
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVacancy formation, stability, and electronic properties of nickel on equimolar ceria–zirconia mixed oxide (111) catalyst†
Sanjana
Srinivas
 ab,
George
Yan
b,
Stavros
Caratzoulas
b and
Dionisios G.
Vlachos
*ab
ab,
George
Yan
b,
Stavros
Caratzoulas
b and
Dionisios G.
Vlachos
*ab
aDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy St., Newark, DE 19716, USA. E-mail: vlachos@udel.edu
bDelaware Energy Institute, University of Delaware, 221 Academy St., Newark, DE 19716, USA
First published on 25th April 2025
Abstract
Nickel (Ni) single atoms and small clusters dispersed on Ce0.5Zr0.5O2 (CZO) are promising for the dry reforming of methane (DRM). However, understanding of defects and electronic interactions between Ni motifs and (non) stoichiometric CZO surfaces is limited. Here, we use Density Functional Theory (DFT) and ab initio thermodynamics to investigate vacancy formation and electronic properties of Ni1 and Ni4 on CZO(111). Surface and subsurface oxygen vacancies near Zr4+ ions dominate due to distortion-induced stabilization across DRM-relevant chemical potentials; vacancies prefer to be subsurface. Interestingly, Ni single atoms coordinate with two OZr surface atoms on CZO(111) (NiCe2Zr), unlike three-fold coordinated Ni atoms on hollow sites of CeO2(111). Ni1 does not directly bind to oxygen vacancies due to its oxophilicity and steric hindrance caused by multiple surface Ce3+ ions. Clusters, on the other hand, can bind favorably at a surface oxygen vacancy. Ni adatoms are more stable than Ni4 at trimer defects comprising one Ce and two oxygen vacancies (Cev(Ov)2), at the pristine surface, and at the NiCe2Zr site with a next-nearest neighbor oxygen vacancy due to coordination with more oxygen atoms. The extent of electron transfer from the metal to the surface and, thus, the degree of cationic character of a nickel adatom varies with its location and defect type and correlate positively with its resistance to sintering. We discuss the expected heterogeneity of actual catalysts.
Introduction
The recent shale gas boom in the US,1 the growing need to reduce carbon emissions, and the importance of hydrogen in transitioning to a renewable energy economy2,3 make the dry reforming of methane (DRM) (CH4 + CO2 ↔ 2H2 + 2CO) an attractive route to produce hydrogen. Reducible metal oxides are potential catalyst supports as they allow facile oxygen vacancy formation and replenishment in the Mars–van Krevelen-type mechanism. CeO2 and mixed CeO2–ZrO2 oxides (CexZr1−xO2) have been shown to be excellent supports for this reaction,4 owing to their high oxygen storage capacity (OSC). Liu et al.5 have shown that CexZr1−xO2 is easier to reduce because Zr is smaller than Ce, which introduces lattice distortions and weakens the Ce–O bonds.6,7 Homogeneously dispersed Zr4+ in the CeO2 lattice at a Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr ratio of 1:1 (hereafter referred to as CZO) shows the highest reducibility and OSC.7,8
:Zr ratio of 1:1 (hereafter referred to as CZO) shows the highest reducibility and OSC.7,8
Highly dispersed nickel on CZO is an attractive catalyst for DRM because of nickel's abundance, high activity, and resistance to coking.9–11 The complex electronic structure of this catalyst remains largely unexplored. Different defects (e.g., oxygen and/or cerium vacancies) can be stabilized during DRM's reduction and oxidation half cycles, but the dominant defect configurations and their distribution are unknown. The nuclearity of Ni clusters could influence the relative stability of defects with potential ramifications for catalysis, but a practical catalyst will often contain a broad distribution of Ni cluster nuclearities. These complexities ultimately hinder a detailed mechanistic understanding, as the interpretation of spectroscopic data for detecting defect-Ni interactions hinges on the structural homogeneity of the catalyst. Furthermore, state-of-the-art experimental techniques have yielded limited information about the active site and its local coordination.12,13 First-principles simulations could provide insights into the physicochemical interactions at the atomic scale.
To shed light on the structure of Ni/CZO catalysts, we use density functional theory (DFT) to electronically characterize vacancy formation as well as nickel adsorption on pristine and defected CZO surfaces. By evaluating the vacancy formation and nickel adsorption energies, we gain insights into the relative stability of the nickel single atom and clusters on different surfaces. This paper is organized as follows: first, we identify the location and configuration of stable oxygen vacancies on the CZO(111) surface, then we investigate the possibility of cerium vacancies and discuss the electronic and geometric properties of defected surfaces. To predict surface structure under experimentally relevant environments, we investigate the stability of different defects at finite temperatures and varying oxygen partial pressure. Next, we assess the stability of a Ni adatom and Ni4 cluster on stoichiometric and non-stoichiometric CZO(111) and vacancy formation in the presence of nickel and provide insights into nickel–support electronic interactions. Last, we discuss the interplay between Ni binding and vacancy formation under different oxygen conditions.
