
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the subtle tuneability of cellulose hydrogels: implications for binding of biomolecules demonstrated for CBM 1†
M. A.
Johns
 ab,
A.
Bernardes
c,
E. Ribeiro
De Azevêdo
c,
F. E. G.
Guimarães
c,
J. P.
Lowe
d,
E. M.
Gale‡
ad,
I.
Polikarpov
c,
J. L.
Scott
*ad and
R. I.
Sharma
*ab
ab,
A.
Bernardes
c,
E. Ribeiro
De Azevêdo
c,
F. E. G.
Guimarães
c,
J. P.
Lowe
d,
E. M.
Gale‡
ad,
I.
Polikarpov
c,
J. L.
Scott
*ad and
R. I.
Sharma
*ab
aCentre for Sustainable Chemical Technologies, University of Bath, BA2 7AY, UK. E-mail: j.l.scott@bath.ac.uk; r.sharma@bath.ac.uk
bDepartment of Chemical Engineering, University of Bath, BA2 7AY, UK
cSão Carlos Institute of Physics, University of São Paulo, 13566-590, Brazil
dDepartment of Chemistry, University of Bath, BA2 7AY, UK
First published on 3rd May 2017
Abstract
Cellulose-based hydrogel materials prepared by regeneration from cellulose solutions in ionic liquids, or ionic liquid containing solvent mixtures (organic electrolyte solutions), are becoming widely used in a range of applications from tissue scaffolds to membrane ionic diodes. In all such applications knowledge of the nature of the hydrogel with regards to porosity (pore size and tortuosity) and material structure and surface properties (crystallinity and hydrophobicity) is critical. Here we report significant changes in hydrogel properties, based on the choice of cellulose raw material (α- or bacterial cellulose – with differing degree of polymerization) and regeneration solvent (methanol or water). Focus is on bioaffinity applications, but the findings have wide ramifications, including in biomedical applications and cellulose saccharification. Specifically, we report that the choice of cellulose and regeneration solvent influences the surface area accessible to a family 1 carbohydrate-binding module (CBM), CBM affinity for the cellulose material, and rate of migration through the hydrogel. By regenerating bacterial cellulose in water, a maximum accessible surface area of 33 m2 g−1 was achieved. However, the highest CBM migration rate, 1.76 μm2 min−1, was attained by regenerating α-cellulose in methanol, which also resulted in the maximum affinity of the biomolecule for the material. Thus, it is clear that if regenerated cellulose hydrogels are to be used as support materials in bioaffinity (or other) applications, a balance between accessible surface area and affinity, or migration rate, must be achieved.
Introduction
Cellulose is well established as a raw material supply for environmentally friendly and biocompatible products, including property-determining additives for foods, cosmetics, coatings, and synthetic fibres.1 Recent applications include tissue scaffolds,2 biomimetic 4D printing,3 and membrane ionic diodes.4 A naturally occurring biopolymer, it is biocompatible and is viewed as being almost inexhaustible, with an estimated 28.2 billion tonnes produced via biomass each year.1,5 The use of ionic liquids to dissolve cellulose, followed by regeneration with an anti-solvent, has enabled the development of cellulose-based materials of variable forms, including hydrogels.6–8 More recently, Rinaldi demonstrated that the instantaneous dissolution of cellulose at room temperature is made possible by combining the ionic liquid with a dipolar aprotic co-solvent.9 It has been demonstrated that the choice of anti-solvent, used to regenerate cellulose from an ionic liquid solution can influence the material properties of the resultant hydrogel. For example, it has been reported that less crystalline materials result when alcohol anti-solvents are applied compared with water10 and that enhanced enzymatic hydrolysis is observed for cellulose regenerated in alcohol.11Carbohydrate-binding modules (CBMs) are protein domains found in cellulose-degrading enzymes that are responsible for guiding the appended catalytic domain of the enzyme to the cellulose surface.12 They can be independently expressed via recombinant plasmid cloning,13 enabling their use in bioaffinity attachment without modification, or grafting, of the cellulose substrate. Bioaffinity attachment is of particular interest as it ensures controlled orientation of the active molecule, resulting in improved activity, and is generally reversible despite the attached agent being strongly bound.14 This enables the production of novel biocatalysts and biosorbents with enhanced performance;15–18 support matrices for affinity chromatography and biosensors;19–21 and biocompatible scaffolds for human tissue growth.22–24
Tomme et al. previously reported that CBMs belonging to family 1, such as CBM1 of cellobiohydrolase I from Trichoderma harzianum (ThCBMCBHI), used in this work, reversibly bind to cellulose.25 This provides the opportunity to develop separation and purification applications, or regenerable biosensors, both of which require reversible binding of the active biomolecule.14 In order to assess whether cellulose hydrogels regenerated from ionic liquids are suitable supports for these applications, and to develop an understanding of their subsequent degradation rate, three parameters need to be investigated: (i) the accessible surface area of the hydrogel; (ii) the CBM partition constant; and (iii) the CBM diffusion rate.
It is desirable that the accessible surface area of the hydrogel is high, as this minimizes the mass of cellulose required for the application. It might also be expected to enable a higher rate of hydrolysis. In the same vein, a high CBM partition constant (a measurement that describes the affinity of the CBM for the material) is desirable in order to minimise excess CBM remaining in solution once the CBM has adsorbed onto the material. This is more important than the accessible surface area for bioaffinity applications given that cellulose is inexpensive compared to biomolecules, which require laborious expression, isolation and purification. Finally, a high CBM diffusion rate is desirable, as this will minimize the time required to load the hydrogel with the CBM and aid its subsequent removal. This is of importance at an industrial scale, as time is directly linked to cost.
It is known that family 1 CBMs preferentially bind to surfaces that are crystalline and hydrophobic.12,26,27 Therefore, we investigated hydrogels regenerated from an ionic liquid/co-solvent mixture (organic electrolyte solution) comprised of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 1-ethyl-3-methyl imidazolium acetate:dimethyl sulfoxide ([EMIm][OAc]:DMSO) by weight using either methanol, or water, as the anti-solvent. Such materials are designated regenerated in methanol (rM), or regenerated in water (rW), respectively. The effect of the cellulose degree of polymerization (DP) on the resulting pore structure was evaluated using α-cellulose (AC), DP: 500–1300,28 and bacterial cellulose (BC), DP: 2000–6000.29 Herein, we demonstrate that the crystallinity, hydrophobicity and tortuosity of the regenerated hydrogel is dependent on both the type of cellulose and the anti-solvent used to regenerate the hydrogel, which, in turn affects the affinity of the CBM for the material and also modulates the rate of CBM migration within the hydrogel.
:70 1-ethyl-3-methyl imidazolium acetate:dimethyl sulfoxide ([EMIm][OAc]:DMSO) by weight using either methanol, or water, as the anti-solvent. Such materials are designated regenerated in methanol (rM), or regenerated in water (rW), respectively. The effect of the cellulose degree of polymerization (DP) on the resulting pore structure was evaluated using α-cellulose (AC), DP: 500–1300,28 and bacterial cellulose (BC), DP: 2000–6000.29 Herein, we demonstrate that the crystallinity, hydrophobicity and tortuosity of the regenerated hydrogel is dependent on both the type of cellulose and the anti-solvent used to regenerate the hydrogel, which, in turn affects the affinity of the CBM for the material and also modulates the rate of CBM migration within the hydrogel.
