Semicondutor quantum dots-based metal ion probes
Peng
Wu
a,
Ting
Zhao
b,
Shanling
Wang
a and
Xiandeng
Hou
*ab
aAnalytical & Testing Center, Sichuan University, Chengdu 610064, China. E-mail: houxd@scu.edu.cn
bCollege of Chemistry, Sichuan University, Chengdu 610064, China
First published on 14th October 2013
Abstract
Semiconductor quantum dots (QDs) exhibit unique optical and photophysical properties that offer significant advantages over organic dyes as optical labels for chemo/bio-sensing. This review addresses the methods for metal ion detection with QDs, including photoluminescent, electrochemiluminescent, photoelectrochemical, and electrochemical approaches. The main mechanisms of direct interaction between QDs and metal ions which lead to photoluminescence being either off or on, are discussed in detail. These direct interactions provide great opportunities for developing simple yet effect metal ion probes. Different methods to design the chemically-modified QD hybrid structures through anchoring metal ion-specific groups onto the surface of QDs are summarized. Due to the spatial separation of the luminescence center and analyte recognition sites, these chemically-modified QDs offer greatly improved sensitivity and selectivity for metal ions. Several interesting applications of QD-based metal ion probes are presented, with specific emphasis on cellular probes, coding probes and sensing with logic gate operations.
 Peng Wu | Peng Wu received his BS in 2003, and completed his MS degree in 2006, both in analytical chemistry from Sichuan University under the supervision of Prof. Xiandeng Hou. He then worked at Sichuan University until 2008. From 2008 to 2011, he studied at Nankai University to pursue a PhD degree in analytical chemistry under the supervision of Prof. Xiu-Ping Yan. After completing his PhD degree in 2011, he returned to Sichuan University, where he is currently an associate professor. His current research interests focus on the development of chemo/bio-sensors with optically-active nanomaterials. |
 Xiandeng Hou | Xiandeng Hou is a professor of analytical chemistry at Sichuan University, China. He obtained his Ph.D. in chemistry in 1999 from University of Connecticut, USA (Advisor: Robert G. Michel). He worked as a post-doctoral research associate at Wake Forest University, USA (1999–2001, with Bradley T. Jones) before he joined the faculty of Chemistry at Sichuan University in 2001. He has authored and co-authored more than 120 publications in peer-reviewed international journals. Now, he serves as an associate editor for Microchemical Journal, and editorial (advisory) board member for Journal of Analytical Atomic Spectrometry, Applied Spectroscopy Reviews, and Spectroscopy Letters. His research interests mainly focus on spectral analysis. |
1. Introduction
Heavy metal contamination is one of the most serious concerns to human health because these substances are toxic and retained by the ecological system. Also, some heavy metal ions such as Pb2+, Hg2+, and Cd2+ are not biologically essential and are harmful to organisms even at very low concentration levels, while in small quantities, certain heavy metals (e.g., Fe, Cu, Mn, and Zn) are nutritionally essential for a healthy life. Due to the biological and environmental significance of metal ions, there is an ongoing demand to develop sensitive sensing strategies for tracing heavy metal ions in living systems and the whole environment, and a large number of organic dye-based metal ion probes have thus been developed so far.1–5Semiconductor quantum dots (QDs) have many unique photophysical properties that offer significant advantages as optical labels for chemo/bio-sensing. Compared with organic dyes, the most striking properties of QDs are their size- and component-tunable emission, broad excitation bands, and improved signal brightness, as well as resistance against photobleaching.6 The efficient fluorescence and stability of QDs improves the sensitivity and prolongs their lifetime in their use as optical labels. Additionally , secondary tethering of ligands or receptor units onto the surface of QDs does not cause appreciable fluorescence loss. The yielded QD–ligand conjugates allow a spatial separation of the luminescence center and analyte-recognition sites, which is structurally different from organic dye-based fluorescent probes.1–5
Due to their superior properties, QDs have been used in a wide range of applications, including solar cells, light-emitting devices, photodetectors, field-effect transistors, luminescent biolabels, and bioimaging probes.7–10 The use of QDs for optosensing has also spread from small molecules to proteins and nucleic acids.11–14 For the detection of metal ions, great progress has also been achieved since the pioneering work by Chen and Rosenzweig in 2002, in which luminescent CdS QDs were employed as probes for Cu2+, Zn2+ and Fe3+.15 In this review, we attempt to summarize the progress in the application of QDs in luminescent probes for metal ions. Various sensing strategies, including direct interaction of metal ions with the QDs, specific surface-functionalized QDs for metal ions, and metal ion-triggered signal change of QDs, are discussed in the following sections. Furthermore, other signal transduction modes besides fluorescence, including phosphorescence, electrochemiluminescence and photoelectrochemistry have also been employed for metal ion sensing, and these are also discussed here. Finally, several advanced applications of QD-based metal ion probes, including intracellular imaging, barcoding and logic gate operations are briefly introduced.
2. QDs as metal ion probes
2.1. Use of fluorescence from QDs for detection
The study on the interaction of metal ions with QDs can be dated back to 1987, when Henglein et al. found Cd2+ could increase the PL efficiency of CdS nanoparticles by about 50% in aqueous basic solution.17 This effect is attributed to the formation of a Cd(OH)2 shell on the CdS core, which effectively eliminates the nonradiation recombination of charge carriers. Later, similar passivation effects were also observed when Zn2+ and Mn2+ were added to basic CdS or ZnS colloidal solutions.18,19 Chen et al. found that the sequential introduction of Zn2+ and Mn2+ could in-turn enhance and quench the dopant emission of Mn-doped ZnS QDs.20 The enhancement and quenching were ascribed to the elimination of surface S2− dangling bonds and the formation of Mn–Mn coupling energy migration over a short distance.
The analytical potential of QDs as metal ion probes was not fully recognized until Chen and Rosenzweig pioneered this area in 2002.15 They showed that thioglycerol-capped CdS QDs could be used as a fluorescent probe for Cu2+ in the presence of Zn2+ in aqueous solutions. Although Wang et al. also found hexametaphosphate-capped CdS QDs could be employed as a fluorescent probe for Cu2+,21 the greatest contribution of Chen and Rosenzweig's work was that they found capping agents for QDs also played profound roles in probing metal ions.15,22 This review covers the applications of QDs in the development of novel metal ion sensors, an area of growing interest over the past decade.23–31
The interaction of metal ions with QDs can either lead to quenching or enhancing of the fluorescence of QDs. Generally, the direct interaction of metal ions with QDs is a very complicated process and there are several interaction pathways that lead to fluorescence quenching (Fig. 1).
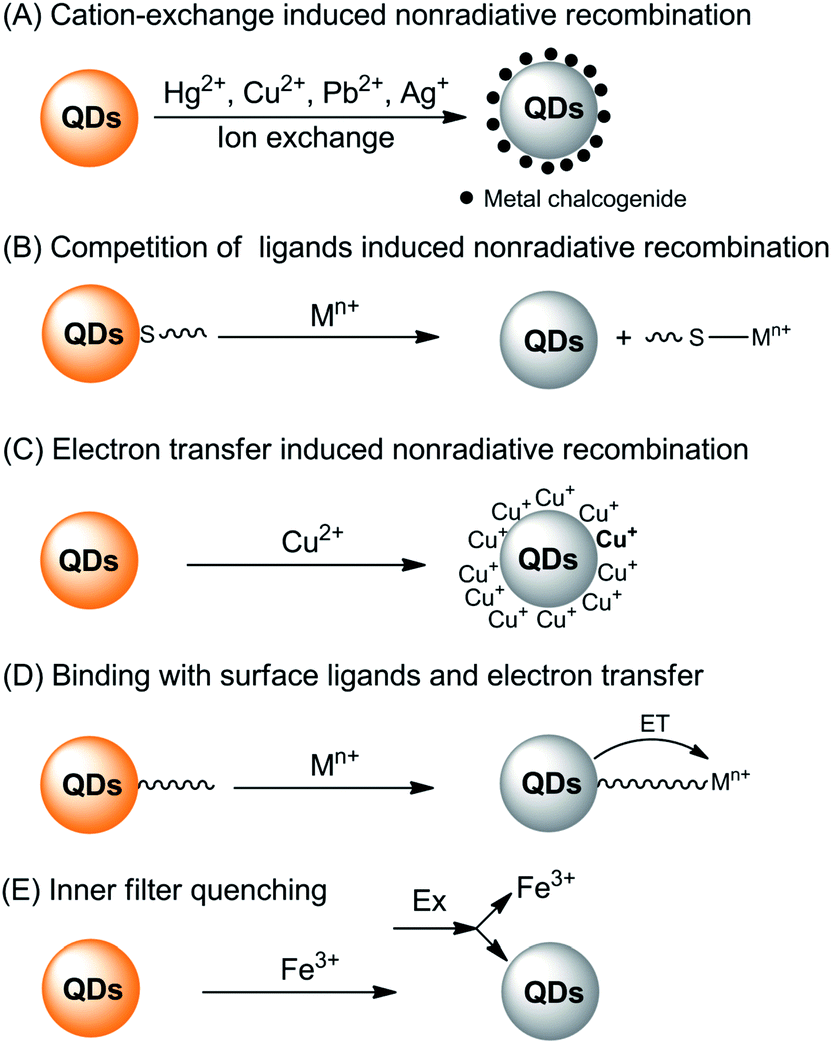 | ||
| Fig. 1 Summary of the fluorescence quenching mechanisms for the detection of metal ions based on direct interaction of the metal ions with QDs. | ||
First of all, typical metal chalcogenide-based QDs (for example, CdS, CdSe and CdTe) are formed via the precipitation of cations with chalcogenide anions. Consequently, those metals with lower Ksp values with corresponding chalcogenide can displace the QD cations via cation-exchange (Fig. 1A). Take sulfides as an example, as shown in Table 1, the pKsp value of CdS is obviously lower than that of HgS, CuS, Ag2S and PbS, giving rise to a potential cation-exchange reaction on the surface of QDs.32 As a result, surface defects are generated, leading to a nonradiative recombination of the excitons and fluorescence quenching. Chemical exchange in bulk solids is limited due to the high activation energy required for the diffusion of atoms in the material. However, the large surface-to-volume ratio of nano-sized QDs favors the diffusion of foreign metal ions. In fact, such cation-exchange is also a very useful strategy for the synthesis of high-quality ionic nanocrystals.33 This may be the basis for a large number of metal ion probes in the literature.34–44 Since the bandgaps of HgS, CuS, Ag2S and PbS are lower than that of CdS (Table 1), typical signs of the cation-exchange are red-shifted in either their absorption or PL wavelengths.36,45,46 Also, the size of the QDs, i.e., the bandgap of QDs, may also influence the quenching selectivity of QDs toward metal ions.47,48 For example, Hg2+ quenched smaller MPA-capped CdTe QDs more efficiently than larger ones, while larger QDs were more markedly quenched by Cu2+,48 probably because of different quenching pathways. As QDs are different from their bulk counterparts, the ligand density has large influence on the cation-exchange reactions.49 A representative exchange pathway, as experimentally confirmed by Dong et al.,50,51 is given in Fig. 2, which takes into account both CdTe QDs and the ligand effect.
| Metal | Electronic configuration | pKsp | Bandgap (eV) |
|---|---|---|---|
| As | 3d10 4s2 4p3 | 21.68 | — |
| Co | 3d7 4s2 | 20.4 (α) | — |
| Cu | 3d10 4s1 | 35.2 (CuS) | — |
| 47.6 (Cu2S) | 1.1 | ||
| Cr | 3d5 4s1 | — | — |
| Fe | 3d6 4s2 | 17.2 (FeS) | 0.1 |
| Mn | 3d5 4s2 | 12.6 | 3.0 |
| Hg | 4f14 5d10 6s2 | 51.8 | 2.0 |
| Pb | 4f14 5d10 6s2 6p2 | 27.1 | 0.37 |
| Ni | 3d8 4s2 | 18.5 (α) | 0.4 |
| Ag | 4d10 5s1 | 49.2 | 0.92 |
| Cd | 4d10 5s1 | 26.1 | 2.42 |
| Zn | 3d10 4s2 | 23.8 (α) | 3.6 |
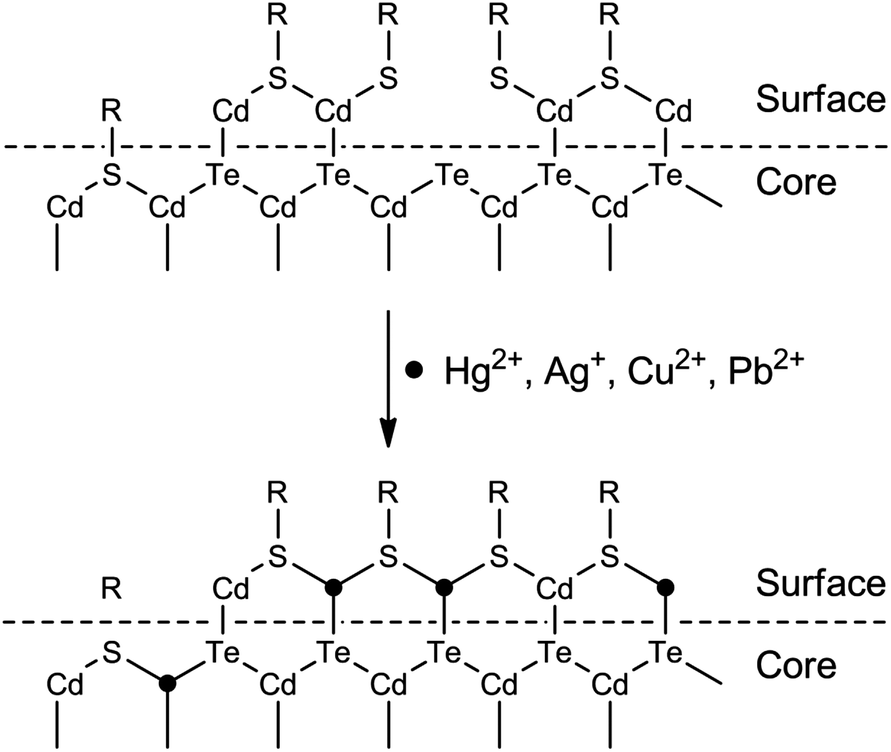 | ||
| Fig. 2 Schematic illustration of the surface structures of CdTe QDs and CdTe QDs quenched by metal ions with the higher pKsp of chalcogenides. | ||
Apart from the lower solubility of corresponding chalcogenide-induced cation-exchange, Hg2+, Cu2+, Ag+ and Pb2+ can also compete with the typical thiol ligands on the surface of QDs because of their higher affinities with thiols (Fig. 1B). As can be seen from Fig. 2, after detaching the ligands from the QDs, surface traps can be generated which can trap electrons from the conduction band, leading to a fluorescence quenching.52,53 The competition between surface ligands and metal ions has been confirmed using electrospray ionization mass spectrometry.54 Normally, the competition of ligands does not change the luminescent center of the QDs, thus the PL and absorption wavelengths would not change. Besides, loss of the surface ligands would result in aggregation of the QDs.52,55
The third quenching pathway, as described by Isarov and Chrysochoos, is the reduction of surface adsorbed metal ions to form nonradiative surface channels (Fig. 1C).56 This case is primarily observed for Cu2+-induced quenching, in which Cu2+ was reduced to diamagnetic Cu+ by surface S2− vacancies in the following way: CdS + Cu2+ → CdS+ + Cu+. The resultant CdS+–Cu+ has a lower energy level than pure CdS QDs, shifting the fluorescence to longer wavelengths, together with fluorescence quenching by Cu+ through a non-radiative recombination of excited electrons in the conduction band and holes in the valence band. Such mechanisms may not be limited to CdS QDs, but also other QDs since the reduction potential of Se2− or Te2− is higher than that of S2−. A recent study harvested the red-shifted fluorescence caused by Cu+ to develop an ultrasensitive probe for Cu2+.57 Cu2+ was firstly reduced to Cu+ by ascorbic acid and the fluorescence wavelength of the resultant Cu2S-covered CdS QDs shifted from 530 nm to 650 nm. Quantification of Cu2+ by the fluorescence change at 650 nm gave rise to a turn-on detection scheme.
For QDs with high density of surface ligands, the attack of the QD core by metal ions may be hindered. However, metal ions can still be adsorbed by QDs either via electrostatic attraction or ligand coordination. Since the metal ions are typically electron-deficient, electron transfer from the conduction band of the QDs to metal ions may occur (Fig. 1D), leading to fluorescence quenching.58–66 In this case, the interaction of metal ions with ligands can be in part predicted with the Pearson acid base concept. For example, the borderline acids Co2+, Ni2+, Cu2+, Pb2+ and Zn2+ prefer binding to ligands containing nitrogen donor atoms, while hard acids such as Ba2+, Al3+, Cr3+ and Mn2+ are prone to coordinate with oxygen-containing ligands. Besides, such Pearson theory-based coordination is generally pH-dependent, thus giving rise to pH-dependent sensitivity.67 Through exploration of the pH-dependent quenching of Mn2+ and pH-independent core-etching (cation exchange) of Cu2+ toward thioglycerol-capped CdTe QDs, a pH-controlled discriminative detection of Mn2+ and Cu2+ was developed.68 It should be noted that normally such electron transfer-induced quenching can be reversible in the presence of ligands with higher affinity toward metal ions, providing new possibilities for sensor fabrication in the indicator-displacement assay format.67,69–72
The last potentiality for fluorescence quenching is a rare case for Fe3+ (Fig. 1E). Chen and Rosenzweig observed the quenching of cysteine capped QDs by Fe3+, which is attributed to an inner filter effect resulting from the strong adsorption by Fe3+ at the excitation wavelength.15
Although all the above interaction pathways have been identified in references, it is still difficult to clearly attribute the interactions. Presumably, the actual interactions between metal ions and QDs may be a combination of several of the above pathways. For example, Zhu et al. observed both ligand detaching and cation exchange when studying the interaction of Hg2+ with glutathione-capped CdTe QDs.54 Yuan et al. explored the Hg2+-induced ligand detaching and surface cation exchange of CdSe QDs for visual detection of Hg2+.73 Accordingly, in the future development of these types of probes, these pathways should be taken into consideration for detailed investigation of the interaction mechanism.
