
Photocatalyst based on a transition metal-Schiff base ligand and V-doped Kegging-type polyoxometalate for efficient and stable CO2 reduction†
Xiao-Yu
Bai
,
Pin-Fang
Yan
,
Jiu-Lin
Zhou
,
Yu
Lv
,
Ji-Lei
Wang
 ,
Hua
Mei
* and
Yan
Xu
*
,
Hua
Mei
* and
Yan
Xu
*
College of Chemical Engineering, State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing 211800, P. R. China. E-mail: yanxu@njtech.edu.cn
First published on 27th May 2025
Abstract
In order to simultaneously control the greenhouse effect and solve the problem of energy depletion, the strategy of converting CO2 into other forms of energy can be used. In this work, two Keggin-type polyoxometalate-based compounds were synthesized via the hydrothermal method: [Co(C11H15N7O2)(H2O)2]4{[Co(C11H15N7O2)(H2O)]2(PWVI11VIVO40)}(PWVI9WV2VIVO40)·14.5H2O (1) and [Fe(C11H15N7O2)(H2O)2]3{[Fe(C11H15N7O2)(H2O)]2(BW12O40)}(BW12O40)·11H2O (2). The structural analysis of compound 1 indicated that an interesting V-doped Keggin-type (PWVI11VIVO40)5− anion is successfully bonded to the two [Co(C11H15N7O2)(H2O)]2+ cations to generate a hybrid [Co(C11H15N7O2)(H2O)]2(PWVI11VIVO40) unit. Through a series of photocatalytic CO2 reduction reaction (CO2RR) experiments, it was found that compound 1 exhibits good photocatalytic performance. The CO generation rate can reach 7081.4 μmol g−1 h−1, and the selectivity was 83.8%. In addition, the stable and efficient photocatalytic activity of compound 1 was verified after four-cycle photocatalytic experiments, which provided a new idea for CO2 photoreduction to CO. Comparatively, the CO production rates was 103.5 μmol g−1 h−1 for compound 2. This is because the adsorption energy of CO on Co is lower than that on Fe. Furthermore, V-modified compound 1 enhances CO2 adsorption to promote CO2 conversion.
Introduction
In recent years, it has triggered negative environmental impacts such as increased consumption of fossil fuel and rising greenhouse gas emissions as the country's industrialisation has progressed smoothly.1–4 Therefore, there is an urgent need to think about how we can simultaneously address the dual challenge of the depletion of non-renewable resources and global warming. One immediate solution is to utilize renewable solar energy for photocatalysis to convert greenhouse gases such as carbon dioxide into needed chemicals or other forms of renewable energy.5 This approach would contribute to the alleviation of the prevailing energy crisis and align with the development of a greener society.Polyoxometalates (POMs) are anionic forms of transition metal oxides with a variety of structures. Different redox potentials and intramolecular charge transfer capabilities can be achieved by selecting POMs with different structures and transition metals (TMs). POMs as a unique organic–inorganic hybrid material have been applied in photocatalysis, electrocatalysis, and magnetic materials.6–16 Nowadays, the development of heterogeneous POM-based photocatalysts with high activity, selectivity, and stability has become a research topic in the field of catalysis.17–20 POM-based hybrid materials were modified by organic ligands during POM self-assembly owing to their limitations such as poor stability and strong solubility. Notably, organic ligands enhanced the stability, light adsorption and hydrophobicity of POM-based hybrid materials.21–23 Moreover, the introduction of TM into POMs can modulate their electronic structure and improve their physicochemical properties to facilitate the reaction. Among numerous POMs, Keggin-type POMs are typical representatives of the most commonly synthesized and well-studied POMs, which stand out for their easy-to-manufacture, economic, green and non-corrosive properties.24–29 In 2006, Prof. Francis Sécheresse's group synthesized the mixed clusters of V-doped Keggin-type heteropolytungstates for the first time. Then, the study on these materials began to enter the public vision. The introduction of V atoms enhanced the adsorption of CO2.30–33 Given that the Schiff base ligand 2,6-diacetylpyridinedicarbazide (DAPSC) was employed for the modification of two Keggin-type POMs because it has a similar structure to chlorophyll's, which possesses a planar conjugate structure. The conjugated structures formed by DAPSC and metal coordination accelerate the ligand-to-metal charge transfer (LMCT) process. Therefore, two different Keggin-type POMs with different TM (Co, Fe) serving as catalytic sites were synthesized by hydrothermal method in this work: [Co(C11H15N7O2)(H2O)2]4{[Co(C11H15N7O2)(H2O)]2(PWVI11VIVO40)}(PWVI9WV2VIVO40)·14.5H2O (1), [Fe(C11H15N7O2)(H2O)2]3{[Fe(C11H15N7O2)(H2O)]2(BW12O40)}(BW12O40)·11H2O (2). In particular, compound 1 was successfully V-modified. Compounds 1–2 formed 3D supramolecular structures through hydrogen bonding interactions. Notably, compound 1 exhibits higher activity than compound 2 because the adsorption energy of CO on Co is lower than that on Fe.34 Furthermore, the V-modified derivative of compound 1 enhances CO2 adsorption, thereby promoting CO2 conversion. The experimental results demonstrated that the optimum dosage of compound 1 (3 mg) resulted in the production of 169.9 μmol of CO after eight hours of visible light irradiation. The results show an average CO generation rate of 7081.4 μmol g−1 h−1 and 83.8% selectivity. The maximum rate was found to be 8690.1 μmol g−1 h−1. Compound 1, as a heterogeneous catalyst, maintained its photocatalytic activity after four cycle experiments. The results prove that compound 1 exhibits remarkable photocatalytic performance and structural stability.
Experimental section
Synthesis of the compounds
Results and discussion
Crystal structure
The crystal structures of compounds 1 and 2 were examined by single-crystal X-ray diffraction. Compound 1 belongs to the monoclinic crystal system and crystallizes in the P21/m space group (Table S1†). Two Keggin-type POMs clusters formed by W and V atoms partially in disorder are present in compound 1 as {PWVI11VIV} and {PWVI9WV2VIV}. The doping of V atoms enhances the adsorption of CO2 on {PWVI11VIV} as well as {PWVI9WV2VIV}. The structure of compound 1 (Fig. 1a) consists of two parts. The first part is a typical double-cap structure composed of two cationic fragments [Co(C11H15N7O2)(H2O)]2+ connected to the POMs [PWVI11VIVO40]5− clusters (Fig. 1b). The [Co(C11H15N7O2)(H2O)]2+ fragment consists of Co2+, a DAPSC ligand and a coordinated water molecule. The other part is the Keggin-type heteropolytungstate [PWVI9WV2VIVO40]7− clusters surrounded by four free [Co(C11H15N7O2)(H2O)2]2+ fragments, forming a flower-like cluster (Fig. 1c). The [Co(C11H15N7O2)(H2O)2]2+ fragment consists of Co2+, a DAPSC ligand and two coordinated water molecules. The Co atoms in compound 1 have two coordination environments, both in the seven-coordination pattern, forming a typical double pentagonal cone structure (Fig. S2†). Co1 is described as an example, which is connected to two O atoms (O1W, O2W) on two coordinated water molecules in addition to three N (N3, N4, N3) atoms and two O atoms (O13, O13) from the DAPSC (Fig. S3a†). The Co ions attached to the [PWVI11VIVO40]5− bind three N atoms (N7, N8, N9) and two O atoms (O12, O45) originating from the same DAPSC, an O atom (O8) on [PWVI11VIVO40]5− and an O atom (O3W) in the coordinated water molecule (Fig. S3b†). The hydrogen bonding interactions between these two parts form the isolated structural unit. The Co–O bond lengths (2.111–2.192 Å) and Co–N bond lengths (2.155–2.201 Å) are indicated in Table S2.† In addition, the neighboring isolated structural units through hydrogen bonding formed a 3D supramolecular structure that maintained the stability of the crystal structure (Fig. 1d).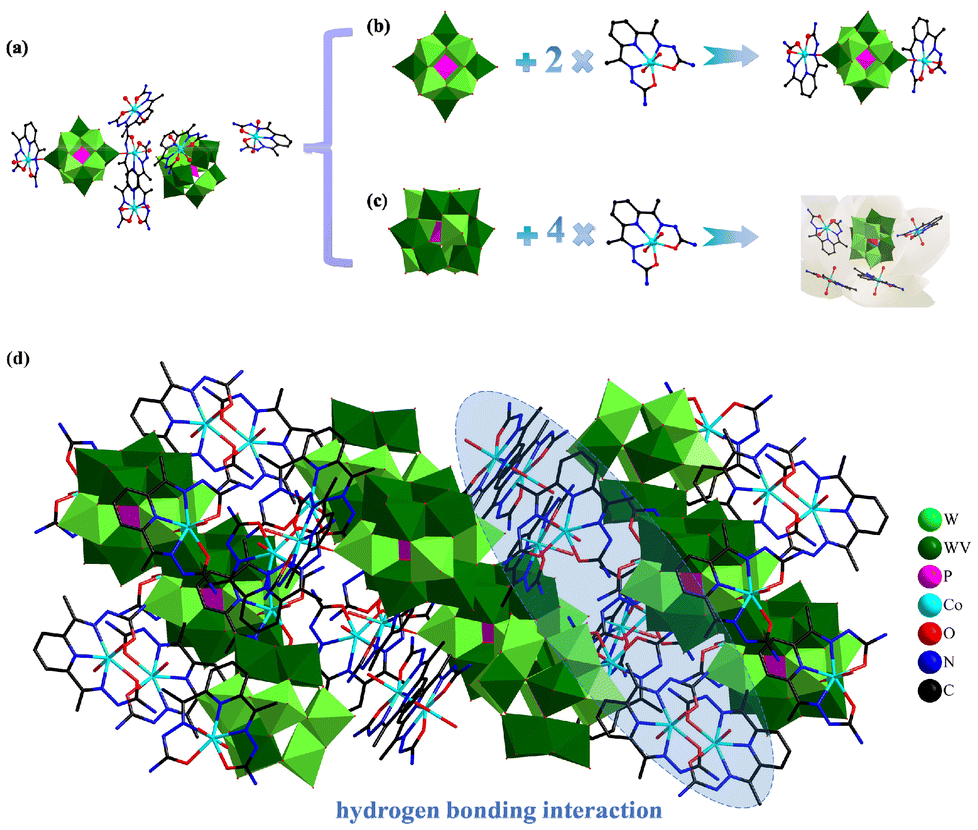 | ||
| Fig. 1 (a) Asymmetric unit of compound 1. (b) Double-cap structure of compound 1. (c) Floral structure of compound 1 (ball-and-stick model). (d) 3D supramolecular framework (polyhedral) of compound 1. | ||
Compound 2 belongs to the triclinic crystal system and crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The individual structure unit (Fig. S5†) of compound 2 consists of a {BW12} cluster bridging two [Fe(C11H15N7O2)(H2O)]2+, a free {BW12} cluster and three free [Fe(C11H15N7O2)(H2O)2]2+ clusters. The asymmetric unit of compound 2 formed a 3D structure through hydrogen bonding (Fig. S6 and S7†).
space group. The individual structure unit (Fig. S5†) of compound 2 consists of a {BW12} cluster bridging two [Fe(C11H15N7O2)(H2O)]2+, a free {BW12} cluster and three free [Fe(C11H15N7O2)(H2O)2]2+ clusters. The asymmetric unit of compound 2 formed a 3D structure through hydrogen bonding (Fig. S6 and S7†).
Characterization
Energy dispersive spectrometer (EDS) spectrum analyzed the elemental compositions of compounds 1 and 2 (Fig. S8 and S9†). All compounds were synthesized with the addition of V, but V was detected only in compound 1. According to the mapping diagrams, the elements Co/Fe, C, W, O, N, and P were uniformly distributed in compound 1 (compound 2), with the exception of V. Their PXRD experimental spectra are unanimity with the practical spectra, which prove that the above compounds are pure phases (Fig. S10 and S11†). The Fourier-transform infrared (FT-IR) spectrogram of compounds 1 and 2 was acquired in the range of 4000 to 400 cm−1 (Fig. S12 and S13†). For example, the broad peak at 3418–3433 cm−1 corresponds to the O–H stretching vibration of free water molecules in compound 1. The peak at 2922 cm−1 indicates the asymmetric stretching vibration of the C–H bond in DAPSC. Peaks at 1627–1664 cm−1, 1445–1533 cm−1, and 1200–1272 cm−1 suggest the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CO, CC, and C–N bonds,35,36 confirming the successful coordination of DAPSC. The absorption near 1053 cm−1 is attributed to the unique P–O bond in the main structural unit of compound 1, while peaks around 955 cm−1 and 792–812 cm−1 correspond to MO (MW, V) and M–O–M bonds37,38 in heteropolytungstate. In compound 2, the peak near 1050 cm−1 is associated with the B–O bond in its primary structure. Bond valence sum (BVS) calculations for the two POM-based anions in compound 1 confirmed the presence of W5+ in the free Keggin-type heteropolytungstate (Tables S4 and S5†). X-ray photoelectron spectroscopy (XPS) was used to characterize the elemental composition of compounds 1 and 2 and their chemical valence states (Fig. 2 and S14†). The experimental XPS spectrum of compound 1 confirmed the presence of Co, W, V, P, C, O and N elements in compound 1 (Fig. 2a). The W 4f XPS spectrum of compound 1 forms four classically overlapping peaks (Fig. 2b). The peaks with binding energies around 35.3 and 37.3 eV are attributed to W6+, while the peaks at 34.6 and 36.7 eV can be assigned to W5+ in compound 1.39 As shown in Fig. 2c, the XPS of V 2p spectra indicate that the V4+ valence in compound 1. In the XPS spectrum of Co (Fig. 2d), the deconvolution peaks with binding energies of 782.6 eV and 798.7 eV are attributed to the 2p3/2 and 2p1/2 of the Co2+ oxidation state, respectively. The XPS spectrum of Fe 2p (Fig. S14b†) indicated that the 2p3/2 and 2p1/2 regions of Fe2+ were at 710.7 and 724.0 eV. In addition, the crystal structures and thermal stability of compounds 1 and 2 were explored by thermogravimetric analysis (TGA) conducted at 25 °C–850 °C under N2 atmosphere (Fig. S15 and S16†). Both compounds 1 and 2 showed two weight loss phases. For compound 1 as an example, the weight loss of compound 1 was 3.57% (calcd 3.28%) at 25–240 °C, attributed to the loss of free and coordinated water molecules. The weight loss of compound 1 during 240–330 °C is attributed to the gradual decomposition of DAPSC.
N, CO, CC, and C–N bonds,35,36 confirming the successful coordination of DAPSC. The absorption near 1053 cm−1 is attributed to the unique P–O bond in the main structural unit of compound 1, while peaks around 955 cm−1 and 792–812 cm−1 correspond to MO (MW, V) and M–O–M bonds37,38 in heteropolytungstate. In compound 2, the peak near 1050 cm−1 is associated with the B–O bond in its primary structure. Bond valence sum (BVS) calculations for the two POM-based anions in compound 1 confirmed the presence of W5+ in the free Keggin-type heteropolytungstate (Tables S4 and S5†). X-ray photoelectron spectroscopy (XPS) was used to characterize the elemental composition of compounds 1 and 2 and their chemical valence states (Fig. 2 and S14†). The experimental XPS spectrum of compound 1 confirmed the presence of Co, W, V, P, C, O and N elements in compound 1 (Fig. 2a). The W 4f XPS spectrum of compound 1 forms four classically overlapping peaks (Fig. 2b). The peaks with binding energies around 35.3 and 37.3 eV are attributed to W6+, while the peaks at 34.6 and 36.7 eV can be assigned to W5+ in compound 1.39 As shown in Fig. 2c, the XPS of V 2p spectra indicate that the V4+ valence in compound 1. In the XPS spectrum of Co (Fig. 2d), the deconvolution peaks with binding energies of 782.6 eV and 798.7 eV are attributed to the 2p3/2 and 2p1/2 of the Co2+ oxidation state, respectively. The XPS spectrum of Fe 2p (Fig. S14b†) indicated that the 2p3/2 and 2p1/2 regions of Fe2+ were at 710.7 and 724.0 eV. In addition, the crystal structures and thermal stability of compounds 1 and 2 were explored by thermogravimetric analysis (TGA) conducted at 25 °C–850 °C under N2 atmosphere (Fig. S15 and S16†). Both compounds 1 and 2 showed two weight loss phases. For compound 1 as an example, the weight loss of compound 1 was 3.57% (calcd 3.28%) at 25–240 °C, attributed to the loss of free and coordinated water molecules. The weight loss of compound 1 during 240–330 °C is attributed to the gradual decomposition of DAPSC.
 | ||
| Fig. 2 (a) Full scan XPS spectrum, (b) W 4f, (c) V 2p, and (d) Co 2p XPS spectra of compound 1. | ||
Photocatalytic CO2 reduction
These two compounds were evaluated through a series of photoelectronic experiments to assess their potential for use in photocatalytic CO2RR. The absorption of visible light by the photocatalyst has a significant impact on the performance. Consequently, compounds 1 and 2 were characterized by solid-state ultraviolet-visible absorption spectroscopy (UV-vis). The results (Fig. 3a) show that compounds 1 and 2 exhibit strong absorption in the visible region. Tauc plots were constructed using the Kubelka–Munk equation (αhν = C(hν − E)2) based on the measured UV-vis spectra, and the bandgap values (Eg) of compounds 1 and 2 were calculated to be 2.02 and 1.73 eV (Fig. 3b and S17†). Subsequently, Mott–Schottky curves were performed at 1000, 1500 and 2000 Hz to explore the energy-band structure and semiconducting properties of compounds 1 and 2. As shown in Fig. 3c and S18,† the positive slopes exhibited indicate that compounds 1 and 2 have n-type semiconductor characteristics. Their flat band potentials (EFB) were −1.36 and −1.09 V vs. Ag/AgCl. The conduction band potentials (ECB) of compounds 1 and 2 were converted to −1.16 and −0.89 V vs. NHE. Their valence band potentials (EVB) were calculated to be 0.86 and 0.84 V vs. NHE in accordance with the equation Eg = EVB − ECB. Then, the band structure diagram40,41 of compound 1 was derived from the above data (Fig. 3d). It can be seen that the lowest unoccupied molecular orbital (LUMO) positions of these two compounds are lower than those of photocatalytic CO2RR (CO2/CO = −0.53 V vs. NHE), which proves that both are theoretically possible for photocatalytic CO2RR. | ||
| Fig. 3 (a) UV-vis spectra of compounds 1 and 2. (b) Tauc plot of (αhν)2vs. hν for compound 1. (c) Mott–Schottky plot for compound 1. (d) Band-structure diagrams of compound 1. | ||
A saturated mixture solution (MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEOA = 4:1, v/v) was designed with [Ru(bpy)3]Cl2·6H2O as photosensitizer (PS) to investigate their catalytic activities in a photocatalytic CO2RR system. The reduction product CO was produced when the two compounds were used separately. As shown in Fig. 4a, compound 1 produced 199.6 μmol of CO, 53.7 μmol of H2, and 0.5 μmol of CH4 after 8 h. The CO, H2, and CH4 yields were 4.48, 5.9, and 0.5 μmol when compound 2 was used. The properties of compounds 1 and 2 are quite different; thus, several factors were considered. First, from the initial step of CO2RR, CO2 + H+ + e− → COOH*, it can determine the ability of the catalysts to activate CO2.42 Research shows that the compounds formed by Co with organic matter have a lower free energy barrier for activation of CO2 molecules than Fe.
:TEOA = 4:1, v/v) was designed with [Ru(bpy)3]Cl2·6H2O as photosensitizer (PS) to investigate their catalytic activities in a photocatalytic CO2RR system. The reduction product CO was produced when the two compounds were used separately. As shown in Fig. 4a, compound 1 produced 199.6 μmol of CO, 53.7 μmol of H2, and 0.5 μmol of CH4 after 8 h. The CO, H2, and CH4 yields were 4.48, 5.9, and 0.5 μmol when compound 2 was used. The properties of compounds 1 and 2 are quite different; thus, several factors were considered. First, from the initial step of CO2RR, CO2 + H+ + e− → COOH*, it can determine the ability of the catalysts to activate CO2.42 Research shows that the compounds formed by Co with organic matter have a lower free energy barrier for activation of CO2 molecules than Fe.
 | ||
| Fig. 4 (a) Yields of CO, H2, and CH4 for compounds 1 and 2. (b) Effect of the dose of compound 1 on the yield of CO. (c) Yield and rate of CO for compound 1 (3 mg). (d) Yield of CO after four cycles of compound 1. | ||
Second, CO can poison the metal active sites; thus, the adsorption barrier of TM for CO also affects the catalytic performance of the catalyst. It was found that CO molecules are more likely to be adsorbed at the TM center through the TM–C bond. The moderate interaction between Co and CO is weaker than Fe. Hence, the CO adsorption energy is lower, and the catalytic performance of Co-POM is improved. Considering the above experimental results, the optimal dosage of compound 1 was investigated in detail. As the catalyst dosage increased, the CO production rate increased and then decreased with time. The CO rate reached a maximum of 7081.4 μmol g−1 h−1 with 3 mg compound 1 (Fig. 4b). When it reached 5 mg, the CO increased to 172.5 μmol. Conversely, the rate dropped sharply to 4311.5 μmol g−1 h−1, which could only reach a little more than half of 3 mg. It was speculated that this phenomenon was attributed to the limitation of the electron transfer dynamics, resulting in too much of the catalyst not being fully utilized. When 3 mg of compound 1 was introduced, the yields of the reduction products CO can reach 169.9 μmol after 8 h. Additionally, the rate of CO reached its highest value of 8690.1 μmol g−1 h−1 and the turnover number (TON) of CO was 450.1 at 6 h (Fig. 4c). The speculation is that the consumption of PS results in not enough photogenerated electrons being transferred to the catalyst to maintain the catalytic reaction. Next, control experiments were performed on compound 1 to demonstrate the indispensability of each reaction component to the CO2RR. According to Fig. S19,† it was found that there was no CO production from the reaction system in the dark and almost no gas production when no PS or catalyst was added.43
Photocatalytic cycling test experiments were conducted to investigate the durability and recyclability to assess the practical application potential of compound 1 (Fig. 4d). The used catalysts were recovered from the reaction solution by centrifugation and dried at 80 °C for 12 h. Four cycles of experiments were carried out over 8 h, with a slight decrease possibly due to a small amount of catalyst loss in the recycling process. In addition, thermal filtration experiments were performed on compound 1, and it was found that the reaction system did not continue to produce CO after filtering it out (Fig. S20†).44 It was found that there was no significant change in their PXRD pattern and IR spectrogram after the cycling experiments (Fig. S21 and S22†), which proved that compound 1 had excellent structural stability and could be reused in at least four cycles.45
Mechanistic study
Photoluminescence (PL) quenching experiments were conducted to investigate the differences in the catalytic properties of the two compounds. The PL spectra of PS and the catalysts (Fig. 5a) revealed that compound 1 possesses a distinct fluorescence quenching effect. This result demonstrates that compound 1 exhibits higher charge transport efficiency than compound 2. Furthermore, compound 1 exhibits a higher transient photocurrent intensity, proving its effective inhibition of photogenerated electron–hole pairs recombination (Fig. 5b). The photoelectrochemical properties of compounds 1 and 2 were further investigated by electrochemical impedance spectroscopy (EIS). The Nyquist plots (Fig. 5c) denote that compound 1 has the smallest arc radius, proving that it has the least charge transfer resistance and higher photocatalytic efficiency.46 The summarized photoelectrochemical experiments have demonstrated the superior charge transfer rate and charge separation ability of compound 1.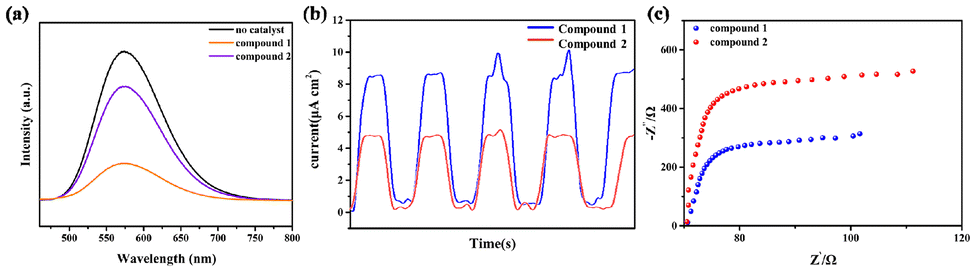 | ||
| Fig. 5 (a) [Ru(bpy)3]Cl2 in MeCN solutions containing catalyst compounds 1 and 2. (b) Transient photocurrent response of compounds 1 and 2. (c) EIS of compounds 1 and 2. | ||
The proposed rational mechanism for the photocatalysis47–49 of CO2RR by compound 1 is based on the described characterizations and experimental results (Fig. 6). Since the conduction band energy level of compound 1 is lower than that of PS, the photoelectrons in the excited state are spontaneously transferred from PS to compound 1. These electrons are transferred to Co-DAPSC, where CO2 is subsequently reduced. Meanwhile, H2O consumes the photoinduced holes and undergoes an oxygen evolution reaction. Finally, TEOA as a sacrificial agent eliminates the photogenerated holes in PS and maintains the catalytic reaction.
 | ||
| Fig. 6 Schematic of the electron transfer energy levels for the proposed mechanism of photocatalytic CO2 reduction over compound 1. | ||
Conclusions
In summary, we successfully synthesized two novel Keggin-type POM-based hybrid materials modified with two different transition metals and Schiff base ligands in this work. Various characteristic methods have demonstrated the presence of V atoms in the structure of compound 1. Photocatalytic CO2RR experiments showed that compound 1 exhibits exceptional photocatalytic activity, achieving a CO rate of 7081.4 μmol g−1 h−1 and selectivity of 83.8% after 8 hours. Cycle tests demonstrated that compound 1 exhibits good structural stability and maintains activity for at least four cycles. The introduction of V atoms into Keggin-type POM may enhance CO2 adsorption. Likewise, the use of DAPSC with the same structure as chlorophyll increased the visible light absorption of the compounds. This innovative approach provides a new perspective for the development of highly efficient photocatalytic CO2RR catalysts.Author contributions
Xiao-Yu Bai: writing – original draft, methodology, data curation, and conceptualization. Pin-Fang Yan: writing – original draft, validation, methodology, formal analysis, and data curation. Jiu-Lin Zhou: software, methodology, and investigation. Yu Lv: review and editing of the manuscript. Ji-Lei Wang: writing – original draft and supervision. Hua Mei: writing, reviewing and editing of the manuscript. Yan Xu: formal analysis, writing, review and editing of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (92161109).References
- L. F. Sanchez and D. I. Stern, Ecol Econ, 2016, 124, 17–24 CrossRef.
- Y. Liu, H. Y. Tang, A. Muhammad and G. Q. Huang, Greenhouse Gases:Sci. Technol., 2019, 9, 160–174 CrossRef CAS.
- N. Apergis and M. T. Majeed, Air Qual., Atmos. Health, 2021, 14, 1289–1300 CrossRef CAS.
- A. Gowrisankar, T. M. C. Priyanka, A. Saha, L. Rondoni, M. K. Hassan and S. Banerjee, Chaos, 2022, 32, 061104 CrossRef CAS PubMed.
- L. Jeffry, M. Y. Ong, S. Nomanbhay, M. Mofijur, M. Mubashir and P. L. Show, Fuel, 2021, 301, 121017 CrossRef CAS.
- A. Li, Q. Cao, G. Zhou, B. V. K. J. Schmidt, W. J. Zhu, X. T. Yuan, H. L. Huo, J. L. Gong and M. Antonietti, Angew. Chem., Int. Ed., 2019, 58, 14549–14555 CrossRef CAS.
- M. Y. Sun, B. H. Zhao, F. P. Chen, C. B. Liu, S. Y. Lu, Y. F. Yu and B. Zhang, Chem. Eng. J., 2021, 408, 127280 CrossRef CAS.
- H. L. Huo, T. Hu, Z. Q. Zhong, C. Zhan, C. X. Huang, Q. Ju, L. Zhang, F. Wu, E. Kan and A. Li, Chem. Sci., 2024, 15, 15134–15144 RSC.
- K. X. Tang, Z. Y. Zhang, D. X. Zhou, J. W. Xu, H. P. Cui, F. Li, X. D. Zhang, J. Q. Lei, L. Tang and N. Liu, Sep. Purif. Technol., 2025, 356, 129952 CrossRef CAS.
- J. Du, Z. L. Lang, Y. Y. Ma, H. Q. Tan, B. L. Liu, Y. H. Wang, Z. H. Kang and Y. G. Li, Chem. Sci., 2020, 11, 3007 RSC.
- Q. Fan, P. F. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Y. Sun, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS.
- Z. H. Wang, X. F. Wang, Z. Tan and X. Z. Song, Mater. Today Energy, 2021, 19, 100618 CrossRef CAS.
- G. Liu, Y. F. Chen, Y. L. Chen, Y. Q. Shi, M. Y. Zhang, G. D. Shen, P. F. Qi, J. K. Li, D. L. Ma, F. Yu and X. Q. Huang, Adv. Mater., 2023, 35, 2304716 CrossRef CAS PubMed.
- D. E. S. Marcano, M. A. Moussawi, A. V. Anyushin, S. Lentink, L. V. Meervelt, I. Ivanović-Burmazović and T. N. Parac-Vogt, Chem. Sci., 2022, 13, 2891–2899 RSC.
- P. Su, X. Zhu, S. M. Wilson, Y. N. Feng, H. Y. Samayoa-Oviedo, C. Sonnendecker, A. J. Smith, W. Zimmermann and J. Laskin, Chem. Sci., 2024, 15, 11825–11836 RSC.
- P. Mialane, C. Mellot-Draznieks, P. Gairola, M. Duguet, Y. Benseghir, O. Oms and A. Dolbecq, Chem. Soc. Rev., 2021, 50, 6152–6220 RSC.
- Y. S. Li, M. X. Liu and L. Chen, J. Mater. Chem. A, 2017, 5, 13757–13762 RSC.
- J. Zhou, W. C. Chen, C. Y. Sun, L. Han, C. Qin, M. M. Chen, X. L. Wang, E. B. Wang and Z. M. Su, ACS Appl. Mater. Interfaces, 2017, 9, 11689–11695 CrossRef CAS.
- C. L. Wang, Z. X. Sun, Y. Zheng and Y. H. Hu, J. Mater. Chem. A, 2019, 7, 865–887 RSC.
- Q. Y. Hu, S. S. Chen, T. Wågberg, H. S. Zhou, S. J. Li, Y. D. Li, Y. L. Tan, W. Q. Hu, Y. Ding and X. B. Han, Angew. Chem., Int. Ed., 2023, 62, e202303290 CrossRef CAS.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC.
- S. Shahebrahimi, E. Rafiee and K. Sadrjavadi, Appl. Organomet. Chem., 2019, 33, e5170 CrossRef CAS.
- M. Mahmoodi, E. Rafiee and S. Eavani, J. Mater. Sci.:Mater. Electron., 2020, 32, 1121–1138 CrossRef.
- A. R. Motz, M. C. Kuo, J. L. Horan, R. Yadav, S. Seifert, T. P. Pandey, S. Galioto, Y. Yang, N. V. Dale, S. J. Hamrock and A. M. Herring, Energy Environ. Sci., 2018, 11, 1499–1509 RSC.
- W. J. Bao, T. T. Huang, C. Z. Wang, S. Y. Ma, H. L. Guo, Y. Pan, Y. Q. Liu, C. G. Liu, D. F. Sun and Y. K. Lu, J. Catal., 2022, 413, 374–387 CrossRef CAS.
- C. A. Chen, Y. Liu and G. Y. Yang, Molecules, 2022, 27, 4295 CrossRef CAS.
- M. Malmir, M. M. Heravi, Z. Yekke-Ghasemi and M. Mirzaei, Sci. Rep., 2022, 12, 11573 CrossRef CAS PubMed.
- P. Sood, A. Joshi and M. Singh, Nanoscale Adv., 2022, 4, 5015–5020 RSC.
- M. Y. Wang, Y. Y. Yuan, Z. Q. Qi, J. Y. Chen, Z. G. Jiang and C. H. Zhan, Chem. Mater., 2022, 34, 10501–10508 CrossRef CAS.
- L. Lisnard, A. Dolbecq, P. Mialane, J. Marrot, E. Rivière, S. A. Borshch, S. Petit, V. Robert, C. Duboc, T. McCormac and F. Sécheresse, Dalton Trans., 2006, 5141–5148 RSC.
- X. Y. Dong, M. L. Wang, Y. Feng, J. Y. Zhang, Y. D. Cao, G. G. Gao, Y. X. Zhang and L. L. Fan, Dalton Trans., 2022, 51, 12876–12882 RSC.
- L. L. Fan, M. L. Wang, X. Y. Dong, G. G. Gao, J. Yu, H. Liu and X. Z. Liu, Chem. Eng. J., 2022, 449, 137819 CrossRef CAS.
- J. Dai, L. L. Yan, W. M. Yang, R. S. Li, Y. Dong and Y. J. Shen, Appl. Catal., B, 2025, 362, 124715 CrossRef CAS.
- J. Du, Y. Y. Ma, H. Q. Tan, Z. H. Kang and Y. G. Li, Chin. J. Catal., 2021, 42, 920–937 CrossRef CAS.
- G. H. Hu, H. Miao, H. Mei, S. Zhou and Y. Xu, Dalton Trans., 2016, 45, 7947–7951 RSC.
- J. H. Wang, H. Li, L. G. Gong, L. M. Dong, Y. H. Gu, M. J. Wang and B. H. Yang, Dalton Trans., 2025, 54, 3456–3466 RSC.
- J. K. Li, J. Dong, C. P. Wei, S. Yang, Y. N. Chi, Y. Q. Xu and C. W. Hu, Inorg. Chem., 2017, 56, 5748–5756 CrossRef CAS PubMed.
- Y. L. Li, Y. J. Zhang, P. F. Wu, C. T. Feng and G. L. Xue, Catalysts, 2018, 8, 639 CrossRef.
- X. Q. Huang, S. Liu, G. Liu, Y. W. Tan, C. R. Wang, Y. L. Zhang, Z. Li, H. W. Wang, Z. Zhou, G. D. Shen, Z. C. Xue and D. Sun, Appl. Catal., B, 2023, 323, 122134 CrossRef CAS.
- Z. M. Dong, Y. H. Zhu, J. L. Zhou, X. Y. Xiang, J. H. Pan, H. Mei and Y. Xu, Inorg. Chem., 2024, 63, 16791–16798 CrossRef CAS.
- R. L. Zhang, G. M. Zhao, J. Hu, P. Lu, S. L. Liu and X. B. Li, Int. J. Hydrogen Energy, 2024, 51, 633–642 CrossRef CAS.
- C. Wang, C. Y. Zhu, M. Zhang, Y. Geng, Y. G. Li and Z. M. Su, J. Mater. Chem. A, 2020, 8, 14807–14814 RSC.
- R. Paul, A. Boruah, R. Das, S. Chakraborty, K. Chahal, D. J. Deka, S. C. Peter, B. K. Mai and J. Mondal, Small, 2023, 20, 2305307 CrossRef.
- N. Das, R. Paul, S. Tomar, C. Biswas, S. Chakraborty and J. Mondal, Inorg. Chem., 2024, 63, 6092–6102 CrossRef CAS PubMed.
- B. Boro, R. Paul, H. L. Tan, Q. T. Trinh, J. Rabeah, C. C. Chang, C. W. Pao, W. Liu, N. T. Nguyen, B. K. Mai and J. Mondal, ACS Appl. Mater. Interfaces, 2023, 15, 21027–21039 CrossRef CAS.
- H. F. Li, H. Kong, J. Z. Guo, B. Li, S. S. Zhang, S. Z. Li, J. W. Zhao and L. Q. Bai, ACS Sustainable Chem. Eng., 2024, 12, 16396–16408 CrossRef CAS.
- H. Xu, Q. Chen, J. L. Wang, Q. Wang, C. Y. Jiao, P. F. Yan, H. Mei and Y. Xu, Inorg. Chem., 2023, 62, 18878–18886 CrossRef CAS.
- J. L. Zhou, X. Y. Xiang, L. T. Xu, J. L. Wang, S. M. Li, Y. T. Yu, H. Mei and Y. Xu, Dalton Trans., 2023, 52, 9465–9471 RSC.
- P. F. Yan, Q. Wang, J. L. Wang, H. Xu, J. B. Yang, Y. H. Zhu, Q. L. Chen, H. Mei and Y. Xu, Chem. Eng. J., 2025, 507, 160789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, details of crystallographic data, structural figures, PXRD patterns, IR spectra, and TGA characterization. CCDC 24492452449246. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01081k |
| This journal is © The Royal Society of Chemistry 2025 |