain-containing monomers (R2 = 0.15). Subsequently, we incorporated both conjugated backbone characteristics, derived from quantum chemistry, and the alkyl-side-chain effects, represented by molecular descriptors, into ML modeling to enhance prediction accuracy. Employing the Eoligomergap of modified oligomers and ECFP6 MF derived from side-chain-containing monomers as input, the resulting model, XGBoost-2, effectively elucidated the structure–property relationship of CPs, achieving an R2 of 0.77 and an MAE of 0.065 eV. To further assess its robustness and transferability in predicting new CP structures beyond the chemical design space of the training set, we manually collected 227 newly synthesized CPs from the literature, categorizing them into two groups based on their electron acceptor units. Group 1 CP structures contain at least one of the five acceptor units existing in the training set, allowing for the evaluation of interpolation performance, while group 2 structures contain at least one of the five acceptor units not present in the training set, aiming for extrapolation performance test. Notably, XGBoost-2 demonstrates excellent interpolation and extrapolation, which stem from the combination of DFT methods and rationally designed oligomer structures that effectively capture the electronic properties of CPs. This study represents the first successful combination of quantum chemistry calculations with ML modeling to accurately predict experimentally measured fundamental properties of CPs (e.g., HOMO, LUMO, and optical gap), facilitating the design and development of next-generation high-performance CPs in photoelectronic and energy conversion applications.Author contributions
B. L. and M. L. initiated this study. B. L. was responsible for conducting the theoretical calculations, training the machine learning models, and analyzing the data. Y. Y. was responsible for data collection, structural generation, and optimization. All authors contributed to the manuscript writing.Data availability
The datasets used and/or analyzed during the current study are available from the GitHub repository at https://github.com/Liu-Group-UF/Machine-Learning-for-Accurate-Optical-Gap-Prediction-in-Conjugated-Polymers.Code availability
The Python code used for Machine Learning model training is available from: https://github.com/Liu-Group-UF/Machine-Learning-for-Accurate-Optical-Gap-Prediction-in-Conjugated-Polymers.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the University of Florida's new faculty start-up funding. The authors acknowledge the University of Florida Research Computing for providing computational resources and support that have contributed to the research results reported in this publication.References
- P. Palani and S. Karpagam, Conjugated polymers–a versatile platform for various photophysical, electrochemical and biomedical applications: a comprehensive review, New J. Chem., 2021, 45, 19182–19209 RSC
.
- A. H. Malik, F. Habib, M. J. Qazi, M. A. Ganayee, Z. Ahmad and M. A. Yatoo, A short review article on conjugated polymers, J. Polym. Res., 2023, 30, 115 CrossRef CAS
- S. Kang, T. W. Yoon, G.-Y. Kim and B. Kang, Review of conjugated polymer nanoparticles: from formulation to applications, ACS Appl. Nano Mater., 2022, 5, 17436–17460 CrossRef CAS
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Recent advances in bulk heterojunction polymer solar cells, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Low bandgap semiconducting polymers for polymeric photovoltaics, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC
- Y. Li, Z. Jia, Q. Zhang, Z. Wu, H. Qin, J. Yang, S. Wen, H. Y. Woo, W. Ma and R. Yang,
et al., Toward efficient all-polymer solar cells via halogenation on polymer acceptors, ACS Appl. Mater. Interfaces, 2020, 12, 33028–33038 CrossRef CAS PubMed
- G. Pace, I. Bargigia, Y.-Y. Noh, C. Silva and M. Caironi, Intrinsically distinct hole and electron transport in conjugated polymers controlled by intra and intermolecular interactions, Nat. Commun., 2019, 10, 5226 CrossRef PubMed
- B. Liu, D. Rocca, H. Yan and D. Pan, Beyond conformational control: effects of noncovalent interactions on molecular electronic properties of conjugated polymers, JACS Au, 2021, 1, 2182–2187 CrossRef CAS PubMed