Direct interaction with metal ions can also lead to a fluorescence enhancement of QDs,74 but there are only a few examples. The interaction mechanisms for fluorescence enhancement can also be divided into three categories (Fig. 3). The first group is the passivation of surface defects by either Zn2+ or Cd2+ (Fig. 3A).17,20 QDs synthesized using aqueous routes often have plenty of surface defects of S2− (or Se2−, Te2−) vacancies, resulting in poor PL efficiency. Zinc-based semiconductors typically have higher bandgaps than any other semiconductors (Table 1). The introduction of Zn2+ not only eliminates surface defects, but also further helps to confine excitons, leading to a PL improvement. For Cd2+, the case is similar to that of Zn2+, but it generally cannot activate zinc-based QDs.
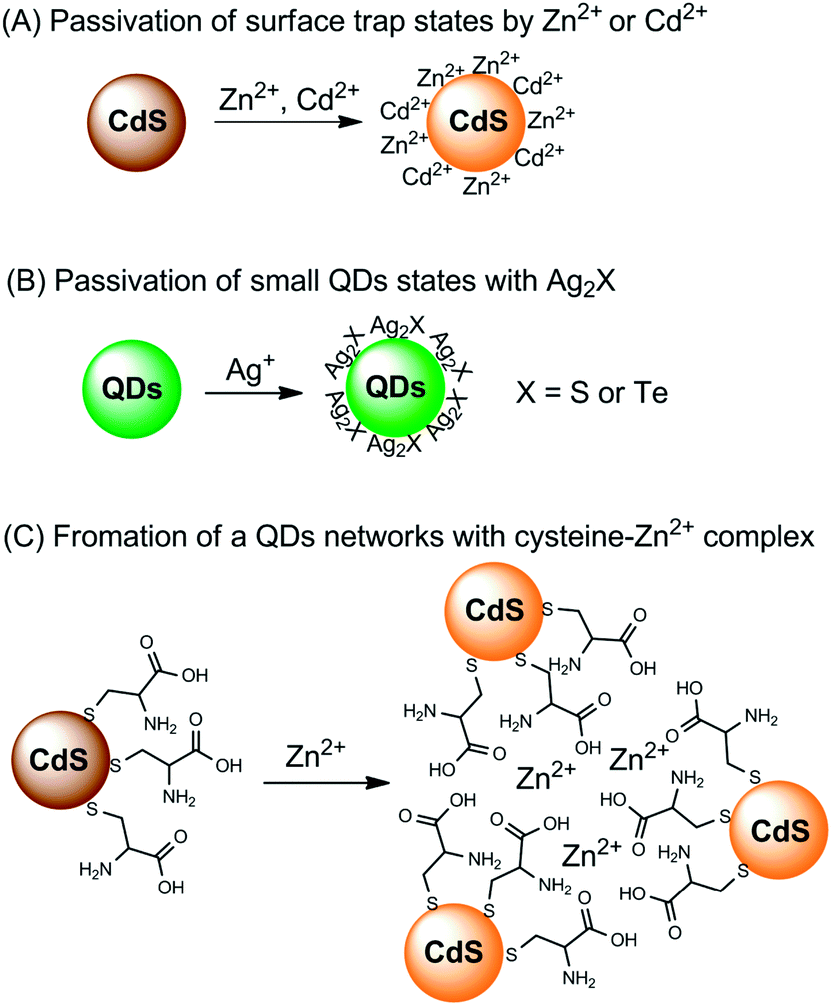 | ||
| Fig. 3 Summary of the fluorescence enhancement mechanisms for the detection of metal ions based on direct interaction of the metal ions with QDs. | ||
It was found that when interacting with QDs, small amounts of Ag+ could first lead to a fluorescence enhancement of CdS QDs and CdSe QDs, together with PL wavelength red-shifting.75,76 However, further increases in the amounts of Ag+ would result in fluorescence quenching. The origin for fluorescence enhancement is the surface traps; the initially adsorbed Ag+ could effectively passivate the surface defects (Fig. 3B). After the initial traps are saturated, the excess Ag+ facilitates nonradiative recombination, resulting in PL quenching. In a mechanism study, Xia et al. prepared two CdTe QDs of different sizes.77 For small particles, the CdTe QDs exhibited PL enhancement in the presence of lower concentrations of Ag+, but showed obvious quenching with further increases of Ag+; while for larger particles, the PL of the CdTe QDs was always quenched upon interaction with Ag+. Small QDs typically have more surface defects than larger ones, and thus surface passivation was observed upon an initial addition of Ag+. On the other hand, larger QDs possess fewer surface defects, thus the nonradiative recombination dominated the whole process.
Chen and Rosenzweig also attributed the Zn2+-induced PL improvement of L-cysteine-caped CdS QDs to the formation of a Zn–cysteine complex (Fig. 3C).15 Such complexes result in the formation of a three dimensional CdS QDs network, which activated the surface states, producing an emission enhancement. Similarly, the selective interaction of Ba2+ with mercaptoethanol-capped CdSe QDs led to fluorescence enhancement of CdSe QDs, with a limit of detection (LOD) of 4.2 nM.78
Although diverse mechanisms have been proposed to explain the interactions between metal ions and QDs, many of the responses still cannot be fully predicted and rely on a speculative mechanistic understanding and missing details. Besides, these metal ion probes based on a direct interaction with QDs also have problems of batch-to-batch variations and less selectivity. The demand for more selective metal ion probes is the driving force for the functionalization of QDs with metal ion-selective receptors.
| Functional group | QDs | Analyte | Detection scheme | Sample media | LOD | Ref. |
|---|---|---|---|---|---|---|
| Peptide, Gly-His-Leu-Leu-Lys | CdS | Cu2+ or Ag+ | Fluorescence quenching | PBS buffer | nM range | 79 |
| Metalloprotein | CdSe@ZnS, InGaP@ZnS | Pb2+ | Fluorescence quenching | Red blood cells | 0.1 nM | 94 |
| Calf-thymus DNA | CdS | Hg2+ | Fluorescence quenching | Waste water | 8.6 nM | 100 |
| p-Sulfonatocalix[4]arene | CdSe@ZnS | Hg2+ | Fluorescence quenching | CH3CN | 15 nM | 87 |
| Calix[6]arene | CdTe@SiO2 | Hg2+ | Fluorescence quenching | Water | 1.6 nM | 101 |

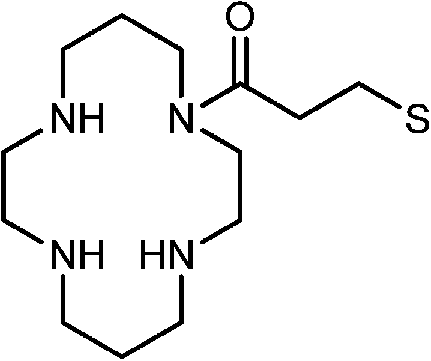
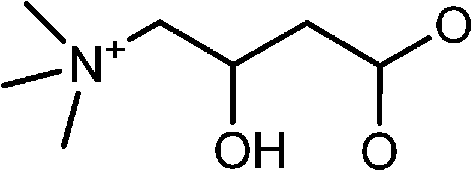
|
CdSe@ZnS | K+ | Ratiometric fluorescence | Water | μM range | 85 |

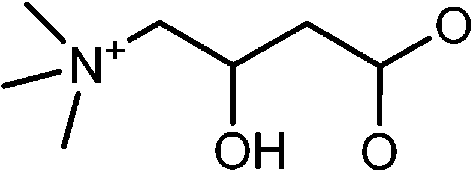
|
Mn-doped CdS | Cd2+ | Fluorescence turn-on | Water | μM range | 83 |
|
CdSe@ZnS | Zn2+ | Fluorescence turn-on | Simulated physiological medium | 2.4 μM | 82 |
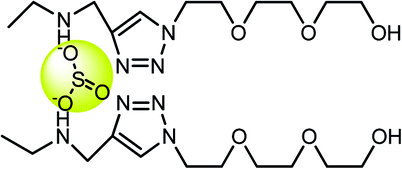
|
CdSe@SiO2 | Cd2+ | Fluorescence turn-on | HeLa cell | 4.9 nM | 102 |
|
Mn-doped ZnS | Ag+ | Fluorescence quenching | Tap water | 0.26 μM | 103 |
|
CdSe@ZnS | Zn2+ | Fluorescence turn-on | Simulated biological fluids | 0.7 μM | 104 |

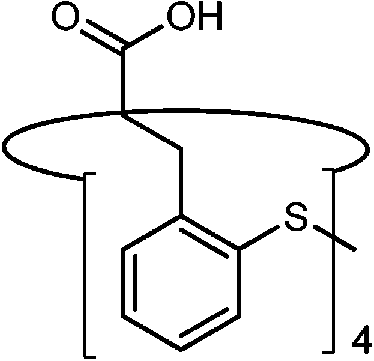
|
CdSe | Zn2+ | Fluorescence enhancement | Dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol = 4:1 :methanol = 4:1 |
0.5 μM | 86 |
|
CdSe | Fe(II) and Cu(II) | Fluorescence quenching | Water | <0.05 nM | 105 |
|
CdSe@ZnS | Hg2+ | Fluorescence quenching | Water | 0.18 μM | 106 |
|
CdSe@ZnS | Cd2+ | Fluorescence enhancement | Water | nM range | 107 |
|
CdSe@ZnS | Cu2+ | Fluorescence quenching | Buffer media | nM range | 88 |
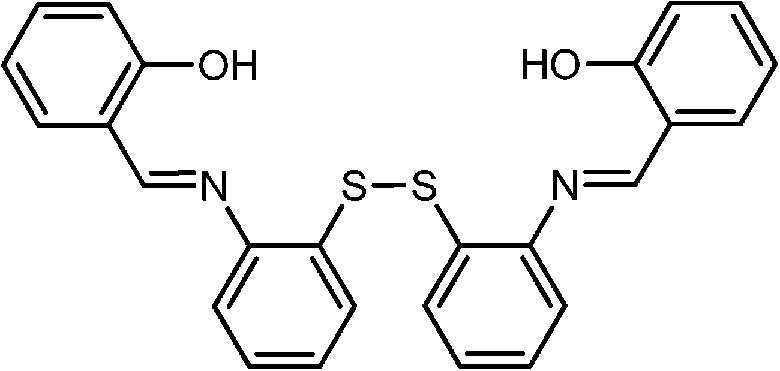
|
CdSe@ZnS | Fe3+, Cu2+ | Color change | 80% THF, 20% HEPES buffer | — | 80 |
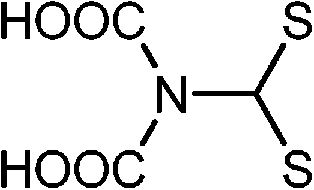
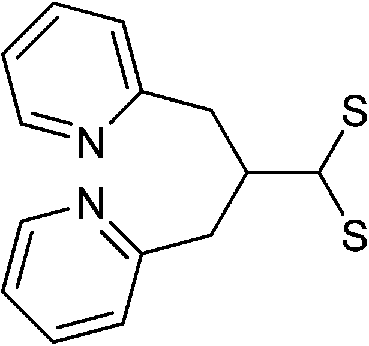
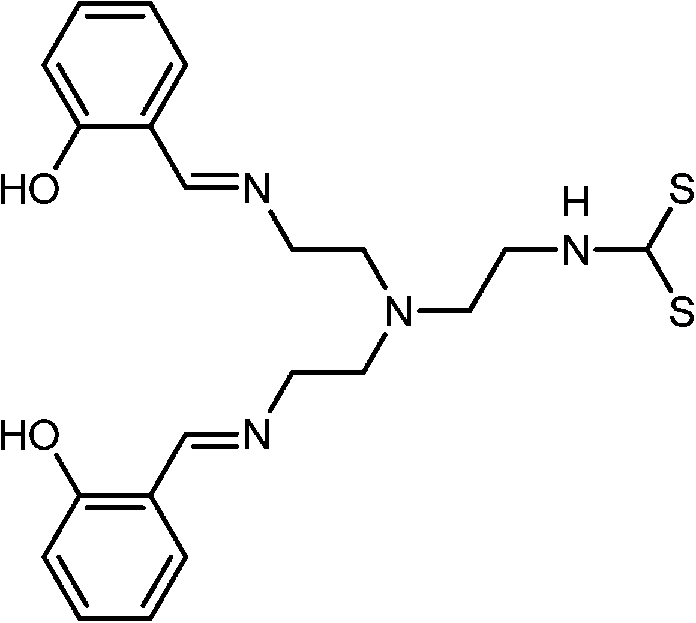
Briefly, the synthetic receptors used to conjugate QDs recognize metal ions through noncovalent interactions, mainly ligand or chelate effects via the affinity of oxygen and nitrogen donor atoms toward metal ions. Of these synthetic receptors, Schiff-base,80,81 azamacrocycle82–84 and suparamolecular receptors with macrocyclic or caged moity85–88 are the most popular choices (Table 2). Metal-bound organic dyes were also employed.89,90 The use of suparamolecular hosts with macrocyclic or caged moity, such as crown ether,85 calixarenes,87,88 or azamacrocycle,82–84 takes advantage of their molecule cavities, or similar hosts, to accept metal ions inside these cavities and bind them selectively. Biological receptors such as protein and DNA offer remarkable advantages in terms of selectivity. DNA receptors typically explore DNA hybridization and the fact that thymine–thymine (T–T) or cytosine–cytosine (C–C) mismatches of complementary sequences can bind Hg2+ or Ag+,91,92 or DNAzymes with divalent metal ions as specific cofactors,93 while protein receptors either exploit the binding affinity of metalloprotein toward Pb2+ (ref. 94) or the selective enzymatic inhibition of Cu2+.95
The signal modulation mechanisms of receptor-functionalized QDs for metal ions are similar to those typically employed in conventional fluorescent sensing,96i.e., photoinduced electron transfer (PET)97 and fluorescence resonance energy transfer (FRET).98 For PET sensing, the attached synthetic receptors act as electron donors and thus turn off the fluorescence of QDs, while coordination with target metal ions results in shifting of the energy level of the ligands, thus cutting off the electron transfer pathway and turning on the fluorescence.82,83 A representative example of PET sensing with QDs for metal ions is given in Fig. 4.84 Through elegant choice of the component and size of the QDs (Fig. 4A), the fluorescence of 3 nm sized CdTe QDs was quenched upon the phenanthroline ligand attachment due to PET from the ligands to QDs. The oxidation potential of the phenanthroline ligand could be adjusted by ion complexation (adjustable hole acceptor). The complexation of Ba2+ ions within the aza crown ether groups shifted the oxidation potential of the ligand across the valence band levels of 3 nm sized CdTe QDs (Fig. 4B), which suppressed the PET process and restored the fluorescence.
 | ||
| Fig. 4 (a) Schematic of fluorescent sensing of metal ions with QDs and an adjustable hole acceptor; and (b) energy levels of CdTe QDs of different sizes and that of the phenanthroline ligand, showing the HOMO level of the ligand across the valence band levels of 3 nm sized CdTe QDs. Reproduced with permission from ref. 84. Copyright 2010 Wiley-VCH. | ||
In fluorescent probes based on specific surface-functionalized QDs, the possibility of direct interaction of the metal ions with the QDs (non-specific interaction) should be minimized to ensure selectivity. Upon understanding the origin of such non-specific interactions, it can be seen that the surface protection of QDs with silica or polymer shell can be an effective way to alleviate and prevent such quenching effects. In fact, in a recent study about the fluorescence quenching of Cu2+ to polyethylene glycol (PEG)-coated CdSeTe@ZnS QDs, the effect of thick PEG shell in protecting the surface of QDs from attacking by Cu2+ has already been demonstrated.99 Besides, adding shells to the QDs may provide increased stability and decreased non-specific interaction, which will also be advantageous for metal ion probes working in biological media.
The first strategy is based on metal-ion triggered FRET between QDs and energy donors. Chen et al. developed a crown-ether (15-crown-5)-functionalized dual QDs system for K+ sensing (Table 3).85 A smaller CdSe@ZnS QD (3.2 nm, QD545) was employed as energy donor and a bigger one (5.6 nm, QD635) as energy acceptor. Upon K+ binding with 15-crown-5, the two QDs were aggregated thus the distance between them was shortened to trigger FRET, resulting in a fluorescence quenching of QD545 and fluorescence enhancement of QD635. However, because of the broad excitation of the QDs, the excitation of the energy donor would also lead to a partial excitation of donors, i.e., a high background. Ruedas-Rama et al. developed zincon-functionalized CdSe@ZnS QDs for differential sensing of Mn2+ and Zn2+.89 Upon binding of zincon with Mn2+, the quenching effect of zincon toward the QDs was deactivated, thereby turning on the fluorescence. When Zn2+ was presented, the absorption of the Zn2+–zincon complex overlapped with the emission of the QDs, which triggered FRET, leading to fluorescence quenching.
| Functional group | QDs | Analyte | Detection scheme | Sample media | LOD | Ref. |
|---|---|---|---|---|---|---|
| Poly T-DNA | Mn-doped ZnS | Hg2+ | Label-free, Hg2+-induced adsorption of dsDNA onto QDs, Ph quenching | Tap and river water | 1.5 nM | 150 |
| Denatured BSA | Mn-doped ZnS | Cr3+ | Electron transfer from QDs to Cr3+, Ph quenching | Water | 3 nM | 149 |
| DNA | Mn-doped CdS@ZnS | Hg2+ | DNA-functionalized QDs and AuNPs, time-gated, turn-on detection | Environmental water | 0.18 nM | 151 |
| DNA | Mn-doped CdS@ZnS | Hg2+ | DNA-functionalized QDs and AuNPs, time-gated, turn-off detection | Environmental water | 0.87 | 152 |
| DNA | CdTe | Pb2+ | Pb2+-induced opening of the hairpin DNA for conjugation of QDs, ECL turn-on | River water and urine | 0.01 nM | 153 |
| G-Quadruplex DNAzyme | CdS | Pb2+ | Pb2+-induced de-activation of G-Quadruplex DNAzyme, photocurrent turn-on | Water | 10 nM | 154 |
| DNAzyme | PbS | Pb2+ | Pb2+-induced cleavage of DNAzyme for labeling with PbS QDs, electrochemical stripping | Environmental water | 0.6 nM | 155 |
| DNAzyme | CdS | Pb2+ | Pb2+-induced cleavage of DNAzyme for subsequent rolling circle amplification, labeling the longed DNA with QDs, electrochemical stripping | River water | 7.8 pM | 156 |
Velu et al. developed a Ca2+/Mg2+-sensitive probe based on aza-crown ether acridinedione (ACEADD)-functionalized Au nanorods (AuNRs) and near-infrared-emitting CdSeTe QDs (Fig. 5A).110 The ACEADD–QDs have dual emission at a visible wavelength of ∼430 nm from the acridinedione (ADD) dye moiety and at a near-infrared (NIR) wavelength of ∼775 nm from the CdTeSe QDs. AuNRs with high extinction in the NIR range (longitudinal plasmon extinction) was judiciously chosen as the energy acceptor for the QDs for maximal spectral overlap and efficient energy transfer. When aza-crown ether (ACE) was conjugated to ADD dye, PET occurred from the nitrogen lone pair of the ACE to the relatively electron deficient ADD moiety, leading to fluorescence quenching of the ADD. In the presence of Ca2+/Mg2+, the cavity of ACE could host Ca2+/Mg2+ and suppress the PET, resulting in fluorescence recovery of ADD (Fig. 5B). Meanwhile, the presence of Ca2+/Mg2+ could also induce the formation of a sandwich complex of QDs and AuNRs (Fig. 1A) and quenching of the fluorescence of QDs through energy transfer (Fig. 5C).
