
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–organic frameworks generated from oligomeric ligands with functionalized tethers†
Hyunyong
Kim
 and
Seth M.
Cohen
*
and
Seth M.
Cohen
*
Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, USA. E-mail: scohen@ucsd.edu
First published on 17th December 2024
Abstract
Metal–organic frameworks (MOFs) can be prepared from oligomeric organic ligands to prepare materials referred to as oligoMOFs. Studies of oligoMOFs are relatively limited, with most existing reports focused on fundamental structure–property relationships. In this report, functional groups, such as terminal alkynes and pyridine groups, are installed on the tether between 1,4-benzene dicarboxylic acid (H2bdc) groups of the dimer ligands. It was found that the position of the pyridine donor atom had pronounced effects on the synthesis and structure of Zn(II)-based isoreticular MOFs (IRMOF-1 analog, oligoIRMOF). Importantly, simple H2bdc ligands with pendant pyridine groups (i.e., not part of an dimeric ligand), were unable to produce crystalline phases or produced new MOF phases, showing the importance of the oligomeric ligand in directing structure. Dimeric ligands with two H2bdc groups and a terminal alkyne produced a crystalline and porous oligoIRMOF that enables postsynthetic modification (PSM) via ‘click’ chemistry to introduce other functional groups into the pores of the material. This oligoIRMOF could be cross-linked using reagents containing multiple azide functional sites. Taken together, these studies are among the first examples of oligoMOFs with functionalized tethers and demonstrate the versatility, utility, and unique opportunities for new chemistry provided by oligoMOFs.
Introduction
Metal–organic frameworks (MOFs) are crystalline hybrid materials with organic linkers and metal clusters.1 The large accessible pores and ability to functionalize the pores of MOFs have garnered interest for applications in gas adsorption, separation, and heterogeneous catalysis.2 The majority of MOFs are made from relatively simple ligands, many of which utilize carboxylic acid donors.3 It was found that incorporating carboxylic acid ligands, specifically 1,4-benzene dicarboxylic acid (H2bdc), into organic polymers produces polymeric ligands that can be used for the preparation of polyMOFs, a type of MOF–polymer hybrid material.4–7 In addition, oligoMOFs, made up of discrete multimers of H2bdc (i.e., dimers, trimers, tetramers, etc.), have been examined as intermediate systems between conventional, molecular MOFs and polyMOFs (Fig. 1).8–10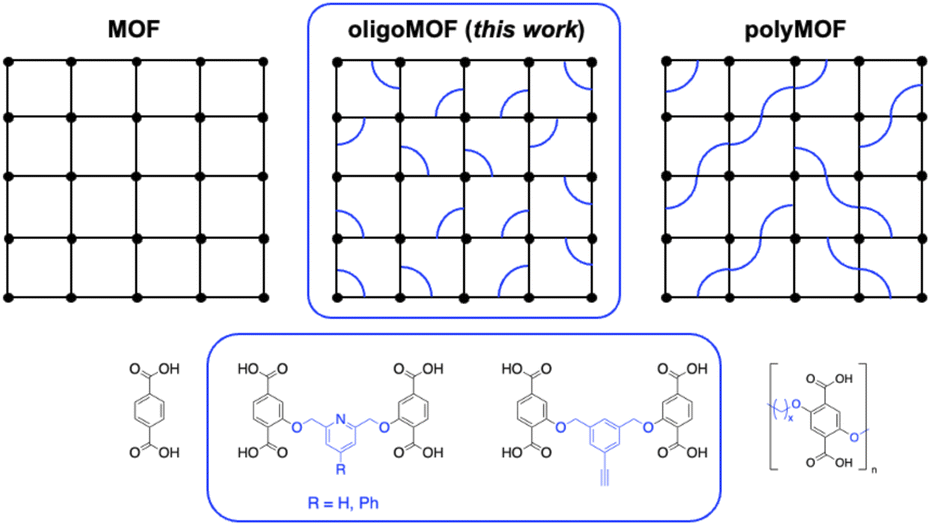 | ||
| Fig. 1 (Top) Schematic representation of conventional/molecular MOFs (left), oligoMOFs (center, in blue box), and polyMOFs (right). (Bottom) Representative organic ligands used for conventional/molecular MOFs, oligoMOFs, and polyMOFs (from left-to-right): terephthalic acid, dimer ligands with pyridine and alkyne functional groups (highlighted in blue box), and polymer ligands. | ||
Oligomeric ligands with two or three H2bdc groups connected by alkyl or xylyl tethers have been investigated for the synthesis of oligoMOFs.8,9 In addition, linear and branched oligomeric linkers possessing four or eight H2bdc groups were reported for the preparation of isoreticular type metal–organic frameworks (oligoIRMOFs).10 The structure, geometry, tether structure, and number of H2bdc groups are important features that govern the crystallinity, porosity, and stability of the resulting oligoMOFs.8–10
A notable gap in the study of oligoMOFs is the introduction of functional groups on the tethering component. Such functionality can be used to expand the properties and utility of oligoMOFs. Xiao and co-workers have published several excellent studies on MOF-74(Mg) derivatives containing isolated dimeric ligands (specifically, 4,4′′-dioxido-[1,1′:4′,1′′-terphenyl]-3,3′′-dicarboxylate) with ester tethers that allow for thermal cleavage of the tether to produce spatially-controlled functional groups.11 The functional sites in these MOF-74(Mg) derivatives could then be metalated to create biomimetic active sites.12 The same group has also reported on oligoMOF derivatives of MIL-53(Al) (using 2′, 5′-dimethyl-[1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylate ligands) that showed improved stability of the open MOF configuration.13 Zhou and co-workers demonstrated postsynthetic modification (PSM) of aldehyde decorated ZIF-8 to install diamine derivatives as cross-linkers, leading to the generation of linked dimer ligands created postsynthetically in ZIF-8.14 These handful of examples are among the few accounts of tethering group in oligoMOFs that contributed added functionality to the framework. Although important contributions, the scope of chemistry described in these studies is quite limited, leaving a substantial opportunity to further explore the role of tether functionalization in MOF structure, properties, and chemical tunability.
