
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalysed hydrogenation of nitrous oxide†
Sophie H.
Dewick
a,
Thomas M.
Hood
 a,
Yisong
Han
b,
Steven
Huband
b and
Adrian B.
Chaplin
*a
a,
Yisong
Han
b,
Steven
Huband
b and
Adrian B.
Chaplin
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
First published on 17th June 2025
Abstract
We report on the discovery of “hidden” heterogeneous catalysis in the hydrogenation of nitrous oxide while assessing the catalytic activity of a rhodium(I) hydride complex supported by a nominally robust phosphine-based pincer ligand. Commercially available [Rh(COD)(OH)]2 was subsequently identified as a more effective catalyst precursor, enabling the hydrogenation of nitrous oxide with an apparent turnover number >3000 at room temeprature.
Nitrous oxide (N2O) is a long-lived gas that accumulates in the atmosphere, contributing to climate change as a potent greenhouse gas and leading to ozone depletion in the stratosphere.1 Although chemical activation is challenging, exponentially increasing anthropogenic emissions of N2O make it imperative that energy efficient methods are developed to remediate point sources of this atmospheric pollutant.2 Direct decomposition into N2 and O2 is encumbered by the formidable kinetic stability of N2O, necessitating temperatures >700 °C at atmospheric pressure.3 While heterogeneous catalysts can promote this reaction (ca. 300–600 °C), variants where sacrificial reducing agents are added to facilitate removal of surface oxygen atoms are capable of operating at lower temperatures and more appealing from a remediation perspective.4 In this context, the hydrogenation of N2O to afford N2 and H2O is a thermodynamically favourable, yet undeveloped transformation, using either heterogeneous or homogeneous catalysts.
Of the limited examples of heterogeneous N2O hydrogenation described in the literature,5 the use of platinum group metal catalysts is outstanding for the mild operating temperatures involved. For instance, ruthenium, rhodium, palladium, and platinum supported on titania promote the hydrogenation of N2O between 50–150 °C, with activity increasing in the order Pd > Rh > Pt > Ru based on measurements made using a flow reactor.6 Rhodium supported on SiO2 and Al2O3 is also active under flow conditions and a TOF of 0.022 s−1 was measured for the former at 72 °C.7 Molecular complexes of the platinum group metals have additionally been investigated as homogeneous catalysts, with seminal work using ruthenium pincer complexes reported by Milstein in 2017 (Fig. 1).8 A mechanism involving O-atom insertion into a Ru–H bond, coupled with bifunctional reactivity of the supporting PNP pincer ligand, was proposed and 417 TONs achieved over 48 h at 65 °C. More productive catalysts have since been identified,9,10 including a remarkable rhodium-based system by Trincado and Grützmacher, during the preparation of this manuscript, which delivered 230![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 apparent TONs after 96 h at 65 °C (Fig. 1).11
000 apparent TONs after 96 h at 65 °C (Fig. 1).11
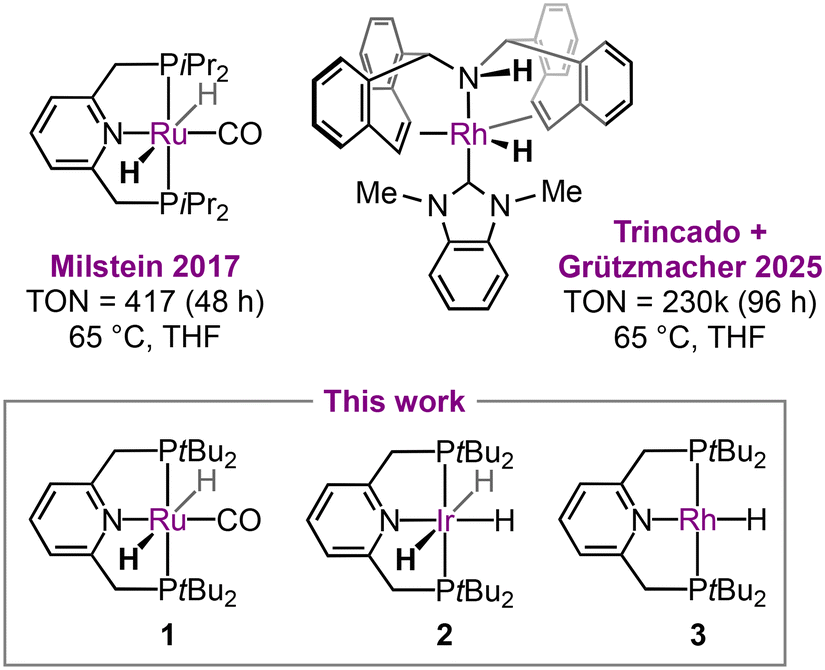 | ||
| Fig. 1 Late transition metal hydride complexes used as catalysts for the hydrogenation of nitrous oxide. | ||
Building upon our work with rhodium pincer complexes, which has included the isolation of well-defined Rh–N2O adducts,12 we became interested in assessing the relative catalytic activity of the homologous series of complexes 1–3 (Fig. 1). Octahedral hydride complexes 1 (generated from [Ru(PNP-tBu)(CO)HCl] and KOtBu) and 2 have previously been assessed by Milstein and Suárez,8,9 and we hypothesised that the component phosphine-based pincer ligand PNP-tBu would be a thermally robust scaffold that would support the homogeneous hydrogenation of N2O using square-planar rhodium(I) hydride 3.13,14
The hydrogenation of N2O was first examined at RT using 5 mM solutions of 1–3 in 2.0 mL THF, stirred within the cold finger of a 100 mL gas bulb pressurised with a ∼1:2 mixture of H2/N2O (3 atm, Table 1). Under these net oxidising conditions, 1 and 2 showed very low catalytic activity, whereas 3 gave 174 apparent TONs over 24 h: as quantified by the formation of water by 1H NMR spectroscopy with the generation of N2 verified by GC-TCD analysis of the head space.
| Entry | Catalyst (additive/variation) | [H2O]/M | TON |
|---|---|---|---|
|
a Conditions: 10 μmol of catalyst/Rh in 2.0 mL of THF placed under ∼1:2 H2/N2O (3 atm) within a 100 mL gas bulb with cold finger (126 mL water volume) and stirred at RT for 24 h. Conversion determined by 1H NMR analysis using a mesitylene internal standard and averaged over duplicate runs.
b No conversion observed in the absence of KOtBu.
c Generation of N2 verified by head space analysis (GC-TCD).
d Hydrogenation of the internal standard was observed. Similar activity is achieved in the absence of the internal standard.
|
|||
| 1 | None | 0.00 | — |
| 2 | [Ru(PNP-tBu)HCl(CO)] (+KOtBu)b | 0.03 | 5 |
| 3 | [Ir(PNP-tBu)H3] 2 | 0.01 | 1 |
| 4 | [Rh(PNP-tBu)H] 3c | 0.90 | 174 |
| 5 | [Rh(PNP-tBu)H] 3 (+Hg) | 0.01 | 2 |
| 6 | [Rh(PNP-tBu)H] 3 (THF→CyH) | <0.01 | <1 |
| 7 | [Rh(PNP-tBu)(OH)] 4c | 0.75 | 146 |
| 8 | [Rh(PNP*-tBu)N2] 5c | 0.75 | 144 |
| 9 | Rh/Cd | 0.24 | 47 |
| 10 | [Rh(COD)(OH)]26c | 1.72 | 318 |
| 11 | [Rh(COD)(OH)]26 (t = 1 h)c | 0.03 | 6 |
Encouraged by the high catalytic activity of 3, we sought to understand the underlying mechanism. To this end, the reaction between 3 (20 mM) and N2O (2 atm) was examined in situ by NMR spectroscopy in d8-THF, revealing quantitative spectroscopic conversion of 3 into a ∼1:1 mixture of the known rhodium(I) hydroxide complex 4 (δ31P 55.6, 1JRhP = 162 Hz) and dearomatized rhodium(I) dinitrogen complex 5 (δ31P 66.6, 1JRhP = 132 Hz; 63.1, 1JRhP = 132 Hz; 2JPP = 269 Hz) within 5 min at RT (Scheme 1).15 This outcome is consistent with activation of N2O by O-atom insertion into the Rh–H bond,16 followed by (partial) bifunctional elimination of water as proposed for 1 by Milstein.8 Although independently isolated 4 and 5 are catalytically competent for the hydrogenation of N2O under the aforementioned conditions (Table 1), they react incompatibly slowly with H2 at RT on a NMR reaction scale and, moreover, do not reform 3 cleanly (Scheme 1). Likewise, whilst 4 eliminated water to give 5 under an atmosphere of N2 and treatment of 5 with excess water gave 4, both reactions are sluggish at RT and partial decomposition was observed during the former. This decomposition is attributed to the instability of 5 and a significant amount of PNP-tBu oxide was produced when a 20 mM solution of 5 in d8-THF was placed under N2O (2 atm; 11% after 24 h at RT by 31P NMR spectroscopy). No reaction with 4 was observed under the same conditions.
 | ||
| Scheme 1 Reactions of isolated 1–3 in d8-THF at RT. | ||
These observations, coupled with the deposition of dark residues on the reactor walls and observation of PNP-tBu oxide by 31P NMR spectroscopy when using 3 in catalysis, led us to question the homogeneous nature of the hydrogenation. The formation of 2.9 ± 0.4 nm rhodium nanoparticles was subsequently confirmed by TEM/EDX analysis of the post-catalysis reaction mixture (Fig. 2A), and their role in catalysis corroborated by a positive mercury drop test, in which addition of mercury almost completely inhibited catalysis using 3 (Table 1, entry 5).17 The N2O hydrogenation observed for 3 is therefore not attributed to homogeneous catalysis as we hypothesized, but instead reconciled by the formation of catalytically-active rhodium nanoparticles from partial decomposition of 5 under the reaction conditions (generated from 3 + N2O or 4 − H2O, Scheme 1). Isolated 5 displays significantly enhanced stability in cyclohexane and, in further support of this conclusion, 3 is an ineffective catalyst for N2O hydrogenation when cyclohexane is used in place of THF as the reaction solvent (Table 1, entry 6).
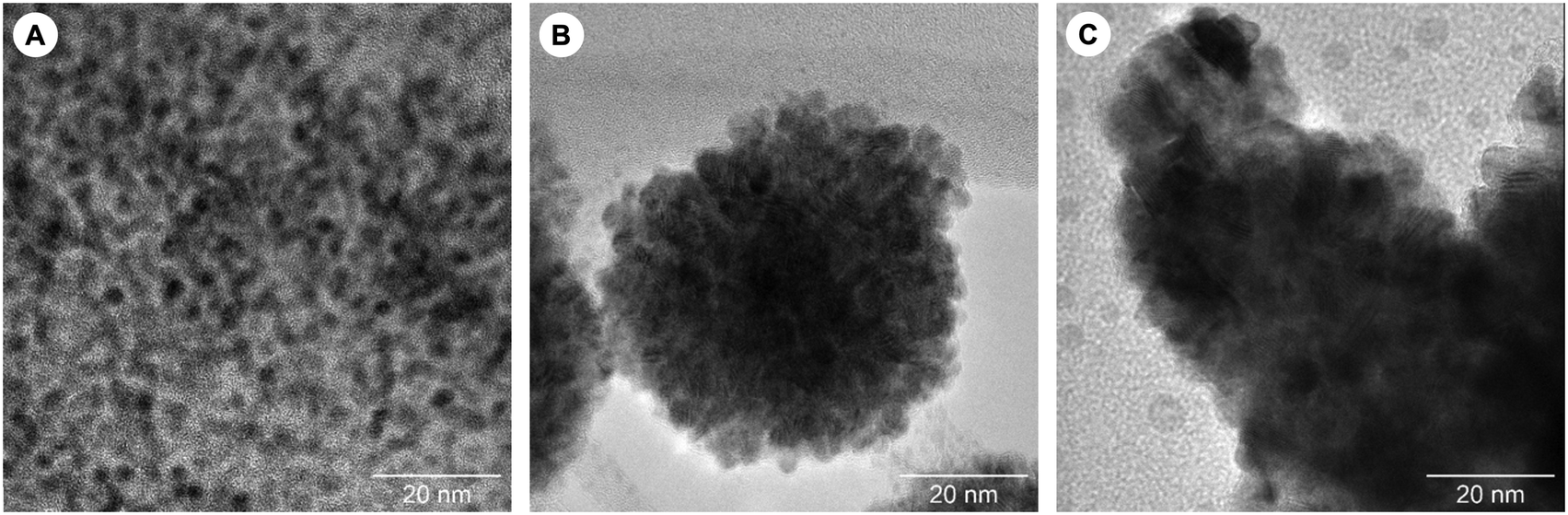 | ||
| Fig. 2 TEM images taken from post catalysis reaction mixtures when using (A) 3, (B) 6 (1 h run) and (C) 6 (24 h run). | ||
Having concluded that 3 operates via heterogeneous catalysis, we sought to identify a more convenient source of rhodium to apply in the hydrogenation of N2O (Table 1). Commercially available Rh/C was first assessed under our conditions but gave only 47 apparent TONs over 24 h. The use of bench stable [Rh(COD)(OH)]2 (6, COD = 1,5-cyclooctadiene) as a nanoparticle precursor was more promising,18 with a catalytic turnover nearly double that of 3 recorded after 24 h. Disproportionately low turnover after 1 h is symptomatic of an induction period for 6 and post catalysis analysis of the different runs by SAXS suggests that activity may correlate with a greater degree of nanoparticle aggregation. For instance, particles of mean radius 26.6 nm were observed after 1 h, while after 24 h the scattering data are best modelled as a mixture containing particles with a mean radius of 58.6 nm (see ESI†). These changes in aggregation are also apparent from TEM/EDX analysis of the samples (Fig. 2B/C).
To further explore the catalytic utility of 6, the hydrogenation reaction was tested on a larger scale using a 250 mL gas bulb, under otherwise unoptimised reaction conditions: 5 mM [Rh(COD)(OH)]2 in 2.0 mL of THF, ∼1:2 H2/N2O (3 atm). After three successive 24 h cycles, where average cumulative apparent TONs of 982, 2055 and 3261 were measured, a total of 16.3 M of water was produced.
In summary, we have discovered “hidden” heterogenous catalysis in the hydrogenation of N2O using a rhodium(I) hydride complex featuring a nominally robust phosphine-based pincer ligand. Although reaction with N2O by O-atom insertion into the Rh–H bond is facile, the ensuing dearomatized rhodium(I) derivative is unstable and partial decomposition into catalytically active rhodium nanoparticles and PNP-tBu oxide was observed during catalysis. Commercially available and bench stable [Rh(COD)(OH)]2 was identified as a more effective catalyst precursor, enabling the hydrogenation of N2O with an apparent turnover number >3000 at RT. We encourage the possible formation of small quantities of catalytically active nanoparticles to be carefully assessed when using molecular catalysts for this reaction.
Data availability
The data supporting this article have been included as part of the ESI.†19Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NERC grants NE/S007350/1 (CENTA2 studentship to SHD) and NE/X018377/1. We also acknowledge funding from the Leverhulme Trust (RPG-2022-214, TMH) and the University of Warwick. TEM and SAXS measurements were made using equipment provided by the University of Warwick Electron Microscopy and X-ray Diffraction Research Technology Platforms.Notes and references
- (a) Climate Change 2023: Synthesis Report, ed. H. Lee and J. Romero, Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2023, DOI:10.59327/ipcc/ar6-9789291691647; (b) A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- H. Tian, R. Xu, J. G. Canadell, R. L. Thompson, W. Winiwarter, P. Suntharalingam, E. A. Davidson, P. Ciais, R. B. Jackson, G. Janssens-Maenhout, M. J. Prather, P. Regnier, N. Pan, S. Pan, G. P. Peters, H. Shi, F. N. Tubiello, S. Zaehle, F. Zhou, A. Arneth, G. Battaglia, S. Berthet, L. Bopp, A. F. Bouwman, E. T. Buitenhuis, J. Chang, M. P. Chipperfield, S. R. S. Dangal, E. Dlugokencky, J. W. Elkins, B. D. Eyre, B. Fu, B. Hall, A. Ito, F. Joos, P. B. Krummel, A. Landolfi, G. G. Laruelle, R. Lauerwald, W. Li, S. Lienert, T. Maavara, M. MacLeod, D. B. Millet, S. Olin, P. K. Patra, R. G. Prinn, P. A. Raymond, D. J. Ruiz, G. R. van der Werf, N. Vuichard, J. Wang, R. F. Weiss, K. C. Wells, C. Wilson, J. Yang and Y. Yao, Nature, 2020, 586, 248–256 CrossRef CAS PubMed.
- P. Glarborg, J. E. Johnsson and K. Dam-Johansen, Combust. Flame, 1994, 99, 523–532 CrossRef CAS.
- X. Wu, J. Du, Y. Gao, H. Wang, C. Zhang, R. Zhang, H. He, G. M. Lu and Z. Wu, Chem. Soc. Rev., 2024, 53, 8379–8423 RSC.
- (a) L. Jacobs, C. Barroo, N. Gilis, S. V. Lambeets, E. Genty and T. V. de Bocarmé, Appl. Surf. Sci., 2018, 435, 914–919 CrossRef CAS; (b) J. Arenas-Alatorre, A. Gómez-Cortés, M. Avalos-Borja and G. Díaz, J. Phys. Chem. B, 2005, 109, 2371–2376 CrossRef CAS PubMed; (c) T. Nobukawa, M. Yoshida, K. Okumura, K. Tomishige and K. Kunimori, J. Catal., 2005, 229, 374–388 CrossRef CAS; (d) A. C. Gluhoi, M. A. P. Dekkers and B. E. Nieuwenhuys, J. Catal., 2003, 219, 197–205 CrossRef CAS; (e) G. Delahay, M. Mauvezin, A. Guzmán-Vargas and B. Coq, Catal. Commun., 2002, 3, 385–389 CrossRef CAS; (f) S. A. Carabineiro and B. E. Nieuwenhuys, Surf. Sci., 2001, 495, 1–7 CrossRef CAS; (g) A. Dandekar and M. A. Vannice, Appl. Catal., B, 1999, 22, 179–200 CrossRef CAS; (h) J. E. Vance and J. K. Dixon, J. Am. Chem. Soc., 1941, 63, 176–181 CrossRef CAS; (i) J. K. Dixon and J. E. Vance, J. Am. Chem. Soc., 1935, 57, 818–821 CrossRef CAS; (j) A. F. Benton and C. M. Thacker, J. Am. Chem. Soc., 1934, 56, 1300–1304 CrossRef CAS; (k) H. Cassel and E. Glückauf, Z. Phys. Chem., Abt. B, 1932, 19, 47–62 CrossRef; (l) C. N. Hinshelwood, Proc. R. Soc. London, Ser. A, 1924, 106, 292–298 CrossRef CAS.
- S. Roy, M. S. Hegde, S. Sharma, N. P. Lalla, A. Marimuthu and G. Madras, Appl. Catal., B, 2008, 84, 341–350 CrossRef CAS.
- (a) N. W. Cant, D. C. Chambers and I. O. Y. Liu, J. Catal., 2011, 278, 162–166 CrossRef CAS; (b) A. Miyamoto, S. Baba, M. Mori and Y. Murakami, J. Phys. Chem., 1981, 85, 3117–3122 CrossRef CAS.
- R. Zeng, M. Feller, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 5720–5723 CrossRef CAS PubMed.
- I. Ortega-Lepe, P. Sánchez, L. L. Santos, P. Lara, N. Rendón, J. López-Serrano, V. Salazar-Pereda, E. Álvarez, M. Paneque and A. Suárez, Inorg. Chem., 2022, 61, 18590–18600 CrossRef CAS PubMed.
- P. Jurt, A. S. Abels, J. J. Gamboa-Carballo, I. Fernández, G. L. Corre, M. Aebli, M. G. Baker, F. Eiler, F. Müller, M. Wörle, R. Verel, S. Gauthier, M. Trincado, T. L. Gianetti and H. Grützmacher, Angew. Chem., Int. Ed., 2021, 60, 25372–25380 CrossRef CAS PubMed.
- S. T. Nappen, J. J. Gamboa-Carballo, E. Tschanen, F. Ricatto, M. D. Wörle, A. Thomas, M. Trincado and H. Grützmacher, Angew. Chem., Int. Ed., 2025, 64, e202502616 CrossRef CAS PubMed.
- M. R. Gyton, B. Leforestier and A. B. Chaplin, Angew. Chem., Int. Ed., 2019, 58, 15295–15298 CrossRef CAS PubMed.
- L. Schwartsburd, M. A. Iron, L. Konstantinovski, E. Ben-Ari and D. Milstein, Organometallics, 2011, 30, 2721–2729 CrossRef CAS.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- (a) S. K. Hanson, D. M. Heinekey and K. I. Goldberg, Organometallics, 2008, 27, 1454–1463 CrossRef CAS; (b) S. M. Kloek, D. M. Heinekey and K. I. Goldberg, Angew. Chem., Int. Ed., 2007, 46, 4736–4738 CrossRef CAS PubMed.
- (a) T. L. Gianetti, S. P. Annen, G. Santiso-Quinones, M. Reiher, M. Driess and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 1854–1858 CrossRef CAS PubMed; (b) J.-H. Lee, M. Pink, J. Tomaszewski, H. Fan and K. G. Caulton, J. Am. Chem. Soc., 2007, 129, 8706–8707 CrossRef CAS PubMed; (c) A. W. Kaplan and R. G. Bergman, Organometallics, 1998, 17, 5072–5085 CrossRef CAS; (d) A. W. Kaplan and R. G. Bergman, Organometallics, 1997, 16, 1106–1108 CrossRef CAS.
- (a) R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed; (b) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- D.-H. Liu, P. M. Pflüger, A. Outlaw, L. Lückemeier, F. Zhang, C. Regan, H. R. Nodeh, T. Cernak, J. Ma and F. Glorius, J. Am. Chem. Soc., 2024, 146, 11866–11875 CrossRef CAS PubMed.
- (a) S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC; (b) B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem., Int. Ed., 2010, 49, 1468–1471 Search PubMed; (c) A. van der Ent, A. L. Onderdelinden and R. A. Schunn, Inorg. Synth., 1990, 28, 90–92 CrossRef CAS; (d) D. Hermann, M. Gandelman, H. Rozenberg, L. J. W. Shimon and D. Milstein, Organometallics, 2002, 21, 812–818 Search PubMed; (e) M. R. Gyton, T. M. Hood and A. B. Chaplin, Dalton Trans., 2019, 48, 2877–2880 Search PubMed; (f) E. M. Pelczar, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2008, 27, 5759–5767 Search PubMed; (g) R. Uson, L. A. Oro, J. A. Cabeza, H. E. Bryndza and M. P. Stepro, Inorg. Synth., 1985, 23, 126–130 CrossRef CAS; (h) P. S. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, 2012, pp. 251–254 Search PubMed; (i) XSACT: X-ray scattering, analysis, and calculation tool (Xenocs, version 2.7), 2023 Search PubMed; (j) J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS; (k) W. Li, J.-H. Xie, H. Lin and Q.-L. Zhou, Green Chem., 2012, 14, 2388–2390 Search PubMed; (l) L. R. Doyle, A. Heath, C. H. Low and A. E. Ashley, Adv. Synth. Catal., 2014, 356, 603–608 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, including analysis of catalytic reactions and characterisation of rhodium nanoparticles by TEM/EDX and SAXS. See DOI: https://doi.org/10.1039/d5cy00490j |
| This journal is © The Royal Society of Chemistry 2025 |