
Understanding and suppressing side reactions in Li–air batteries
Ying
Liu
a,
Liping
Wang
*b,
Lujie
Cao
a,
Chaoqun
Shang
a,
Zhenyu
Wang
a,
Hongen
Wang
 c,
Liqing
He
a,
Jingyi
Yang
b,
Hua
Cheng
a,
Jingze
Li
b and
Zhouguang
Lu
*a
c,
Liqing
He
a,
Jingyi
Yang
b,
Hua
Cheng
a,
Jingze
Li
b and
Zhouguang
Lu
*a
aDepartment of Materials Science & Engineering, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: luzg@sustc.edu.cn
bState Key Laboratory of Electronic Thin Films and Integrated Devices, University of Electronic Science and Technology of China, Chengdu 610054, China. E-mail: lipingwang@uestc.edu.cn
cState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, 122 Luoshi Road, 430070, Wuhan, Hubei, China
First published on 4th September 2017
Abstract
Li–air batteries have attracted intensive recent research attention owing to their extremely high energy density that is ten times that of the conventional Li-ion batteries; however, achieving this energy capability in practical Li–air batteries has been proven to be difficult. A lot of effort has been devoted to improving their stability, energy efficiency and cycle life with a better understanding of the reaction mechanisms. The electrochemical processes associated with the reversible formation/decomposition of Li2O2 are affected by the surrounding conditions, which often involve several side reactions. In this review, we summarize recent progress in Li–air batteries, especially in the side reactions taking place in the electrochemical process, including carbon corrosion in the cathodes, electrolyte degradation, and the shuttle effect of redox mediators in the electrolytes as well as contaminants from the air (CO2, H2O, and N2). The main strategies for suppressing or making full use of these side reactions for enhancing the performance of Li–air batteries are presented. Meanwhile, we also provide perspectives on the development of Li–air batteries.
 Ying Liu | Dr Ying Liu received her BS degree from Nanyang Normal University (2009), MS degree from Central South University (2012), and PhD degree from the City University of Hong Kong (2016). She is currently a Senior Research Fellow in the Department of Materials Science & Engineering, at the Southern University of Science and Technology, Shenzhen, China. Her current research interests mainly focus on designing highly efficient catalysts for Li–air batteries. |
 Liping Wang | Dr Liping Wang is an Associate Professor in the University of Electronic Science and Technology of China. She obtained her PhD degree from the Institute of Physics, Chinese Academy of Sciences, China and Laboratoire de réactivité et chimie des solides, Université de Picardie Jules Verne, France (2008–2011). She worked as a post-doctoral fellow at the Max Planck Institute of Colloids and Interfaces, Germany (2012) and as a Research Associate in Brookhaven National Lab, United States (2013–2014). Her current research interests are the structure–property correlations of electrode materials as energy storage devices. |
 Lujie Cao | Mr Lujie Cao is currently a joint PhD student at the University of Macau and the Southern University of Science and Technology. He received his Master Degree from Liaocheng University in 2010. He has published more than 10 journal papers. His research mainly focuses on the preparation of Metal Organic Framework materials and nanocatalysts which are applied in the energy storage field. |
 Chaoqun Shang | Dr Chaoqun Shang received his BS degree from the Qingdao University of Science and Technology in (2009), MS degree from the Qingdao University of Science and Technology (2012), and PhD degree from the Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (2015). He is currently a postdoc in the Department of Materials Science & Engineering, at the Southern University of Science and Technology, Shenzhen, China. His current research interests mainly focus on designing high-performance electrode materials for energy storage systems. |
 Zhenyu Wang | Mr Zhenyu Wang obtained his BS degree from the Central South University in 2010 and his MS degree from the Beijing University of Technology in 2015. He is now a research assistant in the Department of Materials Science and Engineering, at Southern University of Science and Technology. His research mainly focuses on Li metal anodes and in situ TEM methods to investigate the reaction mechanism of Li/Na-ion batteries. |
 Hongen Wang | Dr Hongen Wang is currently an Associate Professor at the State Key Lab of Advanced Technology for Materials Synthesis and Processing in Wuhan University of Technology. He received his Bachelor degree from the Taiyuan University of Technology (2003), MPhil degree from Central South University (2007) and PhD degree from the City University of Hong Kong (2012). Then, he has worked in the University of Hong Kong, the National University of Singapore, and the University of Washington (Seattle) as a postdoc Research Associate or Visiting Scholar. His current research interest is mainly focused on nanostructured electrode materials for Li/Na ion batteries, Li–S batteries, and solid electrolytes for all-solid-state batteries. He has (co)authored more than 50 SCI papers in refereed international journals with a total number of citations of over 1400 and a H-index of 21. |
 Liqing He | Dr Liqing He graduated from the Nankai University as a bachelor in 2008. He was granted a master's degree by the Technical Institute of Physics and Chemistry, Chinese Academy of Science in 2011. He received his PhD from Kyushu University in 2015. Then he joined the Southern University of Science and Technology of China as a postdoc researcher from 2016. His major research interests involve metal hydride hydrogen storage materials and electrolyte materials for all-solid-state Li/Na-ion battery applications. |
 Jingyi Yang | Miss Jingyi Yang received her bachelor degree in materials science from the Chengdu University of Technology in 2016. She then joined the State Key Laboratory of Electronic Thin Films and Integrated Devices under the supervision of Associate Prof. Liping Wang. Her research focuses on high capacity electrode materials for lithium batteries. |
 Hua Cheng | Dr Hua Cheng is an Associate Professor in the Department of Materials Science and Engineering, at the Southern University of Science and Technology (SUSTech). She obtained her BS degree from the Central South University in 2001 and her MS degree under the join master program between Sichuan University and the Central South University in 2004, and PhD from the City University of Hong Kong in 2011. From 2011 to 2014, she worked as a postdoc researcher at the City University of Hong Kong. Her research interests mainly focus on energy materials. |
 Jingze Li | Dr Jingze Li is a full-time Professor at the University of Electronic Science and Technology of China (UESTC). He graduated from Jishou University in 1990 and received his Master's degree from Sichuan University in 1997. He was awarded a PhD degree of Science from the Institute of Physics, Chinese Academy of Science in 2000. From 2000 to 2007, he worked at Osaka University, Kyoto University, and Tokyo Institute of Technology (JST/JSPS researcher), respectively. His representative research is the field of energy storage including lithium batteries, potassium ion batteries and solid-state batteries. |
 Zhouguang Lu | Dr Zhouguang Lu is now an Associate Professor in the Department of Materials Science and Engineering, at the Southern University of Science and Technology (SUSTech). He obtained his BS degree from the Central South University in 2001 and his MS degree under the joint master's program between Tsinghua University and Central South University in 2004, and a PhD from the City University of Hong Kong in 2009. He was the recipient of a Fulbright Fellowship from the USA Government in 2008–2009 and the Overseas High-Caliber Personnel (Level B) of Shenzhen Government in 2013. His research mainly covers the design and synthesis of nanostructures and their application in energy storage and conversion with focus on lithium/sodium ion batteries, and lithium–air batteries. He has authored more than 100 peer-reviewed journal papers with total citations of more than 2600 and a H-index of 30. |
1. Introduction
As the energy-storage demands driven by the need for portable electronics and the electrification of transportation have continued to increase, the development of electrochemical energy-storage technologies has been intensified during the past few decades.1–4 Li–air batteries have attracted a great deal of interest due to their high theoretical energy density, which is among the highest for known rechargeable lithium batteries.5–8 Combining Li metal as an anode and abundant oxygen from the environment as a cathode, the aprotic Li–air battery delivers a specific energy of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 238 W h kg−1 based on the mass of the lithium metal. If the mass of the oxygen is factored in, its energy density can reach 3608 W h kg−1 which rivals that of gasoline (1700 W h kg−1).9 Taking into account the packaging, the energy density can reach 800 W h kg−1 for real applications with a mileage of 800 km.
238 W h kg−1 based on the mass of the lithium metal. If the mass of the oxygen is factored in, its energy density can reach 3608 W h kg−1 which rivals that of gasoline (1700 W h kg−1).9 Taking into account the packaging, the energy density can reach 800 W h kg−1 for real applications with a mileage of 800 km.
Despite these attractive promises, many scientific and technological limitations hinder the application of high-performance Li–air batteries. The sluggish reaction kinetics on charge and discharge result in short cycle life (<100 cycles), low rate capability (<1 mA cm−2) and poor energy efficiency (<80%).10 To overcome these problems, advanced approaches including catalysts and redox mediators have been demonstrated. However, during the cycling of Li–air batteries, significant side reactions originating from complex interactions such as carbon corrosion, unstable electrolytes, electrolyte additives, and ambient air are detected. These side reactions result in the accumulation of byproducts in Li–air batteries, which increases the resistance and quickly deteriorates the performance of Li–air batteries.
In this review paper, we summarize recent progress in understanding and suppressing side reactions in aprotic Li–air batteries including carbon corrosion, electrolyte degradation, the shuttle effect of electrolyte additives, and contamination from CO2, H2O, and N2.