- H. Bin, Y. Yang, Z. Peng, L. Ye, J. Yao, L. Zhong, C. Sun, L. Gao, H. Huang and X. Li,
et al., Effect of Alkylsilyl Side-Chain Structure on Photovoltaic Properties of Conjugated Polymer Donors, Adv. Energy Mater., 2018, 8, 1702324 CrossRef
- X. Chen, S. Hussain, Y. Hao, X. Tian and R. Gao, Recent advances of signal amplified smart conjugated polymers for optical detection on solid support, ECS J. Solid State Sci. Technol., 2021, 10, 037006 CrossRef CAS
- Y. Liu, V. R. Feig and Z. Bao, Conjugated polymer for implantable electronics toward clinical application, Adv. Healthcare Mater., 2021, 10, 2001916 CrossRef CAS PubMed
- J. H. Luong, T. Narayan, S. Solanki and B. D. Malhotra, Recent advances of conducting polymers and their composites for electrochemical biosensing applications, J. Funct. Biomater., 2020, 11, 71 CrossRef CAS PubMed
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Recent advances in conjugated polymers for visible-light-driven water splitting, Adv. Mater., 2020, 32, 1907296 Search PubMed
- M. C. Scharber and N. S. Sariciftci, Low band gap conjugated semiconducting polymers, Adv. Mater. Technol., 2021, 6, 2000857 CrossRef CAS
- A. D. Laurent and D. Jacquemin, TD-DFT benchmarks: a review, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef CAS
- H. Sun and J. Autschbach, Electronic energy gaps for π-conjugated oligomers and polymers calculated with density functional theory, J. Chem. Theory Comput., 2014, 10, 1035–1047 CrossRef CAS PubMed
- E. O. Pyzer-Knapp, G. N. Simm and A. A. Guzik, A Bayesian approach to calibrating high-throughput virtual screening results and application to organic photovoltaic materials, Mater. Horiz., 2016, 3, 226–233 RSC
- S. J. Yang, S. Li, S. Venugopalan, V. Tshitoyan, M. Aykol, A. Merchant, E. D. Cubuk and G. Cheon, Accurate Prediction of Experimental Band Gaps from Large Language Model-Based Data Extraction, arXiv, 2023, preprint, arXiv:2311.13778.
- D. Chaudhuri and C. Patterson, TDDFT versus GW/BSE Methods for Prediction of Light Absorption and Emission in a TADF Emitter, J. Phys. Chem. A, 2022, 126, 9627–9643 CrossRef CAS PubMed
- N. Meftahi, M. Klymenko, A. J. Christofferson, U. Bach, D. A. Winkler and S. P. Russo, Machine learning property prediction for organic photovoltaic devices, npj Comput. Mater., 2020, 6, 166 CrossRef CAS
- E. O. Pyzer-Knapp, K. Li and A. Aspuru-Guzik, Learning from the harvard clean energy project: The use of neural networks to accelerate materials discovery, Adv. Funct. Mater., 2015, 25, 6495–6502 CrossRef CAS
- B. Mazouin, A. A. Schöpfer and O. A. von Lilienfeld, Selected machine learning of HOMO–LUMO gaps with improved data-efficiency, Mater. Adv., 2022, 3, 8306–8316 RSC
- M. McGibbon, S. Shave, J. Dong, Y. Gao, D. R. Houston, J. Xie, Y. Yang, P. Schwaller and V. Blay, From intuition to AI: evolution of small molecule representations in drug discovery, Briefings Bioinf., 2024, 25, bbad422 CrossRef PubMed
- S. Raghunathan and U. D. Priyakumar, Molecular representations for machine learning applications in chemistry, Int. J. Quantum Chem., 2022, 122, e26870 CrossRef CAS
- D. A. Winkler and T. C. Le, Performance of deep and shallow neural networks, the universal approximation theorem, activity cliffs, and QSAR, Mol. Inf., 2017, 36, 1600118 CrossRef PubMed
- K. Wu, N. Sukumar and N. Lanzillo,
et al., Prediction of polymer properties using infinite chain descriptors (ICD) and machine learning: Toward optimized dielectric polymeric materials, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2082–2091 CrossRef CAS
- S. Nagasawa, E. Al-Naamani and A. Saeki, Computer-aided screening of conjugated polymers for organic solar cell: classification by random forest, J. Phys. Chem. Lett., 2018, 9, 2639–2646 CrossRef CAS PubMed
-
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson, H. Nakatsuji, et al., GAUSSIAN16. Revision C. 01, Gaussian Inc., Wallingford, CT, USA, 2016 Search PubMed
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, Open Babel: An open chemical toolbox, J. Cheminf., 2011, 3, 1–14 Search PubMed
- Avogadro, https://avogadro.cc/.
