
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanism of nitrogenase: formation and release of the second NH3 and completion of the cycle†
Ian
Dance

School of Chemistry, UNSW Sydney, NSW 2052, Australia. E-mail: i.dance@unsw.edu.au
First published on 30th May 2025
Abstract
The enzyme nitrogenase catalyses the reaction N2 + 8e− + 8H+ → 2NH3 + H2. Two prior papers in this series report a computed mechanism for the first and second phases of this catalysis. In the first phase H2 is formed, the H2/N2 exchange occurs, and N2 is captured in a concerted step forming the bound HNHH intermediate. The second phase breaks the N–N bond and converts this intermediate to bound NH plus the first NH3, which dissociates. This third paper describes the final phase, which forms and then releases the second NH3, and recovers the resting state. The mechanism is supported by density functional calculations with a 483+ atom quantum model of the active site, FeMo-co, and relevant surrounding amino acids and water. Calculated reaction trajectories and potential energy profiles generate five mechanistic pathways through this NH3 formation phase of the reaction. These pathways are evaluated with incorporation of entropic components and possible kinetic contributions by H atom tunneling, leading to the identification of the most favourable pathway for generation of NH3 and its subsequent dissociation. The steps regenerating the resting state and completing the mechanism cycle are described. All steps in this third and final phase of the mechanism are thermodynamically and kinetically feasible. Atom S2B of FeMo-co, whose retention during enzyme turnover is experimentally controversial, remains intact as a bridge between Fe2 and Fe6 and is an essential H transfer agent in the proposed mechanism. The architecture of the active site and its surrounds that promote the chemical choreography of nitrogenase in its performance space are outlined, and key features and principles of the proposed complete mechanism are summarised.
1. Introduction
Nitrogenase, the enzyme that has evolved as the natural source of nitrogen for the earth's biosphere,1 catalyses the conversion of inert N2 to NH3 under ambient conditions which contrast markedly with the extreme pressure and temperature conditions required for the Haber–Bosch industrial manufacture of ammonia.2 How does the enzyme achieve a mild catalysis that chemists are unable to replicate?3,4 Extensive experimental investigation involving reaction kinetics,5,6 mutations of amino acids surrounding the active site,7–22 vibrational and spin resonance spectroscopy,23–31 spectro-electrochemistry,32 use of abiological electron sources,33,34 many crystal structures,35,36 cryo-electron-microscopy,4,37–39 and density functional simulations,40–51 still leave many aspects of the chemical mechanism unresolved. There is a fundamental experimental impediment to the isolation and direct investigation of intermediates because unavoidable protons are a substrate of the enzyme and mixtures of transient intermediates occur.36,52 Furthermore, the overall chemical reaction eqn (1) implies a mechanistic cycle with at least 27 steps: eight introductions of a proton, eight additions of an electron, N2 binding, breaking the N–N bond, formation of six N–H bonds, two dissociations of NH3 and formation of one H–H bond.| N2 + 8e− + 8H+ → 2NH3 + H2 | (1) |
The protein domain in which this catalysis occurs is shown in Fig. 1. The active site is the iron-molybdenum cofactor, FeMo-co, a CFe7MoS9 cluster with homocitrate (HCA) and His442 coordination to Mo, and Cys275 coordination at Fe1: throughout this paper amino acids are numbered according to crystal structure PDB 3U7Q of Mo-nitrogenase species Azotobacter vinelandii (Av). Mutagen-reactivity experiments show that the active domain of FeMo-co is the front face, Fe2–S2B–Fe6–S3B–Fe7 enclosed by Val70 and Arg96.8,10–12,17,19,53–55,56,57 There is a significant hydrogen bond between Nε of His195 and S2B. Protons are supplied along a conserved water chain, and the four penultimate water molecules of the proton supply chain are shown on Fig. 1, with hydrogen bonds to O3, O6 and O5 of homocitrate, and the final proton directed towards S3B. Protons are translocated along the complete chain (not shown) by a Grotthuss mechanism.58 Electrons reach FeMo-co from a separate P-cluster.59–61
 | ||
| Fig. 1 Structure of the active site of Mo-nitrogenase, with FeMo-co at the centre. Significant surrounding amino acids and water molecules are included, with labels for Av1 protein, crystal PDB 3U7Q. Homocitrate C atoms are dark green, and hydrogen bonds are striped. The reaction zone is the Fe2–Fe3–Fe6–Fe7 face, under the side chain of Val70 and bounded by the side chain of Arg96. The proposed N2 entry pocket is between His195 and Ser278, approaching Fe2. The four penultimate water molecules of the proton supply chain58 are shown, with hydrogen bonds to O3, O6 and O5 of homocitrate, and with the final proton directed towards S3B. | ||
I have developed a complete chemical mechanism by which Mo-nitrogenase effects catalysis of the reaction in eqn (1). The previously reported components of this mechanism are outlined in Scheme 1. The labelling system for intermediates includes the incorporated moieties N2x, H, H2, HNNH, HNNH2, NH2, NH3, and the locations where they are bound, being S2B, Fe2, Fe6, S3B. Suffixes br = bridging, x = exo coordination position, n = endo coordination position are included in the labels: Fig. 2 defines coordination positions, and the labelling of the six configuration positions for H bonded to S3B. At each intermediate in Scheme 1 the numbers of electrons and protons added beyond the resting state are shown in the red status boxes, as En mH+.
 | ||
| Scheme 1 Stages of the proposed mechanism preceding the stages described in the current paper. The green enclosure describes the introduction and migration of H atoms to the more stable locations on FeMo-co around the reaction domain. The yellow enclosure includes the formation of H2, the H2/N2 exchange step, and then the concerted capture of N2 to form the intermediate with bound HNNH, Fe2-brNH-NH-Fe6. Then the lavender enclosure shows the pathways during which another H is added, the N–N bond breaks, NH3 is formed, and then dissociates. The boxes show the number of added electrons and protons at each intermediate. | ||
 | ||
| Fig. 2 (A) Locations of the exo and endo coordination positions at Fe2 and Fe6. Ligand bridging Fe and Fe6 supplants their endo coordination positions. (B) Labels used for the configurations of H bonded to S3B.62 | ||
The green enclosure in Scheme 1 shows the resting state and the sequence of preparatory additions of protons and electrons, generating H atoms first at S2B, then at the exo and endo coordination positions of Fe6, and then to the endo position of Fe7, forming intermediate S2BH-Fe6HxHn-3b6. Calculations indicate that proton addition to S3B from the proton supply chain is triggered by electron addition to FeMo-co.63 The reaction energies and barriers for configurational interchange at S3BH (Fig. 2) and for migration of each H atom from S3B to other locations on FeMo-co are relatively small and have been reported.42,62 The N2 that binds at the exo-coordination position of Fe2 (labelled N2x) during this sequence is not reducible because it is not surrounded by H atom donors. The thermodynamics of Fe2–N2x binding from the N2 entry pocket (Fig. 1) are calculated to be variable, and the occurrence of N2x in intermediates during the course of the mechanism has been described and rationalised.64
The yellow enclosure in Scheme 1 describes crucial steps.65 First H2 is formed at the endo position of Fe6, then a fifth H is added at endo-Fe7 (intermediate pre-capture). At this point the experimentally characterised H2/N2 exchange reaction can occur, with diffusion of reducible N2 from the N2 entry pocket, past non-reducible N2x,66 and into the reaction space to reach intermediate N2/H2exchange. Dissociation of H2 is followed by capture of N2 in concert with double hydrogenation of N2, to form Fe2-brNH-NH-Fe6 containing HNNH bound and bridging between Fe2 and Fe6. This N2 capture step, described in detail,65 is proposed to be the way in which the enzyme bypasses the inherently difficult first hydrogenation of inert N2. This concerted double hydrogenation and binding of N2 at two Fe atoms differs radically from the conventional assumption of sequential Fe–N2 binding and hydrogenation.
The lavender enclosure in Scheme 1 shows pathways through which the capture intermediate, Fe2-brNH-NH-Fe6, proceeds to form NH3 in concert with severance of the N–N bond. The product of this sequence, Fe2-brNH-Fe6-S2BH, has released this first NH3. Previously reported calculations67 indicate that the most likely pathway in the lavender phase is through Fe2-brNH-NH2-Fe6Hx-S2BH and Fe2-brNH-Fe6NH3-S2BH. Intermediates shown in the green and yellow enclosures of Scheme 1 have a status where the number of added protons is one larger than the number of added electrons. Although it has been generally assumed that proton addition is coupled with electron addition, there is no experimental evidence for equal numbers of added protons and electrons at each stage.4 There are two parts to the rationale for the additional proton status in this sequence of intermediates. First the mechanism is compliant with the accumulated experimental evidence5,6,68,69 that the N2/H2 exchange equilibrium occurs after addition of four electrons. Secondly, in addition to the proton on HisNε, five H are required at the pre-capture intermediate: two leave as H2, two form HNNH, and exo-Fe6H is required for the N–N breaking step. At completion of the sequence in the lavender enclosure (Fe2-brNH-Fe6-S2BH) H is required on S2B as a donor for the next steps to be described below, and it cannot be obtained from S3BH because the bound HNNH entity blocks H migration. It is sourced from the proton reservoir His195NεH+ which originated in the resting state. At completion of the lavender phase of the mechanism intermediate Fe2-brNH-Fe6-S2BH has status E6 6H+.
In this paper I report computational results for addition of the seventh and eighth electrons and protons, with the steps that form the second NH3, then its dissociation, and finally recovery of the resting state and completion of the mechanism cycle.
2. Methodology
2.1 Protein model
The computed protein model is a 483+ atom extract from crystal PDB 3U7Q, including all relevant amino acids. This is my standard model for simulations of nitrogenase reactions and reactivity.43,45,64–67,70 This model includes nine of the ten active-site residues that are conserved across all analyzed extant nitrogenases:71 the exception, Gly424, is outside the reaction zone. Details, and the rationale for inclusion of amino acids and truncation of uninvolved side chains, are provided in the ESI.† Investigations of the protonation state of homocitrate using crystal structures at various pH (summarised in ref. 72), together with vibrational circular dichroism spectroscopy,28 QM/MM calculations,72,73 and quantum refinement,73 indicate that the coordinated alcoholate O7 atom is protonated and hydrogen bonded to O1, and this is included in the computed model. Some constraints on the protein structure are required during optimization calculations because the modelled protein is incomplete and the influences of the complete protein outside the computational model are absent. The strategy for constraints and the 28 distance constraints are described in the ESI.†As previously explained45 trial calculations on the modes of diffusion and coordination of N2/H2 led to the conclusion that the front chain near Val70 should move slightly away from FeMo-co. This is consistent with experimental data on mutants of amino acid 70 with smaller and larger side chains.8,54,57 In the present investigation of NH3 formed in the reaction zone, and diffusing away between the side chains of Val70 and Arg96, it became apparent that a small additional separation of these sidechains should occur, by a libratory movement of the sidechain of Arg96, without changing the resting state hydrogen bond from NH2 of Arg96 to S5A. This is shown in Fig. S1 of the ESI.† The Cα(Val70)⋯Cc and Cα(Val70)⋯CZ(Arg96) distances were fixed, at 8.2 and 5.18 Å respectively.
The charge on the [CFe7MoS9] core of FeMo-co is −1, in agreement with experimental and computational studies.43,72,74,75
2.2 Density functional procedures
My density functional (DF) calculations use the DMol methodology of Delley,76–81 with accurate DNP (double numerical plus polarisation) basis sets.79 The gradient-corrected functional PBE82 is used because validation tests demonstrate that when used with the numerical basis sets of DMol it is more accurate than other commonly used functionals.83 See the ESI† for validation information. The conductor-like screening model (COSMO)84–86 is used with a dielectric constant of 5. The dispersion components of the non-bonding intermolecular interactions are treated effectively with the numerical basis sets used here: see the ESI† for additional information. Constraints on interatomic distances use the Lagrange Multiplier Algorithm. Control of electronic states is via input specifications of spin populations for Fe atoms (usually Fe1, Fe3, Fe4, Fe5 and Fe7) subsequently optimised during the scf procedure.2.3 Electronic states
The electronic structure of the FeMo-co cluster is complex, with many electronic states, which are usually described with the signs and magnitudes of the spin densities on the seven Fe atoms. Investigation of postulated reaction intermediates and reaction trajectories in the enzyme mechanism requires understanding and control of these electronic states. I have expounded a general principle, that maximisation of the weak bonding influences of opposite spin signs on each axial Fe pair (i.e. Fe2–Fe6, Fe3–Fe7, Fe4–Fe5) is a significant stabilising influence, proportional to the magnitudes of the spin densities.87 Ligation of the Fe2 and/or Fe6 atoms, as in the intermediates described here, usually diminishes the magnitude of spin density on the ligated Fe atom, sometimes almost to zero, and so the stabilisation due to opposite spins on Fe2 and Fe6 is similarly diminished. Therefore, opposite spin signs and larger spin densities on the Fe3–Fe7 axial pair or the Fe4–Fe5 axial pair maximise stability, and this prediction is consistent with the findings and procedures of other authors.40,41,46–49,72,88–90 Therefore the two most favourable electronic states have the spin sign combinations −Fe3 +Fe4 −Fe5 +Fe7, or +Fe3 −Fe4 +Fe5 −Fe7. Because electronic states are labelled with the numbers of the Fe atoms with negative spin density, these states are labelled ‘35’ and ‘47’ respectively.2.4 Mapping potential energy surfaces
This investigation comprehensively mapped the potential energy topology in the reaction space, to locate energy minima and then to locate the energy saddle points between them. Explorations deployed the relevant stereochemical principles, as elaborated in the Results. The shape of the potential energy surface was monitored throughout, and the magnitude of the displacement in each energy minimisation step was adjusted to maintain regular small energy changes.Transition states (TS) and intrinsic reaction coordinates were determined by the procedure described previously43,58,91,92 and explained in detail in the ESI.† Variations of Fe spin densities occur during reactions, and continuity of the electronic state throughout each reaction trajectory was checked.