2. Reaction mechanisms of Li–air batteries
The reaction mechanisms of Li–air batteries have been investigated in aprotic electrolytes with a Li metal anode and a porous carbon cathode (Fig. 1). It is generally established that the operation of Li–air batteries is dependent on the oxygen reduction reaction (ORR) generating solid Li2O2 during the discharging process and the reversible oxygen evolution reaction (OER) evolving O2 during the charging process. However, the presence of intermediates (O2−, LiO2, etc.) through in situ technologies reveals more complicated reaction mechanisms for both ORR and OER. The discharging and charging processes of Li–air batteries are not a simple one-step reaction for the formation/decomposition of Li2O2, but are multi-step reactions, involving the emergences of metastable intermediates that are reversibly formed/decomposed in the electrochemical reactions.11,12 | ||
| Fig. 1 Schematic illustration of the electrochemical reactions in a rechargeable aprotic Li–air battery. | ||
2.1. ORR mechanism
The ORR that occurs on discharge is found to contain two steps. First, O2 undergoes one electron reduction to O2−, successively combining with Li+ ions in the electrolyte to form LiO2 species.13 Then, the LiO2 intermediates undergo a second electron reduction or go on to disproportionate giving Li2O2. The solubility of LiO2 plays an important role in the models of O2 reduction on discharge. In weak Li+ solvation solutions, the Li+ ions react with O2 to form LiO2* intermediates, which are adsorbed on the electrode surface, suggesting a surface-mediated mechanism (reactions (1)–(3)). In strong Li+ solvation solutions, the produced LiO2 intermediates are dissolved in the electrolyte solution, promoting the solution-mediated mechanism (reactions (4)–(6)).ORR on discharge mechanism I:13
| O2(sol) + e− + Li+ → LiO2* | (1) |
| LiO2* + Li+ + e− → Li2O2* | (2) |
| 2LiO2* → Li2O2* + O2 | (3) |
ORR on discharge mechanism II:13
| O2(sol) + e− + Li+(sol) → LiO2(sol) | (4) |
| LiO2(sol) + Li+(sol) + e− → Li2O2(sol) | (5) |
| 2LiO2(sol) → Li2O2(sol) + O2(sol) | (6) |
2.2. OER mechanism
The OER that is related to Li2O2 oxidation into O2 on charging has been reported to include three types of reaction mechanisms. In the first charging mechanism, the discharge product Li2O2 is first decomposed into Li+ ions and LiO2 species, and then the removal of Li+ ions continues to release O2 (reactions (7) and (8)). The LiO2 species are charging intermediates. In the second charging mechanism, there is two-stage oxidation to evolve O2 that is driven by Li-deficient phase Li2−xO2 solid solution reaction (reactions (9) and (10)).14 During the first stage, the noncrystalline Li2O2 phase is oxidized in the potential range of 2.8–3.4 V, which is accompanied by some Li+ vacancies at the interface, resulting in a Li+-deficient Li2−xO2 phase. During the second stage, the oxidization of crystalline Li2O2 to evolve O2 is driven by a Li2−xO2 solid solution reaction in the potential range of 3.4–3.9 V. In the third charging mechanism, in the absence of LiO2 intermediates during the charging process, the oxidation of Li2O2 occurs at the Li2O2/electrolyte interface (reaction (11)), where O2 and Li+ are generated from the Li2O2 surface.9OER on charging mechanism I:14
| 2Li2O2 → LiO2 + Li+ + e− | (7) |
| LiO2 → Li+ + e− + O2 | (8) |
OER on charging mechanism II:14
| Li2O2 → Li2−xO2 + xLi+ + xe− | (9) |
| Li2−xO2 → Li2−xO2 + (2 − x)Li+ + (2 − x)e− | (10) |
OER on charging mechanism III:9
| 2Li2O2 → LiO2 + Li+ + e− | (11) |
2.3. Properties of discharge product, Li2O2
Understanding the physicochemical properties of Li2O2 is crucial to clarify the Li–air battery performance. The thermodynamic equilibrium voltage for 2Li + O2 → Li2O2 is 2.96 V, which is calculated based on the thermodynamic data of the homogenous bulk Li2O2.15 However, the value in the real case is diverse based on surface structures.16 In the LixOy family, the most thermodynamically stable compounds are Li2O and Li2O2.17 They are semiconductors or insulators with a band gap of 4.9 eV.18–22 With such a high band gap it is hard to carry out the electrochemical decomposition of Li2O2. However, recent studies reported that nanoscale Li2O2 with vacancies and defects on the surface exhibited a decreased band gap of ∼3.0 eV, leading to an increase in the conductivity of Li2O2.22,23 Herein, four reported defects in Li2O2 are shown (Fig. 2).24 The amorphous Li2O2 phase ((1) in Fig. 2) is commonly detected since the ORR occurs at room temperature.25,26 The phase structures of Li2O2 in different forms (crystalline or amorphous) have various transport properties. Based on first principles theory,27 it is reported that the amorphous Li2O2 has an ionic conductivity of 2 × 10−7 S cm−1 and an electronic conductivity of 2 × 10−16 S cm−1. As for the crystalline Li2O2, the electronic conductivity and ionic conductivity is 10−19 S cm−1 and 10−20 S cm−1, respectively. Recently, Zhang et al.28 prepared amorphous Li2O2via a facile chemical reaction and found that, compared to its crystalline counterpart, amorphous Li2O2 was a more desirable discharge phase as it had enhanced charge-transfer properties and increased electronic-oxidation kinetics.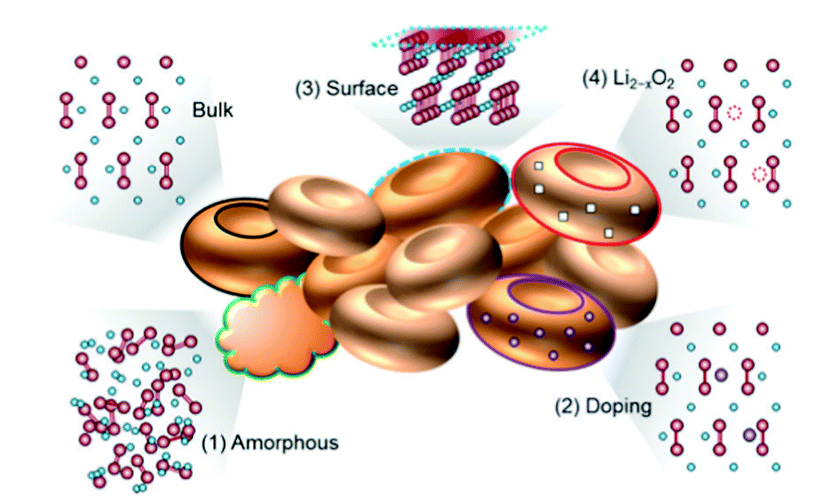 | ||
| Fig. 2 Atomic structures of the reported defects in Li2O2, which can enhance the electronic/ionic conductivities. Oxygen, lithium, and metal dopant elements are designated as red, blue, and purple balls, respectively. Reprinted with permission from ref. 24. | ||
The introduction of heteroatoms into Li2O2 produces defects, as presented in (2) of Fig. 2. When using metal oxides as cathode catalysts, the metal ions could be doped into Li2O2 due to the direct contact between the catalysts and Li2O2. Radin et al.29 showed that the in situ doping of trace Co into Li2O2 could significantly enhance the charge transport through hole polarons and/or Li-ion vacancies. The surface of Li2O2 is found to possess defects due to its metallic nature ((3) in Fig. 2). Lau et al.30 suggested that the surface of Li2O2 has a superoxide structure using density functional theory calculations, which can promote the reversible decomposition and enhance the surface conductivity of Li2O2. Lu et al.31 confirmed the existence of the superoxide structure using a superconducting quantum interference device. The presence of lithium deficiency in Li2O2 (Li2−xO2) is regarded as one of the most common defects in Li2O2 oxidization ((4) in Fig. 2).32 Ganapathy et al.14 provided the first evidence to show the formation of the Li2−xO2 phase in the charging process by using operando XRD. Kang et al.33 reported that the Li2−xO2 phase is beneficial for achieving a low overpotential of ∼0.3 V and the Li1.5O2 phase (x = 0.5) has higher stability when compared to Li2O2.
It is believed that these defects in Li2O2 have a significant influence on the performance of Li–air batteries. The defects in Li2O2 can promote fast charge transport in the decomposition of Li2O2, reducing the charging overpotential. However, the O22− and O2− radical species arising from the defects in Li2O2 and the reactive oxygen species LiO2 induce side reactions with cell components including the carbon electrode, electrolytes, electrolyte additives, and Li metal, degrading the performance of the Li–air batteries.