In this report, the ability to richly functionalize the tether in oligoMOFs with Lewis basic sites or reactive alkynes is described. Further, the impact of installing Lewis basic sites on the tether on the formation of oligoMOF formation are described. Specifically, dimeric ligands with tethers containing pyridine groups (L1–L5) or alkyne groups are described (L6, Fig. 2). Interestingly, for the pyridine-functionalized tethers, the position of the nitrogen donor atom in the tether greatly impacts the ability to generate oligoMOFs. The dimeric linker with alkyne groups on a xylyl-bridged spacer (L6) was shown to produce highly crystalline and porous oligoIRMOFs with terminal alkynes within the oligoMOF pores. PSM of oligoIRMOF-1-L6 was performed via Cu(I) catalyzed ‘click’ reaction to introduce added functionality at the xylyl bridge. Taken together, the findings here show that oligoMOFs are versatile materials, where the tethering groups present tremendous opportunities for pore and framework functionalization.
 | ||
| Fig. 2 Ligands used in this study. Dimeric (L1–L6) and monomeric H2bdc derived ligands (bottom row) possessing similar pyridine-derived functional substituents. Three linkers (L1, L3, L6) generated the desired IRMOF-1 lattice (highlighted in black box). | ||
Results and discussion
Synthesis of pyridine (L1–L5) and alkyne (L6) oligomeric ligands
The synthesis of dimeric ligands with functionalized tethers is summarized in Fig. S1 and S2.† Williamson ether synthesis between the bromoaryl linkers (2, 7, 13, 19, 24) and diethyl 2-hydroxyterephthalate (3) produced the corresponding ester-protected ligands (4, 8, 14, 20, 25). These polyesters were then hydrolysed to produce the desired dimeric linkers (L1–L5) with pyridine tethers. To prepare the alkyne functionalized dimeric linker, a similar synthetic strategy was used (Fig. S2†) to obtain the desired product (L6).In addition to these dimeric ligands, monomeric ligands containing pyridine substituents were synthesized for comparative studies (vide infra), as shown in Fig. S3.† For the synthesis of one ligand (H2bdc-3-py), a nucleophilic substitution reaction between 3-(bromomethyl)pyridine hydrobromide (31) and diethyl 2-hydroxyterephthalate (3) led to the generation of the ester-protected version of the monomeric ligand with a pendant pyridine group (32). The ester group was removed by hydrolysis to generate H2bdc-3-py. The synthesis of the second monomeric ligand (H2bdc-3-ph-4-py) began with a Suzuki-coupling reaction between diethyl 2-((3-bromobenzyl)oxy)terephthalate (36) and pyridin-4-ylboronic acid (18) in the presence of tetrakis(triphenylphosphine)palladium. This produced intermediate 37, which after hydrolysis gave the desired ligand H2bdc-3-ph-4-py.
Synthesis and characterization of oligoIRMOFs from pyridine derivatives (L1–L5, H2bdc-3-py, H2bdc-3-ph-4-py, H2bdc-2-py)
Generally, the direct introduction of heteroatoms (e.g., nitrogen, oxygen) onto the H2bdc ligand scaffold results in MOF structures that do not recapitulate those obtained with H2bdc alone. For example, combining pyridine-2,5-dicarboxylic acid with Zn(II) sources produced a 3-dimensional framework with a nov topology and a Brunauer–Emmett–Teller (BET) surface area of 330 m2 g−1,15 which is much lower than that of the IRMOF systems. Other pyridine dicarboxylate ligands (e.g., pyridine-2,6-dicarboxylic acid16 or pyridine-2,4-dicarboxylic acid17) also produce dense phases with Zn(II), distinct from the IRMOF series and with comparably low surface areas.It was postulated that dimeric linkers with pyridine derivatives incorporated into the tether (L1–L5) could overcome the challenges of including this heterocycle into an IRMOF-type lattice. Various reaction conditions (e.g., solvents such as DMF, DEF; reaction temperatures from 80 °C to 100 °C) were investigated with Zn(II) nitrate hexahydrate as the metal source (Fig. S4–S12†). In the case of L1, utilization of DMF as a solvent and heating at 100 °C for 72 h gave crystalline products (oligoIRMOF-1-L1) with sharp, intense reflections in powder X-ray diffraction (PXRD) studies compared to the other conditions tested (Fig. S4†). Scanning electron microscopy (SEM) images (Fig. S5†) confirm the generation of cubic crystals (∼200 μm). PXRD patterns of the crystals were well-matched with simulated IRMOF-1 (Fig. 3).