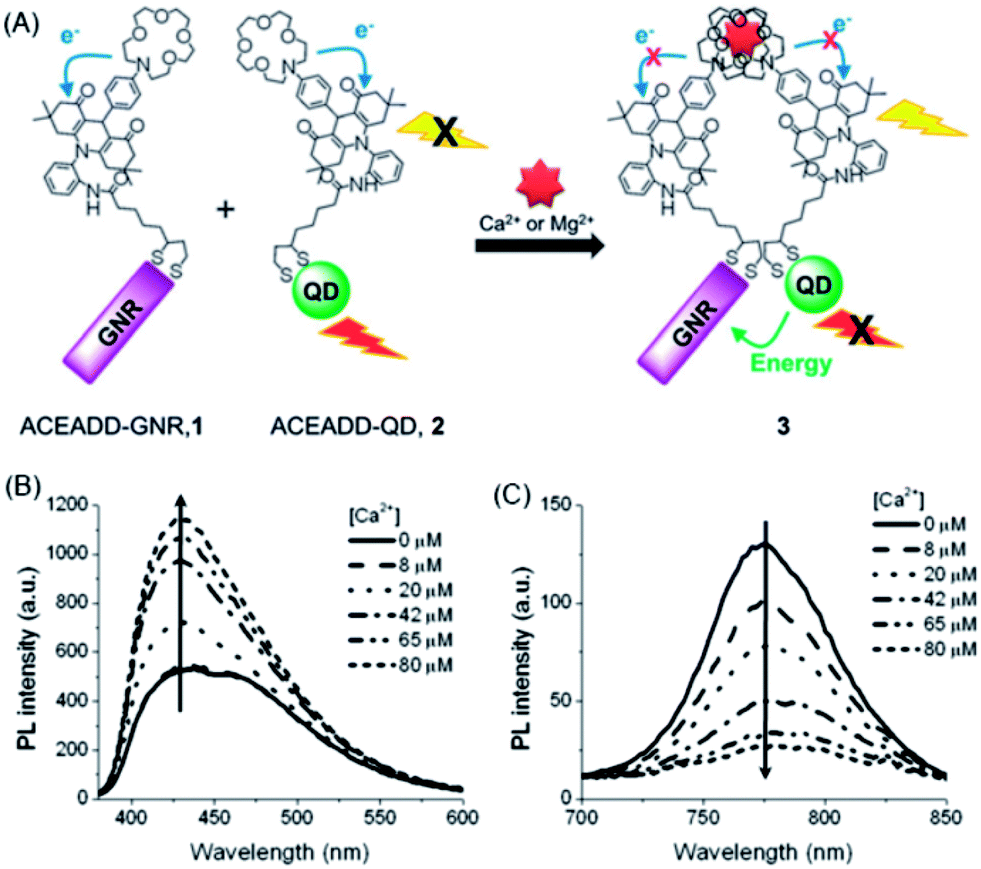 | ||
| Fig. 5 Schematic structures of aza-crown ether acridinedione functionalized AuNRs and aza-crown ether acridinedione-functionalized NIR QDs and Ca2+/Mg2+-triggered FRET between these two pairs (A), and fluorescence emission spectra of the acridinedione moieties (B, λex = 360 nm) and the CdTeSe QDs (C, λex = 500 nm) upon the addition of 0–80 μM Ca2+ ions in acetonitrile. The concentrations of AuNRs and QDs were set at 460 and 920 pM, respectively. Reproduced with permission from ref. 110. Copyright 2012 Royal Society of Chemistry. | ||
A new paradigm of a ratiometric sensing scheme for Hg2+ was developed based on Hg2+-triggered FRET between CdSe@ZnS QDs and rhodamine B.111 As shown in Fig. 6, polymer-encapsulated CdSe@ZnS QDs (λmax = 550 nm) were employed as the energy donor, while a Hg2+-sensitive “turn-on” dye (thiosemicarbazide-functionalized rhodamine B) was utilized as the energy acceptor. The hydrophobic nature of the rhodamine B derivative leads to non-specific adsorption to the surface of QDs. Upon binding with Hg2+, thiosemicarbazide-functionalized rhodamine B experiences a ring-opening reaction through Hg2+-induced de-sulfurization, giving rise to an absorption peak centered at 550 nm. In this manner, the emission of CdSe@ZnS QDs perfectly overlaps the absorption of rhodamine B, thus FRET occurs. The proposed ratiometric sensor exhibited a detection limit of 79 ppb for Hg2+. Recently, a similar sensor design involving FRET from QDs to rhodamine B for Hg2+ has been reported.112 To minimize the direct interaction of Hg2+ and QDs, the QDs were encapsulated inside a silica shell. Considering the versatility of various sensor designs based on spiro-ring opening of xanthenes and related derivatives3 and the color-tunable emission of QDs, such a sensing strategy could be promising for the design of QD-based probes for other metal ions.113
 | ||
| Fig. 6 Ratiometric sensing of Hg2+ based on CdSe@ZnS QDs and rhodamine B. Reproduced with permission from ref. 111 Copyright 2011 Royal Society of Chemistry. | ||
The second strategy is based on metal ion cleavage of FRET between QDs and energy donors. Similar to Page et al.,111 a Hg2+ sensor was developed based on FRET between CdTe QDs and butyl–rhodamine B.114 After fluorescence quenching of CdTe QDs by Hg2+, the FRET was blocked, leading to fluorescence quenching of both the CdTe QDs and butyl–rhodamine B. Yang et al. constructed a FRET assay for Cu2+ based on the energy transfer between CdTe QDs and green luminescent phenol formaldehyde resin (PFR) NPs.115 The CdTe QDs were appended onto the surface of PFR NPs through layer-by-layer functionalization. In the absence of Cu2+, the QDs-PFR NPs nanocomposite gave red fluorescence upon excitation. When Cu2+ was presented, owing to the sensitive quenching effect of Cu2+ ions on CdTe QDs, the FRET was disrupted and the green luminescence of the PFR NPs dominated. Accordingly, the as-prepared FRET nanocomposites could be utilized to monitor Cu2+ with optical visual detection.
A sensitive multiplexed metal ion assay was developed by Wu et al. through the exploration of the FRET between QDs and a metal ion-specific DNAzyme-labeled quencher (Fig. 7A).93 Because of the excitation and narrow emission, differently colored QDs can be excited by a single excitation source, which makes it much easier to achieve multiplexed detection than with traditional dyes that require the excitation light source be tuned into their respective narrow absorption bands. By covalent conjugation of quencher-labeled Cu2+- or Pb2+-specific DNAzymes onto the surface of carboxyl-silanized QDs, the fluorescence of QDs was quenched effectively. In the presence of Cu2+ or Pb2+, the emission was restored due to the cleavage of DNAzymes (Fig. 6A), with detection limits of 0.5 and 0.2 nM for Cu2+ and Pb2+, respectively. Through simple mixing of Cu2+–DNAzyme- and Pb2+–DNAzyme-functionalized QDs (QD530 and QD625) and excitation at 480 nm (Fig. 7B), Cu2+ and Pb2+ can be simultaneously and quantitatively detected with virtually no cross-talk between the 530 nm channel and the 625 nm channel.
 | ||
| Fig. 7 Quencher labeled-DNAzyme-functionalized QDs for multiplex sensing of metal ions. (a) Schematic illustration of the sensor principle; and (b) multiplex detection of Cu2+ and Pb2+ with Cu2+- and Pb2+-specific DNAzymes functionalized QDs (QD530 and QD625). Reproduced with permission from ref. 93 Copyright 2010 American Chemical Society. | ||
Wang et al. constructed a FRET assay for Pb2+ based on Pb2+-modulated donor absorption change.116 A FRET system between cysteamine-capped positively charged CdTe QDs (λmax = 550 nm) and 1-mercaptoundecanoic acid (MUA)-capped negatively charged Au NPs was prepared. Pb2+ can induce the aggregation of MUA-capped Au NPs,117 which changes the state and absorption of Au NPs, leading to inhibited FRET and the fluorescence restoration of the QDs. The fluorescence response was linearly proportional to the concentration of Pb2+ in the range 0.22–4.51 ppm with a detection limit of 30 ppb.
 | ||
| Fig. 8 Schematic illustration of the NSET-based probe for turn-on detection of Pb2+. Reproduced with permission from ref. 120. Copyright 2013 Elsevier. | ||
 | ||
| Fig. 9 A simple chemical etching strategy to create “ion-imprinted” sites on the surface of CdTe QDs for selective fluorescence turn-on detection of Cd2+. Selectivity of MPA–CdTe QDs (60 nM) and 6 μM EDTA-etched MPA–CdTe QDs (60 nM) toward various transition metal ions (1 μM). All solutions were prepared in 10 mM Tris–HCl buffer of pH 9.0 except for the investigation of Ag+ (BT buffer). Reproduced with permission from ref. 121. Copyright 2010 Royal Society of Chemistry. | ||
As discussed in Section 2.1.1, the surface defects of typical II–VI QDs generally originate from unsaturated S2− (or Se2− and Te2−) sites or dangling bonds. The addition of Zn2+ or Cd2+ can eliminate some of these defects, resulting in fluorescence enhancement. Recently, such a mechanism was further engineered to develop a Zn2+ and Cd2+ probe. Through amplifying the surface defects by the introduction of S2−, Xu et al. developed S2−-modified CdTe QDs for fluorescent turn-on detection of Zn2+ and Cd2+ (Fig. 10A).124 When adding S2− into the solution of the CdTe QDs, the fluorescence of the QDs was quenched, further addition of Zn2+ and Cd2+ could restore the fluorescence. As shown in Fig. 10B, the S2−-modified CdTe QDs exhibited good selectivity for Zn2+ and Cd2+ over other metal ions. The selectivity can also be explained from the viewpoint of the bandgap. Since CdTe QDs were employed as the probe and S2− as the modifier, only ZnS and CdS can be used as shells around the CdTe core and passivate the defects.
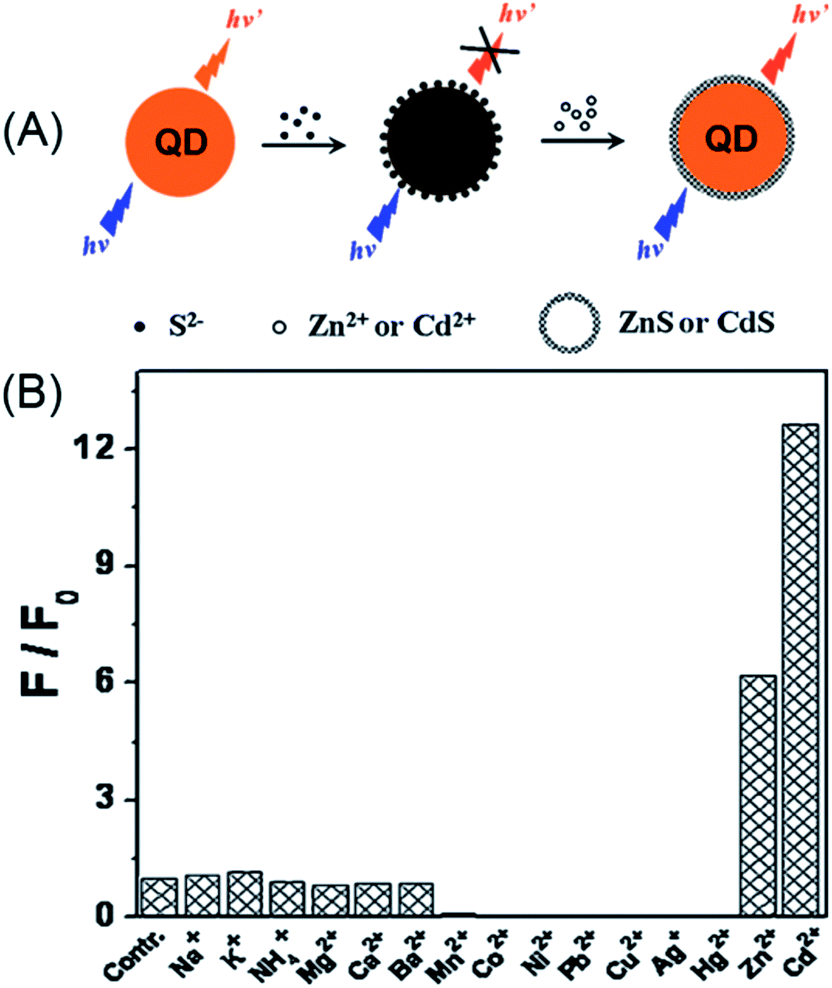 | ||
| Fig. 10 Schematic illustration of the detection of Zn2+ or Cd2+ with S2−-modified QDs (A) and the selectivity of this assay (B). Reproduced with permission from ref. 124. Copyright 2011 Elsevier. | ||
From the view point of the bandgap, the selectivity of S2−-modified CdTe can be well-understood. In semiconductor metal sulfides, only ZnS (3.6 eV) and CdS (2.6 eV) have higher bandgaps than CdTe (2.2 eV), thus only Zn2+ and Cd2+ can eliminate the S2−-induced surface defects.125 In this respect, choosing higher bandgap semiconductor QDs, i.e., ZnS or ZnSe QDs, may eliminate the interference of Cd2+ on Zn2+. Such a design was later realized with Mn-doped ZnS QDs for turn-on detection of Zn2+.126,127 In order to simplify the sensor design, Ren et al. employed a silica coating for the immobilization and enrichment of the S2− quencher.126 In this manner, the sensor fabrication can be finished in the synthetic stage, which guarantees its long-term stability. The developed sensor showed excellent selectivity toward Zn2+, with a detection limit of 80 nM. The silica-coated, S2−-enriched Mn-doped ZnS QDs were successfully explored as a cell-imaging probe for intracellular Zn2+, which demonstrated the advantage of the quencher immobilization.
To enhance the emission of the QDs in the thin films, a plasmon-enhancing strategy was employed through alternate deposition of plasmonic noble metal nanoparticles and QDs on the substrate (layer-by-layer assembly). For example, the emission of the CdSe QDs was enhanced by a factor of 2.5 by coupling with the surface plasmon of the Ag nanoprisms.51 Additionally, the sensitivity and linear range for Cu2+ detection were also substantially improved as compared to pure QDs in solution. A similar surface plasmon-coupled emission (SPCE) of a CdTe QD-based quenchometric sensor was also developed for Hg2+ detection.134 An enlarged linear range and improved sensitivity were the advantages brought by SPCE due to the high light-collection efficiency of SPCE.
A visual detection protocol for heavy metal ions exploring the color change of ZnS QDs during heavy metal ion-induced cation exchange was developed for Hg2+, Ag+ and Pb2+.135 As shown in Fig. 11, a ZnS QDs-impregnated chitosan film was fabricated via evaporation of the solvent. Upon drop-casting of heavy metal ions, characteristic color developments were showed for different metal ions, with Hg2+ being exceptionally identifiable due to the visible bright yellow color formation, while brown coloration was observed for other metal ions. The difference in the solubility of HgS, Ag2S, PbS and ZnS induced cation exchange between Hg2+, Ag+ and Pb2+ and the ZnS QDs. Due to different colors of the resultant metal sulfides, these metal ions can be easily visualized at the ppm level.