3. Issues of side reactions in Li–air batteries
The side reactions occurring in Li–air batteries have been widely reported to significantly influence the performance of the batteries. We discuss the side reactions associated with unstable battery components and operating conditions, such as carbon corrosion in the cathode, electrolyte degradation, the shuttle effect of the redox mediator, and contamination from CO2, H2O, and N2.3.1. Carbon corrosion in the cathode
Cathode catalysts are of critical importance in accelerating the kinetics of the ORR and OER occurring at the triple-phase interface and have been extensively discussed in the early developments of Li–air batteries. Owing to the desirable advantages of carbon materials including large surface areas, high electronic conductivity, and low-cost, carbon materials, such as carbon nanofibers, graphenes, and carbon nanotubes, have been reported to be used in the construction of cathodes, which serve as substrates for the deposition of Li2O2 during the discharging process.34,35 However, it is found that the deposited Li2O2 with thin-films or toroid morphologies can lead to a serious volume change in carbon materials, and then cause structure degradation and a poor cycling performance of Li–air batteries. McCloskey et al.36 employed XPS and isotope labeling coupled with differential electrochemical mass spectrometry (DEMS) to study the oxidation of carbon cathodes in a highly oxidizing environment (Fig. 3). On using a 13C-carbon black cathode, roughly 50% of 13CO2 evolved in the high potential peak, indicating the presence of side reactions from the carbon cathode. The charge product, 13CO2, mainly arises from Li213CO3, which is due to the thermo-chemical reactions of Li2O2 with 13C (Li2O2 + C + 1/2O2 → Li2CO3 and 2Li2O2 + C → Li2O + Li2CO3). Itkis et al.37 reported that carbon with activated double bonds or aromatics, such as reduced GO, is quickly attacked by the superoxide radical intermediate produced during discharge to form epoxy groups and carbonates, which degrades the rechargeability of Li–O2 batteries. They suggested that carbon materials with few functional groups and defects showed more excellent stability than carbon materials with a large number of defects, which keeps the carbon will-o’-the-wisp lit for Li–air batteries. Belova et al.38 studied the Li oxygen reduction reaction (Li-ORR) mechanism of different carbon electrodes, such as highly oriented pyrolytic graphite (HOPG), glassy carbon (GC), and basal and edge planes of pyrolytic graphite, and found that a defective GC electrode has a much higher amount of Li2CO3 and carboxylic functionalities generated during the discharging process when compared to the HOPG electrode in a 0.1 M LiClO4/DMSO electrolyte. The increased defect concentrations on the carbon electrode surface hinder superoxide-to-peroxide conversion via disproportionation, assuming that LiO2 is preferably trapped by the defects on the surface of the carbon and then is involved in the formation of Li2CO3.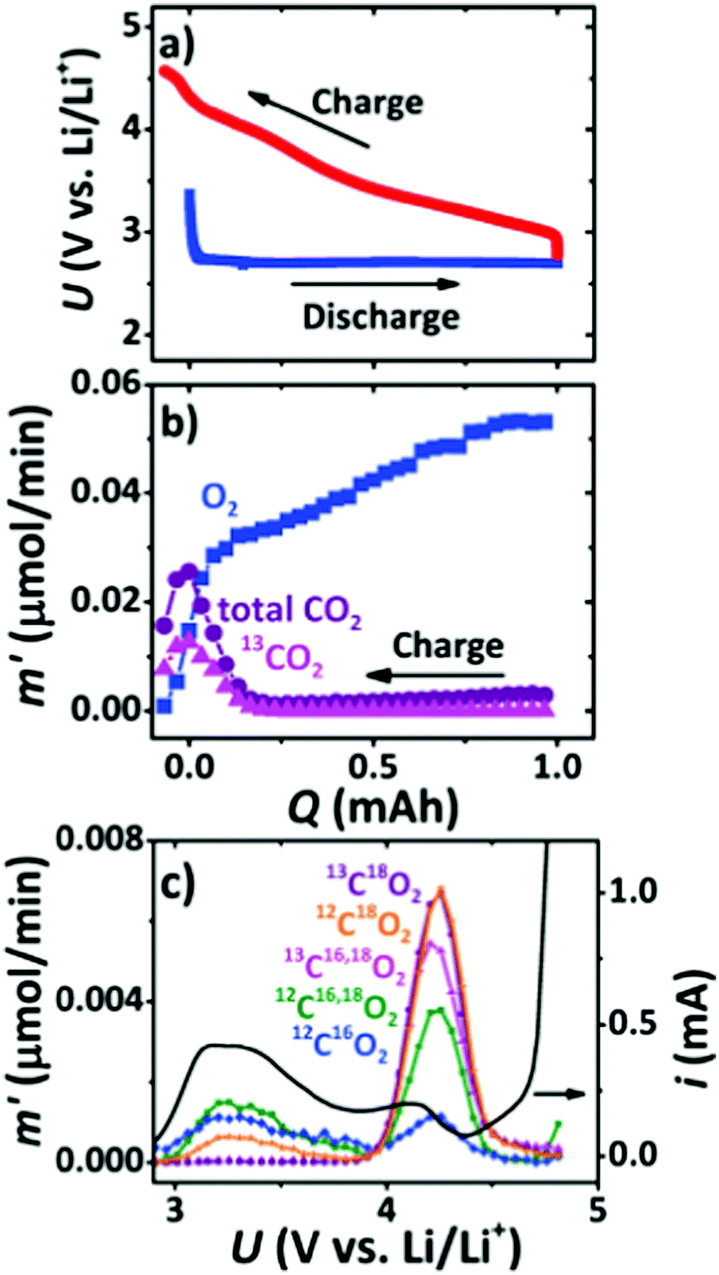 | ||
| Fig. 3 (a) Charge/discharge curves for Li–O2 batteries with a 99% 13C cathode. (b) Gas evolution during charging in a 1 M LiTFSI/DEM electrolyte measured using DEMS. (c) The evolution rate of various CO2 isotopes as a function of charging potential after a 1 mA h discharge at 1.2 bar of 18O2. Reprinted with permission from ref. 36. | ||
To alleviate the side reactions from carbon decomposition, metal carbides have been demonstrated to be alternative cathode catalysts in Li–air batteries. TiC is found to be oxidized to form a thin layer of conductive TiO2−x, ultimately hindering the further oxidation of TiC.39 Unfortunately, not all carbides may provide stable cycling for the cell. For a Mo2C electrode, the formation of soluble LixMoO3 may weaken the catalytic effect on the decomposition of Li2O2.40 Coating metal oxides (MnO2, RuO2, etc.) on carbon surfaces has been regarded as a strategy for the suppression of carbon cathode degradation. However, most of the carbon and carbon supported cathodes employed in Li–O2 batteries have certain reactions with the discharge product Li2O2 due to the negative Gibbs free energy.41 The formed Li2CO3 can be decomposed at a potential above 4.0 V vs. Li+/Li, causing electrolyte decomposition.42,43 Besides, several processes occurring at carbon cathodes, which involve pore clogging induced by the deposition of insulating Li2O2, O2 transport limitations and charge-transfer limitations, are the potential causes of the poor capacity of batteries.11,44
For cathodes using a binder, a series of studies have been reported showing that the binders undergo dehydrofluorination reactions that generate LiF and LiOH, passivating the electrode surface. Papp et al.45 demonstrated the degradation of poly(vinylidene fluoride) (PVDF) when small water impurities were present. This leads to Raman shifts at ∼1133 and 1525 cm−1, which are identical to those of LiO2. Thus, the role of a binder in carbon cathodes should be reconsidered.
3.2. Electrolyte degradation
Exploring high stability electrolytes is an important challenge facing Li–air batteries. Superoxide radicals (O2−) with strong nucleophilicity are generated in the discharging process. It is worth noting that O2− can react with a carbon atom of the solvent molecule to form a [solvent–O2]− complex, leading to electrolyte degradation.46 In-depth studies on the reactivity of O2− towards organic solvents have been carried out to understand the stability of solvents. Carbonate-based electrolytes, such as ethylene carbonate and propylene carbonate, undergo nucleophilic attack to form LiOH, Li2CO3 and other organic carbonates that act as the main discharge products. Alternatively, sulfone-, ether-, and amide-based solvents have been applied, which may effectively promote the formation of discharge products such as Li2O2. However, the noncarbonate-based electrolytes result in unfavorable byproducts through hydrogen atom abstraction.47,48 This is because an acid–base reaction occurs between O2− and a solvent molecule (HA), where the O2− acts as a strong base and the hydrogen atom of the solvent molecule acts as an acid.49,50 The resulting HO2 and A− accelerate the decomposition of the electrolyte, generating numerous byproducts on the cathode surface. The reactivity of the acid–base reaction is associated with the acidity of the solvent and the concentration of O2−. Electrolytes consisting of high donor number (DN) and high acceptor number (AN) solvents possess a high concentration of O2−, inducing the acid-based reaction. Table 1 shows the DN and AN of different electrolytes used in Li–air batteries, which may predict the concentration of O2−.24,51 DME-based electrolytes with low DN could remain stable against the attack of superoxide radicals for over one week. DMSO-based electrolytes with high DN are reported to react with O2− to generate a hydroperoxy radical nucleophile that may attack the S atom of DMSO, consuming an electrolyte and yielding LiOH.52| Solvent | DN | AN |
|---|---|---|
| Ethylene carbonate (EC) | 16.4 | — |
| Propylene carbonate (PC) | 15.1 | 18.3 |
| Dimethoxyethane (DME) | 20.0 | 10.2 |
| Tetraethylene glycol dimethyl ether (TEGDME) | 16.6 | 11.7 |
| Dimethyl acetamide (DMA) | 27.8 | 13.6 |
| N-Methyl-2-pyrrolidenone (NMP) | 27.3 | 13.3 |
| Dimethylformamide (DMF) | 26.6 | 16.0 |
| Dimethyl sulfoxide (DMSO) | 29.8 | 19.3 |
| Acetonitrile (AN) | 14.1 | 18.9 |
| Water | 18.0 | 54.8 |
The properties of the electrolytes alter the electrochemical mechanism of Li–air batteries. Whether the formation of Li2O2 occurs in solution or on the cathode surface relies on the solubility of the intermediate LiO2 in the electrolyte. In a low DN electrolyte, the intermediates exhibit much weaker solvation ability and are adsorbed on the cathode surface, leading to the formation of a Li2O2 film via the surface-mediated mechanism. The film-like Li2O2 shows poor crystallinity when compared to the toroidal-shaped Li2O2, which is ascribed to the shorter lifetime of LiO2 adsorbed on the cathode surface and a fast second reduction to Li2O2.50 Such an insulating film causes passivation of the cathode surface and significantly increases the resistance of Li–air batteries, resulting in low discharge capacity and low rate performance. In a high DN electrolyte, Li+ and Li-containing intermediates (LiO2) are strongly dissolved in the solution and promote a solution route to disproportionate LiO2. The precipitation of Li2O2 driven by a solvation-mediated mechanism induces the formation of toroidal-shaped particles in a high DN electrolyte. The passivation of the electrode surface is less detected in the continuous electrochemical reaction. Thus, it is found that a much higher discharge capacity is yielded in Li–air batteries.
