
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnassisted visible-light–driven NADH regeneration based on a dual-photoelectrode system†
Koya
Kano
a,
Masanobu
Higashi
 *b and
Yutaka
Amao
*ac
*b and
Yutaka
Amao
*ac
aGraduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
bInstitutional Advancement and Communications, Kyoto University, Yoshida-Honcho, Sakyo-ku, Kyoto 606-8501, Japan
cResearch Centre for Artificial Photosynthesis (ReCAP), Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: amao@omu.ac.jp
First published on 30th May 2025
Abstract
NADH regeneration is crucial for biocatalytic processes. One promising example is visible-light–driven NADH regeneration using water as an electron source. Here, we demonstrate for the first time, to the best of our knowledge, visible-light–driven electrochemical NADH regeneration using water as an electron source without the need for an external bias. This is achieved by combining an IrOx/TaON (or RhOx/TaON) photoanode, CdS/CuInS2 photocathode and [Cp*Rh(bpy)(H2O)]2+, as a catalyst for regioselective reduction of NAD+. Furthermore, application of this system to the production of L-lactate from pyruvate using lactate dehydrogenase was attempted.
Reduced nicotinamide adenine dinucleotide (NADH) is widely used as a cofactor functioning as an electron donor in various reactions catalysed by oxidoreductases (i.e., redox enzymes). Efficiently regenerating consumed cofactors (e.g., reducing NAD+ to NADH) is crucial for achieving a broad application of oxidoreductases because of their high cost, low stability, and the stoichiometric amount that needs to be supplied. Various ways of regenerating NADH, for example using biocatalytic, chemical,1–3 electrochemical,4–7 photochemical,8–16 homogeneous catalytic,17 and heterogeneous catalytic18 methods, have been investigated. Among these methods, photochemistry has attracted particularly considerable attention in recent years because clean and abundant solar energy can be converted into chemical energy. It is therefore desirable to develop a visible-light–driven NADH-regeneration system. However, in the process of regenerating NADH, isomers (1,2- or 1,6-NADH) and dimeric NAD+ are produced in addition to 1,4-NADH. Unlike 1,4-NADH, these isomers and dimeric NAD+ have no function as a coenzyme for oxidoreductase, suppression of their production is an important issue for developing an NADH-regeneration system. Rh complex [Cp*Rh(bpy)(H2O)]2+ (Cp* = pentamethylcyclopentadienyl, bpy = 2,2′-bipyridyl) acting as a proton- and electron-transfer catalyst has been widely used and studied for the regiospecific reduction of NAD+ to only 1,4-NADH.19–22 Many NADH-regeneration systems using photocatalytic dye with [Cp*Rh(bpy)(H2O)]2+ have been reported.11–16 However, most light-driven NADH-regeneration systems require a sacrificial electron donor, such as triethanolamine (TEOA); so, it is desirable to develop an NADH-regeneration system that uses water as an electron source. Moreover, there are few stable semiconductor photocatalysts capable of catalyzing both water oxidation and [Cp*Rh(bpy)(H2O)]2+ reduction under visible-light irradiation. With the aim of achieving visible-light-driven NADH regeneration using water as an electron source, a Z-scheme photoelectrochemical system using two semiconductor-based photoelectrodes (a photoanode and a photocathode) and [Cp*Rh(bpy)(H2O)]2+, as shown in Fig. 1 was devised.
 | ||
| Fig. 1 Z-scheme mechanism of NADH regeneration using a system including a photoanode, a photocathode and [Cp*Rh(bpy)(H2O)]2+ under visible-light irradiation. | ||
In this system, the reaction is broken up into two stages: one for reduction of [Cp*Rh(bpy)H2O]2+ and the other for water oxidation. For [Cp*Rh(bpy)H2O]2+ reduction system, the photoexcited electrons reduce [Cp*Rh(bpy)H2O]2+, and the photogenerated holes received the photoexcited electrons from the water oxidation system. Conversely, for water oxidation system, the photogenerated holes oxidize water to O2, and photoexcited electrons are injected into the valence band of the [Cp*Rh(bpy)H2O]2+ reduction system. Note that the [Cp*Rh(bpy)H2O]2+ reduction and water oxidation systems do not require water oxidation and [Cp*Rh(bpy)H2O]2+ reduction, respectively. Therefore, various semiconductor materials can be used in the Z-scheme system.
In this study, CuInS2 and TaON were employed as a photocathode and a photoanode, respectively. CuInS2 has a small bandgap (1.5 eV), allowing visible-light absorption, and a relatively high (negative) conduction band level and therefore is used for various photocatalytic and photoelectrochemical reactions.23–25 We recently reported that a CdS-modified CuInS2 photocathode can reduce CO2 to formate when combined with a biocatalyst system using formate dehydrogenase as the enzyme and methyl viologen as an artificial coenzyme, instead of NADH.26 However, oxidation of water using CuInS2 is not thermodynamically favored. Conversely, TaON, in addition to having a small bandgap (2.4 eV) allowing visible-light absorption, also has a valence band level for water oxidation. We reported photoelectrochemical water oxidation over a TaON photoanode under visible-light irradiation by loading an appropriate water oxidation cocatalyst, such as CoOx, RhOx and IrOx.27–29 In the current work, visible-light–driven electrochemical NADH regeneration using water as an electron source, without an external bias, was achieved by combining a TaON photoanode, a CuInS2 photocathode, and [Cp*Rh(bpy)(H2O)]2+. Its application to a subsequent biocatalytic reaction involving the production of L-lactate from pyruvate using the regenerated NADH and lactate dehydrogenase (LDH) were demonstrated here for the first time, to the best of our knowledge. The CuInS2 and TaON electrodes were fabricated according to previously reported methods.29,30 The CuInS2 samples were modified with CdS using the chemical bath deposition method.31 The resulting electrode was calcined at 673 K for 30 min under N2 gas flow, with the obtained electrode denoted as CdS/CuInS2. Water oxidation cocatalysts (5 wt% CoOx, 0.7 wt% RhOx and 1 wt% IrOx, calculated as metal; optimal loadings for CoOx28 and RhOx29) were each loaded onto TaON using the impregnation method, with the obtained samples denoted as CoOx/TaON, RhOx/TaON, and IrOx/TaON, respectively. The electrochemical cell used for the photocurrent measurements consisted of a prepared electrode, Pt counter electrode, Ag/AgCl reference electrode, and phosphate buffer solution (pH = 7.0). In some cases, [Cp*Rh(bpy)(H2O)]2+ (1.0 mM) was added to the solution. The potential of the working electrode was controlled using a potentiostat. The solution was purged with Ar gas for more than 20 min prior to the measurement. The electrodes were irradiated using a 300 W Xe lamp fitted with an L-42 cut-off filter. The detailed procedures are described in ESI.† Reduction properties of [Cp*Rh(bpy)(H2O)]2+ over a CdS/CuInS2 photocathode were investigated, as shown in Fig. 2. Without [Cp*Rh(bpy)(H2O)]2+, the CdS/CuInS2 photocathode exhibited a negative cathodic photoresponse at a potential more negative than approximately −0.1 V vs. Ag/AgCl. This photocurrent was probably derived from the reduction of H+. In the case of the phosphate buffer solution to which [Cp*Rh(bpy)(H2O)]2+ was added, the photocurrent over the CdS/CuInS2 photocathode significantly increased, strongly suggesting that the CdS/CuInS2 photocathode reduced [Cp*Rh(bpy)(H2O)]2+.
 | ||
| Fig. 2 Current–potential curves for the CdS/CuInS2 photocathode in phosphate buffer solution (pH = 7.0) with and without [Cp*Rh(bpy)(H2O)]2+ under chopped visible-light irradiation. | ||
Next, reduction of NAD+ to NADH was performed via [Cp*Rh(bpy)(H2O)]2+ at −0.2 V vs. Ag/AgCl using a two-compartment cell divided by a Nafion membrane. [Cp*Rh(bpy)(H2O)]2+ (1.0 mM) and NAD+ (2.0 mM) were added to the solution of the cathode side. Generally, although it has been reported that 1,2-, 1,4-, 1,6-NADH isomers and NAD2 are produced in the NAD+ reduction, high-performance liquid chromatography analysis revealed that NAD+ was selectively reduced to 1,4-NADH in our developed system, as shown in Fig. S1.† Hereafter, 1,4-NADH is simply denoted as NADH. Fig. 3 shows the time courses of photocurrent density and the amount of NADH produced over the CdS/CuInS2 photocathode under visible-light irradiation.
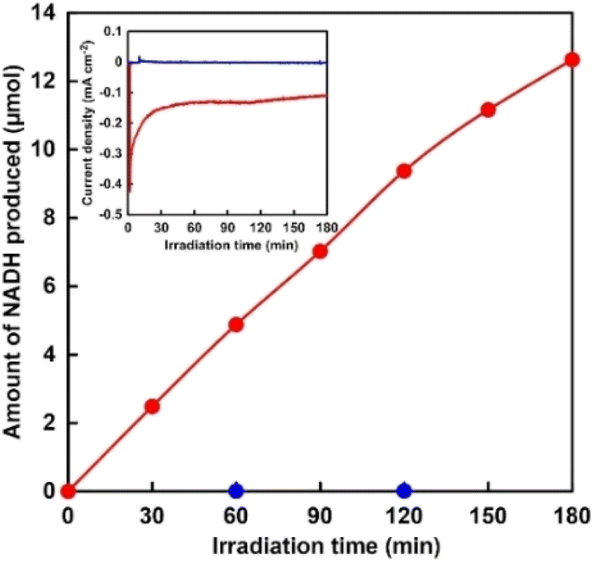 | ||
| Fig. 3 Time dependence of NADH production with the CdS/CuInS2 photocathode at −0.2 V vs. Ag/AgCl under visible-light irradiation. Inset: time dependence of photocurrent density. Red: in the presence of [Cp*Rh(bpy)(H2O)]2+; blue: in the absence of [Cp*Rh(bpy)(H2O)]2+. | ||
Photocurrent density decreased from −0.41 to −0.18 mA cm−2 upon 30 min of irradiation and then remained almost constant for the next 150 min, as shown in the inset of Fig. 3. The amount of NADH produced increased linearly with time. The yield of NADH from NAD+ was estimated to be 32% at 180 min of irradiation. No NADH production was observed in the absence of [Cp*Rh(bpy)(H2O)]2+, indicating that the CdS/CuInS2 photocathode cannot directly reduce NAD+ to NADH. These results indicated that reduction of NAD+ to NADH proceeded via [Cp*Rh(bpy)(H2O)]2+ accepting the photoexcited electrons from the CdS/CuInS2 photocathode. Photoelectrochemical water oxidation performance over the TaON photoanode was found to strongly depend on the kind of water oxidation cocatalyst used. Specifically, the influence of the three cocatalysts (i.e., CoOx, RhOx, and IrOx) on the photoelectrochemistry of the TaON photoanode was investigated. As shown in Fig. S2,† although all electrodes showed clear photoresponses in a wide range of potentials, different onset potentials of the photocurrent were observed. Specifically, the onset potentials of the CoOx/TaON, RhOx/TaON and IrOx/TaON photoanodes were estimated to be approximately −0.45, −0.55, and −0.60 V vs. Ag/AgCl, respectively, with these values considerably more negative than that of the CdS/CuInS2 photocathode (0.03 V vs. Ag/AgCl, as shown in Fig. 2). This result implied that the combination of cocatalyst-loaded TaON photoanode and CdS/CuInS2 photocathode can enable photoelectrochemical NADH regeneration using water as an electron source without an external bias. Here, NADH regeneration using a photoelectrochemical cell consisting of an IrOx/TaON photoanode and CdS/CuInS2 photocathode was attempted. Fig. 4 shows the time dependence of NADH production with the photoelectrochemical cell consisting of an IrOx/TaON photoanode and CdS/CuInS2 photocathode.
 | ||
| Fig. 4 Time dependence of NADH production using a system including a combination of a CdS/CuInS2 photocathode, IrOx/TaON photoanode and [Cp*Rh(bpy)(H2O)]2+ under visible-light irradiation without an external bias. Inset: time dependence of photocurrent density. | ||
Photocurrent density decreased from −0.19 to −0.15 mA cm−2 during the first 2 min of irradiation and then remained almost constant for 120 min, as shown in the inset of Fig. 4. As shown in Fig. S3,† using the system including a combination of CdS/CuInS2 photocathode and IrOx/TaON photoanode in the presence of [Cp*Rh(bpy)(H2O)]2+, a current of approximately −0.15 mA cm−2 was predicted to be observed without any external bias, consistent with the experimentally observed current value shown in Fig. 4. The amount of NADH produced linearly increased with irradiation time when using this system. The yield of NADH from NAD+ was estimated to be 8% at 120 min of irradiation. Here, the irradiation intensity from the light source onto the electrode surface was ca. 680 mW cm−2 (Fig. S4†). The light energy conversion efficiency for NADH production after 120 min of irradiation was estimated to be ca. 0.013%. Thus, NADH regeneration using a photoelectrochemical cell consisting of IrOx/TaON photoanode and CdS/CuInS2 photocathode was accomplished.
Finally, photoelectrochemical production of L-lactate via in situ NADH regeneration was performed by deploying a combination of a cocatalyst-loaded TaON photoanode and a CdS/CuInS2 photocathode without an external bias. The photocurrent over this combination was found to depend on the onset potential of photocurrent over the TaON photoanode. As shown in Fig. S5,† all combinations showed relatively stable photocurrents without an external bias for 2 h of irradiation, while the photocurrent was quite low in the case of the CoOx/TaON photoanode, which showed the most positive onset potential. The IrOx/TaON photoanode, which showed the most negative onset potential, exhibited the highest photocurrent. Fig. 5 shows the time courses of the amounts of products over the system including a combination of the IrOx/TaON photoanode and CdS/CuInS2 photocathode under visible-light irradiation without an external bias. L-Lactate and O2 were produced simultaneously during the 2 h of irradiation. The induction period observed for the O2 evolution was mainly due to a time lag caused by the analysis method used. The amount of evolved H2 was negligibly low (0.2 μmol for 2 h irradiation). Neither production of L-lactate nor of O2 was observed under dark conditions, and when only the IrOx/TaON photoanode was irradiated. Faradaic efficiencies for L-lactate and O2 productions were estimated to be 92% and 93%, respectively, indicating that most of the photogenerated holes in TaON were consumed for water oxidation and most of the photoexcited electrons in CdS/CuInS2 were consumed for L-lactate production through a pathway involving [Cp*Rh(bpy)(H2O)]2+, the NAD+/NADH redox couple and LDH. The results of the experiments using, respectively, the CoOx/TaON and RhOx/TaON photoanodes are shown in Fig. S6.† The photocurrent values obtained were consistent with those predicted from the current–potential curve for the respective photoanode and cathode. The case of the RhOx/TaON or CoOx/TaON photoanode showed simultaneous production of L-lactate and O2, although the production rate using the RhOx/TaON photoanode was lower than that when using IrOx/TaON. From these results, visible-light–driven electrochemical production of L-lactate via NADH regenerated in situ and using water as an electron source was accomplished by deploying a combination of the IrOx/TaON (or RhOx/TaON) photoanode and CdS/CuInS2 photocathode without an external bias. Using the combination of the IrOx/TaON photoanode and CdS/CuInS2 photocathode, NADH regeneration was achieved without the need for a sacrificial reductant under conditions without external bias, but a gradual decrease in photocurrent was observed with irradiation for a long period of time. It has been reported that a stable photocurrent can be obtained in a reduction system using methyl viologen as an electron mediator and a CdS/CuInS2 photocathode.26 In contrast, TaON-based photoanodes have been reported to become gradually deactivated due to factors such as changes in the pH of the solution.29 It is expected that a future development of a TaON-based photoanode that is durable against light irradiation will lead to the construction of a stable and sustainable NADH regeneration system.
 | ||
| Fig. 5 Time dependence of amounts of L-lactate and O2 produced over a system including a combination of a CdS/CuInS2 photocathode, IrOx/TaON photoanode and [Cp*Rh(bpy)(H2O)]2+ under visible-light irradiation without an external bias. | ||
In summary, visible-light–driven electrochemical NADH regeneration using water as an electron source, enabled by a combination of an IrOx/TaON (or RhOx/TaON) photoanode and CdS/CuInS2 photocathode without an external bias was demonstrated for the first time to the best of our knowledge. The CdS/CuInS2 photocathode exhibited a cathodic photocurrent derived from reduction of [Cp*Rh(bpy)(H2O)]2+ at a potential more negative than 0.03 V vs. Ag/AgCl. Reduction of NAD+ to NADH proceeded via reduction of [Cp*Rh(bpy)(H2O)]2+ over the CdS/CuInS2 photocathode at a potential of −0.2 V vs. Ag/AgCl under visible-light irradiation. This NADH regeneration system can be applied to production of L-lactate from pyruvate using LDH. L-Lactate was stably produced over an extended period (30 h), reaching a total amount of 78 μmol, which exceeded the amounts of [Cp*Rh(bpy)(H2O)]2+, NAD+ and LDH that were used in the system. The introduction of the IrOx/TaON (or RhOx/TaON) photoanode enabled the visible-light–driven electrochemical production of L-lactate via NADH regenerated in situ using water as an electron source without an external bias.
Most photoelectrochemical NADH generation systems consist of a photocathode and a counter electrode such as platinum, and require an external bias32,33 or a sacrificial reagent such as TEOA34,35 to produce NADH. The system including the combination of the IrOx/TaON photoanode and CdS/CuInS2 photocathode in this study is a Z-scheme process similar to oxygenic natural photosynthesis using photosystems I and II, and has an advantage over other systems in that it uses water as an electron donor and can regenerate NADH without requiring an external bias. Thus, this study has made available a system including a combination of photoanode and photocathode for sustainable NADH regeneration and subsequent biocatalytic reactions.
Data availability
The authors confirm that the data supporting the findings of this manuscript are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Japan Association for Chemical Innovation (JACI), Institute for Fermentation, Osaka (IFO) (G-2023-3-050), a Grant-in-Aid for Specially Promoted Research (23H05404), Scientific Research (B) (22H01872), (22H01871), Scientific Research (c) (21K05245), and the Fund for the Promotion of Joint International Research (Fostering Joint International Research (B)) (19KK0144, 20KK0116).Notes and references
- K. E. Taylor and J. B. Jones, J. Am. Chem. Soc., 1976, 98, 5689 CrossRef PubMed.
- J. B. Jones, D. W. Sneddon, W. Higgins and A. J. Lewis, J. Chem. Soc. Chem. Commun., 1972, 856 RSC.
- A. Dibenedetto, P. Stufano, W. Macyk, T. Baran, C. Fragale, M. Costa and M. Aresta, ChemSusChem, 2012, 5, 373 CrossRef.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762 CrossRef CAS.
- S. Immanuel, R. Sivasubramanian, R. Gul and M. A. Dar, Chem.–Asian J., 2020, 15, 4256 CrossRef CAS PubMed.
- E. Siu, K. Won and C. B. Park, Biotechnol. Prog., 2007, 23, 293 CrossRef CAS PubMed.
- I. Ali, T. Khan and S. Omanovic, J. Mol. Catal. A:Chem., 2014, 387, 86 Search PubMed.
- J. Kiwi, J. Photochem., 1981, 16, 193 CrossRef CAS.
- H. Y. Lee, J. Ryu, J. H. Kim, S. H. Lee and C. B. Park, ChemSusChem, 2012, 5, 2129 CrossRef CAS PubMed.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486 Search PubMed.
- K. T. Oppelt, E. Wöβ, M. Stiftinger, W. Schöfberger, W. Buchberger and G. Knör, Inorg. Chem., 2013, 52, 11910 Search PubMed.
- D. Yadav, R. K. Yadav, A. Kumar, N. J. Park and J. O. Baeg, ChemCatChem, 2016, 8, 3389 CrossRef CAS.
- R. K. Yadav, J. O. Baeg, G. H. Oh, N. J. Park, J. J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455 CrossRef.
- R. K. Yadav, G. H. Oh, N. J. Park, A. Kumar, K. J. Kong and J. O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728 CrossRef PubMed.
- Y. Zhang, Y. Zhao, R. Li and J. Liu, Sol. RRL, 2021, 5, 2000339 CrossRef.
- J. Huang, M. Antonietti and J. Liu, J. Mater. Chem. A, 2014, 2, 7686 Search PubMed.
- S. Fukuzumi, Y. M. Lee and W. Nam, J. Inorg. Biochem., 2019, 199, 110777 CrossRef PubMed.
- X. Wang and H. H. P. Yiu, ACS Catal., 2016, 6, 1880 Search PubMed.
- H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Angew. Chem., Int. Ed., 1999, 38, 1429 CrossRef.
- C. L. Pitman, O. N. L. Finster and A. J. M. Miller, Chem. Commun., 2016, 52, 9105 RSC.
- R. Ruppert, S. Herrmann and E. Steckhan, J. Chem. Soc. Chem. Commun., 1988, 1150 RSC.
- E. Steckhan, S. Herrmann, R. Ruppert, J. Thömmes and C. Wandrey, Angew. Chem., Int. Ed., 1990, 29, 388 CrossRef.
- A. Aljabour, D. H. Apaydin, H. Coskun, F. Ozel, M. Ersoz, P. Stadler, N. S. Sariciftci and M. Kus, ACS Appl. Mater. Interfaces, 2016, 8, 31695 Search PubMed.
- S. Ikeda, T. Nakamura, S. M. Lee, T. Yagi, T. Harada, T. Minegishi and M. Matsumura, ChemSusChem, 2011, 4, 262 Search PubMed.
- I. Tsuji, H. Kato and A. Kudo, Chem. Mater., 2006, 18, 1969 Search PubMed.
- T. Toyodome, Y. Amao and M. Higashi, New J. Chem., 2021, 45, 14803 RSC.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828 CrossRef PubMed.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968 Search PubMed.
- M. Higashi, Y. Kato, Y. Iwase, O. Tomita and R. Abe, J. Photochem. Photobiol., A, 2021, 419, 113463 CrossRef.
- S. M. Lee, S. Ikeda, T. Yagi, T. Harada, A. Ennaoui and M. Matsumura, Phys. Chem. Chem. Phys., 2011, 13, 6662 RSC.
- H. Kumagai, T. Minegishi, Y. Moriya, J. Kubota and K. Domen, J. Phys. Chem. C, 2014, 118, 16386 CrossRef.
- N. Li, J. You, L. Huang, H. Zhang, X. Wang, L. He, C. Gong, S. Lin and B. Zhang, Green Chem., 2023, 25, 5247 RSC.
- W. S. Choi, S. H. Lee, J. W. Ko and C. B. Park, ChemSusChem, 2016, 9, 1559 CrossRef.
- J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Angew. Chem., Int. Ed., 2012, 51, 517 CrossRef PubMed.
- E. J. Son, Y. W. Lee, J. W. Ko and C. B. Park, ACS Sustainable Chem. Eng., 2019, 7, 2545 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Additional data. See DOI: https://doi.org/10.1039/d5se00676g |
| This journal is © The Royal Society of Chemistry 2025 |