- M. B. Goldey, D. Reid, J. de Pablo and G. Galli, Planarity and multiple components promote organic photovoltaic efficiency by improving electronic transport, Phys. Chem. Chem. Phys., 2016, 18, 31388–31399 RSC
- RDKit: Open-Source Cheminformatics Software, https://www.rdkit.org/.
- RDKit Descriptors module, https://rdkit.org/docs/source/rdkit.Chem.Descriptors.html.
- MACCSkeys module, https://rdkit.org/docs/source/rdkit.Chem.MACCSkeys.html.
- D. Rogers and M. Hahn, Extended-connectivity fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed
- A. Guryanov , Analysis of Images, Social Networks and Texts: 8th International Conference, AIST 2019, Kazan, Russia, July 17–19, 2019, Revised Selected Papers 8, 2019, pp. 39–50.
- J. H. Friedman, Stochastic gradient boosting, Comput. Stat. Data Anal., 2002, 38, 367–378 CrossRef
- G. Ke, Q. Meng, T. Finley, T. Wang, W. Chen, W. Ma, Q. Ye and T.-Y. Liu, Advances in Neural Information Processing Systems, 2017, 30, 3146–3154 Search PubMed
- T. Chen and C. Guestrin, Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining, 2016, pp. 785–794.
- D. P. Solomatine and D. L. Shrestha, 2004 IEEE international joint conference on neural networks (IEEE Cat. No. 04CH37541), 2004, pp. 1163–1168.
- A. Liaw and M. Wiener, R News, 2002, 2(3), 18–22 Search PubMed
- J. Wei, X. Chu, X.-Y. Sun, K. Xu, H.-X. Deng, J. Chen, Z. Wei and M. Lei, Machine learning in materials science, InfoMat, 2019, 1, 338–358 CrossRef CAS
- D. Morgan and R. Jacobs, Opportunities and challenges for machine learning in materials science, Annu. Rev. Mater. Res., 2020, 50, 71–103 CrossRef CAS
- Scikit-learn: Machine Learning in Python, https://scikit-learn.org/stable/.