Methodology
Periodic electronic structure calculations
All electronic structure calculations were performed within the spin-polarized DFT framework, using the Vienna ab initio simulation package (VASP),14,15 version 5.4.1. The exchange–correlation energy was calculated using the Perdew–Burke–Ernzerhof (PBE)16 functional within the generalized gradient approximation (GGA). The core electrons of each atom were described by the projector-augmented wave function (PAW)17,18 method, and the VASP-recommended pseudopotentials (v54) were used for all elements. The one-electron wave functions were developed on a plane-wave basis set with an energy cutoff of 500 eV. The Ce 4f, 5s, 5p, 5d, and 6s electrons; the O 2s and 2p electrons; the Ni 3d and 4s electrons; and the Zr 4s, 4p, 4d, and 5s electrons were treated as valence electrons. The Brillouin zone was sampled at the gamma point. A maximum force convergence criterion of 0.05 eV Å−1 was used, and each self-consistency loop was iterated until reaching a convergence level of 10−6 eV. It is well known that the GGA method incorrectly reproduces the band gaps of CeO2 and ZrO2 and fails to accurately describe the localization of Ce 4f states upon oxygen vacancy formation in CeO2, on account of the electrons being strongly correlated in the Ce 4f state. To remedy this, we adopt the DFT+U formalism,19,20 which has been extensively benchmarked and shown great success in describing vacancy formation, dopant behavior, and catalytic activity on CeO2. Informed by the vast literature on CeO2 and less so on ZrO2, we select U = 5 eV for the Ce 4f states21 and U = 4 eV for the Zr 4d states.22 Similar to the approach presented by Livraghi et al.,23 our work also employs the DFT+U formalism for Zr in CZO, as GGA functionals incorrectly reproduce the bandgap for early transition metal oxides like ZrO2. For comparison, we compute the surface and subsurface vacancy formation energy on a p(4 × 4) CeO2(111) surface and the bulk vacancy formation energy in a (2 × 2 × 2) supercell of bulk CeO2 to ensure similar oxygen vacancy concentrations, using the same settings. Since multiple local minima with different Ce3+ configurations can exist upon reduction or nickel adsorption, we explored the thermodynamically preferred arrangements of Ce3+ sites, focusing on those nearest and next-nearest to the adsorbed nickel species. To generate these configurations, we employed a two-step structural optimization: we first biased the reduction by substituting the target Ce atoms with La, followed by restoring Ce and performing a final relaxation to obtain the desired Ce3+ configuration.24Bulk and surface models of CZO
Two types of bulk phases are often considered in modeling CZO: the κ-Ce2Zr2O8 phase, which has segregated blocks of CeO2 and ZrO2 units as viewed along [001], and the t-Ce2Zr2O8 phase, which has uniformly distributed Ce4+ and Zr4+ ions, as viewed along [001]. The high OSC of the κ-Ce2Zr2O8 phase with its electronic and geometric origins has been widely studied by DFT.25,26 While segregation has been suggested at high temperatures under oxidizing/reducing environments by EXAFS studies,8,27 its extent, dependence on the environment and temperature, and the synthesis procedure remain elusive. Computational studies of bulk CZO have shown low energy gains from phase segregation compared to the random cation arrangement, which can exhibit multiple local minima.28 Therefore, in this work, we assume uniform ordering of Ce4+ and Zr4+ cations in bulk and investigate surfaces cut from the less studied t-phase. To model the CZO unit cell, we substitute Zr atoms for two Ce atoms in the unit cell of cubic-fluorite CeO2. The bulk structure, which is a (2 × 2 × 2) supercell of the cubic unit cell is shown in Fig. S1.† Bulk oxygen vacancy formation energies are computed using this structure. The structure is only slightly distorted from the cubic fluorite to a tetragonal geometry (c/a = 1.005),28 resulting in uniform surface cuts.From the aforementioned bulk structure, we carved out the perfect crystal's most stable (111) and (110) surfaces. We constructed a 9-layer slab (3 O–Ce–O tri-layers) with the 3rd tri-layer frozen during optimization for the (111) surface and a 6-layer slab with the bottom two layers fixed during optimization for the (110) surface. For both surfaces, we used a p(2 × 2) slab and a 20 Å thick vacuum layer. The (111) surface is 10.2 J m−2 more stable than the (110), like in stoichiometric CeO2.29 The lower energy of the (111) termination can be attributed to the Ce and Zr atoms each having one less bond with lattice O (7-coordinate surface atoms) whereas, on the (110) surface, each metal atom has two less bonds with O (6-coordinate surface atoms) (Fig. S2†). All subsequent analysis on vacancy formation and Ni adsorption was done on the (111) surface. The top-view of the CZO(111) surface is shown in Fig. 1a.
 | ||
| Fig. 1 (a) Top-view of the p(2 × 2) CZO(111) surface, and (b) CZO(111) surface showing the four types of oxygen atoms involved in vacancy formation. (O, red; Ce, yellow; Zr, green). | ||
Results and discussion
Formation, structure, and stability of O and Ce vacancies without Ni
The (111) surface of CZO has two types of surface O atoms: O coordinated to two Ce and one Zr atom (hereafter OsCe), and O coordinated to two Zr and one Ce atom (hereafter OsZr). The subsurface O atoms are coordinated to two Ce and two Zr atoms; three atoms lie in the same tri-layer as the oxygen, and one in the layer below it. Therefore, subsurface oxygens are of two types as well: one, where the same tri-layer atoms comprise two Ce and one Zr atom (hereafter OsubCe) and the other with two Zr and one Ce atom in the same layer (hereafter OsubZr). The bulk oxygen atoms are uniform. Fig. 1b shows the types of oxygen atoms. Visual inspection shows that the OsubZr and OsZr atoms are displaced downward along the surface-normal compared to the OsubCe and OsCe atoms (Table S1†), resulting in elongated Ce–OZr bonds (by ∼0.06 Å).Assuming a neutral oxygen vacancy ( , in Kröger–Vink notation), its formation energy is independent of the Fermi level and given by eqn (1)
, in Kröger–Vink notation), its formation energy is independent of the Fermi level and given by eqn (1)
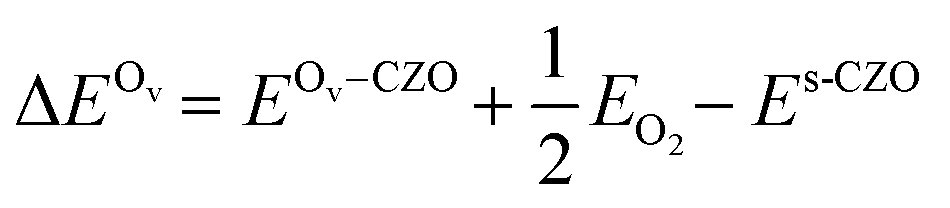 | (1) |
| ΔEOv (eV) | OCe–CZO | OZr–CZO | O–CeO2 |
|---|---|---|---|
| Bulk | 2.69 | 2.69 | 2.79 |
| Subsurface | 1.81 | 1.22 | 1.98 |
| Surface | 2.06 | 1.42 | 2.02 |
Energetically speaking, OZr vacancies are easier to form than OCe vacancies on both the surface and subsurface (Table 1). Among the OZr vacancies, the subsurface O vacancies are more stable than those on the surface, and the bulk vacancies are the least stable, similar to what has been observed for CeO2(111).30 Comparing the oxygen vacancy formation energies in Table 1, Zr doping clearly facilitates their formation in CZO(111) compared to CeO2(111). The preference of an oxygen vacancy in the subsurface near a Zr cation has been reported previously on Zr-doped CeO2(111) surfaces.31 The relative stability trends between OZr and OCe vacancies remain the same across different U values for the Ce f-orbitals (Table S2†).
Next, we consider the energetics for cerium vacancy formation on CZO(111) and compare with previous studies of cerium vacancies on CeO2(111), particularly their interactions with Au and Ir single atoms,21,32 as well as with the Ni-substituted surface examined in this work. Assuming a neutral defect (Ce×Ce + O2 → CeO2(s) + V×Ce), its formation energy is given by eqn (2)
 | (2) |
 is the total energy of bulk ceria per chemical formula. With ΔECev = 4.1 eV, V×Ce formation in CZO(111) is more facile than in undoped CeO2(111) by 0.6 eV but still unlikely compared to oxygen vacancy formation.
is the total energy of bulk ceria per chemical formula. With ΔECev = 4.1 eV, V×Ce formation in CZO(111) is more facile than in undoped CeO2(111) by 0.6 eV but still unlikely compared to oxygen vacancy formation.
The data raises the question: how does Zr facilitate oxygen vacancy formation in CZO(111)? Upon forming an O vacancy, the surface distorts, and two electrons are left localized in two Ce f orbitals, forming polarons.30,33 Here, a polaron is characterized by electron localization at a Ce site and a radially outward relaxation of O surrounding the site. Therefore, following the formalism of Wang et al.,34 we decompose the oxygen vacancy formation energy (ΔEOv) into the energy required to remove oxygen from the fixed CZO(111) structure (ΔEbond) and the energy gained from relaxing the atoms upon oxygen removal (ΔEstrain). The energies are given by eqn (3) and (4):
 | (3) |
 | (4) |
| Oxygen atom removed | ΔEbond (eV) | ΔEstrain (eV) |
|---|---|---|
| OsubZr | 4.07 | −2.85 |
| OsZr | 5.08 | −3.66 |
| OsubCe | 4.34 | −2.53 |
| OsCe | 4.29 | −2.22 |
We now turn to electronic and geometric analyses to further understand the role of Zr in stabilizing oxygen vacancy formation. Fig. 2 shows non-stoichiometric CZO(111) surface structures with the single oxygen vacancy and the corresponding projected density of states (pDOS) on the Ce f orbitals. The cerium atoms stabilizing polarons are highlighted. The stability of multiple possible configurations of Ce3+ ions around the vacancy is evaluated, following the procedure in Otero et al.31 The peaks just below the Fermi level in the pDOS arise from the reduction of Ce4+ to Ce3+. Fig. 2a shows that for the vacancy at OsubZr, the electrons are localized on dissimilar Ce atoms: one is a 6-coordinated surface atom adjacent to the vacancy and the other is a 7-coordinated surface atom away from the vacancy (although one of the Ce–O bonds is elongated to 2.8 Å, so it is not entirely 7-coordinated). The higher-energy f-orbital belongs to the 7-coordinate Ce3+. This is because the reduction of Ce4+ with a higher number of oxygen neighbors weakens the Ce–O ionic bond more. Similarly, for the vacancy at OsZr, the electrons localize on Ce atoms with a seemingly less pronounced dissimilarity due to both electrons having the same spin; the Ce atom away from the vacancy also has one of its Ce–O bonds elongated to 2.8 Å, making it closer to a 6-fold coordination (Fig. 2b). The vacancies at OsubCe and OsCe are accompanied by electron localization on indistinguishable surface Ce atoms, and the f-states are nearly degenerate (Fig. 2c and d). The Ce f orbitals all have similar energies, making it challenging to determine the role of Zr in oxygen vacancy formation. Hence, we turn to geometric analysis to probe the ΔEstrain noted above.
 | ||
| Fig. 2 Defective CZO(111) surface showing the vacancy and reduced cerium atoms and Ce pDOS on its f states for (a) OsubZr vacancy, (b) OsZr vacancy, (c) OsubCe vacancy, and (d) OsCe vacancy. (O, red; Ce, yellow; Zr, green). Left structures; right pDOS. The vertical gray dashed line indicates the Fermi level referenced to the vacuum level. | ||
Visual inspection reveals significant distortions around the OsubZr vacancy. The highlighted surface oxygen (Fig. 2a) shifts downward along the surface normal by 0.33 Å. Distortion-based stabilization of oxygen vacancies around Zr ions has been discussed for Zr-doped CeO2(111) surfaces; however, it has only been quantified in bulk CZO.35 Here, we compute the root-mean-square of displacements (RMSD) between oxygen atoms in the relaxed and unrelaxed geometry to quantify the extent of surface distortion accompanying vacancy formation eqn (5):34
 | (5) |
| RMSD (Å) | OCe | OZr |
|---|---|---|
| Surface (first shell) | 0.28 | 0.4 |
| Surface (extended) | 0.08 | 0.09 |
| Subsurface (first shell) | 0.22 | 0.25 |
| Subsurface (extended) | 0.1 | 0.1 |
OZr vacancies at the surface and subsurface distort the structure more than OCe vacancy. This agrees with the view of distortion-induced stabilization of oxygen vacancies around Zr atoms previously noted in bulk CZO.25 Greater structural relaxation upon vacancy formation helps accommodate the larger Ce3+ ions in the lattice. Similar ΔEstrain trends between OZr and OCe vacancies were noted in Table 2, showing that the RMSDs upon vacancy formation correlate with the ΔEstrain like in bulk CZO.
All the ΔEv values reported so far have been computed at T = 0 K and μO (T = 0 K, p) = 0 and are not representative of vacancy formation under reaction conditions. Therefore, we examine the stability of the defected-CZO(111) surfaces at finite temperatures and different O2 partial pressures. It is useful to understand vacancy stability under oxidant-rich and oxidant-poor conditions, especially for Mars–van Krevelen reaction mechanisms, which invoke oxidation and reduction half-cycles involving lattice O. Assuming the surface vibrational contribution to the free energy does not affect trends, we write the free energy of vacancy formation as in eqn (6)
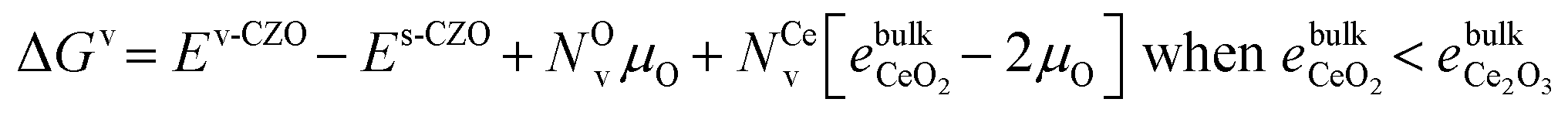
 | (6) |
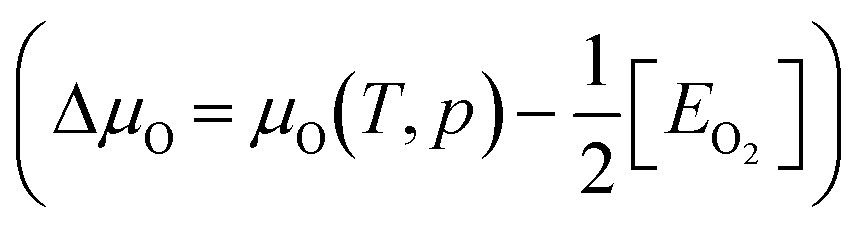 . Based on the DFT+U formation energies of the bulk oxides,
. Based on the DFT+U formation energies of the bulk oxides,  at ΔμO = −2.12 eV per atom.
at ΔμO = −2.12 eV per atom.
Under DRM conditions where lattice O abstraction by CO and O vacancy replenishment by CO2 are quasi-equilibrated, the effective O chemical potential is defined as: μO = μCO2 − μCO. Here, rather than the partial pressure of O2, μO under reaction conditions depends on the ratio of CO2 and CO partial pressures. We follow Zhang et al.'s methodology to compute the reaction-relevant range,21 and find that for T/K ∈ [800, 1100], ΔμO(T, p)/eV per atom ∈ [−3.78, −3.03]. Since ΔμO = −3.15 eV per atom when pCO2/pCO is 0.5, ΔμO values between −3.15 and −3.78 eV per atom correspond to increasingly reducing conditions and between −3.15 and −3.03 eV per atom to increasingly oxidizing conditions.
The upper bound of μO(T, p) can be extended to the energy of an isolated O2 molecule at 0 K, namely

Fig. 3 shows the relative stability of different types of defects on CZO(111). In addition to oxygen and cerium vacancies, we have investigated a cerium vacancy with one oxygen vacancy (Cev, Ov) and with two oxygen vacancies (Cev, (Ov)2). Under reducing conditions, oxygen vacancy is favored, with a more negative ΔGv than on CeO2(111)21 consistent with the reported higher reducibility of CZO.7 Increasing μO(T, p) favors cerium vacancy creation, but even under strongly oxidizing conditions, an oxygen vacancy is favorable over a cerium vacancy. Overall, under DRM conditions, oxygen vacancies are most likely in the CZO surface. The defect stability trends on the CZO(111) surface resemble CeO2(111). In the next section, we discuss how Ni single atoms and clusters alter the stability of the vacancies.
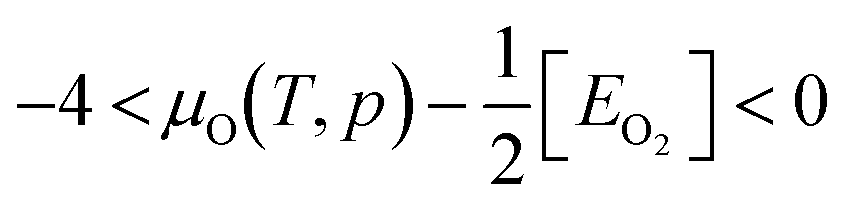
 | ||
| Fig. 3 Vacancy formation energy (ΔGv) as a function of oxygen chemical potential (ΔμO) with the most stable cerium oxide phase as a reference (bulk CeO2 and bulk Ce2O3 to the right and left, respectively, of the vertical dotted line at ΔμO = −2.12 eV per atom). The left end of the graphs corresponds to reducing conditions, and the right end to oxidizing conditions. The region between the vertical dashed lines (ΔμO(T, p) ∈ [−3.78, −3.03]) represents typical DRM reaction conditions. | ||
Structure, electronic properties, and stability of Ni anchored on CZO surfaces
While Ni/CZO is an attractive catalyst for DRM, nickel's interactions with the CZO support under reaction conditions remain largely unexplored. In this section, we investigate the stability and electronic properties of Ni and Ni4 structures on stoichiometric and non-stoichiometric CZO(111) surfaces and how nickel affects support reducibility.First, we investigate nickel adsorption on the stoichiometric CZO(111) surface. Nickel binding energy on the surface per metal atom 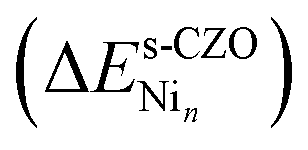 is computed using eqn (7)
is computed using eqn (7)
 | (7) |
 is the total energy of the stoichiometric CZO slab with a Nin cluster on it, n is the nuclearity of the Ni cluster, and ebulkNi is the total energy of bulk nickel per atom.
is the total energy of the stoichiometric CZO slab with a Nin cluster on it, n is the nuclearity of the Ni cluster, and ebulkNi is the total energy of bulk nickel per atom.  measures the stability of a Nin cluster relative to the bulk: large, positive values indicate a strong preference for Ni to stay in the bulk phase.
measures the stability of a Nin cluster relative to the bulk: large, positive values indicate a strong preference for Ni to stay in the bulk phase.
We considered six locations to anchor Ni adatoms: atop a Ce atom (NiCe), atop a Zr atom (NiZr), atop OZr (NiOZr), atop OCe (NiOCe), the hollow site formed by a Ce and two Zr atoms (NiZr2Ce) and the hollow site formed by a Zr and two Ce atoms (NiCe2Zr) (Fig. S4†). The adsorption energies are shown in Table 4. Ni is most stable at the hollow site formed by a Zr and two Ce atoms (NiCe2Zr). With a ΔEs-CZONi of 0.04 eV, the nickel adatom is as stable as it would be in the bulk. Interestingly, Ni adsorption on CeO2(111) has a  of 0.45 eV, which is closer in energy to the NiZr2Ce site. Analysis of the geometries reveals distinct coordination environments. In the NiCe2Zr site, nickel coordinates to two OZr atoms adjacent to the hollow site in a bridge geometry. The OZr atoms are significantly displaced by ∼0.8 Å out of the plane Fig. 4a). In contrast, at the less stable NiZr2Ce site, nickel adopts a three-fold coordination with the adjacent oxygen atoms (Fig. 4b). Similarly, on CeO2(111), nickel binds at the hollow site with a three-fold coordination to the surface oxygen atoms, resulting in a binding energy comparable to that of the NiZr2Ce site (Fig. 4c). The notably stronger binding on CZO(111) can be attributed to the substantial structural distortion of the OZr atoms, which facilitates nickel's bridging mode. Interestingly, Mao et al.36 found that Ni binds to the 〈110〉-type step edge of CeO2(111) in a similar O–Ni–O bridging geometry and is more stable in this configuration than as bulk Ni. This similarity shows the importance of lattice flexibility in creating stable binding sites for single atoms. In summary, the CZO(111) terrace facilitates the dispersion of Ni single atoms much more effectively than the CeO2(111) terrace.
of 0.45 eV, which is closer in energy to the NiZr2Ce site. Analysis of the geometries reveals distinct coordination environments. In the NiCe2Zr site, nickel coordinates to two OZr atoms adjacent to the hollow site in a bridge geometry. The OZr atoms are significantly displaced by ∼0.8 Å out of the plane Fig. 4a). In contrast, at the less stable NiZr2Ce site, nickel adopts a three-fold coordination with the adjacent oxygen atoms (Fig. 4b). Similarly, on CeO2(111), nickel binds at the hollow site with a three-fold coordination to the surface oxygen atoms, resulting in a binding energy comparable to that of the NiZr2Ce site (Fig. 4c). The notably stronger binding on CZO(111) can be attributed to the substantial structural distortion of the OZr atoms, which facilitates nickel's bridging mode. Interestingly, Mao et al.36 found that Ni binds to the 〈110〉-type step edge of CeO2(111) in a similar O–Ni–O bridging geometry and is more stable in this configuration than as bulk Ni. This similarity shows the importance of lattice flexibility in creating stable binding sites for single atoms. In summary, the CZO(111) terrace facilitates the dispersion of Ni single atoms much more effectively than the CeO2(111) terrace.
| Binding site |
|
|---|---|
| NiCe | 1.51 |
| NiZr | 1.16 |
| NiOCe | 2.39 |
| NiOZr | 1.99 |
| NiCe2Zr | 0.04 |
| NiZr2Ce | 0.31 |
| Ni/CeO2 | 0.45 |

 | ||
| Fig. 4 Nickel binding sites (a) NiCe2Zr/CZO(111), (b) NiZr2Ce/CZO(111) and (c) Ni/CeO2(111) (O, red; Ce, yellow; Zr, green; Ni, blue). | ||
A similar exploration was carried out to identify the most stable binding geometry of a Ni4 cluster (Fig. S5†); the top view of the stable structure is shown in Fig. S6.† Mao et al.36 showed that Ni4 prefers the pyramidal geometry over planar on CeO2(111). On the CZO surface too, the most stable Ni4 cluster assumes pyramidal geometry atop a Zr atom. With a  of 0.68 eV, the nickel cluster is less stable at the surface than the adatom. Upon closely inspecting the geometries, we see that the three Ni atoms at the base of the pyramid interact with the surface oxygen atoms and displace them, but by a smaller amount than the single atom. The average Ni–O bond length for the adatom is 1.71 Å, while for the cluster it is 1.79 Å. This indicates that the Ni–O bonding is weaker in the cluster due to competing Ni–Ni bonds, and results in weaker interactions with the support.
of 0.68 eV, the nickel cluster is less stable at the surface than the adatom. Upon closely inspecting the geometries, we see that the three Ni atoms at the base of the pyramid interact with the surface oxygen atoms and displace them, but by a smaller amount than the single atom. The average Ni–O bond length for the adatom is 1.71 Å, while for the cluster it is 1.79 Å. This indicates that the Ni–O bonding is weaker in the cluster due to competing Ni–Ni bonds, and results in weaker interactions with the support.
Next, we investigate how anchored nickel affects surface reducibility. Unlike Au and Pd, Ni is unstable when anchored to oxygen vacancies, as evidenced by nickel heat of adsorption experiments on CeO2−x(111) surfaces.36 This instability arises from nickel's pronounced oxophilicity, which favors coordination with oxygen atoms rather than binding to or nucleating at defects. To investigate how nickel affects surface reducibility compared to bare CZO(111), we compute the energetics for oxygen vacancy formation on Ni/CZO(111) and Ni4/CZO(111) using eqn (8):
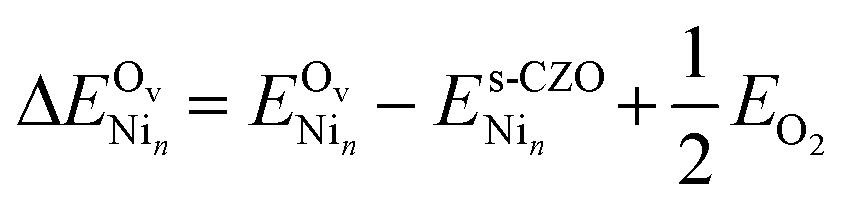 | (8) |
 is the energy of the CZO(111) slab with an oxygen vacancy and a Nin cluster on it. We consider surface oxygen vacancies in the same OZr row (denoted OZr_NN), as the oxygens coordinated to nickel, and in the adjacent OZr row (denoted OZr_NNN). While surface oxygen vacancies are kinetically relevant for the DRM reaction, there is a thermodynamic driving force to create the more stable subsurface oxygen vacancies. At DRM-relevant temperatures of 1000 K, oxygen mobility should allow vacancy diffusion to the subsurface. Therefore, we also evaluated an adjacent subsurface oxygen vacancy formation. Table 5 shows the vacancy formation energies as per eqn (8); Fig. S7 and S8† show the structures.
is the energy of the CZO(111) slab with an oxygen vacancy and a Nin cluster on it. We consider surface oxygen vacancies in the same OZr row (denoted OZr_NN), as the oxygens coordinated to nickel, and in the adjacent OZr row (denoted OZr_NNN). While surface oxygen vacancies are kinetically relevant for the DRM reaction, there is a thermodynamic driving force to create the more stable subsurface oxygen vacancies. At DRM-relevant temperatures of 1000 K, oxygen mobility should allow vacancy diffusion to the subsurface. Therefore, we also evaluated an adjacent subsurface oxygen vacancy formation. Table 5 shows the vacancy formation energies as per eqn (8); Fig. S7 and S8† show the structures.
| Vacancy formation energy (eV) | Surface | Subsurface |
|---|---|---|

|
1.86 | 1.85 |

|
1.4 | 1.2 |
|
— | 1.35 |

|
1.7 | 1.2 |
ΔEOv in Table 1 and  in Table 5 indicate that nickel on the CZO(111) surface generally destabilizes OZr vacancies, especially surface vacancies with larger Ni clusters.
in Table 5 indicate that nickel on the CZO(111) surface generally destabilizes OZr vacancies, especially surface vacancies with larger Ni clusters.
For Ni/CZO, the OZr vacancy formation energy reduces to that on the pristine surface further away from the Ni single atom. Interestingly, upon optimization, a surface OZr vacancy near the nickel cluster on Ni4/CZO converges to the thermodynamically favored subsurface OZr vacancy, reflecting nickel's high oxophilicity. In addition to the structures in Table 5, we also computed oxygen vacancy formation energy at the foot of the Ni single atom and cluster (denoted OZr_Ni), as these vacancies may be more kinetically favored.  and
and  are 3.57 eV and 2.7 eV, respectively, further evidencing nickel's high oxophilicity. The destabilization of oxygen vacancies near nickel contrasts with less oxophilic metals, such as Au, which can undergo partial reduction upon interacting with supports like CeO2 and TiO2.37 We next explored the mechanism of OZr vacancy destabilization. Comparing ΔEstrain values in Tables 2 and S3,† we see that OZr vacancy formation away from the Ni single atom has similar strain effects as the pristine surface. In contrast, the nearest neighbor OZr vacancy benefits less from structural relaxation effects (Table S3†). These findings indicate that the enhanced performance of the Ni/CZO catalyst for DRM cannot be attributed to the thermodynamics of vacancy formation, as Ni's oxophilicity and the reduced lattice flexibility due to Ni–O bonding destabilizes surrounding O vacancies. Instead, the improvement arises from improved kinetics of breaking H–H or C–H bonds over Ni sites and the subsequent vacancy formation at the Ni/CZO interface.
are 3.57 eV and 2.7 eV, respectively, further evidencing nickel's high oxophilicity. The destabilization of oxygen vacancies near nickel contrasts with less oxophilic metals, such as Au, which can undergo partial reduction upon interacting with supports like CeO2 and TiO2.37 We next explored the mechanism of OZr vacancy destabilization. Comparing ΔEstrain values in Tables 2 and S3,† we see that OZr vacancy formation away from the Ni single atom has similar strain effects as the pristine surface. In contrast, the nearest neighbor OZr vacancy benefits less from structural relaxation effects (Table S3†). These findings indicate that the enhanced performance of the Ni/CZO catalyst for DRM cannot be attributed to the thermodynamics of vacancy formation, as Ni's oxophilicity and the reduced lattice flexibility due to Ni–O bonding destabilizes surrounding O vacancies. Instead, the improvement arises from improved kinetics of breaking H–H or C–H bonds over Ni sites and the subsequent vacancy formation at the Ni/CZO interface.
Under the highly reducing conditions of DRM reaction or the oscillating reducing/oxidizing conditions during CH4/CO2 half-cycles of chemical looping DRM, we expect the redox-active CZO surface to be defect-rich. It is then worth checking whether Ni might preferentially bind to surface defects rather than fully oxidized CZO(111) under reaction conditions. Beginning with oxidizing conditions, Jenkins and coworkers32 have suggested surface Ce vacancies for the Au/CeO2(111) catalyst in such environments. However, for the Ni/CZO(111) catalyst, Ni cannot assume a Ni4+ configuration upon Ce vacancy formation due to unstable dangling bonds on surface oxygen. We, therefore, introduced two oxygen vacancies adjacent to a Ce vacancy (Cev(Ov)2), to prevent dangling oxygen bonds. It is worth noting that nickel adsorption at the cerium vacancy is equivalent to Ni2+ doping the CZO(111) surface, which favors the creation of two oxygen vacancies for charge compensation. We define the binding energy of nickel to the pristine and Cev(Ov)2–CZO surfaces by eqn (9):
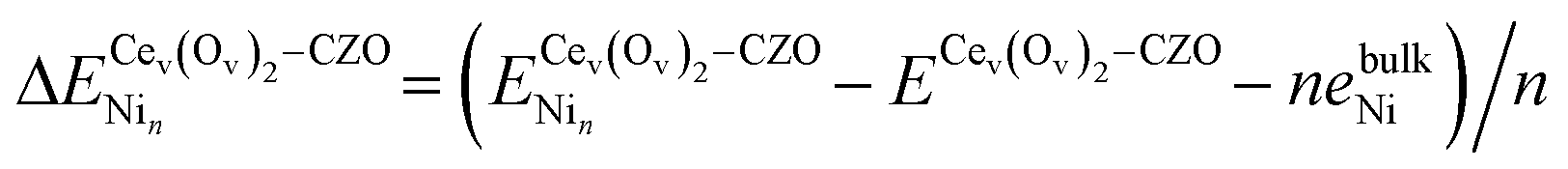 | (9) |
 is the energy of the slab with a Cev(Ov)2 defect and a Nin cluster. In this definition of binding energy, the energy change depends only on Ni adsorption, as defects are expected to be naturally present.32
is the energy of the slab with a Cev(Ov)2 defect and a Nin cluster. In this definition of binding energy, the energy change depends only on Ni adsorption, as defects are expected to be naturally present.32
From Table 6, we see that the Ni adatom is very stable at the Cev(Ov)2 defect, more so than on the pristine surface. Examination of the geometry (Fig. S9c†) reveals that the Ni adatom forms a stable square-planar complex with two surface and two subsurface oxygens. In contrast, Ni coordinates with two surface oxygen atoms on the pristine surface. This enhanced stability of the Ni adatom on the Cev(Ov)2–CZO surface can be attributed to its ability to coordinate with more oxygen atoms than on the pristine surface.
| Energy (eV)/defect | s-CZO | Cev(Ov)2–CZO |
|---|---|---|
| ΔEsurfaceNi | 0.04 | −2.4 |

|
0.69 | 0.12 |
While nickel atoms are highly oxophilic and stabilized by coordinating with oxygens, they can also be stabilized through interactions with other nickel atoms during sintering. We expect this to be the primary mode of Ni stabilization once all Ce vacancies are filled or under reducing reaction conditions. By computing the nucleation energy per Ni atom (ΔEsnuc) from eqn (10), we find that on the pristine surface, the surface with an oxygen vacancy at the OZr_NNN site, and the Cev(Ov)2 defect site, nickel remains dispersed as single atoms. In contrast, Ni single atoms are highly unstable at the oxygen vacancy site. The thermodynamic driving force for nucleation strongly correlates with the number of coordinating oxygen atoms around the nickel adatom: the lowest driving force is observed at the Cev(Ov)2 defect site, where Ni coordinates with four oxygens, while the highest is at the oxygen vacancy site, where Ni coordinates with no oxygen atom (Table 7).
 | (10) |
| Surface | Nucleation energy (ΔEsnuc) in eV |
|---|---|
| s-CZO | 0.65 |
| OZr_Ni–CZO | −1.18 |
| OZr_NNN–CZO | 0.75 |
| Cev(Ov)2–CZO | 2.56 |
| Surface | q Ni | q Ni4 |
|---|---|---|
| s-CZO | 0.7 | 0.3, 0.3, 0.3, 0 |
| OZr_Ni–CZO | −0.02 | 0.15, 0.15, 0.35, 0 |
| OZr_NN–CZO | 0.7 | 0.3, 0.3, 0.3, 0 |
| OZr_NNN–CZO | 0.7 | 0.3, 0.3, 0.3, 0 |
| Cev(Ov)2–CZO | 0.9 | 0.3, 0.5, 0.7, 0 |
Oxygen vacancies adjacent to nickel, while kinetically relevant, are energetically unfavorable due to nickel's pronounced oxophilicity. Nevertheless, understanding the stability of such surface defects adjacent to nickel under realistic oxygen partial pressures is crucial for elucidating the catalytic behavior of Ni/CZO systems. Therefore, we plot  (defined below) as a function of μO (Fig. 5), in the spirit of the analysis presented in Fig. 3. This definition of
(defined below) as a function of μO (Fig. 5), in the spirit of the analysis presented in Fig. 3. This definition of 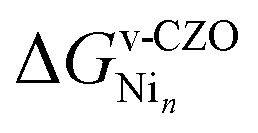 computes the Ni binding energy to the non-stoichiometric surface but also includes a correction for the formation of the defect, as given in eqn (11).
computes the Ni binding energy to the non-stoichiometric surface but also includes a correction for the formation of the defect, as given in eqn (11).

 | (11) |
 | ||
| Fig. 5 Nickel (a) single atom and (b) cluster (Ni4) adsorption energy on the defected and pristine-CZO surfaces as a function of oxygen chemical potential (μO). The most stable cerium oxide phase is used as reference (bulk Ce2O3 and bulk CeO2 to the left and right, respectively, of the vertical dotted line at ΔμO = −2.12 eV). The left end of the graphs corresponds to reducing conditions, and the right end to oxidizing conditions. The region between the two vertical dashed lines (ΔμO(T, p) ∈ [−3.78, −3.03]) represents the typical DRM reaction conditions. | ||
Conclusions
We employed DFT and ab initio thermodynamics to investigate the energetics, stability, and electronic properties of vacancy formation and nickel adsorption on CZO(111). Subsurface and surface oxygen vacancies are favored near Zr4+ ions due to distortion-induced stabilization, and the latter are more favorable across DRM-relevant conditions. Ni single atoms coordinate with two OZr surface atoms as Ni2+ motifs, unlike three-fold coordinated Ni atoms on hollow sites of CeO2(111). Interestingly, Ni dislikes direct binding to oxygen vacancies compared to a pristine surface due to its oxophilicity as well as electrostatic repulsion from multiple surface Ce3+ ions. The enhanced performance of the Ni/CZO DRM catalyst stems probably from the kinetics of H–H or C–H bond activation, vacancy formation, or oxygen diffusion rather than thermodynamics. Further work is needed to delineate the mechanisms.Ni adatoms are more stable than Ni4 at trimer defects comprising one Ce and two oxygen vacancies (Cev(Ov)2), at the pristine surface, and at the NiCe2Zr site with a next-nearest neighbor oxygen vacancy due to coordinating with more oxygen atoms. Thus, defects exposing oxygen atoms, e.g., (Cev(Ov)2), will stabilize Ni single atoms, but single oxygen surface vacancies would not directly bind Ni1. Clusters, on the other hand, can bind favorably at a surface oxygen vacancy due to Ni–Ni interactions compensating for the missing surface oxygen. We expect that high Ni loadings will lead to Ni1 adatoms anchoring next to oxygen vacancies, divacancies, and pristine areas and Ni clusters stabilized by oxygen vacancies at their perimetry or underneath. The extent of electron transfer from the metal to the surface and, thus, the degree of cationic character of a nickel adatom will vary with its location and defect type and correlate positively with its resistance to sintering: the highest charge transfer is on Cev(Ov)2, followed by the pristine surface and surfaces with a single oxygen vacancy in the next nearest neighbor position. In an actual catalyst, we thus expect Ni atoms with varying metallic character and nuclearity to participate in the chemistry.
These findings provide valuable insights into the Ni/CZO DRM catalysts, emphasizing the importance of nickel's electronic interactions with defect sites of the CZO surface.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the US Department of Energy (DOE), award number DE-SC0024085. The computations were partially supported by the high-performance computing resources at the University of Delaware. The authors thank Dr. Jeffrey Frey and Rajas Mehendale for installing the software on the high-performance computing cluster. The authors are grateful to Dr. Stavros Caratzoulas, who sadly passed away while preparing this work.References
- Q. Wang, X. Chen, A. N. Jha and H. Rogers, Natural gas from shale formation – The evolution, evidences and challenges of shale gas revolution in United States, Renewable Sustainable Energy Rev., 2014, 30, 1–28 CrossRef.
- H. Ishaq, I. Dincer and C. Crawford, A review on hydrogen production and utilization: Challenges and opportunities, Int. J. Hydrogen Energy, 2022, 47(62), 26238–26264 CrossRef CAS.
- O. J. Guerra, J. Eichman, J. Kurtz and B.-M. Hodge, Cost Competitiveness of Electrolytic Hydrogen, Joule, 2019, 3(10), 2425–2443 CrossRef.
- C. Wang, S. Sourav, K. Yu, Y. Kwak, W. Zheng and D. G. Vlachos, Green Syngas Production by Microwave-Assisted Dry Reforming of Methane on Doped Ceria Catalysts, ACS Sustainable Chem. Eng., 2023, 11(36), 13353–13362 CrossRef CAS.
- Z. Liu, D. C. Grinter, P. G. Lustemberg, T.-D. Nguyen-Phan, Y. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola, J. Zhou, J. Carrasco, H. F. Busnengo, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Dry Reforming of Methane on a Highly-Active Ni-CeO2 Catalyst: Effects of Metal-Support Interactions on C−H Bond Breaking, Angew. Chem., Int. Ed., 2016, 55(26), 7455–7459 CrossRef CAS PubMed.
- Z. Yang, T. K. Woo and K. Hermansson, Effects of Zr doping on stoichiometric and reduced ceria: A first-principles study, J. Chem. Phys., 2006, 124(22), 224704 CrossRef PubMed.
- H.-F. Wang, H.-Y. Li, X.-Q. Gong, Y.-L. Guo, G.-Z. Lu and P. Hu, Oxygen vacancy formation in CeO2 and Ce1−xZrxO2 solid solutions: electron localization, electrostatic potential and structural relaxation, Phys. Chem. Chem. Phys., 2012, 14(48), 16521–16535 RSC.
- Y. Nagai, T. Yamamoto, T. Tanaka, S. Yoshida, T. Nonaka, T. Okamoto, A. Suda and M. Sugiura, X-ray absorption fine structure analysis of local structure of CeO2–ZrO2 mixed oxides with the same composition ratio (Ce/Zr=1), Catal. Today, 2002, 74(3), 225–234 CrossRef CAS.
- F. Zhang, Z. Liu, X. Chen, N. Rui, L. E. Betancourt, L. Lin, W. Xu, C.-j. Sun, A. M. M. Abeykoon, J. A. Rodriguez, J. Teržan, K. Lorber, P. Djinović and S. D. Senanayake, Effects of Zr Doping into Ceria for the Dry Reforming of Methane over Ni/CeZrO2 Catalysts: In Situ Studies with XRD, XAFS, and AP-XPS, ACS Catal., 2020, 10(5), 3274–3284 CrossRef CAS.
- A. Kambolis, H. Matralis, A. Trovarelli and C. Papadopoulou, Ni/CeO2-ZrO2 catalysts for the dry reforming of methane, Appl. Catal., A, 2010, 377(1), 16–26 CrossRef CAS.
- S. Xu and X. Wang, Highly active and coking resistant Ni/CeO2–ZrO2 catalyst for partial oxidation of methane, Fuel, 2005, 84(5), 563–567 CrossRef CAS.
- G. S. Parkinson, Single-Atom Catalysis: How Structure Influences Catalytic Performance, Catal. Lett., 2019, 149(5), 1137–1146 CrossRef CAS PubMed.
- C. Freysoldt, B. Grabowski, T. Hickel, J. Neugebauer, G. Kresse, A. Janotti and C. G. Van de Walle, First-principles calculations for point defects in solids, Rev. Mod. Phys., 2014, 86(1), 253–305 CrossRef.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal--amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)], Phys. Rev. Lett., 1997, 78(7), 1396–1396 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Band theory and Mott insulators: Hubbard U instead of Stoner I, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44(3), 943–954 CrossRef CAS PubMed.
- A. Rohrbach, J. Hafner and G. Kresse, Electronic correlation effects in transition-metal sulfides, J. Phys.: Condens. Matter, 2003, 15(6), 979 CrossRef CAS.
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, Anchoring Sites for Initial Au Nucleation on CeO2{111}: O Vacancy versus Ce Vacancy, J. Phys. Chem. C, 2009, 113(16), 6411–6417 CrossRef CAS.
- H.-Y. T. Chen, S. Tosoni and G. Pacchioni, Adsorption of Ruthenium Atoms and Clusters on Anatase TiO2 and Tetragonal ZrO2(101) Surfaces: A Comparative DFT Study, J. Phys. Chem. C, 2015, 119(20), 10856–10868 CrossRef CAS.
- S. Livraghi, M. C. Paganini, E. Giamello, G. Di Liberto, S. Tosoni and G. Pacchioni, Formation of Reversible Adducts by Adsorption of Oxygen on Ce–ZrO2: An Unusual η2 Ionic Superoxide, J. Phys. Chem. C, 2019, 123(44), 27088–27096 CrossRef CAS.
- G. E. Murgida, V. Ferrari, M. V. Ganduglia-Pirovano and A. M. Llois, Ordering of oxygen vacancies and excess charge localization in bulk ceria: A $\mathrm{DFT}+U$ study, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90(11), 115120 CrossRef.
- H.-F. Wang, Y.-L. Guo, G.-Z. Lu and P. Hu, Maximizing the Localized Relaxation: The Origin of the Outstanding Oxygen Storage Capacity of κ-Ce2Zr2O8, Angew. Chem., Int. Ed., 2009, 48(44), 8289–8292 CrossRef CAS PubMed.
- Y. Peng, X.-L. Si, C. Shang and Z.-P. Liu, Abundance of Low-Energy Oxygen Vacancy Pairs Dictates the Catalytic Performance of Cerium-Stabilized Zirconia, J. Am. Chem. Soc., 2024, 146(15), 10822–10832 CrossRef CAS PubMed.
- P. Fornasiero, E. Fonda, R. Di Monte, G. Vlaic, J. Kašpar and M. Graziani, Relationships between Structural/Textural Properties and Redox Behavior in Ce0.6Zr0.4O2 Mixed Oxides, J. Catal., 1999, 187(1), 177–185 CrossRef CAS.
- J. C. Conesa, Computer Modeling of Local Level Structures in (Ce, Zr) Mixed Oxide, J. Phys. Chem. B, 2003, 107(34), 8840–8853 CrossRef CAS.
- M. Nolan, J. E. Fearon and G. W. Watson, Oxygen vacancy formation and migration in ceria, Solid State Ionics, 2006, 177(35), 3069–3074 CrossRef CAS.
- J. Paier, C. Penschke and J. Sauer, Oxygen Defects and Surface Chemistry of Ceria: Quantum Chemical Studies Compared to Experiment, Chem. Rev., 2013, 113(6), 3949–3985 CrossRef CAS PubMed.
- G. S. Otero, P. G. Lustemberg, F. Prado and M. V. Ganduglia-Pirovano, Relative Stability of Near-Surface Oxygen Vacancies at the CeO2(111) Surface upon Zirconium Doping, J. Phys. Chem. C, 2020, 124(1), 625–638 CrossRef CAS.
- C. J. Owen and S. J. Jenkins, Comparative study of single-atom gold and iridium on CeO2{111}, J. Chem. Phys., 2021, 154(16), 164703 CrossRef CAS PubMed.
- D. García Pintos, A. Juan and B. Irigoyen, Oxygen vacancy formation on the Ni/Ce0.75Zr0.25O2(111) surface. A DFT+U study, Int. J. Hydrogen Energy, 2012, 37(19), 14937–14944 CrossRef.
- H.-F. Wang, X.-Q. Gong, Y.-L. Guo, Y. Guo, G. Z. Lu and P. Hu, A Model to Understand the Oxygen Vacancy Formation in Zr-Doped CeO2: Electrostatic Interaction and Structural Relaxation, J. Phys. Chem. C, 2009, 113(23), 10229–10232 CrossRef CAS.
- Z. Yang, Y. Wei, Z. Fu, Z. Lu and K. Hermansson, Facilitated vacancy formation at Zr-doped ceria(111) surfaces, Surf. Sci., 2008, 602(6), 1199–1206 CrossRef CAS.
- Z. Mao, P. G. Lustemberg, J. R. Rumptz, M. V. Ganduglia-Pirovano and C. T. Campbell, Ni Nanoparticles on CeO2(111): Energetics, Electron Transfer, and Structure by Ni Adsorption Calorimetry, Spectroscopies, and Density Functional Theory, ACS Catal., 2020, 10(9), 5101–5114 CrossRef CAS.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies, ACS Catal., 2017, 7(10), 6493–6513 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00133a |
| This journal is © The Royal Society of Chemistry 2025 |