 | ||
| Fig. 3 (Top) Schematic representation of the synthesis of oligoIRMOF-1-L1 and oligoIRMOF-1-L3. (Middle left) PXRD patterns of simulated IRMOF-1 (black), oligoIRMOF-1-L1 (red), oligoIRMOF-1-L3 (blue). (Middle right) Nitrogen isotherm of oligoIRMOF-1-L1 (squares) and oligoIRMOF-1-L3 (circles). The filled and unfilled squares correspond to adsorption and desorption, respectively. (Bottom left) Crystal structure of oligoIRMOF-1-L1. (Bottom right) Crystal structure of oligoIRMOF-1-L3. Disordered zinc atoms were omitted for clarity. The pyridine-containing tethers could not be located via SCXRD due to structural disorder. Color scheme: carbon (gray), oxygen (red), zinc (cyan). | ||
Single-crystal X-ray diffraction (SCXRD) structure determination confirmed formation and connectivity of an IRMOF lattice (Fig. 3). The repeating unit of oligoIRMOF-1-L1 shows three dimeric linkers bounded to the expected Zn4O cluster (identical connectivity to IRMOF-1).18 Because of the structural disorder and high symmetry of this crystal system, the position of pyridine tether parts could not be located in the crystal structure of oligoIRMOF-1-L1. However, 1H NMR of a solution of oligoIRMOF-1-L1 digested in a mixture of DMSO-d6 and DCl showed a resonance at 5.6 ppm corresponding to the methylene tether and additional resonances at 8.0 ppm and 8.5 ppm corresponding to the pyridine part of the tether (Fig. S6†), indicating that L1 is indeed intact in these materials. Gas sorption experiments showed that oligoIRMOF-1-L1 gave a BET surface area of 1819 ± 64 m2 g−1 (Fig. 3).
By contrast, the dimer ligand with the pyridine nitrogen atom pointing on the opposite side of the aryl bridge (L2) was unable to produce an oligoIRMOF. Several reaction conditions were explored, including variations in solvent (DMF, DEF) and temperature (80 °C, 100 °C), but none could be found to generate an IRMOF lattice from L2 (Fig. S7†). Microcrystalline powders were obtained from some conditions with L2, but their PXRD patterns (Fig. S7†) did not match that of the IRMOFs and SCXRD data was unavailable because of the small crystallite size.
To further explore the relationship between ligand and oligoMOF structure, other dimer ligands with pyridine tethers were synthesized and studied. Use of a pyridine-phenyl tether (L3) in DMF as a solvent and heating at 100 °C gave a sharp and intense PXRD pattern which was well-matched with simulated IRMOF-1 (Fig. S8†). SEM image of oligoIRMOF-1-L3 displayed cubic-shaped crystals with 200 μm size (Fig. S9†). As shown in Fig. 2, SCXRD analysis of oligoIRMOF-1-L3 confirmed the similar connectivity of IRMOF-1 and oligoIRMOF-1-L1. As expected, the repeating unit in the crystal structure of oligoIRMOF-1-L3 consisted of three functionalized dimers and Zn4O secondary building unit (SBU). Similar to the crystal structure of oligoIRMOF-1-L1, the pyridine tether could not be visualized in the crystal structure due to the positional disorder in this highly symmetric crystal structure. For further verification of the presence of pyridine tether, the crystals of oligoIRMOF-1-L3 were digested in a mixture of DMSO-d6 and DCl, which 1H NMR showed all the expected resonances for an intact L3 (Fig. S10†). Nitrogen isotherms of oligoIRMOF-1-L3 showed reasonable porosity (1410 ± 52 m2 g−1, Fig. 3), which is slightly less than the surface area of oligoIRMOF-1-L1, likely due to the added phenyl group in L3.
The reaction of the pyridine isomer L4 (Fig. 2) with Zn(II) nitrate hexahydrate under various conditions generated a microcrystalline powder. Similar to L2 linker, PXRD patterns of the resulting material did not match with IRMOF-1 lattice (Fig. S11†), indicating the generation of other phases that could not be resolved by SCXRD analysis due to the small crystallite size. Moreover, another pyridine tether isomer (L5) gave only amorphous solids under the various reaction conditions investigated (Fig. S12†).