Experimental
Materials
Glucose, yeast extract, peptone, anhydrous disodium phosphate, citric acid monohydrate, sodium hypochlorite, 1-ethyl-3-methylimidazolium acetate, dimethyl sulfoxide, methanol, methylene blue, trifluoroacetic acid, acetonitrile, kanamycin sulfate, chloramphenicol, isopropyl-β-D-thiogalactopyranoside, Trizma® base, 2-mercaptoethanol, glycerol, imidazole and α-cellulose were purchased from Sigma Aldrich. Alexa Fluor 488 C5 Maleimide dye was purchased from Invitrogen.Bacterial cellulose production
Cellulose-producing bacteria from an acetobacter containing culture§ were grown at 25 °C in Hestrin & Schramm medium (distilled water supplemented with 2 wt% glucose, 0.5 wt% yeast extract, 0.5 wt% peptone, 0.27 wt% anhydrous disodium phosphate, and 0.15 wt% citric acid monohydrate).30 The resulting bacterial cellulose pellicle was treated with a solution of 10 wt% sodium hypochlorite for 1 h before being washed three times with copious amounts of distilled water and lyophilised using a mini lyotrap (LTE Scientific) freeze dryer.CBM mutagenesis, expression and purification
The cellobiohydrolase I carbohydrate-binding module gene from Trichoderma harzianum (ThCBM1CBHI) was cloned as described by Mello & Polikarpov:31 the CBM gene was cloned into a pSMT3 expression vector, which encodes a 6His-SUMO N-terminal tag.32 A CBM site direct-mutagenesis S19C was performed, introducing a free cysteine residue, to ensure efficient protein labelling with fluorescent probes. DNA single mutation was introduced using QuickChange II Site-Directed Mutagenesis Kit (Stratagene) as per the manufacturer's protocol. For this approach, PCR amplification of an entire plasmid was performed using primers with mutation 5′-TGCGCTTGCGGCTACACTTG-3′ (forward) and 5′-TCTGGTAGGTCCGGTCCAGCC-3′ (backward). The mutagenesis product was transformed into E. coli DH5α, and plasmid mini-preparations were used in the single colonies that grew in presence of kanamycin. The coding regions in the mutant plasmid were verified by DNA sequencing and then chemically transformed into E. coli Rosetta (DE3) strain for protein expression.Transformed E. coli cells were cultured in LB broth containing kanamycin (50 μg mL−1) and chloramphenicol (34 μg mL−1) at 37 °C. After the medium absorbance at 600 nm reached 0.8, protein expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside and cells incubated for 16 h at 18 °C. The cells were harvested by centrifugation and resuspended in buffer A (20 mM Tris pH 7.5, 300 mM NaCl, 5% glycerol, 1 mM phenylmethanesulfonylfluoride and 4.3 M 2-mercaptoethanol). The sample was sonicated to disrupt the cells and centrifuged at 14000 rpm for 40 min. The soluble fraction of ThCBM1CBHI + SUMO was submitted to Ni2+ affinity purification. Buffer A was used to wash the set proteins/resin and protein elution was achieved with an imidazole gradient through a gradual increase of buffer B (20 mM Tris pH 7.5, 150 mM NaCl, 300 mM imidazole, 5% glycerol, and 4.3 M 2-mercaptoethanol). All the eluted samples were analyzed by 15% (wt/wt) SDS-PAGE.
Cellulose hydrogel generation
Cellulose (4 wt%, α-, or bacterial) was dissolved in a solution of 30:70 wt% [EMIm][OAc]:DMSO at 25 °C overnight on a roller table in order to achieve complete dissolution. The solutions were then tape cast using an Elcometer 4340 Automatic Film Applicator with a distance of 500 μm between the blade and glass surface. The resulting film was regenerated by immersion in the chosen anti-solvent for 20 min and washed twice with copious amounts of distilled water to remove excess solvent before being stored in a solution of 20 wt% MeOH in water in order to inhibit bacterial growth.
Powder X-ray diffraction (pXRD)
Pieces of the cellulose hydrogels were frozen using liquid nitrogen and ground to powders using a pestle and mortar. These were analyzed using a flat plate BRUKER D8-Advance (Cu Kα, λ 1.5418 Å radiation) powder X-ray diffractometer over the 2θ range 4–60° with a step size of 0.016° and step time of 0.8 s. The curve-fitting software, Fityk, was used to deconvolute the raw pXRD data: Gaussian curves were fitted to the signals in the amorphous and crystalline regions.33 The crystallinity index was calculated as the ratio between the sum of the crystalline peak areas and the total peak area.NMR cryoporometry
The cellulose samples were soaked in water overnight, excess surface water removed, and the samples placed in individual NMR tubes and sealed using damp absorbent paper to maintain high humidity. The 1H NMR signal was recorded on a 400 MHz Bruker Avance spectrometer equipped with a 5 mm BBO probe, running with the boil-off from liquid nitrogen as cooling gas, and a BVT3200 temperature control unit with precision of ±0.1 K. Actual versus recorded temperatures had previously been calibrated using a methanol sample.34 A simple spin echo pulse sequence was used, with an echo time of 2.2 ms, to ensure minimal suppression of signal from liquid water and complete suppression of signals from both cellulose and frozen water.35,36 Measurements were performed by decreasing the temperature to 218 K in order to completely freeze the sample, followed by increasing the temperature stepwise by 5 K up to 258 K, then in 1 K steps to 267 K, 0.2 K steps to 271 K, and finally to bulk melting temperature using a temperature step of 0.1 K. At each increment signals were recorded after establishment of thermal equilibrium, achieved by a waiting time of 20 min.The melting point depression, ΔT, is related to the pore radius, r, via the bulk properties of the probe liquid, P, as described by the Gibbs–Thomson equation:
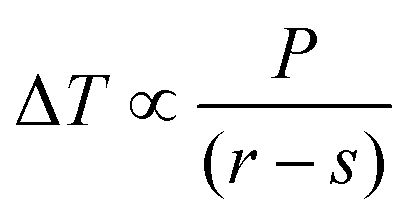
NMR relaxometry
Transverse relaxation times, T2, were measured via Carr–Purcell–Meiboom–Gill (CPMG) echo train acquisition using a Bruker Minispec MQ-20 spectrometer operating with a magnetic field of 0.5 T (1H Larmor frequency of 20 MHz) at 40 °C. 10000 Echoes were acquired with echo (relaxation delay) times (tE) of 1 ms and total recycle delays of 15 s. In fluids in the fast diffusion regime, there is a close relation between the T2 values and the surface-to-volume ratio of pores.35,37 In this particular case, the magnetic field is not highly heterogeneous; there are no paramagnetic, or magnetic, impurities on the pore surface; and relatively short echo times were used. Under these conditions the relation between T2 and the surface to volume ratio is:where ρ2 is the relaxivity constant of the fluid in the pores of the material, and S and V are the pore surface area and volume respectively. As the relaxivity constant of the fluid used to fill the cellulose pores, i.e. water in PBS, is unknown, it is not possible to obtain the surface to volume ratio of the pores, but the trends of T2 may be directly associated with variation in the sizes of regions filled with the solution. Larger T2 values correspond to larger pores.38
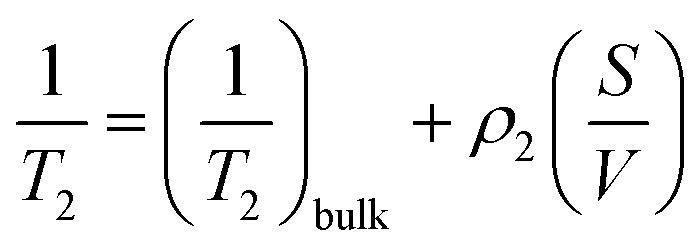
Besides the averaged T2 values, the measurement of the decay of the CPMG echo train, CPMG decay SCPMG(t), T2 distribution profiles, g(T2), associated with distribution of pore sizes and variations in the water mobility within the pores, can be obtained using a non-negative least square procedure also known as a numerical inverse Laplace transform (ILT) to fit the SCPMG(t) curves.39,40 In this case, an ILT method implemented in Matlab was used.
Scanning electron microscopy
Samples of the cellulose hydrogels were dried using a mini lyotrap (LTE Scientific) freeze dryer. The dense skin layer was peeled off with adhesive tape to reveal the core pore structure. Samples were sputter coated with gold for 5 min in an Edwards S150B sputter coater and specimens imaged with a JEOL SEM6480LV operating at 5 kV.Molecular probe adsorption
Methylene blue (MB) depletion isotherms were constructed by incubating five 134.2 mm2 pieces of the cellulose hydrogels with various concentrations (5–1000 μM) of MB in 5 mL of distilled water with 0.01 wt% trifluoroacetic acid. MB controls without cellulose were included and all experiments were carried out in triplicate. Samples were incubated at 4 °C for 24 h, after which time the concentration of the bound MB was calculated from the difference in the original and final MB concentrations in the supernatant: 1 mL of the supernatant was removed and spectrophotometrically analysed in a 1260 Infinity Series (Agilent Technologies) HPLC (flow rate: 1 mL min−1, injection volume: 1 μL, absorbance wavelength: 660 nm, column: Poroshell 120 EC-C18 2.7 μm 4.6 × 50 mm (Agilent Technologies), mobile phase: 50:50 water:acetonitrile with 0.01 wt% trifluoracetic acid). A calibration curve of absorbance against MB concentrations was obtained by using MB solutions of known concentrations (Fig. S1, ESI†).
Carbohydrate-binding module (CBM) depletion isotherms were constructed by incubating a 78.5 mm2 piece of the produced cellulose hydrogels with various concentrations (6.25–200 μM) of ThCBMCBHI in 100 μL of 50 mM PBS pH 7.0. Controls without cellulose were included and all experiments were conducted in triplicate. Samples were incubated at 4 °C for 24 h with agitation (roller table); 2 μL of the supernatant were removed and the concentration of free protein calculated by the absorbance measured at 280 nm using a NanoDrop 2000 Spectrophotometer (Thermos Fisher Scientific). The concentration of the bound ThCBMCBHI was calculated from the difference in initial and final ThCBMCBHI concentration in the supernatant.
Partition constants were obtained from the depletion isotherms (plot of final concentration versus mass of molecular probe adsorbed per gram of cellulose) after fitting of the raw data to a Langmuir-type adsorption model:
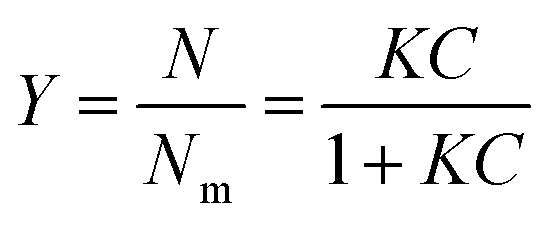
Rearrangement of the equation enabled calculation of Nm and K (Table S4, ESI†) for the molecular probes by plotting C/N versus C (Fig. S2 and S3, ESI†):
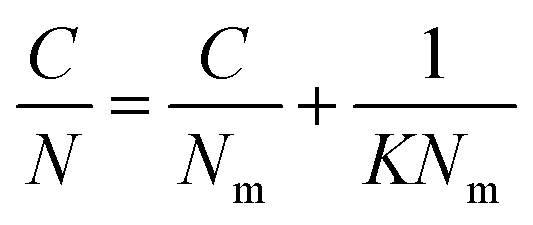
| S = NmaNAv |
Confocal microscopy bleaching
The cysteine residue inserted into ThCBM1CBHI by mutation was specifically labelled with Alexa Fluor 488 C5 Maleimide (Invitrogen) dye, following the manufacturer's instructions. Briefly, 100 μM of ThCBM1CBHI + SUMO in 50 mM phosphate buffer, pH 7.0, was incubated with excess dye (1:10 mole ratio of protein:dye) overnight at 10 °C. Size exclusion chromatography was used to remove excess dye from the labelled protein: the sample was passed through a GE Superdex 75 10/300 GL column, connected to a GE Healthcare ÄKTAprime system, pre-equilibrated with 50 mM phosphate buffer, pH 7.0, and 150 mM NaCl. Fractions collected as the labelled protein eluted were combined and the concentration of labelled protein, and the degree of labelling, were estimated by absorbance measurements at 280 nm and 494 nm (dye absorbance maximum) using a NanoDrop 2000 Spectrophotometer (Thermos Fisher Scientific).
Cellulose hydrogel samples, 134.2 mm2, were incubated with fluorescein-modified ThCBMCBHI at a concentration of 10 nM for 19 h at 16 °C. The samples were then washed twice with PBS before being analysed using a Zeiss LSM 780 confocal microscope. Images were taken using a 405 nm diode laser at 17% power output, an excitation wavelength of 488 nm and absorption wavelength of 525 nm. An area of the sample was selectively bleached by multiple passes of the laser operating at 100% power until the fluorescence intensity of the area had halved (Fig. S5, ESI†). Images of the sample were then taken every minute for 1 h to measure recovery.
Theoretical fluorescence recovery curves based on the equation developed by Axelrod et al.41 were applied to the raw data in order to calculate the maximum fluorescence intensity recovery and the recovery half-life, i.e. the time required for the intensity of the bleached area to recover its intensity to half that of the final intensity value:
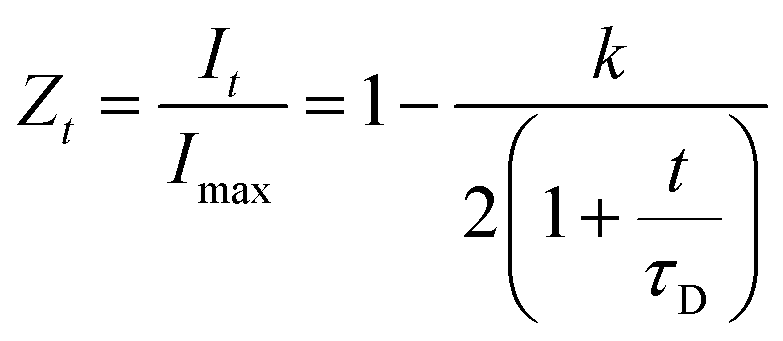
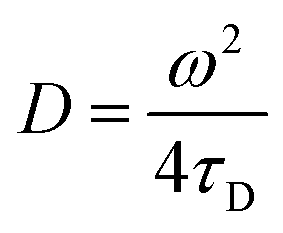
Results and discussion
The dissolution and regeneration of cellulose results in highly porous hydrogels that contain very little cellulosic material (Table 1); densities were ca. 20 kg m−3 compared to the absolute density of pure cellulose of 1592 kg m−3.42 Analysis of pXRD patterns revealed that both cellulose and anti-solvent type affect the overall crystallinity of the regenerated hydrogel (Fig. 1). Both untreated BC and AC are type I cellulose, indicated by pXRD peaks around 2θ = 14.8, 16.8, 22.0 and 22.5°, whilst all the regenerated samples are type II, indicated by peaks around 2θ = 12.1, 13.0, 19.8 and 20.6°.43–45 Dissolution and regeneration of cellulose resulted in hydrogels with reduced crystallinity compared to the starting materials – the crystallinity indices for untreated BC and AC were 0.88 and 0.45 respectively (Table 1). Regeneration in water resulted in a higher degree of crystallinity than regeneration in methanol for both cellulose types with an average increase of 59%.:micro pore ratio
| Sample | Density [kg m−3] | Porosity [%] | Crystallinity index | Modal nanopore diametera [nm] | Median micropore diameterb [nm] | Nano:micro pore ratioc |
|---|---|---|---|---|---|---|
| a Determined from NMR cryoporometry. b Determined from SEM micrographs. c Determined from NMR relaxometry studies. | ||||||
| ACrM | 17 ± 1 | 99 | 0.18 | 50 | 226 | 10:1 |
| ACrW | 22 ± 2 | 99 | 0.27 | 36 | 224 | 12:1 |
| BCrM | 18 ± 1 | 99 | 0.19 | 54 | 190 | 5:1 |
| BCrW | 17 ± 1 | 99 | 0.32 | 44 | 206 | 6:1 |
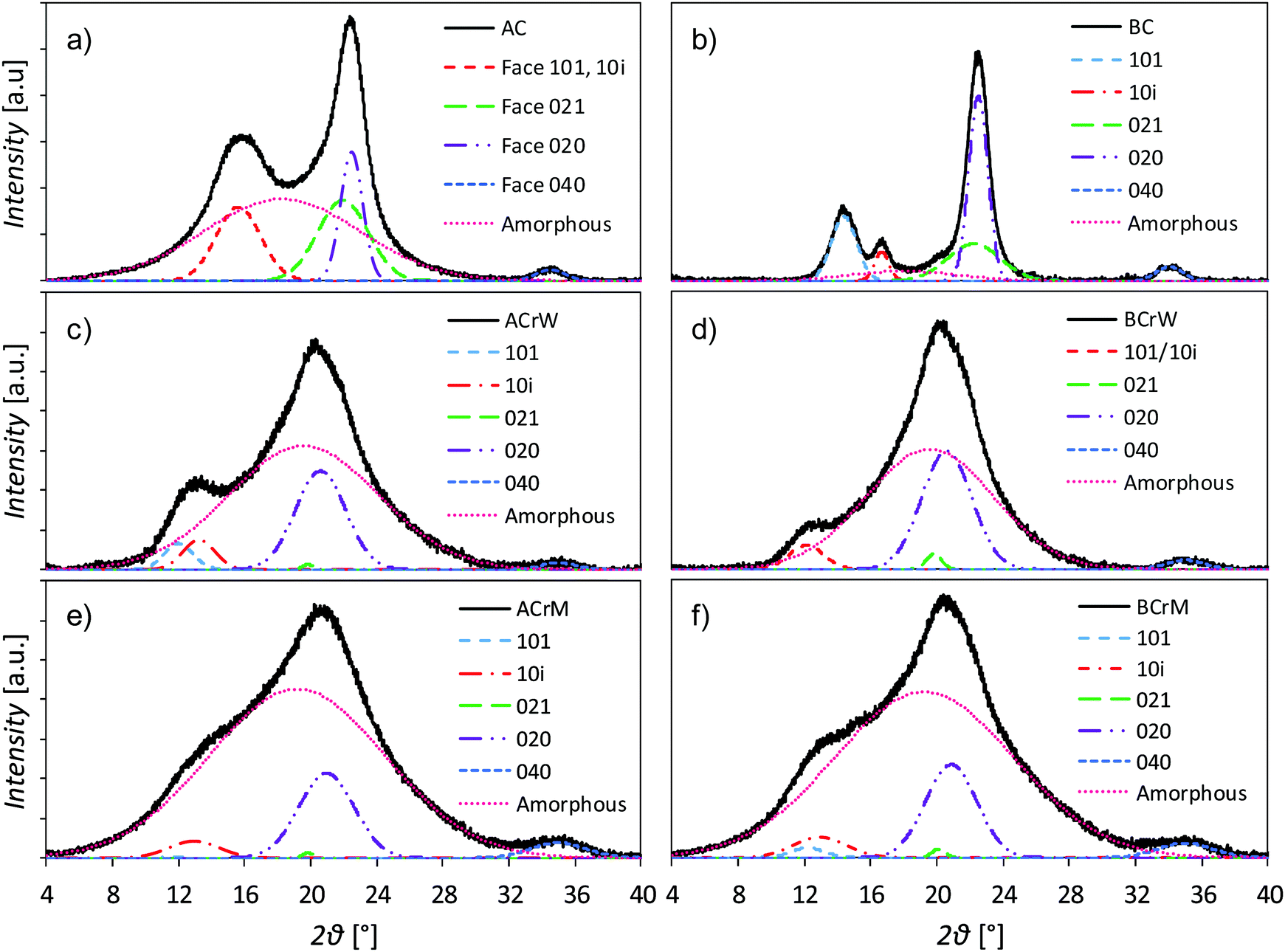 | ||
| Fig. 1 pXRD patterns of samples with crystalline and amorphous peaks fitted: (a) unprocessed α-cellulose (AC); (b) unprocessed bacterial cellulose (BC); (c) α-cellulose regenerated in water (ACrW); (d) bacterial cellulose regenerated in water (BCrW); (e) α-cellulose regenerated in methanol (ACrM); and (f) bacterial cellulose regenerated in methanol (BCrM). Dissolution and regeneration of cellulose results in type II crystalline material with more amorphous material formed after regeneration using methanol compared to water. | ||
To probe the pore structure of the hydrogels without drying, two NMR techniques were employed: relaxometry and cryoporometry. In the former the dependence of proton transverse relaxation time (T2) and surface to volume ratio of pores was exploited. To complement this, and overcome difficulties in defining pore diameters,38 cryoporometry was used to probe specific pore size (<100 nm).46,47
The ILT of the CPMG decays obtained for the hydrogels revealed a T2 distribution profile with three distinct peaks. The peak at the highest T2 value is ascribed to the bulk water in the PBS, which is justified by the comparison with the T2 distribution profile obtained for the pure solution (Fig. 2C, inset).
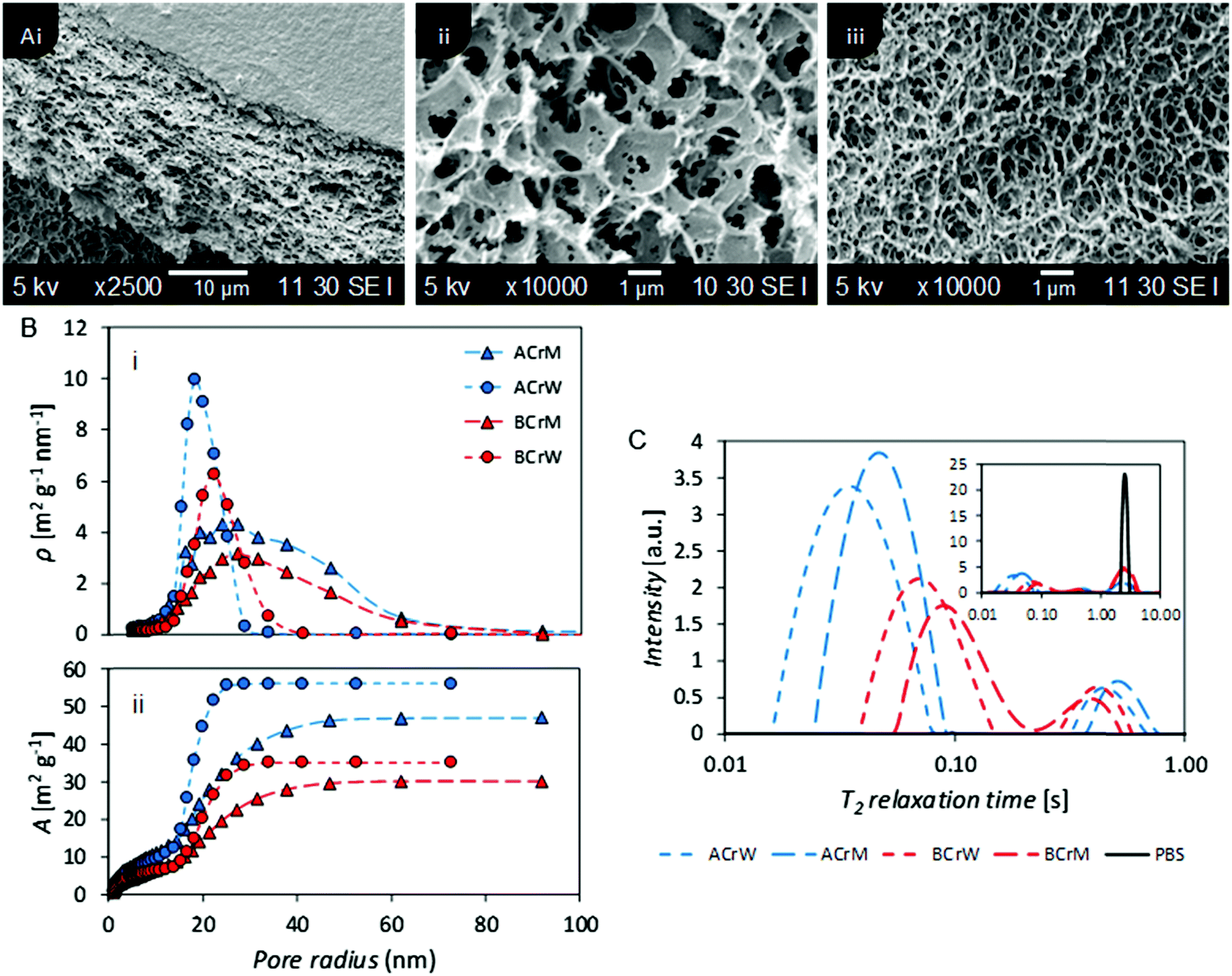 | ||
| Fig. 2 (A) (i) SEM micrograph of a lyophilised cellulose hydrogel showing the porous structure below a dense skin layer – all hydrogels exhibited this bulk structure, (ii) and (iii) SEM micrographs of ACrM and BCrW samples showing differences in micropore size. (B) Nanometer pore size distribution of hydrogels based on NMR cryoporometry: (i) specific pore surface area density (ρ), and (ii) specific cumulative pore surface area. Both choice of cellulose and anti-solvent have an effect on the overall pore structure of the hydrogels in agreement with relaxometry studies. (C) NMR T2 relaxation distribution curves for regenerated cellulose hydrogels providing relative surface areas for nano- and micropores. Both choice of cellulose and anti-solvent have an effect on the overall pore structure of the hydrogels. Bulk PBS measurement included in inset. | ||
The absence of a precise value for the relaxivity constant of water (PBS) inside cellulose pores frustrates association of T2 distributions with specific pore diameters. However, based on previously published data48 and on the averaged T2 values reported for pure water filling pores of known dimensions in cellulose,49–52 the relaxivity of pure water inside cellulose pores can be estimated to be in the range 10−6–10−7 nm s−1. Thus, using the relation between pore dimension T2 and relaxivity constant previously reported,48 the T2 range from 10–100 ms was ascribed to pores with dimensions in the range 10–100 nm and the T2 range from 100 ms to 1 s to pores with dimensions in the range 100 nm to a few μm. Thus, in Fig. 2C, the peaks at the shorter T2 values reflect nanopores, while the peaks with intermediate T2 reflect micropores. The peak corresponding to nanopores for AC reflected a smaller median pore diameter than that for BC, whilst hydrogels regenerated in methanol exhibited larger pores than those regenerated in water. NMR cryoporometry confirmed these observations, yielding a modal pore diameter of 36 nm for AC regenerated in water and 54 nm for BC regenerated in methanol (Fig. 2B and Table 1). It is of interest to note that Östlund et al. reported a pore radius distribution between 2–15 nm, based on NMR cryoporometry for cellulose samples regenerated from [EMIm][OAc] only,10 whilst the distribution in these hydrogels is between 10–70 nm, suggesting that the ratio of ionic liquid to co-solvent in the organic electrolyte solution used could influence the pore size of the resultant regenerated hydrogels, providing further opportunities to tune porosity.
NMR relaxometry studies revealed that micropores (above the range that can be probed with NMR cryoporometry) in AC exhibited a slightly larger median diameter than those in BC, as confirmed by analysis of SEM micrographs (Fig. 2A and Table 1). Significantly more nanopores were present in AC, as reflected in the ratio between the integrated nano- and micropore peak areas in the relaxometry data: that of AC was double that of BC (Table 1). In both cases, hydrogels regenerated in water show a 20% increase in nano- to micropore ratio versus those regenerated in methanol. To validate these results, and to discern whether such differences were important at a molecular scale, passive MB adsorption (used to characterize cotton fibers)53 was conducted. This enabled calculation of the cellulose surface area in the never-dried hydrogels, whilst providing a molecular probe small enough to access pores inaccessible to the larger CBM. (MB has an occupied surface area of 197.2 Å2 whilst ThCBMCBHI has a maximum occupied area of approximately 985 Å2, assuming a globular structure.54,55)
MB adsorption isotherms (Fig. 3a) revealed that BCrM had a lower surface area than ACrW (Table 2), in accordance with the observation that ACrW has a higher population of nanopores with smaller diameters (Table 1). The choice of cellulose influences the surface area by a factor of 1.6 (totalAC/totalBC), whilst the choice of anti-solvent affects the surface area by a factor of 1.2 (totalrW/totalrM).
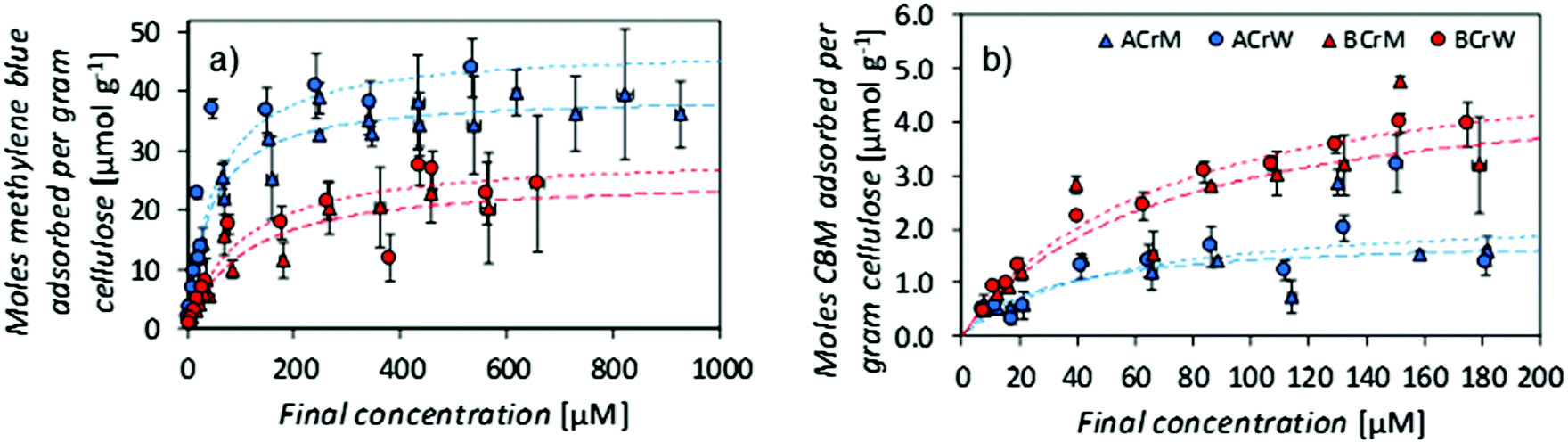 | ||
| Fig. 3 (a) Methylene blue adsorption isotherms on regenerated cellulose scaffolds, (b) ThCBMCBHI adsorption isotherms on regenerated cellulose scaffolds. Error bars ± 1 standard error. Whilst a greater surface area is accessible to methylene blue in α-cellulose samples, a greater area is accessible to ThCBMCBHI in bacterial cellulose samples. Water regenerated samples have a greater accessible area in all cases. | ||
| Sample | MB specific surface area [m2 g−1] | MB partition constant [×10−3 L g−1] | MB partition constant [×10−3 L m−2] | CBM specific surface area [m2 g−1] | CBM partition constant [×10−3 L g−1] | CBM partition constant [×10−3 L m−2] |
|---|---|---|---|---|---|---|
| ACrM | 47 | 855 | 18.3 | 14 | 53 | 3.8 |
| ACrW | 56 | 874 | 15.5 | 16 | 43 | 2.7 |
| BCrM | 30 | 288 | 9.5 | 29 | 70 | 2.4 |
| BCrW | 35 | 236 | 6.8 | 33 | 76 | 2.3 |
The surface area available for CBM attachment was also affected by the cellulose and anti-solvent type: rW hydrogels had a 1.2 increase in the ThCBMCBHI accessible surface area, due either to the higher proportion of nanopores with smaller diameters, or to the increase in crystallinity, compared to rM hydrogels. However, the use of AC resulted in a reduction of the surface area accessible to ThCBMCBHI by a factor of 0.4. It is hypothesised that a significant proportion of the pores in the AC samples accessible to MB are inaccessible to ThCBMCBHI (due to its larger size). Of the pore surface accessible to MB, 95% is accessible to ThCBMCBHI in hydrogels prepared from BC, whilst only 24% is accessible in hydrogels prepared from AC. This is supported by the nanopore:micropore ratios derived from the NMR relaxometry experiments (Table 1).
It is apparent that both the cellulose type (differing by DP only) and anti-solvent identity influenced the partition constants of MB (Table 2). MB partition constants measured for AC derived hydrogels are higher than for BC derived hydrogels and this may reflect differences in processing of the raw materials used.¶ CBM partition constants expressed in units of L m−2 (which more accurately reflects the surfaces available for adsorption of the probe molecules) gave distinctly larger values for rM samples versus rW samples when MB was used as the probe molecule. It has been reported that the partition constant for monosaccharide adsorption increased with the hydrophobicity index of the monosaccharide.56 Considering this to reflect in the reverse, i.e. adsorption onto polysaccharide surfaces, this might suggest greater hydrophobicity of surfaces in rM samples versus rW samples. This reflects previously published data pertaining to crystallinity: Östlund et al. argued that faster rates of demixing in water ‘trap’ the methylhydroxyl groups in the gauche–trans formation that is found in cellulose II.10 This results in a higher crystallinity for samples regenerated in water than those regenerated in methanol, where the methylhydroxyl groups adopt the more energetically favourable gauche–gauche confirmation, demonstrated computationally by Liu et al.57 If samples regenerated in methanol are more hydrophobic – due either to the conformation of the methylhydroxyl groups, or to the specific crystalline faces exposed – this would account for the observed difference. No such differences are noted in binding of ThCBMCBHI, with partition constants ranging between 3.8 and 2.3 mL m−2, reflecting the inaccessibility of the smaller pores to the large biomolecule (Table 2).
It is also of note that the partition constants expressed per unit mass, reported here for a family 1 CBM, are two orders of magnitude lower than those reported previously: a partition constant of 1.0 × 105 M−1 was reported for T. reesei CBMCBHI on native BC,25 and 4.9 L g−1 on microcrystalline cellulose;58 compared to 1.5 × 103 M−1 and 7.6 × 10−2 L g−1 on BCrW in this work. This reflects the lower degree of crystallinity in these regenerated samples, although, as previously discussed in the literature, these values do not take into account the accessible surface area.59
To test the hypothesis that some pores are inaccessible to CBMs, the rates of diffusion of fluorescently tagged ThCBMCBHI in hydrogels were determined from rates of recovery of fluorescent intensity after bleaching, using confocal microscopy. Recovery of the fluorescence intensity after bleaching is observed (Fig. 4), confirming that ThCBMCBHI is reversibly bound to the cellulose, in agreement with previous reports.25 The differences in the recovery half-lives and subsequent diffusion coefficients of the samples can be attributed to three different effects: (i) the pore structure of the hydrogels – a structure with fewer pores accessible to the CBM is more tortuous, resulting in a lower CBM diffusion rate and thus a longer recovery period; (ii) the affinity of the CBM to the cellulose, a higher partition constant resulting in a higher diffusion rate; and (iii) the hydrophobicity of the sample – it has been reported, based on computational simulations, that CBM could diffuse from hydrophilic to hydrophobic surfaces, but that the reverse transition was not observed in 43 ms of simulation,27 suggesting that a slower diffusion rate would be observed along surfaces with more hydrophilic character.
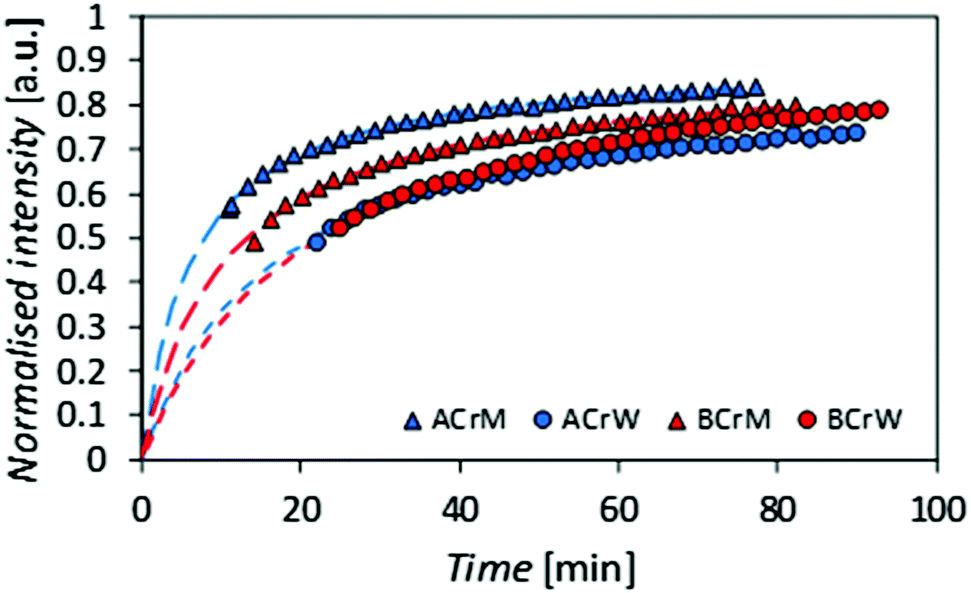 | ||
| Fig. 4 Recovery of fluorescence intensity for regenerated cellulose hydrogels after bleaching. A more rapid recovery is observed in the methanol regenerated samples and those generated from α-cellulose. | ||
Given that the diffusion coefficient increases by a factor of 1.5 for AC compared to BC (Table 3), it is apparent that the affinity of the CBM for the cellulose surfaces is more important than the hydrogel tortuosity (assumed to be proportional to the difference in accessible surface area for ThCBMCBHI compared to MB). With regards to the anti-solvent type, rM hydrogels increase the diffusion coefficient by a factor of 2.2 over rW hydrogels. In this instance, the tortuosity and presumed hydrophobicity of the hydrogel are more important than the CBM affinity.
| Sample | I max | k [min−1] | τ D [min] | t 1/2 [min] | D [μm2 min−1] |
|---|---|---|---|---|---|
| ACrM | 0.91 | 2.00 | 6.3 | 7.8 | 1.76 |
| ACrW | 0.86 | 2.01 | 15.3 | 21.6 | 0.73 |
| BCrM | 0.90 | 2.00 | 10.4 | 13.2 | 1.07 |
| BCrW | 0.96 | 2.01 | 20.3 | 22.5 | 0.55 |
Conclusions
Cellulose hydrogels regenerated from solutions containing ionic liquids have been used in a plethora of applications, but, in many cases, the materials have primarily been characterised using techniques requiring drying. Here, careful “wet” characterisation of never-dried cellulose hydrogels has revealed that subtle changes in choice of raw material and regeneration solvents can result in significant differences in the hydrogel products. This has wide ramifications for many cellulose hydrogel and material applications.Specifically, the choice of cellulose starting material (with different DP) and anti-solvent used in generating cellulose hydrogels from solutions containing ionic liquids influence the crystallinity and pore structure of the resulting hydrogel. In turn, these affect the tortuosity and hydrophobicity of the porous material and, thus, the affinity of molecules adsorbing onto and migrating through, the hydrogel. Small molecules, such as the widely used probe methylene blue, do not yield information that can be extrapolated to larger probes, such as proteins, including CBMs. It is also clear that compromises may need to be made between the maximum accessible surface area for the CBM, here 33 m2 g−1 for bacterial cellulose regenerated in water, and maximum CBM partition constant per unit area and CBM diffusion coefficient, here 3.8 mL m−2 and 1.76 μm2 min−1 for α-cellulose regenerated in methanol. Thus, cellulose hydrogels optimized for various CBM-based bioaffinity applications may be prepared by manipulating the cellulose DP and anti-solvent. For example, the use of methanol to regenerate the hydrogel will lead to a higher enzymatic hydrolysis rate due to the increased CBM migration rate. In addition, bacterial cellulose has been widely posited as a biocompatible material and many potential applications of such hydrogels will rely on hydrogel structure.
Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the following funding: PhD studentship funding for M. A. J. from the UK Engineering and Physical Sciences Research Council (EPSRC) via the EPSRC Doctoral Training Centre in Sustainable Chemical Technologies, University of Bath (EP/G03768X/1); the São Paulo State Research Foundation (FAPESP) (grant 09/52840-7); the Brazilian National Council for Scientific and Technological Research (CNPq) (grants 490022/2009-0 and 312852/2014-2); and the British Council via the Global Innovation Initiative programme, which facilitated UK/Brazilian collaboration. All data are available from the University of Bath data archive at http://doi.org/10.15125/BATH-00316.Notes and references
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- J. C. Courtenay, M. A. Johns, F. Galembeck, C. Deneke, E. M. Lanzoni, C. A. Costa, J. L. Scott and R. I. Sharma, Cellulose, 2017, 24, 253–267 CrossRef CAS.
- A. Sydney Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef PubMed.
- B. D. B. Aaronson, D. He, E. Madrid, M. A. Johns, J. L. Scott, L. Fan, J. Doughty, M. A. S. Kadowaki, I. Polikarpov, N. B. McKeown and F. Marken, ChemistrySelect, 2017, 2, 871–875 CrossRef CAS.
- C. B. Field, M. J. Behrenfeld, J. T. Randerson and P. Falkowski, Science, 1998, 281, 237–240 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- J.-I. Kadokawa, M.-A. Murakami and Y. Kaneko, Carbohydr. Res., 2008, 343, 769–772 CrossRef CAS PubMed.
- O. Aaltonen and O. Jauhiainen, Carbohydr. Polym., 2009, 75, 125–129 CrossRef CAS.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- Å. Östlund, A. Idström, C. Olsson, P. Larsson and L. Nordstierna, Cellulose, 2013, 20, 1657–1667 CrossRef.
- X. Geng and W. A. Henderson, RSC Adv., 2014, 4, 31226–31229 RSC.
- A. B. Boraston, D. N. Bolam, H. J. Gilbert and G. J. Davies, Biochem. J., 2004, 382, 769–781 CrossRef CAS PubMed.
- M. A. Goldstein, M. Takagi, S. Hashida, O. Shoseyov, R. H. Doi and I. H. Segel, J. Bacteriol., 1993, 175, 5762–5768 CrossRef CAS PubMed.
- J. Credou and T. Berthelot, J. Mater. Chem. B, 2014, 2, 4767–4788 RSC.
- Z. Xu, W. Bae, A. Mulchandani, R. K. Mehra and W. Chen, Biomacromolecules, 2002, 3, 462–465 CrossRef CAS PubMed.
- A. Fishman, I. Levy, U. Cogan and O. Shoseyov, J. Mol. Catal. B: Enzym., 2002, 18, 121–131 CrossRef CAS.
- S. Hwang, J. Ahn, S. Lee, T. Lee, S. Haam, K. Lee, I.-S. Ahn and J.-K. Jung, Biotechnol. Lett., 2004, 26, 603–605 CrossRef CAS PubMed.
- H. Park, J. Ahn, J. Lee, H. Lee, C. Kim, J.-K. Jung, H. Lee and E. G. Lee, Int. J. Mol. Sci., 2012, 13, 358–368 CrossRef CAS PubMed.
- W. Lewis, E. Keshavarz-Moore, J. Windust, D. Bushell and N. Parry, Biotechnol. Bioeng., 2006, 94, 625–632 CrossRef CAS PubMed.
- Y. Cao, Q. Zhang, C. Wang, Y. Zhu and G. Bai, J. Chromatogr. A, 2007, 1149, 228–235 CrossRef CAS PubMed.
- A. Karpol, L. Kantorovich, A. Demishtein, Y. Barak, E. Morag, R. Lamed and E. A. Bayer, J. Mol. Recognit., 2009, 22, 91–98 CrossRef CAS PubMed.
- A. Wierzba, U. Reichl, R. F. B. Turner, R. A. J. Warren and D. G. Kilburn, Biotechnol. Bioeng., 1995, 46, 185–193 CrossRef CAS PubMed.
- F. K. Andrade, S. M. G. Moreira, L. Domingues and F. M. P. Gama, J. Biomed. Mater. Res., Part A, 2010, 92, 9–17 CrossRef CAS PubMed.
- R. Pértile, S. Moreira, F. Andrade, L. Domingues and M. Gama, Biotechnol. Prog., 2012, 28, 526–532 CrossRef PubMed.
- P. Tomme, A. Boraston, B. McLean, J. Kormos, A. L. Creagh, K. Sturch, N. R. Gilkes, C. A. Haynes, R. A. J. Warren and D. G. Kilburn, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 715, 283–296 CrossRef CAS.
- J. Lehtiö, J. Sugiyama, M. Gustavsson, L. Fransson, M. Linder and T. T. Teeri, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 484–489 CrossRef PubMed.
- M. R. Nimlos, G. T. Beckham, J. F. Matthews, L. Bu, M. E. Himmel and M. F. Crowley, J. Biol. Chem., 2012, 287, 20603–20612 CrossRef CAS PubMed.
- K. B. Pal, H. Chatterjee and P. B. Sarkar, Nature, 1952, 169, 845–846 CrossRef CAS.
- É. Pecoraro, D. Manzani, Y. Messaddeq and S. J. L. Ribeiro, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2007, ch. 17, pp. 369–383 DOI:10.1016/B978-0-08-045316-3.00017-X.
- M. Schramm and S. Hestrin, Microbiology, 1954, 11, 123–129 CrossRef CAS PubMed.
- B. L. Mello and I. Polikarpov, AMB Express, 2014, 4, 1–6 CrossRef CAS PubMed.
- E. Mossessova and C. D. Lima, Mol. Cell, 2000, 5, 865–876 CrossRef CAS PubMed.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- S. Berger and S. Braun, 200 and More NMR Experiments, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630–638 CrossRef CAS.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- K. R. Brownstein and C. E. Tarr, Phys. Rev. A: At., Mol., Opt. Phys., 1979, 19, 2446–2453 CrossRef.
- J. Mitchell, S. C. Stark and J. H. Strange, J. Phys. D: Appl. Phys., 2005, 38, 1950–1958 CrossRef CAS.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 229–242 CrossRef.
- G. C. Borgia, R. J. S. Brown and P. Fantazzini, J. Magn. Reson., 1998, 132, 65–77 CrossRef CAS PubMed.
- D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson and W. W. Webb, Biophys. J., 1976, 16, 1055–1069 CrossRef CAS PubMed.
- L. Y. Mwaikambo and M. P. Ansell, J. Mater. Sci. Lett., 2001, 20, 2095–2096 CrossRef CAS.
- M. Ago, T. Endo and T. Hirotsu, Cellulose, 2004, 11, 163–167 CrossRef CAS.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- N. Terinte, R. Ibbett and K. C. Schuster, Lenzinger Ber., 2011, 89, 118–131 CAS.
- O. V. Petrov and I. Furó, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 54, 97–122 CrossRef CAS.
- E. Shiko, K. J. Edler, J. P. Lowe and S. P. Rigby, J. Colloid Interface Sci., 2012, 385, 183–192 CrossRef CAS PubMed.
- C. Zhang, P. Li, Y. Zhang, F. Lu, W. Li, H. Kang, J.-F. Xiang, Y. Huang and R. Liu, Polymer, 2016, 98, 237–243 CrossRef CAS.
- D. Capitani, N. Proietti, F. Ziarelli and A. L. Segre, Macromolecules, 2002, 35, 5536–5543 CrossRef CAS.
- N. Proietti, D. Capitani, E. Pedemonte, B. Blümich and A. L. Segre, J. Magn. Reson., 2004, 170, 113–120 CrossRef CAS PubMed.
- C. Felby, L. G. Thygesen, J. B. Kristensen, H. Jørgensen and T. Elder, Cellulose, 2008, 15, 703–710 CrossRef CAS.
- J. E. Tsuchida, C. A. Rezende, R. de Oliveira-Silva, M. A. Lima, M. N. d’Eurydice, I. Polikarpov and T. J. Bonagamba, Biotechnol. Biofuels, 2014, 7, 127 Search PubMed.
- C. Kaewprasit, E. Hequet, N. Abidi and J. Paul, J. Cotton Sci., 1998, 2, 164–173 Search PubMed.
- D. Graham, J. Phys. Chem., 1955, 59, 896–900 CrossRef CAS.
- H. P. Erickson, Biol. Proced. Online, 2009, 11, 32–51 CrossRef CAS PubMed.
- K. Miyajima, K. Machida, T. Taga, H. Komatsu and M. Nakagaki, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2537–2544 RSC.
- H. Liu, G. Cheng, M. Kent, V. Stavila, B. A. Simmons, K. L. Sale and S. Singh, J. Phys. Chem. B, 2012, 116, 8131–8138 CrossRef CAS PubMed.
- M. Linder and T. T. Teeri, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12251–12255 CrossRef CAS.
- R. Pinto, S. Moreira, M. Mota and M. Gama, Langmuir, 2004, 20, 1409–1413 CrossRef CAS PubMed.
- O. N. Reva, I. E. Zaets, L. P. Ovcharenko, O. E. Kukharenko, S. P. Shpylova, O. V. Podolich, J.-P. de Vera and N. O. Kozyrovska, AMB Express, 2015, 5, 35, DOI:10.1186/s13568-015-0124-5.
Footnotes |
| † Electronic supplementary information (ESI) available: Methylene blue HPLC calibration curve, C versus C/N plots for methylene blue and ThCBM1CBHI adsorption isotherms, Nm and K values for methylene blue and ThCBM1CBHI adsorption isotherms, bleaching images demonstrating fluorescent recovery. See DOI: 10.1039/c7tb00176b |
| ‡ Current address: School of Experimental Psychology, University of Bristol, BS8 1TH, UK. |
| § While BC is often ascribed to production by a particular strain, it can be more productive to use mixed cultures, such as those used in production of the drink ‘kombucha’, followed by appropriate purification, particularly where the material is then dissolved so removing any structural features. Reva et al., have reported that the core of such communities are comprised of acetobacteria of two genera, Komagataeibacter (formerly Gluconacetobacter) and Gluconobacter.60 |
| ¶ AC materials may be subjected to an acid wash during purification potentially leading to introduction of a very small number of acid groups on the surfaces that would bind a positively charged probe, such as MB, strongly. |
| This journal is © The Royal Society of Chemistry 2017 |