
Synchronous dimension-crystallization engineering enables highly efficient 2D/3D tin perovskite solar cells†
Ziyong
Kang
a,
Peng
Feng
b,
Kun
Wang
 *c,
Lu
Zhang
d,
Rui
Meng
a,
Yali
Chen
a,
Jiandong
Wu
a,
Feng
Yang
a,
Xuewen
Zhang
b,
Tianxiang
Li
a,
Jingzhi
Shang
b,
Yu
Tong
*a and
Hongqiang
Wang
*a
*c,
Lu
Zhang
d,
Rui
Meng
a,
Yali
Chen
a,
Jiandong
Wu
a,
Feng
Yang
a,
Xuewen
Zhang
b,
Tianxiang
Li
a,
Jingzhi
Shang
b,
Yu
Tong
*a and
Hongqiang
Wang
*a
aState Key Laboratory of Solidification Processing, Center for Nano Energy Materials, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi’an 710072, China. E-mail: yutong@nwpu.edu.cn; hongqiang.wang@nwpu.edu.cn
bFrontier Science Center for Flexible Electronics, Xi’an Institute of Flexible Electronics, Northwestern Polytechnical University, 127 Youyi West Road, Xi’an, Shaanxi 710072, P. R. China
cSchool of microelectronics, Northwestern Polytechnical University, Xi’an 710072, China. E-mail: kunwang@nwpu.edu.cn
dKey Laboratory of Applied Surface and Colloid Chemistry Ministry of Education, Shaanxi Key Laboratory for Advanced Energy Devices, Shaanxi, Engineering Lab for Advanced Energy Technology, School of Materials Science Engineering Shaanxi Normal University, Xi’an 710119, P. R. China
First published on 15th March 2025
Abstract
Tin perovskite films with two-dimensional/three-dimensional (2D/3D) heterostructures show promise for high performance lead-free perovskite solar cells (PSCs); however, they face challenges due to the undesirable carrier transport caused by intrinsic multi-quantum wells, and susceptible crystallization kinetics caused by the introduction of organic spacer cations. We herein propose and validate a strategy that could simultaneously address these challenges based on synchronous dimension-crystallization engineering in 2D/3D tin perovskite films. Different from the conventional dimension engineering that relies on precise n-phase control, the employed 4-guanidinium benzoate hydrochloride (GBAC) in the present work has an unforeseen desorption effect between phenylethylamine (PEA) and the perovskite, which leads to a direct transition from the 2D to the 3D phase. It is also found that introducing GBAC results in the formation of elongated organic–inorganic hybrid chains, which improves the crystallization process of the films by accelerating both nucleation and growth rates. By virtue of these merits, the resulting tin PSCs achieve a champion power conversion efficiency of 15.02%, together with exceptional long-term stability with 87% remaining after 4000 h and 80% after 400 h under working at the maximum power point.
Broader contextRealizing efficient, low-cost and eco-friendly solar cells is crucial for harvesting solar energy and promoting the sustainable development of society. Tin perovskites with 2D/3D heterostructures show huge promise for high-performance solar cells, while the high proportion of multi-quantum wells leads to quantum confinement and dielectric confinement effects. Additionally, the undesirable crystallization process results in a higher density of defects in 2D/3D tin films. Manipulating crystallization and modulating dimensions have thus become crucial for enhancing the efficiency and stability of tin PSCs. This research introduces GBAC molecules to induce a desorption effect between PEA and the perovskite, promoting the transition from the n = 2 phase to the n = ∞ phase. Meanwhile, the formation of an elongated DMF–GBAC–SnI2 chain increases both the nucleation and crystal growth rates, resulting in a film with high uniformity and compactness. Benefitting from the dimension-crystallization engineering which optimizes the film quality and phase distribution, a champion power conversion efficiency of 15.02% is achieved, with excellent long-term and operational stability. We anticipate that the dimension-crystallization engineering strategy employed will provide guidelines for the development of efficient and stable tin PSCs to promote their further applications. |
Introduction
Metal halide perovskites are considered as one of the most promising materials for realizing highly efficient solar cells to harvest solar energy. Over the past decade, the power conversion efficiency (PCE) of lead perovskite solar cells (PSCs) has remarkably increased from 3.8% to recent 27%.1–9 However, the toxicity of water-soluble lead ions has become a critical challenge that cannot be ignored in the commercialization of perovskite photovoltaics.10–14 Consequently, numerous eco-friendly alternatives have been developed for fabricating lead-free PSCs. Among them, tin perovskites have gained considerable attention due to their low toxicity and exceptional optoelectronic properties, including an ideal optical bandgap, high absorption coefficient, and excellent carrier mobility.15–20Although tin perovskites display outstanding optical and electronic properties, the efficiency and stability of tin PSCs have been lagging far behind their lead counterparts. One important reason is that Sn2+ is more susceptible to being oxidized to Sn4+, leading to numerous traps or defects in the polycrystalline film.21–23 Specifically, Sn4+/Sn2+ has a significantly smaller standard reduction potential (+0.15 V) than Pb4+/Pb2+ (+1.67 V), causing easy oxidation of Sn2+, even in the presence of a trace amount of oxygen. The oxidation of Sn2+ makes the tin perovskite film undergo severe p-type self-doping and causes energy level misalignment and non-radiative recombination, which bring huge challenges for charge carrier transport and extraction.24,25 The other reason is that the crystallization kinetics of tin perovskites are less controllable due to the higher Lewis acidity of Sn2+ compared to Pb2+, along with the stronger Sn–I bond (1.69 eV vs. 1.64 eV for Pb–I), which may aggravate the difficulty to regulate the nucleation and growth rates, producing tin perovskite films with an inferior morphology and numerous defects.26–31 To address these issues, tremendous strategies have been explored, such as additive engineering and post-treatment techniques.32,33 Among all the strategies for optimizing tin PSCs, introducing large organic cations to form 2D/3D structures has proven to be an effective way that can regulate crystallization and suppress Sn2+ oxidation.34–39 However, the undesired 2D/3D distribution and the resultant multiple quantum-well heterostructures could bring difficulty for the transport of electrons and holes.40–42 Meanwhile, inadequate crystallization can cause undesirable film quality with abundant surface voids and large gaps between grains, severely constraining the performance of 2D/3D tin PSCs.31,43,44
To address the issue of multi-quantum wells in 2D/3D tin PSCs, efforts are primarily focused on regulating the orientation or thickness of 2D phases.45–48 For example, Yuan and co-workers introduced ammonium chloride (NH4Cl) into quasi-2D AVA2FAn−1SnnI3n+1 perovskites to yield highly vertically oriented tin perovskite films.49 Ning's group used phenethylammonium thiocyanate (PEASCN) to modulate the crystallization of 2D/3D tin perovskites, promoting the formation of highly oriented quasi-2D structures that enhance carrier diffusion lengths and lifetimes.50 The above dimensional modulation strategy causes the transformation from a thinner 2D phase (n = 1, n refers to the number of [SnI6]4− octahedral monolayers) to a thicker 2D phase (n ≤ 5), while numerous quantum wells due to varying n phases may still hinder efficient charge transport. In addition to the dimensional modulation, substantial studies have focused on regulating the perovskite crystallization process and reveal that well-crystalized quasi-2D perovskite films generally result from fast nucleation and slow growth rates.51,52 For instance, Meng et al. used 3-aminopyrrolidine dihydroiodate (APDI2) to adjust the zeta potential (φd) and realized a fast nucleation rate during film formation, resulting in significantly improved tin perovskite films with a pinhole-free morphology and excellent crystallinity.53 Hao et al. utilized the coordination interaction between formamidinium acetate (FAAc) and Sn2+ to slow down the crystallization process of tin halide perovskite films, which leads to an improvement in the films’ crystallinity.54 The above efforts have confirmed the significance of dimensional modulation and crystallization optimization in achieving efficient 2D/3D tin PSCs, yet both approaches still demand additional enhancement. Therefore, it is vital to investigate refined dimensional engineering and crystallization control techniques, as well as explore the highly desirable synchronous dimension-crystallization engineering of 2D/3D tin perovskite films, thereby further improving the device performance.
The present work proposes an efficient strategy of simultaneously modulating the dimension and improving the crystallization of 2D/3D tin-based perovskite films. Specifically, we use GBAC to induce the desorption between PEA and the perovskite, contributing to a transition from the 2D (n = 2) to the 3D (n = ∞) phase, which reduces the quantum confinement effect and improves charge carrier transport. Additionally, we discover that GBAC can interact with both the solute and solvent to form elongated hybrid chains (DMF–GBAC–SnI2), boosting nucleation and growth processes during film formation, thereby also improving the quality of tin perovskite films. Consequently, the inverted 2D/3D tin PSCs show significantly improved PCEs from 11.77 to 15.02%, with a remarkable enhancement in the open-circuit voltage (VOC) from 0.78 to 0.88 V, short-circuit current density (JSC) from 21.78 to 23.20 mA cm−2, and fill factor (FF) from 69.32 to 73.72%. Moreover, both the shelf and operational stability of the tin PSCs are dramatically enhanced, which is beneficial for promoting their future applications.
Results and discussion
Dimension modulation and mechanism study
The possible effects of the GBAC additive on modulating 2D phases and crystallization of 2D/3D tin perovskite films are schematically shown in Scheme 1. To verify the modulation of the 2D/3D phases, UV-visible absorption spectra were measured. Fig. S1 (ESI†) clearly exhibits the absorption feature of 3D phases in both the control and GBAC optimized samples, and the bandgap remains consistent at 1.44 eV for both samples. However, no 2D phase peaks are observed, which is likely due to the relatively low proportion of the 2D phases compared to the 3D phase. Then we raised the PEA content to 50 mol%, and the UV-visible absorption spectra are displayed in Fig. 1(a). The 2D phase shows absorption peaks at ∼560 nm (n = 1) and ∼660 nm (n = 2) in control films, but GBAC-treated films display notably weaker absorption—especially at ∼560 nm—and a slight increase in 3D phase absorption. Meanwhile, the X-ray diffraction (XRD) results show consistency with the absorption spectra (Fig. 1(b)), where the intensity of the peaks corresponding to the 2D phase is markedly diminished, while the intensity of the 3D diffraction peaks is enhanced in the GBAC-containing films, further confirming the effects of GBAC on regulating 2D phases. | ||
| Scheme 1 Schematic illustration of synchronous dimension-crystallization (D&C) engineering. | ||
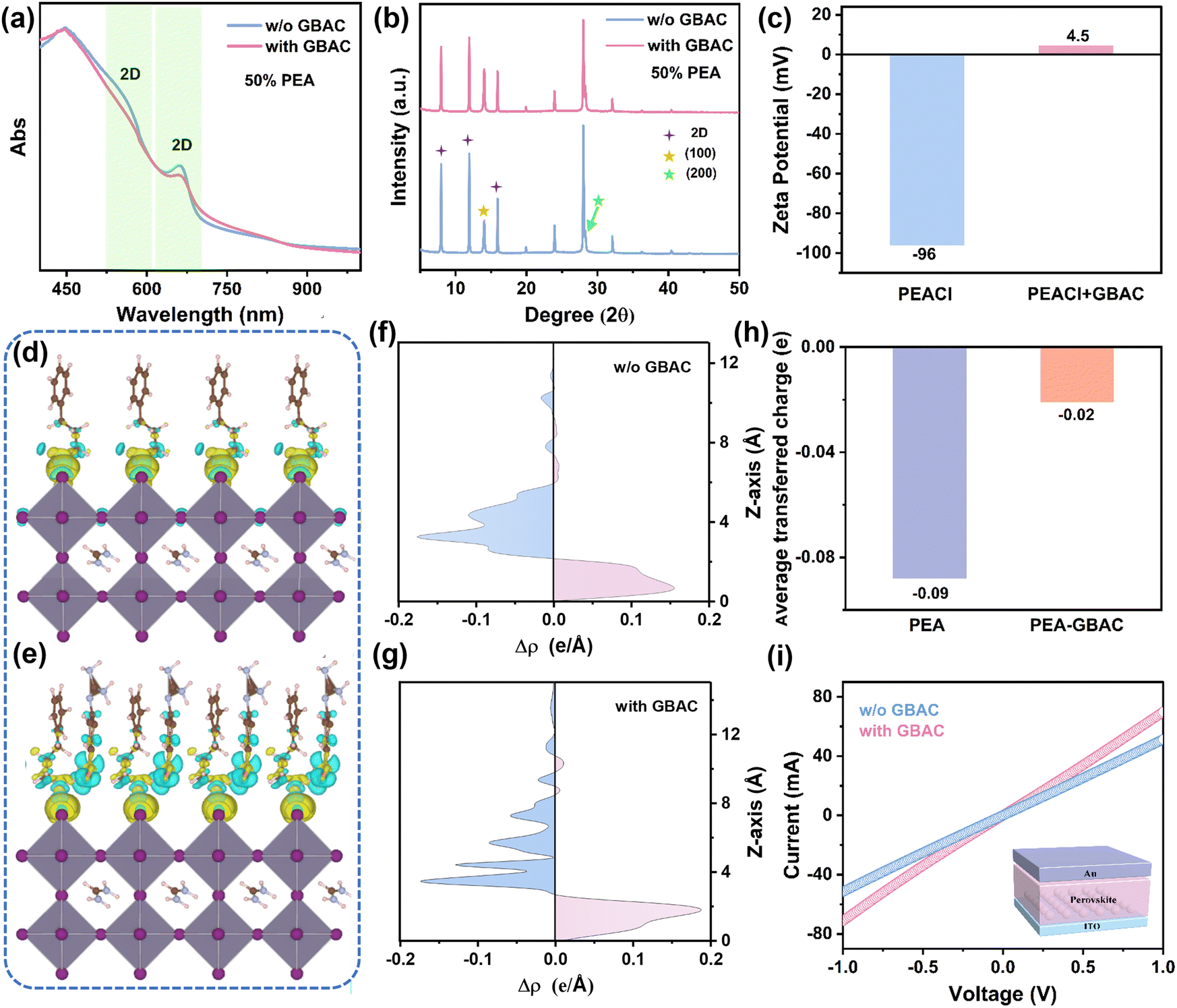 | ||
| Fig. 1 (a) UV-vis absorption spectra of tin perovskite films without and with GBAC. (b) XRD patterns of films without and with GBAC. (c) Zeta potentials of PEA solutions with and without GBAC. The electron density distribution of FASnI3 interacting with (d) PEA and (e) PEA–GBAC. (f) and (g) The charge transfer differences along the Z-axis, and (h) the average transferred charge. (i) The current–voltage (I–V) curves of devices without and with GBAC. | ||
Since the 2D phases are induced by the PEA molecules, it is essential to explore potential interactions between GBAC and PEA molecules in the solution. We first conducted zeta potential (φd) measurements on the PEA–GBAC solution. As shown in Fig. 1(c), after introducing 1 mol% GBAC in the 15 mol% PEA solution, the average zeta potential of PEA–GBAC colloidal particles dramatically changes from −96 mV to 4.5 mV. As proposed by the classical Stern model theory,55 when colloidal particles have a high absolute zeta potential, they are more likely to repel one another. Conversely, when the absolute zeta potential is low, the energy barrier to aggregation decreases, promoting particle clustering. Plausibly, the zeta potential change can be ascribed to the strong chemisorption between the GBAC and PEA. We propose that GBAC may induce an unexpected desorption effect between the organic PEA and inorganic layers ([SnI6]4− octahedra). To verify the changes in adsorption energy (Eads), we conducted density functional theory (DFT) calculations to determine the Eads of PEA and PEA–GBAC on the surface of FASnI3. Fig. S2 (ESI†) reveals that PEA exhibits stronger affinity for the perovskite surface (Eads = −2.434 eV) than the PEA–GBAC complex (Eads = −1.123 eV), attributed to the interaction of its amino groups. Fig. 1(d) and (e) show the calculated electron density distribution to describe the interaction between the organic spacer cation and FASnI3. According to electron density calculations, Fig. 1(f) and (g) show the charge transfer differences along the Z-axis between the inorganic and organic layers. The average charge transfer between PEA and FASnI3 is about 4.5 times that of PEA–GBAC and FASnI3 (Fig. 1(h)), which is due to the charge transfer between PEA and GBAC. If PEA–GBAC acts as a spacer cation to form a continuous 2D layer, the 2D phase would not be reduced, and charge transfer would be suppressed, which contradicts the results of the XRD and absorption results. Additionally, Fig. S3 (ESI†) shows no charge transfer between GBAC and the perovskite, indicating that GBAC does not form a 2D structure. The above results collectively suggest that GBAC–PEA effectively inhibits PEA from adsorbing onto the perovskite surface, preventing the formation of 2D phases. This reduction in quantum confinement significantly facilitates charge carrier transport. To validate this, we measured the current–voltage (I–V) characteristic curves (Fig. 1(i)). Conductivity can be calculated using supporting equation (S1) (ESI†). The I–V curve becomes steeper upon introducing GBAC, indicating that GBAC increases the conductivity of perovskite films (rising from 2.49 × 10−3 mS cm−1 to 3.39 × 10−3 mS cm−1), which can be ascribed to the optimized 2D/3D phases as discussed above.
To gain further insight into the impacts of GBAC on the phase distribution of the 2D/3D tin perovskite films, grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were carried out. The characterization was conducted at various incident angles to obtain information at different depths for the control and GBAC treated perovskite films (Fig. 2(a) and (b)). It is observed that a diffraction spot at qr = 0.32 Å−1 (2D phases, n = 2) exists at different depths for both control and GBAC-containing films, which indicates that the 2D phase is distributed throughout the perovskite film. The more intensive scattering peak at qr = 1 Å−1 is attributed to the (100) planes of the 3D (n = ∞) bulk perovskite with an orthorhombic phase, in agreement with previous reports.56 Since the diffraction spot reflections are predominantly oriented along the qz direction, the crystallites orient with their planes parallel to the substrate, implying that the inorganic slabs are oriented horizontally (Scheme 1). To see the scattering intensity evolution of 2D and 3D structures, the azimuthal integration patterns are shown in Fig. 2(c). The decreased intensity ratio between 2D and 3D along with increasing the incident angle for both the control and GBAC-containing films indicates the preference for 2D structures at the film surface, which is beneficial for in situ growth of a protection layer to improve the film stability. Notably, the GBAC-containing perovskite film exhibits a reduced scattering intensity at qr = 0.32 Å−1 as the incident angle increases from 0.2° to 1.5°, whereas the control sample shows an opposite changing trend. This implies the bulk perovskite film with GBAC has a reduced proportion of the 2D phase compared to the control film. Furthermore, the increased intensity of the scattering peak at qr = 1 Å−1 in the film with GBAC compared to the control film suggests a transformation from the 2D phase to the 3D phase. This transformation could help reduce the number of 2D/3D heterostructures, which in turn enhances charge transport in the tin perovskite film.
 | ||
| Fig. 2 GIWAXS patterns at different depths for tin perovskite films (a) without and (b) with GBAC. (c) Azimuthal integration plots of (a) and (b). (d) Pseudocolor transient absorption (TA) spectra and (e) kinetics of GSB2D and GSB3D from TA spectra for the perovskite films without and with GBAC. | ||
To gain deeper insights into the ultrafast charge carrier dynamics within the 2D/3D tin perovskite films, we conducted femtosecond transient absorption spectroscopy (fs-TA) measurements for the control and GBAC-containing films deposited on glass (Fig. 2(d)). The contour plots of the TA spectra for both the control and GBAC-containing films show two distinct ground-state bleaching (GSB) peaks at approximately ∼660 nm and ∼835 nm, corresponding to the signals of the 2D and 3D phases, respectively. The GSB intensity reflects the photogenerated carrier density, allowing 2D to 3D charge transfer monitoring via quenching rates.57 Compared to the GSB signal intensity in the control film, the GBAC film exhibits a reduced intensity for the GSB2D signal but a markedly increased intensity for the GSB3D signal. Such evolution is ascribed to the increased 3D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2D phase ratio in the films, as well as the promoted energy transfer from 2D to 3D phases. Additionally, compared to the control film, the GBAC film shows weaker 2D and stronger 3D GSB peak intensities at the same delay times (Fig. S4, ESI†), which aligns well with the above results from the contour plots of the TA spectra. To examine the energy transfer and carrier recombination dynamics in the films, we plotted the decay curves of the GSB2D and GSB3D signals for the control and GBSA-containing films (Fig. 2(e)). By fitting the decay curves, the decay times for the GSB2D (τ2D) and the GSB3D (τ3D) signals of the control perovskite film are 208.63 ps and 166.50 ps, respectively, while the τ2D and τ3D values change to 158.4 ps and 180.96 ps after GBAC modification. The decreased τ2D infers an accelerated energy or charge transfer from the 2D phase to the 3D phase, and the increased τ3D value implies an optimized carrier diffusion length in the film. These results further support that GBAC induces desorption between PEA and the perovskite, which facilitates the transformation from 2D to 3D phases and promotes charge transport in tin perovskite films.
:2D phase ratio in the films, as well as the promoted energy transfer from 2D to 3D phases. Additionally, compared to the control film, the GBAC film shows weaker 2D and stronger 3D GSB peak intensities at the same delay times (Fig. S4, ESI†), which aligns well with the above results from the contour plots of the TA spectra. To examine the energy transfer and carrier recombination dynamics in the films, we plotted the decay curves of the GSB2D and GSB3D signals for the control and GBSA-containing films (Fig. 2(e)). By fitting the decay curves, the decay times for the GSB2D (τ2D) and the GSB3D (τ3D) signals of the control perovskite film are 208.63 ps and 166.50 ps, respectively, while the τ2D and τ3D values change to 158.4 ps and 180.96 ps after GBAC modification. The decreased τ2D infers an accelerated energy or charge transfer from the 2D phase to the 3D phase, and the increased τ3D value implies an optimized carrier diffusion length in the film. These results further support that GBAC induces desorption between PEA and the perovskite, which facilitates the transformation from 2D to 3D phases and promotes charge transport in tin perovskite films.
Modulating crystallization through organic–inorganic hybrid chains
The GBAC molecule with multiple functional groups may interact with the uncoordinated Sn2+ as well as the organic and inorganic defects via the Lewis acid–base interaction, thus significantly affecting the nucleation and growth processes of the perovskite film. The molecular structure and electrostatic potential (ESP) are shown in Fig. S5 (ESI†). The electron-deficient regions (positive charge) are primarily concentrated in the guanidinium group, and the electron-rich regions (negative charge) are primarily located in the carboxyl group.58 The binding energies between GBAC and the perovskite precursor components were estimated using DFT calculations, as illustrated in Fig. 3(a). It can be seen in Fig. 3(b) that there is stronger ionic bonding between GBAC, SnI2, and DMF (−1.444 eV) compared to the Lewis acid–base adducts formed with DMF (−0.524 eV), FA (−0.844 eV), and Sn2+ (−0.981 eV), indicating that the elongated organic–inorganic hybrid chain (DMF–GBAC–SnI2) is readily formed in the precursor. To investigate the strong coordination between the perovskite and the GBAC molecules, X-ray photoelectron spectroscopy (XPS) measurements were carried out to analyze the surface chemistry of the perovskite (Fig. 3(c) and Fig. S6, ESI†). It can be clearly seen that the binding energies of N 1s, Sn 3d and I 3d signals exhibit shifts compared with the control perovskite film (Table S1, ESI†), indicating the formation of hydrogen bonds and electrostatic interaction between GBAC and the component of the perovskite. Meanwhile, we observe a shift of the C–O signals to a higher binding energy level in the O 1s spectrum, suggesting that the GBAC interacts with DMF to form a hydrogen bond. We then conducted Fourier-transform infrared (FTIR) spectroscopy measurements to investigate the interactions between GBAC and the perovskite precursor components in solution (Fig. S7, ESI†). The results show that the N–H stretching peak shifts from 3379.4 cm−1 in GBAC to 3574.1 cm−1 in the GBAC–SnI2 complex; and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak shifts from 1651.4 cm−1 in DMF to 1646.4 cm−1 in DMF–GBAC–SnI2. These results suggest that the guanidium groups and carboxylic acid in GBAC are prone to form electrostatic interactions with Sn2+ and hydrogen bonds with DMF, further validating the existence of the GBAC linker in the precursor. Additionally, the N–H stretching peak shifts from 3343.4 cm−1 in FAI to 3371.4 cm−1 in the GBAC–FAI complex, and the CO stretching peak shifts from 1698.7 cm−1 in GBAC to 1693.8 cm−1 in GBAC–FAI, indicating the interaction between GBAC and FA+. Moreover, the proton nuclear magnetic resonance (1HNMR) analysis was conducted, as shown in Fig. S8 (ESI†). Upon mixing GBAC with SnI2, a noticeable shift in the 1H peak of the guanidinium group is observed. The 1H peak also shows a shift when mixing FAI with GBAC, suggesting the chemical interactions between GBAC and both Sn2+ and FA+. These results indicate that GBAC can effectively reduce uncoordinated Sn2+ and positively/negatively charged cation vacancies.
O stretching peak shifts from 1651.4 cm−1 in DMF to 1646.4 cm−1 in DMF–GBAC–SnI2. These results suggest that the guanidium groups and carboxylic acid in GBAC are prone to form electrostatic interactions with Sn2+ and hydrogen bonds with DMF, further validating the existence of the GBAC linker in the precursor. Additionally, the N–H stretching peak shifts from 3343.4 cm−1 in FAI to 3371.4 cm−1 in the GBAC–FAI complex, and the CO stretching peak shifts from 1698.7 cm−1 in GBAC to 1693.8 cm−1 in GBAC–FAI, indicating the interaction between GBAC and FA+. Moreover, the proton nuclear magnetic resonance (1HNMR) analysis was conducted, as shown in Fig. S8 (ESI†). Upon mixing GBAC with SnI2, a noticeable shift in the 1H peak of the guanidinium group is observed. The 1H peak also shows a shift when mixing FAI with GBAC, suggesting the chemical interactions between GBAC and both Sn2+ and FA+. These results indicate that GBAC can effectively reduce uncoordinated Sn2+ and positively/negatively charged cation vacancies.
 | ||
| Fig. 3 (a) Distributions of the charge density from DFT and (b) binding energies of DMF, FAI, SnI2, and DMF–SnI2 with GBAC, respectively. (c) XPS spectra of Sn, N, and O for films without and with GBAC. (d) Average cluster size and (e) zeta potentials of perovskite precursor solutions without and with GBAC. (f) XRD patterns for tin perovskite films without and with GBAC. | ||
The above results imply that GBAC interacts with the solute and solvent in the precursor solution to form an elongated organic–inorganic hybrid chain, which will increase the nanocluster size. To assess the impact of the GBAC additive on the colloidal properties of the tin perovskite precursor solution, the dynamic light scattering (DLS) results are presented in Fig. 3(d). The average cluster size increases from 800 nm to 2000 nm after the introduction of GBAC. Fig. S9 (ESI†) illustrates the contact angle measurements of the perovskite precursor on poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS). The contact angle for the original perovskite precursor on the PEDOT: PSS film is ∼11.8°, whereas it reduced to ∼4.2° for the precursor containing GBAC. According to the relationship between the free energy required for heterogeneous nucleation:59
 | (1) |
The classical Stern model theory posits that φd is a key factor governing the nucleation dynamics of colloidal particles. We employ φd to investigate the influence of GBAC on the nucleation dynamics of the modified perovskite. As shown in Fig. 3(e), after introducing 1 mol% GBAC in the pristine 0.9 M FASnI3 precursor solution (to avoid the impact of GBAC–PEA adsorption on results), the φd value of perovskite colloidal particles significantly changed from −40 mV to 22 mV. According to Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, the relationship between the critical concentration required for nucleation (Cmin) and the φd value of colloidal particles is given by the following equation:60
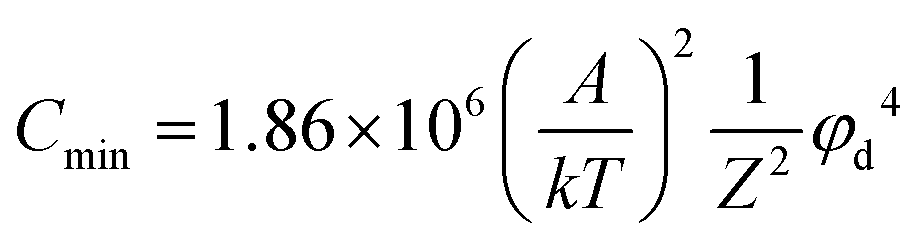 | (2) |
 of the perovskite precursor. Supersaturation of the solution increases the nucleation rate (Jn), as described using the following equation:61
of the perovskite precursor. Supersaturation of the solution increases the nucleation rate (Jn), as described using the following equation:61| Jn = Dexp(−B(lnS)−2) | (3) |
To further explore the effects of GBAC on the formation kinetics of tin perovskite films, we used in situ photoluminescence (PL) spectroscopy to track the film evolution. During the spin coating process (Fig. 4(a)), in stage I (approximately 21–25 s), the antisolvent drop (t = 21 s) triggers the immediate emergence of an intense and broad PL peak centered at around 801 nm (Fig. S12a and b, ESI†). It is reported that the PL emission arises from the instantly formed nanocrystals, with a polydisperse size distribution of luminescing moieties responsible for the broad full width at half maximum (FWHM) of the PL emissions. Within the next second, the peak position remains constant, and the FWHM of the PL peak shows significant narrowing from t = 22 s to t = 25 s. This could be attributed to the cluster coalescence of the nanoparticles, which have highly correlated surface and interfacial energies. The high surface and interfacial energies are possibly strong driving forces for the cluster coalescence of the nucleated species due to their high surface-to-volume ratios. Coalescence results in size homogenization of the particles, which results in the narrowing of the PL spectra. Notably, the film containing GBAC shows stronger and broader PL peak compared to the control film, implying that GBAC accelerates the nucleation rate and produces nuclei, consistent with the DLS and φd results. Furthermore, the PL peak remains centered at 804 nm with no significant reduction in intensity (Fig. 4(b)), which might be attributed to the increased number of nuclei. In stage II (approximately 25–40 s), the nuclei further grow into larger crystals and a gradual redshift in the PL emission peak is observed, implying the increase of the nuclei size (Fig. S12c and d, ESI†). Meanwhile, the PL intensity also gradually increases, and by fitting the linear region of the PL intensity with the Avrami model, a crystallization growth rate constant k1 can be obtained. The k1 value of the GBAC-containing film is higher than that of the control film, indicating that GBAC accelerates the growth rate in spin-coating.
 | ||
| Fig. 4 (a) In situ photoluminescence spectra of tin perovskite films without and with a GBAC additive during spin-coating and annealing processes. (b) Evolution of the PL intensity during different film formation stages for perovskite films without and with GBAC. (c) XRD patterns of perovskite films without and with GBAC at different annealing times. (d) SEM images of perovskite films without and with GBAC at different annealing times. (e) KPFM images of perovskite films without and with GBAC and the corresponding potential distribution. (f) PL intensity mapping images of the perovskite films without and with GBAC and the corresponding statistical distribution PL wavelength. | ||
In the subsequent annealing process, a second bright PL response emerges between 785 and 865 nm in stage III (approximately 0–8 s), with its center at around 834 nm (Fig. S12b, ESI†). This signature is attributed to the co-existence of disparate nanocrystallite sizes with high radiative efficiency. The non-Gaussian peak shape comes from a superposition of luminescence signals with disparate intensity contributions. Beyond 2 s, there is a slow decrease in the intensity of the solvent-complex (Fig. 4(b)) up to 8 s, which is attributed to the removal of the solvent complex from the deeper parts of the thin film, and a longer annealing time is required for complete solvent removal. The crystallization in stage III of both films is almost the same, which could be explained by that the quick applied high temperature dominates the crystallization in this stage. In stage IV (>8 s), the small crystallites further grow and merge into larger ones to reduce the overall grain boundaries, resulting in an increased PL intensity again in both perovskite films with a stable PL emission peak position at ∼840 nm. The initial slow rise in the PL intensity suggests that both crystal reconstruction and solvent evaporation occur in this phase. Compared to the control film, the GBAC-containing film shows a higher growth rate k2 and a longer solvent evaporation time, which can be attributed to more thorough crystal reconstruction induced by the DMF–GBAC–SnI2 intermediate. Subsequently, the rapid increase in PL intensity marks the transition into the Ostwald-ripening stage. The parameter k3 represents the growth rate during Ostwald ripening, and the k3 value of the GBAC-modified film is higher than that of the control. The findings substantiate that accelerated nucleation and crystal growth can help enhance the film quality, as schematically illustrated in Scheme 1.
To further compare the formation process of the tin perovskite film without and with GBAC, the XRD measurements were carried out on samples with different annealing times (Fig. 4(c)). The XRD peak intensity of the control film decreases initially before rising again, whereas the diffraction peak intensity of the GBAC-containing film steadily increases with prolonged annealing time. To investigate this change, we then compared the SEM images for the control and GBAC-containing films with and without annealing. As displayed in Fig. 4(d), before annealing, the GBAC-containing film shows a much smoother surface compared to the control film. After annealing for 60 s, the GBAC-containing film still maintains a compact film morphology, whereas the control film shows obvious pinholes, which is likely due to lower crystallization, and a higher ratio of 2D phases. Kelvin probe force microscopy (KPFM) characterization was conducted to measure the potential distribution of the perovskite films. As can be seen in Fig. 4(e), the GBAC-containing film exhibits a higher and more uniform surface potential compared to the control film, indicating that the perovskite films with GBAC have a more n-type surface and fewer surface defects, which is in line with the SEM and AFM results. A more n-type perovskite surface induces a downward bending of the energy band, which could benefit the transfer of electrons to the ETL. To investigate the electronic band structure of the Sn-based perovskite films, we conducted ultraviolet photoelectron spectroscopy (UPS) measurements (Fig. S13, ESI†). The work functions (WF) of the control and GBAC perovskite films are −4.85 eV and −4.77 eV, respectively, while the valence band maxima (VBM) are −5.09 eV and −5.06 eV, respectively (Fig. S14, ESI†). This indicates that the conduction band minimum (CBM) shifts upward by 0.03 eV relative to the Fermi level (EF), suggesting that the GBAC treatment generates a more n-type perovskite surface by inhibiting the oxidation of Sn2+ (Fig. S15, ESI†), which aligns with the KPFM findings.
To better understand how the GBAC affects the optoelectronic properties of the tin perovskite films, we performed a series of optical characterization studies. The PL mapping and time-resolved photoluminescence (TRPL) were measured, and the results are given in Fig. 4(f), Fig. S16 and Table S2 (ESI†). A remarkably elevated emission peak intensity and a more concentrated emission peak distribution are observed. Furthermore, compared to the control film with an average carrier lifetime (τave) of 21.65 ns, the GBAC-modified perovskite films exhibit a dramatically longer τave value of 41.48 ns. The photoluminescence quantum yield (PLQY) of control and GBAC-containing perovskite films (0.9 M) was measured and found to be 0.48% and 0.76% (Fig. S17, ESI†). These results indicate that the introduction of GBAC effectively suppresses non-radiative recombination. Additionally, we investigated the defect state density for the control and GBAC-containing perovskites using the space-charge-limited current (SCLC) method. The electron-only devices with the ITO/SnO2/perovskite/PCBM/Ag structure and hole-only devices with the ITO/PEDOT:PSS/perovskite/spiro-OMeTAD/Ag structure were fabricated for current–voltage measurements (Fig. S18, ESI†). The trap density (Nt) is calculated from the onset voltage of the trap-filled limit region (VTFL). The electron trap density reduces from 1.99 × 1015 to 1.37 × 1015 cm−3 in devices with GBAC, while the hole trap density decreases from 4.10 × 1015 cm−3 to 2.79 × 1015 cm−3 (Table S3, ESI†). These results further confirm the favorable effects of GBAC on reducing the defect density and suppressing the nonradiative recombination, which would contribute to improving the performance of tin PSCs.
Photovoltaic performance of 2D/3D tin PSCs
To evaluate the device performance, tin PSCs were fabricated with a device configuration of ITO/PEDOT: PSS/perovskite/PCBM/BCP/Ag (Fig. 5(a)). The J–V curves of the champion devices for the control and GBAC-containing ones are shown in Fig. 5(b). The control tin PSC exhibits a PCE of 11.77% with a VOC of 0.78 V, an FF of 69.32%, and a JSC of 21.78 mA cm−2. In contrast, the GBAC-containing PSCs display a dramatically enhanced PCE of 15.02%, with a VOC value of 0.88 V, an FF of 73.72%, and a JSC value of 23.20 mA cm−2. Table S4 (ESI†) shows the statistical distribution of photovoltaic parameters for both the control and GBAC modified tin PSCs. Clearly, all parameters of the GBAC-based devices are higher than those of the control devices, with particularly significant improvements in VOC and JSC. This performance makes our device one of the best-performed tin PSCs reported so far (Table S5, ESI†). The enhancement in device performance can be attributed to the optimized 2D/3D phase distribution and film quality as discussed above. Additionally, the external quantum efficiency (EQE) measurements, as shown in Fig. 5(c), indicate that the integrated photocurrent from the EQE spectrum closely aligns with the values obtained from J–V measurements. | ||
| Fig. 5 (a) Schematic illustration of the device structure for tin PSCs. (b) J–V curves of the champion tin PSCs without and with GBAC. (c) EQE spectra of the corresponding tin PSCs. (d) Open-circuit voltage depends on the light intensity. (e) Dark current and (f) EIS results of tin PSCs without and with GBAC. (g) Long-term shelf stability and (h) operational stability of tin PSCs without and with GBAC (in a N2 environment). | ||
The charge transport and carrier recombination dynamics of the devices were further investigated. Transient photovoltage decay (TPV) and transient photocurrent decay (TPC) measurements were conducted, with the longer photovoltage decay lifetime and shorter photocurrent decay lifetime in GBAC devices, suggesting the suppressed charge recombination and improved charge extraction (Fig. S19, ESI†). The plots of VOC dependence on light intensity are analyzed, as shown in Fig. 5(d). The slope of VOCversus the natural logarithm of light intensity of the control device (1.60kBT/q) decreases after introducing GBAC (1.27kBT/q), indicating that trap-induced non-radiative recombination of carriers is suppressed in the device with GBAC. Moreover, Fig. 5(e) presents the dark current measurement results, demonstrating a remarkably reduced dark current of the GBAC-modified device compared with the control one, which can be attributed to the improved tin perovskite film quality as well as the interfacial contact, as further corroborated by the electrochemical impedance spectroscopy (EIS) results in Fig. 5(f).
Finally, to investigate the effects of GBAC on the stability of tin PSCs, we monitored the evolution of both devices stored in a nitrogen atmosphere. As shown in Fig. 5(g), the PCE of the control device drops to below 80% after 2100 hours of storage. In contrast, the device containing GBAC retains 87% of its initial PCE after 4000 hours of storage. Subsequently, we evaluated the operational stability of both devices by tracking the maximum power point (MPP) performance under continuous light illumination. As depicted in Fig. 5(h), the PCE of the control device falls below 80% of its initial value after ∼200 hours of illumination, while the device optimized by GBAC maintains 80% of its PCE even after 400 hours of illumination. The significantly enhanced long-term shelf and operational stability can be attributed to the optimized 2D/3D phase distribution and improved film quality due to GBAC incorporation.
Conclusion
In summary, we utilize a multifunctional molecule GBAC to regulate the 2D/3D phases and crystallization of tin perovskite films, achieving high-efficiency and stable tin PSCs. Experimental results and theoretical calculations reveal that the GBAC can induce a desorption effect between PEA and the perovskite, effectively achieving a direct transition from the n = 2 phase to the n = ∞ phase. Meanwhile, elongated organic–inorganic hybrid chains (DMF–GBAC–SnI2) form between the GBAC and the precursor components, promoting the nucleation and growth processes during film formation. As a result, the reduction of the quantum confinement effect in the film, along with improvements in the film morphology and crystallinity, leads to more efficient charge transport and significantly reduced defects. Consequently, the 2D/3D tin PSCs show remarkable enhancement in PCE from 11.78% to 15.02%, along with notably improved device stability. This work demonstrates a simple and effective way to simultaneously improve the morphology and promote charge transport in 2D/3D tin perovskite films, which can provide key guidelines for designing efficient and stable tin PSCs.Author contributions
Z. Kang conceived the idea. Z. Kang, P. Feng, L. Zhang, R. Meng, Y. Chen, J. Wu, F. Yang, X. Zhang, T. Li, and J. Shang conducted the experiments and analyses. Z. Kang, Y. Tong, K. Wang, and H. Wang wrote the manuscript. All authors contributed to the writing of the paper.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI† files.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 52402128), the Natural Science Basic Research Program of Shaanxi (program no. 2024JC-YBQN-0443), the Basic Research Programs of Taicang 2024 (TC2024JC02) and the Natural Science Foundation of Chongqing, China (CSTB2022NSCQ-MSX0926 and CSTB2022NSCQ-MSX1335). The authors acknowledge Prof. Peter Müller-Buschbaum for helping with GIWAXS measurement and analysis.References
- Best Research-Cell Efficiency Chart (NREL), https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.pdf (accessed: Feb 2025).
- H. Wang, S. Su, Y. Chen, M. Ren, S. Wang, Y. Wang, C. Zhu, Y. Miao, C. Ouyang and Y. Zhao, Nature, 2024, 634, 1091–1095 CAS.
- Y. Yang, H. Chen, C. Liu, J. Xu, C. Huang, C. D. Malliakas, H. Wan, A. S. Bati, Z. Wang and R. P. J. S. Reynolds, Science, 2024, 386, 898–902 CAS.
- J. Chen, X. Wang, T. Wang, J. Li, H. Y. Chia, H. Liang, S. Xi, S. Liu, X. Guo, R. Guo, Z. Jia, X. Yin, Q. Zhou, Y. Wang, Z. Shi, H. Zhou, D. Lai, M. Zhang, Z. Xing, W. R. Leow, W. Yan and Y. Hou, Nat. Energy, 2024, 1–10 Search PubMed.
- Y. Zheng, Y. Li, R. Zhuang, X. Wu, C. Tian, A. Sun, C. Chen, Y. Guo, Y. Hua, K. Meng, K. Wu and C.-C. Chen, Energy Environ. Sci., 2024, 17, 1153–1162 CAS.
- Z. Ni, S. Xu, H. Jiao, H. Gu, C. Fei and J. Huang, Sci. Adv., 2022, 8, eabq8345 CAS.
- D. Gao, B. Li, Q. Liu, C. Zhang, Z. Yu, S. Li, J. Gong, L. Qian, F. Vanin and K. J. S. Schutt, Science, 2024, 386, 187–192 CAS.
- J. Du, J. Chen, B. Ouyang, A. Sun, C. Tian, R. Zhuang, C. Chen, S. Liu, Q. Chen, Z. Li, X. Wu, J. Cai, Y. Zhao, R. Li, T. Xue, T. Cen, K. Zhao and C.-C. Chen, Energy Environ. Sci., 2025 10.1039/d4ee05849f.
- J. Jin, Z. Zhang, S. Zou, F. Cao, Y. Huang, Y. Jiang, Z. Gao, Y. Xu, J. Qu, X. Wang, C. Chen, C. Xiao, S. Ren and D. Zhao, Adv. Energy Mater., 2024, 2403718 CrossRef.
- X. Jiang, H. Li, Q. Zhou, Q. Wei, M. Wei, L. Jiang, Z. Wang, Z. Peng, F. Wang and Z. Zang, J. Am. Chem. Soc., 2021, 143, 10970–10976 CrossRef CAS PubMed.
- J. Chen, J. Luo, E. Hou, P. Song, Y. Li, C. Sun, W. Feng, S. Cheng, H. Zhang, L. Xie, C. Tian and Z. Wei, Nat. Photonics, 2024, 18, 464–470 CrossRef CAS.
- M. Lyu, J. H. Yun, P. Chen, M. Hao and L. Wang, Adv. Energy Mater., 2017, 7, 1602512 CrossRef.
- M. Yang, Y. Tan, G. Yang, X. Chang, T. Tian, W. G. Li, Y. Fang, J. Shen, S. Yang and W. Q. Wu, Angew. Chem., Int. Ed., 2025, 64, e202415966 CrossRef CAS PubMed.
- X. Xiao, N. Xu, X. Tian, T. Zhang, B. Wang, X. Wang, Y. Xian, C. Lu, X. Ou, Y. Yan, L. Sun, F. You and F. Gao, Nature, 2025, 638, 670–675 CrossRef CAS.
- S. Wang, C. Wu, T. Wu, H. Yao, Y. Hua and F. J. A. E. L. Hao, ACS Energy Lett., 2024, 9, 1168–1175 CrossRef CAS.
- S. Zou, S. Ren, Y. Jiang, Y. Huang, W. Wang, C. Wang, C. Chen, X. Hao, L. Wu, J. Zhang and D. Zhao, Energy Environ. Mater., 2023, 6, e12465 CrossRef CAS.
- B. Chang, L. Wang, H. Li, L. Pan, Y. Wu, Z. Liu, Y.-N. Zhang, E. Guo and L. J. A. E. L. Yin, ACS Energy Lett., 2024, 9, 363–372 CrossRef CAS.
- P. Chen, D. He, X. Huang, C. Zhang and L. Wang, ACS Nano, 2024, 18, 67–88 CrossRef CAS.
- Z. Kang, Y. Tong, K. Wang, Y. Chen, P. Yan, G. Pan, P. Müller-Buschbaum, L. Zhang, Y. Yang, J. Wu, H. Xie, S. Liu and H. Wang, ACS Mater. Lett., 2023, 6, 1–9 Search PubMed.
- M. Yang, T. Tian, Y. Fang, W.-G. Li, G. Liu, W. Feng, M. Xu and W.-Q. J. N. S. Wu, Nat. Sustain., 2023, 6, 1455–1464 CrossRef.
- Z. Zhang, X. Tian, C. Wang, J. Jin, Y. Jiang, Q. Zhou, J. Zhu, J. Xu, R. He, Y. Huang, S. Ren, C. Chen, P. Gao, R. Long and D. Zhao, Energy Environ. Sci., 2022, 15, 5274–5283 RSC.
- L. Lanzetta, T. Webb, N. Zibouche, X. Liang, D. Ding, G. Min, R. J. E. Westbrook, B. Gaggio, T. J. Macdonald, M. S. Islam and S. A. Haque, Nat. Commun., 2021, 12, 2853 CrossRef CAS PubMed.
- Y. Chen, Y. Tong, F. Yang, T. Li, W. Li, H. Qi, Z. Kang, H. Wang and K. Wang, Nano Lett., 2024, 24, 5460–5466 CrossRef CAS PubMed.
- M. Ma, X. Jiang, Z. Zang, X. Wen, W. Zhou, H. Wu, S. Peng, Y. Liu, H. Li and D. J. A. F. M. Yu, Adv. Funct. Mater., 2024, 34, 2407095 CrossRef CAS.
- J. Zhu, Y. Xu, Y. Luo, J. Luo, R. He, C. Wang, Y. Wang, K. Wei, Z. Yi and Z. J. S. A. Gao, Sci. Adv., 2024, 10, eadl2063 CrossRef CAS PubMed.
- X. Ding, M. Yan, C. Chen, M. Zhai, H. Wang, Y. Tian, L. Wang, L. Sun and M. J. A. C. Cheng, Angew. Chem., Int. Ed., 2024, 136, e202317676 CrossRef.
- K. Dey, D. Ghosh, M. Pilot, S. R. Pering, B. Roose, P. Deswal, S. P. Senanayak, P. J. Cameron, M. S. Islam and S. D. Stranks, Energy Environ. Sci., 2024, 17, 760–769 RSC.
- J. Zhou, S. Fu, S. Zhou, L. Huang, C. Wang, H. Guan, D. Pu, H. Cui, C. Wang, T. Wang, W. Meng, G. Fang and W. Ke, Nat. Commun., 2024, 15, 2324 CrossRef CAS PubMed.
- J. Zeng, J. Wang, J. Wang, J. Li, J. Chen, F. Wei, J. Zhang, W. Song and X. Fan, ACS Nano, 2024, 18, 31390–31400 CrossRef CAS PubMed.
- G. Liu, G. Yang, W. Feng, H. Li, M. Yang, Y. Zhong, X. Jiang and W. Q. J. A. M. Wu, Adv. Mater., 2024, 36, 2405860 CrossRef CAS PubMed.
- H. Rao, Y. Su, G. Liu, H. Zhou, J. Yang, W. Sheng, Y. Zhong, L. Tan and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202306712 CrossRef CAS PubMed.
- C. Gao, X. Wang, Q. Yang, X. Wang, C. Gao and X. Liu, Adv. Mater. Technol., 2024, 9, 2302029 CrossRef CAS.
- Y. Chen, H. Qi, K. Wang, Z. Kang, G. Pan, C. R. Everett, P. Müller-Buschbaum, Y. Tong and H. Wang, Small Methods, 2023, 8, 202300029 Search PubMed.
- Y. Wang, R. Lin, C. Liu, X. Wang, C. Chosy, Y. Haruta, A. D. Bui, M. Li, H. Sun, X. Zheng, H. Luo, P. Wu, H. Gao, W. Sun, Y. Nie, H. Zhu, K. Zhou, H. T. Nguyen, X. Luo, L. Li, C. Xiao, M. I. Saidaminov, S. D. Stranks, L. Zhang and H. Tan, Nature, 2024, 635, 867–873 CrossRef PubMed.
- Z. Kang, K. Wang, L. Zhang, Y. Yang, J. Wu, Y. Tong, P. Yan, Y. Chen, H. Qi, K. Sun, P. Müller-Buschbaum, X. Zhang, J. Shang and H. Wang, Small, 2024, 202402028 Search PubMed.
- G. Li, Z. Su, M. Li, F. Yang, M. H. Aldamasy, J. Pascual, F. Yang, H. Liu, W. Zuo, D. Di Girolamo, Z. Iqbal, G. Nasti, A. Dallmann, X. Gao, Z. Wang, M. Saliba and A. Abate, Adv. Energy Mater., 2021, 11, 2101539 CrossRef CAS.
- B. B. Yu, Z. Chen, Y. Zhu, Y. Wang, B. Han, G. Chen, X. Zhang, Z. Du and Z. He, Adv. Mater., 2021, 33, 2102055 CrossRef CAS PubMed.
- G. Feng, H. L. Loi, T. Wang, W. Deng, Z. Guan, Q. Wei, J. He, M. Li, C. S. Lee, J. Wang, Q. Zhang and F. Yan, Angew. Chem., Int. Ed., 2024, 137, e202413584 CrossRef.
- C. Liu, Y. Yang, H. Chen, I. Spanopoulos, A. S. R. Bati, I. W. Gilley, J. Chen, A. Maxwell, B. Vishal, R. P. Reynolds, T. E. Wiggins, Z. Wang, C. Huang, J. Fletcher, Y. Liu, L. X. Chen, S. De Wolf, B. Chen, D. Zheng, T. J. Marks, A. Facchetti, E. H. Sargent and M. G. Kanatzidis, Nature, 2024, 633, 359–364 CrossRef CAS PubMed.
- Z. Guo, Y. Liang, D. Ni, L. Li, S. Liu, Y. Zhang, Q. Chen, Q. Zhang, Q. Wang and H. Zhou, Adv. Mater., 2023, 35, e2302711 CrossRef PubMed.
- H. Chen, S. Teale, B. Chen, Y. Hou, L. Grater, T. Zhu, K. Bertens, S. M. Park, H. R. Atapattu, Y. Gao, M. Wei, A. K. Johnston, Q. Zhou, K. Xu, D. Yu, C. Han, T. Cui, E. H. Jung, C. Zhou, W. Zhou, A. H. Proppe, S. Hoogland, F. Laquai, T. Filleter, K. R. Graham, Z. Ning and E. H. Sargent, Nat. Photonics, 2022, 16, 352–358 CrossRef CAS.
- H. Min, N. Wang, N. Chen, Y. Tong, Y. Wang, J. Wang, J. Liu, S. Wang, X. Wu, P. Yang, H. Shi, C. Zhuo, Q. Chen, J. Li, D. Zhang, X. Lu, C. Zhu, Q. Peng, L. Zhu, J. Chang, W. Huang and J. Wang, Nat. Nanotechnol., 2024, 19, 632–637 CrossRef CAS PubMed.
- K. Zhang, Z. Wang, G. Wang, J. Wang, Y. Li, W. Qian, S. Zheng, S. Xiao and S. Yang, Nat. Commun., 2020, 11, 1006 CrossRef CAS PubMed.
- P. Liu, X. Li, T. Cai, W. Xing, N. Yang, H. Arandiyan, Z. Shao, S. Wang and S. Liu, Nano-Micro Lett., 2024, 17, 35 CrossRef PubMed.
- Q. Cao, P. Li, W. Chen, S. Zang, L. Han, Y. Zhang and Y. J. N. T. Song, Nano Today, 2022, 43, 101394 CrossRef CAS.
- T. Wang, L. Bi, L. Yang, Z. Zeng, X. Ji, Z. Hu, S.-W. Tsang, H.-L. Yip, Q. Fu, A. K. Y. Jen and Y. Liu, J. Am. Chem. Soc., 2024, 146, 7555–7564 Search PubMed.
- A. Zanetta, V. Larini, Vikram, F. Toniolo, B. Vishal, K. A. Elmestekawy, J. Du, A. Scardina, F. Faini, G. Pica, V. Pirota, M. Pitaro, S. Marras, C. Ding, B. K. Yildirim, M. Babics, E. Ugur, E. Aydin, C.-Q. Ma, F. Doria, M. A. Loi, M. De Bastiani, L. M. Herz, G. Portale, S. De Wolf, M. S. Islam and G. Grancini, Nat. Commun., 2024, 15, 9069 CrossRef CAS PubMed.
- R. Azmi, E. Ugur, A. Seitkhan, F. Aljamaan, A. S. Subbiah, J. Liu, G. T. Harrison, M. I. Nugraha, M. K. Eswaran and M. J. S. Babics, Science, 2022, 376, 73–77 CrossRef CAS PubMed.
- H. Xu, Y. Jiang, T. He, S. Li, H. Wang, Y. Chen, M. Yuan and J. Chen, Adv. Funct. Mater., 2019, 29, 1807696 CrossRef CAS.
- H. Li, Z. Zang, Q. Wei, X. Jiang, M. Ma, Z. Xing, J. Wang, D. Yu, F. Wang, W. Zhou, K. S. Wong, P. C. Y. Chow, Y. Zhou and Z. Ning, Sci. China: Chem., 2023, 66, 459–465 CrossRef CAS.
- Z. Xing, X. Meng, D. Li, T. Hu, X. Hu and Y. Chen, Sci. Bull., 2022, 67, 561–564 Search PubMed.
- K. Yan, M. Long, T. Zhang, Z. Wei, H. Chen, S. Yang and J. Xu, J. Am. Chem. Soc., 2015, 137, 4460–4468 CrossRef CAS PubMed.
- J. Wang, J. Huang, M. Abdel-Shakour, T. Liu, X. Wang, Y. Pan, L. Wang, E. Cui, J. S. Hu, S. Yang and X. Meng, Angew. Chem., Int. Ed., 2024, 63, e202306712 Search PubMed.
- S. Wang, H. Yao, W. Zhu, C. Wu, Z. Tang, J. Liu, L. Ding and F. Hao, Adv. Funct. Mater., 2023, 33, 2215041 CrossRef CAS.
- J. Wu, Chem. Rev., 2022, 122, 10821–10859 CAS.
- F. Wang, X. Jiang, H. Chen, Y. Shang, H. Liu, J. Wei, W. Zhou, H. He, W. Liu and Z. J. J. Ning, Joule, 2018, 2, 2732–2743 CAS.
- G. Liu, X. Jiang, W. Feng, G. Yang, X. Chen, Z. Ning and W. Q. Wu, Angew. Chem., Int. Ed., 2023, 62, e202305551 CAS.
- F. Li, X. Deng, Z. Shi, S. Wu, Z. Zeng, D. Wang, Y. Li, F. Qi, Z. Zhang, Z. Yang, S.-H. Jang, F. R. Lin, S. W. Tsang, X.-K. Chen and A. K. Y. Jen, Nat. Photonics, 2023, 17, 478–484 CAS.
- X. Chen, F. Yang, L. Yuan, S. Huang, H. Gu, X. Wu, Y. Shen, Y. Chen, N. Li, H.-J. Egelhaaf, C. J. Brabec, R. Zhang, F. Gao, Y. Li and Y. Li, Joule, 2024, 8, 2265–2282 CAS.
- H. Al-Hashim, A. Kasha, W. Abdallah and B. Sauerer, Energy Fuels, 2018, 32, 1644–1656 CAS.
- J.-P. Hsu, S.-H. Lin and S. Tseng, J. Phys. Chem. B, 2004, 108, 4495–4500 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee06142j |
| This journal is © The Royal Society of Chemistry 2025 |