3. Results
The results are organised as follows. First I outline the scope of the results, defining and classifying the reaction intermediates and their labels, and the reaction steps. In section 3.2 I present and discuss some normal and abnormal electronic states. Then, because the minimal skeletal diagrams used in the schemes are minimal and uninformative about geometry, section 3.3 contains pictures of representative intermediates. A key component of this investigation is reaction trajectory analysis, and aspects of this are elaborated in section 3.4. After these preliminaries the calculated reaction potential energies and potential energy barriers for all possible steps are presented in section 3.5. From these it is possible to construct sequences of reaction steps, presented as reaction pathways in section 3.6. Because some of the H atom transfer steps could occur with H atom quantum tunneling, section 3.7 describes additional details of reaction trajectories in attempt to assess the occurrence of tunneling in this mechanism.3.1 Scope
The scope of the results reported here is shown in Scheme 2, which displays all intermediates and transformations investigated, and defines the labels for intermediates. Labels now explicitly include the non-reducible N2x when present, because dissociations/associations of this ligand are part of the mechanism explored. The first step, continuing from the terminus of Scheme 1, is H transfer from S2B to generate the Fe2–NH2–Fe6 bridge (grey enclosure 1). Then the seventh H atom is introduced as 3b5, followed by reconfiguration around S3B to 3b3 (salmon enclosure 2) and then to 3b2 at the top of the blue enclosure 3. There are three pathways from this pivotal intermediate N2x-Fe2-brNH2-Fe6-3b2: (i) 3b2 to 6Hx in the blue enclosure 3, (ii) dissociation of 2N2x yielding the corresponding 3b2 and 6Hx in the green enclosure 4, or (iii) formation of NH3 at the endo-Fe2 position (N2x-Fe2NH3) in the buff enclosure 5. The buff enclosure contains the intermediates with bound NH3, and the intermediates after dissociation of NH3 are in the turquoise enclosure 6. Completion of the mechanism is shown in the pink 7, lilac 8 and yellow 9 enclosures. Scheme 2 also shows the numbers of electrons and protons added since the beginning of the mechanism cycle. In the top grey enclosure 1 the status is 6 electrons and 6 protons. After addition of an H atom (salmon enclosure 2) all intermediates through to the turquoise enclosure 6 are E7 7H+, with even spin S. Finally an electron and proton are added to recover the His195NεH+ resting state. | ||
| Scheme 2 Intermediates and reaction steps investigated in this report, and the labels used. | ||
3.2 Electronic states and Fe spin densities
Understanding and control of the electronic states of the intermediates and of structures during the reaction trajectories is essential. Section 2.3 describes the rationale for selection of energetically favourable electronic states, and explanation of the two states labelled ‘35’ and ‘47’. Control of the electronic state is by input specification of Fe spin densities which are optimised during the scf calculation. In this investigation the ‘35’ and ‘47’ electronic states were tested, with specified orbital occupancies yielding several total spins S. In general states with smaller values of S are calculated to be more stable. Electronic states for reported structures with spin S are labelled as S, 35 or S, 47. Electronic states were monitored throughout by examination of the set of seven Fe calculated spin densities.Electronic states and Fe spin densities are listed (with atomic coordinates) for all intermediates and transition states in the ESI.† The variable coordination chemistry at Fe2 and Fe6 throughout the reaction sequences results in considerable variation in the spin densities at these and other Fe atoms, illustrated with the selected results in Table 1. The first section, N2x-Fe2NH3, contains typical results for the S = 1, 356 and S = 1, 476 states (here Fe atoms with spin densities more negative than −1 are added to the state label). Note that the Fe6 spin density is not small, because Fe6 is not ligated, that the signs only on Fe3, Fe4, Fe5 and Fe7 are reversed, and that the two states are almost equi-energetic. The second section, N2x-Fe2-brNH2-Fe6H, shows how a reduction in total spin S from 1 to 0 is associated with a decrease in the spin density at Fe1, and decrease also in this example at Fe3, without significant energy change. Four electronic states for Fe2NH3 are shown in Table 1. Two (356, 235) are nominally S = 1 ‘35’ and equi-energetic, but have spin densities differing appreciably at Fe1, Fe2 and Fe6. This example of electronic state isomerism is occasionally seen in other related systems. The slightly less stable S = 0, 47 and S = 1, 47 states of Fe2NH3 also have different spin densities at Fe1, Fe2 and Fe6. Note that in the S = 1, 47 state the spin density at Fe6 is negligible even though this atom is not ligated: this is abnormal in the general pattern of spin densities for the electronic states of FeMo-co. In the ‘47’ states of Fe6NH3 the difference between S = 0 and S = 1 is primarily a flip in the spin sign at Fe6. The results for N2x-Fe2-brNH2-Fe6H and Fe2NH3 in Table 1 are unusual, but they demonstrate the importance of monitoring electronic states via Fe spin densities during calculations of reaction trajectories.
| N2x-Fe2NH3 | ||
|---|---|---|
| S = 1, 356 | S = 1, 476 | |
| Fe1 | 3.16 | 3.15 |
| Fe2 | 0.01 | −0.04 |
| Fe3 | −2.70 | 2.68 |
| Fe4 | 2.70 | −2.76 |
| Fe5 | −2.25 | 2.36 |
| Fe6 | −1.70 | −1.66 |
| Fe7 | 2.29 | −2.12 |
| Relative energy | +2 | 0 |
| N2x-Fe2-brNH2-Fe6H | ||
|---|---|---|
| S = 1, 47 | S = 0, 47 | |
| Fe1 | 3.03 | 1.50 |
| Fe2 | −0.49 | −0.15 |
| Fe3 | 2.70 | 1.97 |
| Fe4 | −2.53 | −2.49 |
| Fe5 | 2.20 | 2.16 |
| Fe6 | −0.85 | −0.78 |
| Fe7 | −2.30 | −2.21 |
| Relative energy | 0 | +2 |
| Fe2NH3 | ||||
|---|---|---|---|---|
| 356 S = 1 | 235 S = 1 | S = 0, 47 | S = 1, 47 | |
| Fe1 | 3.21 | 1.80 | 2.89 | 1.78 |
| Fe2 | 0.21 | −1.16 | −1.11 | −0.01 |
| Fe3 | −2.52 | −2.48 | 2.70 | 2.46 |
| Fe4 | 2.60 | 2.57 | −2.57 | −2.41 |
| Fe5 | −2.47 | −2.37 | 2.35 | 2.25 |
| Fe6 | −2.06 | 1.59 | −2.29 | 0.11 |
| Fe7 | 2.45 | 1.95 | −2.30 | −2.15 |
| Relative energy | +1 | 0 | +7 | +4 |
| Fe6NH3 | ||
|---|---|---|
| S = 0, 47 | S = 1, 47 | |
| Fe1 | 2.87 | 2.93 |
| Fe2 | −2.62 | −2.33 |
| Fe3 | 2.74 | 2.78 |
| Fe4 | −2.50 | −2.39 |
| Fe5 | 2.22 | 2.36 |
| Fe6 | −0.88 | 0.85 |
| Fe7 | −2.36 | −2.28 |
| Relative energy | 0 | +2 |
3.3 Pictures
Fig. 3 illustrates geometrical properties of the reaction site in representative intermediates, with significant dimensions marked (all for the electronic state S = 1, 35). N2x-Fe2-brNH2-Fe6-3b5 and N2x-Fe2-brNH2-Fe6-3b3 possess standard characteristics, extension of S3B–Fe6 and S3B–Fe7 respectively, with extension of Fe2–Cc due to exo-Fe2–N2x. When the H atom on S3B is conformed as 3b2, there are isomers with long and short S3B–Fe6 distances. The short 3b2 isomer (Fig. 3C) is effectively an S3B–H–Fe6 bridge. When 2N2x is dissociated (Fig. 3E) the Fe2–Cc distance decreases from 2.40 to 1.95 Å, and the S3B–H–Fe6 bridge elongates slightly. The Fe2–N and Fe6–N bonds of the NH2 bridge are effectively invariant, 1.98 ± 0.03 Å. Fig. 3F and G illustrate how N2x influences coordination geometry at Fe2, and indirectly at Fe6.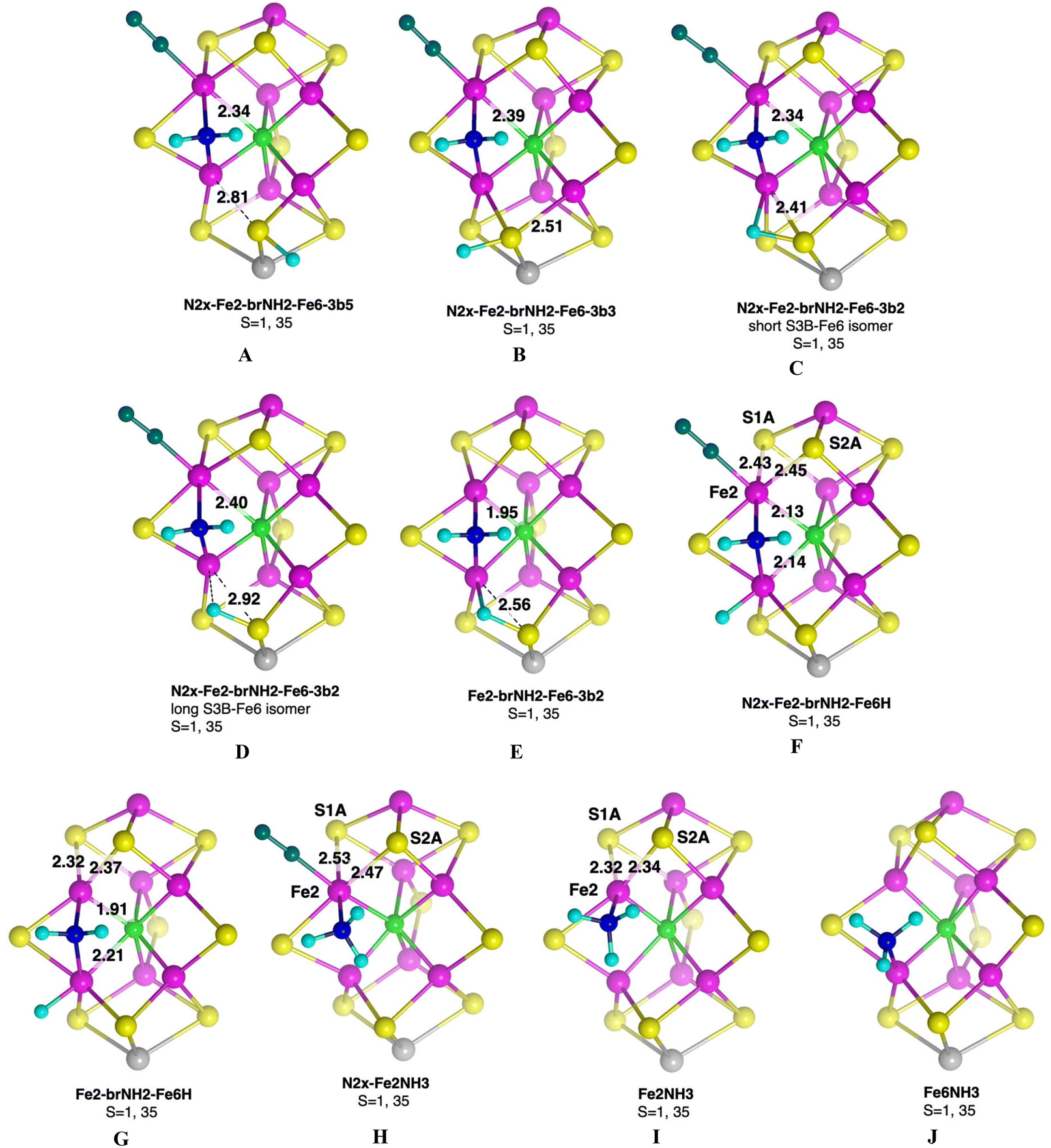 | ||
| Fig. 3 The core structures of representative intermediates, with significant dimensions (Å) marked. Exo-Fe2–N2x is dark turquoise. | ||
There are two postulated intermediates with NH3 bound at the endo position of Fe2. In N2x-Fe2NH3 where Fe2 is near octahedrally coordinated there is a distinctive elongation of Fe2–S1A trans to the Fe2–NH3, and the Fe2–S2A bond is also lengthened. When N2x is absent, Fe2NH3, Fe2 possesses more regular square pyramidal coordination (Fig. 3I), and similar square pyramidal coordination at Fe6 occurs in Fe6NH3.
3.4 Reaction trajectory analyses
Potential energy topologies between the energy minima presented in Scheme 2 were explored to find the lowest energy pathways between them. The pragmatic procedure for calculation of transition states is described in detail in the ESI.† Some of the transformations involve multiple geometrical variables, which need to be recognised and understood during determination of the transition state and reaction trajectory. The following outlines of reaction trajectories refer to the atom labels in Fig. 4. | ||
| Fig. 4 Atom labels used in descriptions of reaction trajectories. | ||
The formation of NH3 is by transfer of H, from either 3b2 or 6Hx to Nbr of the bridging NH2, involving multiple changes: extension then breaking of the S3B–H bond or of the Fe–H bond, formation of the H–Nbr bond, and extension then breaking of one of the two Nbr–Fe bonds. In the 3b2 to Fe2NH3 reaction step (N2x-Fe2-brNH2-Fe6-3b2 → N2x-Fe2NH3 and Fe2-brNH2-Fe6-3b2 → Fe2NH3), at the transition state the H⋯Nbr and S3B⋯H distances are both ca. 1.55 Å, S3B is closing towards Fe6, the transferring H is 1.9–2.0 Å from Fe6, and the originally equal Fe–Nbr distances are only slightly differentiated by 0.1–0.2 Å, towards Fe2–Nbr. All of these dimensions are monitored and adjusted during the trajectory analysis. The influence of N2x is small. The formation of NH3 by transfer of H at 3b2 (i.e. from N2x-Fe2-brNH2-Fe6-3b2 or from Fe2-brNH2-Fe6-3b2) generated only Fe2–NH3. The possibility that H transfer from 3b2 to the NH2 bridge would first form NH3 bonded to Fe6 was thoroughly tested, and no pathway from 3b2 to generate NH3 at Fe6 could be found.
In the alternative formation of NH3 by transfer of 6Hx (instead of 3b2) to Nbr the stereochemistry at Nbr is an additional factor. In the reactant the angles 6Hx–Nbr–Ha and 6Hx–Nbr–Hb are unequal (ca. 80, 126°) and their equalisation during the reaction step needs to be incorporated in the trajectory analysis. The formation of Fe–NH3 by transfer of 6Hx to bridging NH2 is also affected by electronic state. When the electronic state is 35 and S = 0 or 1, the product of H transfer is NH3 bound at the endo position of Fe2, but in electronic state 47, S = 0 or 1, the product NH3 is bound instead at endo Fe6.
NH3 formed at the endo position of Fe2 or Fe6 dissociates from them along a pathway that leads first between the sidechains of Val70 and Arg96, before reaching the hydrophilic mouth of the NH3 egress pathway.93 The space between Val70 and Arg96 is relatively tight, and I have postulated a minor expansion achieved by a libratory movement of the sidechain of Arg96. This is shown in Fig. 5. Small reconformations in the CH2CH2CH2 arm of Arg96 effect this movement, which involves no change in the hydrogen bond from the terminal NH2 of Arg96 to S5A. The 70Cα–96CZ separation, 3.66 Å in the resting crystal PDB-3U7Q, was constrained to 5.18 Å in the modelling.
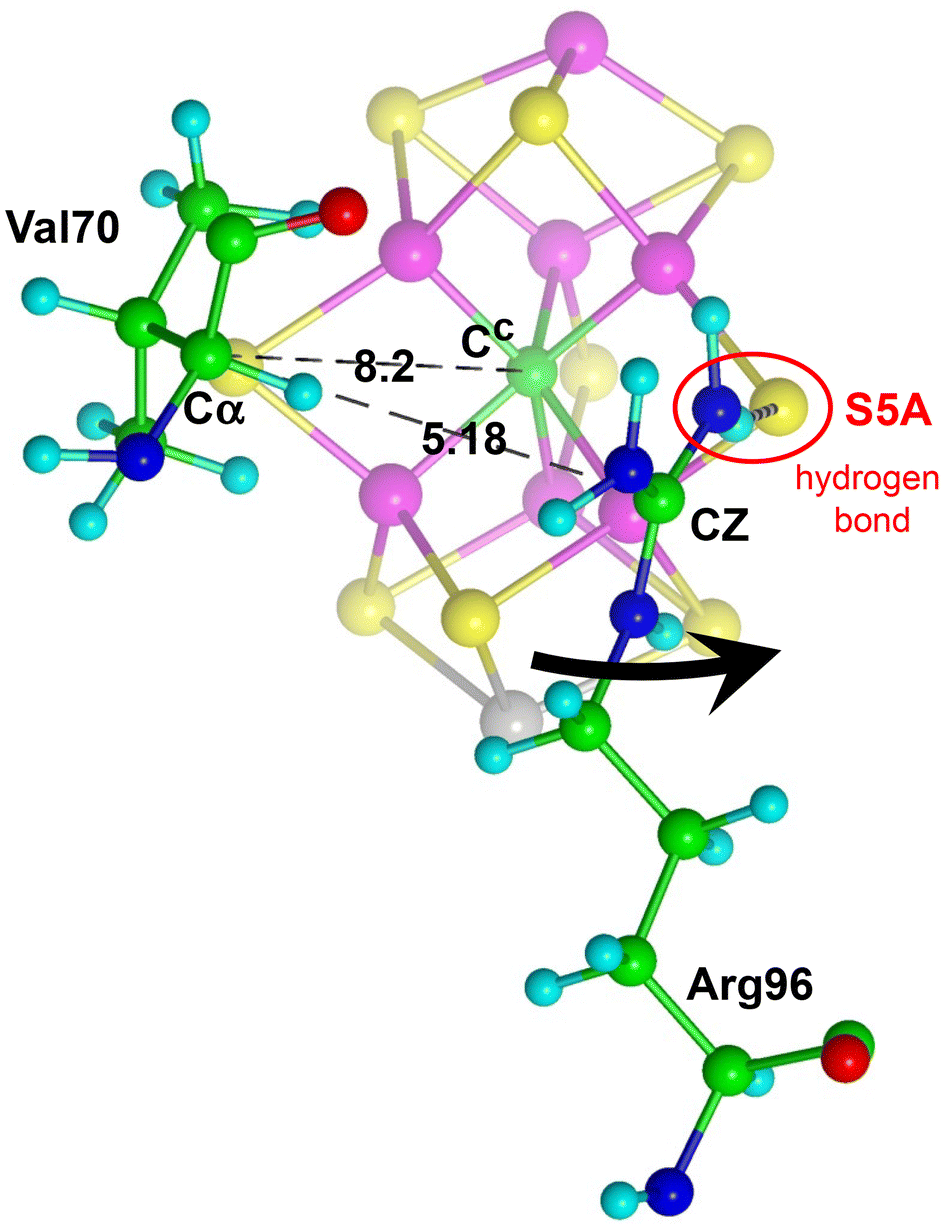 | ||
| Fig. 5 Expansion of the space between the sidechains of Val70 and Arg96 by a libratory movement of the sidechain of Arg96, black arrow. The hydrogen bond from terminal NH2 of Arg96 to S5A is unaffected. | ||
Dissociating NH3 can wiggle through this space. Fig. 6 shows examples, one (A) at the transition state (Fe2⋯N 2.79 Å), one (B) with NH3 dissociated from Fe2, and two (C and D) with NH3 dissociated from Fe6. These four are representative of the variety of orientations possible for dissociating NH3, which are quantified by the three Fe⋯N–H angles recorded on Fig. 6. The dotted lines identify the closest contacts, which in almost all instances are longer than the destabilisation regime of the N⋯H dispersion energy potential.94 Dissociating NH3 can also orient to form a bent hydrogen bond with S3B (Fig. 6A) but this interaction does not appear to be advantageous.
 | ||
| Fig. 6 Representations of typical positions and orientations of NH3 on its dissociation pathway. Orientations are quantified with the three Fe⋯N–H angles marked. Dotted lines locate the closest contacts with the sidechains of Val70 and Arg96. | ||
3.5 Reaction energies
For each of the transformations in Scheme 2 the potential energy profile was calculated, by first determining the transition state (TS), and then following (by small step energy minimisations) the trajectories from TS to reactant and TS to product. The comprehensive results are presented in Scheme 3, with symbols comprising the reaction energy (red), the reaction barrier (black), and the electronic state. The uncertainty level is 1 kcal mol−1. Results where larger calculational uncertainty arose, mainly due to anomalous electronic structure, are not included. Panel A shows the preparatory steps from N2x-Fe2-brNH-Fe6-S2BH (the final intermediate in the previous phase, Scheme 1) to N2x-Fe2-brNH2-Fe6-3b2 (the grey and salmon enclosures of Scheme 2). Characteristically the transfer of H from S2B to N forming N2x-Fe2-brNH2-Fe6 is strongly exergonic. Then a new H introduced at 3b5 surmounts a barrier of ca. 10 kcal mol−1 to reach N2x-Fe2-brNH2-Fe6-3b2. Panel B describes the three possible reactions of N2x-Fe2-brNH2-Fe6-3b2. The energy profiles for these three steps are quite different: dissociation of N2x is endergonic, as usual;64 formation of NH3 at Fe2 is very strongly exergonic, with moderate barriers; conversion to Fe2-brNH2-Fe6H is exergonic with very small barriers. Panel C shows the two reactions of N2x-Fe2-brNH2-Fe6H: slightly endergonic dissociation of N2x with small barriers, and exergonic formation of NH3 at Fe2 with large barriers. Panel D contains the steps in which NH3 is formed in the absence of N2x, i.e. the green 4 to buff 5 transformations in Scheme 2. All pathways are strongly exergonic, and the barriers are variable, being unusually small for the Fe2-brNH2-Fe6-3b2 → Fe2NH3 step. No pathway could be found for the formation of NH3 at Fe6 from Fe2-brNH2-Fe6-3b2. Panel E describes the energy profiles for dissociation of NH3 from Fe2 and Fe6. In the absence of N2x this dissociation is exergonic with relatively small barriers, but with N2x present dissociation of NH3 to form N2x-core is appreciably endergonic. Panel F shows recovery of the resting state, after addition of the final electron + proton pair. | ||
| Scheme 3 Calculated reaction potential energies (red) and reaction barriers (black), in kcal mol−1. Electronic states are marked, as S,35 or S,47. Panel A describes the preparatory steps forming N2x-Fe2-brNH2-Fe6-3b2 from N2x-Fe2-brNH2-Fe6-S2BH. Panel B describes the three reactions of N2x-Fe2-brNH2-Fe6-3b2 and panel C describes the two reactions of N2x-Fe2-brNH2-Fe6-6Hx. Panel D contains the steps that form NH3 after prior dissociation of N2x. Panel E describes the NH3 dissociation steps from intermediates with and without N2x, and also the two N2x dissociation steps with and without bound NH3. Panel F shows recovery of the resting state. | ||
Four different NH3 formation steps have been calculated. All are strongly exergonic. The barriers are 13 or 19 kcal mol−1 for H transfer from 3b2 when N2x is present, 22 or 23 kcal mol−1 from 6Hx when N2x is present, 5 or 7 kcal mol−1 for H transfer from 3b2 when N2x is absent, and 21, 22, or 25 kcal mol−1 for transfer from 6Hx when N2x is absent. The smaller barriers when H is transferred from 3b2 reflect the positional variability of 3b2 (see Fig. 3) and the slight stereochemical awkwardness of the 6Hx to Nbr transfer (section 3.4).
Non-reducible N2x occurs in three structure types, all with a ligand at the endo position of Fe2, and all with a positive potential energy for dissociation of N2x. This is consistent with the generalisations previously described.64 In the intermediate with NH2 bridge and 6Hx (N2x-Fe2-brNH2-Fe6H) the N2x dissociation barrier (7 to 9 kcal mol−1) and endergonicity (2 to 6 kcal mol−1) are smaller than the barrier (range 12 to 16 kcal mol−1) and endergonicity (range 8 to 12 kcal mol−1) for N2x dissociation from N2x-Fe2-brNH2-Fe6-3b2 and N2x-Fe2NH3.
Further discussion of these results is deferred until they are incorporated into possible reaction sequences, in the next section.
3.6 Reaction pathways
I have constructed the possible reaction pathways from N2x-Fe2-brNH2-Fe6-3b2 to core, and the potential energy profiles for these are charted in Chart 1. Note that the variations in energy due to different electronic and spin states are relatively small, and there is consistency in each sequence of ridges and valleys. Paths P1, P2 and P3 continue from the three different reactions of the precursor N2x-Fe2-brNH2-Fe6-3b2 (see Panel B of Scheme 3). Paths P1 and P2 involve formation of NH3 from 3b2, preceding or following dissociation of N2x. Paths P3 and P4 shift 3b2 to 6Hx which is then used to generate NH3, with dissociation of N2x after (P3) or before (P4) the formation of NH3. Path P5 is the variant of P4 in which NH3 is formed at Fe6 rather than Fe2. This variation is dependent on the electronic state (illustrated by the black/red difference in P4/P5 at the NH3 formation step). | ||
| Chart 1 Potential energy profiles for five reaction pathways from N2x-Fe2-brNH2-Fe6-3b2 to core. Circles signify S = 0, squares S = 1, black = 35, red = 47. Discontinuities in the traces occur where a linked intermediate or transition state was not calculated due to electronic structure uncertainties. | ||
Referring to Chart 1, in path P1 the first intermediate, N2x-Fe2NH3, is a deep energy well, followed by endergonic NH3 dissociation with a barrier of ca. 22 kcal mol−1, disfavouring this path. Path P2 first dissociates N2x (barrier ca. 14 kcal mol−1) and then forms NH3 with a barrier (ca. 6 kcal mol−1) considerably smaller than the NH3 formation barrier in path P1. Again there is a deep energy well at N2x-Fe2NH3, from which, in state S = 0, 47, the final exergonic (−12 kal mol−1) dissociation of NH3 encounters a barrier of only 6 kcal mol−1. The overall reaction energy is ca −40 kcal mol−1. For path P3 the first substantial barrier, ca. 22 kcal mol−1, is the formation of NH3. This intermediate, N2x-Fe2NH3, is again in an energy well of ca −40 kcal mol−1, close to the energy of the final product core. This low energy of N2x-Fe2NH3, and the two subsequent energy barriers for dissociation of NH3 and N2x, appear to disfavour completion of this reaction pathway.
Paths P4 and P5 do not include the very stable intermediate N2x-Fe2NH3, and possess a different overall energy profile. The first two steps in both paths, to N2x-Fe2-brNH2-Fe6H and Fe2-brNH2-Fe6H, possess small energy changes and barriers. NH3 formation, at either Fe2 (P4) or Fe6 (P5) involves barriers 21 to 23 kcal mol−1, and is exergonic at −25 kcal mol−1. Final dissociation of NH3 is also exergonic, −6 to −12 kcal mol−1.
In summary at this point, pathways P1 and P3 appear to be unfavourable, P2 is feasible with no large barriers, and pathways P4 and P5 are feasible except for the relatively large barrier to NH3 formation. However, another factor is to be considered in the kinetic analysis, namely the possibility of H atom tunnelling. This is investigated in the following section.
3.7 H atom tunneling possibilities
Quantum mechanical tunneling through a reaction barrier is maximized for the smallest atom encountering the barrier, hydrogen. Tunneling is often evaluated in terms of the de Broglie wavelength, which for a hydrogen atom with ambient (300 K) thermal energy of 2.5 kcal mol−1 is ca. 0.5 Å: the basic concept is that as the H atom ascends a barrier it can tunnel through it when the barrier width is of similar dimension.95–99 Many of the steps in the mechanism of nitrogenase involve H atom transfer, and I have suggested that H atom tunneling might occur in some H transfer steps, and should be investigated.43In a previous trajectory analysis of an S3B–H to N (N2) hydrogen atom transfer in nitrogenase100 I noted similarities with the trajectories for proton transfer in the enzyme aromatic amine dehydrogenase, where a detailed investigation concluded that the reaction is dominated by quantum tunneling over a distance of ca. 0.6 Å, at ∼10 kcal mol−1 below the top of the classical potential energy barrier.101 Experimental and theoretical investigation of hydrogen atom tunneling is complicated, even for reaction systems much more conventional than those in the mechanism of nitrogenase. In the present context the assessment of possible tunneling adopts the basic criterion that the potential energy profile for H transfer be symmetrical about the classical transition state, for a narrow (∼0.6 Å) section of the H atom transfer coordinate, during which heavier atoms do not move. I am not aware of other reports of H tunneling involving S–H or Fe–H bonds.
First I examine the 6Hx → NH2 component of NH3 formation, which occurs in reaction paths P3, P4 and P5. Fig. 7 plots the relevant geometrical properties in the vicinity of the transition state (TS) on the potential energy surface for the Fe2-brNH2-Fe6H → Fe6NH3 step, in the S = 1, 47 electronic state. The calculation used energy minimisation with small energy intervals from the TS towards reactant and towards product. The 6Hx atom moves by swinging from the exo-Fe6 position towards Nbr (Fig. 4), with hardly any change in the H–Fe6 distance until beyond the TS. As the potential energy climbs towards the TS there is only one major geometry change, the shortening of the H–N distance, from near 2.5 Å to 1.5 Å at the TS. The two Fe–N bonds of the NH2 bridge are essentially unchanged near the TS, and differentiate in favour of Fe6–N only after the H–N bond and NH3 are almost fully formed and the H–Fe6 bond is weakening. This reaction is atypical of other H atom transfer reactions that are believed to involve quantum tunneling, because here the geometry bracketing the TS involves a change mainly in only one H–X distance. The majority of H atom transfer reactions involve synchronous bond breaking and bond making. Conventionally understood H atom tunneling systems are usually intermolecular, between distinctly separate H donor and acceptor atoms, and tunneling occurs when there is a shortening of the distance between them:98,102 in the present intramolecular reaction the H atom donor (Fe6) and acceptor (N) atoms are instead strongly bonded.
 | ||
| Fig. 7 Potential energy profile near the transition state for the Fe2-brNH2-Fe6H → Fe6NH3 reaction step in the S = 1, 47 electronic state. The ‘goal posts’ symmetrically bracket the TS energy potential, and encompass movement of H by ca. 0.5 Å. | ||
To assess of the possibility of a tunneling contribution to this Fe2-brNH2-Fe6H → Fe6NH3 step, consider the ‘goal posts’ on Fig. 7. They are placed to bracket symmetrically the energy potential about the TS (as required for quantum tunneling), and to include an H atom displacement of ∼0.5 Å. These goal posts cross the potential energy trace ∼4 kcal mol−1 below the TS, and so suggest that H atom tunneling here could occur ∼4 kcal mol−1 below the classical barrier. In a different electronic state, S = 1 35, Fe2-brNH2-Fe6H forms NH3 at Fe2 instead of Fe6 (Scheme 3 panel D). During this step the potential energy profile as it extends 10 kcal mol−1 either side of the TS is very similar to that shown in Fig. 7. Therefore the involvement of H quantum tunneling is similarly indicated for the Fe2-brNH2-Fe6H → Fe2NH3 step. The differentiation of products, Fe2–NH3 in the 35 electronic state and Fe6–NH3 in the 47 electronic state, appears late in both trajectories, after the H–N bond has formed. Results for the formation of NH3 by H transfer from 6Hx but with N2x present, N2x-Fe2-brNH2-Fe6H → N2x-Fe2NH3 resemble those in Fig. 7.
The alternative step forming NH3 by H transfer from 3b2 to N, is different because the S3B⋯Fe6 distance is variable, and movements of S3B during passage over the potential energy barrier for H transfer could obviate quantum tunneling by H. In the reaction trajectory for N2x-Fe2-brNH2-Fe6-3b2 → N2x-Fe2NH3 the S3B⋯Fe6 distance is initially long, ca. 2.9 Å, decreases to ca. 2.6 Å at the transition state, and continues towards 2.3 Å in the product. Although not analysed in detail it appears that this reaction is not a candidate for possible H tunneling. However, the trajectory for corresponding reaction with N2x absent, Fe2-brNH2-Fe6-3b2 → Fe2NH3, is different because the S3B–Fe6 separation is almost invariant through most of the H transfer. The trajectory plot (see ESI Fig. S4†) shows minor heavy atom movement through the TS, and suggests that there could be H tunneling about 2 kcal mol−1 below the TS. For this reaction step (in path P2) the classical potential energy barrier is small, 5–7 kcal mol−1, and so H any tunneling would have minor kinetic influence.
Note that although these putative tunneling systems appear to be complex, a small 22 atom model such as that of Fig. 3G embodies essential features of Fe2-brNH2-Fe6H → Fe6NH3 and could be amenable to higher level calculations of the tunneling characteristics.103
4. Discussion
Prior reports described how the active site of nitrogenase can capture N2 using a propitious array of H atoms bound at S2B and Fe7, to generate an intermediate with bound HNNH, Fe2-brNH-NH-Fe6.65 Then, in a second phase, two sequential hydrogenations can convert this HNNH first to HNNH2, then to NH and NH3, after which this first NH3 is released.67 These two phases of the reaction are reviewed in outline in Scheme 4. The results presented in this report, the third and final phase, describe how the intermediate at the end of the second phase, Fe2-brNH-Fe6-S2BH with status E6 6H+, can continue by adding the seventh electron + proton, generate and release the second NH3, and after adding the eighth electron + proton complete the cycle and revert to the resting state. | ||
| Scheme 4 | ||
At the beginning of this third phase the transfer of S2BH to bridging NH (N2x-Fe2-brNH2-Fe6), and introduction and placement of the seventh H at the 3b2 position, intermediate N2x-Fe2-brNH2-Fe6-3b2, are straightforward and energetically accessible (Scheme 3, panel A). Thereafter five pathways can proceed from this intermediate, forming and then releasing NH3, finally reaching the intermediate core. There is no change in the electron–proton status of the system during this sequence. These pathway possibilities (Chart 1) are now evaluated according to their potential energy profiles, the possibility of H atom tunneling, and inclusion of entropic effects.
4.1 Entropy contributions
This last phase of the mechanism involves two dissociations of small molecules, N2x from the exo position of Fe2, and NH3 from the endo position of Fe2 or Fe6. My previous assessment of the entropic component of N2x binding at exo-Fe2 leads to the conclusion here that TΔS for dissociation is ca +4 kcal mol−1 and no more than +6 kcal mol−1.64 This estimate was based on experimental entropy measurement for the binding of N2 in comparable model complexes in solution, experimental measurements of the entropy of dissolution of N2, and an assumption that the environments, solution vs. protein, are similar in their encapsulation of small molecules.Estimation of the entropic component of NH3 dissociation from Fe is less certain. Despite abundant knowledge of metal-ammonia complexes and measurements of ligand substitution reactions in aqueous solution, I have not been able to find experimental entropy data for dissociation of NH3 from a suitable model complex into a medium comparable with the non-hydrogen bonding and weakly polar domain between Val70 and Arg96. There are gas phase data for adduct formation between trimethylgallium (and trimethylindium) and ammonia: ΔSassociation = −32.4 cal mol−1 K−1. Despite an apparent dissimilarity of this model and the enzyme, Me3Ga is chemically comparable with endo-Fe in FeMo-co: both are fully coordinated except for one vacant position, both have soft coordination, and Fe and Ga are similar size. This ΔSassociation is for NH3 in the gas phase: adjustment to the liquid phase with the standard entropy of vaporisation of NH3 (+23 cal mol−1 K−1), results in eqn (2).
 | (2) |
In search of entropy data for Fe–NH3 → Fe + NH3 as it occurs in proteo, another approximation is NH3(s) → NH3(l), i.e., immobilised NH3 to fluid NH3. The standard entropy of fusion of NH3 is 6.92 cal mol−1 K−1. In the absence of more direct data I average this and the numerically similar eqn (2), to yield 8 cal mol−1 K−1, or TΔS = +2.4 kcal mol−1 for the NH3 dissociation steps in the nitrogenase mechanism.
4.2 Quantum tunneling contributions
The analyses described in section 3.7 indicate that three steps Fe2-brNH2-Fe6H → Fe6NH3, Fe2-brNH2-Fe6H → Fe2NH3, and N2x-Fe2-brNH2-Fe6H → N2x-Fe2NH3, could involve some H atom tunneling, up to 4 kcal mol−1 below the classical barrier. The steps that involve hydrogen atom transfer between S2B and Nε of His195, occurring at N2x-Fe2-brNH-Fe6-S2BH → N2x-Fe2-brNH2-Fe6 (Scheme 2, grey enclosure) and core-S2BH → resting (Scheme 2, lavender-yellow enclosures), and Fe2-brNH2-Fe6-3b2 → Fe2NH3, appear to fit the criteria for intermolecular H atom tunneling, but a detailed analysis is not available. These tunneling contributions are included in the following analyses.4.3 Corrected energy profiles
The reaction pathways described in section 3.6 are described as their potential energy profiles. These can now be adjusted, to include the favourable entropy contributions to the dissociation of N2x (4 kcal mol−1) and of NH3 (2.4 kcal mol−1), and the predicted H atom tunneling kinetic advantage (4 kcal mol−1) when NH3 is formed by transfer of 6Hx. These adjustments have been applied to pathways P2, P3, P4 and P5, and the results, P2*, P3* and P5*, are shown in Chart 2. Because the dependence of potential energy profile on electronic and spin state is small (Chart 1), the potential energies at each intermediate and TS are averaged in Chart 2 to yield a single energy profile. The adjusted path P4* is very similar to P5* and is not shown.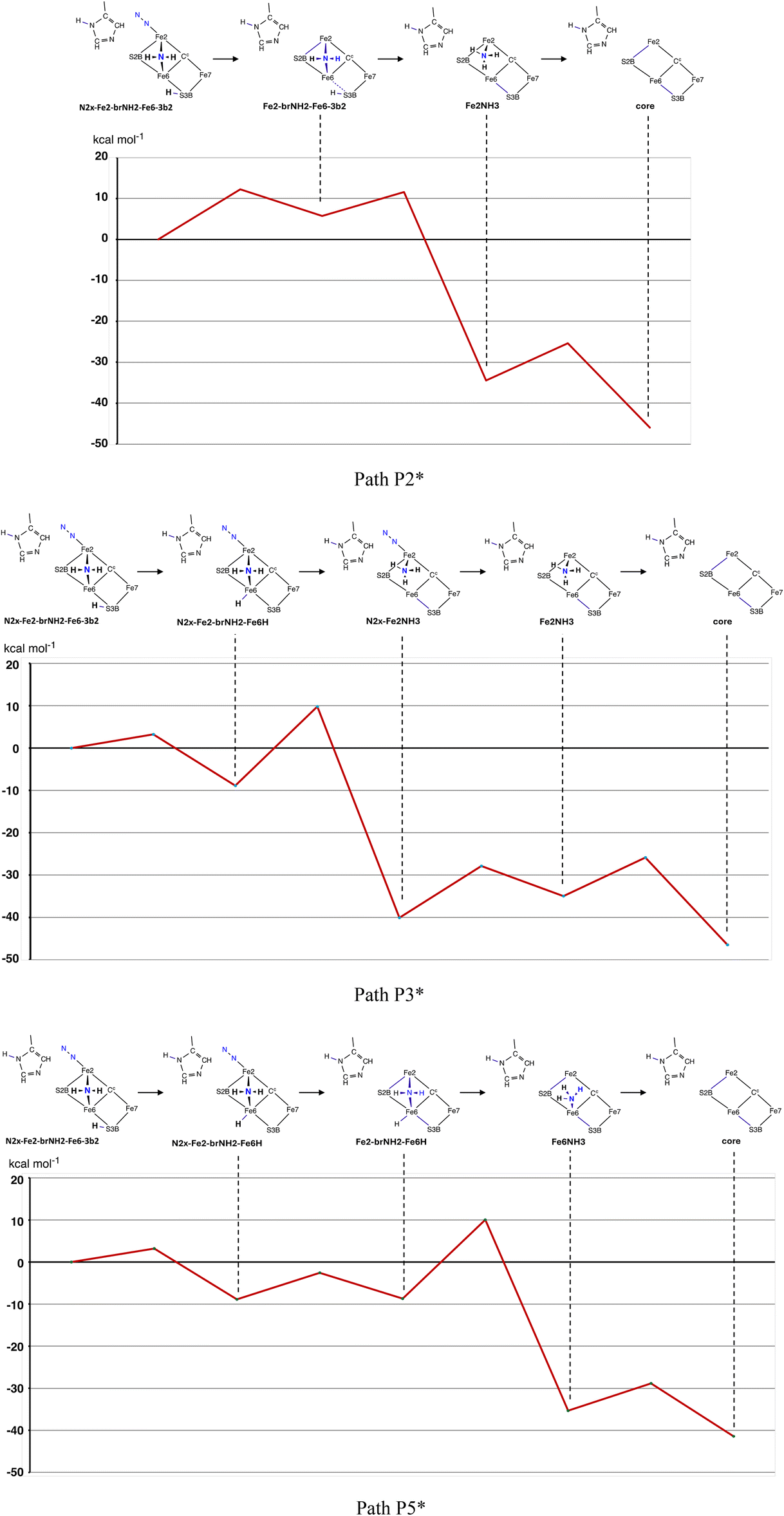 | ||
| Chart 2 Energy plots for three pathways, with the estimated TΔS entropy component applied to the potential energies in the steps where N2x or NH3 dissociate, and with the estimated tunneling contribution included to decrease the reaction barrier in the steps where 6Hx forms NH3. | ||
Each of the pathways P2*, P3*, P4* and P5* is thermodynamically and kinetically feasible. They are exergonic by more than 40 kcal mol−1, and kinetic barriers do not exceed 20 kcal mol−1. However there is a significant differentiating datum, which is the substantially lower barrier, ca. 5 kcal mol−1, for formation of NH3 at Fe2 from 3b2 when 2N2x is absent, in path P2*. Barriers for the other NH3 formation steps at Fe2 or Fe6 are larger, 13 to 18 kcal mol−1. As described in section 3.7 (and Fig. S4†) the Fe2–NH3 formation step in P2* probably benefits from H-tunneling. Pathway P2* encounters an initial barrier of ca. 12 kcal mol−1 for dissociation of 2N2x, followed by barriers of ca. 5 and 8 kcal mol−1 in two strongly exothermic steps, and is the favoured pathway from intermediate N2x-Fe2-brNH2-Fe6-3b2 to intermediate core.
4.4 Completion of the mechanism
The final part of the mechanism is from intermediate core to the resting state, shown on the salmon, lavender and yellow enclosures of Scheme 2. The migration of H from core-3b5, and its terminus on S2B (intermediate core-S2BH) as the most stable location have been described previously.42,62,104 The final step, core-S2BH to resting, is exergonic with a small barrier (Scheme 3, Panel F). This concludes description of the third and final phase of the complete mechanism, from N2x-Fe2-brNH2-Fe6-3b2 back to resting.4.5 Overall kinetics
Harris et al. reported steady-state kinetic studies on Mo-nitrogenase, monitoring formation of H2 and of NH3 using a full range of variables, and deriving key kinetic parameters.6 These results revealed a previously unrecognised slow step in the route from HNNH (labelled E4(N2; 2H) in ref. 6) to product NH3. Specifically, the rate constant for conversion of the HNNH intermediate to NH3 was less than the rate of electron accumulation during these steps. The rate of N2 binding and formation of the HNNH intermediate could not be explicitly determined. However, the conclusion was that a slow step followed the formation of the HNNH intermediate. In the previous paper I provided an explanation of this.67 In my calculated mechanism the formation of the HNNH intermediate (Fe2-brNH-NH-Fe6 in Scheme 1) involved classical potential energy barriers of 19 to 29 kcal mol−1, while the later N–N breaking step in which HNNH2 converts to NH + NH3 (lavender enclosure in Scheme 1) faces a larger potential energy barrier in the range 30 to 36 kcal mol−1. Therefore I proposed that the post-HNNH slow step identified in the kinetic analysis by Harris et al. is the N–N breaking step. This argument depends on the absence of larger barriers in all of the subsequent steps to the conclusion of the mechanistic cycle, and the present report confirms this and the validity of the overall kinetic interpretation.4.6 The role of S2B
The complete mechanism that I propose involves a catalytic site, FeMo-co, that has some structural plasticity without major disruption of intra-cluster bonding. In recent years the structural integrity of FeMo-co during catalysis has been questioned by the crystal structures and reactivities of Mo-nitrogenase isoforms in which S2B has been replaced by Se,105 or CO,106,107 or, controversially, by N2.108–111 The mechanistic relevance of these findings is unresolved, because the apparent displacement or replacement of S2B could reflect different dynamics between the time scales of substrate reduction and crystal growth.4 CryoEM diminishes this limitation. A recent cryoEM investigation of Mo-nitrogenase under pH 9.5 turnover conditions in the presence of acetylene recorded time dependent depletion of EM density in the vicinity of S2B, and changes near the S5A–Fe7 bond.39 Hopefully further experiments will provide greater certainty about the presence or absence of S2B during turnover.In the mechanism I propose S2B is retained within intact FeMo-co, and in that position has crucial roles in which an H atom on S2B is used to form an H–N bond. One role is the N2 capture step forming Fe2-brNH-NH-Fe6, and the second is the Fe2-brNH-Fe6-S2BH → Fe2-brNH2-Fe6 step. In both steps S2BH is needed to form an H–N bond on the His195 side of intermediates in the reaction zone, a space which cannot be accessed by H atoms coming from the S3B source. Throughout the mechanism His195 functions as proton reservoir and proton buffer, using a hydrogen bond with S2B at a distance (ca. 3.2 Å) very similar to that in the resting state. Key components of the proposed mechanism would be invalidated if S2B were absent or unable to function as H donor.
The cryoEM images of Warmack and Rees mentioned above also showed other substantial changes in FeMo-co, interpreted as HCA depletion and His442 rearrangement.39 There is clear distinction between any mechanism involving disruptive chemistry (S2B displaced, cofactor distorted, HCA rearranged or displaced) and the conservative chemistry of FeMo-co underlying the mechanism I have constructed.
5. Conclusions
5.1 The final phase of the mechanism
This report describes the third and final phase of the mechanism, starting at the intermediate with NH bridging Fe2 and Fe6 and with H on S2B, then proceeding to form and dissociate the second NH3, and return the catalytic site to its resting state. First S2BH is used to form NH2 bridging Fe2 and Fe6. This is strongly exergonic, in part reflecting the greater strength of an N–H bond relative to S–H. Here the system has E6 6H+ status. The seventh electron + proton is used to generate NH3, bound at the endo coordination position of either Fe2 or Fe6. Calculated reaction trajectories and potential energy profiles generate five mechanistic pathways through this NH3 formation phase of the reaction. These pathways are evaluated with incorporation of entropic components and possible kinetic contributions by H atom tunneling, leading to identification of P2* as the most favourable pathway for generation of NH3 and its subsequent dissociation. Addition of the eighth electron + proton straightforwardly regenerates the resting state. All steps in this third and final phase of the mechanism are thermodynamically and kinetically feasible.5.2 The complete mechanism
In conjunction with the first65 and second67 phases, this report completes a trilogy describing a total mechanism for the intrinsic chemistry of Mo-nitrogenase. The mechanism contains mainstream steps and auxiliary steps. Mainstream steps involve the generation and release of H2, the H2/N2 exchange, the concerted capture and hydrogenation of N2 to form the central HNNH intermediate, further hydrogenation to H2NNH followed by concerted breaking of the N–N bond and generation of the first NH3, then after release of this NH3 further hydrogenation to form the second NH3, and its dissociation. The auxiliary steps include introduction of protons, triggered by electronation,63 and migration of the resulting H atoms to positions on FeMo-co where they are required for the mainstream steps. These auxilliary steps replenish the H atom gallery, and occur without influential energy barriers.43,70,112,1135.3 Architecture and function
This proposed chemical mechanism is founded on the architecture of Mo-nitrogenase as the performance space for the choreography of catalysis by nitrogenase. Fig. 8 shows the macro-structural components that support the mechanism, and Fig. 9 shows the functional components in and adjacent to the reaction domain. With reference to these cartoons and the two prior papers of the trilogy,65,67 the key features and principles of the complete mechanism are summarised here.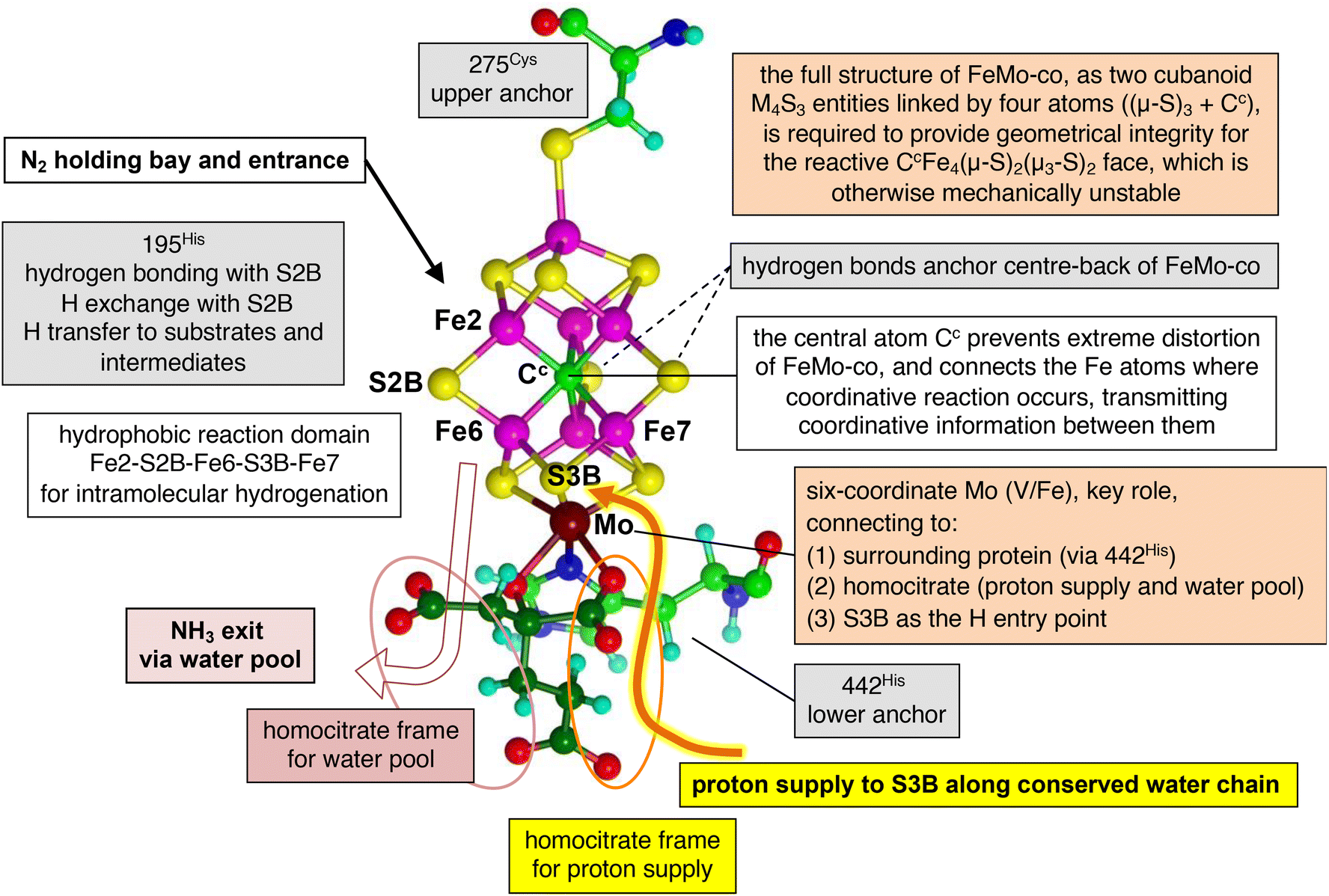 | ||
| Fig. 8 Structural components of the nitrogenase active site, and their mechanistic roles supporting the proposed mechanism. | ||
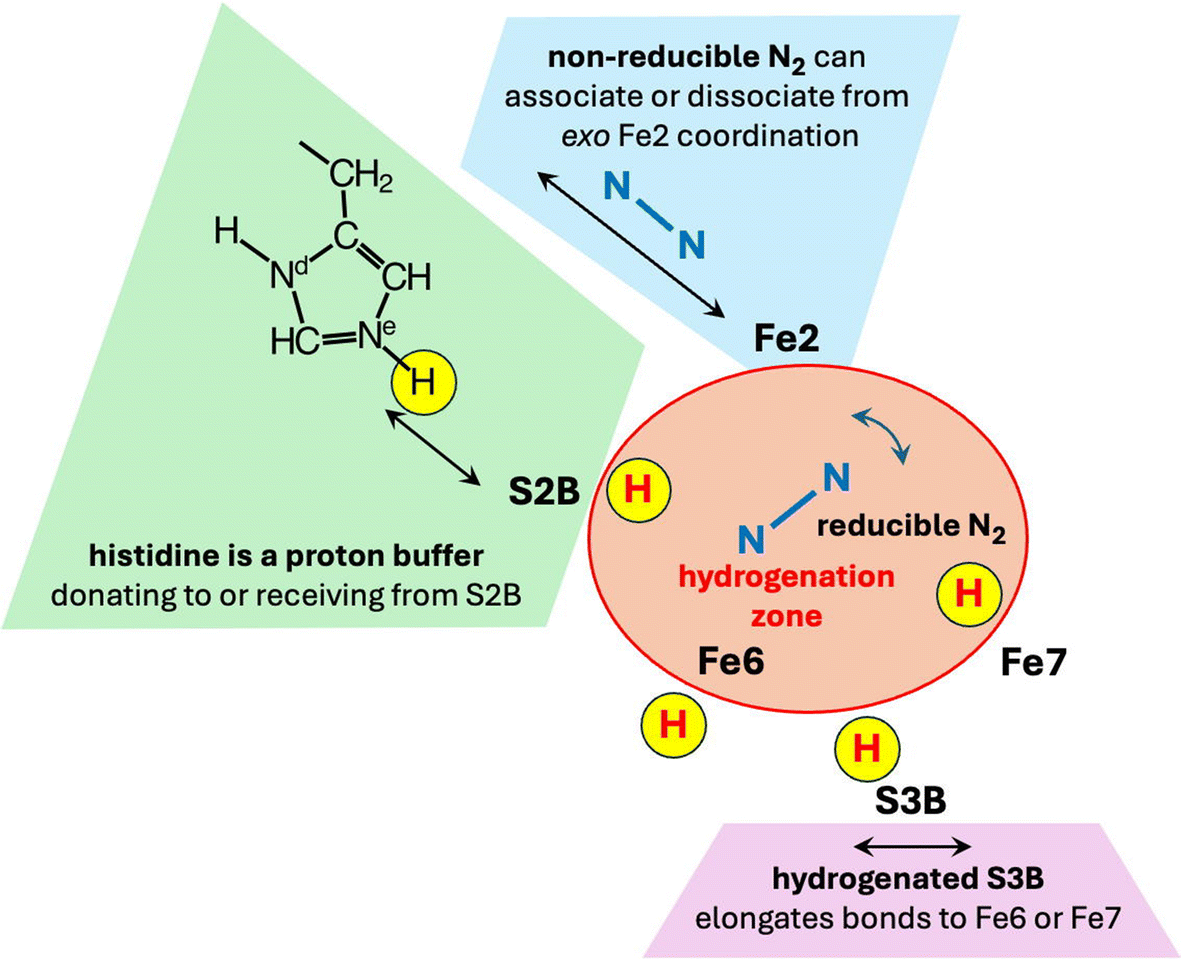 | ||
| Fig. 9 Functional components of the nitrogenase mechanism. The red H atom sites, S2B-H, exo-Fe6-H, S3B–H and endo-Fe7–H, effect the intramolecular hydrogenation of N2 and of subsequent intermediates in the hydrogenation zone. | ||
1. All eight protons in the reaction are derived from water at the protein surface and supplied by a Grotthuss proton wire along the conserved water chain to S3B.58,114 This could be triggered by electron addition to FeMo-co.63 In all stages except one at the beginning, proton addition to FeMo-co is accompanied by electron addition. The proton + electron combination occurs on FeMo-co as a slightly polarised H atom.115
2. An H atom on S3B can migrate to other sites on FeMo-co, with reaction barriers generally <10 kcal mol−1.58
3. From an early stage in the mechanism, until status E6 6H+ after N2 capture, the number of added protons is one greater than the number of added electrons.
4. All hydrogenation steps are intramolecular, enabled by a strategic gallery of H atoms located at S2B, exo-Fe6, S3B or endo-Fe7.
5. The sidechain of His195 acts as a proton buffer, via Nε, donating to S2B or receiving from S2B. This is influenced (perhaps controlled) by a hydrogen bonding network that extends from Nδ of His195 through water molecules to the protein surface.116
6. There is no hydrogenation of intermediates directly from His195: S2B is the intermediary transfer agent between His195Nε and the N atoms of mechanism intermediates.
7. Resting state His195 is protonated at Nε and Nδ. After use of this NεH via S2B it is replenished via S2B to regenerate the resting state.
8. Homocitrate has an essential role in maintaining a framework for the penultimate sections of the water chain that constitutes the proton wire, and in supporting the water pool that absorbs product NH3 at the commencement of its egress along a defined pathway.93
9. The distinctive CH2CH2COOH arm of homocitrate maintains a necessary separation of the acidic chemistry of the proton wire and the basic chemistry of departing NH3.
10. H2 is formed at the endo position of Fe6, from which it dissociates, providing space for reducible N2 to enter. This is the H2/N2 exchange property of the enzyme.
11. Reducible N2 entering the reaction zone is able to tumble freely (ca. 4 kcal mol−1 (ref. 65)) and orient obliquely to Fe2 and Fe6, favouring the N2 capture step.
12. The N2-capture step is a concerted double hydrogenation combined with bonding to both Fe2 and Fe6. H atoms at S2B and endo-Fe7 are the key reactants. Two N–H bonds and three Fe–N bonds are formed in one reaction trajectory, producing bound HNNH.
13. The singular N2-capture step allows the enzyme to bypass the notoriously difficult initial hydrogenation of unreactive N2.65 It is different from the conventional view of the activation of N2 by metal systems, in which the binding of N2 initiates activation, with subsequent hydrogenation.
14. The orientation of bound HNNH oblique to Fe2–Fe6 provides excellent stereochemistry at N for subsequent hydrogenation to form bound HNNH2.67
15. H at exo-Fe6 is well placed to break the N–N bond in the HNNH2 intermediate and form Fe-bound NH3, in a single step.67 This step is proposed as the slow step identified in the kinetic analysis of Harris et al.6
16. All intermediates bond to Fe2 and Fe6 only at their endo positions, except the first NH3 which is formed at exo-Fe6, from which it dissociates.
17. Two of the hydrogenation steps (capture and formation of bridging NH2) are, necessarily, from the left side of the reaction zone. The first uses S2BH, and then HisNεH replenishes S2BH for the second.
18. S2B must be in its position bridging Fe2 and Fe6 to effect its two hydrogenation steps. There is ample space between Fe2 and Fe6 for both the S2B bridge and bridging intermediates.
19. Non-reducible N2, sliding easily between the N2 entry pocket (see Fig. 1) and ligation of Fe2 at the exo position,64 modulates the coordination capacity of Fe2 and the dynamics of NH3 dissociation from Fe2.
20. Hydrogenation steps that involve very little movement of atoms heavier than H might benefit kinetically from H atom tunneling. Further investigation is required.
21. To facilitate some reaction steps the reaction space is expanded slightly by ca. 1.4 Å movements of Val70 and Arg96, consistent with the secondary structure around Val70,65 the hydrogen bond to S5A by Arg96 (Fig. S1†), and experimental reactivities of Val70 mutants.8,54,57
The crucial test of this proposed mechanism is its ability to accommodate and account for all experimental data, and this will be elaborated in a future publication.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was undertaken with the aid of resources from the National Computational Infrastructure (NCI Australia), an NCRIS enabled capability supported by the Australian Government. My research is funded by UNSW Sydney.References
- A. K. Garcia, D. F. Harris, A. J. Rivier, B. M. Carruthers, A. Pinochet-Barros, L. C. Seefeldt and B. Kaçar, Nitrogenase resurrection and the evolution of a singular enzymatic mechanism, eLife, 2023, 12, e85003, DOI:10.7554/eLife.85003.
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, MIT Press, Cambridge, Massachusetts, 2001 Search PubMed.
- S. D. Threatt and D. C. Rees, Biological nitrogen fixation in theory, practice, and reality: a perspective on the molybdenum nitrogenase system, FEBS Lett., 2022, 597, 45–58, DOI:10.1002/1873-3468.14534.
- R. A. Warmack and D. C. Rees, Nitrogenase beyond the Resting State: A Structural Perspective, Molecules, 2023, 28, 7952, DOI:10.3390/molecules28247952.
- R. N. F. Thorneley and D. J. Lowe, in Molybdenum enzymes, ed. T. G. Spiro, Wiley Interscience, New York, 1985, ch. 5, pp. 221–284 Search PubMed.
- D. F. Harris, A. Badalyan and L. C. Seefeldt, Mechanistic Insights into Nitrogenase FeMo-Cofactor Catalysis through a Steady-State Kinetic Model, Biochemistry, 2022, 61, 2131–2137, DOI:10.1021/acs.biochem.2c00415.
- B. M. Barney, R. Y. Igarashi, P. C. Dos Santos, D. R. Dean and L. C. Seefeldt, Substrate Interaction at an Iron-Sulfur Face of the FeMo-cofactor during Nitrogenase Catalysis, J. Biol. Chem., 2004, 279, 53621–53624 CrossRef CAS PubMed.
- R. Sarma, B. M. Barney, S. Keable, D. R. Dean, L. C. Seefeldt and J. W. Peters, Insights into substrate binding at FeMo-cofactor in nitrogenase from the structure of an a-70Ile MoFe protein variant, J. Inorg. Biochem., 2010, 104, 385–389 CrossRef CAS PubMed.
- D. J. Scott, H. D. May, W. E. Newton, K. E. Brigle and D. R. Dean, Role for the nitrogenase MoFe protein alpha-subunit in FeMo-cofactor binding and catalysis, Nature, 1990, 343, 188–190 CrossRef CAS PubMed.
- C. H. Kim, W. E. Newton and D. R. Dean, Role of the MoFe protein alpha subunit histidine-195 residue in FeMo-cofactor binding and nitrogenase catalysis, Biochemistry, 1995, 34, 2798–2808 CrossRef CAS PubMed.
- M. J. Dilworth, K. Fisher, C. H. Kim and W. E. Newton, Effects on substrate reduction of substitution of histidine-195 by glutamine in the alpha-subunit of the MoFe protein of Azotobacter vinelandii nitrogenase, Biochemistry, 1998, 37, 17495–17505 CrossRef CAS PubMed.
- K. Fisher, M. J. Dilworth and W. E. Newton, Differential Effects on N2 Binding and Reduction, HD Formation, and Azide Reduction with a-195His- and a-191Gln-Substituted MoFe Proteins of Azotobacter Vinelandii, Nitrogenase, Biochemistry, 2000, 39, 15570–15577 CrossRef CAS PubMed.
- Z.-Y. Yang, K. Danyal and L. C. Seefeldt, Mechanism of Mo-Dependent Nitrogenase, Methods Mol. Biol., 2011, 766, 9–29 CrossRef CAS PubMed.
- K. Fisher, M. J. Dilworth, C.-H. Kim and W. E. Newton, Azotobacter Vinelandii, Nitrogenases Containing Altered MoFe Proteins with Substitutions in the FeMo-Cofactor Environment: Effects on the Catalyzed Reduction of Acetylene and Ethylene, Biochemistry, 2000, 39, 2970–2979 CrossRef CAS PubMed.
- J. Christiansen, V. L. Cash, L. C. Seefeldt and D. R. Dean, Isolation and characterisation of an acetylene-resistant nitrogenase, J. Biol. Chem., 2000, 275, 11459–11464 CrossRef CAS PubMed.
- J. Christiansen, L. C. Seefeldt and D. R. Dean, Competitive Substrate and Inhibitor Interactions at the Physiologically Relevant Active Site of Nitrogenase, J. Biol. Chem., 2000, 275, 36104–36107 CrossRef CAS PubMed.
- Z. Maskos, K. Fisher, M. Sorlie, W. E. Newton and B. J. Hales, Variant MoFe proteins of Azotobacter vinelandii: effects of carbon monoxide on electron paramagnetic resonance spectra generated during enzyme turnover, J. Biol. Inorg. Chem., 2005, 10, 394–406 CrossRef CAS PubMed.
- B. M. Barney, D. Lukoyanov, T.-C. Yang, D. R. Dean, B. M. Hoffman and L. C. Seefeldt, A methyldiazene (HN=N-CH3)-derived species bound to the nitrogenase active-site FeMo cofactor: Implications for mechanism, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17113–17118 CrossRef CAS PubMed.
- P. C. Dos Santos, S. M. Mayer, B. M. Barney, L. C. Seefeldt and D. R. Dean, Alkyne substrate interaction within the nitrogenase MoFe protein, J. Inorg. Biochem., 2007, 101, 1642–1648 CrossRef CAS PubMed.
- K. Fisher, D. J. Lowe, P. Tavares, A. S. Pereira, B. H. Huynh, D. Edmondson and W. E. Newton, Conformations generated during turnover of the Azotobacter vinelandii nitrogenase MoFe protein and their relationship to physiological function, J. Inorg. Biochem., 2007, 101, 1649–1656 CrossRef CAS PubMed.
- L. B. Gee, A. D. Scott, C. H. Dapper, W. E. Newton and S. P. Cramer, Carbon monoxide binding to α-R277H Mo-nitrogenase – Evidence for multiple pH-dependent species from IR-monitored photolysis, J. Inorg. Biochem., 2022, 232, 111806, DOI:10.1016/j.jinorgbio.2022.111806.
- C. Cadoux, D. Ratcliff, N. Maslać, W. Gu, I. Tsakoumagkos, S. Hoogendoorn, T. Wagner and R. D. Milton, Nitrogen Fixation and Hydrogen Evolution by Sterically Encumbered Mo-Nitrogenase, JACS Au, 2023, 3, 1521–1533, DOI:10.1021/jacsau.3c00165.
- A. D. Scott, V. Pelmenschikov, Y. Guo, L. Yan, H. Wang, S. J. George, C. H. Dapper, W. E. Newton, Y. Yoda, Y. Tanaka and S. P. Cramer, Structural Characterization of CO-Inhibited Mo-Nitrogenase by Combined Application of NRVS, EXAFS, and DFT: New Insights into the Effects of CO Binding and the Role of the Interstitial Atom, J. Am. Chem. Soc., 2014, 136, 15942–15954, DOI:10.1021/ja505720m.
- M. Maiuri, I. Delfino, G. Cerullo, C. Manzoni, V. Pelmenschikov, Y. Guo, H. Wang, L. B. Gee, C. H. Dapper, W. E. Newton and S. P. Cramer, Low frequency dynamics of the nitrogenase MoFe protein via femtosecond pump probe spectroscopy—Observation of a candidate promoting vibration, J. Inorg. Biochem., 2015, 153, 128–135, DOI:10.1016/j.jinorgbio.2015.07.005.
- L. B. Gee, H. Wang and S. P. Cramer, in Methods in Enzymology, ed. S. S. David, Academic Press, 2018, vol. 599, pp. 409–425 Search PubMed.
- V. Hoeke, L. Tociu, D. A. Case, L. C. Seefeldt, S. Raugei and B. M. Hoffman, High-Resolution ENDOR Spectroscopy Combined with Quantum Chemical Calculations Reveals the Structure of Nitrogenase Janus Intermediate E4(4H), J. Am. Chem. Soc., 2019, 141, 11984–11996, DOI:10.1021/jacs.9b04474.
- C. Van Stappen, L. Decamps, G. E. Cutsail, R. Bjornsson, J. T. Henthorn, J. A. Birrell and S. DeBeer, The Spectroscopy of Nitrogenases, Chem. Rev., 2020, 120, 5005–5081, DOI:10.1021/acs.chemrev.9b00650.
- L. Deng, H. Wang, C. H. Dapper, W. E. Newton, S. Shilov, S. Wang, S. P. Cramer and Z.-H. Zhou, Assignment of protonated R-homocitrate in extracted FeMo-cofactor of nitrogenase via vibrational circular dichroism spectroscopy, Commun. Chem., 2020, 3, 145, DOI:10.1038/s42004-020-00392-z.
- A. Pérez-González, Z.-Y. Yang, D. A. Lukoyanov, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Exploring the Role of the Central Carbide of the Nitrogenase Active-Site FeMo-cofactor through Targeted 13C Labeling and ENDOR Spectroscopy, J. Am. Chem. Soc., 2021, 143, 9183–9190, DOI:10.1021/jacs.1c04152.
- D. A. Lukoyanov, D. F. Harris, Z.-Y. Yang, A. Pérez-González, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, The One-Electron Reduced Active-Site FeFe-Cofactor of Fe-Nitrogenase Contains a Hydride Bound to a Formally Oxidized Metal-Ion Core, Inorg. Chem., 2022, 61, 5459–5464, DOI:10.1021/acs.inorgchem.2c00180.
- D. A. Lukoyanov, Z. Y. Yang, A. Perez-Gonzalez, S. Raugei, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, C-13 ENDOR Characterization of the Central Carbon within the Nitrogenase Catalytic Cofactor Indicates That the CFe6 Core Is a Stabilizing “Heart of Steel”, J. Am. Chem. Soc., 2022, 144, 18315–18328, DOI:10.1021/jacs.2c06149.
- K. Sengupta, J. P. Joyce, L. Decamps, L. Kang, R. Bjornsson, O. Rüdiger and S. DeBeer, Investigating the Molybdenum Nitrogenase Mechanistic Cycle Using Spectroelectrochemistry, J. Am. Chem. Soc., 2025, 147, 2099–2114, DOI:10.1021/jacs.4c16047.
- S. Koh, Y. Choi, I. Lee, G. M. Kim, J. Kim, Y. S. Park, S. Y. Lee and D. C. Lee, Light-Driven Ammonia Production by Azotobacter vinelandii Cultured in Medium Containing Colloidal Quantum Dots, J. Am. Chem. Soc., 2022, 144, 10798–10808, DOI:10.1021/jacs.2c01886.
- L. M. Pellows, G. E. Vansuch, B. Chica, Z.-Y. Yang, J. L. Ruzicka, M. A. Willis, A. Clinger, K. A. Brown, L. C. Seefeldt, J. W. Peters, G. Dukovic, D. W. Mulder and P. W. King, Low-temperature trapping of N2 reduction reaction intermediates in nitrogenase MoFe protein–CdS quantum dot complexes, J. Chem. Phys., 2023, 159, 235102, DOI:10.1063/5.0170405.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor, Science, 2011, 334, 940 CrossRef CAS PubMed.
- O. Einsle and D. C. Rees, Structural Enzymology of Nitrogenase Enzymes, Chem. Rev., 2020, 120, 4969–5004, DOI:10.1021/acs.chemrev.0c00067.
- H. L. Rutledge, B. D. Cook, H. P. M. Nguyen, M. A. Herzik and F. A. Tezcan, Structures of the nitrogenase complex prepared under catalytic turnover conditions, Science, 2022, 377, 865–869, DOI:10.1126/science.abq7641.
- R. A. Warmack, B. B. Wenke, T. Spatzal and D. C. Rees, Anaerobic cryoEM protocols for air-sensitive nitrogenase proteins, Nat. Protoc., 2024, 19, 2026–2051, DOI:10.1038/s41596-024-00973-5.
- R. A. Warmack and D. C. Rees, Structural evolution of nitrogenase states under alkaline turnover, Nat. Commun., 2024, 15, 10472, DOI:10.1038/s41467-024-54713-0.
- S. Raugei, L. C. Seefeldt and B. M. Hoffman, Critical computational analysis illuminates the reductive-elimination mechanism that activates nitrogenase for N2 reduction, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10521–E10530, DOI:10.1073/pnas.1810211115.
- A. T. Thorhallsson, B. Benediktsson and R. Bjornsson, A model for dinitrogen binding in the E4 state of nitrogenase, Chem. Sci., 2019, 10, 11110–11124, 10.1039/C9SC03610E.
- I. Dance, Survey of the geometric and electronic structures of the key hydrogenated forms of FeMo-co, the active site of the enzyme nitrogenase: principles of the mechanistically significant coordination chemistry, Inorganics, 2019, 7, 8, DOI:10.3390/inorganics7010008.
- I. Dance, Computational Investigations of the Chemical Mechanism of the Enzyme Nitrogenase, ChemBioChem, 2020, 21, 1671–1709, DOI:10.1002/cbic.201900636.
- L. Cao and U. Ryde, What Is the Structure of the E4 Intermediate in Nitrogenase?, J. Chem. Theory Comput., 2020, 16, 1936–1952, DOI:10.1021/acs.jctc.9b01254.
- I. Dance, Structures and reaction dynamics of N2 and H2 binding at FeMo-co, the active site of nitrogenase, Dalton Trans., 2021, 50, 18212–18237, 10.1039/d1dt03548g.
- A. T. Thorhallsson and R. Bjornsson, The E2 state of FeMoco: Hydride Formation versus Fe Reduction and a Mechanism for H2 Evolution, Chem. – Eur. J., 2021, 27, 16788–16800, DOI:10.1002/chem.202102730.
- H. Jiang and U. Ryde, Thermodynamically Favourable States in the Reaction of Nitrogenase without Dissociation of any Sulfide Ligand, Chem. – Eur. J., 2022, 28, e202103933, DOI:10.1002/chem.202103933.
- Y. Pang and R. Bjornsson, The E3 state of FeMoco: one hydride, two hydrides or dihydrogen?, Phys. Chem. Chem. Phys., 2023, 25, 21020–21036, 10.1039/D3CP01106B.
- Y. Pang and R. Bjornsson, Understanding the Electronic Structure Basis for N2 Binding to FeMoco: A Systematic Quantum Mechanics/Molecular Mechanics Investigation, Inorg. Chem., 2023, 62, 5357–5375, DOI:10.1021/acs.inorgchem.2c03967.
- H. Jiang and U. Ryde, Putative reaction mechanism of nitrogenase with a half-dissociated S2B ligand, Dalton Trans., 2024, 53, 11500–11513, 10.1039/D4DT00937A.
- P. E. M. Siegbahn, Final E5 to E8 Steps in the Nitrogenase Mechanism for Nitrogen Fixation, J. Phys. Chem. B, 2024, 128, 9699–9705, DOI:10.1021/acs.jpcb.4c04331.
- R. N. F. Thorneley and D. J. Lowe, Nitrogenase - Substrate Binding and Activation, J. Biol. Inorg. Chem., 1996, 1, 576–580 CrossRef CAS.
- B. M. Barney, H.-I. Lee, P. C. Dos Santos, B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Breaking the N2 triple bond: insights into the nitrogenase mechanism, Dalton Trans., 2006, 2277–2284, 10.1039/b517633f.
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Mechanism of Mo-Dependent Nitrogenase, Annu. Rev. Biochem., 2009, 78, 701–722 CrossRef CAS PubMed.
- S. M. Keable, J. Vertemara, O. A. Zadvornyy, B. J. Eilers, K. Danyal, A. J. Rasmussen, L. De Gioia, G. Zampella, L. C. Seefeldt and J. W. Peters, Structural characterization of the nitrogenase molybdenum-iron protein with the substrate acetylene trapped near the active site, J. Inorg. Biochem., 2018, 180, 129–134, DOI:10.1016/j.jinorgbio.2017.12.008.
- P. M. C. Benton, J. Christiansen, D. R. Dean and L. C. Seefeldt, Stereospecificity of Acetylene Reduction Catalyzed by Nitrogenase, J. Am. Chem. Soc., 2001, 123, 1822–1827 CrossRef CAS PubMed.
- P. C. Dos Santos, R. Igarashi, H.-I. Lee, B. M. Hoffman, L. C. Seefeldt and D. R. Dean, Substrate Interactions with the Nitrogenase Active Site, Acc. Chem. Res., 2005, 38, 208–214 CrossRef CAS PubMed.
- I. Dance, The pathway for serial proton supply to the active site of nitrogenase: enhanced density functional modeling of the Grotthuss mechanism, Dalton Trans., 2015, 44, 18167–18186, 10.1039/C5DT03223G.
- S. M. Keable, O. A. Zadvornyy, L. E. Johnson, B. Ginovska, A. J. Rasmussen, K. Danyal, B. J. Eilers, G. A. Prussia, A. X. LeVan, S. Raugei, L. C. Seefeldt and J. W. Peters, Structural characterization of the P1+ intermediate state of the P-cluster of nitrogenase, J. Biol. Chem., 2018, 293, 9629–9635, DOI:10.1074/jbc.RA118.002435.
- H. L. Rutledge, J. Rittle, L. M. Williamson, W. A. Xu, D. M. Gagnon and F. A. Tezcan, Redox-Dependent Metastability of the Nitrogenase P-Cluster, J. Am. Chem. Soc., 2019, 141, 10091–10098, DOI:10.1021/jacs.9b04555.
- H. L. Rutledge, M. J. Field, J. Rittle, M. T. Green and F. A. Tezcan, Role of Serine Coordination in the Structural and Functional Protection of the Nitrogenase P-Cluster, J. Am. Chem. Soc., 2022, 144, 22101–22112, DOI:10.1021/jacs.2c09480.
- I. Dance, The stereochemistry and dynamics of the introduction of hydrogen atoms onto FeMo-co, the active site of nitrogenase, Inorg. Chem., 2013, 52, 13068–13077, DOI:10.1021/ic401818k.
- I. Dance, What triggers the coupling of proton transfer and electron transfer at the active site of nitrogenase?, Dalton Trans., 2024, 53, 7996–8004, 10.1039/d4dt00474d.
- I. Dance, Understanding non-reducible N2 in the mechanism of Mo-nitrogenase, Dalton Trans., 2025, 3013–3026, 10.1039/d4dt03146f.
- I. Dance, The activating capture of N2 at the active site of Mo–nitrogenase, Dalton Trans., 2024, 53, 14193–14211, 10.1039/d4dt01866d.
- I. Dance, The binding of reducible N2 in the reaction domain of nitrogenase, Dalton Trans., 2023, 52, 2013–2026, 10.1039/D2DT03599E.
- I. Dance, The mechanism of Mo-nitrogenase: from N2 capture to first release of NH3, Dalton Trans., 2024, 53, 19360–19377, 10.1039/d4dt02606c.
- D. Lukoyanov, N. Khadka, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Reductive Elimination of H2 Activates Nitrogenase to Reduce the N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme, J. Am. Chem. Soc., 2016, 138, 10674–10683, DOI:10.1021/jacs.6b06362.
N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme, J. Am. Chem. Soc., 2016, 138, 10674–10683, DOI:10.1021/jacs.6b06362. - D. Lukoyanov, N. Khadka, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Reversible Photoinduced Reductive Elimination of H2 from the Nitrogenase Dihydride State, the E4(4H) Janus Intermediate, J. Am. Chem. Soc., 2016, 138, 1320–1327, DOI:10.1021/jacs.5b11650.
- I. Dance, The HD Reaction of Nitrogenase: a Detailed Mechanism, Chem. – Eur. J., 2023, 29, e202202502, DOI:10.1002/chem.202202502.
- A. K. Garcia, H. McShea, B. Kolaczkowski and B. Kaçar, Reconstructing the evolutionary history of nitrogenases: Evidence for ancestral molybdenum-cofactor utilization, Geobiology, 2020, 18, 394–411, DOI:10.1111/gbi.12381.
- B. Benediktsson and R. Bjornsson, QM/MM Study of the Nitrogenase MoFe Protein Resting State: Broken-Symmetry States, Protonation States, and QM Region Convergence in the FeMoco Active Site, Inorg. Chem., 2017, 56, 13417–13429, DOI:10.1021/acs.inorgchem.7b02158.
- L. Cao, O. Caldararu and U. Ryde, Protonation States of Homocitrate and Nearby Residues in Nitrogenase Studied by Computational Methods and Quantum Refinement, J. Phys. Chem. B, 2017, 121, 8242–8262, DOI:10.1021/acs.jpcb.7b02714.
- T. Spatzal, J. Schlesier, E.-M. Burger, D. Sippel, L. Zhang, S. L. A. Andrade, D. C. Rees and O. Einsle, Nitrogenase FeMoco investigated by spatially resolved anomalous dispersion refinement, Nat. Commun., 2016, 7, 10902, DOI:10.1038/ncomms10902.
- R. Bjornsson, F. Neese and S. DeBeer, Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge, Inorg. Chem., 2017, 56, 1470–1477, DOI:10.1021/acs.inorgchem.6b02540.
- B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, in Modern density functional theory: a tool for chemistry, ed. J. M. Seminario and P. Politzer, Elsevier, Amsterdam, 1995, vol. 2, pp. 221–254 Search PubMed.
- J. Baker, A. Kessi and B. Delley, The generation and use of delocalized internal coordinates in geometry optimization, J. Chem. Phys., 1996, 105, 192–212, DOI:10.1063/1.471864.
- B. Delley, From molecules to solids with the DMol3 approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. Andzelm, R. D. King-Smith and G. Fitzgerald, Geometry optimization of solids using delocalized internal coordinates, Chem. Phys. Lett., 2001, 335, 321–326, DOI:10.1016/S0009-2614(01)00030-6.
- T. Todorova and B. Delley, Molecular Crystals: A Test System for Weak Bonding, J. Phys. Chem. C, 2010, 114, 20523–20530, DOI:10.1021/jp1049759.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- I. Dance, Evaluations of the Accuracies of DMol3 Density Functionals for Calculations of Experimental Binding Enthalpies of N2, CO, H2, C2H2 at Catalytic Metal Sites, Mol. Simul., 2018, 44, 568–581, DOI:10.1080/08927022.2017.1413711.
- J. Andzelm, C. Kolmel and A. Klamt, Incorporation of solvent effects into density functional calculations of molecular energies and geometries, J. Chem. Phys., 1995, 103, 9312–9320 CrossRef CAS.
- A. Klamt, V. Jonas, T. Burger and J. C. W. Lohrenz, Refinement and Parametrization of COSMO-RS, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- B. Delley, The conductor-like screening model for polymers and surfaces, Mol. Simul., 2006, 32, 117–123 CrossRef CAS.
- I. Dance, Calculating the chemical mechanism of nitrogenase: new working hypotheses, Dalton Trans., 2022, 51, 12717–12728, 10.1039/d2dt01920e.
- L. Cao and U. Ryde, Influence of the protein and DFT method on the broken-symmetry and spin states in nitrogenase, Int. J. Quantum Chem., 2018, 118, e25627, DOI:10.1002/qua.25627.
- H. Jiang, O. K. G. Svensson and U. Ryde, QM/MM Study of Partial Dissociation of S2B for the E2 Intermediate of Nitrogenase, Inorg. Chem., 2022, 61, 18067–18076, DOI:10.1021/acs.inorgchem.2c02488.
- H. Jiang and U. Ryde, N2 binding to the E0–E4 states of nitrogenase, Dalton Trans., 2023, 52, 9104–9120, 10.1039/D3DT00648D.
- I. Dance, A pragmatic method for location of transition states and calculation of reaction paths., Mol. Simul., 2008, 34, 923–929 CrossRef CAS.
- I. Dance, A pragmatic method for location of transition states and calculation of reaction paths: erratum, Mol. Simul., 2011, 37, 257 CrossRef CAS.
- I. Dance, A molecular pathway for the egress of ammonia produced by nitrogenase, Sci. Rep., 2013, 3, 3237, DOI:10.1038/srep03237.
- I. Dance, Intermolecular Embraces and Intermolecular Energies, Mol. Cryst. Liq. Cryst., 2005, 440, 265–293 CrossRef CAS.
- A. Kohen and J. P. Klinman, Enzyme Catalysis: Beyond Classical Paradigms, Acc. Chem. Res., 1998, 31, 397–404 CrossRef CAS.
- A. Kohen and J. P. Klinman, Hydrogen tunneling in biology, Chem. Biol., 1999, 6, R191–R198 CrossRef CAS PubMed.
- M. J. Sutcliffe and N. S. Scrutton, A new conceptual framework for enzyme catalysis. Hydrogen tunnelling coupled to enzyme dynamics in flavoprotein and quinoprotein enzymes, Eur. J. Biochem., 2002, 269, 3096–3102 CrossRef CAS PubMed.
- Z. D. Nagel and J. P. Klinman, Tunneling and Dynamics in Enzymatic Hydride Transfer, Chem. Rev., 2006, 106, 3095–3118 CrossRef CAS PubMed.
- J. P. Layfield and S. Hammes-Schiffer, Hydrogen Tunneling in Enzymes and Biomimetic Models, Chem. Rev., 2013, 114, 3466–3494, DOI:10.1021/cr400400p.
- I. Dance, The chemical mechanism of nitrogenase: hydrogen tunneling and further aspects of the intramolecular mechanism for hydrogenation of η2-N2 on FeMo-co to NH3, Dalton Trans., 2008, 5992–5998, 10.1039/B806103C.
- L. Masgrau, A. Roujeinikova, L. O. Johannissen, P. Hothi, J. Basran, K. E. Ranaghan, A. J. Mulholland, M. J. Sutcliffe, N. S. Scrutton and D. Leys, Atomic Description of an Enzyme Reaction Dominated by Proton Tunneling, Science, 2006, 312, 237–241, DOI:10.1126/science.1126002.
- G. Tresadern, J. P. McNamara, M. Mohr, H. Wang, N. A. Burton and I. H. Hillier, Calculations of hydrogen tunnelling and enzyme catalysis: a comparison of liver alcohol dehydrogenase, methylamine dehydrogenase and soybean lipoxygenase, Chem. Phys. Lett., 2002, 358, 489–494 CrossRef CAS.
- A. Nandi, G. Molpeceres, P. K. Gupta, D. T. Major, J. Kastner, J. M. Martin and S. Kozuch, in Comprehensive Computational Chemistry, ed. M. Yanez and R. J. Boyd, Elsevier, 2022, pp. 713–734 Search PubMed.
- L. Cao, O. Caldararu and U. Ryde, Protonation and Reduction of the FeMo Cluster in Nitrogenase Studied by Quantum Mechanics/Molecular Mechanics (QM/MM) Calculations, J. Chem. Theory Comput., 2018, 14, 6653–6678, DOI:10.1021/acs.jctc.8b00778.
- T. Spatzal, K. A. Perez, J. B. Howard and D. C. Rees, Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor, eLife, 2015, 4, e11620, DOI:10.7554/eLife.11620.
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase, Science, 2014, 345, 1620–1623, DOI:10.1126/science.1256679.
- T. M. Buscagan, K. A. Perez, A. O. Maggiolo, D. C. Rees and T. Spatzal, Structural Characterization of Two CO Molecules Bound to the Nitrogenase Active Site, Angew. Chem., Int. Ed., 2021, 60, 5704–5707, DOI:10.1002/anie.202015751.
- W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase, Science, 2020, 368, 1381–1385, DOI:10.1126/science.aaz6748.
- J. W. Peters, O. Einsle, D. R. Dean, S. DeBeer, B. M. Hoffman, P. L. Holland and L. C. Seefeldt, Comment on “Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase”, Science, 2021, 371, abe5481, DOI:10.1126/science.abe5481.
- W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Response to Comment on “Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase”, Science, 2021, 371, abe5856, DOI:10.1126/science.abe5856.
- C. C. Lee, W. Kang, A. J. Jasniewski, M. T. Stiebritz, K. Tanifuji, M. W. Ribbe and Y. Hu, Evidence of substrate binding and product release via belt-sulfur mobilization of the nitrogenase cofactor, Nat. Catal., 2022, 5, 443–454, DOI:10.1038/s41929-022-00782-7.
- I. Dance, The Hydrogen Chemistry of the FeMo-co Active Site of Nitrogenase, J. Am. Chem. Soc., 2005, 127, 10925–10942 CrossRef CAS PubMed.
- I. Dance, The chemical mechanism of nitrogenase: calculated details of the intramolecular mechanism for hydrogenation of η2-N2 on FeMo-co to NH3, Dalton Trans., 2008, 5992–5998, 10.1039/B806100A.
- I. Dance, The controlled relay of multiple protons required at the active site of nitrogenase, Dalton Trans., 2012, 41, 7647–7659, 10.1039/C2DT30518F.
- I. Dance, Misconception of reductive elimination of H2, in the context of the mechanism of nitrogenase, Dalton Trans., 2015, 44, 9027–9037, 10.1039/C5DT00771B.
- I. Dance, New insights into the reaction capabilities of His195 adjacent to the active site of nitrogenase, J. Inorg. Biochem., 2017, 169, 32–43, DOI:10.1016/j.jinorgbio.2017.01.005.
Footnote |
| † Electronic supplementary information (ESI) available: Description of the protein model and the structural constraints used, including movement of Val70 and the contiguous chain, movement of Arg96, a description of the density functional procedures and their validation, a description of procedures for determination of transition states and reaction trajectories, and an additional potential energy profile. Electronic states and atomic coordinates for reactants, transition states and products are listed. See DOI: https://doi.org/10.1039/d5dt00658a |
| This journal is © The Royal Society of Chemistry 2025 |