 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled synthesis of conjugated random copolymers in a droplet-based microreactor†
James H.
Bannock
a,
Mohammed
Al-Hashimi
ab,
Siva H.
Krishnadasan
ac,
Jonathan J. M.
Halls
c,
Martin
Heeney
*a and
John C.
de Mello
*a
aCentre for Plastic Electronics, Department of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AY, UK. E-mail: m.heeney@imperial.ac.uk; j.demello@imperial.ac.uk
bDepartment of Chemistry, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar
cSolar Press, 2 Royal College Street, London NW1 0NH, UK
First published on 10th October 2013
Abstract
We report the highly controlled synthesis of conjugated random copolymers in a droplet-based microfluidic reactor. Using two optically distinct polymers, poly(3-hexylthiophene) (P3HT) and poly(3-hexylselenophene) (P3HS), a series of highly regioregular random copolymers is generated with physical properties intermediate to those of the parent homopolymers. Analysis by 1H nuclear magnetic resonance spectroscopy reveals the co-polymerisation process to follow ideal Bernoullian behavior.
Semiconducting polymers are attracting widespread interest due to their potential applications in lighting, displays and solar cells, and for their compatibility with high volume manufacturing on flexible plastic substrates.1–4 However, with the latest generations of semiconducting polymers now nearing application-viable levels of performance,1 an urgent need has arisen for new quality-assured production methods that can scale to industrially relevant quantities, while providing reliable batch-to-batch reproducibility in terms of stoichoimetry, molecular weight and structural order.
We recently described a new strategy for the scalable synthesis of high quality semiconducting polymers based on the use of droplet microfluidics.5 In brief, by injecting monomer feedstock into a fast-flowing stream of immiscible carrier fluid, a series of near-identical sub μL droplets is generated inside the carrier fluid, each of which acts as a self-contained microscale reaction vessel. The small droplet size ensures rapid equilibration of composition and temperature, and so provides a highly uniform environment for polymerisation. Importantly, since materials throughput can be raised independently of droplet volume (by ramping up the rate of droplet generation while keeping the droplet size fixed), production levels can in principle be scaled-up indefinitely without detriment to yield or product quality. To validate the droplet approach we previously used the Grignard metathesis (GRIM) route to synthesise device-grade poly(3-hexylthiophene) (P3HT) with number average molecular weights up to 50 kDa. The materials had high regioregularities (RR > 97%) and low polydispersity indices (PDI < 1.7), with production rates of up to 60 g per day being readily achievable without leakage or fouling. Importantly the droplet approach was found to facilitate the synthesis of high molecular weight polymers, which are challenging to synthesise in conventional single-phase flow reactors where precipitation of product on the reactor walls can lead to blockage.