To further understand the role of the tether and position of the pyridine group in MOF synthesis, a monomeric H2bdc ligand with a pendant pyridine group (H2bdc-3-py) in the same position as L2 was employed under various MOF reaction conditions (solvents: DMF, DEF; reaction temperatures: 80 °C, 100 °C). From among these experiments, the use of DEF as a solvent and heating to 100 °C generated block-shaped crystals (∼100 μm) suitable for the SCXRD analysis (Fig. S13†). The resulting material, designated Zn(H2bdc-3-py) is a new 3-dimensional (3D) MOF. Unlike oligoIRMOF-1-L1 and oligoIRMOF-1-L3 discussed in this study, the Zn SBU adopted in Zn(H2bdc-3-py) is a dinuclear 4-fold symmetric Zn(II) paddlewheel (Fig. 3). The equatorial positions of the paddlewheel SBU are occupied by bridging carboxylate groups from bdc-3-py2− ligands, while the axial sites are occupied by the nitrogen atoms of the 3-pyridyl arms of bdc-3-py2−. Each ligand connects the equatorial positions of two adjacent paddle wheel SBUs with the axial position of an additional paddle wheel SBU, resulting in an overall (6,3)-connected 3D structure with an anatase (ant) topology (Fig. 4 and S13†). Approximately 56% of the cell volume is void space (1.2 Å probe radius), originating from rhomboidal channels (ca. 10 Å × 14 Å) formed along the crystallographic c-axis. The pillaring pyridine rings also form π–π stacking interactions (pyridine ring–ring distance of 3.58 Å) (Fig. 4). The structure of the bulk material was confirmed by PXRD, which was well-matched to the simulated PXRD pattern derived from the SCXRD structure of Zn(H2bdc-3-py) (Fig. S14†). These control experiments with H2bdc-3-py show that constraining the pyridine group via a tether in the dimeric ligands plays an important role in the resulting oligoMOF material.
 | ||
| Fig. 4 (Top left) Crystal structure of Zn(H2bdc-3-py) along (010) plane. (Top right) Packing structure of Zn(H2bdc-3-py) along the crystallographic a-axis (100). (Bottom) Packing structure of Zn(H2bdc-3-py) along the c-axis (001). Color scheme: carbon (gray), nitrogen (blue), oxygen (red), zinc (cyan). | ||
In addition, a monomer ligand with pyridine in the same position as L4 (H2bdc-3-ph-4-py) was prepared and explored for MOF synthesis under similar conditions. With H2bdc-3-ph-4-py, only polycrystalline powders were obtained, which were also not suitable for SCXRD analysis. PXRD patterns of the products formed with H2bdc-3-ph-4-py do not match the IRMOF-1, or those of Zn(H2bdc-3-py) (Fig. S15†). Another monomeric ligand with a pyridine tether (H2bdc-2-py) in the same position as L1 was prepared. Similar to H2bdc-3-ph-4-py, Materials generated using H2bdc-2-py with a Zn(II) source under various reaction conditions gave PXRD patterns with rather broad peaks (Fig. S16†) that did not match either simulated IRMOF-1 or Zn(H2bdc-3-py). Consistent with its dimer analog, H2bdc-2-py did not produce an IRMOF-1 lattice, which further indicate that the position, geometry, and constraints of the pyridine group impact the structure of the resulting MOFs. Collectively, these results highlight the importance of the appropriate position of nitrogen atom in the tether for producing crystalline MOFs. Of the various pyridine isomers studied (L1–L5, H2bdc-3-py, H2bdc-3-ph-4-py, H2bdc-2-py), only L1 and L3, which share the feature of the 2,6-pyridine motif, were found to be suitable for generating highly porous and crystalline oligoIRMOF structures.
Synthesis and characterization of oligoIRMOF-1-L6
The introduction of alkyne groups in the organic linkers of MOFs has enable many studies in PSM. For example, Hupp and Nguyen were among the earliest to demonstrate copper(I) catalyzed azide–alkyne click chemistry on a MOF.19,20 By using a Zn(II)-based MOF consisting of 1,2,4,5-tetrakis(4-carboxy-phenyl) benzene and 3-[(trimethylsilyl)ethynyl]-4-[2-(4-pyridinyl)ethenyl]pyridine linkers, click PSM reactions with benzyl or ethidium bromide azide reagents could be performed.21 Similarly, Shao and co-workers described H2bdc ligands with one or two alkynes could form UiO-66 MOFs22,23 that could act as ligands upon PSM metalation with Ni(PPh3)2Cl2 and Co(PPh3)2Cl2. Sada and co-workers described an IRMOF-16 derivative containing 2′,5′-bis(azidomethyl)-[1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylic acid (AzTPDC) as an organic linker to perform PSM with alkyne derivatives such as propagyl alcohol, methyl propiolate, and pentaerythritoltetrapropargyl ether bearing four alkyne groups as a cross-linker.24,25 PSM in this system results in cross-linking of the MOF ligands, that can subsequently be demetallated to obtain unique polymer gel monoliths that maintain some characteristics of the crystalline MOF precursor.25For the preparation of oligoIRMOFs with an alkyne group in the tether, L6 (Fig. 2) was combined with Zn(II) nitrate hexahydrate in DMF at 100 °C to give cubic crystals of oligoIRMOF-1-L6 (Fig. S17†). PXRD of the resulting material was well matched with simulated IRMOF-1 (Fig. 5). Crystals of oligoIRMOF-1-L6 suitable for SCXRD confirmed the isostructural nature of oligoIRMOF-1-L6 with IRMOF-1 (Fig. 5). Again, it should be noted that the xylyl tether units could not be located in the SCXRD structure, which is typical of substituted IRMOF materials.8,26 However, the ether oxygen atoms of the tethers, which are directly connected to bdc2− units, could be modelled in the SCXRD of oligoIRMOF-1-L6 with positional disorder.8,26,27 Activated oligoIRMOF-1-L6 displayed a BET surface area of 1852 ± 72 m2 g−1 (Fig. S17†). To confirm the integrity of the alkyne group in oligoIRMOF-1-L6, the crystals were digested in a mixture of DMSO-d6 and DCl (35% solution in D2O). 1H NMR of the digested solution showed resonances at 5.2 ppm and 4.2 ppm corresponding to the methylene bridge and terminal alkyne in L6, respectively (Fig. S18†). In addition, infrared spectroscopy (FTIR) of oligoIRMOF-1-L6 showed a characteristic alkyne stretching band at 3285 cm−1 (Fig. S19†).