3.3. Shuttle effect of electrolyte additives
Electrolyte additives, like redox mediators (RMs), have a significant influence on the performance of Li–air batteries.9,53–56 Notably, with soluble redox mediators in the electrolyte, the discharge product, Li2O2, can be readily decomposed at the liquid–solid interface by a solvation-mediated mechanism (Fig. 4). In the whole process, the redox mediator functions as an electron carrier moving from the cathode surface to Li2O2. During the charging process, the RM firstly diffuses into the cathode surface and is electrochemically oxidized to RM+, which, then, in turn, oxidizes Li2O2 to evolve O2 and the RM. The resultant alternative pathways for electron transportation can effectively reduce the overpotential on charging, enhancing the capability of Li–air batteries. | ||
| Fig. 4 Proposed OER mechanism with a suitable redox mediator in the Li–air battery. Reprinted with permission from ref. 60. | ||
A variety of redox mediators have been demonstrated, such as Li iodide (LiI),10,57,58 tetramethylpiperidinyloxyl (TEMPO),59–61 5,10-dihydro-5,10-dimethylphenazine (DMPZ)53,62 and iron phthalocyanine (FePc),63,64 in Li–air batteries since tetrathiafulvalene (TTF) was first applied by Chen et al.55 All of them accelerate the reversible formation/decomposition of Li2O2. However, using redox mediators could induce some side reactions, owing to their high solubility in the electrolyte.60 The oxidized RMs generated at the carbon cathode can diffuse out through the separator and can be chemically reduced at the Li anode, and then diffuse back into the carbon cathode.65 Such a shuttle effect makes the RMs less efficient in the oxidation of Li2O2, resulting in an increased polarization of Li–air batteries. Therefore, the shuttle effect of redox mediators has been considered as one of the dominant factors for the increase in overpotential during cycling. Besides, an internal ionic short circuit from the direct contact between the Li anode and oxidized redox mediator is inevitable, causing Li metal corrosion. Lee et al.62 reported a severe morphological change in the Li metal after the 1st cycle with 0.2 M DMPZ, which indicates the presence of side reactions on the surface of the Li metal during cycling. These byproducts verified by XPS are lithium salts (LiOH or Li2CO3). There is no degradation of Li metal without DMPZ, suggesting that the formation of byproducts is related to the side reactions of the Li metal caused by DMPZ. H2O has been regarded as a redox mediator in Li–air batteries to achieve a large discharge capacity and excellent energy efficiency, where the discharge product is LiOH and not Li2O2. Nevertheless, when water is added into the electrolyte, the Li metal anode is attacked by the concomitant H2O, leading to cell failure and safety issues. With only 5000 ppm of H2O in the electrolyte, the corrosion of the Li metal can be detected, and a large amount of LiOH is deposited on the surface of the Li metal (Fig. 5).66 The Li+ ion generation and transfer are blocked. Another critical issue of Li–air batteries is that they are an open system, because of which water is evaporated after many cycles. Deep investigation should be carried out to better understand the mechanism of side reactions of the Li metal with a redox mediator. Minimizing the side reactions between the RM and Li metal remains a challenge.
 | ||
| Fig. 5 (a) Schematic illustrations of Li–O2 cells with a general configuration in a G4–H2O–H2O2 electrolyte. (b) Discharge–charge profiles of Li–O2 cells with 5000 ppm of H2O in the electrolyte (current density: 500 mA gKB−1). (c) The XRD pattern of the Li metal anode after cycling in an electrolyte containing 5000 ppm of H2O. (d) A photo of a Li metal anode after cycling in an electrolyte containing 5000 ppm of H2O. Reprinted with permission from ref. 66. | ||
In addition, the quantity relationship between a redox mediator and Li2O2 should be studied. A relatively low RM concentration results in low charge potential in the initial stage, but the overpotential on charging gradually increased to a value higher than 4.0 V. High RM concentrations can contribute to the capacity, especially when using LiI as a redox mediator, which changes the electrochemical mechanism of batteries. Theoretically, the redox mediator must meet a number of requirements, including fast and reversible electron transfer kinetics, fast reaction with Li2O2 to generate O2, a higher redox potential than 2.96 V vs. Li+/Li, and high chemical and electrochemical stability in the presence of O2, O2−, LiO2, and Li2O2. However, if the redox potential of RM is too high (>4.0 V), the charging of Li–O2 batteries needs a higher potential, where the degradation of the electrolyte occurs, and some unwanted byproducts may form.
3.4. Effect of contamination (CO2, H2O, and N2)
Normally, a Li–air battery is an open system operated under an ambient atmosphere including N2 (78%), O2 (21%) and CO2 (∼0.03%) and other gases. Thus, it can easily undergo side reactions with CO2, H2O, and N2. The plausible reactions are listed as follows:10,67,68For CO2,
| 4Li + O2 + 2CO2 → 2Li2CO3 | (12) |
| Li2O + CO2 → Li2CO3 | (13) |
| 2Li2O2 + 2CO2 → O2 + 2Li2CO3 | (14) |
| 2LiOH + CO2 → Li2CO3 + H2O | (15) |
For H2O,
| 2Li + 2H2O → 2LiOH + H2 | (16) |
| LiOH + H2O → LiOH·H2O | (17) |
| Li2O2 + H2O → LiOH + H2O2 | (18) |
| 4Li + O2 + 2H2O → 4LiOH | (19) |
| 2Li2O2 + 2H2O → 4LiOH + O2 | (20) |
| Li2O2 + H2O → LiOOH + LiOH | (21) |
| LiOH + H2O2 → LiOOH + H2O | (22) |
For N2,
| 6Li + N2 → 2Li3N | (23) |
| Li3N + 3H2O → 3LiOH + NH3 | (24) |
Inspired by the reversible decomposition of Li2CO3, further research has been performed by utilizing pure CO2 gas.78 Archer et al.79,80 initially proposed the concept of a Li–CO2 primary battery and the battery exhibited a significant performance enhancement at high temperature (100 °C). Zhang et al.81 improved the reversibility of Li–CO2 batteries at room temperature by introducing graphene as the cathode material. The electrochemical reaction of reversible Li–CO2 batteries is based on the formation/decomposition of Li2CO3 (4Li + 3CO2 ↔ 2Li2CO3 + C). However, Li2CO3 is a wide bandgap insulator. The continuous accumulation of Li2CO3 increases the battery resistance, inducing huge polarization on charging. Thus, poor cycling performance of Li–CO2 batteries is still a big challenge. By using Ru nanoparticles as the cathode catalyst, Yang et al.82 demonstrated reduced charge overpotential in Li–CO2 batteries. Hou et al.83 employed an efficient Mo2C/CNT catalyst to stabilize the intermediate reduction product of CO2 during the discharging process, which may suppress the formation of Li2CO3. It can decompose the amorphous Li2C2O4–Mo2C discharge product below 3.5 V on charging. Wang et al.84 fabricated a gel polymer electrolyte with a polymer matrix and tetraglyme-based liquid electrolyte to alleviate the contact between the Li metal and CO2. These strategies for improving Li–CO2 batteries may open a new avenue to capture and utilize CO2.
 | ||
| Fig. 6 (a) Discharge–charge profiles of Li–air batteries under three atmospheres (pure CO2, pure O2, and a 10:90 CO2:O2 mixture). (b) FTIR of discharged cathodes extracted from batteries in a pure O2 and a 10:90 CO2:O2 mixture. Reprinted with permission from ref. 76. | ||
 | ||
| Fig. 7 (a) The 1st discharge–charge profiles of a Li–O2 cell and a Li–O2/CO2 cells in a DMSO-based electrolyte. (b) XRD patterns of the Li–O2/CO2 cell after discharge and charge. (c) The proposed mechanism of Li–O2/CO2 discharged in different electrolytes. Reprinted with permission from ref. 74. | ||
 | ||
| Fig. 8 (a) Schematic illustration of Li–O2 under a humid atmosphere. (b) The 1st discharge–charge profiles of Li–O2 batteries in different RHs at 500 mA g−1 with a limited capacity of 1000 mA h g−1. Reprinted with permission from ref. 85. | ||
Water influences the electrochemical mechanism of Li–air batteries. The moisture contaminant can produce LiOH and LiOOH, influencing the growth and morphology of Li2O2 and even dominating the whole reaction to take the place of Li2O2.67,87,88 Aetukuri et al.54 found that the addition of water was beneficial for the formation of Li2O2 toroids via a solution-mediated mechanism since H2O is a strong electron acceptor that may promote the solubility of LiO2 into the solution; the parasitic reactions affected by water are detected by combining DEMS with a Li2O2 titration. Usually, the formation of H2O2 intermediates arising from water-induced superoxide disproportionation is responsible for the decreased amount of Li2O2 on the cathode surface with an increasing H2O content in the electrolyte. The H2O concentrations are found to influence the charging potential and cycling performance of Li–air batteries. Schwenke et al.89 reported that, with 1% H2O in a HClO4-based electrolyte, the initial charge voltage plateau was fairly high, which was attributed to the water-induced changes in the discharge product Li2O2 in terms of size, morphology, and crystal structure. Li et al.90 showed that an excess amount of H2O in the electrolyte produced disk-shaped LiOH as a discharge product. The use of MnO2/Ru/SP catalysts could promote the reaction of Li2O2 with H2O to form LiOH and H2O2. Liu et al.57 showed the reversible formation and decomposition of LiOH by the use of LiI and H2O as additives in Li–O2 batteries. A series of studies suggested that the introduction of H2O into Li–O2 batteries has a marked impact on the properties of the discharge products and its concentration should be carefully controlled.
4. Approaches for achieving high-performance Li–air batteries
In recent years, much effort has been devoted to alleviating these side reactions for achieving high-performance Li–air batteries. These strategies include designing a carbon-free cathode, promoting the Li2CO3 byproduct, protecting the Li metal anode, and using an oxygen-permeable membrane.4.1. Carbon-free cathode
As is generally known, carbon corrosion from the side reactions of carbon materials is found to be one of the primary origins of the limited cycle life of Li–air batteries. A few studies based on carbon-free cathodes, such as Au,92 Ru,93 TiN,94 and TiO2,95,96 have shown that they largely suppress the accumulation of irreversible byproducts.Liu et al.97 fabricated a carbon-free O2 cathode with Ru nanoparticles grown on porous ultralight Ni foam (UNF@Ru) to eliminate the side reactions of carbon and the binder; the resulting Li–air batteries show stable cycling over 100 cycles without voltage decay. It should be noted that no CO2 evolution is detected by DEMS, indicating that the UNF@Ru cathode can effectively catalyze the ORR and OER of Li–air batteries. Kim et al.94 designed mesoporous titanium nitride (m-TiN) with a 2D hexagonal structure and large pores (>30 nm) using a block copolymer with tunable chain lengths as a template; the carbon-free cathode contributes to a robust cycling performance for over 100 cycles under potential cut-off conditions (Fig. 9). The well-aligned pore structure of m-TiN could result in spatial restriction of the growth of Li2O2, preventing the detachment of Li2O2 from the cathode, which was essential for achieving the long-term cyclability of Li–air batteries. They reported that, by employing a polyurethane separator to protect the Li metal from corrosion and by adding LiI solution as a redox mediator, the cycle life of batteries was extended to more than 280 cycles under a limited capacity of 430 mA h g−1. Kang et al.95 prepared oxygen deficient TiO2−x with an ordered macro-mesoporous structure on Ni mesh (HOP-bTiO2) to serve as a carbon-free and binder-free cathode in Li–air batteries. Owing to the high electrical conductivity arising from oxygen vacancies or Ti3+ and the porous structure that provides large space for the accommodation of Li2O2, the resulting Li–air batteries demonstrated an excellent round-trip density over 300 cycles (Fig. 10). The HOP-bTiO2 electrode can promote the solution-mediated growth of discharge products based on the presence of toroidal-shaped Li2O2 during the discharging process. Interestingly, these Li2O2 particles are deposited on the TiO2 with an ordered separation, and the diffusion channels of O2 and the electrolyte could not be blocked during cycling.
 | ||
| Fig. 9 (a) The 1st discharge–charge profiles of the Li–air batteries with an m-TiN electrode at 50 mA g−1, along with schematic illustrations showing the m-TiN morphology change during discharge and charge. (b) The 1st discharge–charge profiles of Li–air batteries with different cathodes (OMC, m-TiN, OMC + LiI, m-TiN + LiI, and m-TiN + LiI + PU). (c) The cycling performances of the same five Li–air batteries. Reprinted with permission from ref. 94. | ||
 | ||
| Fig. 10 (a) Schematic illustration showing the effect of electrode architecture on the electrochemical mechanism. (b) Cycling performance of HOP-bTiO2 without and with LiI at 500 mA g−1. Reprinted with permission from ref. 95. | ||
4.2. Promoting byproduct decomposition
The sluggish reaction kinetics of Li–air batteries require high overpotential on charge, which induces the degradation of battery components, leading to the accumulation of byproducts in the pores of the cathode. These byproducts are difficult to remove and block the pathways of O2 and ions. One of the most effective strategies to tackle cathode passivation/clogging is to decompose these byproducts (Li2CO3 and LiOH) generated during the discharging and charging processes. Song et al.98 presented that the Ir/B4C cathode in an ether-based electrolyte could completely decompose Li2CO3 under a relatively low charge voltage of only 4.37 V due to the synergistic effect of Ir and B4C. The affinity of Ir to adsorb oxygen species may significantly reduce the energy barrier for the electrochemical oxidation of Li2CO3, and B4C possesses more excellent chemical and electrochemical stability when compared with the carbon-based electrode. Peng et al.99 reported a strongly solvating hexamethylphosphoramide (HMPA) electrolyte solvent with high donor number (DN = 38.8), where the Li2O2, Li2CO3 and LiOH are highly dissolved. The solvated Li2O2 could not passivate or clog the O2 cathode during the discharging process and is completely decomposed on charging. The O2 recovery efficiency tested by DEMS for cycles 1, 20, 50, and 100 reaches as high as 100%, indicating that the side reaction may be neglected. As a result, the HMPA-based Li–air batteries show a much lower overpotential gap on charge and discharge, suggesting better O2 reaction kinetics.Liang et al.100 studied the effect of a redox mediator (LiBr) on the charging stability of Li–air batteries and found that the parasitic gas evolution may be reduced when charging with a redox mediator, LiBr. They demonstrated that when charging without LiBr, the electrochemical oxidation of Li2O2 produces reactive intermediates (Li2−xO2) that attack the electrode and electrolyte, leading to their decomposition. When charging with LiBr, the Li2O2 and Li2CO3 are oxidized by Br3−, which avoids the formation of reaction intermediates, suppressing the side reactions during the charging process. At high charging rates, the fast charge-transfer kinetics of the redox mediator is beneficial for the prevention of side reactions, delivering a low charging overpotential for Li–air batteries.
4.3. Protection of the Li metal anode
Lithium metal is the ultimate choice as an anode in Li–air batteries because of its high specific capacity (3860 mA h g−1) and low potential (−3.04 V vs. standard hydrogen electrode).101–104 Unlike the intercalation/de-intercalation of Li+ ions in the traditional Li-ion batteries, Li metal is involved in O2 oxidation via solvated O2, causing severe corrosion of the Li metal anode. Besides, the discharge intermediates, such as O2− and O22−, and H2O from the air may react with Li metal to generate some byproducts. During cycling, dendritic deposition of Li metal is a common phenomenon. The use of a porous glass fiber separator could not block the dissolved O2 and discharge intermediates. Thus, the design of a high chemical stability protective film towards Li metal is attractive.105,106Xu et al.102 proposed the fabrication of a stable tissue-directed/reinforced bifunctional separator/protection film (TBF) on the surface of the lithium metal anode, showing durable and full range protection. After being exposed to air for 20 min, the Li sheet covered with TBF still retains its primary features. The TBF films made of a lithium-exchanged Nafion (LN) ionomer and a tissue can effectively prevent the corrosion and morphology change of the surface of the Li metal. The increase of anodic reversibility (300 cycles) enhances the cycle stability of Li–air batteries (106 cycles). They believed that the protection strategy could be applied to the next-generation energy storage devices including Li–S batteries and Na–O2 batteries.
Lee et al.65 presented the integration strategy of a RM-containing electrolyte with a Al2O3–PVDF–HFP protective layer (CPL) on a Li anode for addressing the self-discharge of the electrochemically oxidized RM; batteries with improved cycling stability and high coulombic efficiency are achieved. The key to the success of this strategy is that the CPL can suppress the redox reaction of RMs at the Li metal anode, ensuring the stability of the Li anode for the RM-containing electrolyte and providing a sustainable redox oxidation for Li–air batteries. When combined with the CPL-coated anode, the growth of the Li dendrite may be suppressed. The concept of Li anode protection to prevent the shuffle effect of RM offers a new possibility for the application of Li–air batteries.
In addition to the commercialized Li conducting ceramic film of LiPON and polymer-based composite layer, a LiF layer on the Li anode fabricated by immersing a Li sheet in N,N-dimethyltrifluoroacetamide (DMTFA) solution is reported to prevent Li anode corrosion during cycling.107 The synergic effect between the LiF protective layer and DMTFA additives contributes to the excellent electrochemical performance of Li–air batteries, including a lower overpotential on charge and discharge and improved cycle life.
Zhang et al.108 combined TTF+ with LiCl in the electrolyte to prepare an organic conductor TTF+Clx− on the surface of the cathode; an outstanding performance and prolonged cycle life were achieved with the aid of the TTF+Clx− layer (Fig. 11). The resultant precipitate is found to limit TTF+ movement around the cathode, minimizing the direct contact between TTF+ and the Li metal anode. The use of TTF as a redox mediator causes electron shuffling in the electrolyte and severe corrosion of the Li metal anode is observed. In TTF + LiCl electrolyte, there is no noticeable degradation of the Li metal anode. The TTF+Clx− layer not only protects the Li anode, but also enhances the electrical connections of insulating Li2O2 from the top of the TTF+Clx− layer to the bottom of the carbon layer, which offers a fast solid–solid pathway to transfer electrons.
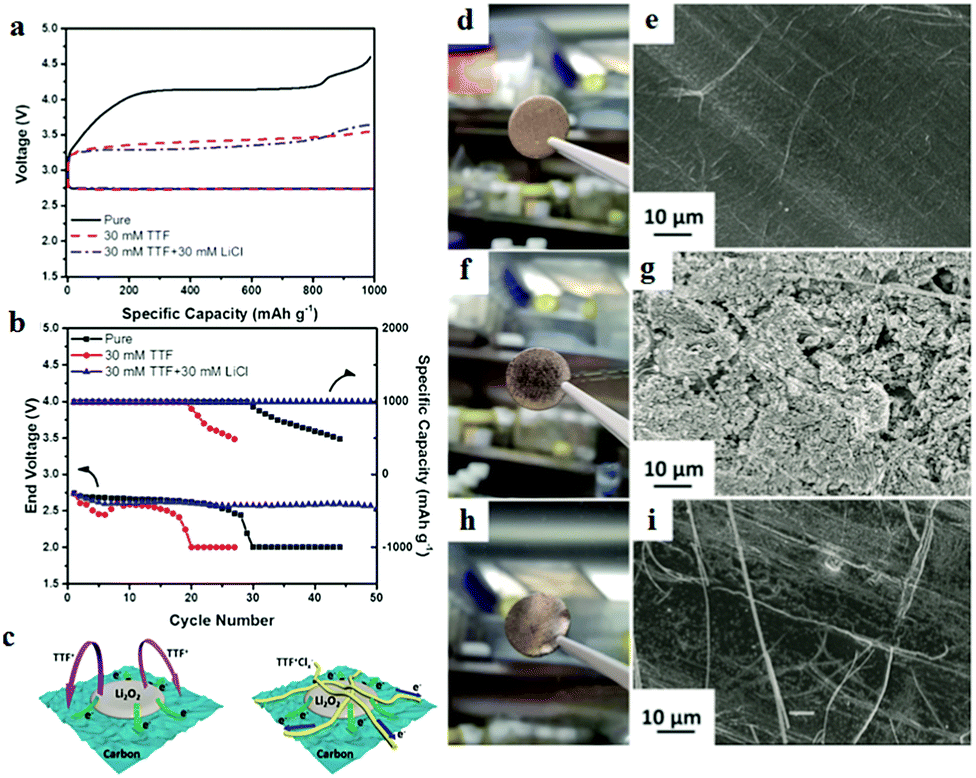 | ||
| Fig. 11 (a) The discharge and charge profiles of Li–O2 batteries at 200 mA g−1. (b) The cycling performances of Li–O2 batteries at 200 mA g−1. (c) Schematic illustration of the mechanism of TTF+Clx− facilitating the decomposition of Li2O2. (d) Digital photo and (e) SEM image of a pristine Li anode. (f) Digital photo and (g) SEM image of the Li anode after 5 cycles in 30 mm TTF electrolyte. (h) Digital photo and (i) SEM image of the Li anode after 5 cycles in 30 mm TTF + 30 mm LiCl electrolyte. Reprinted with permission from ref. 108. | ||
Liu et al.109 presented that the cycling performance of Li–O2 batteries was affected by salt concentration in DME-based electrolytes. They found that highly concentrated electrolytes (3 M) could mitigate the decomposition of electrolyte components and suppress the severe corrosion of the Li metal anode.
4.4. Oxygen-permeable membranes
Li–air batteries are unique in that the cathode material is not stored in the batteries, but is absorbed from air.110 However, it is possible that impurities like H2O, CO2, and N2 are involved in the interface reactions, significantly affecting the mechanism of the ORR and OER in Li–air batteries. One effective method to alleviate these side reactions of Li–air batteries exposed to air is to use a suitable membrane covering the outer surface of the cathode.111,112 An oxygen-permeable membrane is regarded as an interphase barrier to protect the battery components, which can separate O2 from air allow it to diffuse into Li–air batteries.113 To realize the maximum theoretical energy density of Li–air batteries, the membrane should meet a number of requirements, such as high oxygen permeability, high O2 selectivity, and high chemical and mechanical stability. Cao et al.114 designed a novel mixed matrix membrane (MMM) by incorporating polydopamine-coated metal organic framework (MOF) crystals of CAU-1-NH2 into a PMMA (polymethylmethacrylate) matrix (Fig. 12); thus, the Li–air batteries could work well at an RH value of 30%. The pre-coating of polydopamine on the CAU-1-NH2 particles can suppress CO2 molecules due to its abundant –OH groups and the rich-NH2 on the CAU-1-NH2 framework can adsorb CO2 molecules. Membranes for gas selectivity are modified by using inorganic fillers or organic fillers, resulting in enhanced cycling stability of Li–air batteries.71,113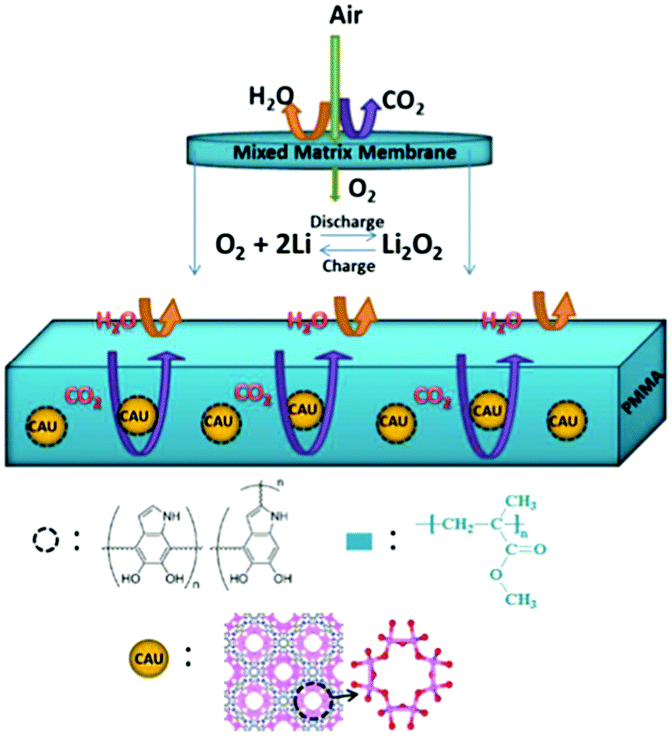 | ||
| Fig. 12 A schematic illustration of the MMM based on CAU-1-NH2@PDA (PDA = polydopamine) and the PMMA polymer for repelling H2O and CO2 molecules. The framework of CAU-1-NH2 viewed along the c-axis (f) and symbols: Al, pink; O, red; C, gray; H, blue. Reprinted with permission from ref. 114. | ||
In addition, an alternative way of employing a solid-state electrolyte to remove the parasitic reactions from CO2 and H2O has attracted much attention. Zhou et al.72 combined a solid Li-ion conductor of Li1.35T1.75Al0.25P2.7Si0.3O12 (LATP) and a gel cathode together in a Li–air cell, which sustained repeated cycling in ambient air for 100 cycles (∼78 days), with a limited discharge capacity of 2000 mA h g−1. This architecture can afford a soft interface with the solid ceramic conductor, providing a valid approach to solve the short-circuit issues from the growth of the Li dendrite and avoid the side reactions between the Li metal and air. Wu et al.115 demonstrated a superhydrophobic H2O-preventing quasi-solid electrolyte (SHQSE) with high thermal stability, which effectively prevents the permeation of H2O and thus the operation of Li–O2 under a humid atmosphere is achieved. However, it should be noted that the introduction of the oxygen-permeable membrane and the solid-state electrolyte increases the resistance of Li–air batteries and leads to energy loss.
5. Summary and perspectives
In this review, we summarize the most recent progress of Li–air batteries with a focus on the side reactions from battery components including the carbon cathode, electrolytes, electrolyte additives and the effect of CO2, H2O, and N2 on the Li metal anode (Fig. 13). It is believed that the use of carbon materials in the cathode results in carbon corrosion due to the side reaction of carbon and the discharge product Li2O2. The resultant Li2CO3 byproduct are very hard to remove, passivating the electrode and leading to high overpotential and early cell death. The properties of the electrolytes significantly affect the stability against oxygen radicals and determine the growth mechanism of Li2O2 through the regulation of either a surface-mediated process or solution-mediated process. An electrolyte with high DN shows a strong solvation effect towards superoxide intermediates, which is beneficial for the solution-mediated reaction and delivers large discharge capacity. However, the stabilization effect may result in an increase in the concentration of O2−, inducing the decomposition of the electrolyte. Soluble redox mediators, such as electrolyte additives, can reduce the overpotential on charge, but the shuttle effect causes Li metal anode degradation. For operation in ambient air, parasitic reactions of the Li metal with CO2, H2O, and N2 are inevitable, which changes the composition and structure of the discharge products. All these could complicate the electrochemical reaction mechanism in Li–air batteries, involving the reversible formation/decomposition of Li2O2, Li2CO3, LiOH, and Li3N. Thus, novel batteries, such Li–CO2 batteries and Li–N batteries have been proposed.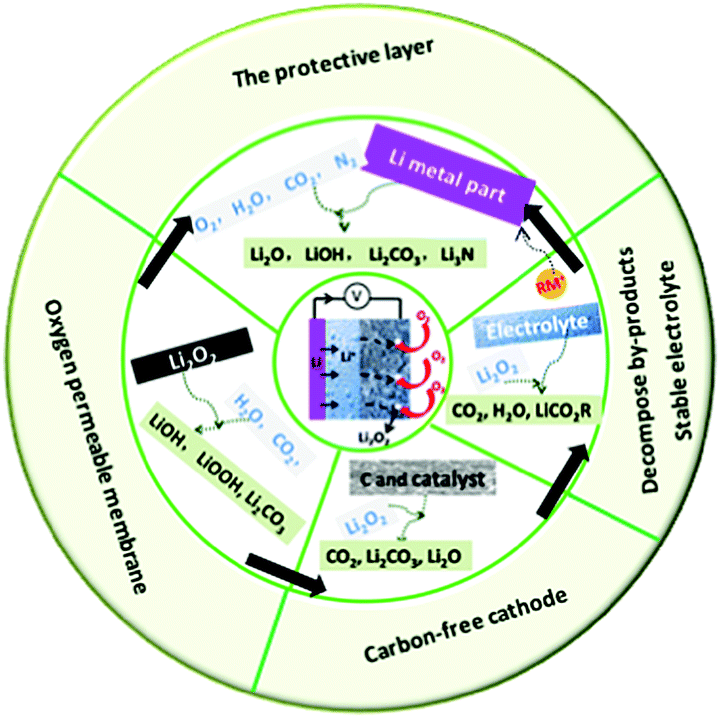 | ||
| Fig. 13 Summary of side reactions that occur in Li–air batteries and strategies for suppressing these side reactions in Li–air batteries. | ||
In addition, the strategies used for suppressing side reactions in Li–air batteries, including designing a carbon-free cathode, promoting byproduct decomposition, protecting the Li anode, and using an oxygen-permeable membrane, have been discussed. Although considerable achievements have been made in Li–air batteries, it is hard to predict how long it will take for the real application of Li–air batteries to be achieved. Many issues remain, particularly in preventing the side reactions due to carbon materials, the electrolyte, electrolyte additives, and CO2, H2O and N2 contaminants. Possible strategies for developing high-performance Li–air batteries include the following:
(1) Constructing new carbon-free cathodes for both ORR and OER catalytic activities. The ideal cathode should be a carbon-free and binder-free material with high conductivity and porous structure that can facilitate the fast transport of Li+ ions and electrons during the formation/decomposition of Li2O2. Carbon-free materials with oxygen deficiency should attract much attention, which may first adsorb the intermediates (O2−, LiO2, etc.) on their surface, significantly reducing the overpotential. The space limitation of Li2O2 growth may suppress the detachment of Li2O2 from the cathode surface by using carbon-free materials with an ordered architecture.
(2) Exploring more stable electrolytes with high donor number to increase the solubility of discharge products and intermediates. The solution growth of Li2O2 dominates the whole electrochemical reaction of Li–air batteries, which is beneficial for obtaining large discharge capacity, high rate capability, and long cycle life. The high DN electrolyte could decompose the byproducts, avoiding their accumulation on the electrode, but the protection of the Li metal anode needs to be carried out. High DN electrolytes are susceptible to nucleophilic attack by superoxide radical species.
(3) Designing an ultrathin composite protective film on the Li metal surface to prevent attack from the electrolyte additives. This protective film not only needs to increase the reversibility of Li metal, but could also provide sustainable redox mediation for Li–air batteries when adding redox mediators to the electrolyte. Understanding the reaction kinetics of the redox mediator for Li2O2 decomposition is very meaningful for realizing the reversibility of Li–air batteries.
(4) Developing new oxygen selective membranes with high O2 solubility and diffusion rates to permit the entry of O2 into Li–air batteries. It should be noted that better oxygen selective membranes can transport O2 at a high current density, which is crucial for the operation of high-rate Li–air batteries.
In achieving high efficiency, the design of battery components that can control the desired reaction pathway appears to be critical. Also, future work on Li–air batteries should focus on deeper understanding of the side reactions upon discharge and charge by using advanced in situ characterization techniques and quantitative analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21671096, 21603094, and 51502032), the Natural Science Foundation of Shenzhen (No. JCYJ20170412153139454, JCYJ20150630145302231, and JCYJ20150331101823677), and the Fundamental Research Funds for the Central Universities, China (No. ZYGX2016J044).Notes and references
- Y. C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 CAS
.
- Z. Jian, P. Liu, F. Li, P. He, X. Guo, M. Chen and H. Zhou, Angew. Chem., Int. Ed., 2014, 53, 442–446 CrossRef CAS PubMed
- Z. L. Wang, D. Xu, J. J. Xu, L. L. Zhang and X. B. Zhang, Adv. Funct. Mater., 2012, 22, 3699–3705 CrossRef CAS
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed
- R. Black, B. Adams and L. Nazar, Adv. Energy Mater., 2012, 2, 801–815 CrossRef CAS
- Z. Guo, D. Zhou, X. Dong, Z. Qiu, Y. Wang and Y. Xia, Adv. Mater., 2013, 25, 5668–5672 CrossRef CAS PubMed
- J. Shui, F. Du, C. Xue, Q. Li and L. Dai, ACS Nano, 2014, 8, 3015–3022 CrossRef CAS PubMed
- L. Luo, B. Liu, S. Song, W. Xu, J. G. Zhang and C. Wang, Nat. Nanotechnol., 2017, 12, 535–539 CrossRef CAS PubMed
- Y. G. Zhu, Q. Liu, Y. Rong, H. Chen, J. Yang, C. Jia, L. J. Yu, A. Karton, Y. Ren, X. X. Xu, S. Adams and Q. Wang, Nat. Commun., 2017, 8, 14308–14315 CrossRef CAS PubMed
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128–16138 CrossRef CAS
- Z. Peng, S. A. Freunberger, L. J. Hardwick, Y. Chen, V. Giordani, F. Bardé, P. Novák, D. Graham, J. M. Tarascon and P. G. Bruce, Angew. Chem., 2011, 123, 6475–6479 CrossRef
- Y. Qiao, S. Wu, J. Yi, Y. Sun, S. Guo, S. Yang, P. He and H. Zhou, Angew. Chem., Int. Ed., 2017, 56, 4960–4964 CrossRef CAS PubMed
- S. Ganapathy, B. D. Adams, G. Stenou, M. S. Anastasaki, K. Goubitz, X. F. Miao, L. F. Nazar and M. Wagemaker, J. Am. Chem. Soc., 2014, 136, 16335–16344 CrossRef CAS PubMed
- I. Kowalczk, J. Read and M. Salomon, Pure Appl. Chem., 2007, 79, 851–860 CrossRef CAS
- J. Hummelshøj, A. Luntz and J. Nørskov, J. Chem. Phys., 2013, 138, 034703 CrossRef PubMed
- K. C. Lau, L. A. Curtiss and J. Greeley, J. Phys. Chem. C, 2011, 115, 23625–23633 CAS
- O. Gerbig, R. Merkle and J. Maier, Adv. Mater., 2013, 25, 3129–3133 CrossRef CAS PubMed
- M. D. Radin, J. F. Rodriguez, F. Tian and D. J. Siegel, J. Am. Chem. Soc., 2012, 134, 1093–1103 CrossRef CAS PubMed
- J. M. Garcia-Lastra, J. D. Bass and K. S. Thygesen, J. Chem. Phys., 2011, 135, 121101 CrossRef CAS PubMed
- J. Chen, J. S. Hummelshoj, K. S. Thygesen, J. S. G. Myrdal, J. K. Norskov and T. Vegge, Catal. Today, 2011, 165, 2–9 CrossRef CAS
- S. P. Ong, Y. Mo and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 081105 CrossRef
- R. R. Mitchell, B. M. Gallant, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 2952–2958 CAS
- H. D. Lim, B. Lee, Y. Bae, H. Park, Y. Ko, H. Kim, J. Kim and K. Kang, Chem. Soc. Rev., 2017, 46, 2873–2888 RSC
- H. G. Jung, H. S. Kim, J. B. Park, I. H. Oh, J. Hassoun, C. S. Yoon, B. Scrosati and Y. K. Sun, Nano Lett., 2012, 12, 4333–4335 CrossRef CAS PubMed
- J. Lu, Y. Lei, K. C. Lau, X. Luo, P. Du, J. Wen, R. S. Assary, U. Das, D. J. Miller and J. W. Elam, Nat. Commun., 2013, 4, 2383–2392 CrossRef PubMed
- F. Tian, M. D. Radin and D. J. Siegel, Chem. Mater., 2014, 26, 2952–2959 CrossRef CAS
- Y. Zhang, Q. Cui, X. Zhang, W. C. McKee, Y. Xu, S. Ling, H. Li, G. Zhong, Y. Yang and Z. Peng, Angew. Chem., Int. Ed., 2016, 55, 10717–10721 CrossRef CAS PubMed
- M. D. Radin, C. W. Monroe and D. J. Siegel, Chem. Mater., 2015, 27, 839–847 CrossRef CAS
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed
- J. Lu, H. J. Jung, K. C. Lau, Z. Zhang, J. A. Schlueter, P. Du, R. S. Assary, J. Greeley, G. A. Ferguson, H. H. Wang, J. Hassoun, H. Iddir, J. Zhou, L. Zuin, Y. Hu, Y. K. Sun, B. Scrosati, L. A. Curtiss and K. Amine, Chemsuschem, 2013, 6, 1196–1202 CrossRef CAS PubMed
- Y. C. Lu and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 4, 93–99 CrossRef PubMed
- S. Kang, Y. Mo, S. P. Ong and G. Ceder, Chem. Mater., 2013, 25, 3328–3336 CrossRef CAS
- B. Sun, S. Chen, H. Liu and G. Wang, Adv. Funct. Mater., 2015, 25, 4436–4444 CrossRef CAS
- L. Wang, W. Jia, X. Liu, J. Li and M. M. Titirici, J. Energy Chem., 2016, 25, 566–570 CrossRef
- B. McCloskey, A. Speidel, R. Scheffler, D. Miller, V. Viswanathan, J. Hummelshøj, J. Nørskov and A. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed
- D. M. Itkis, D. A. Semenenko, E. Y. Kataev, A. I. Belova, V. S. Neudachina, A. P. Sirotina, M. Hävecker, D. Teschner, A. Knop-Gericke and P. Dudin, Nano Lett., 2013, 13, 4697–4701 CrossRef CAS PubMed
- A. I. Belova, D. G. Kwabi, L. V. Yashina, Y. Shao-Horn and D. M. Itkis, J. Phys. Chem. C, 2017, 121, 1569–1577 CAS
- B. D. Adams, R. Black, C. Radtke, Z. Williams, B. L. Mehdi, N. D. Browning and L. F. Nazar, ACS Nano, 2014, 8, 12483–12493 CrossRef CAS PubMed
- D. Kundu, R. Black, B. Adams, K. Harrison, K. Zavadil and L. F. Nazar, J. Phys. Chem. Lett., 2015, 6, 2252–2258 CrossRef CAS PubMed
- F. Li, D. M. Tang, Y. Chen, D. Golberg, H. Kitaura, T. Zhang, A. Yamada and H. Zhou, Nano Lett., 2013, 13, 4702–4707 CrossRef CAS PubMed
- L. Wang, H. Li and X. Huang, Prog. Nat. Sci., 2012, 22, 207–212 CrossRef
- R. Wang, X. Yu, J. Bai, H. Li, X. Huang, L. Chen and X. Yang, J. Power Sources, 2012, 218, 113–118 CrossRef CAS
- J. Wandt, P. Jakes, J. Granwehr, H. A. Gasteiger and R. A. Eichel, Angew. Chem., 2016, 55, 6892–6895 CrossRef CAS PubMed
- J. K. Papp, J. D. Forster, C. M. Burke, H. W. Kim, A. C. Luntz, R. M. Shelby, J. J. Urban and B. D. McCloskey, J. Phys. Chem. Lett., 2017, 8, 1169–1174 CrossRef CAS PubMed
- V. S. Bryantsev, V. Giordani, W. Walker, M. Blanco, S. Zecevic, K. Sasaki, J. Uddin, D. Addison and G. V. Chase, J. Phys. Chem. A, 2011, 115, 12399–12409 CrossRef CAS PubMed
- C. P. Andrieux, P. Hapiot and J. M. Saveant, J. Am. Chem. Soc., 1987, 109, 3768–3775 CrossRef CAS
- C. M. Collins, C. Sotiriou-Leventis, M. T. Canalas and N. Leventis, Electrochim. Acta, 2000, 45, 2049–2059 CrossRef CAS
- F. S. Gittleson, R. E. Jones, D. K. Ward and M. E. Foster, Energy Environ. Sci., 2017, 10, 1167–1179 CAS
- J. Hassoun, F. Croce, M. Armand and B. Scrosati, Angew. Chem., Int. Ed., 2011, 50, 2999–3002 CrossRef CAS PubMed
- L. Wang, Y. Zhang, Z. Liu, L. Guo and Z. Peng, Green Energy Environ., 2017, 1–18 Search PubMed
- M. J. Gibian and T. Ungermann, J. Org. Chem., 1976, 41, 2500–2502 CrossRef CAS
- H. D. Lim, B. Lee, Y. Zheng, J. Hong, J. Kim, H. Gwon, Y. Ko, M. Lee, K. Cho and K. Kang, Nat. Energy, 2016, 1, 16066–16074 CrossRef CAS
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489–494 CrossRef CAS PubMed
- B. D. McCloskey and D. Addison, ACS Catal., 2017, 7, 772–778 CrossRef CAS
- T. Liu, M. Leskes, W. Yu, A. J. Moore, L. Zhou, P. M. Bayley, G. Kim and C. P. Grey, Science, 2015, 350, 530–533 CrossRef CAS PubMed
- J. Han, G. Huang, Y. Ito, X. Guo, T. Fujita, P. Liu, A. Hirata and M. Chen, Adv. Energy Mater., 2017, 7, 1601933 CrossRef
- B. J. Bergner, A. Schürmann, K. Peppler, A. Garsuch and J. R. Janek, J. Am. Chem. Soc., 2014, 136, 15054–15064 CrossRef CAS PubMed
- N. Feng, P. He and H. Zhou, ChemSusChem, 2015, 8, 600–602 CrossRef CAS PubMed
- N. Feng, X. Mu, X. Zhang, P. He and H. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 3733–3739 CAS
- S. H. Lee, J. B. Park, H. S. Lim and Y. K. Sun, Adv. Energy Mater., 2017, 1602417 CrossRef
- I. Gunasekara, M. N. Ates, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. Abraham, J. Electrochem. Soc., 2017, 164, A760–A769 CrossRef CAS
- D. Sun, Y. Shen, W. Zhang, L. Yu, Z. Yi, W. Yin, D. Wang, Y. Huang, J. Wang and D. Wang, J. Am. Chem. Soc., 2014, 136, 8941–8946 CrossRef CAS PubMed
- D. J. Lee, H. Lee, Y. J. Kim, J. K. Park and H. T. Kim, Adv. Mater., 2016, 28, 857–863 CrossRef CAS PubMed
- S. Wu, Y. Qiao, S. Yang, M. Ishida, P. He and H. Zhou, Nat. Commun., 2017, 8, 15607–15615 CrossRef CAS PubMed
- N. Feng, P. He and H. Zhou, Adv. Energy Mater., 2016, 6, 1502303 CrossRef
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2011, 159, R1–R30 CrossRef
- Z. Xie, X. Zhang, Z. Zhang and Z. Zhou, Adv. Mater., 2017, 29, 1605891 CrossRef PubMed
- T. Zhang and H. Zhou, Angew. Chem., 2012, 124, 11224–11229 CrossRef
- D. Geng, N. Ding, T. Hor, S. W. Chien, Z. Liu, D. Wuu, X. Sun and Y. Zong, Adv. Energy Mater., 2016, 6, 1502164 CrossRef
- T. Zhang and H. Zhou, Nat. Commun., 2013, 4, 1817–1824 CrossRef PubMed
- W. Yan, Z. Guo, H. Xu, Y. Lou, J. Chen and Q. Li, Mater. Chem. Front., 2017, 1, 1324–1330 RSC
- H. K. Lim, H. D. Lim, K. Y. Park, D. H. Seo, H. Gwon, J. Hong, W. A. Goddard III, H. Kim and K. Kang, J. Am. Chem. Soc., 2013, 135, 9733–9742 CrossRef CAS PubMed
- N. Mahne, O. Fontaine, M. M. O. Thotiyl, M. Wilkening and S. A. Freunberger, Chem. Sci., 2017 Search PubMed
- S. R. Gowda, A. Brunet, G. M. Wallraff and B. D. McCloskey, J. Phys. Chem. Lett., 2013, 4, 276–279 CrossRef CAS PubMed
- K. Takechi, T. Shiga and T. Asaoka, Chem. Commun., 2011, 47, 3463–3465 RSC
- Q. C. Zhu, S. M. Xu, Z. P. Cai, M. M. Harris, K. X. Wang and J. S. Chen, Energy Storage Mater., 2017, 7, 209–215 CrossRef
- S. Xu, S. K. Das and L. A. Archer, RSC Adv., 2013, 3, 6656–6660 RSC
- S. Xu, S. Lau and L. A. Archer, Inorg. Chem. Front., 2015, 2, 1070–1079 RSC
- Z. Zhang, Q. Zhang, Y. Chen, J. Bao, X. Zhou, Z. Xie, J. Wei and Z. Zhou, Angew. Chem., Int. Ed., 2015, 54, 6550–6553 CrossRef CAS PubMed
- S. Yang, Y. Qiao, P. He, Y. Liu, Z. Cheng, J. J. Zhu and H. Zhou, Energy Environ. Sci., 2017, 10, 972–978 CAS
- Y. Hou, J. Wang, L. Liu, Y. Liu, S. Chou, D. Shi, H. Liu, Y. Wu, W. Zhang and J. Chen, Adv. Funct. Mater., 2017, 27, 1700564 CrossRef
- Y. Wang, C. Li, Z. Guo, B. Yang, Y. Liu and Y. Xia, Angew. Chem., Int. Ed., 2017, 56, 9126–9130 CrossRef PubMed
- S. Wu, J. Tang, F. Li, X. Liu, Y. Yamauchi, M. Ishida and H. Zhou, Adv. Funct. Mater., 2016, 26, 3291–3298 CrossRef CAS
- P. Tan, W. Shyy, T. S. Zhao, R. H. Zhang and X. B. Zhu, Appl. Energy, 2016, 182, 569–575 CrossRef CAS
- D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, N. Ortiz-Vitoriano, P. Hammond, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 2850–2856 CrossRef CAS PubMed
- D. G. Kwabi, T. P. Batcho, S. Feng, L. Giordano, C. V. Thompson and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2016, 18, 24944–24953 RSC
- K. U. Schwenke, M. Metzger, T. Restle, M. Piana and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, A573–A584 CrossRef CAS
- F. Li, S. Wu, T. Zhang, P. He, A. Yamada and H. Zhou, Nat. Commun., 2015, 6, 7843–7849 CrossRef CAS PubMed
- J. L. Ma, D. Bao, M. M. Shi, J. M. Yan and X. B. Zhang, Chem, 2017, 2, 525–532 CAS
- D. Su, S. Dou and G. Wang, NPG Asia Mater., 2015, 7, e155–e167 CrossRef CAS
- K. Liao, T. Zhang, Y. Wang, F. Li, Z. Jian, H. Yu and H. Zhou, ChemSusChem, 2015, 8, 1429–1434 CrossRef CAS PubMed
- B. G. Kim, C. Jo, J. Shin, Y. Mun, J. Lee and J. W. Choi, ACS Nano, 2017, 11, 1736–1746 CrossRef CAS PubMed
- J. Kang, J. Kim, S. Lee, S. Wi, C. Kim, S. Hyun, S. Nam, Y. Park and B. Park, Adv. Energy Mater., 2017, 1700814 CrossRef
- G. Zhao, R. Mo, B. Wang, L. Zhang and K. Sun, Chem. Mater., 2014, 26, 2551–2556 CrossRef CAS
- Z. Liu, N. Feng, Z. Shen, F. Li, P. He, H. Zhang and H. Zhou, ChemSusChem, 2017, 10, 2714–2719 CrossRef CAS PubMed
- S. Song, W. Xu, J. Zheng, L. Luo, M. H. Engelhard, M. E. Bowden, B. Liu, C. M. Wang and J. G. Zhang, Nano Lett., 2017, 17, 1417–1424 CrossRef CAS PubMed
- B. Zhou, L. Guo, Y. Zhang, J. Wang, L. Ma, W. H. Zhang, Z. Fu and Z. Peng, Adv. Mater., 2017, 1701568 CrossRef PubMed
- Z. Liang and Y. C. Lu, J. Am. Chem. Soc., 2016, 138, 7574–7583 CrossRef CAS PubMed
- M. H. Ryou, Y. M. Lee, Y. Lee, M. Winter and P. Bieker, Adv. Funct. Mater., 2015, 25, 834–841 CrossRef CAS
- J. J. Xu, Q. C. Liu, Y. Yu, J. Wang, J. M. Yan and X. B. Zhang, Adv. Mater., 2017, 29, 1606552 CrossRef PubMed
- Q. Yun, Y. B. He, W. Lv, Y. Zhao, B. Li, F. Kang and Q. H. Yang, Adv. Mater., 2016, 28, 6932–6939 CrossRef CAS PubMed
- L. Wang, L. Zhang, Q. Wang, W. Li, B. Wu, W. Jia, Y. Wang, J. Li and H. Li, Energy Storage Mater., 2017 Search PubMed
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed
- L. Wang, Q. Wang, W. Jia, S. Chen, P. Gao and J. Li, J. Power Sources, 2017, 342, 175–182 CrossRef CAS
- E. Yoo and H. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 21307–21313 CAS
- J. Zhang, B. Sun, Y. Zhao, K. Kretschmer and G. Wang, Angew. Chem., Int. Ed., 2017, 56, 8505–8509 CrossRef CAS PubMed
- B. Liu, W. Xu, P. Yan, X. Sun, M. E. Bowden, J. Read, J. Qian, D. Mei, C. M. Wang and J. G. Zhang, Adv. Funct. Mater., 2016, 26, 605–613 CrossRef CAS
- Y. Shao, F. Ding, J. Xiao, J. Zhang, W. Xu, S. Park, J. G. Zhang, Y. Wang and J. Liu, Adv. Funct. Mater., 2013, 23, 987–1004 CrossRef CAS
- J. Zhang, W. Xu and W. Liu, J. Power Sources, 2010, 195, 7438–7444 CrossRef CAS
- J. Zhang, W. Xu, X. Li and W. Liu, J. Electrochem. Soc., 2010, 157, A940–A946 CrossRef CAS
- U. Farooqui, A. Ahmad and N. Hamid, Renewable Sustainable Energy Rev., 2016, 77, 1114–1129 CrossRef
- L. Cao, F. Lv, Y. Liu, W. Wang, Y. Huo, X. Fu, R. Sun and Z. Lu, Chem. Commun., 2015, 51, 4364–4367 RSC
- S. Wu, J. Yi, K. Zhu, S. Bai, Y. Liu, Y. Qiao, M. Ishida and H. Zhou, Adv. Energy Mater., 2017, 7, 1601759 CrossRef
| This journal is © the Partner Organisations 2017 |