
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conquering the impossible: mechanochemistry as a tool for tackling coordination chemistry challenges
Huanxin
Zhang
,
Nathan
Davison
* and
Erli
Lu
 *
*
School of Chemistry, University of Birmingham. Edgbaston, Birmingham, B15 2TT, UK. E-mail: n.davison@bham.ac.uk; e.lu@birmingham.ac.uk; Web: https://www.birmingham.ac.uk/staff/profiles/chemistry/lu-erli Web: https://x.com/erli_lu
First published on 19th March 2025
Abstract
Organic solvents are ubiquitously used in synthetic coordination chemistry, but these solution-based synthetic methods have severe drawbacks, such as the incapability of isolating highly reactive species which react with the solvent molecules, less-controllable heterogeneous reactions when using insoluble substrate(s), and unfavourable solvent molecule coordination. In recent years, coordination chemists have started employing mechanochemical methods (e.g., ball mill) to overcome these drawbacks. Herein, we offer our perspective about how mechanochemical methods have enabled coordination chemists to achieve what would otherwise be impossible. It should be noted that, this perspective should not be treated as a comprehensive review of mechanochemistry in coordination chemistry, instead, a “stepping stone” aiming at inspiring further endeavours. Also, due to the research background of the authors, the selection of examples herein may appear biased towards main-group chemistry, which we feel necessary to remind our readers.
1. Introduction
Organic solvents are used in almost all chemical reactions – there is no exception for coordination chemistry. Yet, it is not unusual that the organic solvents used give rise to undesired outcomes, such as product decomposition and formation of unfavoured by- or side-products. For example, tetrahydrofuran (THF), one of the most-used organic solvents, is well-known to undergo deprotonation and C–O bond cleavage, induced by strong organometallic Brønsted bases and/or nucleophiles.1 While the ethereal solvents (including THF) are not inert towards C–H and C–O activations,2 arene solvents, especially benzene and toluene, are also known to be activated via multiple pathways, such as toluene C(sp3)–H deprotonation,3 arene reduction,4 and transition-metal mediated dearomatisation activation.5 Even the most inert aliphatic hydrocarbon solvents, such as the linear hexanes6 and cyclo-hexane,7 were reported to undergo C–H activation with highly reactive organometallic species.8,9 On one hand, these reactivities are a blessing as they open up new territories in small molecule activation. On the other hand, they are a curse which may hamper the isolation and characterisation of targeted highly reactive coordination complexes.Another inherent challenge of the solution-based synthetic methods is the heterogeneity caused by poorly soluble reactants. A classic example is reductions using potassium graphite (KC8), Na/K alloy, K or Na mirror, or sodium over sodium chloride (Na/NaCl). The heterogeneity presents kinetic challenges: on the solution-solid surface (where the reaction takes place), the reactant![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :reductant stoichiometric ratio could differ substantially from the ideal ratio. To overcome the challenge, very fine highly dispersed reductants and vigorous stirring are usually needed, such as demonstrated in divalent lanthanide chemistry.10 A similar challenge presents in reactions involving zero-valent metals (as reductant in most cases), which are insoluble in organic solvents. The metal vapour deposition (MVD) method was used to tackle the challenge, such as in the synthesis of the classic samarocenes [(C5Me5)2Sm(THF)2]11/[(C5Me4Et)2Sm(THF)2],11 their non-solvated analogue [(C5Me5)2Sm],12 and large-scale synthesis of dysprosium and neodymium diiodides.13
:reductant stoichiometric ratio could differ substantially from the ideal ratio. To overcome the challenge, very fine highly dispersed reductants and vigorous stirring are usually needed, such as demonstrated in divalent lanthanide chemistry.10 A similar challenge presents in reactions involving zero-valent metals (as reductant in most cases), which are insoluble in organic solvents. The metal vapour deposition (MVD) method was used to tackle the challenge, such as in the synthesis of the classic samarocenes [(C5Me5)2Sm(THF)2]11/[(C5Me4Et)2Sm(THF)2],11 their non-solvated analogue [(C5Me5)2Sm],12 and large-scale synthesis of dysprosium and neodymium diiodides.13
A solvent-free synthetic method would be ideal for tackling these solvent-related challenges. Mechanochemistry, defined by IUPAC as “chemical reaction[s] that [are] induced by the direct absorption of mechanical energy”14 and regarded as the “fourth way” of synthetic chemistry alongside solvothermal, electrochemical, and photochemical methods, offers a prime candidate.15 Moreover, the unique energy transfer mode of mechanochemistry (e.g. impact forces for milling), opens exciting opportunities for the exploration of entirely new complexes, which are unable to be synthesised via traditional solution-state methods.
In this perspective, from a viewpoint of synthetic coordination chemists, we will categorise and analyse the unique advantages offered by mechanochemical methods, which have unlocked new horizons in synthetic coordination chemistry.16 In other words, instead of a comprehensive review of the applications of mechanochemical methods in synthetic coordination chemistry, this perspective focuses on the cases which “achieve the impossible”, i.e., cases where mechanochemistry succeeds, while other methods fail. We believe that the capability of “achieving the impossible” is a unique value of mechanochemistry, beside the sustainability merits of being solvent-free and energy-efficient.
2. Isolating highly reactive species
2.1 Low-valent main-group complexes
The synthesis, structure, and reactivity studies of low-valent complexes, especially those of main-group elements (s-17 and p-blocks18), lanthanide,19 and actinide20 metals have emerged as a hot topic of modern chemistry, not only because of their roles in expanding our knowledge boundaries, but also because of their potential in inert small molecule and chemical bond activations.21 Due to their rich electron density in the valence shell orbitals, these low-valent complexes usually feature high reactivity, which often lead to reactions with solvents such as tetrahydrofuran (THF) and benzene.22A classic example is magnesium: Mg-containing species have attracted interest in extraterrestrial, organic synthesis and biological systems.23 Mg(I) radical species have long been postulated to exist as fleeting species and reaction intermediates, which are important for understanding life processes in Earth and extraterrestrial24 environments and also play crucial roles in the formation of Grignard reagents.25 They have been spectroscopically detected in harsh conditions, for example, ˙MgF as a gaseous molecule is generated at 2300 °C and trapped in an inert-gas gas matrix26 and ˙MgCH3 was produced by a laser ablation/photodissociation technique.27 Calculations have showed that Mg2Cl2 and other Mg–Mg compounds disproportionate exothermically into solid Mg(0) and the corresponding Mg(II) compound, which further indicated the difficulty of isolation of a Mg(I) monomeric radical species.28
The first stable and isolable Mg(I) complexes were Mg(I)–Mg(I) dimers, isolated in 2007 by Jones and Stasch,29 which are thermodynamically stabilised by forming a Mg(I)–Mg(I) bond, and kinetically protected by bulky Priso or Nacnac ligands (Priso = [(DippN)2CNiPr2], (Dipp = 2,6-diisopropylphenyl); Nacnac = [ArNC(Me)2CH]). Since then, this field has emerged as one of the most fruitful areas of main group chemistry. In the 2010s and early 2020s, Mg(I) dimers have developed significantly from a niche area into a library of soluble, selective, stoichiometric and safe reducing agents for organic and inorganic synthesis.30 Despite the flourishing status quo, almost all the reported Mg(I) complexes are dimers: the Mg(I)–Mg(I) intermetallic bond plays an underpinning role in their stabilisation.
The defining structural feature of these Mg(I) dimers is the aforementioned 2-electron Mg(I)–Mg(I) σ-bond. In contrast, a hypothetical monomeric Mg(I) complex would feature an unpaired Mg-based single electron, effectively creating a Mg(I) radical. Such a monomeric radical would likely exhibit distinct, and potentially enhanced, reactivity compared to the dimeric Mg(I)–Mg(I) complexes.31 Consequently, the pursuit of such monomeric Mg(I) radicals has been a longstanding challenge in the main-group chemistry community for over a decade.
One possible strategy to isolate a Mg(I) monomeric complex is increasing the steric repulsion between the two Mg(I) centres in the dimer until the Mg(I)–Mg(I) bond homolyses and generates the desired Mg(I) monomer. Great efforts have been devoted to this route: several research groups adopted sterically bulky ligands, and external Lewis bases to increase the steric repulsion.22 But these efforts only led to limited successes. In most of the cases, Mg(I)–Mg(I) bond elongation was observed upon increasing the steric repulsion. The Mg(I)–Mg(I) bond is fairly “elastic” and can adopt a wide range of bond lengths (2.808 Å (ref. 32) to 3.196 Å (ref. 33)) without cleavage. Increasing the steric repulsion by increasing the steric bulkiness of the ligands can also lead to a change of the ligand coordination mode and hapticity (e.g., for β-diketiminateo ligands, from κ2-N, N to κ1-N) to release the steric repulsion, instead of forming the desired Mg(I) monomer.34
In 2021, a bulky bis-fluorenyl flanked ligand, namely hydrindacene, was introduced into Mg chemistry by the Tan group. Reducing the Mg(II)-hydrindacene precursor led to a ligand-reduced product, instead of the desired Mg(I) monomer.35 It is worth noting that the hydrindacene ligand family has succeeded in isolating a number of low-valent mononuclear p-block complexes,36 emphasising the challenge of isolating a Mg(I) monomer.
The first evidence of Mg(I)–Mg(I) homolysis emerged in 2021 by using the photochemical method. The Jones group used blue/UV light to activate Mg(I) dimers containing bulky ligands ([{(ArNacnac)Mg}2] (ArNacnac = [HC-(MeCNAr)2]; Ar = Dipp or 2,4,6-tricyclohexylphenyl (TCHP))) to generate transient Mg(I) radical intermediates, which underwent fast reactions with aromatic solvents, such as benzene, toluene and xylenes37 (Fig. 1). The presence of Mg(I) radical intermediates was supported by both computational studies and their observed enhanced reactivity compared with the corresponding Mg(I) dimers, such as [{(ArNacnac)Mg}2], (Ar = Dipp, mesityl, 2,6-xylyl), reported earlier.32 The dimeric complexes do not react with these arenes without photolysis. The exceedingly reactive nature of the putative Mg(I) monomer intermediates emphasises the need for a solvent-free or solvent-less synthetic protocol for isolating the fleeting Mg(I) monomers.
 | ||
| Fig. 1 Photo-activated Mg(I)–Mg(I) homolysis.37 | ||
Traditionally, in coordination/organometallic chemistry, solvent-free synthesis is achieved by the chemical vapour deposition (CVD) method.38,39 However, mechanochemistry offers an easier-to-operate and more scalable solvent-free approach to tackle the challenge.
In 2022, the Harder group adopted mechanochemistry to synthesise the first isolated and well-characterised Mg(I) monomeric radical complex, which was stabilised by a cyclic (alkyl)(amino)carbene (cAAC) ligand.40 A Mg(II) iodide precursor 3 (in Fig. 2) was ball-milled with excess K/KI for two hours at room temperature to produce a deep purple solid. The unusual colour is in sharp contrast with the Mg(II) precursor 3, which is pale yellow. The solid was extracted with cold pentane and kept at −40 °C overnight to yield bluish black crystals of 4. The single crystal X-ray diffraction (SCXRD) study of 4 revealed the structure to be a cAAC-coordinated Mg(I) monomer. Spin- and charge-density calculations suggested a non-trivial electron density back-donation from the formal Mg(I) centre to the carbene carbon (calculated NPA charges in +1.73 for Mg and −0.82 for cAAC). It is important to note that the same reaction was also attempted in solution, but with no success. When 3 was treated with slightly excess K/KI (1.1 eq.) in toluene, the reaction solution turned deep purple intermediately, indicating that the Mg(I) radical species formed in the system. However, the purple colour faded rapidly in toluene, indicating decomposition and/or reacting with toluene (though the exact reaction time for the colour to fade off was not reported). While the reaction product decomposed rapidly in solution, it is ‘frozen’ from further decomposition in the solid-state reaction and could be isolated.40 This work clearly demonstrates the unique synthetic merit of the mechanochemical method in isolating highly reactive species which are incompatible with solvents.
 | ||
| Fig. 2 Synthesis of monomeric magnesium radical by ball-milling.40 | ||
The π-acidic cAAC ligand clearly played a key role in the isolation of 4, however the oxidation state of cAAC-stabilised Group-2 complexes has recently been subject to debate.41 Later in 2022, via a ball-milling reduction of a β-diketiminate (BDI)-supported Mg(II) precursors 5, the Harder group attempted to remove the cAAC ligand and synthesise a cAAC-free Mg(I) monomeric complex.42 As shown in Fig. 3, Mg(II) iodide precursors 5 with three different BDI ligands of varying steric profiles were reduced with K/KI or Na/NaCl using ball-milling. After ball-milling, deep purple powders were obtained from all the three reactions, indicating the formation of Mg(I) radicals. The deep purple solids were not characterised in the solid-state, hence their exact nature remains unclear, but the colour putatively suggests the presence of Mg(I) radical species. The deeply-coloured solids from ball-milling were treated with d6-benzene, H2, and triphenyl benzene, producing [(BDI)Mg(C6D6)Mg(BDI)] (7) containing a dearomatised puckered benzene dianion, [{(BDI)MgH}2] (8), and a binuclear Mg complex containing a Ph3C6H3-dianion (9), respectively.
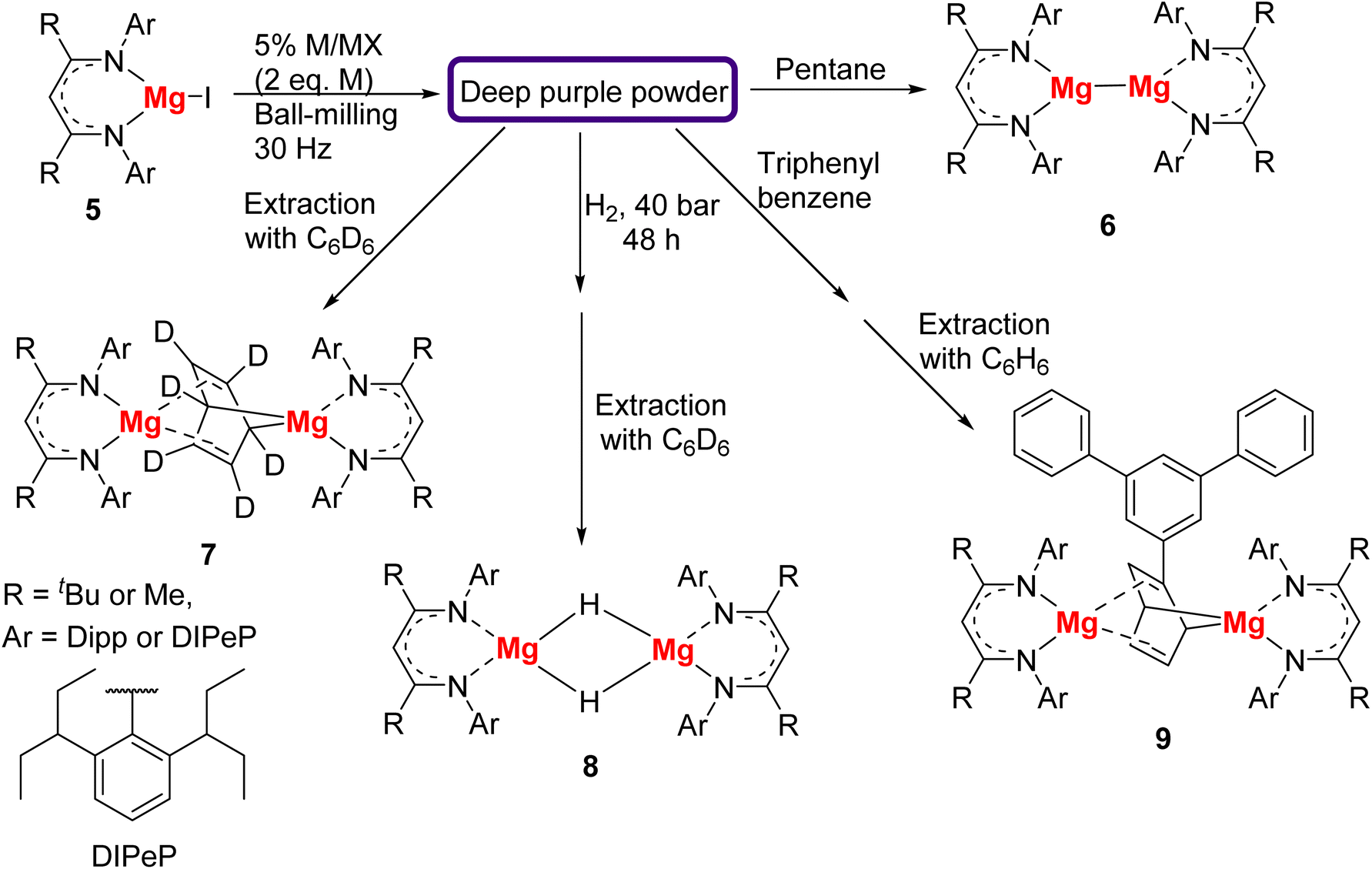 | ||
| Fig. 3 Synthesis of low-valent Mg(I) complexes by ball-milling.42 | ||
After extraction with pentane, instead of monomeric Mg(I) complexes, these deep purple solids produced corresponding yellow Mg(I) dimer 6 in excellent yields. It was observed that during the extraction, the deep colours faded, suggesting the loss of the open-shell Mg(I) monomeric structures. This work offered a convenient gateway to the Mg(I) dimers 6 with significantly enhanced yields and reduced reaction times compared with the previous solution-based syntheses.43
As evidenced by the successes in Mg(I) monomer chemistry, mechanochemistry has potential to offer a more direct and efficient synthetic approach for other low-valent or highly reactive species.
While Mg(I) chemistry is flourishing, monovalent heavier Group-2 metal chemistry develops much slower. Over the past two decades, only one example of a calcium(I) complex was reported in 2009,44 namely a formal Ca(I) invert sandwich complex [(THF)3Ca{μ-C6H3-1,3,5-Ph3}Ca(THF)3]. However, the oxidation state of calcium in this instance remains a topic of debate.45 A possible challenge for pursuing Ca(I) complexes is the weak hypothetical Ca(I)–Ca(I) bond: theoretical studies indicate that the strength of monovalent metal–metal bonds in Group-2 metals significantly decreases descending the group.46,47 Hence, unlike most of the Mg(I) complexes which are stabilised by the Mg(I)–Mg(I) bond, the marginal thermodynamic advantage of a weak Ca(I)–Ca(I) bond could only offer a limited stabilisation effect. This is reflected in the previous unsuccessful attempts: the same ligands and synthetic conditions that succeeded for the synthesis of the Mg(I)–Mg(I) complexes failed for the Ca(I) complexes, which favours disproportionation to Ca(0) and Ca(II).32
Nevertheless, multiple groups of main-group chemists are determined to overcome the challenge and to synthesise the first Ca(I) complex. Though the target has not be achieved yet, substantial progress has been made, especially since the 2020s, and benefitted from the use of mechanochemistry. First, inspired by their successful isolation of the Mg(I) monomer 4, in 2023 the Harder group adapted the mechanochemical synthesis into Ca chemistry. A Ca(II) precursor 10 and K/KI were ball-milled to produce a purple solid. Like the Mg(I) monomer 4, the deep colour of the solid likely links to the formation of a radical species. While the purple solid was not characterised in solid state, it was subjected to reaction with benzene to yield a biphenyl dianion complex 11, [(DIPPBDI)Ca(THF)]2(biphenyl) (Fig. 4). The production of 11 is postulated to proceed via a Ca(I) radical species, ˙Ca(DIPPBDI)(THF), which could react with benzene to give the coupling products.48 In 2024, in another attempt to use mechanochemistry to reduce Ca(II) to Ca(I), a new Ca-based room temperature stable electride (RoSE) was obtained, instead of the Ca(I) complexes.49 This Ca-based RoSE will be discussed in the following electride Chapter.
 | ||
| Fig. 4 Syntheses of Ca biphenyl complexes.48 | ||
From the abovementioned cases, it is obvious that the mechanochemical ball-milling method has exhibited unique capability, especially in isolating solvent-incompatible species (whether due to reaction with solvents or solubility issues). Though still in its infancy, successes such as the first, and so far, the only Mg(I) monomeric complex should encourage more synthetic main-group chemists to adopt mechanochemistry into their work.
2.2 Electrides
An electride is a type of chemical compound in which electrons serve as the anion.50,51 In electrides, these electrons are localised in voids, cavities, or channels within the structure, such as in crystals, molecular clusters, or between atomic layers, instead of occupying atomic/molecular orbitals. The electrons are not associated with any specific atom but are instead confined within spaces created by their surrounding structures (e.g., crystal lattice).52 Though it should be noted electride phase does not necessarily only exist in crystalline materials, but also in amorphous and liquid materials (e.g., Na metal dissolved in liquid ammonia53). Such properties make electrides ideal for a wide range of applications. In synthesis, they serve as highly effective reductants or catalysts, facilitating processes such as organic synthesis, ammonia production, and CO2 splitting.54 Beyond synthesis, electrides also play crucial roles in materials science, functioning as electron emitters, superconductors, battery anodes, and components in optics, lamps, and even radioactive waste storage.55Since 1983, when the Dye group first reported the electride Cs+(18-C-6)2e− (18-C-6: 18-crown-6), the synthesis of electrides has proven to be exceptionally challenging.56 For instance, the initial synthesis of Cs+(18-C-6)2e− required dissolving caesium metal in dimethyl ether–methylamine mixture, with a 2:1 stoichiometric amount of the crown ether to metal, all conducted at −30 °C until crystals were formed.57 Since the 1983 inaugural report, the Dye group synthesised eight “organic electrides”, with a general structure AM+(crown ether/cryptand)ne−, where AM+ is a Group-1 alkali metal cation.58 All the organic electrides were synthesised via a similar cryogenic low-boiling-point solvent dissolved metal process, which is challenging, if not impossible, for a conventional synthetic chemistry laboratory. Most of Dye's organic electrides are unstable at room temperature. It wasn't until 2005 that the first room-temperature-stable organic electride was successfully synthesised.59 This breakthrough was achieved by employing a specially designed aza-cryptand ligand, in order to avoid the C–O cleavage decomposition pathway, and employed a week-long, meticulously controlled process in liquid ammonia at −78 °C, performed in a helium glovebox, underscoring the extreme difficulty of obtaining these materials.
In 2022, the Martin group reported a second room-temperature-stable organic electride, namely a magnesium electride [(THF)4Mg4(μ2-bipy)4][(THF)6Mg2(μ2-bipy)(Cl)], (bipy = 2,2′-bipyridine) which was synthesised as a serendipitous product during their attempt to reduce a bipyridine-Ni(II) dichloride.60 Indeed, the two organic RoSE from the Dye and Martin group are tremendous synthetic achievements, but their need for a specialised aza-cryptand ligand, impractical synthetic protocol, or their serendipitous nature, prevent their wider application as a general gateway to a library of organic RoSEs.
In parallel with the organic electrides, there are several “inorganic electrides”, which are derived from binary or ternary inorganic materials AxBy or AxByCz.61 In 2003, the Hosono group achieved a milestone by synthesising the first air- and room-temperature-stable electride, [Ca24Al28O64]4+(4e−).62 However, this synthesis was not straightforward. The electride was derived from 12CaO·7Al2O3 crystals, which were obtained by melting the starting material (melting point 1415 °C), followed by a careful reduction process, involving heating in a hydrogen atmosphere. Similarly, the synthesis of the first water-stable electride, Y5Si3, involved melting yttrium and silicon ingots in a precise stoichiometric ratio.63 The resulting silver-coloured ingot was then finely ground using an agate mortar under a strictly controlled argon atmosphere.
Both organic and inorganic electrides are typically synthesised under harsh and demanding conditions, underscoring the pressing need for a milder, more accessible synthetic method. In this context, mechanochemistry presents a tempting candidate. But it was not introduced into this area until very recently.
In 2023, the Lu group first utilised the mechanochemical method for electride synthesis, successfully producing a RoSE, namely K+(LiHMDS)e− (13) (HMDS = 1,1,1,3,3,3-hexamethyldisilazide), using commercial accessible starting materials via the mechanochemical ball-milling (Fig. 5).64 A 1:1 molar ratio mixture of the starting materials, LiHMDS and potassium metal, were ball-milled at room temperature for 15 minutes, resulting in the formation of the electride 13 as a blue coloured powder. Remarkably, this method could be scaled up to 20 mmol without significant deterioration in yield, showcasing the efficiency and scalability of mechanochemistry in synthesising such reactive species. 13 is stable at room temperature under an inert atmosphere (N2 or Ar) for several months, therefore, 13 is a RoSE by definition. As a highly reactive amorphous material, characterisation of 13 is inherently challenging. By combining electron paramagnetic resonance (EPR), magnetometry, DFT calculations and ab initio random structure searching 13 was proved to feature a unique three-dimensional helical delocalised electron density (Fig. 6).
 | ||
| Fig. 5 Synthesis of a room-temperature-stable, Li/K heterobimetallic electride K+(LiHMDS)e− by ball-milling.64 | ||
 | ||
| Fig. 6 The AIRSS calculated structure of electride 13, showing the helical anionic electron density topology.64 | ||
13 exhibited versatile reactivity, including: (i) C–H activation and C–C coupling of benzene and pyridine; (ii) solvent-free Birch reductions, highlighting its potential as a strong reducing agent in synthetic chemistry. Moreover, upon treating with external ligands, the anionic electron in 13 can selectively re-combine with the Li+ or K+, achieving the first selective chemical reduction of Group-1 metal cations.65
In 2024, a new calcium-based RoSE 15, K[{Ca[N(Mes)(SiMe3)]3(e−)}2K3] (Mes = 2,4,6-trimethylphenyl), was synthesised by the mechanochemical ball-milling method (Fig. 7).49 This process involved ball-milling a calcium tris-amide 14, [Ca{N(Mes)(SiMe3)}3K], with potassium metal at 30 Hz for 30 min (mixer mill) or 4000 rpm for 3 h (planetary mill). It is worth to mention that in a previous work, the reduction of 14 with KC8 was attempted both with 18-C-6 in THF, and in benzene without any sequestering agent, however no reaction was observed.66 Also, in a control reaction, the precursor complex 14 was observed to undergo direct degradation in toluene, with no formation of the electride 15. This observation clearly indicates that, in the presence of a solvent, electride 15 cannot be obtained. 15 reacted with pyridine and benzene, confirming its potential as a strong reductant.
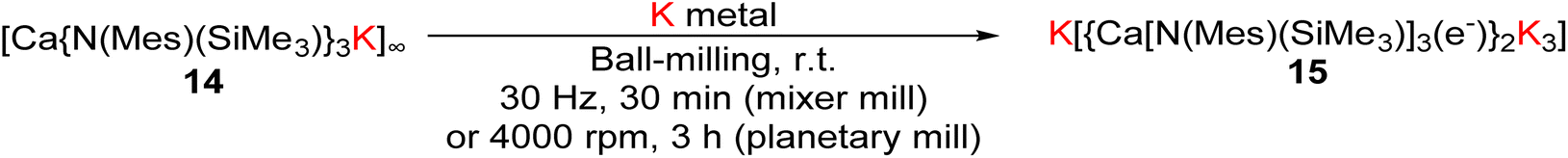 | ||
| Fig. 7 Synthesis of a room-temperature-stable, Ca-based electride by mechanochemistry methods.49 | ||
2.3 Alkalides
The alkali metals are the most electropositive elements of the periodic table, with their Pauling-scale electronegativities spanning from 0.98 (Li) to 0.79 (Cs) (cf. C 2.55, Mg 1.31, H 2.20).67 Hence, the zero-valent alkali metal atoms AM(0) easily lose one electron to form the corresponding AM(+1) cations. However, another possibility is accommodating one more electron into their valence-shell s-orbitals, resulting in AM(−1) anions with a fully occupied electron-paired ns2 close-shell structure. Indeed, alkalides are a unique class of compounds where the alkali metals (such as sodium, potassium) exist as anions (e.g., Na−, K−).68 In alkalides, the zero-valent alkali metal atoms gain an extra electron, resulting in a negatively charged species. This unusual electronic configuration makes alkalides highly reactive.69Despite being predicted in the 1960s,70–72 the first definite proof of alkali metal anions came in 1974 when Dye and co-workers isolated the first alkalide, a sodide (Na−), and by 1979 the group had reported Na−, K−, Rb− and Cs−.73 Their unique electronic properties, exceptional reducing power, and the potential to advance coordination chemistry and electron transfer mechanisms have generated significant interest.74 However, like the electrides, the synthesis of alkalides is exceptionally challenging, which limits their reactivity studies. Their high sensitivity to air, moisture, and in some cases, light, necessitates that the synthesis and storage must be conducted under rigorously controlled inert atmospheres, often requiring specialised techniques and equipment. However, the synthesis itself is not the only difficulty. While alkalides achieve some degree of thermodynamic stability with fully occupied s-orbitals, their negatively charged metal anions in solution can undergo comproportionation with the cations, i.e., AM(–1) + AM(+1) → 2 AM(0). Additionally, equilibria between the alkali metal and ligands in solution mean that electrides and alkalides can coexist, further complicating their isolation.75 Moreover, trace impurities that are difficult to eliminate can readily trigger decomposition, adding another layer of difficulty to an already complicated synthesis process.
The synthesis of alkalides was developed by the Dye group, requiring stringent purification of all reagents to remove trace impurities that could induce product decomposition. These synthetic protocols commonly demand meticulous attention to detail to ensure their success. Typically, the synthesis of alkalides is achieved by dissolving appropriate ratios of the alkali metal and the macrocyclic ligands, such as cryptands or crown ether, in low-boiling point solvents, such as dimethyl ether or methylamine, at low temperatures. The vast majority of alkalides synthesised contain macrocyclic ligands. Indeed, the macrocyclic effect is a major driving force for the isolation of alkalides, in which the alkali metal cation is sequestered and kinetically kept away from the alkalide anion, preventing comproportionation. Dissolving the alkali metal often takes a long time. For instance, it takes about 3 h of constant agitation at −30 °C to dissolve enough Na–K alloy in 10 mL of C222-isopropylamine solution to produce a 0.04 M solution of K+C222Na−.76 For Na+(C222)Na− (16), sodium metal is dissolved in ethylamine or methylamine with [2.2.2]-cryptand (C222) at −78 °C.77 Although higher temperatures might accelerate the dissolving and reaction, they also significantly increase the risk of comproportionation and decomposition.
Once the reaction is completed, most of the solvent needs to be removed by evaporation, and a suitable co-solvent or mixture is introduced to promote crystallisation. The resultant crystalline powder or crystals are then repeatedly washed with diethyl ether and dried via residual ether evaporation, which stabilises the samples at low temperatures. However, a vacuum seal-off near the preparation bulb presents additional risk, as sealing under dynamic vacuum, even with cooling from liquid nitrogen, can cause rapid, irreversible decomposition.78 Consequently, it is crucial to transfer the resultant powder products to a clean vessel before final sealing, even for the relatively stable Na+(C222)Na−.77 To meet these rigorous synthesis requirements, specially designed glassware, as illustrated in Fig. 8, is employed.76,79 Although the Dye group has also explored Direct Vapor Deposition (shown in Fig. 8(c)) as an alternative alkalide synthetic method, this approach has achieved only modest improvements regarding its synthetic efficiency (e.g., improving the yield) and easiness of handling.80
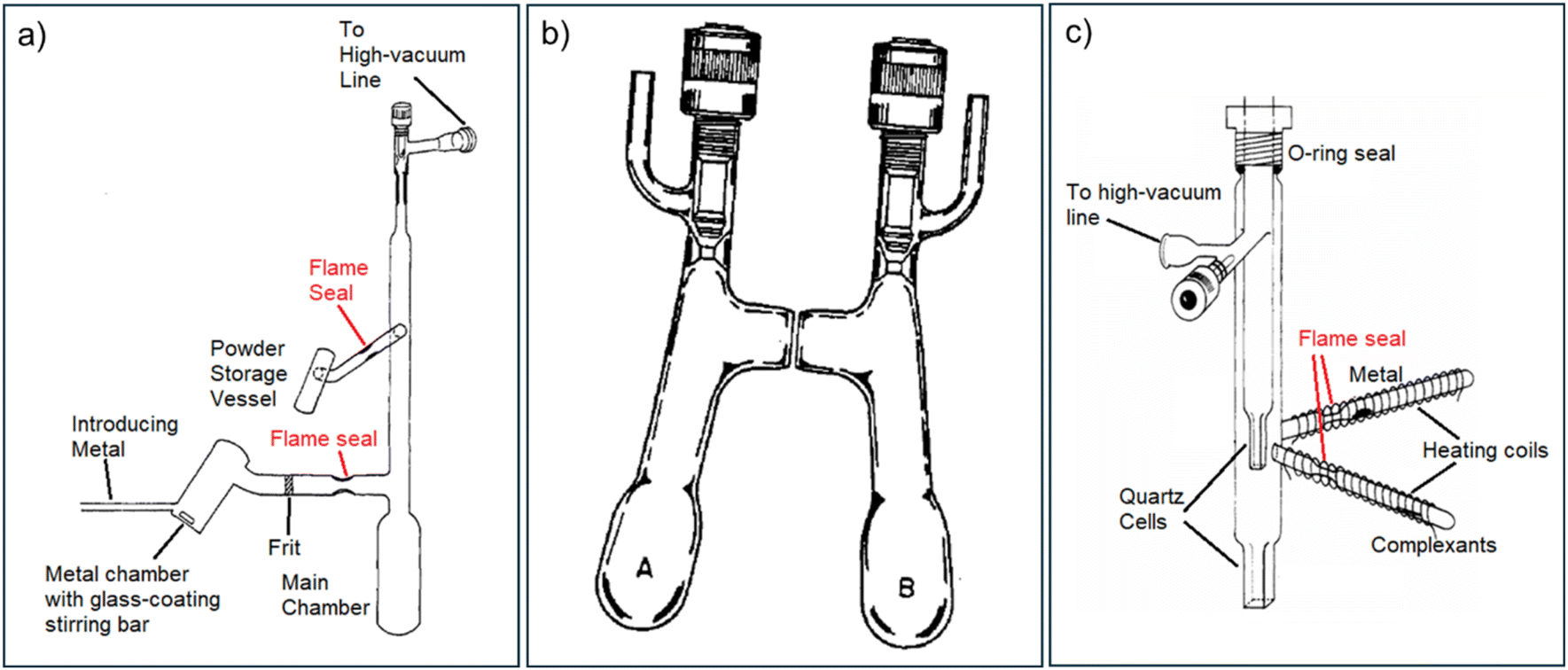 | ||
| Fig. 8 Experiment set-up of classic alkalide synthesis.76,78,80 (a) Apparatus for preparing alkalides using flame-sealed tubes and ampoules. This is the old design, used mostly before 1980. Adapted with permission from J. Phys. Chem. 1980, 84, 1084. Copyright {1980} American Chemical Society. (b) An H-cell with J.-Young taps, used with high-vacuum line. In between the cells is a sinter filter pad. This is the new design, used after 1980 until the 2000s. A similar K-cell was used as well. Reprinted with permission from J. Phys. Chem. 1984, 88, 3842. Copyright {1984} American Chemical Society. (c) Pyrex-quartz apparatus for the preparation of alkalides by vapor deposition. This was used when the volatile solvents need to be avoided. Adapted with permission from J. Phys. Chem. 1982, 86, 7. Copyright {1982} American Chemical Society. | ||
It is quite obvious that the alkalide synthesis is very challenging, which limits a full exploration of the alkalide chemistry. An accessible and scalable synthetic route is needed. Mechanochemistry presents a promising candidate: the absence of solvents could effectively eliminate equilibria and comproportionation issues.
In 2024, the Lu group introduced the mechanochemical ball-milling method into alkalide chemistry, reporting the first facile and scalable synthesis of an alkalide.81 The prototypical alkalide, the sodide Na+(C222)Na− (16), was synthesised by a simple 10 minutes ball-milling reaction between 2 equivalents of Na metal and 1 equivalent of [2.2.2]-cryptand at room temperature and under argon atmosphere (Fig. 9). 16 can be obtained in 48% yield as a crystalline solid from its dry THF solution at −35 °C. The sodide 16 features two distinct signals on its magic-angle spinning (MAS) solid-state 23Na NMR for Na+ (−11 ppm) and Na−1 (–62 ppm), respectively.
 | ||
| Fig. 9 Synthesis of sodide complex Na+(2.2.2-Cryptand)Na− by ball-milling.81 | ||
Compared to the classical alkalide synthesis methods mentioned earlier, this mechanochemical method offers multiple advantages. Firstly, the ball-milling method employs commercially available materials: both the Na metal and the [2.2.2]-cryptand were used without further purification from their commercial sources; especially, removal of the oxide layer on the Na metal surface is not necessary, which makes the synthesis substantially easier. Secondly, it operates at room temperature without stringent temperature control. Additionally, the reaction time is drastically shortened to 10 minutes, a substantial improvement over the previous method.79 Last but not least, the Lu protocol can be scaled up, which is essential for the following reactivity studies. With such an accessible entry to the sodide 16, the authors investigated the sodide reactivity. 16 delivers 2-electron reductive cleavages of C–N bonds in primary amines, and 1-electron reduction of azobenzene (Fig. 10).81
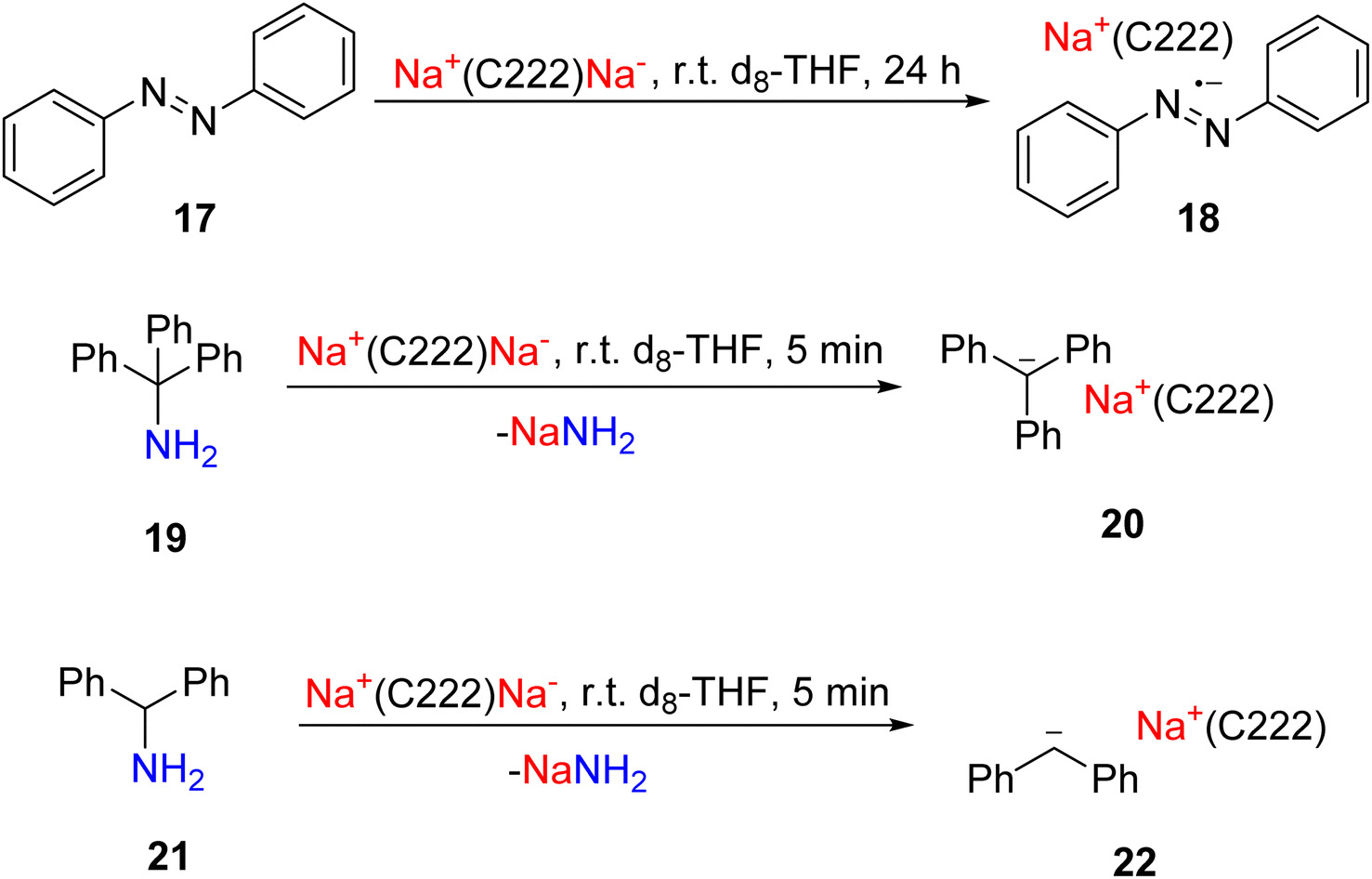 | ||
| Fig. 10 Reactivity of the sodide (16) towards amines and azobenzene.81 | ||
2.4 Miscellaneous
The Nobel Prize-winning metallocene is an essential class of organometallic complexes for all metals across the periodic table. The centre of the metallocene chemistry is the coordination hapticity between the cyclopentadienyl (Cp) ligand (and its derivatives) and the metal cation.As a part of the early metallocene “gold rush”, Fischer and co-workers reported the beryllocene, [BeCp2], in 1959.82 Four decades later, Carmona and co-workers reported the Cp* version, [BeCp*2], in 2000.83 It is interesting that the sterically less bulky [BeCp2] adopts an η5:η1 hapticity for its two Cp ligands, but the bulkier [BeCp*2] adopts an η5:η5 hapticity. While indenyl complexes had previously been reported for all other non-radioactive s-block metals, no such beryllium indenyl complex had been isolated. To investigate such a complex, in 2021 Hanusa and co-workers set out to synthesise the indenyl (Ind) version of beryllocene, namely [Be(Ind)2].84 However, the authors found that the conventional solution synthesis between K[Ind] and BeBr2 in THF yielded an intractable mixture.84 The authors then turned to the mechanochemical ball-milling method, which led to the successful isolation of the desired complex 23 (Fig. 11). Noting that in 23, one indenyl ligand coordinates in η5-mode, while the other in η1-mode. In this case, the mechanochemical method appeared superior to the conventional synthesis in solution, though the exact reason(s) for this phenomenon remains unclear.
 | ||
| Fig. 11 Synthesis of di(indenyl)beryllium.84 | ||
3. Reactions involving reactants with limited solubility
Traditional reactions are mostly conducted in solution. However, solution-state reactions are often challenging when one or more reactants are poorly soluble, particularly in heterogeneous systems. Under such conditions, limited surface interaction between the reactants can lead to sluggish reaction rates and incomplete conversion. Mechanochemical methods offer a promising solid-state alternative to the traditional solution-state synthesis, successfully facilitating reactions with no solvent required, thereby providing a pathway to bypass the limitations in heterogenous reactions.In 2023, the Gouverneur and Hayward groups reported a remarkable example of mechanochemistry's transformative potential in facilitating reactions involving insoluble reactants, in this case is fluorspar (calcium fluoride CaF2).85 All fluorochemicals originate from naturally occurring fluorspar.86 However, CaF2, a white solid with a high melting point at around 1420 °C, is poorly soluble in water (0.016 g L−1 at 20 °C) and insoluble in organic solvents, making it unsuitable for direct use in fluorochemical synthesis. Traditionally, the production of fluorochemicals relies on the process first reported by C. W. Scheele in 1771.87 involving the generation of hydrogen fluoride (HF) via the reaction of acid-grade (>97%) fluorspar with sulfuric acid (>98%) at 100–300 °C.88 Given the toxicity and corrosive nature of the HF and the concentrated sulfuric acid, a method directly utilising CaF2 and circumventing the HF stage is highly desirable.
The Gouverneur and Hayward groups reported such a HF-free direct CaF2 utilisation by using mechanochemistry.85 Acid-grade fluorspar (CaF2, 1 eq.) was milled with anhydrous K2HPO4 (>98%, 2.5 eq.), giving a fluorinating reagent named fluoromix. The fluoromix, a white powder, was prepared via a stepwise process: 1 eq. of CaF2 and 1 eq. of K2HPO4 were milled at 35 Hz for 3 hours, followed by the addition of another 1 eq. of K2HPO4 for a further 3 hours of milling, and finally, 0.5 eq. of K2HPO4 was added and milled for further 3 hours (Fig. 12). The resulting fluoromix 25, obtained after 9 hours of milling. Even after storing under inert atmosphere at room temperature for 9 months, the fluoromix was found still active.
 | ||
| Fig. 12 The synthesis of fluoromix.85 | ||
As a nucleophilic fluorination reagent, the fluoromix showed impressive versatility, enabled a number of fluorination reactions listed in Fig. 13. Mechanistic studies revealed that the fluoromix contains crystalline constituents, including K3(HPO4)F and K2−XCaY(PO3F)α(PO4)β, which serve as fluorinating agents. Studies to confirm its composition include 31P and 19F NMR for soluble species, PXRD analysis for crystalline species, and control experiments for amorphous species. These species facilitate the synthesis of sulfonyl fluorides, α-fluoroketones, fluoroesters, α-fluoroamides, benzylic fluorides, alkyl fluorides, and (hetero)aryl fluorides.
 | ||
| Fig. 13 Summary of C–F and S–F bonds formation.85 | ||
In this work, the mechanochemical method unlocked the direct utilisation of the insoluble CaF2 as a fluoro-building block, circumventing the conventional HF stage, which is undesirable from the aspects of cost, health and safety and sustainability.
Very recently, the Gouverneur group further developed the mechanochemical CaF2 activation methodology by reporting a one-step synthesis of alkali metal fluorides (AMF, AM = Li, Na, K) from CaF2.89 This was achieved by ball-milling CaF2 with 2.0 equivalents of AMOH and 1.0 equivalent of TiO2 at 35 Hz, room temperature, for 3 hours. Byproducts are CaTiO3 and H2O. Further fluorination reactions using AMF were not reported in this work, though.
Zero-valent metals are good electron sources for reduction reactions. The ability to use zero valent metals directly and easily is undoubtedly desirable. However, due to their poor solubility in organic solvents, the direct use of zero valent metals is often difficult in traditional solution-state reactions. Zero-valent metals can be accessed as variety of forms (e.g. powder, lumps, etc.) and typically have an oxide coating on the outer layer, which needs to be removed to allow the reaction to happen. Typically, the zero valent metals must be activated prior to the reaction to increase the surface area, for example, using highly dispersed metal suspensions.90 Reactions involving zero-valent metals are often difficult to predict and are sometimes difficult to reproduce, since the surface area, a determining factor, is difficult to control and could be different from batch to batch. In recent years, mechanochemistry offers an alternative pathway to use zero-valent metals, such as Li,91 Mg, Mn, Zn, Ag, and Bi, as reductants.92
A demonstration of mechanochemistry's potential is a manganese metal-mediated dimerisation of electron-poor alkenes reported by the Browne group in 2021.93 Ball-milling arylidene malonates 32 with manganese pieces, LiCl, and a minimal amount of THF (1 eq., liquid-assisted grinding, LAG)94 yielded the dimerised products 33 (Fig. 14). The precise reaction mechanism remains unclear – whether the manganese metal participates as a concerted two-electron reduction or two sequential single-electron reductions. Nevertheless, control experiments unequivocally proved the necessity of the ball-milling method: stirring the reactants and manganese metal (powder or pellets) in bulk organic solvents did not work.
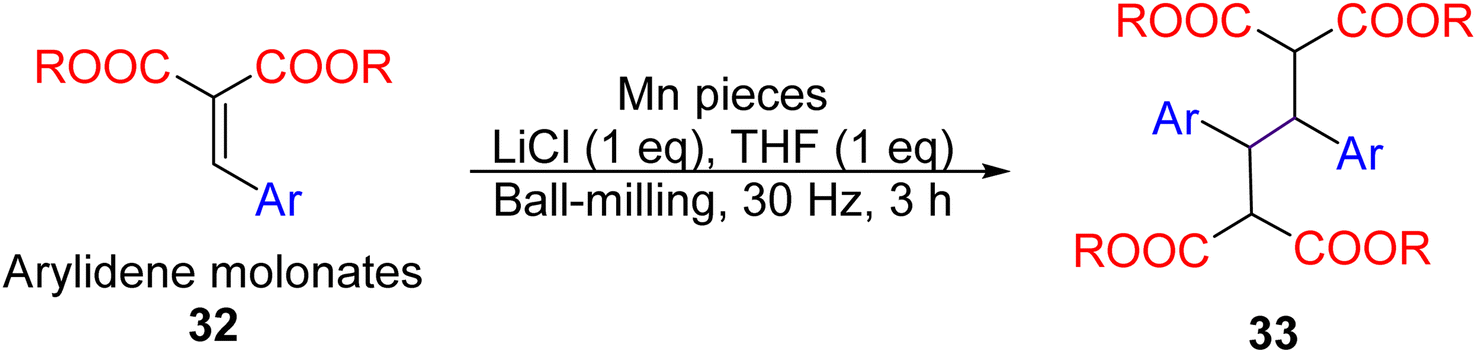 | ||
| Fig. 14 Synthesis manganese mediated reductive dimerisation of arylidene malonates by ball-milling.93 | ||
The Birch reduction has been widely used by organic chemists for over half a century, enabling the dearomatisation of arenes into 1,4-cyclohexadiene derivatives.95 Traditionally, this transformation relies on alkali metals dissolved in liquid ammonia under cryogenic conditions. However, these classical procedures are fraught with operational challenges, including the need of liquid ammonia, which has to be slowly evaporated and carefully exhausted after the reaction. The restricted solubility of alkali metals limits the scope of solvents available, prompting significant interest in alternative, more practical methodologies. In recent years there has been an increasing interest in ammonia-free Birch reduction, including reports utilising electrochemical,96 photochemical,97 and enzymatic catalytic methods,98 as well as employing lithium metal in an ammonia-free system.99 However, all these methods still need bulk organic solvent, which is a sustainability culprit.
In 2023, the Lu group reported the first mechanochemical solvent-free Birch reduction by using the newly developed RoSE reagent, K+(LiHMDS)e− (13),64 previously discussed in the electrides chapter. As the first solvent-free Birch reduction, the Lu group's work was a conceptual breakthrough, but there are two technical drawbacks: (i) the reductant, K+(LiHMDS)e−, is still a specialised reagent; (ii) the reactions need to operate under argon.
Shortly after, the Ito/Kubota group reported a Li metal mediated mechanochemical Birch–Benkeser reaction, which operates under air, overcoming the technical drawbacks.100 As shown in Fig. 15, arenes 34 were ball-milled with Li metal and amine additives at room temperature under air for only 1 min, yielding the corresponding Birch-type reduction products 35 with good to excellent yield. The scope of substrates covers from carboxylic acids to electron-rich aromatic ethers, arene-bearing alcohols and several bioactive molecules.
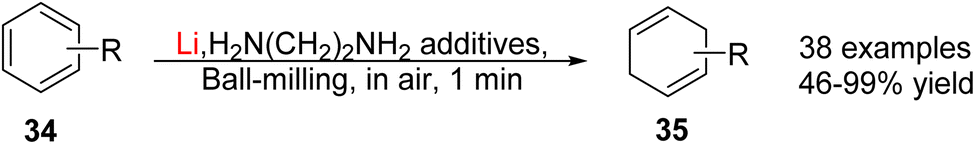 | ||
| Fig. 15 Lithium-based Birch reduction by ball-milling.100 | ||
This approach circumvents the need to dissolve lithium in liquid ammonia or other solvents. More importantly, the reaction is fast, air-tolerant, requires no specialised equipment or handling of cryogenic or hazardous materials, and demonstrated good functional group compatibility.
Following these works, several reports on mechanochemical Birch-type reductions emerged, underscoring the growing interest in this area. These include a Birch reduction by sodium lumps101 and calcium metal102 (the Ito/Kubota group), as well as calcium- and magnesium-metal mediated Birch reductions103 (the Kananovich group). It appears that the mechanochemical Birch reduction is turning into a hot area.
Beside Birch reduction, Group-1 metals have also been used in other mechanochemical reactions. In 2023, the Lu group reported mechanochemical reduction and Li-ion doping of transition metal oxides, by ball-milling Li metal and nickel and cobalt oxides under argon (Fig. 16).104
 | ||
| Fig. 16 Ball-milling reactions between Li metal and transition metal oxides, leading to the transition metal reduction and Li-ion doping. | ||
In 2025, the Ito/Kubota group reported mechanochemical synthesis of organolithium reagents, by ball-milling Li metal and organo-halides in air (Fig. 17).105 It is noteworthy that Et2O is needed to facilitate the organolithium formation, through a LAG mechanism. The organolithiums were not isolated, instead, they were used for further mechanochemical reactions with electrophiles (E+), also in air. This method is significantly easier and more practical in comparison to the traditional method to make organolithiums, i.e., refluxing Li metal pieces and organo-halides in solution under inert gas atmosphere.
 | ||
| Fig. 17 Mechanochemical synthesis of organolithium reagents.105 | ||
Mechanochemical methods not only offer new and versatile reaction conditions for the direct utilisation of metals but also provide immediate and significant improvements for substrates with poor solubility in conventional solvents.
In recent years, the Ito/Kubota group has reported several mechanochemical Suzuki–Miyaura cross-coupling reactions.106–111 A key benefit of the mechanochemical cross-couplings is their capability to activate insoluble electrophiles, especially the aryl halides112 (Fig. 18). In this work, the use of 1,5-cod as a dispersant and a LAG reagent is also crucial.
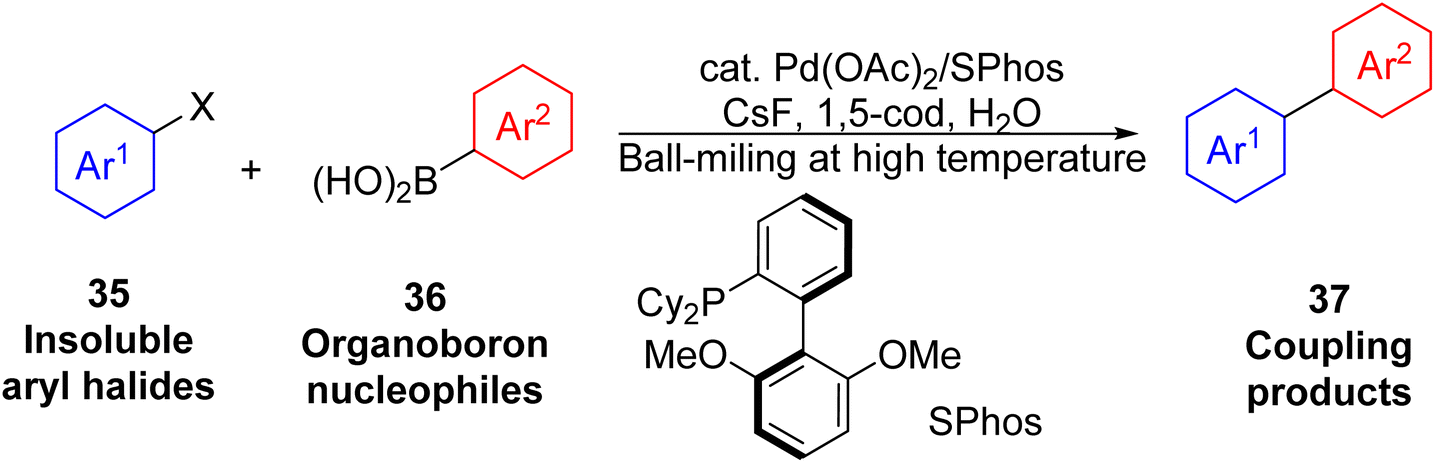 | ||
| Fig. 18 Cross-coupling reactions with insoluble aryl halides.112 | ||
In Fig. 19, aryl halides are classified by their solubility based on US Pharmacopoeia, 10−2 to 10−3 M as ‘slightly soluble’, 10−3 to 10−5 M as ‘very slightly soluble’ and 10−5 to 10−6 M as ‘practically insoluble’.112 For aryl halides in different catalogues, they compared the results of the mechanochemical method (5–90 minutes at 250 °C) with solution-based methods (24 hours, 120 °C). Despite the much longer reaction time, the solution-state reactions afforded significantly lower yields. Of particular note is the successful reaction of “practically insoluble” (class 3) alkyl halides which gave no reaction in solution. The mechanochemistry strategy offers the first practical method for the cross-coupling of insoluble aryl halides. While the need for high temperature ball-milling may be a drawback, this method provides new opportunities to expand the structural diversity of organic molecules. In 2023, the Ito/Kubota group managed to lower the reaction temperature to 45 °C by using a bespoke phosphine ligand.113
 | ||
| Fig. 19 Classification of aryl halides based on their solubility.112 (Reprinted with permission from J. Am. Chem. Soc., 2021, 143, 6165. Copyright {2021} American Chemical Society.) | ||
Beyond metals, minerals,114 and organic compounds, the mechanochemical method has also been used to activate chalcogen elements. Element selenium is a cheap, commercially available, stable, odourless, and easy to handle starting material for organic selenation reactions.115 However, elemental selenium is fairly inert, has low solubility in organic solvent, and has a tendency to form transition-metal selenium clusters, which make using it directly challenging.
In 2024, the Wei group reported a ball-milling-enabled C–Se bond formation from accessible organic halides, Mg metal and elemental selenium, with a wide substrate scope and good functional group tolerance.116 After ball-milling aryl halides 39, Mg, LiCl, Se under LAG117 conditions (THF), the corresponding diselenides 40 were obtained in good yields (Fig. 20). If excess Mg was used, a magnesium-based selenium nucleophile intermediate forms, with the corresponding unsymmetrical monoselenides 41 forming after N-phenylacrylamide was added (Fig. 20). Notably, this mechanochemical method also works for thiolation and telluration. The formation of 40 and 41 share the same magnesium selenide intermediate. Though not clearly pointed out in Wei's paper, we hypothesise that the reaction can proceed in two ways: (1) in air, the Mg selenide oxidises into selenium radical and dimerise into 40; (2) with N-phenylacrylamide, the Mg selenide undergoes a magnesium-selenation towards the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, which is then protonated to form 41. Another possibility is that, Se reacts with the Grignard reagents ArMgX, through a Mg–C bond selenation mechanism,118 to yield the Mg selenide intermediate.
C bond, which is then protonated to form 41. Another possibility is that, Se reacts with the Grignard reagents ArMgX, through a Mg–C bond selenation mechanism,118 to yield the Mg selenide intermediate.
 | ||
| Fig. 20 The synthesis of symmetrical diselenides and unsymmetrical monochalcogenides.116 | ||
Grignard reagents are some of the most widely used organometallic reagents in synthetic laboratories. Recent advancements in mechanochemistry have not only enhanced the synthesis of traditional magnesium-based Grignard reagents but also expanded the scope to heavier Group-2 elements such as calcium, strontium, and barium.119 From the textbook, higher atomisation energies and larger first ionisation energies (for alkaline earth metal, the second ionisation) is needed to prepare organometallics from the alkaline earth metals. As a result, the heavier alkaline earth metals need elevated temperatures or specialised activation methods to initiate the reactions to form their Grignard-analogues. But these heavier Grignard reagents are not only chemical curiosities, but could offer enhanced reactivity from their more ionic, more reactive metal–carbon bond.120
Since the activation of the insoluble metals (Ca, Sr, Ba) is the barrier, mechanochemistry offers a prime solution. In 2024, the Ito/Kubota group demonstrated a direct arylation of alkyl fluorides through in situ generated Ca-based heavy Grignard reagents via mechanochemical ball-milling.121 In this work, aryl iodides 42 and alkyl fluorides 43 were ball-milled with commercially available calcium metal and a small amount of tetrahydropyran (THP) in air for 60 minutes at 80 °C, giving the C(sp3)-F bond arylation production 44 in good to high yields (Fig. 21).
 | ||
| Fig. 21 Arylation of alkyl fluorides using mechanochemically generated calcium-based heavy Grignard reagents.121 | ||
This mechanochemical method not only simplified the generation of reactive Grignard species but also bypassed the need for transition metal catalysts. The direct arylation in Fig. 21 of alkyl fluorides is particularly noteworthy. Additionally, the reaction conditions were air-tolerant and conducted at 80 °C (internal), making the process far more practical than traditional methods that require inert atmospheres and highly controlled environments.
In 2024, the Ito/Kubota group reported the mechanochemical generation of aryl barium nucleophiles from inactive barium metal.122 Barium metal is usually inert – its activation was reported via metal vapour synthesis.123 However, in Ito/Kubota group's study, ball-milling commercial Ba metal and the substrates (45–48) yielded the silylation and nucleophilic addition products, 47 and 49, respectively (Fig. 22). These reactions proceed via the barium organometallics ArBaI, which subsequently react with the electrophiles.
 | ||
| Fig. 22 Reactions via direct generation of various aryl barium nucleophiles from commercially available inactivated barium metal and aryl halides.122 | ||
There are more examples of using the mechanochemical strategy for Mg-based Grignard reagents.124,125 Advantages of this strategy include faster reaction rates, reduction in safety hazards and increasing reactivity of metal.
An interesting example of a mechanochemical preparation of bis(n-propyltetramethylcyclopentadienyl)strontium (SrCp′2, 51, Cp′ = n-propyltetramethylcyclopentadiene) was reported by Peters and Blair in 2014.126 The common strontium starting material, SrI2, is poorly soluble in hydrocarbon and ethereal solvents. By using a bespoke reactor modified from commercial glass flasks, SrI2 and Cp′K were made into a suspension with stirring from six 12.7 mm stainless steel balls for 24 hours, to afford the Et2O adduct 50 (Fig. 23). After a vacuum distillation of 50, the solvent-free 51 was produced. Though this case is more akin to a classic suspension reaction, the authors did categorise it into a mechanochemical reaction.
 | ||
| Fig. 23 The synthesis of unsolvated Sr(Cp′)2.126 | ||
4. Elimination of solvent coordination
Coordinated solvent molecules are ubiquitous in coordination chemistry. Solvent molecules can bond strongly to metal centres and influence the structure and reactivity. On one hand, they can stabilise reactive species by masking the exposed metal centre. But on the other hand, they could also block the reactive centre. The coordination solvent effects are particularly pronounced in cationic polymerisations, which are rather sensitive to the coordination number and electronic environments of the metal centre.127 The elimination of solvent coordination is also particularly important when the aim is to isolate highly reactive, base-free, or low-coordinate metal complexes.128 The mechanochemical methods have inherent advantages in avoiding the coordination of solvent molecules.Early in 2014, Hanusa and co-workers reported a mechanochemical synthesis of unsolvated tris(allyl)aluminum complexes, [1,3-(SiMe3)2C3H3]3Al, 54, and their scandium analogue, [1,3-(SiMe3)2C3H3]3Sc, 55 (Fig. 24).129 In their study, attempts to synthesise the base-free tris(allyl)aluminum complex, 54, in the presence of solvents such as THF or diethyl ether resulted in unidentified complex mixtures. However, by employing ball-milling, aluminum/scandium halides (AlX3, X = Cl, Br, I; ScCl3) were successfully reacted with the potassium allyl salt 52 (1,3-bis(trimethylsilyl)allyl potassium) in solid-state, yielding the unsolvated 54 and 55, respectively. The high reactivity of the unsolvated complex, 54, is demonstrated by its immediate reaction with benzophenone in hexanes at −78 °C. In contrast, the THF adduct [(C3H5)3Al(THF)] required a longer reaction time of 10 minutes at room temperature, despite the larger steric bulk of 1,3-(SiMe3)2C3H3 compared to C3H5.129
 | ||
| Fig. 24 Synthesis of unsolvated [1,3-(SiMe3)2C3H3]3(Al, Sc).129 | ||
In 2018, the Hanusa group reported a transformation in unsolvated organotin complexes driven by mechanochemistry.130 In this work, both unsolvated tris(allyl) stannate 56 and tetra(allyl)tin species 57 are synthesised by ball-milling. The Hanusa group first ball-milled potassium allyl salt 52 ([1,3-(SiMe3)2C3H3]K) with SnCl2 in a 2:1 ratio for 5 minutes in a planetary mill, generating a partially hexane soluble brown powder, tris(allyl) stannate 56, {Sn[1,3-(SiMe3)2C3H3]3K}∞ (Fig. 25). To further prove the necessity of the mechanochemical method, the potassium allyl salt 52 and SnCl2 was stirred in hexane with no reaction occurring. In addition, a ball-milling-triggered disproportionation reaction between 52 and SnCl2 resulted in the formation of a novel unsolvated tetra(allyl)tin(IV) species, 57. It is worth to mention that the tetra(allyl)tin(IV) species 57 will decompose in solution in 1 hour but is stable in solid-state for several months. Furthermore, during the synthesis of 57, the stereochemical control of products can be realised without solvent, which shows possibility of stereochemical control during mechanochemical reaction. These unsolvated complexes could serve as strong Lewis acid catalysts or be utilised in metal-exchange reactions.130
 | ||
| Fig. 25 Synthesis of unsolvated {Sn[1,3-(SiMe3)2C3H3]3K}∞ and Sn[1,3-(SiMe3)2C3H3]4.130 | ||
In 2024, the Yamashita group reported the synthesis of a non-solvated dialkylalumanyl anion, [K(AlR2)]2 (60) via the mechanochemical method (Fig. 26).13160 is an example of monovalent aluminium anionic complex, i.e., an aluminyl anion, which is a recent topical area in p-block chemistry.132 The corresponding toluene adduct, [R2Al–K(tol)2] (60-tol), was reported by the same group in 2020,133 which is highly reactive but quite unstable. Though 60-tol was characterised by SCXRD and UV-vis spectroscopy,133 its instability prevented its characterisation using X-ray photoelectron spectroscopy (XPS): a nuclear-specific characterisation to elucidate oxidation state. Unlike its toluene adduct, 60 is stable at room temperature, which allowed collecting its Al 2p XPS spectra, unequivocally proving its Al(I) oxidation state and the Al 2p-electron binding energies.
 | ||
| Fig. 26 Synthesis of non-solvated dialkylalumanyl anion, [K(AlR2)]2via mechanochemical method.131 | ||
5. Conclusion and outlook
As a closing remark, we summarise a few situations where we believe that mechanochemical methods could offer unique synthetic advantages:(1) The reactants/products are highly reactive and react with solvent molecules;
(2) One or more reactants are insoluble, resulting in slow or no reaction and which causes the stoichiometric ratio to deviate from the designed value, and lead to unwanted side reactions;
(3) A non-solvated product is desired, but the reaction has to be conducted in coordinative solvents.
Though not reported yet, we envisage that the mechanochemical methods could also be used in the following directions:
(1) Trap highly unstable intermediate. A unique benefit of mechanochemistry is, once the ball-milling stops, the reaction is “frozen” and allows a number of solid-state characterisations. Fuelled by recent advances in the instrument (precise temperature-control cryogenic ball mill is now commercially available), we are sure that in future, the low-temperature ball-milling method will allow coordination chemists to isolate and characterise more elusive species;
(2) In situ monitoring of mechanochemical reactions has been recently widely used in organic reactions.134 But this invaluable tool has seen less use in coordination chemistry. The major spectroscopic handles currently used in the in situ studies are vibrational spectroscopies (IR and THz Raman135), and powder X-ray diffraction.136 It can be envisaged that more analytical probes will be developed. We are particularly interested in electron paramagnetic resonance (EPR) and metal-nuclear solid-state NMR – there are works underway in our group on these regards.
(3) Combining mechanochemistry with other energy inputs. While mechano-photochemistry137 and mechano-piezoelectric chemistry138 have been reported in organic reactions, these transformative tools have not been introduced into synthetic coordination chemistry yet. We are very interested in seeing how these tools will enable coordination chemists to achieve more and more impossibles—which we believe is only a matter of time.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Leverhulme Trust (RPG-2022-231 and RPG-2023-159) and the University of Birmingham for their generous financial support.References
- For representative examples of THF fragmentation, see: (a) J. Clayden and S. A. Yasin, New J. Chem., 2002, 26, 191–192, 10.1039/B109604D; (b) A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706–708, DOI:10.1126/science.1178165; (c) R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588–591, DOI:10.1038/nchem.667; (d) E. Crosbie, P. García-Álvarez, A. R. Kennedy, J. Klett, R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2010, 49, 9388–9391, DOI:10.1002/anie.201005119; (e) E. Tanuhadi, A. S. Bair, M. Johnson, P. Fontaine and J. Klosin, Chem. Sci., 2025, 16, 280–287, 10.1039/D4SC05983B.
- (a) M. H. Holthausen, T. Mahdi, C. Schlepphorst, L. J. Hounjet, J. J. Weigand and D. W. Stephan, Chem. Commun., 2014, 50, 10038–10040, 10.1039/C4CC01922A; (b) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097, 10.1039/C4CS00206G.
- For recent representative examples, see: (a) D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498, DOI:10.1002/anie.202218498; (b) N. Davison, P. G. Waddell, C. Dixon, C. Wills, T. J. Penfold and E. Lu, Dalton Trans., 2022, 51, 10707–10713, 10.1039/D1DT03532K.
- For recent representative examples, see: (a) S. K. Thaakur, N. Roig, R. Monreal-Corona, J. Langer, M. Alonso and S. Harder, Angew. Chem., Int. Ed., 2024, 63, e202405229, DOI:10.1002/anie.202405229; (b) Y. Wang, Y. Zhang, J. Liang, B. Tan, C. Deng and W. Huang, Chem. Sci., 2024, 15, 8740–8749, 10.1039/D4SC02491E; (c) K. R. McClain, A. H. Vincent, A. Rajabi, D. X. Ngo, K. R. Meihaus, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2024, 146, 32708–32716, DOI:10.1021/jacs.4c12278; (d) G. M. Richardson, T. Rajeshkumar, F. M. Burke, S. A. Cameron, B. D. Nicholls, J. E. Harvey, R. A. Keyzers, T. Butler, S. Granville, L. Liu, J. Langley, L. F. Lim, N. Cox, N. F. Chilton, J. Hicks, N. J. L. K. Davis, L. Maron and M. D. Anker, Nat. Chem., 2025, 17, 20–28, DOI:10.1038/s41557-024-01688-6.
- For a recent example, see: M. Jakoobi, Y. Tian, R. Boulatov and A. G. Sergeev, J. Am. Chem. Soc., 2019, 141, 6048–6053, DOI:10.1021/jacs.9b01562.
- (a) n-Hexane C–H activation: H. M. Hoyt, F. E. Michael and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 1018–1019, DOI:10.1021/ja0385944; (b) iso-hexane C–H activation: J. K. Hoyano and W. A. G. Graham, J. Am. Chem. Soc., 1982, 104, 3723–3725, DOI:10.1021/ja00377a032.
- A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1982, 104, 352–354, DOI:10.1021/ja00365a091.
- Even methane can undergo C–H activation with highly reactive coordination complexes, see a recent example: Y. Liu, W. Dong, Z. H. Li and H. Wang, Chem, 2021, 7, 1843–1851, DOI:10.1016/j.chempr.2021.03.010.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514, DOI:10.1038/417507a.
- For a recent review of divalent lanthanide chemistry, see: S. Schäfer, S. Kaufmann, E. S. Rösch and P. W. Roesky, Chem. Soc. Rev., 2023, 52, 4006–4045, 10.1039/D2CS00744D.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, Organometallics, 1985, 4, 112–119, DOI:10.1021/om00120a019.
- W. J. Evans, L. A. Hughes and T. P. Hanusa, J. Am. Chem. Soc., 1984, 106, 4270–4272, DOI:10.1021/ja00327a037.
- W. J. Evans, N. T. Allen, P. S. Workman and J. C. Meyer, Inorg. Chem., 2003, 42, 3097–3099, DOI:10.1021/ic0300316.
- Mechano-chemical reaction, in IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry, 2025, 5th edn, Online version 5.0.0, DOI:10.1351/goldbook.MT07141.
- L. E. Wenger and T. P. Hanusa, Chem. Commun., 2023, 59, 14210, 10.1039/d3cc04929a.
- J. F. Reynes, F. Leon and F. García, ACS Org. Inorg. Au, 2024, 4, 432–470, DOI:10.1021/acsorginorgau.4c00001.
- M. Arrowsmith, H. Braunschweig, M. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890, DOI:10.1038/nchem.2542.
- S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486, DOI:10.1002/cctc.201501015.
- A. Rajabi, R. Grotjahn, D. Rappoport and F. Furche, Dalton Trans., 2024, 53, 410, 10.1039/d3dt03221c.
- N. Tsoureas and I. Vagiakos, Inorganics, 2024, 12, 275, DOI:10.3390/inorganics12110275.
- R. Akhtar, K. Gaurav and S. Khan, Chem. Soc. Rev., 2024, 53, 6150, 10.1039/d4cs00101j.
- B. Rösch and S. Harder, Chem. Commun., 2021, 57, 9354, 10.1039/D1CC04147A.
- S. Petrie, Aust. J. Chem., 2003, 56, 259, DOI:10.1071/CH03006.
- M. A. Anderson and L. M. Ziurys, Astrophys. J., Lett., 1995, 439, 25, DOI:10.1086/187736.
- T. Kruczyński, N. Pushkarevsky, P. Henke, R. Kçppe, E. Baum, S. Konchenko, J. Pikies and H. Schnçckel, Angew. Chem., Int. Ed., 2012, 51, 9025, DOI:10.1002/anie.201204997.
- L. B. Knight Jr, W. C. Easley, W. Weltner Jr and M. Wilson, J. Chem. Phys., 1971, 54, 322, DOI:10.1063/1.1674610.
- R. Rubino, J. M. Williamson and T. A. Miller, J. Chem. Phys., 1995, 103, 5964, DOI:10.1063/1.470476.
- R. Köppe, P. Henke and H. Schnöckel, Angew. Chem., Int. Ed., 2008, 47, 8740, DOI:10.1002/anie.200802960.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754, DOI:10.1126/science.1150856.
- A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659, 10.1039/c0dt01831g.
- C. Jones, Commun. Chem., 2020, 3, 159, DOI:10.1038/s42004-020-00408-8.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938, DOI:10.1002/chem.200902425.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079, DOI:10.1002/anie.200803960.
- B. Rösch, T. X. Gentner, J. Eyselein, A. Friedrich, J. Langer and S. Harder, Chem. Commun., 2020, 56, 11402, 10.1039/d0cc05200k.
- J. Pan, L. Zhang, Y. He, Q. Yu and G. Tan, Organometallics, 2021, 40, 3365, DOI:10.1021/acs.organomet.1c00512.
- (a) Q. Yu, L. Zhang, Y. He, J. Pan, H. Li, G. Bian, X. Chen and G. Tan, Chem. Commun., 2021, 57, 9268, 10.1039/D1CC03448K; (b) M. Janssen, T. Frederichs, M. Olaru, E. Lork, E. Hupf and J. Beckmann, Science, 2024, 385, 318–321, DOI:10.1126/science.adp496; (c) D. Wang, W. Chen, R. Sun, J.-L. Liu, B. Wang, S. Ye, G. Tan and S. Gao, Chem, 2025, 11, 102392, DOI:10.1016/j.chempr.2024.102392.
- D. D. L. Jones, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 7087, DOI:10.1002/anie.202017126.
- L. Sun, G. Yuan, L. Gao, J. Yang, M. Chhowalla, M. H. Gharahcheshmeh, K. K. Gleason, Y. S. Choi, B. H. Hong and Z. Liu, Nat. Rev. Methods Primers, 2021, 1, 5, DOI:10.1038/s43586-020-00005-y.
- C. E. Knapp and C. J. Carmalt, Chem. Soc. Rev., 2016, 45, 1036, 10.1039/c5cs00651a.
- D. Jędrzkiewicz, J. Mai, J. Langer, Z. Mathe, N. Patel, S. DeBeer and S. Harder, Angew. Chem., Int. Ed., 2022, 61, e202200511, DOI:10.1002/anie.202200511.
- M. Gimferrer, S. Danés, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2022, 13, 6583, 10.1039/d2sc01401g.
- D. Jędrzkiewicz, J. Langer and S. Harder, Z. Anorg. Allg. Chem., 2022, 648, e202200138, DOI:10.1002/zaac.202200138.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810, DOI:10.1021/acs.organomet.8b00803.
- S. Krieck, H. Görls, L. Yu, M. Reiher and M. Westerhausen, J. Am. Chem. Soc., 2009, 131, 2977, DOI:10.1021/ja808524y.
- W. Huang and P. L. Diaconescu, Dalton Trans., 2015, 44, 15360, 10.1039/c5dt02198g.
- Y. Xie, H. F. Schaefer and E. D. Jemmis, Chem. Phys. Lett., 2005, 402, 414–421, DOI:10.1016/j.cplett.2004.11.106.
- J. Mai, J. Maurer, J. Langer and S. Harder, Nat. Synth., 2024, 3, 368, DOI:10.1038/s44160-023-00451-y.
- J. Mai, M. Morasch, D. Jędrzkiewicz, J. Langer, B. Rösch and S. Harder, Angew. Chem., Int. Ed., 2023, 62, e202212463, DOI:10.1002/anie.202212463.
- A. W. J. Bowles, J. A. Quirk, Y. Liu, G. H. Morritt, M. Freitag, G. F. S. Whitehead, A. W. Woodward, A. Brookfield, C. A. P. Goodwin, D. Collison, F. Tuna, C. L. McMullin, J. A. Dawson, E. Lu and F. Ortu, J. Am. Chem. Soc., 2024, 146, 28914–28924, DOI:10.1021/jacs.4c09408.
- J. L. Dye, Science, 1990, 247, 663, DOI:10.1126/science.247.4943.663.
- J. L. Dye, Acc. Chem. Res., 2009, 42, 1564, DOI:10.1021/ar9000857.
- C. Liu, S. A. Nikolaev, W. Ren and L. A. Burton, J. Mater. Chem. C, 2020, 8, 10551, 10.1039/d0tc01165g.
- T. Buttersack, P. E. Mason, R. S. McMullen, H. C. Schewe, T. Martinek, K. Brezina, M. Crhan, A. Gomez, D. Hein, G. Wartner, R. Seidel, H. Ali, S. Thürmer, O. Marsalek, B. Winter, S. E. Bradforth and P. Jungwirth, Science, 2020, 1086, DOI:10.1126/science.aaz7607.
- H. Hosono and M. Kitano, Chem. Rev., 2021, 121, 3121, DOI:10.1021/acs.chemrev.0c01071.
- X. Zhang and G. Yang, J. Phys. Chem. Lett., 2020, 11, 3841, DOI:10.1021/acs.jpclett.0c00671.
- A. Ellaboudy and J. L. Dye, J. Am. Chem. Soc., 1983, 105, 6490, DOI:10.1021/ja00359a022.
- S. B. Dawes, D. L. Ward, R. H. Huang and J. L. Dye, J. Am. Chem. Soc., 1986, 108, 3534, DOI:10.1021/ja00272a073.
- J. L. Dye, Acc. Chem. Res., 2009, 42, 1564, DOI:10.1021/ar9000857.
- M. Y. Redko, J. E. Jackson, R. H. Huang and J. L. Dye, J. Am. Chem. Soc., 2005, 127, 12416, DOI:10.1021/ja053216f.
- C. S. Day, C. D. Do, C. Odena, J. Benet-Buchholz, L. Xu, C. Foroutan-Nejad, K. H. Hopmann and R. Martin, J. Am. Chem. Soc., 2022, 144, 13109–13117, DOI:10.1021/jacs.2c01807.
- W. Meng, J. Wang, X. Wang, W. Wang, X. Zhang, Y. Bandoe and Z. Cheng, J. Mater. Chem. A, 2024, 12, 2583, 10.1039/d3ta06546d.
- S. Matsuishi, Y. Toda, M. Miyakawa, K. Hayashi, T. Kamiya, M. Hirano, I. Tanaka and H. Hosono, science, 2003, 301, 626, DOI:10.1126/science.1083842.
- Y. Lu, J. Li, T. Tada, Y. Toda, S. Ueda, T. Yokoyama, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2016, 138, 3970, DOI:10.1021/jacs.6b00124.
- N. Davison, J. A. Quirk, F. Tuna, D. Collison, C. L. McMullin, H. Michaels, G. H. Morritt, P. G. Waddell, J. A. Gould, M. Freitag, J. A. Dawson and E. Lu, Chem, 2023, 9, 576, DOI:10.1016/j.chempr.2022.11.006.
- N. Davison, P. G. Waddell and E. Lu, J. Am. Chem. Soc., 2023, 145, 17007, DOI:10.1021/jacs.3c06066.
- A. W. J. Bowles, Y. Liu, M. P. Stevens, I. J. Vitorica-Yrezabal, C. L. McMullin and F. Ortu, Chem.–Eur. J., 2023, 29, e202301850, DOI:10.1002/chem.202301850.
- M. Weller, T. Overton, J. Rourke, and F. Armstrong, Inorganic Chemistry, OUP, Oxford, 2018 Search PubMed.
- J. L. Dye, J. Chem. Educ., 1977, 54, 332, DOI:10.1021/ed054p332.
- J. L. Dye, Philos. Trans. R. Soc., A, 2015, 373, 2037, DOI:10.1098/rsta.2014.0174.
- E. Clementi, A. D. McLean, D. L. Raimondi and M. Yoshimine, Phys. Rev., 1964, 133, A1274, DOI:10.1103/PhysRev.133.A1274.
- S. Matalon, S. Golden and M. Ottolenghi, J. Phys. Chem., 1969, 73, 3098, DOI:10.1021/j100843a052.
- S. Golden, C. Guttman and T. R. Tuttle Jr, J. Am. Chem. Soc., 1965, 87, 135, DOI:10.1021/ja01079a036.
- J. L. Dye, C. W. Andrews and J. M. Ceraso, J. Phys. Chem., 1975, 79, 3076, DOI:10.1021/j100593a058.
- J. L. Dye, Annu. Rev. Phys. Chem., 1987, 38, 271, DOI:10.1146/annurev.pc.38.100187.001415.
- S. G. Dale and E. R. Johnson, Phys. Chem. Chem. Phys., 2017, 19, 12816, 10.1039/C7CP00882A.
- J. L. Dye, J. Phys. Chem., 1980, 84, 1084, DOI:10.1021/j100447a003.
- J. L. Dye, Angew Chem. Int. Ed. Engl., 1979, 18, 587, DOI:10.1002/anie.197905871.
- J. L. Dye, J. Phys. Chem., 1984, 88, 3842, DOI:10.1021/j150661a031.
- J. L. Dye, Sci. Am., 1977, 237, 92, DOI:10.1038/scientificamerican0777-92.
- L. D. Le, D. I sea, B. V. Eck and J. L. Dye, J. Phys. Chem., 1982, 86, 7, DOI:10.1021/j100390a004.
- N. Davison, J. M. Hemingway, C. Wills, T. Stolar, P. G. Waddell, C. M. Dixon, L. Barron, J. A. Dawson and E. Lu, Inorg. Chem., 2024, 63, 15247, DOI:10.1021/acs.inorgchem.4c02914.
- E. O. Fischer and H. P. Hofmann, Chem. Ber., 1959, 92, 482–486, DOI:10.1002/cber.19590920233.
- M. del M. Conejo, R. Fernández, E. G. Puebla, Á. Monge, C. Ruiz and E. Carmona, Angew. Chem., Int. Ed., 2000, 39, 1949–1951, DOI:10.1002/1521-3773(20000602)39:11<1949::AID-ANIE1949>3.0.CO;2-0.
- R. F. Koby, N. D. Schley and T. P. Hanusa, Angew. Chem., Int. Ed., 2021, 60, 21174, DOI:10.1002/anie.202107980.
- C. Patel, E. André-Joyaux, J. A. Leitch, X. M. Irujo-Labalde, F. Ibba, J. Struijs, M. A. Ellwanger, R. Paton, D. L. Browne, G. Pupo, S. Aldridge, M. A. Hayward and V. Gouverneur, Science, 2023, 381, 302, DOI:10.1126/science.adi1557.
- V. Gouverneur and K. Seppelt, Chem. Rev., 2015, 115, 563–565, DOI:10.1021/cr500686k.
- C. W. Scheele, Kongl. Vetensk. Acad. Handl., 1771, 32, 120 Search PubMed.
- J. Aigueperse, P. Mollard, D. Devilliers, M. Chemla, R. Faron, R. Romano, and J. P. Cuer, Fluorine compounds, inorganic, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- T. Schlatzer, C. A. Goult, M. A. Hayward and V. Gouverneur, J. Am. Chem. Soc., 2025, 147(8), 6338–6342, DOI:10.1021/jacs.4c16608.
- P. Lei, Y. Ding, X. Zhang, A. Adijiang, H. Li, Y. Ling and J. An, Org. Lett., 2018, 20, 3439, DOI:10.1021/acs.orglett.8b00891.
- K. Fujishiro, Y. Morinaka, Y. Ono, T. Tanaka, L. T. Scott, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 8163, DOI:10.1021/jacs.3c01185.
- A. C. Jones, J. A. Leitch, S. E. Raby-Buck and D. L. Browne, Nat. Synth., 2022, 1, 763, DOI:10.1038/s44160-022-00106-4.
- W. I. Nicholson, J. L. Howard, G. Magri, A. C. Seastram, A. Khan, R. R. A. Bolt, L. C. Morrill, E. Richards and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 23128, DOI:10.1002/anie.202108752.
- (a) T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwel, Angew. Chem., Int. Ed., 2006, 45, 7546, DOI:10.1002/anie.200603235; (b) T. Friščić, A. V. Trask, W. D. S. Motherwel and W. Jones, Cryst. Growth Des., 2008, 8, 1605, DOI:10.1021/cg700929e; (c) T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418, 10.1039/B815174A.
- A. J. Birch, J. Chem. Soc., 1944, 430, 10.1039/JR9440000430.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838, DOI:10.1126/science.aav5606.
- J. P. Cole, D. Chen, M. Kudisch, R. M. Pearson, C. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573, DOI:10.1021/jacs.0c05899.
- B. Thiele, O. Rieder, B. T. Golding, M. Müller and M. Boll, J. Am. Chem. Soc., 2008, 130, 14050, DOI:10.1021/ja805091w.
- J. Burrows, S. Kamo and K. Koide, Science, 2021, 374, 741, DOI:10.1126/science.abk3099.
- Y. Gao, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2023, 62, e202217723, DOI:10.1002/anie.202217723.
- K. Kondo, K. Kubota and H. Ito, Chem. Sci., 2024, 15, 4452, 10.1039/d3sc06052g.
- K. Kubota, Y. Fukuzawa, K. Kondo, Y. Gao and H. Ito, Chem. Lett., 2024, 53, 60, DOI:10.1093/chemle/upae060.
- J. V. Nallaparaju, R. Satsi, D. Merzhyievskyi, T. Jarg, R. Aav and D. G. Kananovich, Angew. Chem., Int. Ed., 2024, 63, e202319449, DOI:10.1002/anie.202319449.
- N. Davison, T. Khatun, I. Arce-Garcia, J. A. Gould, J. A. Dawson and E. Lu, Eur. J. Inorg. Chem., 2023, e202300344, DOI:10.1002/ejic.202300344.
- K. Kondo, K. Kubota and H. Ito, Nat. Synth., 2025 DOI:10.1038/s44160-025-00753-3.
- K. Kubota, K. Kondoa, T. Seoa and H. Ito, Synlett, 2022, 33, 898, DOI:10.1055/a-1748-3797.
- R. Takahashi, T. Seo, K. Kubota and H. Ito, ACS Catal., 2021, 11, 14803, DOI:10.1021/acscatal.1c03731.
- T. Seo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2020, 142, 9884, DOI:10.1021/jacs.0c01739.
- K. Kubota, K. Kondo, T. Seo, M. Jin and H. Ito, RSC Adv., 2023, 13, 28652, 10.1039/d3ra05571j.
- T. Seo, T. Ishiyama, K. Kubota and H. Ito, Chem. Sci., 2019, 10, 8202, 10.1039/c9sc02185j.
- K. Kubota, T. Seo, K. Koide, Y. Hasegawa and H. Ito, Nat. Commun., 2019, 10, 111, DOI:10.1038/s41467-018-08017-9.
- T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165, DOI:10.1021/jacs.1c00906.
- T. Seo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2023, 145, 6823, DOI:10.1021/jacs.2c13543.
- F. Puccetti, C. Schumacher, H. Wotruba, J. G. Hernández and C. Bolm, ACS Sustainable Chem. Eng., 2020, 8, 7262–7266, DOI:10.1021/acssuschemeng.0c02447.
- T. Guo, Z. Li, L. Bi, L. Fan and P. Zhang, Tetrahedron, 2022, 112, 132752, DOI:10.1016/j.tet.2022.132752.
- S. Chen, C. Fan, Z. Xu, M. Pei, J. Wang, J. Zhang, Y. Zhang, J. Li, J. Lu, C. Peng and X. Wei, Nat. Commun., 2024, 15, 769, DOI:10.1038/s41467-024-44891-2.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–4271, DOI:10.1002/adsc.202001245.
- E. Lu, J. Chu, Y. Chen, M. V. Borzov and G. Li, Chem. Commun., 2011, 47, 743, 10.1039/C0CC03212C.
- S. Harder and J. Langer, Nat. Rev. Chem, 2023, 7, 843, DOI:10.1038/s41570-023-00548-0.
- M. Westerhausen, A. Koch, H. Gçrls and S. Krieck, Chem.–Eur. J., 2017, 23, 1456, DOI:10.1002/chem.201603786.
- P. Gao, J. Jiang, Y. Fukuzawa, S. Maeda, K. Kubota and H. Ito, RSC Mechanochem., 2024, 1, 486, 10.1039/D4MR00067F.
- K. Kubota, S. Kawamura, J. Jiang, S. Maeda and H. Ito, Chem. Sci., 2024, 15, 17453, 10.1039/d4sc05361c.
- O. P. E. Townrow, C. Färber, U. Zenneck and S. Harder, Angew. Chem., Int. Ed., 2023, 63, e202318428, DOI:10.1002/anie.202318428.
- J. M. Harrowfield, R. J. Hart and C. R. Whitaker, Aust. J. Chem., 2001, 54, 423, DOI:10.1071/CH01166.
- J. V. Nallaparaju, T. Nikonovich, T. Jarg, D. Merzhyievskyi, R. Aav and D. G. Kananovich, Angew. Chem., Int. Ed., 2023, 62, e202305775, DOI:10.1002/anie.202305775.
- D. W. Peters and R. G. Blair, Faraday Discuss., 2014, 170, 83, 10.1039/c3fd00157a.
- L. Sian, A. Dall'Anese, A. Macchioni, L. Tensi, V. Busico, R. Cipullo, G. P. Goryunov, D. Uborsky, A. Z. Voskoboynikov, C. Ehm, L. Rocchigiani and C. Zuccaccia, Organometallics, 2022, 41, 547–560, DOI:10.1021/acs.organomet.1c00653.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352, 10.1039/c5dt03866a.
- N. R. Rightmire, T. P. Hanusa and A. L. Rheingold, Organometallics, 2014, 33, 5952, DOI:10.1021/om5009204.
- R. F. Koby, T. P. Hanusa and N. D. Schley, J. Am. Chem. Soc., 2018, 140, 15934, DOI:10.1021/jacs.8b09862.
- S. Kurumada, R. Yamanashi, K. Sugita, K. Kubota, H. Ito, S. Ikemoto, C. Chen, T. Moriyama, S. Muratsugu, M. Tada, T. Koitaya, T. Ozaki and M. Yamashita, Chem.–Eur. J., 2024, 30, e202303073, DOI:10.1002/chem.202303073.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldrich, Angew. Chem., Int. Ed., 2020, 60, 1702–1713, DOI:10.1002/anie.202007530.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39, DOI:10.1038/s41557-019-0365-z.
- A. L. Michalchuk and F. Emmerling, Angew. Chem., Int. Ed., 2022, 61, e202117270, DOI:10.1002/anie.202117270.
- T. H. Borchers, F. Topić, M. Arhangelskis, M. Ferguson, C. B. Lennox, P. A. Julien and T. Friščić, Chem, 2025, 11, 102319, DOI:10.1016/j.chempr.2024.09.018.
- (a) T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. Dinnebier, Nat. Chem., 2013, 5, 66–73, DOI:10.1038/nchem.1505; (b) S. T. Emmerling, L. S. Germann, P. A. Julien, I. Moudrakovski, M. Etter, T. Friščić, R. E. Dinnebier and B. V. Lotsch, Chem, 2021, 7, 1639–1652, DOI:10.1016/j.chempr.2021.04.012; (c) N. Y. Gugin, K. V. Yusenko, A. King, D. Al-Sabbagh, J. A. Villajos and F. Emerling, Chem, 2024, 10, 3459–3473, DOI:10.1016/j.chempr.2024.07.033.
- Two recent examples, see: (a) D. M. Baier, C. Spula, S. Fanenstich, S. Grätz and L. Borchardt, Angew. Chem., Int. Ed., 2023, 62, e202218719, DOI:10.1002/anie.202218719; (b) X. Xin, J. Geng, D. Zhang, H. T. Ang, H. Wang, Y. Cheng, Y. Liu, R. W. Toh, J. Wu and H. Wang, Nat. Synth., 2024, 4, 177–187, DOI:10.1038/s44160-024-00681-8.
- For representative examples, see: (a) K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504, DOI:10.1126/science.aay8224; (b) X. Wang, X. Zhang, L. Xue, Q. Wang, F. You, L. Dai, J. Wu, S. Kramer and Z. Lian, Angew. Chem., Int. Ed., 2023, 62, e202307054, DOI:10.1002/anie.202307054 , A review:; (c) E. M. Marrero, C. J. Caprara, C. N. Gilbert, E. E. Blanco and R. G. Blair, Faraday Discuss., 2023, 241, 91–103, 10.1039/D2FD00084A.
| This journal is © The Royal Society of Chemistry 2025 |