Here we report the extension of the droplet method to the preparation of statistical copolymers. Using P3HT and poly(3-hexylselenophene) (P3HS) as parent materials, we demonstrate the controlled synthesis of a series of statistical copolymers, varying in composition from 100% P3HT to 100% P3HS. P3HT is amongst the most widely studied of conjugated polymers due to its formation of semicrystalline thin-films with good hole-transporting properties and broad spectral coverage up to 600 nm – favourable properties for thin-film transistors and organic photovoltaics.6 P3HS, the selenium analogue of P3HT, has comparable hole transporting properties but due to its deeper-lying LUMO level has improved spectral coverage, extending to 760 nm.7,8 Copolymerisation of 3-hexylthiophene and 3-hexylselenophene has previously been found to yield materials with absorption spectra intermediate to those of the two parent polymers, and was selected here as an attractive test reaction for evaluating the levels of synthetic control achievable using droplet-based synthesis.9,10
All materials were prepared here via the Grignard metathesis route of McCullough et al.,11 which provides a simple method for preparing highly regioregular polymers from readily prepared dibrominated monomers (see Scheme 1). Reaction of the monomers with one equivalence of an alkyl Grignard reagent – typically isopropylmagnesium chloride (iPrMgCl) or n-butylmagnesium chloride (n-BuMgCl) – results in a metathesis reaction that affords the more stable thienyl/selenyl Grignard. For asymmetric monomers such as 2,5-dibromo-3-hexylthiophene and 2,5-dibromo-3-hexylselenophene, formation of the Grignard at the 5-position is favoured over formation at the 2-position by an approximate ratio of four to one.7,11 Polymerisation is readily initiated by the addition of a Kumada-type nickel or palladium catalyst, with 1,3-bis[diphenylphosphinopropane]nickel(II) chloride [Ni(dppp)Cl2] being the catalyst of choice due to the high regioregularity of the resultant material.‡ The chain-growth character12,13 of the polymerization yields polymers with well controlled, narrow weight distributions.14–18
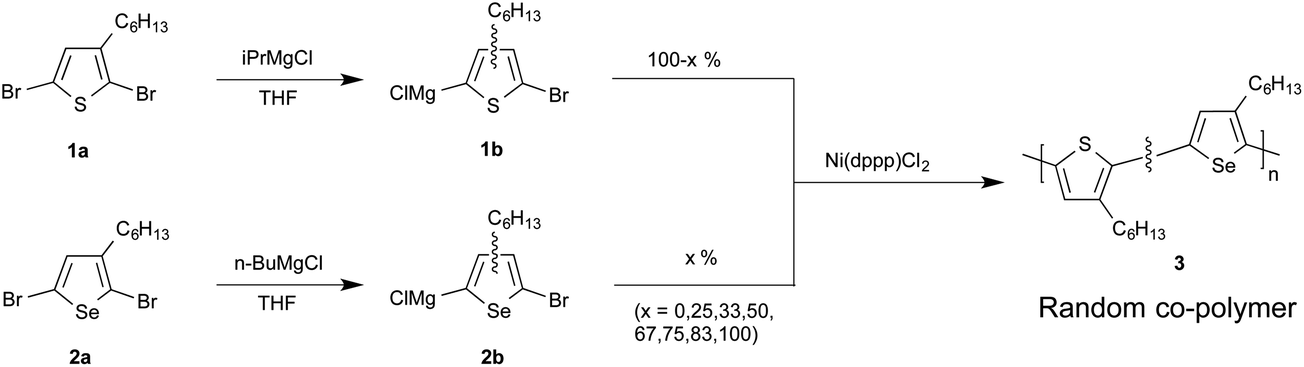 | ||
| Scheme 1 Synthesis of poly(3-hexylthiophene)-co-poly(3-hexylselenophene) [P3HT-co-P3HS] (3). | ||
In adapting the GRIM method to the droplet synthesis of P3HT, we previously dispersed the Ni(dppp)Cl2 catalyst in the carrier fluid since direct dissolution in THF under N2 was found to cause ligand detachment, leading to bleaching of its red coloration and a severe loss of catalytic activity. One drawback of dispersing the catalyst in the carrier fluid is the difficulty of controlling the catalyst-to-monomer ratio since the catalyst enters the reagent phase diffusively via the droplet surface. In the current work, the catalyst was dissolved directly in THF, with the problem of catalyst deactivation being solved by co-dissolving excess dppp ligand alongside the Ni(dppp)Cl2. (A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio of ligand to catalyst was found to be sufficient). Dissolution of the catalyst in the reagent phase provides a more versatile and rigorously defined chemical environment for the polymerisation since the ratio of catalyst to monomer can be precisely controlled by fixing their relative rates of infusion.
:1 weight ratio of ligand to catalyst was found to be sufficient). Dissolution of the catalyst in the reagent phase provides a more versatile and rigorously defined chemical environment for the polymerisation since the ratio of catalyst to monomer can be precisely controlled by fixing their relative rates of infusion.
The catalyst solution was prepared by dissolving 10 mg Ni(dppp)Cl2 (18.44 μmol) and 10 mg dppp (24.25 μmol, 1.31 equiv.) in 10 mL anhydrous THF (a-THF). The catalyst solution and 0.25 M solutions of monomers 1b and 2b in a-THF were loaded into three separate gas-tight syringes, each of which was then mounted on an individual syringe pump. (See ESI† for a description of the synthesis routes for the monomers.) The two monomer solutions were injected from their respective syringes into a static 1.7 μL Y-shaped mixer, Fig. 1a. The selenophene/thiophene mixture from the mixer and the catalyst/ligand solution from the third syringe were fed into a three-input droplet generator (see Fig. 1b and d), using Galden HT-170 perfluorinated polyether (PFPE) (Solvay Solexis) supplied by a fourth syringe pump as an immiscible carrier fluid. The outlet of the droplet generator was connected to a 2.25 m length of polytetrafluoroethylene (PTFE) tubing (1 mm ID, 2 mm OD, Bohlender). The innermost 2 m (1.57 mL) of the PTFE tubing was held in a flat spiral arrangement using a 3D-printed polylactic acid plastic framework and fully immersed in paraffin oil at 55 °C, Fig. 1c and SI4.† The effluent was collected in methanol-filled vials for quenching and precipitation of the product. To ensure a constant heating time of 436 s for all syntheses, the total flow rate was fixed at 216 μL min−1, using flow-rates of 108, 54 and 54 μL min−1 for the carrier, catalyst, and combined (1b + 2b) monomer streams respectively. The composition of the reaction mixture was varied by tuning the ratio of the selenophene and thiophene infusion rates, while holding their combined value constant at 54 μL min−1. Under these flow conditions, the catalyst loading was fixed at 0.74 mol% relative to the total monomer concentration. (The complete reactor setup and droplet generation process is described in the ESI†.)
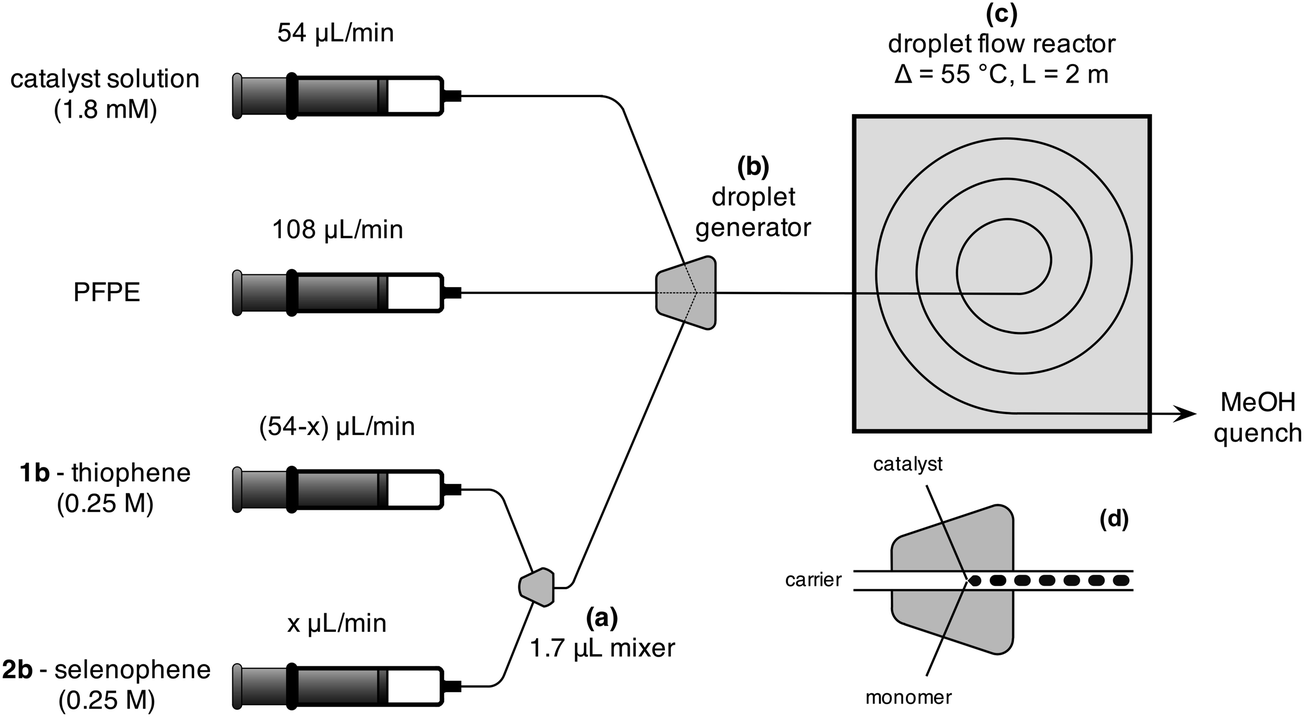 | ||
| Fig. 1 Schematic of droplet reactor, comprising: (a) a static mixer for controlling the selenophene to thiophene ratio; (b) a three-input droplet generator into which the carrier fluid, catalyst solution and premixed monomer solution are injected; and (c) coiled PTFE tubing of inner diameter 1 mm in a temperature-stabilised oil-bath. The stated flow conditions correspond to a residence time of 436 s in the oil-bath and a catalyst loading of 0.74 mol% relative to the monomer. (d) A schematic of the droplet generator. | ||
Eight different pairings of monomer flow-rates were used to prepare the series of copolymers, ranging from 100% thiophene to 100% selenophene. (In the case of the two homopolymers, the Y-mixer was removed and the relevant monomer was injected directly into the PTFE reaction channel from its syringe.) After each change of flow rate, the system was allowed to settle for eight minutes before commencing sample collection. Each sample was collected in a separate methanol-filled vial over a four minute period. Refractive index size-exclusion chromatography (calibrated to polystyrene standards) showed all copolymers to have broadly similar number average molecular weights in the range 36 kDa to 44 kDa, with relatively low polydispersity indices in the range 1.3–1.5 (see Fig. SI and Table SI2†).
1H NMR was used to determine the absolute ratio of selenophene to thiophene in the product, the statistical nature of the copolymerization process, and the structural regioregularity. The chemical environment of the ring proton is affected by the heteroatom in the ring and the heterocycle coupled at the 5-position. Consequently, as seen in the left hand spectra of Fig. 2a, for fully regioregular materials there are four distinct couplings evident in the NMR: Se–S, S–S, Se–Se and S–Se at 6.95, 7.01, 7.16 and 7.22 ppm, respectively. (Note, the unit containing the ring proton is specified here in bold, and signals were corrected for interference by the CHCl3 signal.) The relative frequencies of the four coupling types can be straightforwardly determined from the relative integrals of the coupling peaks.
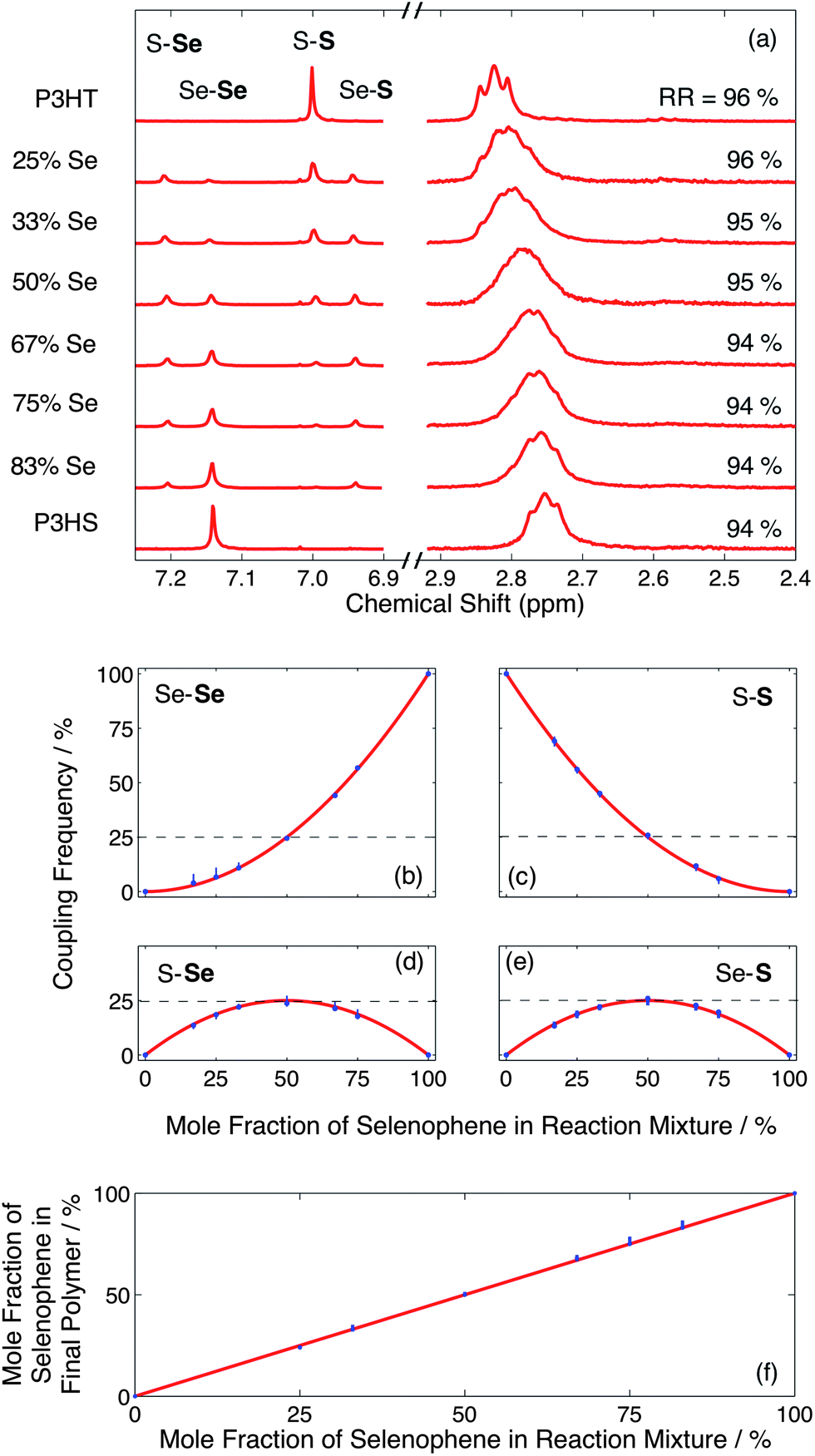 | ||
| Fig. 2 (a) 1H NMR spectra showing the ring proton peaks (left) and α-methylene proton peaks (right) of a series of droplet-synthesised P3HT-co-P3HS statistical copolymers, ranging in composition from 100% P3HT to 100% P3HS; the y-axis labels denote the mole fraction of selenophene in the initial reaction mixture. (b–e) Relative frequencies of the four coupling types versus mole fraction of selenophene in the initial reaction mixture. (f) Mole fraction of selenophene in the final polymer versus mole fraction of selenophene in the initial reaction mixture. The relative coupling frequencies and selenophene content were determined from the areas under the ring proton peaks in (a), correcting the area under the selenophene peak for interference by CHCl3. The blue markers in (b) to (f) indicate experimental data (with the vertical lines denoting extreme error bounds), while the red curves indicate ideal Bernoullian behavior. | ||
For a fully random copolymerization, the two monomers are added to the polymer chain with equal probabilities at all stages of the polymerisation. This results in the formation of a Bernoullian copolymer in which the relative frequency of each coupling type is determined by the product of the mole fractions of the corresponding monomers in the initial reaction mixture.19Fig. 2b–e show for the four coupling types, the experimentally determined coupling frequencies versus the mole fraction of selenophene in the initial reaction mixture. Also shown in Fig. 2f is the mole fraction of selenophene in the final copolymer versus the mole fraction of selenophene in the initial reaction mixture. All experimental data (blue markers) adhered closely to ideal Bernoullian behavior19 (red curves), confirming the random nature of the copolymerisation process. The near-ideal nature of the coupling data demonstrates the very high levels of reaction control that can be attained using the droplet approach, with the stoichiometry of the final copolymers being quantitatively determined in all cases by the relative infusion rates of the two monomer solutions.
As shown in the right hand spectra of Fig. 2a the two homopolymers exhibited clearly resolved head-to-tail α-methylene peaks, corresponding to splitting by the neighbouring aliphatic protons. The copolymers exhibited peaks from both parent polymers, albeit with relatively poor resolution between individual peaks due to overlapping contributions from the heterocouplings. In the case of the 50:50 copolymer, a single broad peak was observed with no discernible fine structure. A steady shift in the peak position of the head-to-tail α-methylene proton from 2.83 to 2.76 ppm was observed in moving through the series of copolymers from pure P3HT to pure P3HS, consistent with the random nature of the copolymerisation. Integration of the head-to-tail and head-to-head/tail-to-tail α-methylene signals20 indicated only a slight change in regioregularity from 96% to 94% in moving from pure P3HT to pure P3HS, indicating a strong preference for head-to-tail coupling in all cases.