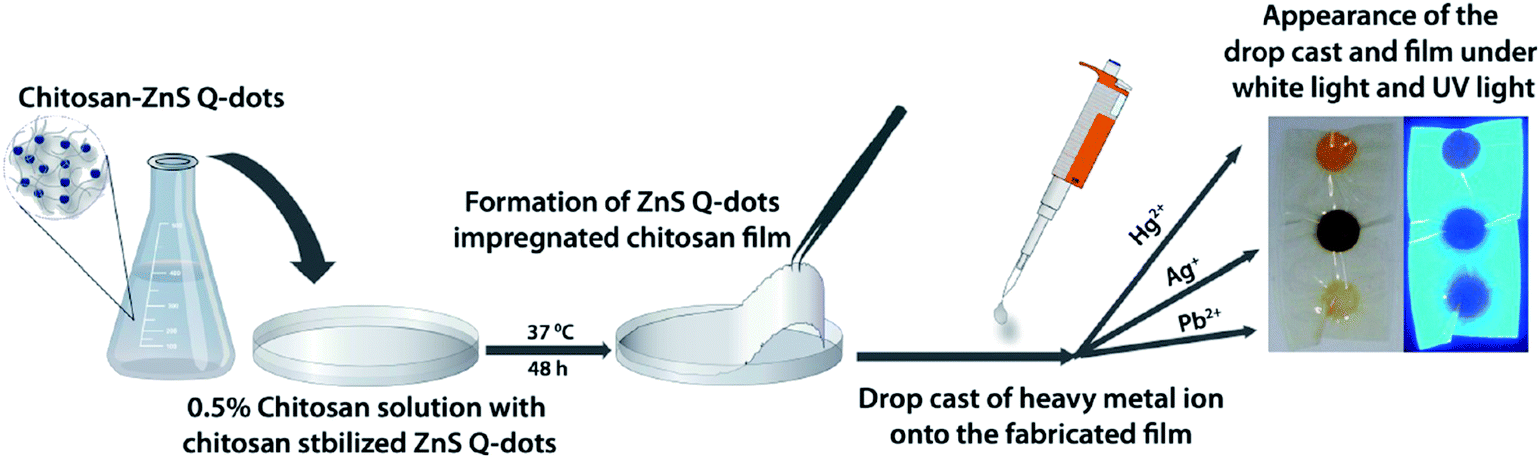 | ||
| Fig. 11 Schematic demonstrating the fabrication of a ZnS QD-impregnated chitosan film along with the appearance of the drop castings of different metal ions onto the fabricated film under visible and UV light. Reproduced with permission from ref. 135. Copyright 2010 American Chemical Society. | ||
Owing to the quite different quantum confinement energies, one dimensional (1D) II–VI nanostructures (nanorods or nanowires) present some novel optical properties different from the zero-dimensional QDs.139 When compared with CdTe QDs, Li et al. found CdTe nanorods with a λmax, em of 575 nm exhibited a similar response to diverse metal ions as CdTe QDs:24 Zn2+ enhanced the fluorescence, while Mn2+, Ni2+, and Co2+ quenched the fluorescence. Accordingly, CdTe nanorods may also have broad applications in metal ion sensing.140,141 A near-IR probe for Cu2+ was developed using CdHgTe nanorods.142 For nanowires, the emission of an ethylenediamine-capped CdS nanowire was reported to be quenched selectively by Cu2+ and Ag+.143 Tang et al. developed a fluorescent probe for Cu2+ based on highly luminescent CdTe nanowires.144
Liu et al. developed a ratiometric sensor for Cu2+ based on CdTe nanorods and a metal bound organic dye (Calcein Blue) (Fig. 12A).90 Upon apposing to Cu2+, the CdTe nanorods experienced a cation-exchange reaction via the incorporation of Cu2+ into the lattice of the CdTe nanorods and releasing Cd2+.33 Meanwhile, the fluorescence of the CdTe nanorods was effectively quenched. Then, the released Cd2+ can bind with Calcein Blue, giving rise to blue emission (Fig. 12B). A ratiometric fluorescent sensing system, which separated the recognition site and the signal transduction unit by employing two different emitting states, was thus developed (Fig. 12B). In this manner, the detection of Cu2+ was amplified via the cation-exchange reaction.145 Such a ratiometric sensor exhibited good selectivity toward Cu2+ (Fig. 12C), with a detection limit of 0.13 nM.
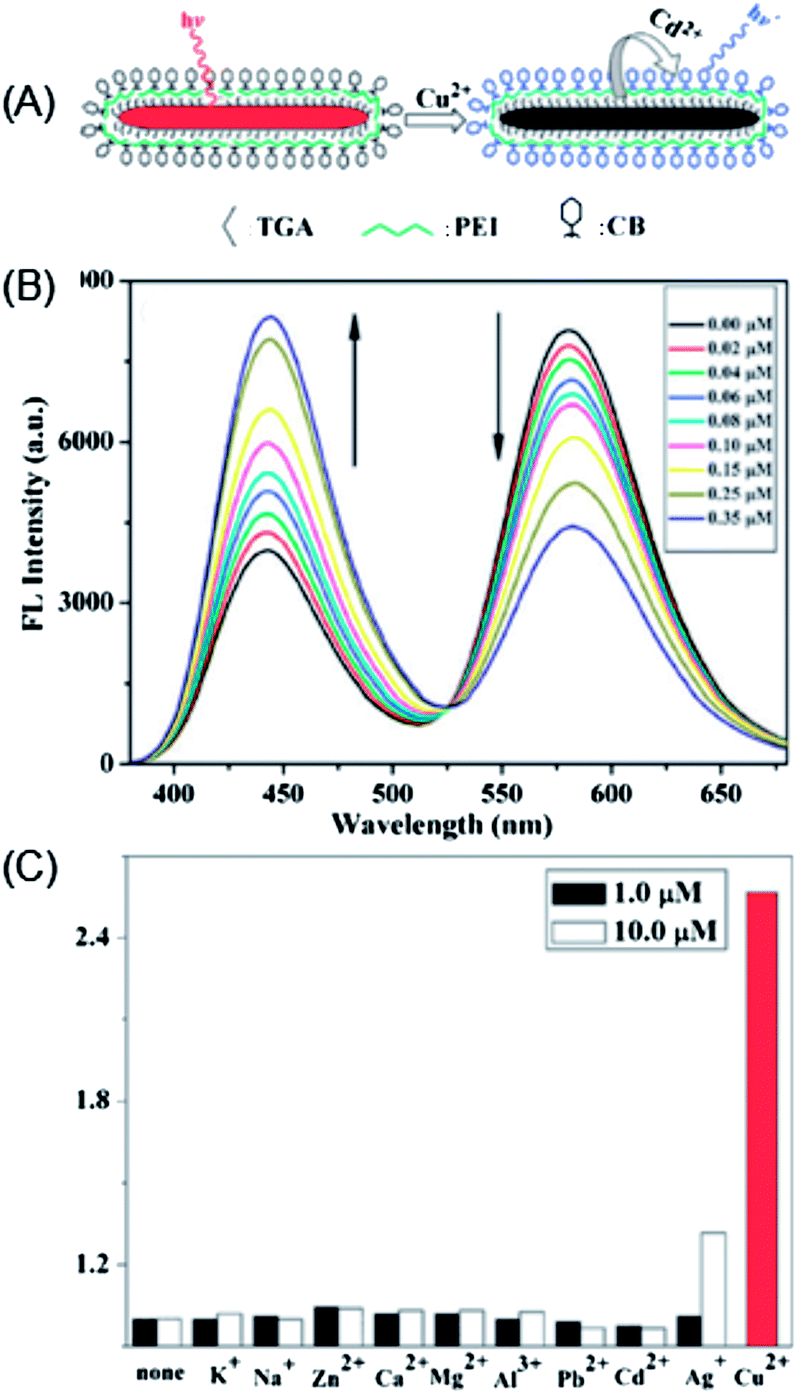 | ||
| Fig. 12 (a) Schematic illustration of the cation-exchange-based signal amplification for ratiometric sensing of Cu2+ based on CdTe nanorods and Calcein Blue; (b) changes in the fluorescence intensity (FL) emission spectra upon addition of different concentrations of Cu2+ to an aqueous solution containing CdTe NRs (0.88 mM), CB (0.5 mM) and KOH (1 mM) (λex = 350 nm); and (c) FL intensity ratio (I442/I581)/(I0442/I0581) response to different concentrations of metal ions (the concentration of Cu(II) was 0.1 μM), including CdTe NRs (0.88 mM), CB (0.5 mM) and KOH (1 mM). Reproduced with permission from ref. 90. Copyright 2010 Royal Society of Chemistry. | ||
2.2. Use of phosphorescence from QDs for detection
Phosphorescence emission of QDs is generally relaxed from the dopant-involved transitions. Upon photoexcitation, energy transfer from the host to the Mn2+ triplet transition (4T1 → 6A1) generates phosphorescence emission centered at 590 nm, with lifetimes on the ms-scale.146 Although the emission mechanism of the phosphorescence from doped QDs is different from that of fluorescent QDs, the sensor strategies for metal ions of these two types of QDs are generally the same;147–149 phosphorescence or time-gated detection allows autofluorescence-free detection. Xie et al. developed a label-free aptamer phosphorescent sensor for Hg2+ based on CTAB-capped Mn-doped ZnS QDs.150 In the absence of Hg2+-specific aptamer, the interaction between Hg2+ and QDs was restricted due to electrostatic hindrance. In the presence of poly-T DNA (T20), Hg2+ and T20 could readily form a T–Hg–T dsDNA configuration, which could bind to positively charged QDs and quench the phosphorescence of the QDs. Huang et al. developed a turn-on time-gated fluorescent sensor for Hg2+ based on DNA-functionalized Mn-doped CdS QDs and AuNPs.151,152 As shown in Fig. 13, Mn-doped CdS@ZnS QDs and AuNPs were functionalized with 33-mer T-rich ssDNA and 10-mer ssDNA. Upon hybridization, AuNPs could quench the phosphorescence of Mn-doped CdS QDs because of energy transfer. The presence of Hg2+ could detach AuNPs from close proximity to QDs and thus restored the fluorescence of QDs. Although successful, it should be noted that the Hg2+-recognition unit was anchored on the surface of the QDs, which could also be a potential electron-transfer quencher to the QDs.91,150 Presumably, the linear range of this sensor may be restricted to a critical concentration of Hg2+, which is a compromise between the fluorescence restoration resulting from detachment of the AuNPs and Hg2+-induced fluorescence quenching to the QDs. Compared with the former turn-off detection,119 turn-on detection with long-lived Mn-doped CdS QDs provided both a signal background and sample fluorescence background.151 The details of the surface-functionalized phosphorescent QDs for metal ions detection are summarized in Table 3.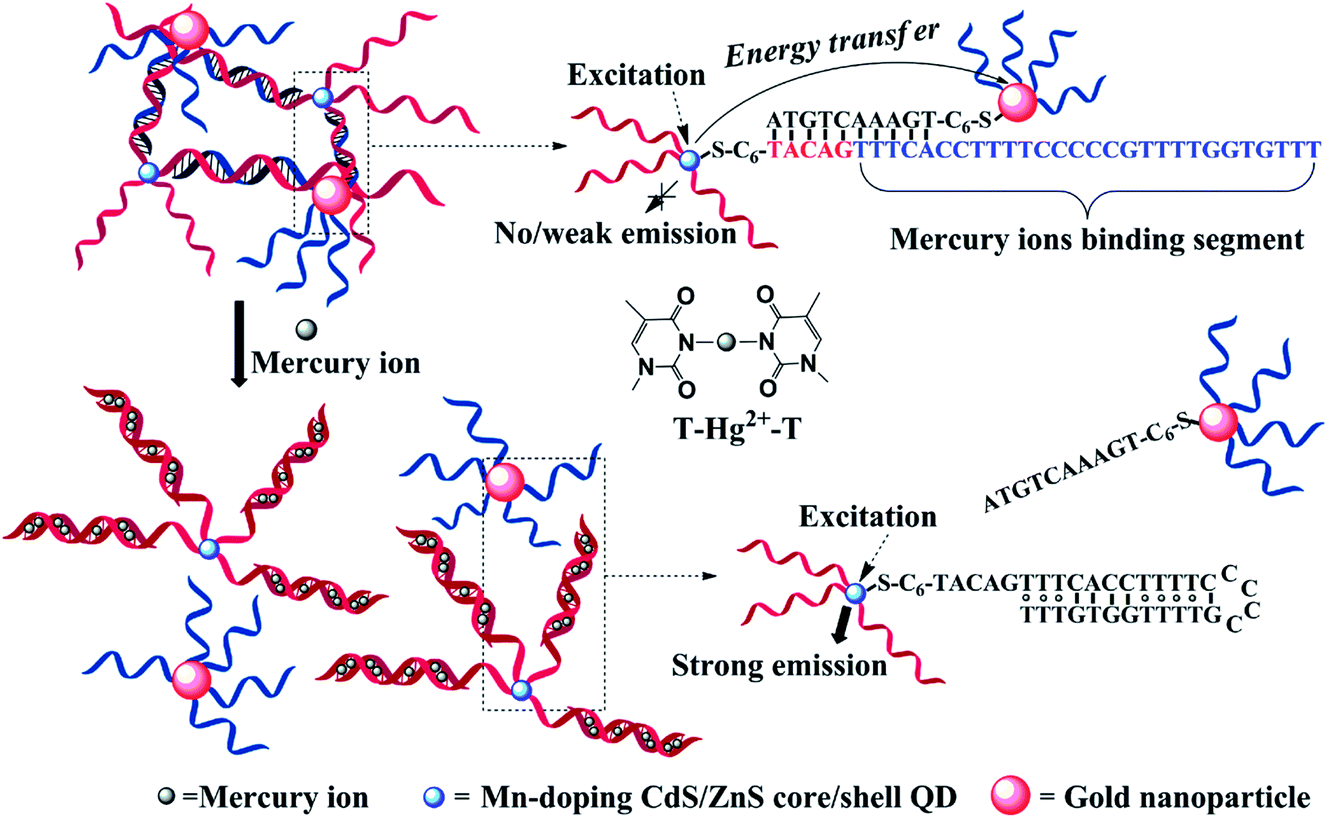 | ||
| Fig. 13 DNA-functionalized Mn-doped CdS@ZnS QDs and AuNPs assembly for turn-on detection of Hg2+. In the absence of Hg2+, the fluorescence of QDs was quenched due to energy transfer between QDs and AuNPs, while in the presence of Hg2+, it could mediate the formation of DNA duplexes and detach AuNPs from close proximity to QDs, restoring the fluorescence. Reproduced with permission from ref. 151. Copyright 2010 American Chemical Society. | ||
2.3. Use of electrochemiluminescence from QDs for detection
Apart from photoluminescence, the electrochemiluminescence (ECL) from semiconductor QDs is also very popular for analyte sensing,157,158 as ECL has the unique advantage of lack of background from photoexicitation over PL. The details of the surface-functionalized QDs for ECL detection of metal ions are summarized in Table 3. The first QD-ECL sensor for Cu2+ was reported by Zhang et al. in 2008.159 The system is based on the removal of the mercaptosuccinic acid from the surface of QDs by Cu2+, followed by the further ECL quenching of the CdTe QDs, with detection limit of 30 nM. Exploration of dual ligands (GSH and TGA) for synthesis of CdTe QDs could greatly enhance the ECL intensity, thereby improve the sensitivity for Pb2+.160 Introduction of co-reactant tripropylamine could participate in the ECL process of QDs and lower the onset potential, and greatly improve the ECL intensity thus better sensitivity for Cu2+.161To decrease the cathodic ECL potential and improve the immobilization of QDs on the electrode surface, a bidentate ligand, meso-2,3-dimercaptosuccinic acid (DMSA), was employed for synthesis of CdTe QDs.162 The DMSA capped CdTe QDs exhibited an intensive ECL emission peak around −0.87 V with an onset potential of −0.64 V, compared to typical ECL peak potential of CdTe QDs at −2.0 V. Cu2+ could compete the ligand with QDs, thereby quenching the ECL of QDs, with a detection limit of 3.0 nM.
The chemiluminescence from CdTe QDs is also sensitive to the presence of metal ions.163 Kanwal et al. found that the CdTe QDs could activate the pyrogallol–H2O2 chemiluminescence system, and the presence of Cr(VI) could further selectively enhance the chemiluminescence in the concentration range of 20 pM to 30 μM, with a detection limit of 6 pM.164 This system represented one of the most sensitive approach for metal ion detection based on QDs.
2.4. Use of QDs for photoelectrochemical detection
Photoelectrochemical (PEC) detection is a reversed process of ECL, in which light is used to excite active species on the electrode, and current is obtained as the detection signal.165 PEC sensors combine the advantages of both optical methods and electrochemical sensors. The strategies for PEC sensors for metal ions based on QDs are generally inherited from the knowledge of direct interaction between metal ions with QDs.166 The details of the surface-functionalized QDs for PEC detection of metal ions are summarized in Table 3. Wang et al. firstly explored the PEC from CdS QDs for Cu2+ detection with a detection limit of 10 nM.167 A cathode PEC sensor for Cu2+ based on Cu2+-induced formation of exciton trapping was developed.168 As shown in Fig. 14, Cu2+ can compete the thiol ligands on the surface of the CdTe QDs, leading to partial decomposition of the QDs and formation of Cu+ as exciton trapping sites. These trapping sites decreased the cathode photocurrent, giving rise to a sensitive method for trace Cu2+ detection with an LOD of 5.9 nM. Shen et al. explored ZnO/CdS hierarchical nanospheres as photoelectrochemical-active moiety for Cu2+ sensing.169 The light scattering of ZnO nanospheres and the heterointerface between CdS and ZnO provided significant advantages for enhanced light absorption and charge separation, thus resulting in an improvement in the photocurrent intensity. The formation of Cu2S on the surface of CdS in the presence of Cu2+ inhibited the photochemical process and detection of Cu2+ was thus achieved with an LOD of 0.01 μM. Recently, the improved photoelectrochemical performance of CdZnS alloyed QDs upon compositing with reduced graphene oxide has been reported.170 Cu2+ could induce selective quenching of the photoelectrochemical current, with a detection limit of 6.7 nM.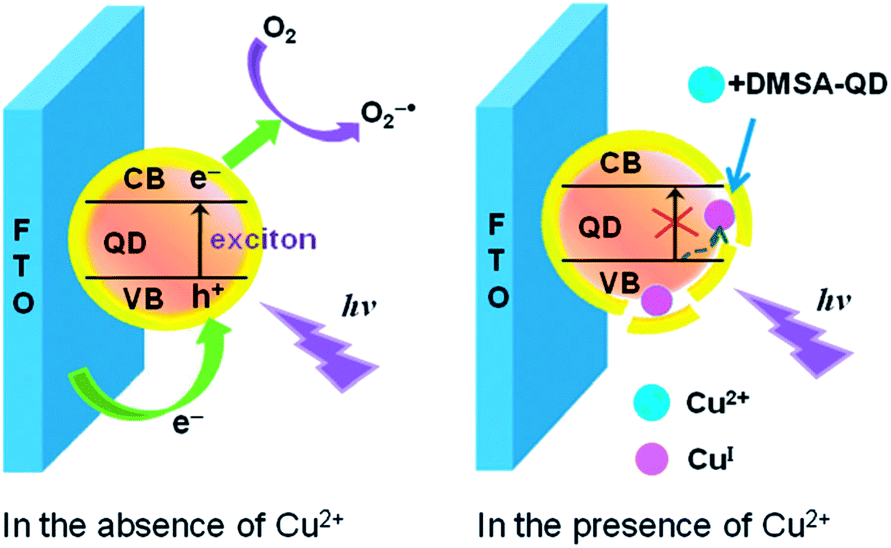 | ||
| Fig. 14 Schematic illustration of the exciton trapping mechanism and cathode photoelectrochemistry for sensing of Cu2+. Reproduced with permission from ref. 168. Copyright 2012 Royal Society of Chemistry. | ||
2.5. Use of QDs as electrochemical labels for metal ion detection
The dimensions of QDs and their facile surface modification with biomolecules make them ideal electrochemical tracers for bio-recognition. Subsequent acid-induced solubilization of the QDs labels and the electrochemical stripping off of the released ions provide quantitative readout signals for the labeled analytes. Zhang et al. developed a highly sensitive DNAzyme-based electrochemical sensor for monitoring of Pb2+ based on Pb2+-induced cleavage of the DNAzyme.155 As shown in Fig. 15, in the presence of Pb2+, the biotin-modified substrate strand in DNAzyme was cleaved into two DNA fragments by the catalytic DNA strand at the cleavage site. The biotin-modified fragments were used as signaling probes to hybridize with the complementary thiolated DNA strands assembled on the surface of the Au electrode. Layer-by-layer assembled PbS QDs with a surface streptavidin coating were employed as signal labels and introduced to interact with the hybridized biotin-signaling probes through biotin–avidin affinity interactions. The captured PbS QDs were then dissolved by nitric acid and monitored with electrochemical stripping. A dynamic range from 1 to 1000 nM with a detection limit of 0.6 nM was obtained under the optimal conditions.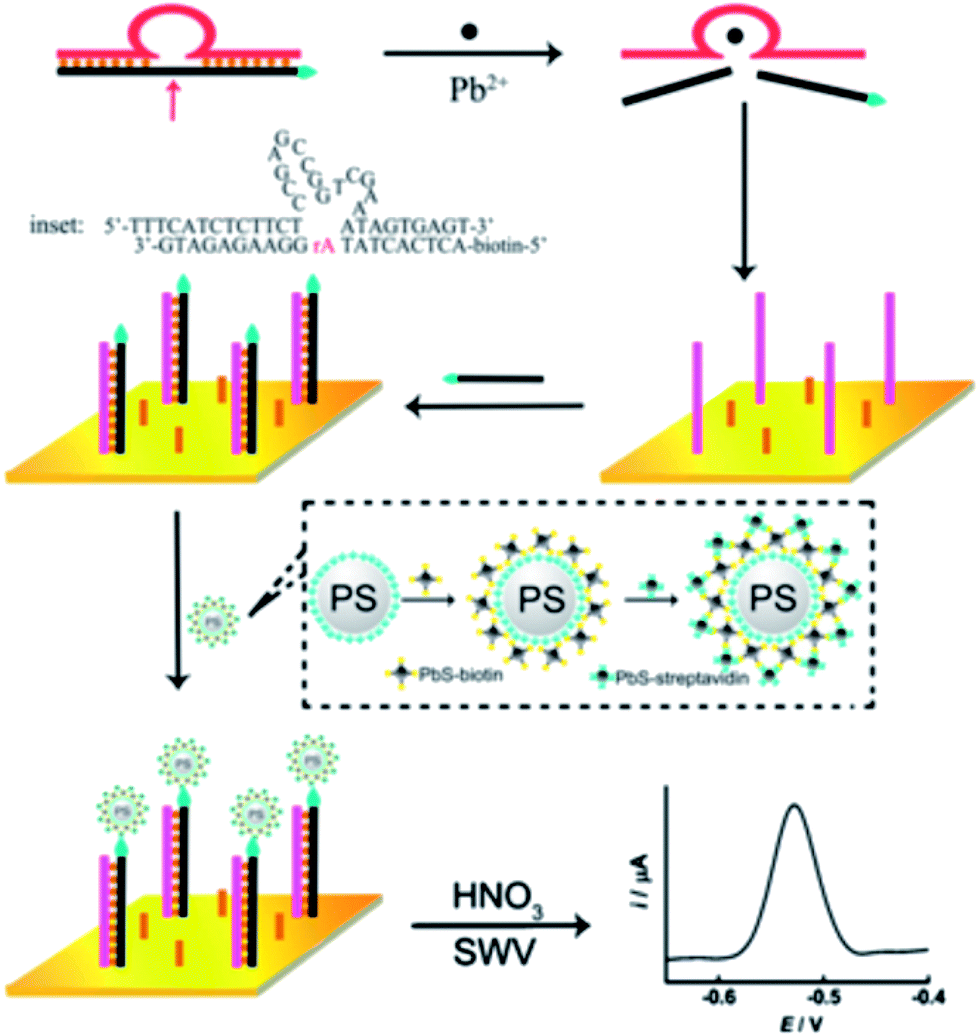 | ||
| Fig. 15 Mechanistic illustration of using QDs as electrochemical labels combined with a DNAzyme (“8–17” DNAzyme, Pb2+-specific)-based assay for Pb2+ detection. Reproduced with permission from ref. 155. Copyright 2011 Elsevier. | ||
The electrochemical stripping strategy can also be coupled with signal amplifying strategies. In order to further improve the sensitivity for Pb2+ detection, Zhang et al. introduced the rolling circle amplification (RCA) process as a signal amplification tool.156 In this strategy, the DNAzyme catalytic strands were firstly immobilized onto the surface of magnetic beads and then hybridized with substrate strands. In the presence of Pb2+, the DNAzyme could be activated to cleave the substrate strand into two DNA fragments. After the RCA reaction, a long ssDNA product with repeating sequences was obtained. Subsequently, CdS QD modified ssDNA was used as a signaling probe to hybridize with the long ssDNA product. Due to the dramatic signal amplification by the numerous QDs and the low background signal by magnetic separation, an ultra-low level (7.8 pM) of Pb2+ was detected.
2.6. Comparison of QD-based PL, ECL, PEC and electrochemical stripping methods for metal ion detection
The PL from QDs has long been recognized and the physics and chemistry of the PL process has been thoroughly studied. The PL-based sensors are by far the most popular choices for research. ECL and PEC from QDs are relatively new signal transduction modes. Electrochemical stripping is actually based on the elemental composition of the QDs, which transfers the target-induced biomolecular recognition into the amount of QDs for stripping on the electrode. Many of the sensing strategies from PL sensors can be transferred to ECL and PEC systems, especially for metal ions. However, differences between PL, ECL and PEC do exist. Mechanistically, PL and PEC are both arising from photo-excited excitons, while ECL is based on the redox chemistry of QDs on the electrode. Meanwhile, ECL and PEC are both electrode-involved and coreactants (ECL) or electron/hole donors (PEC) are needed, while PL can work in homogenous solution, without any extra assistance. Accordingly, ECL and PEC may harvest from coreactants (ECL) or electron/hole donors-involved sensing strategies, which are not possible in PL process.The selectivity of the above signal transduction modes for metal ion detection can be modulated according to their mechanisms. The selectivity of the modulated strategies can be further improved through surface shelling of the QDs and conjugation with metal ion-selective receptors. For ECL, PEC and electrochemical stripping, the immobilization of QDs on the electrode permits more convenient conjugation of biomolecules (DNA) capable of selective recognition of metal ions with QDs. Accordingly, the selectivity of these methods will be largely dependent on modern DNA technology. For further development of metal ion probes based on QDs, these signal transduction modes can be elegantly chosen with better understanding of the physics and chemistry inside these processes.
3. Other analytical applications of QD-based metal ion probes
3.1. Cellular probes
Although plenty of QD-based nanosensors have been developed for various metal ions, applications of these nanoprobes for detection of cellular metal ions have been seldom reported. Duback et al. developed an ion-selective nano-optodes containing QDs (ISQDs) for cellular sodium ion measurements.171 The construction of ISQDs was through the assembling of QDs, an ion-selective polymer and a biocompatible lipid-modified poly-ethylene glycol (PEG). The polymer matrix contains a light absorption pH indicator (with absorption overlapping with the emission of QDs) in conjugation with a non-absorbing, specific ion-binding moiety. The absorption of the pH indicator overlapped with the emission of QDs, and thus quenched the fluorescence of the QDs through an inner filter effect. Upon Na+ binding, the polymer released H+ into solution and the absorption spectra shifted to a low wavelength range, minimizing the overlap range and turning on the fluorescence of the QDs. The incorporation of QDs substantially improved the sensitivity of the nanosensor to a Na+ resolution of 80 μM, compared to 370 μM from that without QDs.172 Besides, such ISQDs displayed a selectivity of 2.3 decades for Na+ over K+.For the cellular imaging of metal ions, ratiometric sensors possess the advantage of higher sensitivity and built-in correction for environmental effects over the first-generation intensity probes. In a similar design as described in Fig. 6,111 Hu et al. developed a ratiometric fluorescence sensor for imaging of Hg2+ in live HeLa cells based on FRET from CdTe QDs to Rhodamine 6G derivative-mercury conjugate (R6G-D-Hg).173 Hg2+ could annihilate the fluorescence of CdTe QDs at 508 nm and meanwhile interact with the R6G derivative to form a fluorescent conjugate giving rise to emission at 554 nm. FRET from CdTe QDs to R6G-D-Hg was triggered by Hg2+, resulting in a concentration-dependent variation of fluorescence ratio F508/F554. Cellular Hg2+ could be imaged with this ratiometric probe, with a dual-channel (green and yellow) readout. To increase the fluorescence color difference, Zhu et al. integrated carbon dots (λem, max = 485 nm) and CdSe@ZnS QDs (λem, max = 644 nm) for ratiometric imaging of Cu2+ in living cells.174 As shown in Fig. 16A, carbon dots were hybridized with CdSe@ZnS QDs to form a dual-emission fluorophore, in which CdSe@ZnS QDs was embedded in silica shells (CdSe@SiO2). Meanwhile, an organic molecule specific for Cu2+ (named AE-TPEA), was anchored onto the surface of the CdSe@C nanohybrid. In the presence of Cu2+, the blue emission of the carbon dots was selectively quenched, while the red emission of CdSe@ZnS QDs that were inert to Cu2+ served as reference signal. Consequently, variation of the two fluorescence intensities (I485/I644) displayed continuous color changes upon the addition of Cu2+ ions, resulting in a ratiometric fluorescent sensor for Cu2+ ions. Such a ratiometric sensor was successfully employed for intracellular imaging of Cu2+ in HeLa cells. As shown in Fig. 16B, the confocal fluorescence images of HeLa cells before and after exogenous Cu2+ source treatment can be easily visualized via the fluorescence color (from green-yellow (b) to red (c)). Meanwhile, the green fluorescence image from the carbon dots became dimmer after addition of the Cu source and incubation for 2 h (e and g), while the red reference channel from the CdSe@ZnS QDs showed almost no change (f and h).
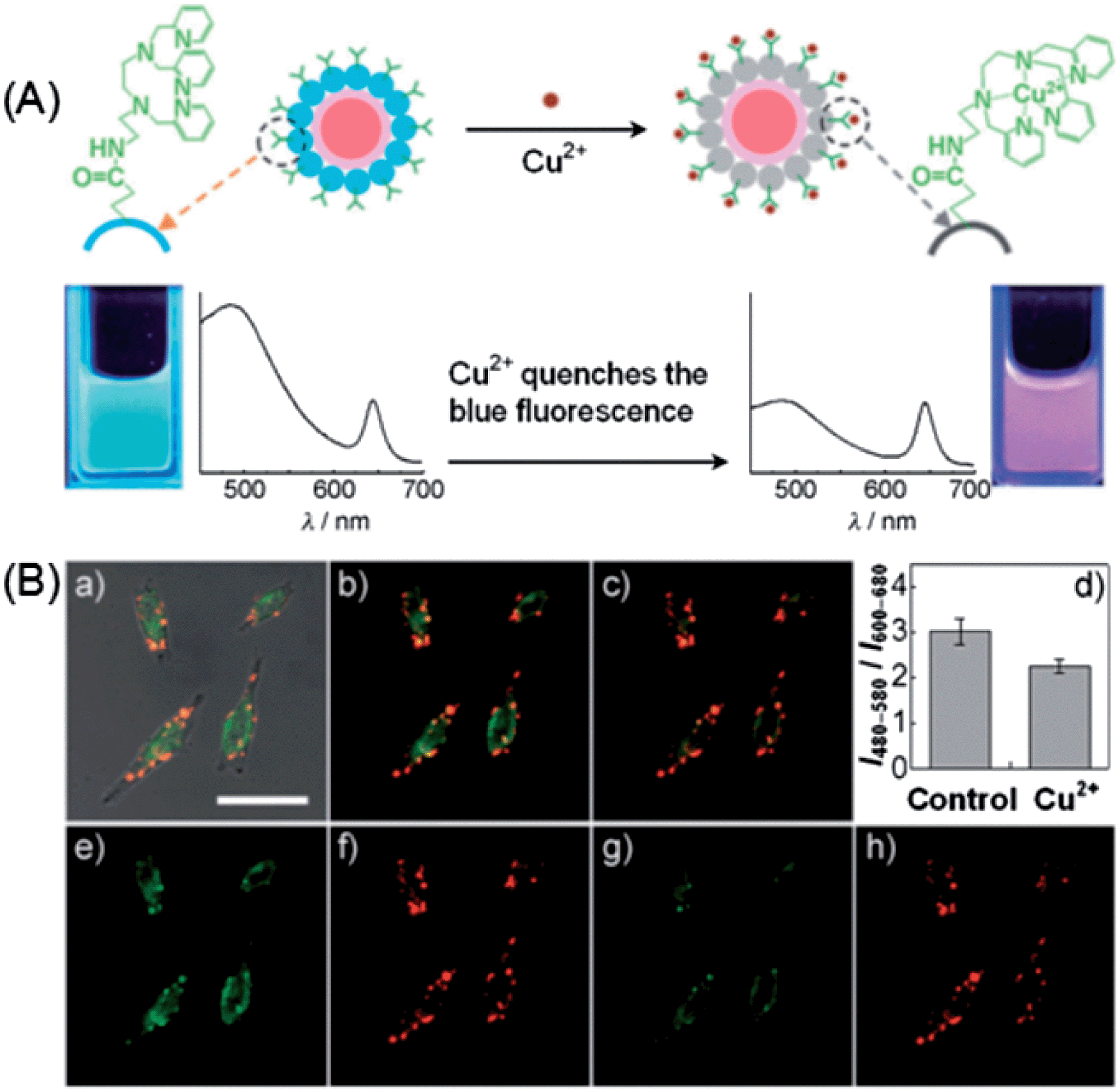 | ||
| Fig. 16 (A) Schematic illustration of the dual-emission fluorescent probe for Cu2+ based on a CdSe@carbon dot-TPET nanohybrid. (B) Ratiometric imaging of Cu2+ in HeLa cells: (a) the overlay of bright-field and fluorescence images of HeLa cells incubated with 0.05 mg mL−1 CdSe@C-TPEA and 0.1 μM PDBu in PBS (pH 7.4) for 2 h at 310 K and 5% CO2. Panels (b) and (c) are the confocal fluorescence images of HeLa cells before (b) and after (c) the exogenous Cu2+ source treatment (addition of 100 μM CuCl2 and 100 mm PDTC to the Petri dish). (d) A bar graph showing the integrated intensity from 480–580 nm over the integrated fluorescence intensity from 600–680 nm. Values are the mean ratio generated from the intensity from three randomly selected fields in both channels. The error bars represent standard error measurement (s.e.m). Panels (e) and (g) are the confocal fluorescence images obtained from 480–580 nm before and after the exogenous Cu source treatment, whereas panels (f) and (h) are the confocal fluorescence images obtained from 600–680 nm. Scale bar: 25 mm. Reproduced with permission from ref. 174. Copyright 2012 Wiley-VCH. | ||
For cellular probes, the cytotoxicity is an important issue that should be considered.175 To prevent heavy metal ion leakage, a silica coating is frequently employed. By embedding CdSe QDs in a silica container, Li et al. developed a quantum dot-based fluorescent sensor for imaging of cellular Cd2+.102 The Cd2+-recognition unit, triethylene-glycol triazole, was appended onto the surface of a silica nanosphere containing CdSe quantum dots (CdSe@SiO2–TGT) via a “click” chemistry route. A sulfite anion was firstly introduced as a fluorescence quencher via a PET quenching mechanism. Then the presence of Cd2+ resulted in fluorescence turn-on of QDs with high sensitivity and selectivity. The as-prepared composite quantum dot (CdSe@SiO2–TGT)-based fluorescent sensor successfully worked in imaging of Cd2+ in HeLa cells.102 Through the bridging effect of S2−, Ren et al. developed a silica-coated S2−-enriched Mn-doped ZnS QDs probe for imaging intracellular Zn2+ ions (Fig. 17).126 The fluorescence of Mn-doped ZnS QDs was firstly quenched by S2−, and then turned on by Zn2+. To better immobilize S2− for a stable probe, Mn-doped ZnS QDs after adsorbing of S2− were further shelled with a thin layer of silica. The SiO2–S–Mn–ZnS QDs showed low cytotoxicity, good sensitivity and selectivity for Zn2+, and were successfully explored for imaging of Zn2+ in HepG2 cells.
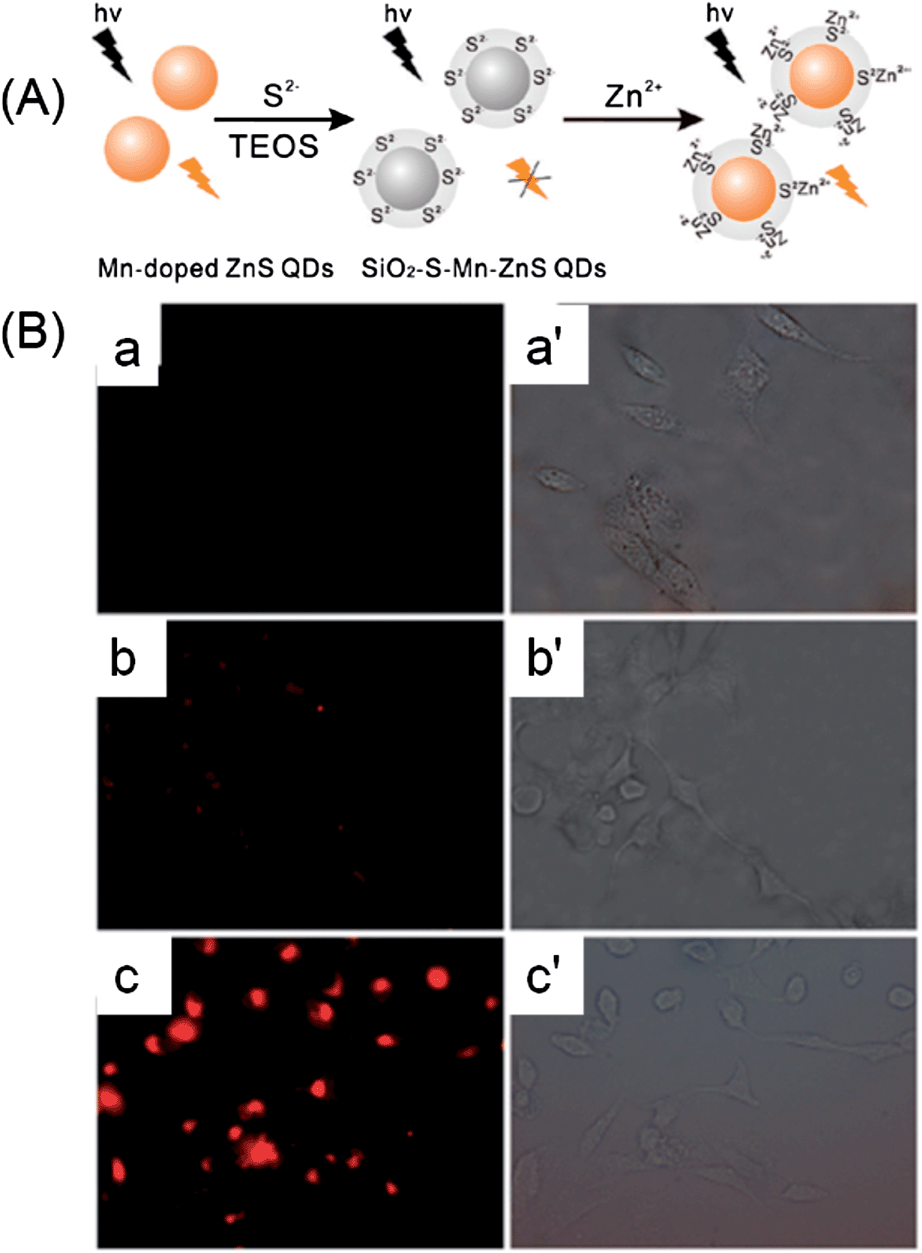 | ||
| Fig. 17 (A) Schematic illustration for the fabrication and application of SiO2–S–Mn–ZnS QDs as a turn-on fluorescent probe for Zn2+. (B) Fluorescence images (a–c) and their corresponding brightfield transmission images (a′–c′; ×400): (a and a′), HepG2 cells were incubated with SiO2–S–Mn–ZnS QDs (0.8 g L−1) in a fresh serum-free medium (i.e., serum starvation condition) for 4 h and then in a fresh serum-containing medium (i.e., without serum starvation) for 12 h; (b and b′), HepG2 cells were incubated with Zn2+ (100 μM) in a fresh serum-containing medium for 20 h; the collected Zn2+-uptaken HepG2 cells were then incubated with SiO2–S–Mn–ZnS QDs (0.8 g L−1) in a fresh serum-containing medium for 16 h; (c and c′) HepG2 cells were incubated with Zn2+ (100 μM) in a fresh serum containing medium for 20 h; the collected Zn2+-uptaken HepG2 cells were then incubated with SiO2–S–Mn–ZnS QDs (0.8 g L−1) in a fresh serum-free medium (i.e., serum starvation condition) for 4 h and then in a fresh serum-containing medium (i.e., without serum starvation) for 12 h. Reprinted with permission from ref. 126. Copyright 2011 American Chemical Society. | ||
3.2. Coding-based sensing of metal ions with QDs
An important ratiometric sensing strategy was based on introducing a reference background light together with signal transduction light, i.e., encoding. Compared to fluorescence encoding with absolute intensity, ratiometric encoding is much more reliable because the ratio values do not suffer from simultaneous drifts or fluctuations of individual signals. QD-decorated dye-doped silica NPs were constructed by Zhao et al. for post-encoding sensing of Cu2+.176 Fluorescein isothiocyanate (FITC, λem = 510 nm)-doped silica NPs was firstly synthesized via the well-known Stöber method. CdTe QDs (λem = 615 nm) were then appended onto the surface of amine-terminated silica NPs through electrostatic assembly, followed by coating a further silica shell. Upon apposing to Cu2+, the red emission of the QDs was quenched effectively, while that of the FITC was almost unchanged, giving rise to a ratiometric signal for Cu2+. Chen et al. employed a similar strategy by employing green-emitting CdTe QDs and Ru(bpy)3Cl2 with red emission peaking at 600 nm.177 To construct the coding sensing moiety, glass slides were firstly immobilized with Ru(bpy)3Cl2via the assistance of polyacrylonitrile–dimethylsulfoxide and then inserted into the solution of CdTe QDs. Cu2+ caused efficient fluorescence quenching toward CdTe QDs (green emission), while the fluorescence of Ru(bpy)3Cl2 was kept unchanged. For visual detection, the color of the glass slide was recorded with a CCD camera upon excitation with a 396 nm LED source. A gradual color change from green to red was observed upon increasing the Cu2+ concentration from 1 × 10−8 to 1 × 10−2 M.The encoding can also be achieved by using one type of QDs (one color) as the signal code and another type of QDs (another color) as the reference code. Since QDs have broad absorption spectra and the emission colors can be facilely tuned via bandgap and particle size, so such an encoding strategy provides more versatility than QD-dye hybrid systems. Sun et al. prepared a dual-emission nanocomposite of CdS QDs–chitosan–CdTe QDs as a ratiometric sensor for Hg2+.178 CdS QDs (λem, max = 450 nm) inside the nanocomposite were employed as an internal standard, while the CdTe QDs (λem, max = 590 nm) were used as a signal probe for Hg2+. The presence of Hg2+ resulted in fluorescence quenching of the CdTe QDs, while the fluorescence of the CdS QDs remained unchanged. In a similar approach, a ratiometric probe with blue-emitting carbon dots as the standard and red-emitting CdSe@ZnS QDs as the signal probe for Hg2+ was developed.179 To increase the color resolution of the ratiometric probe, Yao et al. used highly luminescent green- and red-emitting CdTe QDs as the signal and reference codes, respectively, for the fluorescent visual detection of Cu2+.180 As shown in Fig. 18A, the ratiometric probe was synthesized via first embedding the red CdTe QDs into silica, then the green CdTe QDs were loaded onto the surface of the silica particles through covalent conjugation. In the presence of Cu2+, the fluorescence of the green QDs was gradually quenched, while the fluorescence of the red QDs embedded inside silica remained constant, resulting in a fluorescent color change of the probe from green to red (Fig. 18B).
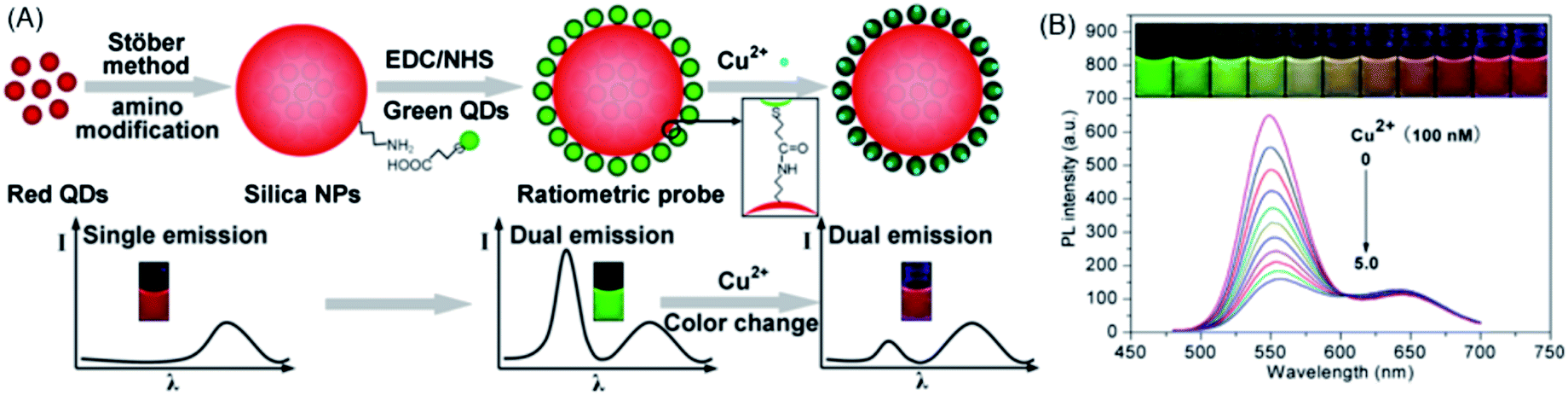 | ||
| Fig. 18 (A) Schematic illustration of the synthetic procedure and visual principle of the ratiometric probe for Cu2+. (B) The fluorescence colors and the corresponding fluorescence spectra (λex = 365 nm) of the ratiometric probe upon the exposure to different concentrations of Cu2+. Reproduced with permission from ref. 180. Copyright 2013 American Chemical Society. | ||
The broad absorption spectra of QDs and the pairing of their narrow emission spectra with the absorption spectra of acceptor dyes allow facile construction of sensors with multi-ion sensing capabilities. Hall et al. developed a sub-micro polyacrylic sensor containing two independent ion sensing systems for coding sensing of K+ and Cl−.181 CdSe@ZnS QDs (λem, max = 540 nm), Cl−-sensitive lucigenin (λem, max = 483 nm), K+-selective valinomycin and chromoionophore I (above pKa: λabs/em, max = 540 nm/660 nm; below pKa: weakly fluorescent, λabs, max = 610, 660 nm) were embedded into the polyacrylic matrix. Upon excited at 400 nm, both lucigenin and the QDs could be excited. Meanwhile, dual-emission peaks at 540/648 nm confirmed FRET from the QDs to deprotonated chromoionophore I. In the presence of KCl (K+ and Cl−), the emission from lucigenin was quenched, while the dual-emission peaks exhibited a systematic change due to K+-induced proton exchange of chromoionophore I, giving rise to coding type-sensing of K+ and Cl−. A double-walled polyelectrolyte capsule barcoded with QDs was developed for the multiplexed sensing of protons, Na+ and K+.182 Via a layer-by-layer approach, the microcapsules were fabricated by embedding of an ion-sensitive dye, i.e., FITC (H+), SBFI (sodium-binding benzofuran isophthalate) or PBFI (potassium-binding benzofuran isophthalate), and a reference dye (Dy647) into polyelectrolyte networks.183 For spatial separation of dyes and QDs, a second polyelectrolyte wall with the integrated QD barcode (orange QD–FITC, green QD–SBFI, and yellow QD–PBFI), was formed around the first one, whereby both walls were clearly spaced. As shown in Fig. 19A, after adding of the mixed capsules to eight different solutions permutations of low/high H+ (c(H+) = pH 9/pH 5), low/high Na+ (c(Na+) = 5 mM/140 mM), and low/high K+ (c(K+) = 5 mM/140 mM), the three different types of capsules can be clearly distinguished by their QD barcode in the outer wall. Additionally, the concentrations of H+, Na+, and K+ could be measured with their corresponding blue green to red ratios. Low and high H+ concentrations (pH = 5, pH = 9) could be distinguished with high precision regardless of the Na+ and K+ concentration (Fig. 19B and C). Although crosstalk existed with high pH in the case of SBFI and PBFI filled capsules, errors due to crosstalk between Na+ and K+ were significantly lower than the signal to be detected. In other words, they could distinguish between low and high Na+ and K+ concentration (5 mM/140 mM), despite the respective K+ and Na+ concentration (Fig. 19D and E), allowing discrimination of Na+ and K+ in parallel.
 | ||
| Fig. 19 Multiplexed measurements of ions with barcoded polyelectrolyte sensor capsules. (A) Three different types of capsules have been synthesized. (B) Fluorescence image of a mixture of the three different types of capsules in two solutions with different ion concentrations; via the fluorescent barcode the type of each capsule can be clearly identified. By changing from a low Na+ [c(Na+) = 5 mM, c(K+) = 5 mM, pH = 5] to a high Na+ condition [c(Na+) = 140 mM, c(K+) = 140 mM, pH = 9] the ISBFI/IDy647 ratio of the Na+ responsive capsules is raised, and in the false color fluorescence image the capsule cavities appear more blue-green compared to the more reddish appearance at low Na+ concentration. (C–E) By variation of the pH from low (5) to high (9), the Na+ concentration from low (5 mM) to high (140 mM), and the K+ concentration from low (5 mM) to high (140 mM) eight different combinations of ion concentrations with defined ion mixtures were generated. From left to right: the IFITC/IDy647, ISBFI/IDy647, and IPBFI/IDy647 read-outs of the different capsules in these buffers were determined and are plotted here (data points represent mean values as determined from 30 capsules and standard deviations). The IFITC/IDy647 read-out of the pH sensitive capsules clearly allows for discrimination between low and high pH in all solutions, regardless the Na+ and K+ concentration. The red dotted line shows the threshold for the IFITC/IDy647 read-out distinguishing between low and high pH. The ISBFI/IDy647 and IPBFI/IDy647 read-outs of Na+ and K+ sensitive capsules allowed for discrimination between low and high Na+ and K+ concentrations at high pH, and a threshold (red dotted line) can be given. At low pH, SBFI as well as PBFI interfere with pH. Reprinted with permission from ref. 182. Copyright 2011 American Chemical Society. | ||
3.3. Use of metal ion-sensitive QDs for logic operations
Besides metal ion sensing, QD-based metal ion probes are also capable of logic gate operations. Taking the advantage of the recognition of Hg2+ and Ag+ by T- and C-rich DNA, respectively, Willner et al. developed a multiplexed assay for Hg2+ and Ag+ based on DNA-functionalized QDs (Fig. 20A).91 Thymine-rich DNA (DNA 1) was functionalized with QD560, and cytosine-rich DNA (DNA 2) for QD620. In the presence of Hg2+ and Ag+, DNA 1 and DNA 2 can form a hairpin structure, respectively, which brings the metal ions and QDs into close proximity and induces electron transfer quenching toward QDs. They further engineered such DNA-functionalized QDs to two “AND” and “OR” logic gates, with Hg2+ and Ag+ as inputs. As shown in Fig. 20B, the mixture of the two QDs yielded an “AND” gate. Fluorescence quenching at either 560 nm or 620 nm was defined as a “0” output, while both emissions being quenched yielded a “1” output. Only in the presence of the two inputs, Ag+ and Hg2+, were both emission quenched (“True” output, or “1”), i.e., the “AND” gate. By functionalization of each of the QDs (QD560 and QD620) with both DNAs, an “OR” gate was developed (Fig. 20C). The fluorescence of QDs was quenched by either of the inputs Hg2+ or Ag+, giving rise to the “OR” logic gate.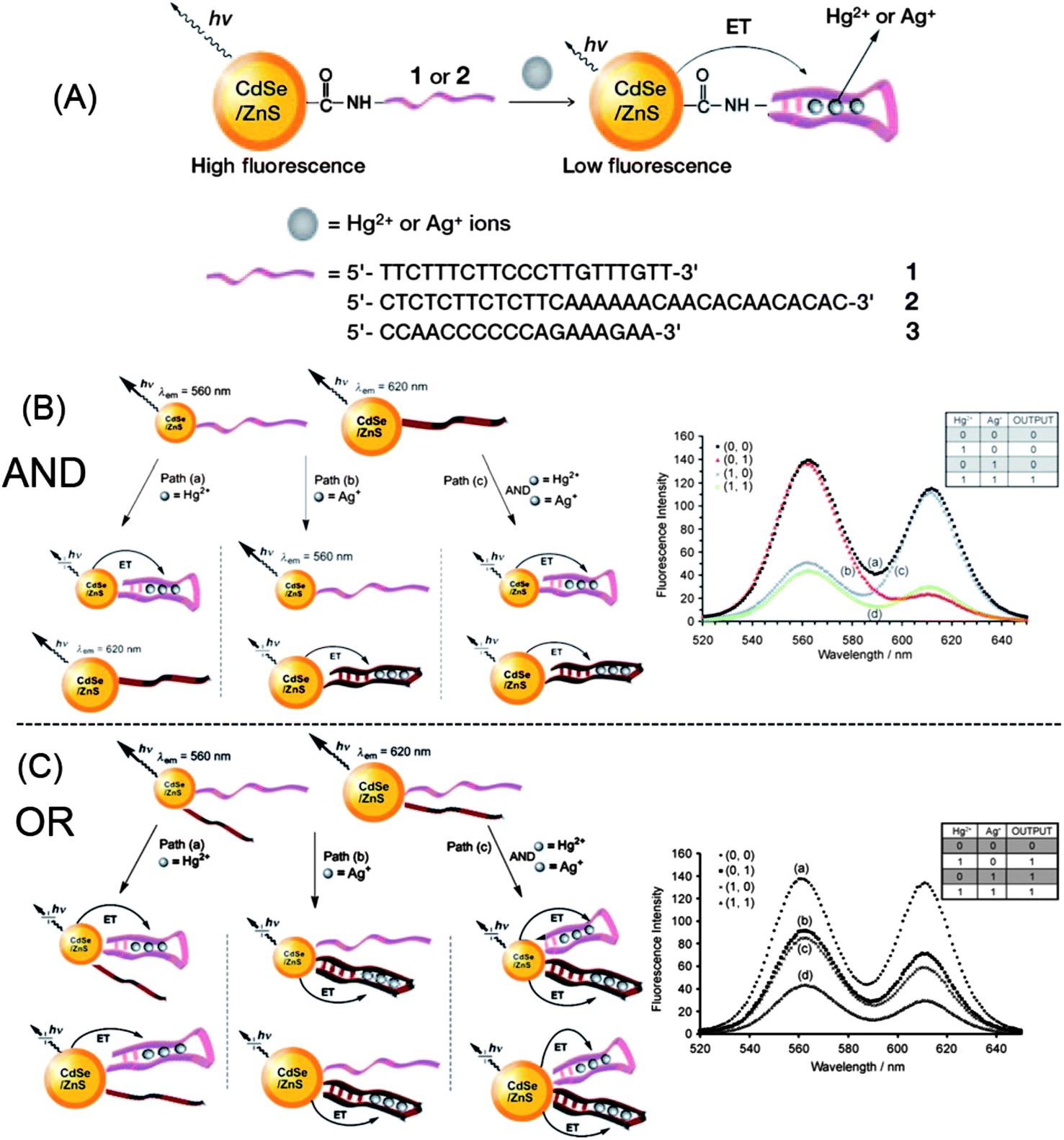 | ||
| Fig. 20 Development of “AND” and “OR” logic gates with DNA-functionalized CdSe@ZnS QDs. Reprinted with permission from ref. 91. Copyright 2009 Wiley-VCH. | ||
An “AND” logic gate was developed with CdSe@ZnS QDs functionalized with mercaptoaniline (H+-sensitive) and crown ether [N-(omethoxyphenyl) aza-15-crown-5, Na+-sensitive] receptors.184 H+ and Na+ were used as inputs and the emission of QDs at 560 nm as output. Because of PET from the receptors to the excited QDs, the fluorescence of the QDs was quenched and couldn't be activated with either H+ or Na+ (“0” state). In the presence of both H+ and Na+, the fluorescence of the QDs was restored (“1” state).