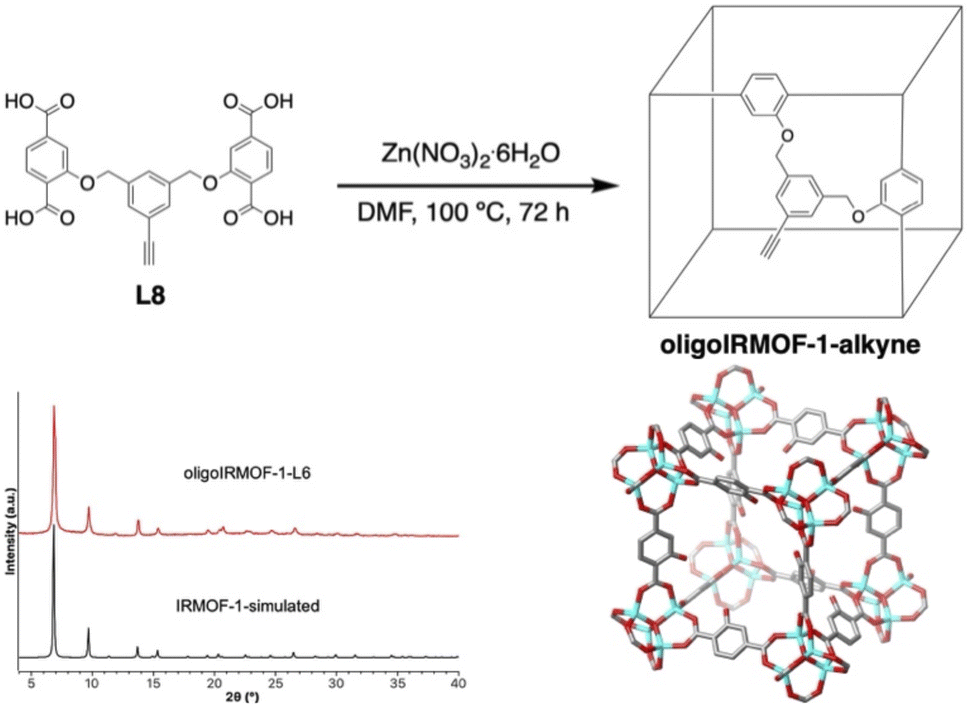 | ||
| Fig. 5 (Top) Synthesis of oligoIRMOF-1-L6. (Bottom left) PXRD patterns of simulated IRMOF-1 (black) and oligoIRMOF-1-L6 (red). (Bottom right) SCXRD structure of oligoIRMOF-1-L6. Only one position of the disordered ether oxygen is shown on each bdc2− ligand. Color scheme: carbon (gray), oxygen (red), zinc (cyan). | ||
PSM of oligoIRMOF-1-L6
To demonstrate the utility of the alkyne-functionalized tether for pore tuning, PSM of oligoIRMOF-1-L6 with benzyl azide was performed in the presence of CuBr at different time intervals (6, 12, 24, and 48 h, Fig. 6 and S21†). The conversion for this PSM reaction was monitored by 1H NMR of digested solutions of the MOF crystals (digested in a mixture of DMSO-d6 and DCl, Fig. S21†). Full PSM conversion was achieved at 80 °C after 48 h. | ||
| Fig. 6 (Top) Postsynthetic modification (PSM) of oligoIRMOF-1-L6 with benzyl azide, 2-(azidomethyl)pyridine, and 4-(azidomethyl)-1,1′-biphenyl. (Bottom left) PXRD patterns of simulated IRMOF-1 (black), oligoIRMOF-1-L6 (red), oligoIRMOF-1-benzyltriazole (blue), oligoIRMOF-1-pyridinetriazole (green), oligoIRMOF-1-biphenyltriazole (orange). (Bottom right) Nitrogen isotherm of oligoIRMOF-1-L6 (red squares), oligoIRMOF-1-benzyltriazole (blue circles), oligoIRMOF-1-pyridinetriazole (green triangles), oligoIRMOF-1-biphenyltriazole (orange diamonds). The filled and unfilled squares correspond to adsorption and desorption, respectively. | ||
The PSM click reaction products with benzyl azide were characterized by FTIR spectroscopy of the resulting crystals (oligoIRMOF-1-benzyltriazole), where the alkyne stretch at 3285 cm−1 was no longer present (Fig. S22†). OligoIRMOF-1-benzyltriazole was digested in a mixture of DMSO-d6 and DCl and analysed by 1H NMR and ESI-MS (Fig. S23†). The 1H NMR spectrum of the digested solution showed the resonance corresponding to the terminal alkyne at 4.2 ppm was no longer present and new resonances related to the triazole proton at 8.6 ppm and the methylene bridge adjacent to the triazole ring at 5.6 ppm were observed. ESI-MS analysis produced a molecular ion peak corresponding to the triazole compound (m/z 622.24). In addition, the crystallinity and porosity of the resulting oligoIRMOF-1-benzyltriazole were retained (Fig. 6), with a BET value of 1467 ± 53 m2 g−1, which is only slightly lower than the precursor material (oligoIRMOF-1-L6, 1852 ± 72 m2 g−1).