- J.-L. Bredas, Mind the gap!, Mater. Horiz., 2014, 1, 17–19 RSC
- S. M. Falke, C. A. Rozzi, D. Brida, M. Maiuri, M. Amato, E. Sommer, A. De Sio, A. Rubio, G. Cerullo and E. Molinari,
et al., Coherent ultrafast charge transfer in an organic photovoltaic blend, Science, 2014, 344, 1001–1005 CrossRef CAS PubMed
- B. Liu, P. C. Chow, J. Liu and D. Pan, Polarized local excitons assist charge dissociation in Y6-based nonfullerene organic solar cells: a nonadiabatic molecular dynamics study, J. Mater. Chem. A, 2024, 12, 15974–15983 RSC
- R. E. Larsen, Simple extrapolation method to predict the electronic structure of conjugated polymers from calculations on oligomers, J. Phys. Chem. C, 2016, 120, 9650–9660 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
- Z.-G. Zhang and J. Wang, Structures and properties of conjugated donor–acceptor copolymers for solar cell applications, J. Mater. Chem., 2012, 22, 4178–4187 RSC
- M. H. Chua, Q. Zhu, T. Tang, K. W. Shah and J. Xu, Diversity of electron acceptor groups in donor–acceptor type electrochromic conjugated polymers, Sol. Energy Mater. Sol. Cells, 2019, 197, 32–75 CrossRef CAS
- R. Hildner, A. Köhler, P. Müller-Buschbaum, F. Panzer and M. Thelakkat, π-Conjugated Donor Polymers: Structure Formation and Morphology in Solution, Bulk and Photovoltaic Blends, Adv. Energy Mater., 2017, 7, 1700314 CrossRef
- W. Sun, Y. Zheng, K. Yang, Q. Zhang, A. A. Shah, Z. Wu, Y. Sun, L. Feng, D. Chen and Z. Xiao,
et al., Machine learning–assisted molecular design and efficiency prediction for high-performance organic photovoltaic materials, Sci. Adv., 2019, 5, eaay4275 CrossRef CAS PubMed
- S. Kesornsit, C. Direksilp, K. Phasuksom, N. Thummarungsan, P. Sakunpongpitiporn, K. Rotjanasuworapong, A. Sirivat and S. Niamlang, Synthesis of highly conductive poly (3-hexylthiophene) by chemical oxidative polymerization using surfactant templates, Polymers, 2022, 14, 3860 CrossRef CAS PubMed
- P. Aruna and C. Joseph, Optical and photosensing properties of gold nanoparticles doped poly (3-hexylthiophene-2, 5-diyl) thin films, Mater. Lett., 2021, 295, 129726 CrossRef CAS
- I. H. Jung, C. T. Hong, U.-H. Lee, Y. H. Kang, K.-S. Jang and S. Y. Cho, High thermoelectric power factor of a diketopyrrolopyrrole-based low bandgap polymer via finely tuned doping engineering, Sci. Rep., 2017, 7, 44704 CrossRef CAS PubMed
- S. S. Prasad, G. Divya and K. S. Kumar, 2021 2nd International Conference on Advances in Computing, Communication, Embedded and Secure Systems (ACCESS), 2021, pp. 5–8.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu,
et al., For the bright future-bulk heterojunction polymer solar cells with power conversion efficiency of 7.4%, Adv. Mater., 2010, 22, E135 CrossRef CAS PubMed
- F. Bencheikh, D. Duché, C. M. Ruiz, J.-J. Simon and L. Escoubas, Study of optical properties and molecular aggregation of conjugated low band gap copolymers: PTB7 and PTB7-Th, J. Phys. Chem. C, 2015, 119, 24643–24648 CrossRef CAS
- T. Basel, U. Huynh, T. Zheng, T. Xu, L. Yu and Z. V. Vardeny, Optical, electrical, and magnetic studies of organic solar cells based on low bandgap copolymer with spin 1/2 radical additives, Adv. Funct. Mater., 2015, 25, 1895–1902 CrossRef CAS
- Y. He, L. Huo and B. Zheng, Advances of batch-variation control for photovoltaic polymers, Nano Energy, 2024, 109397 CrossRef CAS
- Q. Shi, J. Wu, X. Wu, A. Peng and H. Huang, Perylene Diimide-Based Conjugated Polymers for All-Polymer Solar Cells, Chem. – Eur. J., 2020, 26, 12510–12522 CrossRef CAS PubMed
- N. Zhou and A. Facchetti, Naphthalenediimide (NDI) polymers for all-polymer photovoltaics, Mater. Today, 2018, 21, 377–390 CrossRef CAS
- J. Cao, L. Yi, L. Zhang, Y. Zou and L. Ding, Wide-bandgap polymer donors for non-fullerene organic solar cells, J. Mater. Chem. A, 2023, 11, 17–30 RSC
- J. J. Intemann, E. S. Hellerich, M. D. Ewan, B. C. Tlach, E. D. Speetzen, R. Shinar, J. Shinar and M. Jeffries-El, Investigating the impact of conjugation pathway on the physical and electronic properties of benzobisoxazole-containing polymers, J. Mater. Chem. C, 2017, 5, 12839–12847 RSC
- M. Kataria, H. D. Chau, N. Y. Kwon, S. H. Park, M. J. Cho and D. H. Choi, Y-series-based polymer acceptors for high-performance all-polymer solar cells in binary and non-binary systems, ACS Energy Lett., 2022, 7, 3835–3854 CrossRef CAS
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng and P. A. Johnson,
et al., Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core, Joule, 2019, 3, 1140–1151 CrossRef CAS
- T. Jia, J. Zhang, W. Zhong, Y. Liang, K. Zhang, S. Dong, L. Ying, F. Liu, X. Wang and F. Huang,
et al., 14.4% efficiency all-polymer solar cell with broad absorption and low energy loss enabled by a novel polymer acceptor, Nano Energy, 2020, 72, 104718 CrossRef CAS
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao and K. Sun,
et al., 18% Efficiency organic solar cells, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed
- S. Riniker and G. A. Landrum, Similarity maps-a visualization strategy for molecular fingerprints and machine-learning methods, J. Cheminf., 2013, 5, 1–7 Search PubMed
- Z.-W. Zhao, M. Del Cueto and A. Troisi, Limitations of machine learning models when predicting compounds with completely new chemistries: possible improvements applied to the discovery of new non-fullerene acceptors, Digital Discovery, 2022, 1, 266–276 RSC
- E. S. Muckley, J. E. Saal, B. Meredig, C. S. Roper and J. H. Martin, Interpretable models for extrapolation in scientific machine learning, Digital Discovery, 2023, 2, 1425–1435 RSC
- J. Jin, S. Faraji, B. Liu and M. Liu, Comparative Analysis of Conventional Machine Learning and Graph Neural Network Models for Perovskite Property Prediction, J. Phys. Chem. C, 2024, 128, 16672–16683 CrossRef CAS
- C. A. Ramezan, T. A. Warner, A. E. Maxwell and B. S. Price, Effects of training set size on supervised machine-learning land-cover classification of large-area high-resolution remotely sensed data, Remote Sens., 2021, 13, 368 CrossRef
- F. Fischer, K. Tremel, A.-K. Saur, S. Link, N. Kayunkid, M. Brinkmann, D. Herrero-Carvajal, J. L. Navarrete, M. R. Delgado and S. Ludwigs, Influence of processing solvents on optical properties and morphology of a semicrystalline low bandgap polymer in the neutral and charged states, Macromolecules, 2013, 46, 4924–4931 CrossRef CAS
- S. S. Zade and M. Bendikov, Twisting of Conjugated Oligomers and Polymers: Case Study of Oligo- and Polythiophene, Chem. – Eur. J., 2007, 13, 3688–3700 CrossRef CAS PubMed
- R. Gutzler, Band-structure engineering in conjugated 2D polymers, Phys. Chem. Chem. Phys., 2016, 18, 29092–29100 RSC
- Y. Li, Molecular design of photovoltaic materials for polymer solar cells: toward suitable electronic energy levels and broad absorption, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed
- J. W. Jung, J. W. Jo, E. H. Jung and W. H. Jo, Recent progress in high efficiency polymer solar cells by rational design and energy level tuning of low bandgap copolymers with various electron-withdrawing units, Org. Electron., 2016, 31, 149–170 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: RDKit descriptor selection. Model training strategy. Model performance metrics. Oligomer structure construction. Exchange–correlation functional test. Statistical analysis of the experimentally measured HOMO, LUMO, and optical band gap values. SHAP analysis. Chemical structures of 18 non-conjugated polymers. The number of data points and the corresponding percentage of each group of conjugated polymers categorized based on donor and acceptor units. The performance metrics of 6 machine learning models. See DOI: https://doi.org/10.1039/d4nr03702b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |