
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable oxidation of methane into formic acid using a polarized bioceramic under mild reaction conditions†
Marc
Arnau
 ab,
Jordi
Sans
*ab,
Pau
Turon
*c and
Carlos
Alemán
*abd
ab,
Jordi
Sans
*ab,
Pau
Turon
*c and
Carlos
Alemán
*abd
aIMEM-BRT Group, Departament d'Enginyeria Química, EEBE, Universitat Politècnica de Catalunya – BarcelonaTech, C/Eduard Maristany, 10-14, Barcelona, 08019, Spain. E-mail: jordi.sans.mila@upc.edu; carlos.aleman@upc.edu
bBarcelona Research Center in Multiscale Science and Engineering, Universitat Politècnica de Catalunya – BarcelonaTech, Barcelona, 08930, Spain
cB. Braun Surgical, S.A.U. Carretera de Terrassa 121, Rubí, Barcelona, 08191, Spain. E-mail: pau.turon@bbraun.com
dInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 10-12, Barcelona, 08028, Spain
First published on 27th May 2025
Abstract
The conversion of methane into useful chemicals has an enormous interest from both economic and social points of view as it is an important feedstock in chemical industry and is the second most important greenhouse gas contributor to climate change. In this work, we report the sustainable and selective conversion of methane into formic acid in batch and continuous-flow conditions using a biocatalyst made of permanently polarized. Formic acid was the only product identified in absence of UV irradiation, while a mixture of formic acid and methanol was obtained under UV light. The reaction pathway was investigated, on the one hand experimentally, by varying the reaction time, temperature and pressure in a batch reactor, in addition to the analysis of gaseous products, which allowed to understand the role of UV light in the change of selectivity, and on the other hand theoretically, using Density Functional Theory (DFT) computer calculations. The reaction pathway was experimentally investigated varying the reaction time, temperature and pressure in a batch reactor, besides gas products analysis, enabling to understand the UV light role in selectivity shift. Continuous-flow reactions using non-irradiated catalysts were conducted at 120 °C, produced a formic acid yield of 4.2 mmol per gram of catalyst and hour.
Introduction
Methane (CH4) is one of the most abundant and low-cost available feedstock.1,2 Currently, CH4 is mainly employed as source of energy and its chemical conversion is limited to the formation of syngas and the following generation of higher molecular weight compounds.3–5 However, CH4 direct oxidation into valuable oxygenated chemicals is of great significance.4–16 In particular, direct conversion of CH4 to formic acid (HCOOH) has addressed an on-going attention worldwide, as formic acid is one of the most promising molecules to develop key processes oriented to H2 storage, fuel cells, industrial chemical processes, and multiple direct uses (as, for example, leather tanning, decalcifier and cleaning product, food additive and textile dyeing).17–22Although selective transformation of CH4 into HCOOH has been reported using semiconductors, such as GaN,11 usually metals are employed as catalysts, such as ruthenium,9 vanadium,5 copper–titanium,13 copper,14 palladium15 or iron.16 Moreover, the photo-induced C–H bond breaking of CH4 and successive C–O bond formation to form other C1 products, such as CH3OH, have recently been reported,23–25 using for example gold–copper,23 gold–platinum,24 and copper.25
In recent studies, we reported a new bioceramic catalyst obtained by applying the thermal stimulation polarization (TSP) process to hydroxyapatite, Ca5(PO4)3OH (HAp).26 The TSP treatment, in which a constant voltage of 500 V is applied at 1000 °C for 2 h, transformed HAp into permanently polarized HAp (p-HAp). The unique properties of p-HAp can be summarized as follows: (1) electrical conductivity much above the intrinsic level of HAp; (2) controlled surface charge accumulation at grain boundaries; and (3) heterogeneous structure at the surface layer in which brushite (CaHPO4·2H2O) coexists with HAp due to the TSP.26–28 The enhanced electrical properties of p-HAp, which are stable in a wide temperature range without any external stimuli, conferred enhanced catalytic properties. Thus, the catalytic activity of p-HAp stable for at least 5 consecutive reaction cycles (the maximum studied) when the reaction temperature was lower than 600 °C.29 It is worth noting that p-HAp was able to trigger the reaction of CO2, CH4, N2 and water, under very mild conditions, yielding alanine and glycine (Scheme 1).30,31 More recently, the capacity of the p-HAp catalyst to convert CO2 in contact with water into ethanol and formic acid32,33 and N2 with water into ammonia,34 was demonstrated (Scheme 1).
 | ||
| Scheme 1 Processes catalyzed by p-HAp in presence of UV irradiation (amino acids and ammonia production) and without irradiation (methanol production). | ||
The high chemical stability of CH4, which is due to the strength of the C–H bond (first bond dissociation energy of 105 kcal mol−1) and its symmetric tetrahedral molecular geometry, results in difficult activation, low reactivity, weak acidity and low polarizability. Nevertheless, the growing interest in the CH4 oxidation to formic acid9,11–16 led us to question whether p-HAp could catalyze such process using mild reaction conditions. It should be noted that the use of mild conditions is a strict requirement in this case, since the use of low-cost, noble metal-free catalysts, such as p-HAp, is only justified when the processes involve low or very low energy requirements. In order to investigate the conversion of CH4 into formic acid using p-HAp, ultraporous catalyst was prepared, characterized and used in both batch and continuous-flow processes. Reactions were conducted using mild conditions with and without UV irradiation, which played a key role in the selectivity. Furthermore, in order to elucidate the mechanism, the batch process was investigated varying the reaction conditions (i.e. time, the pressure and the temperature) and computer Density Functional Theory (DFT) calculations were conducted.
Methods
Synthesis of ultraporous p-HAp
Ultraporous p-HAp was prepared using a previously described method.35 In brief, 60% wt. of Pluronic® F-127 hydrogel was added to previously synthesized grinded HAp powder in a cold room at 4 °C. The resulting homogeneous mixture was modeled using a cold spatula (<4 °C) to obtain 3D ultraporous HAp scaffolds with the desired shape. Then, the mixture was calcined at 1000 °C for 2 h to completely remove the Pluronic® F-127 hydrogel from the modeled structure.Calcined ultraporous HAp structures were catalytically activated by applying the TSP treatment. More specifically, a constant DC voltage of 500 V was applied for 1 h while the temperature was kept at 1000 °C. Details of HAp synthesis, preparation of ultraporous calcined structures and catalytic activation are provided in the ESI.†
p-HAp catalyst characterization
X-ray diffraction (XRD) diffractograms were obtained using a Bruker D8 Advance model along with Bragg–Brentano 2θ configuration and Cu Kα radiation source (λ = 0.1542 nm). Acquisitions within a 2θ range of 20°–60° in steps of 0.02° and scan speed of 2 s, were acquired using a one-dimensional Lynx Eye detector.Raman analyses were performed by means of an inVia Qontor confocal Raman microscope (Renishaw) equipped with a Renishaw Centrus 2957T2 detector and a 532 nm laser. In order to obtain representative data, 32 single point spectra were averaged.
Surface analysis by means of Brunner–Emmett–Teller (BET) was performed with a Micrometrics ASAP 2000 system using N2 and with sample degasification conditions of 1 hour at 90 °C followed by 4 hours at 300 °C. Barrett–Joyner–Halenda (BJH) model, BET model and the desorption isotherm were used for calculating the surface area and the pore size diameter.
Scanning electron microscopy (SEM) images were obtained using a Zeiss Neon40 microscope equipped with a SEM GEMINI column.
Electrochemical impedance spectroscopy (EIS) studies were performed using a Multi Autolab/M101 from Metrohm connected to a conductivity meter cell by means of two stainless steel electrodes AISI 304 isolated by a resin holder. Measurements were performed in the 1 MHz to 100 mHz frequency range and applying a 100 mV sinusoidal voltage. Samples were placed between the two electrodes and EIS measurements were performed. Electrical Equivalent Circuits (EECs) were obtained by fitting the experimental data.
Batch reactions
The reactor consisted of an inert reaction chamber (120 mL) coated with a perfluorinated polymer where both the catalyst and water were placed. All surfaces were coated with a thin film of perfluorinated polymer in order to avoid any contact between the reactants and catalyst with the stainless steel reactor surfaces, which allowed us to discard other catalytic effects. The reactor was equipped with an inlet valve for the entrance of gases and an outlet valve to recover the gaseous reaction products.Both the p-HAp catalyst (30 mg) and de-ionized liquid water (20 mL) were introduced into the reactor. After exhaustive purge with CH4 (Nippon gases, purity 4.5 (99.995%)), the chamber pressure was increased up to desired pressure of CH4 (measured at room temperature). Reactions were conducted considering different CH4 pressures (1, 2 and 4 bar), temperatures (70, 85, 100 and 120 °C) and times (6, 12, 18, 24 and 48 h). All processes were performed in triplicate, demonstrating the reproducibility and consistency of the results.
Continuous reactions
The continuous CH4 conversion into valuable chemicals was carried out using a commercial stainless steel continuous-flow reactor novaLight CUBE 100 (Peschl Ultraviolet GmbH, Germany). All reactions were conducted using a constant CH4 or CO2 flow of 1.84 mmol s−1 and without UV light (i.e. studies using the batch reactor revealed that the highest selectivity and yield of formic acid was achieved without irradiation). Reactions were performed introducing 1 mL of liquid water into the reactor and 95.2 mg of catalyst. All the studied processes were performed in triplicate, demonstrating the reproducibility and consistency of the results.X-ray photoelectron spectroscopy (XPS) analyses were conducted to determine the stability of the catalyst using a SPECS ultra-high vacuum multi-chamber system. The spectrometer was equipped with a high-intensity twin-anode XR-50 X-ray source of Mg/Al (1253 eV/1487 eV) operating with the Al anode at 150 W, positioned perpendicular to the analyzer axis, and utilizing a Phoibos 150 EP hemispherical energy analyzer with a MCD-9 detector. The stage position was digitally controlled to ensure consistency throughout the analysis. The pass energy of the hemispherical analyzer was set at 25 eV, and the energy step for high-resolution spectra was set at 0.1 eV. The pressure in the analysis chamber was maintained below 10−7 Pa, and binding energy (BE) values were referred to the C 1s peak at 284.5 eV. Data were processed using CasaXPS software (Casa Software Ltd, UK).
Inductively Coupled Plasma Mass Spectrometry (ICP-MS) tests were performed employing a 7800 ICP-MS from Agilent Technologies, equipped with a MicroMist Nebulizer sample introduction system, omega lens, collision reaction cell and a high frequency quadrupole mass analyzer.
Analysis of the reaction products
The reaction products dissolved in the liquid water (batch reactions), the reaction products condensed by the cold trap (continuous reaction), and the reaction products adsorbed on the catalyst were analyzed by 1H NMR spectroscopy. In order to desorb the reaction products from the catalyst, samples were dissolved in a solution containing 100 mM of HCl and 50 mM of NaCl with the final addition of deuterated water. All 1H NMR spectra were acquired with a Bruker Avance III-400 spectrometer operating at 400.1 MHz. The chemical shift was calibrated using tetramethylsilane as internal standard. 256 scans were recorded in all cases. In order to compare the different products obtained from the studied reactions, the areas associated to the proton contribution were normalized and calibrated through external references.For a specific catalysis reaction, gas mixture was stored using a Tedlar® bag (screw cap valve, 1 liter, 30272-U) and analyzed by gas chromatography (GC) using a Micro-Chromatograph Agilent 3000 equipped with capillary column and a thermal conductivity detector (TCD). Prior to the measurements, the samples were transferred from the Tedlar® bags to multi-sorbent bed tubes (Carbotrap, Carbopack X and Carboxen 569) to ensure proper storing and manipulation. The injector temperature was set at 100 °C and 250 μL of the sample, with syringe flush time of 30 s, was transferred onto a 30 m × 0.32 mm × 20 μm HP-Plot/Q column (split 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) with a He flow rate of 1.5 mL min−1. The column oven temperature was 40 °C isotherm.
:20) with a He flow rate of 1.5 mL min−1. The column oven temperature was 40 °C isotherm.
DFT calculations
DFT studies were performed using Quantum ESPRESSO (QE) v.7.2 package36 treating exchange correlation energy through generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhoff (PBE) formalism.37 Ultrasoft pseudopotentials (PP) were acquired from standard solid-state pseudopotentials library (SSSP Precision v 1.3.0).38 Kinetic energy cutoff for wavefunctions and charge density were set to 40 Ry and 350 Ry, respectively. Catalytic reaction temperature was simulated applying smearing through a Fermi–Dirac function with broadening consisting of 2.5 × 10−3 Ry (393 K). All the simulations performed were carried out implementing a van der Waals Grimme-D3 correction to account for weak interactions. This functionality is implemented in the Quantum ESPRESSO v.7.2 package.39p-HAp models were designed as hexagonal HAp (space group symmetry P63/m) with experimental cell parameters a = b = 9.432 Å and c = 6.881 Å while generating 50% hydroxyl groups vacancies as reported in previous studies27 while correcting total charge to the number of missing charges. Hydroxyl groups were re-oriented towards z-axis direction and latter relaxed to minimize steric hindrances, mimicking TSP process effects. According to HR-TEM studies reported in the literature, two slab supercells 2 × 1 × 2 and 2 × 2 × 1 for crystallographic planes (101) and (121), respectively, were assembled (168 atoms each) with 30 Å vacuum thickness. It is worth noting that, although this model is ideal, it is also consistent with X-ray diffractograms and Raman data (see below), which reflected that HAp was the only specie detected. Although the synthetic process was thoroughly optimized to obtain highly pure and crystalline HAp,40 in accordance with the requirements for its catalytic activation, we know that the presence of surface defects and adsorbed atmospheric contaminants were not considered in this model.
Gamma point calculation was used to reduce computational time due to slab size. The equation was used for adsorption energy calculation. Reaction pathway was obtained by calculating the adsorption energies following the equation Eads = Eslab+M − Eslab − EM where the energies of the sole slab (Eslab) and the adsorbed molecule (EM) are subtracted from the energy of the combined system (Eslab+M). For reaction pathway representation, methane energy adsorption was subtracted from all energies, thus being adsorbed methane positioned at 0.0 eV.
Results and discussion
Ultraporous HAp was prepared according to a previously reported procedure35 and, subsequently, catalytically activated by applying the TSP treatment. Although HAp can be synthesized using different procedures, such as the sonochemical and microwave methods, the hydrothermal method was used since the reaction parameters (e.g. temperature, solvent and pH) can be easily controlled.29,40 Application of this thermoelectric polarization method to HAp for producing p-HAp results not only in significant structural changes (i.e. the crystallinity increased and a brushite-like domains appeared at the outer surface layer due to the re-orientation of the OH− groups41) but also in an enhancement of the electrical properties, which was attributed to the controlled generation of vacancies, the specific orientation of the remaining OH− groups and the surface charge accumulation.28 Both structural and electrical changes were maximum when p-HAp was obtained using a calcination and polarization temperature of 1000 °C, the material derived from such conditions showing the best performance as catalyst in CO2 conversion reactions. However, the polarization temperature was found to depend on the structure of the material, for example being 150 °C for amorphous silica.42 The TSP has been extensively discussed and compared with other method in recent reviews and perspective articles.26,43The successful transformation of HAp into p-HAp was confirmed by, XRD, Raman spectroscopy and electrochemical impedance spectroscopy (EIS). The diffractogram of p-HAp (Fig. 1a) presents main characteristic peaks at 2θ = 25.9°, 31.7°, 32.8°, 34.0° and 39.8°, which correspond to the (002), (211), (300), (202) and (310) reflections of HAp, respectively (JCPDS card number 9-0077). The Raman spectrum of p-HAp (Fig. 1b) displayed the characteristic active modes of HAp with ν1 = 962 cm−1 (P–O symmetric stretching), ν2 = 400–480 cm−1 (doubly degenerated O–P–O bending), ν3 = 570–625 cm−1 (P–O triply degenerated asymmetric stretching) and ν4 = 1020–1095 cm−1 (triply degenerated O–P–O bending mode).44 Besides, the peaks at 878, 844 and 794 cm−1 were attributed to the formation of brushite (CaHPO4·2H2O) due to the polarization process, thus proving the correct catalytic activation of HAp.41 SEM micrographs of p-HAp (Fig. 1c) did not display significant morphological differences with respect HAp (Fig. S1†).
 | ||
| Fig. 1 Characterization of p-HAp: (a) XRD of HAp and p-HAp; (b) Raman spectra of HAp and p-HAp; and (c) SEM micrograph of p-HAp. | ||
The results of pore volume and porosity of p-HAp catalyst are shown in Fig. S2.† From these results, it was found that the BET surface areas and pore size were 3.85 ± 0.04 m2 g−1 and 142 ± 13 nm, respectively. EIS measurements, which were conducted on dry p-HAp and calcined HAp samples using an adapted electrochemical cell,45 evidenced the success of the TSP treatment (Fig. S3a†). The resistance obtained by adjusting experimental measures to the corresponding equivalent electrical circuits28 (Fig. S3b†) was lower for p-HAp than for calcined HAp (Rb = 175 and 1109 MΩ cm−2), as is shown in Table S1.† On the other hand, SEM micrographs of p-HAp reflected the ultraporous structure of the catalyst with average pore size of 156 ± 32 nm (Fig. 1c). The porosity, which was estimated considering five independent SEM images, was 17% ± 2%.
CH4 (4 bar; 12.26 mmol) and water (20 mL) were introduced together with the p-HAp catalyst in a batch reactor. After reacting at 120 °C for 24 h with and without UV irradiation, the reaction products dissolved in liquid water (hereafter named “supernatant”) and those adsorbed on the catalyst were identified and quantified by 1H NMR using the procedure described in the Methods section.
1H NMR spectra of the supernatant for the reactions without and with UV illumination are shown in Fig. 2 (complete spectra are shown in Fig. S4†), while those of the dissolved catalyst are displayed in Fig. S5.† Formic acid, with a singlet at 8.45 ppm, was identified as the only reaction product for the process without UV illumination (Fig. 2a), while both formic acid and methanol (with the singlet at 3.36 ppm) were identified for the reaction under UV irradiation (Fig. 2b). The singlet at 0.16 ppm, which was observed regardless of the illumination, was attributed to the solubilization of CH4 in the supernatant.
 | ||
| Fig. 2 1H NMR spectra of the supernatant for the reactions catalyzed by p-HAp (a) without and (b) with UV irradiation at 120 °C. The singlets at 8.45, 3.36 and 0.16 ppm correspond to formic acid, methanol and methane. | ||
For quantification of the reaction products, thermally- and UV-induced methane decompositions need to be considered. It is well-known that the methyl radical can be produced by the reaction of methane with hydroxyl radical, as indeed occurs in the troposphere:46
 | (1) |
Although hydroxyl radicals can be generated by the thermal decomposition of water,47 UV irradiation is known to significantly enhance their production.48 Accordingly, in order to correctly quantify the catalytic yield of formic acid and methanol from such decompositions, blank reactions without catalyst and with non-polarized HAp were performed considering 4 bar of CH4 and 20 mL of water at 120 °C with and without UV irradiation. Results were very similar for both blanks.
1H-NMR spectra of the supernatant after 24 h under such conditions (Fig. S6 and S7† for the blanks without catalyst and with non-polarized HAp as catalyst, respectively), evidenced a small amount of formic acid (i.e. around 0.7 μmol) for the reactions without illumination and of both formic acid and methanol (i.e. around 1.6 and 0.4 μmol, respectively) for the reactions with UV irradiation, regardless of the blank. Furthermore, the solubilisation of CH4 was observed for both blanks, which was consistent with the results obtained for the reactions with p-HAp as catalyst.
The yields of formic acid and methanol in the supernatant and in the catalyst (Table S2†), which were obtained by calibrating the 1H-NMR signals using known concentrations of such products, were expressed in terms of μmol per gram of catalyst (μmol gc−1). It is worth mentioning that yields in the supernatant were corrected by subtracting the area of formic acid and methanol peaks obtained in the corresponding blank reaction (i.e. with or without UV irradiation). The total yield for a given product has been defined as the sum of the yield of product adsorbed in the catalyst and the yield in the supernatant once corrected with the corresponding blank.
The total yields, which are listed in Table 1, show that the highest yield of formic acid was achieved for the reaction conducted without UV irradiation (by a factor ×2.6), suggesting that the adsorption of CH4 on the catalyst is disfavoured when an excess of energy is provided by UV illumination. Furthermore, while the reaction without UV was 100% selective towards formic acid in the identified products, the ratio between the yields of formic acid and methanol was 1.8 for the reaction with UV. The conversion efficiency (in %, mmol of product with respect to the initial 12.26 mmol of CH4) was around 1.1% ± 0.1% for the reaction without UV and around 0.7% ± 0.1% for the reaction with UV (Table 1). On the other hand, it is worth mentioning that control reactions in which the p-HAp catalyst was replaced by non-polarized HAp led to results very similar to those described above for the blank reactions (i.e. the yields of reaction products were lower than 4 μmol gc−1 due to the (photo)thermal decomposition of CH4).
| Formic acid | Methanol | |
|---|---|---|
| Without UV | 136.9 ± 8.6 μmol gc−1 (1.12% ± 0.07%) | — |
| With UV | 51.9 ± 8.3 μmol gc−1 (0.42% ± 0.06%) | 28.2 ± 3.3 μmol gc−1 (0.23% ± 0.03%) |
Inspection of values listed in Table S2† shows that the yield of products was around twenty times higher in the supernatant than in the dissolved catalyst. This observation has important implications, such as facilitating removal of reaction products from the reactor and avoiding catalyst poisoning from excess adsorption of products. Additionally, the confirmation of the formic acid origin by isotopic labeling experiment was performed using a 4 bar gas mixture of 13CH4 and 12CH4, (1 and 3 bar respectively). Batch reaction with p-HAp catalyst for 48 hours without UV allowed direct observation of the double peak for labeled formic acid (H13COOH), due to the coupling between 13C and 1H, at 7.92 and 8.45 ppm (Fig. 3), with the latter peak being overlapped with the non-labelled formic acid (H12COOH) peak displayed by its different intensity.49
 | ||
| Fig. 3 1H NMR spectra of the supernatant after the reaction for the isotope labelling experiment without UV irradiation. | ||
The fact that formic acid is the only reaction product in the process without UV indicates that p-HAp promotes the oxidation of CH4, which is mediated through hydroxyl radicals supplied by p-HAp. However, in the process with UV light, the appearance of two reaction products, formic acid and methanol, opens up three possibilities: (1) competition between two oxidation mechanisms for CH4, one leading to the formation of formic acid and the other leading to obtain methanol; (2) oxidation of CH4 to methanol, which in turn is subsequently oxidized to formic acid; and (3) the oxidation of CH4 to formic acid that is, subsequently, reduced to methanol.
The influence of the reaction time, temperature and pressure were examined in order to gather more information about those processes and, also, to clarify what is the reaction mechanism when UV light is used. Results are summarized in Fig. 4. As expected, for those reactions without UV, the yield of formic acid increased rapidly with both time and temperature (Fig. 4a and b). Conversely, the effect of pressure was moderate, indicating that the adsorption of CH4 on the catalyst stabilized at 2 bar (Fig. 4c). On the other hand, acetic acid was observed through CH3 singlet at 2.05 ppm (Fig. S8†) at long times (48 h) for the reaction with UV irradiation (Fig. 4d). Thus, under such conditions, the energy required for C–C bonds formation was reached, giving place to C2 product synthesis. Furthermore, UV illumination resulted in a reduction of formic acid yield when time, temperature and pressure were increased, while increment of methanol yield was observed in all three cases (Fig. 4d–f). Thus, the yield curves for methanol and formic acid allowed to discard the oxidation of CH4 to methanol and of the latter to formic acid as a possible reaction mechanism.
 | ||
| Fig. 4 Total yield of products for the reactions (a)–(c) without and (d)–(f) with UV illumination varying the experimental conditions: (a) and (d) 120 °C and 4 bar, varying the reaction time; (b) and (e) 4 bar and 24 h, varying the temperature; and (c) and (f) 120 °C and 24 h, varying the pressure. | ||
In order to get more information about the mechanism, the gas mixture obtained after 48 h using CH4 (4 bar) and H2O (20 mL) at 120 °C without UV irradiation and p-HAp as catalyst was analyzed by gas chromatography, with the results being displayed in Table 2. It should be emphasized that, before performing this reaction, intensive gas purge of both the reaction chamber and the gas transport tubes was carried out using a vacuum pump to ensure almost no presence of other contaminants such as CO2. In addition, the CH4 gas containers used in this work presented a 99.995% purity and, therefore, CO2 contamination from the precursor gas was insignificant (if any). Unfortunately, N2 contamination was unavoidable since commercial Tedlar® bags used for gas storage were cleaned with dinitrogen.
| Gas mixture (%) | CH4 | CO2 | N2 | O2 |
|---|---|---|---|---|
| 92.92 ± 0.04 | 0.12 ± 0.03 | 5.69 ± 0.06 | 1.27 ± 0.02 |
Gas chromatography revealed a significant yield of CO2 in the gas products (150 ± 38 μmol gc−1) for the 48 h reaction with UV light, suggesting the oxidation of methane to ˙COOH radicals. Those species are adsorbed on the catalyst, later enabling their evolution into formic acid or CO2. Therefore, the noteworthy presence of CO2 and the preferential oxidation towards carboxylic radicals allow discarding the competing CH4 oxidation towards formic acid and methanol. Following this reasoning, UV light acts as an additional energy source, facilitating formic acid reduction to methanol (in addition, promoting C–C bond formation). Also, the proposed mechanism is consistent with the time-increasing yield observed for methanol in Fig. 4d. Despite such consistency, the existence of other possible competing reaction pathways cannot be discarded. It is also worth mentioning that H2 released by CH4 oxidation was not detected, which was mainly attributed to its high volatility and logistic storage limitations due to Tedlar® bags usage.
DFT calculations confirmed the proposed mechanism. While the surface binding sites of p-HAp are displayed in Fig. 5a, the reaction pathways calculated considering both the (101) and (121) crystallographic planes are shown in Fig. 5b and c. Besides, Fig. S9† provides a simpler scheme with improved annotations clarifying all the steps depicted in Fig. 5b and c in terms of chemical species. Thus, the calculated adsorption energies showed that the pathway associated to the oxidation of methane to ˙COOH is the preferred one. The adsorption of such radical is more favored for the (101) plane for the (121) one by 3.8 kcal mol−1. The proposed intermedium specie can evolve towards formic acid, methanol and CO2. While the adsorption of formic acid is the preferred for the (101) plane, the adsorption is the most favored for the (121) plane. In both cases, the adsorption of CO2 was the least favored.
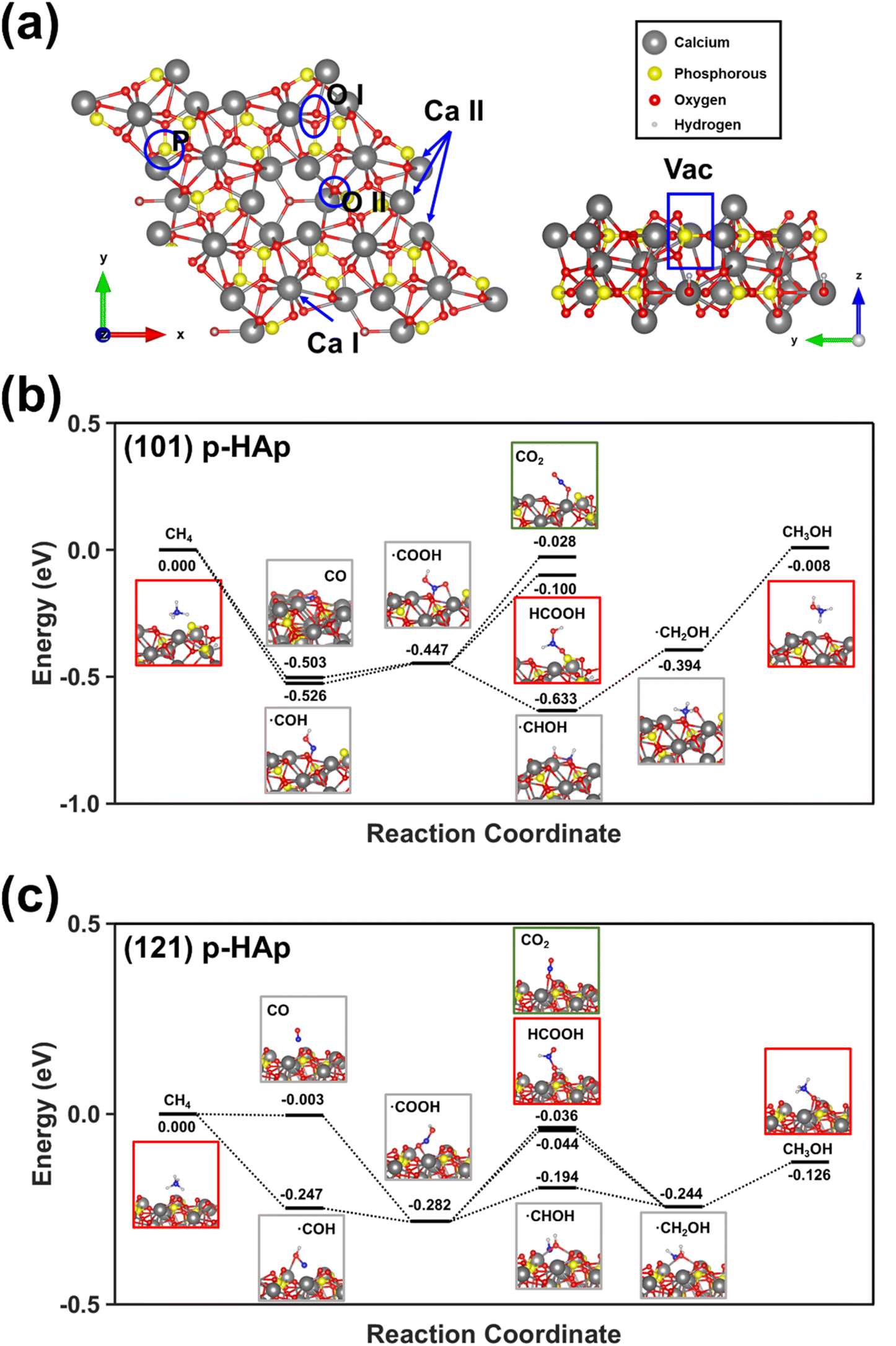 | ||
| Fig. 5 (a) Schematic drawing for a 2 × 2 × 1 p-HAp supercell where the different surface binding sites can be observed. (b) and (c) Reaction pathways calculated considering the (b) (101) and (c) (121) crystallographic planes of p-HAp. | ||
Although only 0.12% of CO2 was detected in the reaction without UV light (Table 2), p-HAp has been reported capable of catalyzing the reduction reaction of CO2 into formic acid.41 Therefore, to discard CO2 conversion to formic acid in reactions without UV light, continuous-flow reaction studies were deemed necessary to corroborate the sole oxidation of CH4.
Although only 0.12% of CO2 was detected in the reaction without UV light (Table 2), p-HAp has been reported capable of catalyzing the reduction reaction of CO2 into formic acid.41 Therefore, to discard CO2 conversion to formic acid in reactions without UV light, continuous-flow reaction studies were deemed necessary to corroborate the sole oxidation of CH4.
On the other hand, although the stability of the catalyst against the temperature and the number of catalytic cycles was explicitly afforded in a recent study,29 in this work we re-examined the effect of the number of cycles on the structure of p-HAp catalyst. The Raman spectra of the p-HAp catalyst as prepared and after five reaction cycles, which are compared in Fig. 6a, show that no lattice structural distortion occurred during the reactions. The morphological changes observed after five reaction cycles, which are displayed in Fig. 6b and c, are consistent with previous studies on the stability of p-HAp.29 Accordingly, the porous structure of as prepared p-HAp (Fig. 6b) transformed into a field of nanorods of micrometric length (Fig. 6c) able to maintain the catalytic activity because of the high surface area of such nanostructures. Thus, the yield of formic in the reaction without UV was preserved after five cycles (i.e. 136.9 ± 8.6 and 132.8 ± 3.2 μmol gc−1 for the first and fifth cycle, respectively).
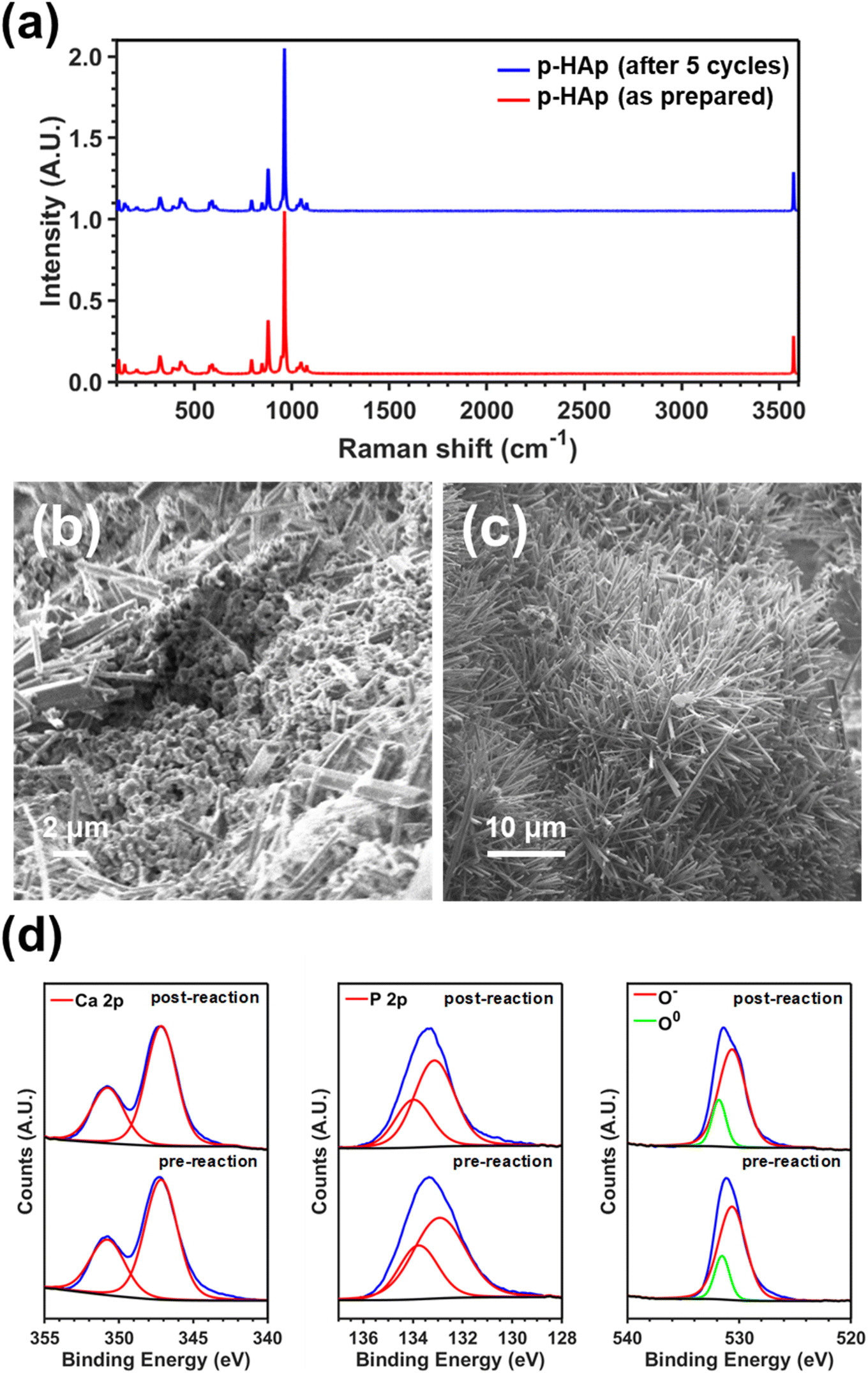 | ||
| Fig. 6 (a) Raman spectra of p-HAp as prepared and after five batch reaction cycles without UV. (b) and (c) SEM micrographs of p-HAp (b) as prepared and (c) after five batch reaction cycles without UV. (d) Ca 2p (left), P 2p (middle) and O 1s (right) high resolution XPS spectra for the p-HAp catalyst before and after batch reaction at 120 °C for 24 h with 4 bar of CH4. The C 1s spectra are displayed in Fig. S10.† | ||
XPS analyses of the p-HAp catalyst before and after the reaction were performed to ascertain any surface modification resulting from methane oxidation. High resolution Ca 2p, P 2p and O 1s spectra are displayed in Fig. 6d. While no shift was observed for Ca 2p and P 2p peaks, which is consistent with the above discussed stability of the catalyst, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbon species were detected post reaction attributed to adsorbed carboxylic group from formic acid/acetic acid. This hypothesis was corroborated by the C 1s high resolution spectra, which are displayed in Fig. S10.† To further confirm the stability of the catalyst, the possible leaching of Ca2+ was measured using ICP-MS after a batch reaction at 120 °C for 48 h using CH4 (4 bar) and H2O (20 mL) without UV irradiation. The amount of Ca2+ in the supernatant was found to be as low as 4.18 ppm (±1.84%).
O carbon species were detected post reaction attributed to adsorbed carboxylic group from formic acid/acetic acid. This hypothesis was corroborated by the C 1s high resolution spectra, which are displayed in Fig. S10.† To further confirm the stability of the catalyst, the possible leaching of Ca2+ was measured using ICP-MS after a batch reaction at 120 °C for 48 h using CH4 (4 bar) and H2O (20 mL) without UV irradiation. The amount of Ca2+ in the supernatant was found to be as low as 4.18 ppm (±1.84%).
The reactor used to explore the continuous-flow transformation of CH4 into formic acid using p-HAp as catalyst is described in Fig. S11.† The reactor was filled with 1 mL of de-ionized water, which was homogeneously distributed, and four specimens of the p-HAp catalyst. The reaction products in the gas phase were condensed in a cold trap that was connected to the outlet valve. The inlet valve was connected to a CH4 cylinder to achieve a constant flow of 1.84 mmol s−1 controlled by a back-pressure regulator. In order to have available enough water, wet CH4 was produced by bubbling the gas in liquid water through a bubbler arranged between the gas cylinder and the inlet valve. The reaction was conducted at 120 °C without UV illumination, and products were analysed by 1H NMR at selected time intervals. Furthermore, in order to stablish a reliable comparison between the conversion of CH4 and CO2 gases, the same reaction was performed under identical experimental conditions using CO2 as sole reagent.
Results, which are displayed in Fig. 7, led to the formation of formic acid with complete selectivity. Table 3 compares the selectivity obtained in the batch and continuous flow reaction. The reaction exhibited the maximum yield of formic acid (expressed as μmol of product per gram of catalyst and min), 95.9 ± 11.1 μmol gc−1 min−1, after 10 min. Then, the yield of formic acid decreased and stabilized at around 70 μmol gc−1 min−1. This observation was attributed to the evaporation of liquid water inside of the reactor that occurred in the first minutes of the reaction, which resulted in an increment of the yield. However, after 10 min the availability of water molecules decreased to only those supplied by continuous feeding of wet CH4 and the yield stabilized. Table S3† compares the formic acid yield recently reported for noble-metal free catalysts with that obtained for p-HAp, considering both batch and continuous reactions. As it can be seen, in spite of its simplicity, the level of efficiency of the latter catalyst is very significant.
 | ||
| Fig. 7 Evolution of the non-accumulated yield of formic acid, which was identified by 1H NMR as the only reaction product, against the reaction time for the continuous process at 120 °C conducted using CH4 or CO2 as reagent (1.84 mmol s−1) and introducing 1 mL of liquid water and the p-HAp catalyst in the reactor. The yields were derived from the 1H NMR spectra of the liquids collected in the cold trap at the displayed time intervals. | ||
| Reaction | Irradiation | Formic acid (%) | Methanol (%) |
|---|---|---|---|
| Batch | With UV | 64.8 | 35.2 |
| Without UV | 100 | 0 | |
| Continuous flow | Without UV | 100 | 0 |
A similar reaction profile was obtained for CO2, even though in this case the production of formic acid at 10 min was of 814 ± 90 μmol gc−1 min−1 (Fig. 3, inset) and the yield stabilized at around 70 μmol gc−1 min−1.50 These values indicate the yield of formic acid from CO2 and CH4 fixation catalysed by p-HAp in reactors able to supply liquid water continuously can reach 49 and 5.7 mmol gc−1 h−1, respectively, at the conditions studied in this work (120 °C and a gas flux of 1.84 mmol s−1), these values decreasing to 4.8 and 4.2 mmol gc−1 h−1, respectively, when wet gases are used.
Overall, this study demonstrates the potential of p-HAp to convert CH4 in valuable chemical products. However, before industrialization several issues related with the p-HAp catalyst, such as the stability, scalability and long-term usage, need to be considered. It should be noted that in an early study we demonstrated that the electrical and structural properties of p-HAp were maintained for, at least, a period of 3 months,51 while in a very recent one we proposed a process to regenerate the catalyst after CO2 fixation processes.29 Furthermore, this work included 5-cycle batch studies to study the p-HAp stability. However, these studies do not guarantee long-term catalytic activity, and it is necessary to investigate the conversion of CH4 to formic acid over long or very long periods of time to evaluate this point. These studies would allow us to accurately determine the economic viability of the catalyst since, although it is a low-cost bioceramic, it must have a useful life greater than a certain threshold for the process to be profitable. This threshold will be defined by the degree of conversion when the process is carried out on a pilot scale. It is important to emphasize that the continuous process presented in this article cannot yet be considered a pre-pilot scale since the characteristics of the reactor (Fig. S11†) do not allow us to control the pressure drop across the catalyst. In this sense, our next step will be to repeat this study on a pre-pilot scale using continuous tubular reactors with a length of 30 cm and a diameter of 1–3 cm.
Conclusions
In summary, we report the conversion of inert methane to formic acid using p-HAp under mild conditions. While formic acid was the only product identified in absence of UV irradiation with a yield of 4.2 mmol gc−1 h−1 in continuous flow without continuous water supply (5.7 mmol gc−1 h−1 with continuous water supply), a mixture of formic acid and methanol is obtained in the process with UV illumination. Although formic acid is only obtained in the first steps of the reaction, its yield decreases over time, evolving towards the formation of methanol, whose yield increases over time. Results suggest a mechanism based on methane oxidation to carboxylic radicals, which evolve to formic acid and, in the process with UV light, to CO2. Future efforts will focus on improving the selectivity towards methanol in reactions with UV by promoting the reduction of formic acid. Thus, the selective production of formic acid or methanol depending on the presence or absence of UV light would enrich the field of C1 chemistry, while it would play a significant role in the renewable production of formic acid and methanol.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Authors declare that the preparation and application of permanently polarized hydroxyapatite as catalyst was patented by the Universitat Politècnica de Catalunya and B. Braun Surgical, S.A.U. (EP16382381, EP16382524, P27990EP00, PCT/EP2017/069437, P58656 EP, P59205 EP, P59091 EP, P59008 EP, P59528 EP, 2021P00074 EP, P 59985 EP).Acknowledgements
Authors acknowledge the Agència de Gestió d'Ajuts Universitaris i de Recerca (2021 SGR 00387) and CEX2023-001300-M funded by MCIN/AEI/10.13039/501100011033/FEDER for financial support. Support for the research of C. A. was also received through the prize “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya. M. A. thanks the Generalitat de Catalunya for a FI Fellowship.References
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- H. D. Gesser, N. R. Hunter and C. B. Prakash, Chem. Rev., 1985, 85, 235–244 CrossRef CAS.
- H. Cruchade, I. C. Medeiros-Costa, N. Nesterenko, J.-P. Gilson, L. Pinard, A. Beuque and S. Mintova, ACS Catal., 2022, 12, 14533–14558 CrossRef CAS.
- E. Kondratenko, T. Peppel, D. Seeburg, V. A. Kondratenko, N. Kalevaru, A. Martin and S. Wohlrab, Catal. Sci. Technol., 2017, 7, 366–381 RSC.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F.-S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- D. Hu, A. Addad, K. B. Tayeb, V. V. Ordomsky and A. Y. Khodakov, Cell Rep. Phys. Sci., 2023, 4, 101277 CrossRef CAS.
- X. Wei, L. Ye and Y. Yuan, J. Nat. Gas Chem., 2009, 18, 295–299 CrossRef CAS.
- J.-T. Han, H. Su, L. Tan and C.-J. Li, iScience, 2023, 26, 105942 CrossRef CAS PubMed.
- K. Ohkubo and K. Hirose, Angew. Chem., Int. Ed., 2018, 57, 2126–2129 CrossRef CAS PubMed.
- A. Prajapati, R. Sartape, N. C. Kani, J. A. Gauthier and M. R. Singh, ACS Catal., 2022, 12, 14321–14329 CrossRef CAS.
- S. A. Yashnik, V. V. Boltenkov, D. E. Babushkin, T. A. Surovtsova and V. N. Parmon, Kinet. Catal., 2022, 63, 555–568 CrossRef CAS.
- F. Li, L. Lei, J. Yi, C. Dou, Z. Meng and P. Wang, Catal. Lett., 2023, 153, 1847–1858 CrossRef CAS.
- L. Lin, G. Zhang, L. Kang, T. Yu, Y. Su, G. Zeng, S. Chu and W. Luo, ChemCatChem, 2023, 15, e202201234 CrossRef CAS.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- H. Kawanami, Y. Himeda and G. Laurenczy, Adv. Inorg. Chem., 2017, 70, 395–427 CrossRef CAS.
- S. Pradeep, M. Sathish, K. J. Sreeram and J. R. Rao, ACS Omega, 2021, 6, 12965–12976 CrossRef CAS PubMed.
- A. K. Khangura, S. Gupta, A. Gulati and S. T. Singh, Int. J. Oral Maxillofac. Pathol., 2021, 25, 463–469 CrossRef PubMed.
- M. Yadav, S. Dhyani, P. Joshi, S. Awasthi, S. Tanwar, V. Gupta, D. K. Rathore and S. Chaudhuri, Front. Microbiol., 2022, 24, 966207 CrossRef PubMed.
- K. Lech and M. Jarosz, Anal. Bioanal. Chem., 2011, 399, 3241–3251 CrossRef CAS PubMed.
- L. Luo, Z. Gong, Y. Xu, J. Ma, H. Liu, J. Xing and J. Tang, J. Am. Chem. Soc., 2022, 144, 740–750 CrossRef CAS PubMed.
- Y. Xu, D. Wu, P. Deng, J. Li, J. Luo, Q. Chen, W. Huang, C. M. Shim, C. Jia, Z. Liu, Y. Shen and X. Tian, Appl. Catal., B, 2022, 308, 121223 CrossRef CAS.
- W. Li, Z. Li, H. Zhang and Z. Zao, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2206619120 CrossRef CAS PubMed.
- J. Sans, M. Arnau, P. Turon and C. Alemán, Mater. Horiz., 2022, 9, 1566–1576 RSC.
- J. Sans, M. Arnau, F. Estrany, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2021, 8, 2100163 CrossRef CAS.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2022, 9, 2101631 CrossRef CAS.
- M. Arnau, J. Sans, J. L. Tamarit, M. Romanini, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2024, 11, 2400422 CrossRef CAS.
- J. Sans, V. Sanz, L. J. del Valle, J. Puiggalí, P. Turon and C. Alemán, J. Catal., 2021, 397, 98–107 CrossRef CAS.
- M. Rivas, L. J. del Valle, P. Turon, C. Alemán and J. Puiggalí, Green Chem., 2018, 20, 685–693 RSC.
- J. Sans, V. Sanz, P. Turon and C. Alemán, ChemCatChem, 2021, 13, 5025 CrossRef CAS.
- J. Sans, G. Revilla, V. Sanz, J. Puiggalí, P. Turon and C. Alemán, Chem. Commun., 2021, 57, 5163–5166 RSC.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Chem. Eng. J., 2022, 446, 137440 CrossRef CAS.
- J. Sans, M. Arnau, J. J. Roa, P. Turon and C. Alemán, ACS Appl. Nano Mater., 2022, 5, 8526–8536 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dl Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebaure, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. MartinSamos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarell, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Prandini, A. Marrazzo, I. E. Castelli, N. Mounet and N. Marzari, npj Comput. Mater., 2018, 4, 72 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Sans, V. Sanz, J. Puiggalí, P. Turon and C. Alemán, Cryst. Growth Des., 2021, 21, 748–756 CrossRef CAS.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Chem. Eng. J., 2022, 433, 133512 CrossRef CAS.
- M. Arnau, I. Teixidó, J. Sans, P. Turon and C. Alemán, Sustainable Energy Fuels, 2024, 8, 5937–5949 RSC.
- J. Sans, M. Arnau, A. Fontana-Escartín, P. Turon and C. Alemán, Chem. Mater., 2023, 35, 3765–3780 CrossRef CAS.
- J. Sans, J. Llorca, V. Sanz, J. Puiggalí, P. Turon and C. Alemán, Langmuir, 2019, 35, 14782–14790 CrossRef CAS PubMed.
- F. Müller, C. A. Ferreira, D. S. Azambuja, C. Alemán and E. Armelin, J. Phys. Chem. B, 2014, 118, 1102–1112 CrossRef PubMed.
- D. S. Stevenson, A. Zhao, V. Naik, F. M. O'Connor, S. Tilmes, G. Zeng, L. T. Murray, W. J. Collins, P. T. Griffiths, S. Shim, L. W. Horowitz, L. T. Sentman and L. Emmons, Atmos. Chem. Phys., 2020, 20, 12905 CrossRef CAS.
- N. K. Srinivasan and J. V. Michael, Int. J. Chem. Kinet., 2006, 38, 211–219 CrossRef CAS.
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS PubMed.
- R. Hudson, R. de Graaf, M. S. Rodin and V. Sojo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32900930 CrossRef PubMed.
- M. Arnau, L. Pielsticker, W. Hetaba, J. Casanovas, P. Turon, C. Alemán and J. Sans, ACS Catal., 2025 DOI:10.1021/acscatal.5c02426.
- M. Rivas, J. J. del Valle, E. Armelin, O. Bertran, P. Turon, J. Puiggalí and C. Alemán, ChemPhysChem, 2018, 17, 1746–1755 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00134j |
| This journal is © The Royal Society of Chemistry 2025 |