4. Conclusions and outlooks
As expected, research in the area of designing and fabricating QD-based metal ion probes has received great attention and remarkable progress has been made so far, and this further confirms the versatility of QDs in chemo/bio-sensor developments. These QD-based fluorescent metal ion probes offer advantages such as high sensitivity, rapidity, and cost-effectiveness. Due to the unique characteristics of QDs, vast potential exists for the development of metal ion sensors based on direct interaction between metal ions and QDs. Such types of probes are rather simple and sensitive, but the selectivity is limited. Besides, the selectivity of these probes cannot be easily predicted because of the artifacts brought about by the synthesis, which makes the rational design of such probes difficult. Specific-surface functionalized QDs, on the other hand, provide new opportunities for the construction of highly selective metal ion probes. These types of metal ion probes will probably be the next research focus, particularly as intracellular probes for studying the cell biology of metal ions as well as cellular metallomics, while thin films of QDs in conjugation of these functionalization strategies may be extremely appealing in developing simple visual type metal ion probes.Compared with organic dyes in metal ion probe development, QDs possess two major advantages. Firstly, the fruitful surface functionalization schemes permit more versatility in probe design.185 Those new metal ion-specific ligands, such as DNA and peptides, can be easily conjugated with QDs to improve the metal ion selectivity of QDs. Secondly, the high photostability of QDs gives researchers a real opportunity to observe a very high sensitivity and stability, as it facilitates orders of magnitude longer measurement integration times than are permissible with organic aromatic dyes. In fact, the photostability of QDs that allows longer measurement times for intracellular Ca2+ over a conventional Ca2+-specific dye has already been demonstrated.186 Such issues may be appealing for studying intracellular metallomics. However, it should be also noted that since the size of organic dyes is typically smaller than QDs and QD-based ensembles, the cell-permeability of dyes may be better and have less influence on the cell viability.
The signal transduction of QDs may be not limited to photoluminescence,187 and in fact, CL, ECL and PEC from QDs have all been explored as methods for sensing metal ions. The signal transduction modes make QDs even more versatile for sensor development. Currently, the sensing strategies for metal ions based on either CL/ECL or PEC are mainly copied from QD-based fluorescent probes. Whether there are more appropriate strategies exactly matching these signal transduction modes needs to be reconsidered, which may pave the way for more efficient metal ion sensors. Besides, the manipulation of fluorescence of QDs is mostly carried out in homogenous solution, while ECL and PEC are presumably signaled at the electrode surface. Such a difference should not be ignored for future applications.
Up to now, although several cellular metal ion probes have been developed with QDs, most of the QD-based metal ion probes have only been demonstrated as proof-of-concept applications in buffer solutions or artificial matrices. Presumably, broad interests in the application of QDs as opto- or electrochemical labels have been received only in the past decade. Understanding the detailed photophysical properties of various QDs are still on the way. Besides, the exact toxicity as well as the metabolic pathway of QDs is still a topic of hot debate. However, this is expected to change in the next few years. One can see from this review that the number of cellular metal ion probes steadily increased in the past 5 years. Given that the stability and non-specific quenching of QDs are resolved, the future of QD-based metal ion probes may be an important tool for advanced cellular application and metallomics.
Last but not least, ongoing development means that nanotechnology can be revolutionized very quickly nowadays. The label “QDs” now is not restricted to conventional semiconductor nanocrystals. Analytical exploration of these new QDs for sensor developments is on the way. For example, noble metal clusters,188–190 carbon dots,191,192 and lanthanide-containing nanoparticles,193 have already been explored as fluorescent metal ion probes. Rational exploration of the unique structural and optical characteristics of these QDs for metal ion probes should be next to be considered. It should be noted that for majority of these probes, the sensitivity may not comparable to that of inductively coupled plasma mass spectrometry, but they hold great potential for fundamental studies of these nanoparticles and for advanced cellular imaging applications.
Acknowledgements
We appreciate the financial support from National Natural Science Foundation of China (no. 21205084), the Fundamental Research Funds for the Central Universities (no. 2013SCU04A10), and the Startup Grant from Sichuan University (no. YJ2011-015).References
- T. Q. Duong and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef PubMed.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- X. Q. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- Z. C. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996–2006 RSC.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- A. J. Nozik, M. C. Beard, J. M. Luther, M. Law, R. J. Ellingson and J. C. Johnson, Chem. Rev., 2010, 110, 6873–6890 CrossRef CAS PubMed.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- R. E. Galian and M. de la Guardia, Trends Anal. Chem., 2009, 28, 279–291 CrossRef CAS PubMed.
- W. R. Algar, M. Massey and U. J. Krull, Trends Anal. Chem., 2009, 28, 292–306 CrossRef PubMed.
- F. A. Esteve-Turrillas and A. Abad-Fuentes, Biosens. Bioelectron., 2013, 41, 12–29 CrossRef CAS PubMed.
- R. Freeman and I. Willner, Chem. Soc. Rev., 2012, 41, 4067–4085 RSC.
- Y. F. Chen and Z. Rosenzweig, Anal. Chem., 2002, 74, 5132–5138 CrossRef CAS.
- X. J. Wang, M. J. Ruedas-Rama and E. A. H. Hall, Anal. Lett., 2007, 40, 1497–1520 CrossRef CAS.
- L. Spanhel, M. Haase, H. Weller and A. Henglein, J. Am. Chem. Soc., 1987, 109, 5649–5655 CrossRef CAS.
- K. Sooklal, B. S. Cullum, S. M. Angel and C. J. Murphy, J. Phys. Chem., 1996, 100, 4551–4555 CrossRef CAS.
- D. E. Moore and K. Patel, Langmuir, 2001, 17, 2541–2544 CrossRef CAS.
- L. Chen, J. H. Zhang, Y. S. Luo, S. Z. Lu and X. J. Wang, Appl. Phys. Lett., 2004, 84, 112–114 CrossRef CAS.
- L. Y. Wang, Y. G. Zhu, C. Q. Zhu, L. Wang, H. Y. Song, S. Z. Li and X. J. Li, Chin. J. Anal. Chem., 2002, 30, 1352–1354 CAS.
- Y. H. Zhang, H. S. Zhang, M. Ma, X. F. Guo and H. Wang, Appl. Surf. Sci., 2009, 255, 4747–4753 CrossRef CAS PubMed.
- B. Chen, Y. Ying, Z. T. Zhou and P. Zhong, Chem. Lett., 2004, 33, 1608–1609 CrossRef CAS.
- J. Li, D. S. Bao, X. Hong, D. Li, J. H. Li, Y. B. Bai and T. J. Li, Colloids Surf., A, 2005, 257–258, 267–271 CrossRef CAS PubMed.
- A. S. Susha, A. M. Javier, W. J. Parak and A. L. Rogach, Colloids Surf., A, 2006, 281, 40–43 CrossRef CAS PubMed.
- Y. Y. Song, X. B. Cao, Y. Guo, P. Chen, Q. R. Zhao and G. Z. Shen, Chem. Mater., 2009, 21, 68–77 CrossRef CAS.
- S. Y. Liu, Y. Y. Li and X. G. Su, Anal. Methods, 2012, 4, 1365–1370 RSC.
- F. D. Wei, H. L. Yu, M. Hu, G. H. Xu, Z. Cai, J. Yang, L. Li and Q. Hu, Anal. Methods, 2012, 4, 1438–1444 RSC.
- G. L. Wang, Y. M. Dong and Z. J. Li, Nanotechnology, 2011, 22, 085503 CrossRef PubMed.
- M. Koneswaran and R. Narayanaswamy, Sens. Actuators, B, 2009, 139, 91–96 CrossRef CAS PubMed.
- M. Koneswaran and R. Narayanaswamy, Sens. Actuators, B, 2009, 139, 104–109 CrossRef CAS PubMed.
- L. K. Leung, N. J. Komplin, A. B. Ellis and N. Tabatabaie, J. Phys. Chem., 1991, 95, 5918–5924 CrossRef CAS.
- D. H. Son, S. M. Hughes, Y. D. Yin and A. P. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- J. G. Liang, X. P. Ai, Z. K. He and D. W. Pang, Analyst, 2004, 129, 619–622 RSC.
- M. T. Fernández-Argüelles, W. J. Jin, J. M. Costa-Fernández, R. Pereiro and A. Sanz-Medel, Anal. Chim. Acta, 2005, 549, 20–25 CrossRef PubMed.
- Z.-X. Cai, H. Yang, Y. Zhang and X.-P. Yan, Anal. Chim. Acta, 2006, 559, 234–239 CrossRef CAS PubMed.
- J. H. Wang, H. Q. Wang, H. L. Zhang, X. Q. Li, X. F. Hua, Y. C. Cao, Z. L. Huang and Y. D. Zhao, Anal. Bioanal. Chem., 2007, 388, 969–974 CrossRef CAS PubMed.
- Y. H. Zhang, H. S. Zhang, X. F. Guo and H. Wang, Microchem. J., 2008, 89, 142–147 CrossRef CAS PubMed.
- J. L. Chen, A. F. Zheng, Y. C. Gao, C. Y. He, G. H. Wu, Y. C. Chen, X. M. Kai and C. Q. Zhu, Spectrochim. Acta, Part A, 2008, 69, 1044–1052 CrossRef PubMed.
- J. F. Callan and R. C. Mulrooney, Phys. Status Solidi C, 2009, 6, 920–923 CrossRef CAS.
- Y. S. Xia and C. Q. Zhu, Analyst, 2008, 133, 928–932 RSC.
- G. X. Liang, H. Y. Liu, J. R. Zhang and J. J. Zhu, Talanta, 2010, 80, 2172–2176 CrossRef CAS PubMed.
- J. Y. Pei, H. Zhu, X. L. Wang, H. C. Zhang and X. R. Yang, Anal. Chim. Acta, 2012, 757, 63–68 CrossRef CAS PubMed.
- K. X. Zhang, Y. X. Yu and S. Q. Sun, Appl. Surf. Sci., 2013, 276, 333–339 CrossRef CAS PubMed.
- H. G. Wang, L. Sun, Y. P. Li, X. L. Fei, M. D. Sun, C. Q. Zhang, Y. X. Li and Q. B. Yang, Langmuir, 2011, 27, 11609–11615 CrossRef CAS PubMed.
- T.-T. Gan, Y.-J. Zhang, N.-J. Zhao, X. Xiao, G.-F. Yin, S.-H. Yu, H.-B. Wang, J.-B. Duan, C.-Y. Shi and W.-Q. Liu, Spectrochim. Acta, Part A, 2012, 99, 62–68 CrossRef CAS PubMed.
- Y. S. Xia and C. Q. Zhu, Talanta, 2008, 75, 215–221 CAS.
- Y. S. Xia, C. Cao and C. Q. Zhu, Chin. J. Chem., 2007, 25, 1836–1841 CrossRef CAS.
- Z. Y. Tang, P. Podsiadlo, B. S. Shim, J. Lee and N. A. Kotov, Adv. Funct. Mater., 2008, 18, 3801–3808 CrossRef CAS.
- C. Q. Dong, H. F. Qian, N. H. Fang and J. C. Ren, J. Phys. Chem. B, 2006, 110, 11069–11075 CrossRef CAS PubMed.
- Y. H. Chan, J. X. Chen, Q. S. Liu, S. E. Wark, D. H. Son and J. D. Batteas, Anal. Chem., 2010, 82, 3671–3678 CrossRef CAS PubMed.
- E. M. Ali, Y. G. Zheng, H. H. Yu and J. Y. Ying, Anal. Chem., 2007, 79, 9452–9458 CrossRef CAS PubMed.
- X. X. Wang, Y. Lv and X. D. Hou, Talanta, 2011, 84, 382–386 CrossRef CAS PubMed.
- X. L. Zhu, Z. Q. Zhao, X. Q. Chi and J. H. Gao, Analyst, 2013, 138, 3230–3237 RSC.
- J. Ke, X. Y. Li, Y. Shi, Q. D. Zhao and X. C. Jiang, Nanoscale, 2012, 4, 4996–5001 RSC.
- A. V. Isarov and J. Chrysochoos, Langmuir, 1997, 13, 3142–3149 CrossRef CAS.
- Y. Q. Hao, L. Liu, Y. F. Long, J. X. Wang, Y. N. Liu and F. M. Zhou, Biosens. Bioelectron., 2013, 41, 723–729 CrossRef CAS PubMed.
- B. Chen and P. Zhong, Anal. Bioanal. Chem., 2005, 381, 986–992 CrossRef PubMed.
- C. Tiseanu, R. Mehra and R. Kho, J. Photochem. Photobiol., A, 2005, 173, 169–173 CrossRef CAS PubMed.
- J. L. Chen, Y. C. Gao, Z. B. Xu, G. H. Wu, Y. C. Chen and C. Q. Zhu, Anal. Chim. Acta, 2006, 577, 77–84 CrossRef CAS PubMed.
- J. L. Duan, X. C. Jiang, S. Q. Ni, M. Yang and J. H. Zhan, Talanta, 2011, 85, 1738–1743 CrossRef CAS PubMed.
- H. H. Wang, Y. P. Li, X. L. Fei, L. Sun, L. G. Zhang, Z. Z. Zhang, Y. Zhang, Y. X. Li and Q. B. Yang, New J. Chem., 2010, 34, 2996–3003 RSC.
- S. G. Ge, C. C. Zhang, Y. N. Zhu, J. H. Yu and S. S. Zhang, Analyst, 2010, 135, 111–115 RSC.
- T. W. Sung and Y. L. Lo, Sens. Actuators, B, 2012, 165, 119–125 CrossRef CAS PubMed.
- C. X. Sui, Y. F. Liu, P. A. Li, D. Zhang and F. Xia, Anal. Methods, 2013, 5, 1695–1701 RSC.
- J. Duan, X. Jiang, S. Ni, M. Yang and J. Zhan, Talanta, 2011, 85, 1738–1743 CrossRef CAS PubMed.
- P. Wu and X.-P. Yan, Biosens. Bioelectron., 2010, 26, 485–490 CrossRef CAS PubMed.
- S. H. Xu, C. L. Wang, H. S. Zhang, Q. F. Sun, Z. Y. Wang and Y. P. Cui, J. Mater. Chem., 2012, 22, 9216–9221 RSC.
- L. Shang, L. H. Zhang and S. J. Dong, Analyst, 2009, 134, 107–113 RSC.
- Z. B. Shang, Y. Wang and W. J. Jin, Talanta, 2009, 78, 364–369 CrossRef CAS PubMed.
- B. Paramanik, S. Bhattacharyya and A. Patra, Chem.–Eur. J., 2013, 19, 5980–5987 CrossRef CAS PubMed.
- P. Wu, C. Y. Xu and X. D. Hou, Appl. Spectrosc. Rev., 2013, 48, 629–653 CrossRef.
- C. Yuan, K. Zhang, Z. P. Zhang and S. H. Wang, Anal. Chem., 2012, 84, 9792–9801 CrossRef CAS PubMed.
- K. Konishi and T. Hiratani, Angew. Chem., Int. Ed., 2006, 45, 5191–5194 CrossRef CAS PubMed.
- J. L. Chen and C. Q. Zhu, Anal. Chim. Acta, 2005, 546, 147–153 CrossRef CAS PubMed.
- A. Sahu, M. S. Kang, A. Kompch, C. Notthoff, A. W. Wills, D. Deng, M. Winterer, C. D. Frisbie and D. J. Norris, Nano Lett., 2012, 12, 2587–2594 CrossRef CAS PubMed.
- Y. S. Xia, C. Cao and C. Q. Zhu, J. Lumin., 2008, 128, 166–172 CrossRef CAS PubMed.
- W. E. Mahmoud, Sens. Actuators, B, 2012, 164, 76–81 CrossRef CAS PubMed.
- K. A. Gattás-Asfura and R. M. Leblanc, Chem. Commun., 2003, 2684–2685 RSC.
- N. Singh, R. C. Mulrooney, N. Kaur and J. F. Callan, Chem. Commun., 2008, 4900–4902 RSC.
- N. Singh, R. C. Mulrooney, N. Kaur and J. F. Callan, J. Fluoresc., 2009, 19, 777–782 CrossRef CAS PubMed.
- M. J. Ruedas-Rama and E. A. H. Hall, Anal. Chem., 2008, 80, 8260–8268 CrossRef CAS PubMed.