To demonstrate the generality and potential utility of this PSM reaction, oligoIRMOF-1-L6 was treated with other organic azides. Pyridine-derived azides were selected because pyridine dicarboxylic acid derivatives frequently are unable to form IRMOF topologies, as described above.15,28–30 As shown in Fig. 6, PSM of oligoIRMOF-1-L6 was achieved with 2-(azidomethyl)pyridine at 80 °C for 48 h. As with benzylazide, the stretching band of alkyne in the FTIR spectrum of the resulting crystals (oligoIRMOF-1-pyridinetriazole) was no longer observed (Fig. S24†). Digested crystals after PSM also showed a loss of the terminal alkyne resonance at 4.2 ppm in the 1H NMR spectrum of the digested sample, and the appearance of new resonances that correspond with the triazole proton in the desired product at 8.8 ppm (Fig. S25†). In addition, ESI-MS of the digested solution also displayed a molecular peak ion at m/z 623.19, which matches the expected value for the PSM modified dimer ligand. As with oligoIRMOF-1-benzyltriazole, the PXRD pattern of oligoIRMOF-1-pyridinetriazole was well-matched with simulated IRMOF-1 (Fig. 6). Finally, the modified oligoMOF also maintained high porosity (1333 ± 42 m2 g−1) after the attachment of pyridine group (Fig. 6).
To test the steric limits of the Cu(I) catalyzed click reaction, oligoIRMOF-1-L6 was treated with 4-(azidomethyl)-1,1′-biphenyl at 80 °C for 48 h (Fig. 6). Again, the FTIR spectrum of the resulting MOF (oligoIRMOF-1-biphenyltriazole) displayed a loss of the alkyne stretching band (Fig. S26†). However, 1H NMR of the digested oligoMOF indicated the presence of unreacted alkyne (Fig. S27†). Based on the integration values of the alkyne resonance at 4.2 ppm and the resonance associated with the methylene bridge adjacent to the triazole ring at 5.7 ppm, the calculated conversion for this reaction is ∼92%. ESI-MS of the digested solution displayed the molecular ion peak corresponding to both the dimer ligand with the biphenyl substituent (m/z 698.24) and the unreacted alkyne ligand (L6, m/z 489.16). Although PSM was incomplete under these conditions, the PXRD pattern of oligoIRMOF-1-biphenyltriazole showed good retention of crystallinity, and a BET surface area of 1263 ± 62 m2 g−1 (Fig. 6). The reduced yield of this PSM reaction is consistent with molecular crowding of these larger substituents within the pores of the oligoMOF.
SCXRD of the PSM click modified oligoMOFs were collected, and all showed the expected framework consisting of bdc2− linkers and Zn4O SBUs consistent with an IRMOF topology (Fig. S23, S25 and S27†). As with the parent oligoMOF, structural disorder and high symmetry prevented observation of the tethering groups or PSM substituents; however, the ether oxygen atoms were located in the electron density maps of all three oligoMOFs. To evaluate the copper content of the oligoMOFs after the Cu(I) catalysed PSM reaction, the three modified oligoMOFs (oligoIRMOF-1-phenyltriazole, oligoIRMOF-1-pyridinetriazole, oligoIRMOF-1-biphenyltrizole) were evaluated by inductively coupled plasma mass spectrometry (ICP-MS). The materials were activated by solvent exchange with CH2Cl2, and dried under vacuum for 18 h, after which the crystals were digested in a mixture of nitric acid and hydrogen peroxide for ICP-MS analysis. The digested solutions of three MOFs showed only small amounts of copper (0.2%, Table S1†), which is only slightly higher compared to those of unmodified oligoIRMOF-1-L6 (0.1%) and unmodified oligoIRMOF-1-alkyne treated with CuBr at 80 °C for 48 h (0.2%). This indicates that most residual copper can be removed by washing these materials after PSM. Taken together, these results demonstrate that oligoIRMOF-1-L6 is a suitable platform for introducing diverse functional groups into the pores of these materials, while maintaining crystallinity and high porosity of the resulting materials.