Fig. 3a shows normalised absorption spectra in chlorobenzene for the eight samples, with each sample showing a single absorption peak that progressively red-shifted and slightly broadened as the mole fraction of selenophene in the initial reaction mixture was raised from 0% to 100%. Plotting the peak wavelength versus selenophene content (Fig. 3b) yielded a linear trend from 455 nm for P3HT to 498 nm for P3HS. The progressive shift in spectra and absence of multiple absorption features are consistent with the formation of statistical as opposed to block copolymers. We note that the ability to finely tune the optical properties of the copolymers through simple adjustments to the monomer infusion rates provides a simple and effective means of optimising the copolymer characteristics for a desired application.
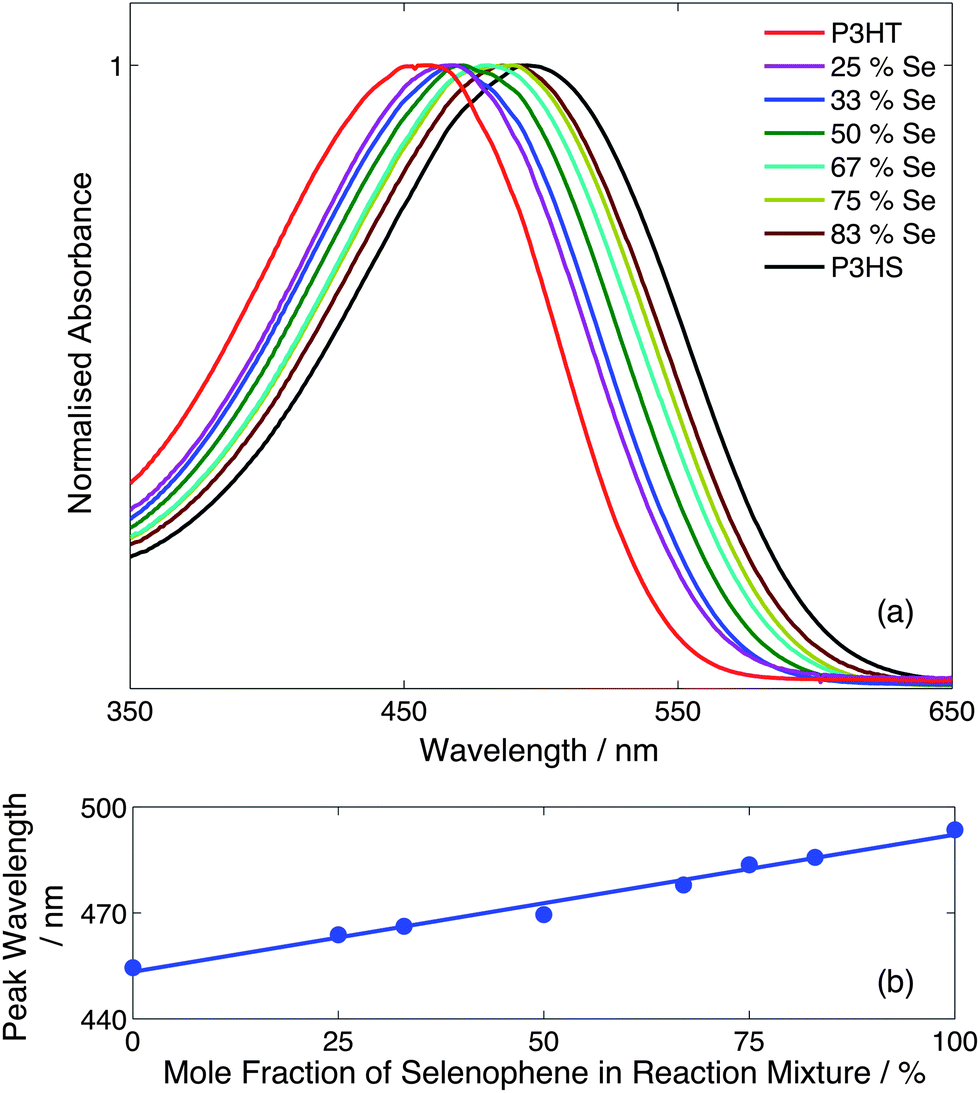 | ||
| Fig. 3 (a) Solution-phase absorption spectra in chlorobenzene for the series of droplet-synthesised P3HT-co-P3HS random copolymers. The legend denotes the mole fraction of selenophene in the initial reaction mixture. (b) Peak absorption wavelength versus mole fraction of selenophene in the initial reaction mixture; the solid line is a guide to the eye. | ||
In conclusion we have extended the droplet method to the synthesis of random copolymers of poly(3-hexylthiophene) and poly(3-hexylselenophene) to produce a series of statistical copolymers, ranging in composition from 100% P3HT to 100% P3HS. The ring proton signals in the 1H NMR indicated a fully random (Bernoullian) copolymerisation in which the two monomers are incorporated into the chain with equal probabilities at all stages of chain growth, resulting in a final stoichiometry that matches the initial composition of the reaction mixture. Analysis of the α-methylene proton region indicated that all copolymers had a strong preference for head-to-tail coupling, with the regioregularity shifting only slightly from 96 to 94% as the selenophene content was increased. The copolymers exhibited composition-dependent absorption spectra intermediate to those of the parent polymers, with the optical band-gap varying linearly from 455 to 498 nm as the mole fraction of selenophene in the feedstock was increased from 0% to 100%. The high degree of reaction control demonstrated here, combined with the excellent stability and ready scalability reported previously, confirms droplet synthesis to be a highly effective method for the controlled production of conjugated polymers with significant advantages over conventional batch methods.
Acknowledgements
JB is funded under an EPSRC Doctoral Training Centre in Plastic Electronics (grant number EP/G037515/1) and holds an Industrial Fellowship with the Royal Commission for the Exhibition of 1851. SK acknowledges financial support from the EPSRC under its Knowledge Transfer Secondment scheme and from the Technology Strategy Board (Grant name CAMPFiRe). JdM acknowledges financial support under the Royal Society Industry Fellowship scheme. The authors acknowledge additional financial support from the EPSRC under grant number EP/G031088/1.References
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- R. Parashkov, E. Becker, T. Riedl, H.-H. Johannes and W. Kowalsky, Proc. IEEE, 2005, 93, 1321–1329 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394–412 CrossRef CAS PubMed.
- J. H. Bannock, S. H. Krishnadasan, A. M. Nightingale, C. P. Yau, K. Khaw, D. Burkitt, J. J. M. Halls, M. Heeney and J. C. de Mello, Adv. Funct. Mater., 2013, 23, 2123–2129 CrossRef CAS.
- A. Marrocchi, D. Lanari, A. Facchetti and L. Vaccaro, Energy Environ. Sci., 2012, 5, 8457 CAS.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061–5063 RSC.
- A. M. Ballantyne, L. Chen, J. Nelson, D. D. C. Bradley, Y. Astuti, A. Maurano, C. G. Shuttle, J. R. Durrant, M. Heeney, W. Duffy and I. McCulloch, Adv. Mater., 2007, 19, 4544–4547 CrossRef CAS.
- J. Hollinger, A. A. Jahnke, N. Coombs and D. S. Seferos, J. Am. Chem. Soc., 2010, 132, 8546–8547 CrossRef CAS PubMed.
- E. F. Palermo and A. J. McNeil, Macromolecules, 2012, 45, 5948–5955 CrossRef CAS.
- R. Loewe, S. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–253 CrossRef CAS.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS.
- E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS PubMed.
- M. C. Stefan, M. P. Bhatt, P. Sista and H. D. Magurudeniya, Polym. Chem., 2012, 3, 1693–1701 RSC.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424 RSC.
- A. Kiriy, V. Senkovskyy and M. Sommer, Macromol. Rapid Commun., 2011, 32, 1503–1517 CrossRef CAS PubMed.
- G. Odian, Principles of Polymerization, Wiley-Interscience, New York, 4th edn, 2004 Search PubMed.
- T. A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mh00066d |
| ‡ The high regioregularity derives from the slow reaction kinetics of the (minority) monomers having the Grignard at the 2 position, which minimises their participation in the polymerisation and so ensures predominantly head-to-tail couplings by the majority monomer.16 |
| This journal is © The Royal Society of Chemistry 2014 |