- S. Banerjee, S. Kara and S. Santra, Chem. Commun., 2008, 3037–3039 RSC.
- J. Völker, X. Y. Zhou, X. D. Ma, S. Flessau, H. W. Lin, M. Schmittel and A. Mews, Angew. Chem., Int. Ed., 2010, 49, 6865–6868 CrossRef PubMed.
- C. Y. Chen, C. T. Cheng, C. W. Lai, P. W. Wu, K. C. Wu, P. T. Chou, Y. H. Chou and H. T. Chiu, Chem. Commun., 2006, 263–265 RSC.
- M. F. Frasco, V. Vamvakaki and N. Chaniotakis, J. Nanopart. Res., 2010, 12, 1449–1458 CrossRef CAS.
- H. B. Li, Y. Zhang, X. Q. Wang, D. J. Xiong and Y. Q. Bai, Mater. Lett., 2007, 61, 1474–1477 CrossRef CAS PubMed.
- T. Jin, F. Fujii, E. Yamada, Y. Nodasaka and M. Kinjo, Comb. Chem. High Throughput Screening, 2007, 10, 473–479 CrossRef CAS.
- M. J. Ruedas-Rama and E. A. H. Hall, Analyst, 2009, 134, 159–169 RSC.
- M. Liu, H. M. Zhao, X. Quan, S. O. Chen and H. T. Yu, Chem. Commun., 2010, 46, 1144–1146 RSC.
- R. Freeman, T. Finder and I. Willner, Angew. Chem., Int. Ed., 2009, 48, 7818–7821 CrossRef CAS PubMed.
- J. Ma, Y. Chen, Z. Hou, W. Jiang and L. Wang, Biosens. Bioelectron., 2013, 43, 84–87 CrossRef CAS PubMed.
- C. S. Wu, M. K. K. Oo and X. D. Fan, ACS Nano, 2010, 4, 5897–5904 CrossRef CAS PubMed.
- V. S. Shete and D. E. Benson, Biochemistry, 2009, 48, 462–470 CrossRef CAS PubMed.
- C. X. Guo, J. L. Wang, J. Cheng and Z. F. Dai, Biosens. Bioelectron., 2012, 36, 69–74 CrossRef CAS PubMed.
- Z. P. Liu, W. J. He and Z. J. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC.
- A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385–2393 RSC.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed.
- G. Beaune, S. Tamang, A. Bernardin, P. Bayle-Guillemaud, D. Fenel, G. Schoehn, F. Vinet, P. Reiss and I. Texier, ChemPhysChem, 2011, 12, 2247–2254 CrossRef CAS PubMed.
- Y. F. Long, D. L. Jiang, X. Zhu, J. X. Wang and F. M. Zhou, Anal. Chem., 2009, 81, 2652–2657 CrossRef CAS PubMed.
- T. Li, Y. Y. Zhou, J. Y. Sun, D. B. Tang, S. X. Guo and X. P. Ding, Microchim. Acta, 2011, 175, 113–119 CrossRef CAS.
- Y. L. Li, J. Zhou, C. L. Liu and H. B. Li, J. Mater. Chem., 2012, 22, 2507–2511 RSC.
- B. H. Zhang, L. Qi and F. Y. Wu, Microchim. Acta, 2010, 170, 147–153 CrossRef CAS.
- H. Xu, Z. P. Wang, Y. Li, S. J. Ma, P. Y. Hu and X. H. Zhong, Analyst, 2013, 138, 2181–2191 RSC.
- R. Ganguly, S. H. Wang and D. J. Huang, Nanosci. Nanotechnol. Lett., 2010, 2, 208–212 CrossRef CAS PubMed.
- H. B. Li, Y. Zhang, X. Q. Wang and Z. N. Gao, Microchim. Acta, 2008, 160, 119–123 CrossRef CAS.
- H. B. Li, Y. Zhang and X. Q. Wang, Sens. Actuators, B, 2007, 127, 593–597 CrossRef CAS PubMed.
- S. Mayilo, J. Hilhorst, A. S. Susha, C. Hohl, T. Franzl, T. A. Klar, A. L. Rogach and J. Feldmann, J. Phys. Chem. C, 2008, 112, 14589–14594 CAS.
- F. Zhang, Z. Ali, F. Amin, A. Feltz, M. Oheim and W. J. Parak, ChemPhysChem, 2010, 11, 730–735 CrossRef CAS PubMed.
- R. Velu, N. Won, J. Kwag, S. Jung, J. Hur, S. Kim and N. Park, New J. Chem., 2012, 36, 1725–1728 RSC.
- L. E. Page, X. Zhang, A. M. Jawaid and P. T. Snee, Chem. Commun., 2011, 47, 7773–7775 RSC.
- B. Liu, F. Zeng, G. Wu and S. Wu, Analyst, 2012, 137, 3717–3724 RSC.
- Q. Zhao, X. L. Rong, H. B. Ma and G. H. Tao, J. Hazard. Mater., 2013, 250, 45–52 CrossRef PubMed.
- J. Li, F. Mei, W. Y. Li, X. W. He and Y. K. Zhang, Spectrochim. Acta, Part A, 2008, 70, 811–817 CrossRef PubMed.
- P. Yang, Y. Zhao, Y. Lu, Q. Z. Xu, X. W. Xu, L. Dong and S. H. Yu, ACS Nano, 2011, 5, 2147–2154 CrossRef CAS PubMed.
- X. Wang and X. Q. Guo, Analyst, 2009, 134, 1348–1354 RSC.
- Y. J. Kim, R. C. Johnson and J. T. Hupp, Nano Lett., 2001, 1, 165–167 CrossRef CAS.
- M. Li, S. K. Cushing, Q. Wang, X. Shi, L. A. Hornak, Z. Hong and N. Wu, J. Phys. Chem. Lett., 2011, 2, 2125–2129 CrossRef CAS.
- M. Li, Q. Y. Wang, X. D. Shi, L. A. Hornak and N. Q. Wu, Anal. Chem., 2011, 83, 7061–7065 CrossRef CAS PubMed.
- M. Li, X. J. Zhou, S. W. Guo and N. Q. Wu, Biosens. Bioelectron., 2013, 43, 69–74 CrossRef CAS PubMed.
- P. Wu and X. P. Yan, Chem. Commun., 2010, 46, 7046–7048 RSC.
- H. Borchert, D. V. Talapin, N. Gaponik, C. McGinley, S. Adam, A. Lobo, T. Moller and H. Weller, J. Phys. Chem. B, 2003, 107, 9662–9668 CrossRef CAS.
- R. Gui, X. An, H. Su, W. Shen, Z. Chen and X. Wang, Talanta, 2012, 94, 257–262 CrossRef CAS PubMed.
- H. Xu, R. Miao, Z. Fang and X. H. Zhong, Anal. Chim. Acta, 2011, 687, 82–88 CrossRef CAS PubMed.
- R. J. Gui, X. Q. An and W. X. Huang, Anal. Chim. Acta, 2013, 767, 134–140 CrossRef CAS PubMed.
- H.-B. Ren, B.-Y. Wu, J.-T. Chen and X.-P. Yan, Anal. Chem., 2011, 83, 8239–8244 CrossRef CAS PubMed.
- Y. Shang, L. Qi and F. Y. Wu, Microchim. Acta, 2012, 177, 333–339 CrossRef CAS.
- S. Q. Liu and Z. Y. Tang, J. Mater. Chem., 2010, 20, 24–35 RSC.
- L. Wang, S. Z. Kang, Z. F. Gu and J. Mu, J. Dispersion Sci. Technol., 2005, 26, 71–74 CrossRef CAS PubMed.
- T. Danieli, N. Gaponik, A. Eychmüller and D. Mandler, J. Phys. Chem. C, 2008, 112, 8881–8889 CAS.
- C. Wang, J. Zhao, Y. Wang, N. Lou, Q. Ma and X. G. Su, Sens. Actuators, B, 2009, 139, 476–482 CrossRef CAS PubMed.
- Q. Ma, E. N. Ha, F. P. Yang and X. G. Su, Anal. Chim. Acta, 2011, 701, 60–65 CrossRef CAS PubMed.
- F. P. Yang, Q. A. Ma, W. Yu and X. G. Su, Talanta, 2011, 84, 411–415 CrossRef CAS PubMed.
- X. Q. Liu, Q. Liu, S. H. Cao, W. P. Cai, Y. H. Weng, K. X. Xie and Y. Q. Li, Anal. Methods, 2012, 4, 3956–3960 RSC.
- A. Jaiswal, S. S. Ghsoh and A. Chattopadhyay, Langmuir, 2012, 28, 15687–15696 CrossRef CAS PubMed.
- P. Wu, Y. Li and X.-P. Yan, Anal. Chem., 2009, 81, 6252–6257 CrossRef CAS.
- H. Q. Chen and J. C. Ren, Analyst, 2012, 137, 1899–1903 RSC.
- W. T. Huang, W. Y. Xie, Y. Shi, H. Q. Luo and N. B. Li, J. Mater. Chem., 2012, 22, 1477–1481 RSC.
- J. Li, X. Hong, D. Li, K. Zhao, L. Wang, H. Z. Wang, Z. L. Du, J. H. Li, Y. B. Bai and T. J. Li, Chem. Commun., 2004, 1740–1741 RSC.
- A. F. Zheng and J. L. Chen, J. Inorg. Mater., 2009, 24, 251–254 CrossRef CAS.
- A. F. Zheng, J. L. Chen, H. J. Li, C. Y. He, G. H. Wu, Y. G. Zhang, H. P. Wei and G. L. Wu, Microchim. Acta, 2009, 165, 187–194 CrossRef CAS.
- M. C. Yu, Y. Q. Jin, X. Qi, L. J. Bao, Z. S. Qian and C. Q. Zhu, Chin. J. Chem., 2010, 28, 1165–1170 CrossRef CAS.
- Y. J. Hsu, S. Y. Lu and Y. F. Lin, Chem. Mater., 2008, 20, 2854–2856 CrossRef CAS.
- B. Tang, J. Y. Niu, C. G. Yu, L. H. Zhuo and J. C. Ge, Chem. Commun., 2005, 4184–4186 RSC.
- J. S. Li, T. R. Zhang, H. P. Ge, Y. D. Yin and W. W. Zhong, Angew. Chem., Int. Ed., 2009, 48, 1588–1591 CrossRef CAS PubMed.
- P. Wu and X.-P. Yan, Chem. Soc. Rev., 2013, 42, 5489–5521 RSC.
- L. Tan, Y. X. Li, Y. W. Tang, C. C. Kang, Z. R. Yu and S. Y. Xu, J. Nanosci. Nanotechnol., 2012, 12, 7788–7795 CrossRef CAS PubMed.
- J. Chen, J. F. Sun, L. Guo, J. W. Xie and G. X. Sun, Chin. J. Anal. Chem., 2012, 40, 1680–1685 CrossRef CAS.
- T. Zhao, X. D. Hou, Y.-N. Xie, L. Wu and P. Wu, Analyst, 2013, 138, 6589–6594 RSC.
- W. Y. Xie, W. T. Huang, H. Q. Luo and N. Li, Analyst, 2012, 137, 4651–4653 RSC.
- D. W. Huang, C. G. Niu, X. Y. Wang, X. X. Lv and G. M. Zeng, Anal. Chem., 2013, 85, 1164–1170 CrossRef CAS PubMed.
- D. W. Huang, C. G. Niu, M. Ruan, X. Y. Wang, G. M. Zeng and C. H. Deng, Environ. Sci. Technol., 2013, 47, 4392–4398 CrossRef CAS PubMed.
- H. Hai, F. Yang and J. P. Li, RSC Adv., 2013, 3, 13144–13148 RSC.
- D.-M. Han, Z.-Y. Ma, W.-W. Zhao, J.-J. Xu and H.-Y. Chen, Electrochem. Commun., 2013, 35, 38–41 CrossRef CAS PubMed.
- H. X. Zhang, B. Y. Jiang, Y. Xiang, J. Su, Y. Q. Chai and R. Yuan, Biosens. Bioelectron., 2011, 28, 135–138 CrossRef CAS PubMed.
- S. R. Tang, P. Tong, H. Li, J. Tang and L. Zhang, Biosens. Bioelectron., 2013, 42, 608–611 CrossRef CAS PubMed.
- H. P. Huang, J. J. Li and J. J. Zhu, Anal. Methods, 2011, 3, 33–42 RSC.
- S. Y. Deng and H. X. Ju, Analyst, 2013, 138, 43–61 RSC.
- L. H. Zhang, L. Shang and S. J. Dong, Electrochem. Commun., 2008, 10, 1452–1454 CrossRef CAS PubMed.
- H. Wang, Q. Chen, Z. Tan, X. Yin and L. Wang, Electrochim. Acta, 2012, 72, 28–31 CrossRef CAS PubMed.
- Y. L. Mei, H. S. Wang, Y. F. Li, Z. Y. Pan and W. L. Jia, Electroanalysis, 2010, 22, 155–160 CrossRef CAS.
- L. X. Cheng, X. Liu, J. P. Lei and H. X. Ju, Anal. Chem., 2010, 82, 3359–3364 CrossRef CAS PubMed.
- X. Z. Li, J. Li, J. L. Tang, J. Kang and Y. H. Zhang, J. Lumin., 2008, 128, 1229–1234 CrossRef CAS PubMed.
- S. Kanwal, X. H. Fu and X. G. Su, Microchim. Acta, 2010, 169, 167–172 CrossRef CAS.
- G. L. Wang, J. J. Xu and H. Y. Chen, Sci. China, Ser. B: Chem., 2009, 52, 1789–1800 CrossRef CAS PubMed.
- F. L. Huang, F. Pu, X. Q. Lu, H. F. Zhang, Y. Xia, W. Huang and Z. L. Li, Sens. Actuators, B, 2013, 183, 601–607 CrossRef CAS PubMed.
- G. L. Wang, J. J. Xu and H. Y. Chen, Nanoscale, 2010, 2, 1112–1114 RSC.
- P. Wang, X. Y. Ma, M. Q. Su, Q. Hao, J. P. Lei and H. X. Ju, Chem. Commun., 2012, 48, 10216–10218 RSC.
- Q. M. Shen, X. M. Zhao, S. W. Zhou, W. H. Hou and J. J. Zhu, J. Phys. Chem. C, 2011, 115, 17958–17964 CAS.
- J. J. Yan, K. Wang, Q. Liu, J. Qian, X. Y. Dong, W. Liu and B. J. Qiu, RSC Adv., 2013, 3, 14451–14457 RSC.
- J. M. Dubach, D. I. Harjes and H. A. Clark, J. Am. Chem. Soc., 2007, 129, 8418–8419 CrossRef CAS PubMed.
- J. M. Dubach, D. I. Harjes and H. A. Clark, Nano Lett., 2007, 7, 1827–1831 CrossRef CAS PubMed.
- B. Hu, L. L. Hu, M. L. Chen and J. H. Wang, Biosens. Bioelectron., 2013, 49, 499–505 CrossRef CAS PubMed.
- A. W. Zhu, Q. Qu, X. L. Shao, B. Kong and Y. Tian, Angew. Chem., Int. Ed., 2012, 51, 7185–7189 CrossRef CAS PubMed.
- W. W. Xiong, G. H. Yang, X. C. Wu and J. J. Zhu, J. Mater. Chem. B, 2013, 1, 4160–4165 RSC.
- C. L. Wu, J. S. Zheng, C. B. Huang, J. P. Lai, S. Y. Li, C. Chen and Y. B. Zhao, Angew. Chem., Int. Ed., 2007, 46, 5393–5396 CrossRef CAS PubMed.
- Q. G. Chen, T. Y. Zhou, C. Y. He, Y. Q. Jiang and X. Chen, Anal. Methods, 2011, 3, 1471–1474 RSC.
- X. Y. Sun, B. Liu and Y. B. Xu, Analyst, 2012, 137, 1125–1129 RSC.
- B. M. Cao, C. Yuan, B. H. Liu, C. L. Jiang, G. J. Guan and M. Y. Han, Anal. Chim. Acta, 2013, 786, 146–152 CrossRef CAS PubMed.
- J. L. Yao, K. Zhang, H. J. Zhu, F. Ma, M. T. Sun, H. Yu, J. Sun and S. H. Wang, Anal. Chem., 2013, 85, 6461–6468 CrossRef CAS PubMed.
- M. J. Ruedas-Rama, X. J. Wang and E. A. H. Hall, Chem. Commun., 2007, 1544–1546 RSC.
- L. L. del Mercato, A. Z. Abbasi, M. Ochs and W. J. Parak, ACS Nano, 2011, 5, 9668–9674 CrossRef CAS PubMed.
- L. L. del Mercato, A. Z. Abbasi and W. J. Parak, Small, 2011, 7, 351–363 CrossRef CAS PubMed.
- N. Kaur, N. Singh, B. McCaughan and J. F. Callan, Sens. Actuators, B, 2010, 144, 88–91 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- J. Z. Xia, Y. Yu, Q. M. Liao, Y. J. Cao, B. X. Lin, X. G. Hu and J. Z. Wu, J. Inorg. Biochem., 2013, 118, 39–47 CrossRef CAS PubMed.
- P. Wu, L.-N. Miao, H.-F. Wang, X.-G. Shao and X.-P. Yan, Angew. Chem., Int. Ed., 2011, 50, 8118–8121 CrossRef CAS PubMed.
- J. P. Xie, Y. G. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- W. B. Chen, X. J. Tu and X. Q. Guo, Chem. Commun., 2009, 13, 1736–1738 RSC.
- W. W. Guo, J. P. Yuan and E. K. Wang, Chem. Commun., 2009, 3395–3397 RSC.
- Y. Q. Dong, R. X. Wang, G. L. Li, C. Q. Chen, Y. W. Chi and G. N. Chen, Anal. Chem., 2012, 84, 6220–6224 CrossRef CAS PubMed.
- L. Zhou, Y. H. Lin, Z. Z. Huang, J. S. Ren and X. G. Qu, Chem. Commun., 2012, 48, 1147–1149 RSC.
- F. M. Xue and H. F. Wang, Talanta, 2012, 99, 1057–1061 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |