
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical synthesis of Pt/TiO2 for enhanced stability in dehydrogenation of methylcyclohexane†
Krista
Kuutti
 a,
Manoj Kumar
Ghosalya
b,
Paavo
Porri
a,
Jacopo
De Bellis‡
c,
Päivi
Jokimies
a,
Harishchandra
Singh
b,
Shubo
Wang
b,
Graham
King
d,
Javier
Fernández-Catalá
be,
Ferdi
Schüth
c,
Kaisu
Ainassaari
f,
Mika
Huuhtanen
f,
Marko
Huttula
b,
Samuli
Urpelainen
*b and
Sari
Rautiainen
*a
a,
Manoj Kumar
Ghosalya
b,
Paavo
Porri
a,
Jacopo
De Bellis‡
c,
Päivi
Jokimies
a,
Harishchandra
Singh
b,
Shubo
Wang
b,
Graham
King
d,
Javier
Fernández-Catalá
be,
Ferdi
Schüth
c,
Kaisu
Ainassaari
f,
Mika
Huuhtanen
f,
Marko
Huttula
b,
Samuli
Urpelainen
*b and
Sari
Rautiainen
*a
aVTT Technical Research Centre of Finland Ltd, Espoo FI-02044, Finland. E-mail: sari.rautiainen@vtt.fi
bNano and Molecular Systems Research Unit, University of Oulu, 90014 Oulu, Finland. E-mail: samuli.urpelainen@oulu.fi
cDepartment of Heterogeneous Catalysis, Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
dCanadian Light Source, 44 Innovation Boulevard, Saskatoon, Saskatchewan, S7N 2V3 Canada
eMaterials Institute and Inorganic Chemistry Department, University of Alicante, Ap. 99, Alicante E-03080, Spain
fEnvironmental and Chemical Engineering Research Unit, University of Oulu, 90014 Oulu, Finland
First published on 17th June 2025
Abstract
Catalytic hydrogenation/dehydrogenation of liquid organic hydrogen carriers (LOHCs), such as methylcyclohexane (MCH), enables versatile and safe transport and storage of hydrogen as a carbon neutral fuel. Supported platinum catalysts are commonly used for the dehydrogenation reaction, however, they often suffer from loss of activity due to coking. Herein, we present mechanochemically synthesised platinum on titania catalyst for the dehydrogenation of MCH, prepared starting only from metallic platinum and titania. Dry mechanochemical catalyst syntheses do not produce waste waters or toxic fumes, which are generated in the deposition of metal precursors by conventional wet synthesis methods. Detailed characterisation of the catalysts revealed that ball milling produced highly dispersed nanoparticles. Furthermore, continuous-flow MCH dehydrogenation experiments showed that the mechanochemically prepared Pt catalyst exhibited improved selectivity and stability compared to a conventional impregnated Pt/TiO2 catalyst. The hydrogen production rate of the novel ball-milled catalyst was among the highest reported for dehydrogenation of methylcyclohexane, 670 mmolH2 gPt−1 min−1.
1 Introduction
Green hydrogen produced via water splitting using renewable energy could serve as a carbon neutral fuel.1 High energy density is essential for a fuel, and H2 possesses the highest gravimetric energy density of all energy carriers, 120 MJ kg−1. For comparison, the corresponding value for gasoline is ∼40 MJ kg−1.2 The volumetric energy density of ambient H2 gas, however, is very low requiring a compaction method for its viable storage and transportation.3 One promising H2 compaction method is chemical bonding to a storage medium, such as liquid organic hydrogen carriers (LOHCs). LOHCs are mainly cycloalkanes/aromatic compounds, that can undergo reversible hydrogenation/dehydrogenation reactions in the presence of suitable catalysts. A well-performing LOHC should have relatively high H2 storage capacity (6–8 wt%) and wide temperature range for the liquid state. The liquid form enables usage of existing pumping systems, fuel containers and transportation vehicles with just minor modifications. Notably, two LOHC systems have already been commercialised, namely methylcyclohexane/toluene by Chiyoda Corporation2 and a technical mixture of benzyltoluenes by Hydrogenious.3 For the present work, the methylcyclohexane/toluene system was selected for the experiments, being one of the most promising LOHC systems given its low toxicity and broad applicability to the conventional fuel distribution infrastructure.4,5Scheme 1 presents the endothermic 47 dehydrogenation reaction of MCH to toluene.
47 dehydrogenation reaction of MCH to toluene.
 | ||
| Scheme 1 MCH dehydrogenation reaction. | ||
Industrially, aromatics are refined from crude oil via catalytic reforming, and cycloalkanes are manufactured by hydrogenating these oil-derived aromatics. In terms of LOHC technologies this means that there are commercial processes and catalysts readily available for aromatic hydrogenation, but cycloalkane dehydrogenation technologies are not comparably established.3 Cycloalkane dehydrogenation is an endothermic reaction that occurs at elevated temperatures (150–400 °C) and relatively low pressure (<10 bar) over a metal catalyst. The choice of catalyst is crucial for the process as it should enable numerous LOHC hydrogenation/dehydrogenation cycles with minimum side products formation.4 While hydrogenation catalysts are well established, viable and stable catalysts for the dehydrogenation reaction are still under development.
Platinum is the most extensively studied metal catalyst for LOHC dehydrogenation due to its high selectivity and superior activity in the reaction.4 Pt is often supported on high surface area alumina owing to the thermal stability of the support and the ability to maintain high dispersion of Pt nanoparticles.6 However, the acid sites on alumina may lead to coking and deactivation of the catalyst. Different strategies have been suggested to reduce the excess acidity, for instance doping the catalyst with alkali metals.7 Yang et al. modified the alumina support by deposition of titania to reduce the acidity, resulting in increased stability and conversion of MCH.8 Another strategy is to use another support material altogether. Nagatake et al. studied MCH dehydrogenation over TiO2 and Al2O3 supported Pt catalysts and detected significantly less coke formation over the TiO2 supported catalyst.5 Both studies suggested strong metal support interactions between Pt and the TiO2 support may induce charge transfer from the support to Pt causing an electron rich state. Similar phenomenon was not detected with the Al2O3 supported Pt. Moreover, Pt/TiO2 catalyst was shown to excel over alumina-supported one in the dehydrogenation of perhydrogenated dibenzyl toluene.9 Interestingly, platinum supported on a mixture of rutile–anatase demonstrated higher activity in the reaction compared to its alumina-supported counterpart or Pt on pure anatase support.
Typically, the catalyst preparation is carried out using solution-based methods, e.g. precipitation and impregnation. These methods produce large amounts of waste and can have challenges in the reproducibility of the preparation. Recently, mechanochemical methods, such as ball milling have gained attention as easily reproducible, scalable and simple methods for heterogeneous catalyst preparation.10 Generally, ball milling is a controllable, efficient, low cost, and green solid-state mechanical powder processing technique that is frequently used to fabricate equilibrium and/or non-equilibrium phases with reduced crystallite size compared to initial ingredients.11 Ball mill catalyst synthesis can be operated fully solvent-free and with high yield.12 In a planetary ball mill, rotating movement of the jar causes the grinding balls to collide and rub against each other and the walls, exerting impact, shear and friction forces on the materials inside the jar. As a result, highly dispersed nano-sized particles can be obtained in a high energy ball mill, starting simply from support material and metal powder.12,13 The harder material (oxide support) acts as a grinding body towards the softer material (metal) that is subjected to activation by deformation, leading to metal application on top of the oxide support.14 Direct deposition of the metal instead of a metal containing precursor, such as nitrate or chloride, omits the need for an energy intensive calcination step that is required for the wet synthesis methods for precursor removal.
Mechanochemically prepared catalysts have been studied in various reactions, and many reports show similar or improved activities compared to conventionally prepared catalysts. For instance, ball-milled gold catalysts were tested in CO oxidation, and their performance was well in line with catalysts prepared by impregnation methods.13 Comparison of ball-milled and impregnated cobalt catalysts in methane dry reforming showed that the mechanochemical synthesis improved the dispersion and stability of the active metal on the support, resulting in more active and stable catalysts compared to the impregnated ones.15 These interesting results prompted us to investigate the use of ball milling for the preparation of platinum catalysts for dehydrogenation of liquid hydrogen carriers, which to our knowledge has not been previously reported.
In this contribution, we show that highly active and stable dehydrogenation catalysts are prepared by solvent-free high energy ball milling by mixing only titania support and metallic platinum. The performance of the catalysts is studied in continuous gas-phase dehydrogenation of methylcyclohexane and compared with conventional incipient wetness impregnated catalyst. Extensive catalyst characterisation is carried out to elucidate the effect of the synthesis method on the catalyst properties, activity and stability.
2 Methods
2.1 Catalyst synthesis
The catalyst solid precursors, including the metallic Pt (325 mesh, ≥99.9%, Thermo Scientific) and oxide support (TiO2 anatase, Sigma-Aldrich, 99.7%) powders, were loaded in the milling jar starting with the precursor with the highest loading mass, i.e., support material. Every time, Pt loading of 1 wt% and preparation scale of 1 g was targeted. The solid mixture was milled for 3 h at 600 rpm. The ball milling parameters were selected based on our previous expertise on noble metal deposition on an oxide carrier with this mill geometry.13 Furthermore, a time series of samples milled for 1, 3 and 6 hours was prepared and screened with XRD (not shown). The XRD patterns showed no significant difference in titania peaks between the samples milled for 3 or 6 hours, which suggests that a steady state was reached already after 3 hours of milling. Several repetitions of the synthesis were necessary to ensure enough material was available for follow-up characterisation and catalysis testing. The catalyst synthetised by ball milling was denoted as BM Pt/TiO2.
2.2 MCH dehydrogenation experiments
 | (1) |
 | (2) |
 | (3) |
2.3 Catalyst characterisation techniques
The in situ ambient pressure X-ray photoelectron spectroscopy (APXPS) measurements were performed at the HIPPIE beamline at the MAX IV Laboratory in Sweden.18 The Pt 4f and Ti 3s spectra were recorded using photon energies of 1200 eV. Prior to the operando measurements, the catalyst was suspended in water and drop-cast onto a Si foil. The spectra were initially recorded for the as-synthesised catalyst. Subsequently, the catalyst was reduced under 1 bar of pure H2 at 350 °C. Finally, the APXPS spectra were measured at 2 mbar MCH pressure and 345 °C and 365 °C. The Pt 4f and Ti 3s spectra were fitted using a Tougaard background with DS line shapes.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12.5. The carbon content of the fresh and spent catalysts was analysed by Leco CS230 analyser.
:12.5. The carbon content of the fresh and spent catalysts was analysed by Leco CS230 analyser.
3 Results & discussion
3.1 Catalyst synthesis and physical properties
Titania-supported platinum catalyst was synthesised by both ball milling and incipient wetness impregnation methods, denoted BM and IWI, respectively. The ball-milled catalyst was prepared by simply milling metallic Pt powder with anatase-phase titania in a planetary ball mill equipped with corundum grinding balls and jars. The program was selected based on our earlier studies, which showed that 3 h was sufficient to disperse the metal on the support surface.13 Furthermore, we expected the high-energy milling to transform the anatase into rutile. Several batches of 1 g scale were made and combined to improve reproducibility.Incipient wetness impregnation method was applied to prepare a more conventional catalyst as a point of reference. To align with the majority of catalysts studied in the scientific works, we selected a commonly used commercial titania P25 support, which is a mixture of anatase and rutile. Based on a previous report, this type of support outperformed pure anatase support in LOHC dehydrogenation.9 According to BET analysis, both catalysts had a specific surface area in similar range, around 50 m2 g−1 (Table 1). The adsorption average pore width by BET analysis was 6 nm for the BM catalyst, and slightly larger 9 nm for the IWI catalyst. Analysis of the metal content by ICP showed that BM Pt/TiO2 contained 0.92 wt% Pt, whereas IWI Pt/TiO2 had a lower Pt content of 0.83 wt%. The nominal concentration was 1 wt% for both catalysts, indicating that the metal is loaded more efficiently using the mechanochemical synthesis compared to IWI, which is beneficial from both economic and environmental aspects.
| Catalyst | S BET (m2 g−1) | Pore size (nm) | Pt content (wt%) |
|---|---|---|---|
| BM Pt/TiO2 | 54 | 6.1 | 0.92 |
| IWI Pt/TiO2 | 50 | 9.0 | 0.83 |
3.2 MCH dehydrogenation experiments
The dehydrogenation of vaporised MCH was carried out in a continuous flow reactor at atmospheric pressure. Liquid MCH was pumped to a heated (150 °C) process line together with nitrogen to facilitate a steady reactant flow through the reactor. The catalyst bed comprised of catalyst powder mixed with quartz sand, and the catalyst was reduced in situ prior to the MCH dehydrogenation. The reaction products were analysed online by gas chromatography. A more detailed process scheme is provided in the experimental section of this paper.Before experimenting with the catalysts, we wanted to have an understanding on the effect of the reaction parameters on MCH conversion and stability using the conventional impregnated catalyst IWI Pt/TiO2. A simple experimental design matrix was used to find the optimal conditions over 0.2 g of IWI Pt/TiO2. The studied factors were the MCH feed rate (0.1–0.2 ml min−1) and temperature (300–350 °C), while other reaction conditions were maintained constant (see ESI† Section S2.1). The minimum value for MCH feed rate was the minimum flow rate of the liquid feeding pump, and the maximum was double the minimum. MCH was co-fed with 50 ml min−1 of N2 to ensure steady gas flow. The minimum temperature 300 °C was chosen based on literature,7 which suggested that lowering the temperature might be beneficial to decrease coke formation and thus inhibit the catalyst deactivation. The maximum temperature 350 °C was the typical MCH dehydrogenation temperature seen in relevant literature.3,7 The reaction parameters were chosen with emphasis on highlighting the possible differences between the two catalysts instead of solely maximising the MCH conversion.
All the MCH dehydrogenation condition screening experiments had a high selectivity to toluene (>99.5%), hence our emphasis on analysis focuses on catalyst activity which was more responsive to the varied process conditions. During the first hour of the experiment, the conversion of MCH was between 20–70% depending on the conditions (Fig. S2†). However, the conversion showed a slight decline over time on stream, which prompted us to calculate the rate of deactivation for the reaction as decrease of the MCH conversion per hour. Interestingly, the rate of deactivation varied depending on the reaction conditions. Based on these observations, we used the conversion and H2 productivity at 4 h along with the rate of deactivation as responses for the statistical analysis (Table S1†). The results were fitted using MODDE software version 13.0.2 (see ESI† for detailed information). Temperature had the biggest positive influence on both conversion and H2 production (Fig. S3 and S4†). However, increasing the temperature also increased the deactivation rate. While increasing the feed rate had a slightly positive effect on H2 production, it also caused increased deactivation and decreased conversion. Based on the analysis, we chose to use 345 °C and MCH feed rate 0.1 ml min−1 for catalyst comparison studies. At lower temperatures (300–325 °C), the H2 productivity was insufficient, whereas the higher MCH feed rate (0.2 ml min−1) caused rapid catalyst deactivation.
Next, we wanted to compare the ball milled and impregnated Pt/TiO2 catalysts at the chosen conditions (345 °C, 0.1 ml min−1 MCH feed rate, 0.2 g catalyst). The experiments were operated for 15 h, or until the catalyst fully deactivated. The MCH conversion of IWI Pt/TiO2 started at around 70% and decreased to around 45% during the 15 h TOS (Fig. 1). The initial MCH conversion of BM Pt/TiO2 was around 50%, thus lower than for its IWI prepared counterpart. However, the deactivation rate of BM Pt/TiO2 was lower than that of the IWI catalyst, and the MCH conversion of BM Pt/TiO2 was around 40% at the end of the experiment (Table S2†). In addition to the experiments performed at 345 °C, we tested the catalysts at a higher temperature of 365 °C in 15 h experiments (Fig. 1). In these conditions, the IWI Pt/TiO2 catalyst completely deactivated in just 9 hours on stream, supporting the observation of the effect of temperature on the deactivation. Remarkably, the ball-milled catalyst showed different behaviour; the increased temperature stabilised the BM Pt/TiO2 catalyst, and the deactivation rate during the 15 h experiment was only 0.2 Δmol% h−1. The MCH conversion remained constant at around 55%. At 15 h TOS, the hydrogen production rate was 670 mmolH2 gPt−1 min−1, which is among the highest reported (Table S3†). To our knowledge, higher rates in MCH dehydrogenation have only been observed with a trimetallic Pt3(Fe0.75Zn0.25)/SiO2 catalyst19 and a single atom Pt1/CeO2,20 757 and 2509 mmolH2 gPt−1 min−1, respectively.
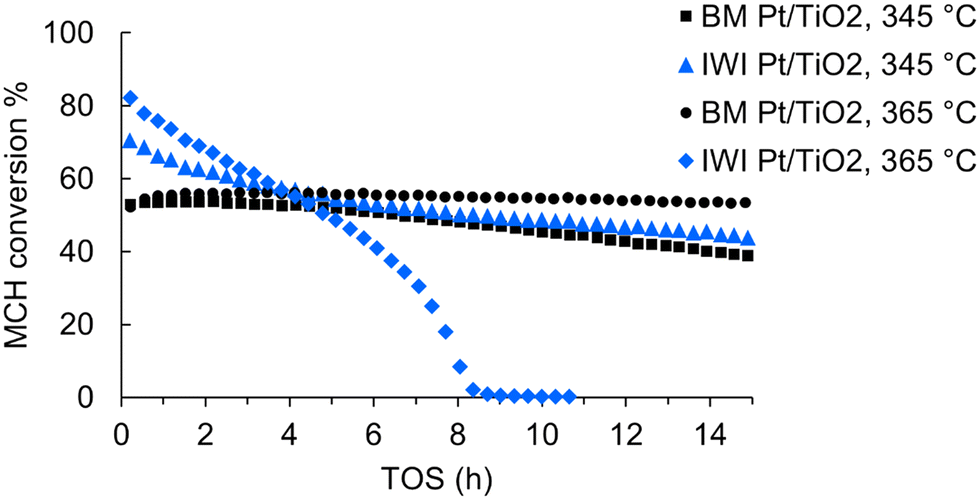 | ||
| Fig. 1 Catalytic performance of the monometallic Pt/TiO2 catalysts in terms of MCH conversion at two different temperatures: 345 and 365 °C. Atmospheric pressure, 0.2 g catalyst, MCH feed rate 0.1 ml min−1. | ||
Analysis of side products in the outlet gas showed that all of the aforementioned experiments had over 99% selectivity to toluene (Fig. S5†). Minor amounts of hydrocarbon side products, which were attributed to partially dehydrogenated intermediate products such as methylcyclohexene, were observed (Fig. S6†). Selectivity to demethylation products i.e., cyclohexane and benzene, varied between 0–0.12% (Fig. S7†). Methane concentrations were measured in the process gas arriving to the online GC (Fig. S5†). No CO or CO2 were observed above the detection limit, which for the TCD-GC setup was about 1000 ppm. The highest amount of side products was detected with IWI Pt/TiO2 at 365 °C; over 1.2% selectivity to hydrocarbon side products and over 50 ppm methane were observed just before the catalyst deactivated. At 345 °C, IWI Pt/TiO2 produced about 20 ppm methane and the side product selectivity was about 0.4%. Remarkably, in case of BM Pt/TiO2, the selectivity to side products decreased from 0.3% to less than 0.2% upon increasing the temperature from 345 to 365 °C and furthermore, no cyclohexane or benzene were detected at 365 °C. It is also important to point out that we could not detect any methane in the experiments with the ball-milled catalyst, which highlights its superior stability and selectivity in MCH dehydrogenation.
In industrial Pt-catalysed processes for hydrocarbons, such as naphtha reforming, a small co-feed of hydrogen is often used to maintain optimal hydrogen pressure and prevent coking.21–23 In our experiments, we chose to study the catalysts without additional hydrogen. Firstly, it allowed to better observe the differences in catalyst activity and deactivation, and secondly, it would be ideal to develop a robust catalyst and omit the need for additional hydrogen. To study if the additional hydrogen stabilises the conversion as efficiently as the temperature elevation to 365 °C, we performed an experiment with a small additional 5 ml min−1 hydrogen feed for BM Pt/TiO2 at 345 °C (Fig. S8†). While the co-fed H2 helped to decrease the catalyst deactivation, increasing the temperature had a more pronounced effect on stability for the BM Pt/TiO2 catalyst. Furthermore, the conversion at 15 h TOS was about 45% in the H2 co-feed experiment at 345 °C, and about 55% in the 365 °C experiment.
3.3 Catalyst characterisation
To better understand the properties and activity of the catalysts, detailed characterisation was carried out for the fresh catalysts before reduction. The spent catalysts were recovered from the reactor as a mixture with quartz sand and the mixture was analysed to investigate the catalyst deactivation.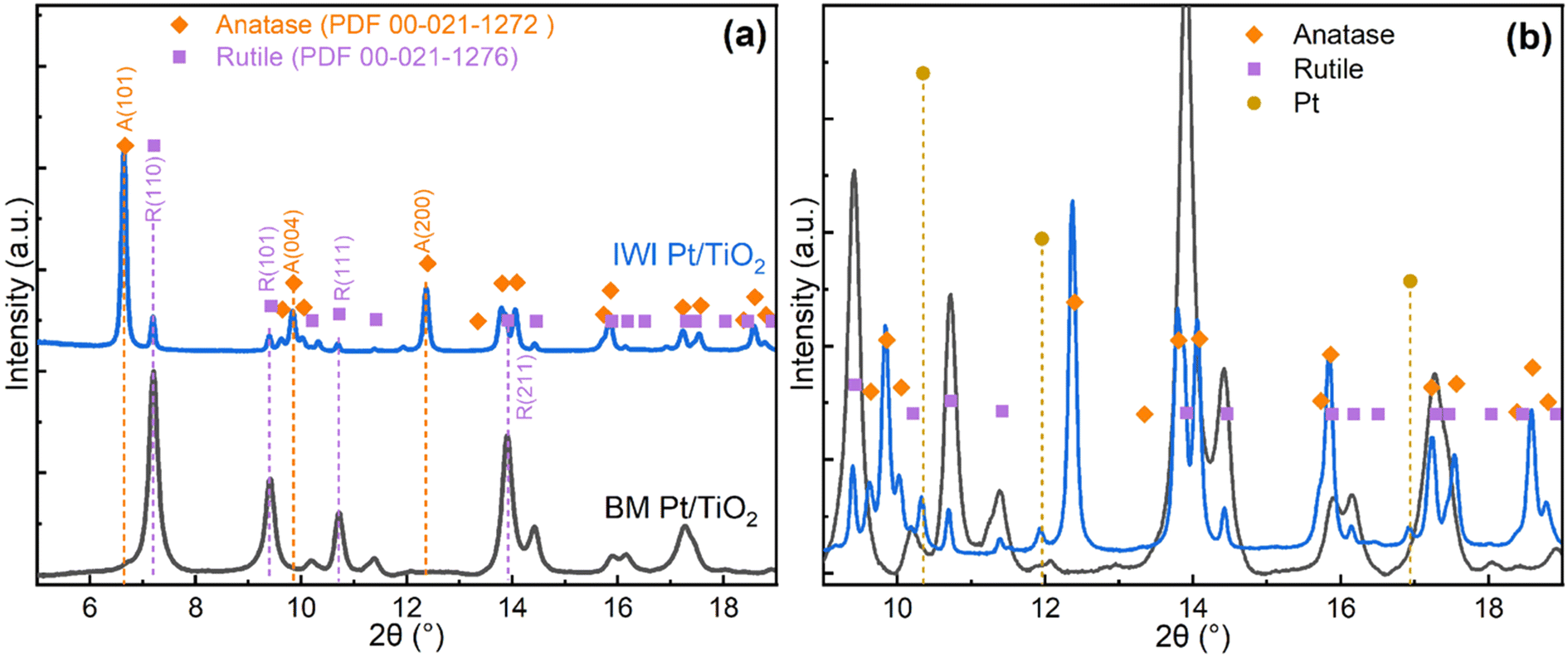 | ||
| Fig. 2 (a) Synchrotron X-ray diffraction profiles and (b) magnified view of the catalyst samples. | ||
The impregnated IWI Pt/TiO2 showed patterns corresponding to a mixture of rutile and anatase (PDF 00-021-1272) with a ratio around 1:9, consistent with earlier reports.9 Pt-related diffraction peaks were only observed in the pattern for IWI Pt/TiO2 in Fig. 2b, while no such features were seen in other patterns due to the low noble metal content and small particle size.
A closer look at the diffraction patterns reveals that the rutile peaks of the ball-milled samples are broader than the anatase peaks in the impregnated samples, indicating smaller crystallite size or higher disorder of the support in the ball-milled catalysts. Mean crystallite sizes of the support for ball-milled catalysts estimated by Scherrer equation from the rutile (110) reflection are about 20 nm. The structural order of the catalysts is also evident from the PDF profiles, which show peaks characteristic to the corresponding titania phases (Fig. S9†).
Fresh catalysts. The nano-to-micro-scale morphology and particle size distribution of the catalysts was investigated with transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The predominant size distribution of titania particles in all samples was concentrated within the range of 10 to 20 nm (Fig. S10†). The average size of the metal particles was determined from the fresh catalysts prior to reduction. In case of BM Pt/TiO2, the Pt particles were uniformly distributed across the titania particles, and the mean Pt particle size was 0.7 nm with a rather large standard deviation at 0.5 nm (Fig. 3a and c, Table S4†). Some larger Pt particles over 10 nm were also observable, however, these were excluded from the particle size calculations due to their minor representation in comparison to the smaller particles. EDS analysis confirms the even distribution of Pt on the support (Fig. S11†).
 | ||
| Fig. 3 HAADF-STEM images and Pt particle size distributions of the fresh catalysts (a and c) BM Pt/TiO2, (b and d) IWI Pt/TiO2. | ||
In case of IWI Pt/TiO2, the Pt particle size varied between 0.6 nm to 2.0 nm with an average of 1.3 nm (Fig. 3d). The Pt particles were non-uniformly distributed on the support, with Pt being selectively present on specific TiO2 particles (Fig. 3b and S11†). Similar observation was reported by Iddir et al. when investigating Pt deposition on P25 titania; Pt particles preferred nucleation on rutile particles rather than anatase particles, which was attributed to the higher number of oxygen vacancies in the rutile phase.25 Compared to its ball-milled counterpart, the impregnated catalyst clearly showed wider particle size distribution and larger average particle size.
Pt dispersion of the catalysts was measured by static CO chemisorption (ESI† Section S3.7). Prior to the measurement, the catalysts were reduced with H2 at 350 °C, similarly to the reduction before the dehydrogenation experiments. Interestingly, the BM Pt/TiO2 catalyst showed only 8.3% dispersion and IWI Pt/TiO2 12.1% dispersion. These values are significantly lower than what can be calculated based on geometry26 and the mean particle size determined by TEM; the IWI catalyst Pt dispersion should be 86% whereas the BM catalyst should have nearly full dispersion. Platinum is known to have strong metal-support interactions (SMSI) with some support materials, for instance titania, which result in encapsulation of the metal particles with a thin layer of the support material and thus reduced chemisorption of CO.27
Spent catalysts. To investigate the changes caused by the MCH dehydrogenation, the catalyst particle size and distribution were characterised after the experiments at 345 °C and 365 °C (Fig. S12 and S13†). After the dehydrogenation at 345 °C, the BM Pt/TiO2 showed a slight increase in the Pt particle size from 0.7 nm to 1.0 nm (Table 2). Increase to 365 °C did not inflict further growth of the metal particles, highlighting the stability of the ball-milled catalyst. Ball milling is known to increase surface defects,10 which has been shown to improve anchoring of nanoparticles to increase the stability.28 In case of IWI Pt/TiO2, the Pt particle size of the catalyst spent at 345 °C was similar to the fresh catalyst, around 1.3 nm. However, at 365 °C the Pt particle size increased slightly from 1.3 nm to 1.7 nm.
| Catalyst | Average Pt metal particle size and standard deviation (nm) | ||
|---|---|---|---|
| Fresh | Spent at 345 °C | Spent at 365 °C | |
| BM Pt/TiO2 | 0.7 ± 0.5 | 1.0 ± 0.5 | 1.1 ± 0.3 |
| IWI Pt/TiO2 | 1.3 ± 0.2 | 1.3 ± 0.3 | 1.7 ± 0.3 |
Fresh catalysts. The XPS measured Pt 4f and Ti 3s spectra of fresh catalysts prior to reduction are presented in Fig. 4. In all the catalysts, the Ti 3s peak was observed at 62.6 eV with two charge transfer satellite peaks at 66.4 and 75.8 eV (red in Fig. 4).29 The BM Pt/TiO2 catalyst exhibited a complex mixture of various Pt oxidation states based on the Pt 4f peak; metallic Pt0 (71.1 eV, blue color),30 Pt2+ (73 eV, navy blue color) and Pt4+ (75.1 eV, green).31 Another peak was observed centred at 72 eV (dark yellow), between the levels of Pt0 and Pt2+, indicating a partial positive charge of Pt. This peak is assigned as Ptn in the figures. Based on quantification of the Pt peak areas, this was the most abundant Pt species in the BM Pt/TiO2 catalyst (Fig. S14a†). Potentially, this peak could originate from the very small Pt particles, as similar positive shifts of about 1 eV have been reported previously for single atom Pt catalysts.32,33 Another possibility is interaction with titania; Pt is known to form SMSI with titania, which can result in an overlayer of titania on the metal partiles.5 Beck et al. assigned a similar peak to Pt–O at the interface of the titania overlayer.34 The observation of SMSI is supported by the chemisorption results which indicate partial encapsulation or coverage of Pt with titania. Furthermore, an additional peak at 460.1 eV was observed in the Ti 2p spectrum, which could originate from charge transfer between the support and Pt (Fig. S15†).32,35,36
 | ||
| Fig. 4 Pt 4f and Ti 3s spectra for fresh catalyst. (a) BM Pt/TiO2 and (b) IWI Pt/TiO2. | ||
The fresh IWI Pt/TiO2 catalyst contained mostly platinum in oxide states Pt4+ and Pt2+, and only a small amount of Pt0 was detected (Fig. 4b). This was expected since no reductive treatment was made during the catalyst preparation. Curiously, no Pt peak at 72 eV was observed in the impregnated sample.
Spent catalysts. To investigate changes in the oxidation states, we analysed the catalysts (as a mixture with quartz sand) after the dehydrogenation experiments. Fig. 5 depicts the Pt 4f spectra of the catalysts used at 345 °C and 365 °C, as denoted in the figures. The relative percentages of the Pt species are presented in Fig. S14b.† In all samples, most of Pt was present as metallic Pt0, discerned by a characteristic peak at 70.9 eV. This is expected after reducing the catalysts with H2 prior to the dehydrogenation experiments. Nevertheless, a noticeable presence of Ptn species at 72.2 eV was observed in the spent BM Pt/TiO2 catalysts (Fig. 5a and c). The relative amount of Ptn increased with increasing the reaction temperature (Fig. S14b†). This indicates that the Ptn peak arises from the SMSI and interaction with titania; the titania overlayer is known to grow during prolonged times at increased temperatures.34 Additionally, a minor peak corresponding to Pt4+ at 75 eV was observed in the BM Pt/TiO2 365 °C sample. In case of IWI Pt/TiO2 catalyst, a new peak at 72.1 eV was detected in the spent IWI Pt/TiO2 catalyst at 365 °C. This peak is assigned to PtOx species in Fig. 5. The PtOx species is likely attributed to partial oxidation of the spent catalyst, caused by prolonged air exposure between the MCH dehydrogenation reaction and the UHV XPS measurements. As explained vide infra, this species was not observed in the APXPS measurements. No significant changes were observed in the Ti 3s spectra compared to the fresh catalyst XPS spectra (not shown).
 | ||
| Fig. 5 The Pt 4f and Ti 3s spectra of spent catalysts after dehydrogenation at different temperatures. (a) BM Pt/TiO2, 345 °C (b) IWI Pt/TiO2, 345 °C (c) BM Pt/TiO2, 365 °C and (d) IWI Pt/TiO2, 365 °C. | ||
Ambient pressure XPS (APXPS). To further investigate the changes in the electronic structure of the catalysts during the experiments, APXPS measurements were carried out at HIPPIE beamline at MAX IV, which enabled in situ reduction and MCH dehydrogenation during the XPS measurement. The Pt 4f and Ti 3s spectra of BM and IWI Pt/TiO2 were initially recorded for the fresh catalyst and, subsequently, after the catalyst was reduced under 1 bar of pure H2 at 350 °C. Finally, the APXPS spectra were measured at 2 mbar MCH pressure at 345 °C (Fig. 6) and 365 °C (Fig. S16†).
 | ||
| Fig. 6 APXPS Pt 4f and Ti 3s spectra recorded for fresh catalyst (bottom), after reduction (middle) and during the dehydrogenation (top) for (a) BM Pt/TiO2 and (b) IWI Pt/TiO2. | ||
The spectra of the fresh catalysts are very similar to those recorded with UHV XPS (Fig. 4vs.6). The ball-milled catalyst contained metallic Pt, various Pt oxides, including Pt2+ and Pt4+, and the Ptn peak at 72 eV attributed to the SMSI and interaction with titania. The spectra of the IWI catalyst also closely resemble those described in the UHV XPS analysis. The Ti 3s spectra are consistent with those observed in UHV measurements, with binding energies similar to those found in UHV XPS.
After reduction of the catalysts at 350 °C, Pt is reduced to Pt0 in both catalysts. However, in the ball-milled catalyst, a partial positive charge on Pt (Ptn), is still observable, indicating more pronounced SMSI in the ball-milled catalyst. In contrast, all Pt oxides are fully reduced to Pt0 in the IWI catalyst. Additionally, Ti is partially reduced to TiOx, as indicated by the orange peak fitted at 60 eV.27 The detection of reduced Ti species in both catalysts, likely arising from high-temperature treatment under H2 atmosphere, is indicative of partial TiO2 reduction. Separately, the observed Ptn peak as well as the decrease in Pt dispersion, as measured by CO chemisorption, is consistent with the manifestation of strong metal–support interactions (SMSI), in agreement with previous reports on Pt/TiO2 systems.
The in situ APXPS observations highlight that the chemical state of Pt during the dehydrogenation is very similar to that of the reduced and spent catalysts, indicating that there is no transient state of the catalyst under MCH exposure. The catalysts were tested for more than 3 hours under operando conditions at 345 °C (Fig. 6) and 365 °C (Fig. S16†), showing no significant changes over time. Both catalysts exhibit rather similar characteristics, indicating comparable surface chemistry and electronic environments of Pt and TiO2 at these temperatures. In both catalysts, Pt0 is the predominant species during the MCH dehydrogenation. Interestingly, the Ptn peak is present in the BM Pt/TiO2 during the dehydrogenation, which corroborates the SMSI in the ball mill-prepared catalyst. In case of IWI Pt/TiO2, only Pt0 species was observed, which suggests that the PtOx peak observed in the UHV XPS measurements of the spent catalyst is likely due to partial oxidation caused by prolonged air exposure between the dehydrogenation experiment and the XPS analysis. This underscores the importance of performing in situ XPS analyses to accurately capture the true oxidation states of active species during catalytic reactions, as ex situ measurements can be misleading due to post-reaction changes.
 | ||
| Fig. 7 Carbon content of the fresh and spent BM Pt/TiO2 and IWI Pt/TiO2 catalysts. | ||
The catalyst samples were further analysed by Raman spectroscopy to elaborate the carbonaceous compounds present. The fresh catalysts were measured as a mixture with quartz sand to enable comparison with the spent catalysts. The fresh BM and IWI catalysts had quite similar spectra in general (Fig. 8). The strong bands in the range from 100 to 800 cm−1 are mainly caused by titania support (marked as TiO2 in the figure) and used quartz sand (marked as Q in the figure).37,38 Characteristic sp2 carbon bands and their composites determined in the spent samples are G band (1580 cm−1) and D band (between 1300–1400 cm−1).39,40 These bands are negligible over the fresh catalyst samples. In the spent ball-milled catalyst samples, both the G and D bands are weakly visible in the BM Pt/TiO2 345 °C sample but not visible in BM Pt/TiO2 365 °C (Fig. 8a). Conversely, the spent IWI Pt/TiO2 samples show much stronger carbon bands; a small increase can already be detected in IWI Pt/TiO2 345 °C, whereas the G band is clearly at its strongest in the IWI Pt/TiO2 365 °C sample. The results gained are well in line with the total carbon content measurements and confirm that the IWI Pt/TiO2 catalyst was deactivated by carbonaceous compounds at 365 °C.
 | ||
| Fig. 8 Raman spectra of fresh and spent BM Pt/TiO2 (left) and IWI Pt/TiO2 (right) catalysts. D and G denote carbonaceous species, Q quartz. | ||
The formation of graphitic carbon is further corroborated by the APXPS measurements. Fig. S17† illustrates the evolution of the C 1s spectra obtained during the in situ dehydrogenation of MCH. Two distinct C 1s peaks are observed in the spectra. The first peak, located at 284.4 ± 0.1 eV, is attributed to adventitious carbon,41 while the second peak, positioned at 285.5 ± 0.1 eV, corresponds to gas-phase MCH carbon. These features are consistent for both the BM and IWI catalysts. Under reaction conditions of 2 mbar and elevated temperatures of 345 °C and 365 °C, the C 1s spectra for the BM catalyst remain unchanged relative to those recorded for the reduced catalyst. This suggests that the surface chemistry of the BM catalyst, with respect to carbon species, remains stable during MCH dehydrogenation, and no significant alteration in binding energy is observed. In contrast, the C 1s peak located at the 284.4 eV for the IWI catalyst exhibit a ∼0.3 eV shift towards lower binding energies to 284 eV when compared to the spectra collected after the reduction process. The shift towards lower binding energy is commonly associated with the formation of graphitic carbon as the carbon atoms become more covalently bonded to the metal catalyst, leading to changes in their electronic environment.42
Previous studies have suggested that deactivation can be caused by adsorption of the produced toluene on the catalysts, which can lead to formation of coke.5,21 A plausible reason for the different coking behaviour of the two catalysts might be their different acidity, which originates from the different titania polymorphs. Anatase is known to be more acidic than rutile, and the acidity of the support is known to increase coke formation.7 To further investigate the catalysts, we measured the acidity by NH3-TPD measurements (Fig. S19†). The quantity of NH3 desorbed within the measured temperature range of 50–650 °C was 138 and 307 μmol g−1 for BM Pt/TiO2 and IWI Pt/TiO2, respectively. The result confirms that the anatase-supported IWI Pt/TiO2 has in total over twice as many acid sites as the rutile-supported BM Pt/TiO2. Interestingly, BM Pt/TiO2 had also a minor amount of strong acid sites (NH3 desorption peak maximum at 500 °C)43 that the IWI catalyst did not present. While NH3-TPD does not separate Lewis acid sites from Brønsted acid sites, reports in the literature have shown that the acid sites on rutile are predominantly Lewis acid sites.44 Brønsted acid sites can induce cracking reactions leading to unwanted products and further to catalyst coking.45 Analysis of the side products of MCH dehydrogenation reveals that the formation of side products and especially methane increased prior to the deactivation of IWI Pt/TiO2 at 365 °C (Fig. S5†). Remarkably, no methane was observed with BM Pt/TiO2 and at 365 °C the catalyst produced no C6 side products resulting from cracking.
In addition to the support acidity, the catalysts differ in the electronic state of the Pt during the dehydrogenation. The partial positive charge of Ptn, which arises from the SMSI, could facilitate the desorption of toluene from the Pt surface; DFT calculations by Tuo et al. showed that electron deficient state of Pt decreased the adsorption energy of the aromatic product leading to enhanced dehydrogenation activity.46 As toluene adsorption on the catalyst has been shown to lead to coke formation,5 the Ptn species observed on BM Pt/TiO2 can play an important role in preventing the coking.
The combination of total carbon, APXPS and Raman results provides a robust confirmation of the IWI catalyst deactivation mechanism through the formation of carbonaceous compounds under the reaction conditions. The NH3-TPD measurements confirm that the ball-milled catalyst benefits from a significantly lower acid site density compared to the IWI prepared catalyst. Furthermore, the SMSI on the BM catalyst facilitates desorption of the produced toluene from the platinum surface. These factors combined ensure the improved stability and resistance towards cracking reactions and coking.
4 Conclusions
Efficient and stable catalysts for the hydrogen release from liquid organic hydrogen carriers are crucially important for the feasibility of LOHCs in storage and transport of hydrogen. In this work, we have shown a waste-free mechanochemical synthesis to produce highly active and stable Pt catalysts for the dehydrogenation of methylcyclohexane into toluene. By mixing simply metallic platinum and anatase-phase titania powder in a high energy ball mill, stable nanometre-sized Pt particles were evenly distributed onto the titania support, and furthermore, phase transformation of anatase to rutile was observed.Comparison of the ball-milled catalyst with a conventionally impregnated platinum catalyst in the dehydrogenation of methylcyclohexane clearly showed the high activity and superior stability of the mechanochemically prepared BM Pt/TiO2 catalyst. The best performance was achieved at 365 °C; the BM Pt/TiO2 catalyst was stable throughout the 15 h experiment. The hydrogen production rate was 670 mmolH2 gPt−1 min−1, which is among the highest values reported for methylcyclohexane dehydrogenation. At the same conditions, the conventionally impregnated IWI Pt/TiO2 fully deactivated already at 9 h TOS due to coking. The ball-milled catalyst also showed higher selectivity towards toluene compared to the impregnated catalyst. Based on our characterisations, the rutile support of the BM Pt/TiO2 catalyst is less acidic compared to the anatase-rich IWI Pt/TiO2, which increases resistance towards coke formation. Furthermore, a partial positive charge on Pt, arising from interaction of the platinum with the support titania, facilitates the desorption of the produced toluene and prevents coking.
Data availability
The authors confirm that the data supporting the manuscript “Mechanochemical synthesis of Pt/TiO2 for enhanced stability in dehydrogenation of methylcyclohexane” by Krista Kuutti et al. are available within the article and its ESI.†Author contributions
KK: methodology, investigation, visualization and writing – original draft. MG: investigation, formal analysis, visualization and writing – review & editing. PP: conceptualization and methodology. JDB: methodology and investigation. PJ: methodology and investigation. HS: investigation, formal analysis and visualization. SW: investigation, formal analysis and visualization. GK: investigation. JFC: investigation. FS: methodology and resources. KA: investigation and analyses. MiH: investigation, analyses, funding acquisition and writing – review & editing. MaH: supervision, funding acquisition and resources. SU: conceptualization, investigation, formal analysis, supervision, project administration and writing – review & editing. SR: conceptualization, formal analysis, supervision, funding acquisition and writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Authors (KK, PP, PJ, MiH, MaH, SU, SR) acknowledge Business Finland for providing funding to perform the research work (funding decisions 50/31/2021, 45774/31/2020, 5667/31/2023). MG, MaH and SU were supported by the NANOCAT project of Kvantum Institute of University of Oulu, University of Oulu & Research Council of Finland Profi 352788 (H2FUTURE). Part of the work was performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan. The authors gratefully acknowledge the Center of Materials Analysis (CMA), University of Oulu for the XPS, carbon content, and TEM measurements. JFC thanks MARSALAS21-09 grant funded by MCIN/AEI/10.13039/501100011033 and European Union NextGeneration EU/PRTR. HS and MaH acknowledge EU/Interreg Aurora/Sustainable Hydrogen project (2023–2025) for funding. We acknowledge MAX IV Laboratory for time on Beamline HIPPIE under Proposal 20221414. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. Päivi Aakko-Saksa is acknowledged for insights in the catalyst preparation and Satu Ojala for the Raman measurements.References
- S. Atilhan, S. Park, M. M. El-Halwagi, M. Atilhan, M. Moore and R. B. Nielsen, Curr. Opin. Chem. Eng., 2021, 31, 100668 CrossRef.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed.
- P. T. Aakko-Saksa, C. Cook, J. Kiviaho and T. Repo, J. Power Sources, 2018, 396, 803–823 CrossRef CAS.
- Y. Sekine and T. Higo, Top. Catal., 2021, 64, 470–480 CrossRef CAS.
- S. Nagatake, T. Higo, S. Ogo, Y. Sugiura, R. Watanabe, C. Fukuhara and Y. Sekine, Catal. Lett., 2016, 146, 54–60 CrossRef CAS.
- J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- Y. Okada, E. Sasaki, E. Watanabe, S. Hyodo and H. Nishijima, Int. J. Hydrogen Energy, 2006, 31, 1348–1356 CrossRef CAS.
- X. Yang, Y. Song, T. Cao, L. Wang, H. Song and W. Lin, J. Mol. Catal., 2020, 492, 110971 CrossRef CAS.
- P. T. Aakko-Saksa, M. Vehkamäki, M. Kemell, L. Keskiväli, P. Simell, M. Reinikainen, U. Tapper and T. Repo, Chem. Commun., 2020, 56, 1657–1660 RSC.
- A. P. Amrute, J. De Bellis, M. Felderhoff and F. Schüth, Chem. – Eur. J., 2021, 27, 6819–6847 CrossRef CAS PubMed.
- C. Suryanarayana, Prog. Mater. Sci., 2001, 46, 1–184 CrossRef CAS.
- Y. Hu, B. Li, C. Yu, H. Fang and Z. Li, Mater. Today, 2023, 63, 288–312 CrossRef CAS.
- H. Schreyer, R. Eckert, S. Immohr, J. De Bellis, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2019, 58, 11262–11265 CrossRef CAS PubMed.
- J. De Bellis, H. Petersen, J. Ternieden, N. Pfänder, C. Weidenthaler and F. Schüth, Angew. Chem., Int. Ed., 2022, 61, e202208016 CrossRef CAS PubMed.
- M. Armengol-Profitós, A. Braga, L. Pascua-Solé, I. Lucentini, X. Garcia, L. Soler, X. Vendrell, I. Serrano, I. J. Villar-Garcia, V. Pérez-Dieste, C. Escudero, N. J. Divins and J. Llorca, Appl. Catal., B, 2024, 345, 123624 CrossRef.
- A. Rahemtulla, G. King, A. Gomez, N. Appathurai, A. Leontowich, R. Castle, N. Burns, C.-H. Kim, B. Moreno and S. Kycia, J. Synchrotron Radiat., 2025, 32, 750–756 CrossRef PubMed.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- S. Zhu, M. Scardamaglia, J. Kundsen, R. Sankari, H. Tarawneh, R. Temperton, L. Pickworth, F. Cavalca, C. Wang, H. Tissot, J. Weissenrieder, B. Hagman, J. Gustafson, S. Kaya, F. Lindgren, I. Kallquist, J. Maibach, M. Hahlin, V. Boix, T. Gallo, F. Rehman, G. D'Acunto, J. Schnadt and A. Shavorskiy, J. Synchrotron Radiat., 2021, 28, 624–636 CrossRef CAS PubMed.
- Y. Nakaya, M. Miyazaki, S. Yamazoe, K.-I. Shimizu and S. Furukawa, ACS Catal., 2020, 10, 5163–5172 CrossRef CAS.
- L. Chen, P. Verma, K. Hou, Z. Qi, S. Zhang, Y.-S. Liu, J. Guo, V. Stavila, M. D. Allendorf, L. Zheng, M. Salmeron, D. Prendergast, G. A. Somorjai and J. Su, Nat. Commun., 2022, 13, 1092 CrossRef CAS PubMed.
- K. Murata, N. Kurimoto, Y. Yamamoto, A. Oda, J. Ohyama and A. Satsuma, ACS Appl. Nano Mater., 2021, 4, 4532–4541 CrossRef CAS.
- M. D. Argyle and C. H. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- R. Alcala, D. P. Dean, I. Chavan, C.-W. Chang, B. Burnside, H. N. Pham, E. Peterson, J. T. Miller and A. K. Datye, Appl. Catal., A, 2023, 658, 119157 CrossRef CAS.
- Y. Mechiche, D. Lofficial, S. Humbert, M. Digne and A. Méthivier, Powder Technol., 2024, 433, 119179 CrossRef CAS.
- H. Iddir, V. Skavysh, S. Öğüt, N. D. Browning and M. M. Disko, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 041403(R) CrossRef.
- G. Bergeret and P. Gallezot, in Handbook of Heterogeneous Catalysis, Wiley-VCH, 2008, vol. 2, pp. 738–765 Search PubMed.
- J. Lee, I. Song and D. H. Kim, ChemCatChem, 2018, 10, 1258–1262 CrossRef CAS.
- S. Yuan, Y. Duan, C. Yu, Z. Xiong, Y. Li, H. Wang, Y. Zhang and Y. Gao, Mol. Catal., 2023, 535, 112830 CrossRef CAS.
- H. Chermette, P. Pertosa and F. M. Michel-Calendini, Chem. Phys. Lett., 1980, 69, 240–245 CrossRef CAS.
- P. Mani, R. Srivastava and P. Strasser, J. Phys. Chem. C, 2008, 112, 2770–2778 CrossRef CAS.
- X. Li, X. Wang, I. I. Sadykov, D. Palagin, O. V. Safonova, J. Li, A. Beck, F. Krumeich, J. A. van Bokhoven and L. Artiglia, ACS Catal., 2021, 11, 13041–13049 CrossRef CAS.
- A. von Weber and S. L. Anderson, Acc. Chem. Res., 2016, 49, 2632–2639 CrossRef CAS PubMed.
- Q. Liu and Z. Zhang, Catal. Sci. Technol., 2019, 9, 4821–4834 RSC.
- A. Beck, X. Huang, L. Artiglia, X. Wang, P. Rzepka, D. Palagin, M.-G. Willinger and J. A. van Bokhoven, Nat. Commun., 2020, 11, 3220 CrossRef CAS PubMed.
- L. Qiu, F. Liu, L. Zhao, W. Yang and J. Yao, Langmuir, 2006, 22, 4457–4884 CrossRef PubMed.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- T. Sekiya, S. Ohta, S. Kamei, M. Hanakawa and S. Kurita, J. Phys. Chem. Solids, 2001, 62, 717–721 CrossRef CAS.
- E. Enríquez, A. del Campo, J. J. Reinosa, G. Konstantopoulos, C. Charitidis and J. F. Fernández, J. Mater. Res. Technol., 2023, 26, 2655–2666 CrossRef.
- Z. Li, L. Deng, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2023, 135, 101089 CrossRef CAS.
- M. Veres, S. Tóth and M. Koós, Diamond Relat. Mater., 2008, 17, 1692–1696 CrossRef CAS.
- M. C. Biesinger, Appl. Surf. Sci., 2022, 597, 153681 CrossRef CAS.
- H. Su, Y. Ye, K.-J. Lee, J. Zeng, B. S. Mun and E. J. Crumlin, J. Catal., 2020, 391, 123–131 CrossRef CAS.
- N. Bou-Orm, A. Iorgu, S. Daniele and N. Guilhaume, Appl. Catal., A, 2013, 467, 414–420 CrossRef CAS.
- H. Li, M. Vrinat, G. Berhault, D. Li, H. Nie and P. Afanasiev, Mater. Res. Bull., 2013, 48, 3374–3382 CrossRef CAS.
- P. Wang, Z. Xu, T. Wang, Y. Yue, X. Baoa and H. Zhu, Catal. Sci. Technol., 2020, 10, 3537–3541 RSC.
- Y. Tuo, Y. Meng, C. Chen, D. Lin, X. Feng, Y. Pan, P. Li, D. Chen, Z. Liu, Y. Zhou and J. Zhang, Appl. Catal., B, 2021, 288, 119996 CrossRef CAS.
- Z. Dong, A. Mukhtar and H. Lin, Top. Catal., 2021, 64, 481–508 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00173k |
| ‡ Present address: Department of Sustainable Process & Energy Systems, Energy & Materials Transition Unit, TNO, Kesslerpark 1, 2288 GS Rijswijk, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2025 |