PSM of oligoIRMOF-1-L6 with diazido cross-linkers
In an attempt to cross-link two alkyne tethers, as well as gain insight into the relative orientation of these groups in the oligoMOF pores, xylyl isomers having two azide groups were used in click reactions with oligoIRMOF-1-L6. PSM of oligoIRMOF-1-L6 was conducted with ortho-diazido xylene at 80 °C for 48 h (Fig. 7). The crystals (oligoIRMOF-1-triazole2-o-xylyl) after the reaction were analyzed by FTIR showed bands corresponding to azide functional groups at 2098 cm−1 and a notable absence of the alkyne stretching band at 3285 cm−1 (Fig. S28†). 1H NMR of digested solution of the PSM reaction showed a mixture of products. One set of resonances corresponded to the desired compound with two triazole groups (oligoIRMOF-1-triazole2-o-xylyl) (Fig. S29†). A second set of resonances corresponded to a partially reacted intermediate (oligoIRMOF-1-triazole-o-xylyl) where only one azide group had undergone PSM. The ratio between the NMR resonances of the two products (specifically resonances at 5.9 ppm and 5.7 ppm versus at 4.7 ppm) indicated 45 ± 1.3% of the desired compound (oligoIRMOF-1-triazole2-o-xylyl) and 54 ± 1.3% of partially converted product (oligoIRMOF-1-triazole2-o-xylyl) were obtained. ESI-MS of the digested products showed peaks related to the intermediate compound (oligoIRMOF-1-triazole-o-xylyl) and the desired oligoIRMOF-1-triazole2-o-xylyl product at m/z 701.22 and 1191.32, respectively. After PSM, the PXRD pattern of the resulting oligoMOF showed that crystallinity was retained, which was further supported by the BET surface area of 1291 ± 30 m2 g−1 (Fig. 7). | ||
| Fig. 7 (Top) PSM of oligoIRMOF-1-L6 with diazido xylene isomers. The contents of each compound in this PSM reaction were determined by solution 1H NMR of the digested reaction products. (Bottom left) PXRD patterns of simulated IRMOF-1 (black), oligoIRMOF-1-L6 (red), oligoIRMOF-1-triazole2-o-xylyl (blue), oligoIRMOF-1-triazole2-m-xylyl (green), oligoIRMOF-1-triazole2-p-xylyl (orange). (Bottom right) Nitrogen isotherm of oligoIRMOF-1-L6 (red squares), oligoIRMOF-1-triazole2-o-xylyl (blue circles), oligoIRMOF-1-triazole2-m-xylyl (green triangles), oligoIRMOF-1-triazole2-p-xylyl (orange diamonds). The filled and unfilled squares correspond to adsorption and desorption, respectively. | ||
Cu(I) catalyzed PSM between oligoIRMOF-1-L6 and meta-diazido xylene at 80 °C for 48 h gave similar results to the reaction with ortho-azido xylene (Fig. S30†). Based on the 1H NMR of the digested products (Fig. S31†), the products of this PSM reaction are 53 ± 1.1% of oligoIRMOF-1-triazole2-m-xylyl and 47 ± 1.1% of oligoIRMOF-1-triazole-m-xylyl. ESI-MS spectrum also supported a mixture of oligoIRMOF-1-triazole2-m-xylyl and oligoIRMOF-1-triazole-m-xylyl, with molecular ion peaks observed at m/z 1191.31 and 701.20, respectively. This oligoMOF also maintained crystallinity and porosity after the PSM reaction (1314 ± 31 m2 g−1) (Fig. 7).
Finally, the PSM reaction of oligoIRMOF-1-L6 with para-diazido xylene also gave similar results to the other isomers (Fig. S32†), with a mixture of products (oligoIRMOF-1-triazole2-p-xylyl) and incomplete cross-linking (oligoIRMOF-1-triazole-p-xylyl). 1H NMR of the digested product indicated that both 44 ± 1.4% of desired compound (oligoIRMOF-1-triazole2-p-xylyl) and 56 ± 1.4% of incomplete product (oligoIRMOF-1-triazole-p-xylyl) were formed (Fig. S33†), which was further confirmed by ESI-MS showing molecular ion peaks of both compounds at m/z 1191.28 and 701.19.
After PSM with ortho-, meta-, and para-diazido xylene isomers, the oligoIRMOFs were analyzed by SCXRD (Fig. S29, S31 and S33†). In all structures, the position of xylyl tether and triazole functional groups could not be located in the electron density map, as observed with oligoIRMOF-1-L6. Overall, these cross-linking PSM reactions show little difference between the ortho-, meta-, and para-xylenes, with perhaps a slightly lower conversion with the para-isomer (Fig. 7). This suggests that there are a variety of tether orientations, and that the alkyne groups within the oligoMOF pores can accommodate the different geometries and distances in these diazide cross-linking reagents. This is consistent with the observed disorder of the bridging units in the SCXRD structure of oligoIRMOF-1-alkyne.
Conclusions
Functional groups such as pyridine and alkyne groups have been introduced into the tethering bridges of oligoIRMOFs. Dimer linkers with pyridine derivatives as tether units with different positions of nitrogen in aromatic rings and variations of the lengths of tether groups (L1–L5) and monomer linker with pyridine (H2bdc-3-py, H2bdc-3-ph-4-py, H2bdc-2-py) were utilized for the preparation of oligoIRMOFs. Among them, L1 and L3 linkers produced highly crystalline oligoIRMOF-1-L1 and oligoIRMOF-1-L3 phases, possessing an IRMOF-1 lattice and connectivity. Interestingly, ligands with other pyridine isomers on the tether were unable to generate and IRMOF-1 like material. A monomeric ligand with pyridine tether (H2bdc-3-py) was shown to generate a new MOF, Zn(H2bdc-3-py), with different connectivity compared to IRMOF-1, in which pyridine spacer in H2bdc-3-py act as ‘built in’ pillaring linkers. In addition, a dimer linker with a terminal alkyne (L6) led to the generation of crystalline and highly porous oligoIRMOF-1 with accessible alkyne groups in the pores of oligoIRMOF-1-L6. PSM was conducted to convert the alkyne decorated oligoIRMOF-1-L6 to the corresponding triazole-bearing MOFs with various functional groups or to cross-link the MOF structure. Cross-linking PSM experiments suggest that there are a variety of relative orientations of the alkyne groups in oligoIRMOF-1-L6. Together, this report demonstrates the utility of the tethering group in oligoMOFs to serve as a location for functional groups that can modulate MOF structure, function, and provide a handle for further modification of oligoMOF properties.Data availability
X-ray data as .cif files and gas sorption data as .aif files are available.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under Award No. DE-FG02-08ER46519. We thank Dr Yongxuan Su for mass spectrometry sample analysis at the Molecular Mass Spectrometry Facility at U. C. San Diego, Dr Jake B. Bailey for helpful discussion of disordered crystal structure at UCSD Crystallography Facility, and Dr Debobroto Sensharma for helpful discussion of topology in crystal structure of Zn(H2bdc-3-py).Notes and references
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- W. G. Lu, Z. W. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. W. Zhang, Q. Zhang, T. Gentle, M. Bosch and H. C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- Z. J. Zhang, H. T. H. Nguyen, S. A. Miller and S. M. Cohen, Angew. Chem., Int. Ed., 2015, 54, 6152–6157 CrossRef PubMed.
- G. E. M. Schukraft, S. Ayala, B. L. Dick and S. M. Cohen, Chem. Commun., 2017, 53, 10684–10687 RSC.
- S. Ayala, Z. J. Zhang and S. M. Cohen, Chem. Commun., 2017, 53, 3058–3061 RSC.
- S. Ayala, K. C. Bentz and S. M. Cohen, Chem. Sci., 2019, 10, 1746–1753 RSC.
- C. A. Allen and S. M. Cohen, Inorg. Chem., 2014, 53, 7014–7019 CrossRef CAS PubMed.
- R. A. Dodson, J. Park, J. Kim, M. J. Cliffe and S. M. Cohen, Inorg. Chem., 2022, 61, 12284–12292 CrossRef CAS PubMed.
- H. Kim and S. M. Cohen, Inorg. Chem., 2024, 63, 1853–1857 CrossRef CAS PubMed.
- J. Geary, A. H. Wong and D. J. Xiao, J. Am. Chem. Soc., 2021, 143, 10317–10323 CrossRef CAS PubMed.
- J. Geary, J. P. Aalto and D. J. Xiao, Chem. Mater., 2024, 36, 3949–3956 CrossRef.
- D. S. Rollins, J. Geary, A. H. Wong and D. J. Xiao, Chem. Commun., 2022, 58, 12361–12364 RSC.
- L. Feng, K. Y. Wang, X. L. Lv, J. A. Powell, T. H. Yan, J. Willman and H. C. Zhou, J. Am. Chem. Soc., 2019, 141, 14524–14529 Search PubMed.
- V. I. Isaeva, E. V. Belyaeva, A. N. Fitch, V. V. Chernyshev, S. N. Klyamkin and L. M. Kustov, Cryst. Growth Des., 2013, 13, 5305–5315 Search PubMed.
- H. L. Gao, L. Yi, B. Zhao, X. Q. Zhao, P. Cheng, D. Z. Liao and S. P. Yan, Inorg. Chem., 2006, 45, 5980–5988 CrossRef PubMed.
- Z. G. Li, G. H. Wang, H. Q. Jia, N. H. Hu and J. W. Xu, Crystengcomm, 2007, 9, 882–887 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef.
- T. Gadzikwa, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 13613–13615 CrossRef PubMed.
- H. Cheng, L. M. Ning, S. Y. Liao, W. Li, S. Y. Tang, J. L. Li, H. X. Chen, X. Liu and L. M. Shao, Appl. Catal., A, 2021, 623, 118216 CrossRef.
- Q. Q. Liang, H. Cheng, C. W. Li, L. M. Ning and L. M. Shao, New J. Chem., 2022, 46, 1210–1221 RSC.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354–14355 CrossRef.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427–5432 CrossRef.
- Z. Q. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368–12369 CrossRef.
- P. Mondal, Z. Neuschuler, D. Mandal, R. E. Hernandez and S. M. Cohen, Angew. Chem., Int. Ed., 2024, 63, e202317062 CrossRef PubMed.
- E. E. Sileo, O. E. Piro, G. Rigotti, M. A. Blesa, A. S. de Araujo and E. E. Castellano, Struct. Chem., 2008, 19, 651–657 CrossRef.
- S. L. Huang, L. Zhang, Y. J. Lin and G. X. Jin, CrystEngComm, 2013, 15, 78–85 RSC.
- E. E. Sileo, A. S. de Araujo, G. Rigotti, O. E. Piro and E. E. Castellano, J. Mol. Struct., 2003, 644, 67–76 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, instrumentation, organic linker, MOF synthesis, characterization detail, crystallographic data, and refinement description. CCDC 2387819–2387828. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06666a |
| This journal is © The Royal Society of Chemistry 2025 |