
Non-covalent molecular tweezer/guest complexation with Pt(II)⋯Pt(II) metal–metal interactions: toward intelligent photocatalytic materials†
Zijian
Li
,
Yifei
Han
,
Zongchun
Gao
,
Tengfei
Fu
and
Feng
Wang
 *
*
CAS Key Laboratory of Soft Matter Chemistry, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: drfwang@ustc.edu.cn
First published on 18th October 2017
Abstract
A new organoplatinum(II) molecular tweezer with a stimuli-responsive 2,2′:6′,2′′-terpyridine spacer has been synthesized, which undergoes “U”- to “W”-shaped conformational transition upon adding Zn2+ ions. The molecular tweezer receptor displays a high binding affinity toward the complementary guests, with the implementation of Pt(II)⋯Pt(II) metal–metal interactions for the non-covalent tweezering systems. MMLCT absorbance simultaneously emerges in the visible-light region (470–620 nm), which can be sensitized by O2 and thereby utilized for photo-catalyzed reactions in organic and aqueous media. Moreover, temporally “on-demand” photo-catalytic efficiency can be accomplished, via the successive addition of Zn(OTf)2 and a competitive ligand to the supramolecular tweezering photosensitizers. Hence, the current study opens up new avenues toward intelligent photocatalytic materials via elaborate supramolecular engineering.
 Feng Wang | Feng Wang got his bachelor's degree from Hefei University of Technology in 2003. He then received his PhD in Chemistry from Zhejiang University under the supervision of Prof. Feihe Huang in 2009. He joined Prof. E. W. Meijer's group at Eindhoven University of Technology as a postdoctoral fellow. He joined the University of Science and Technology of China as an associate professor in 2011, and was promoted to full professor in 2016. His current research interests are focused on transition metal-based supramolecular assemblies for biomedical, optoelectronic, and catalytic applications. |
Introduction
Visible-light photosensitization represents a sustainable method for organic transformation under mild conditions.1,2 Up to now, a variety of photosensitizers capable of undergoing energy transfer between their triplet excited states and oxygen molecules (O2) have been developed, leading to the formation of reactive oxygen species for effective catalysis. However, the inherent photo-bleaching issues of the visible-light photosensitizers hamper their long-term use and storage with the threat of photo-degradation. In this regard, it is appealing to endow “on/off” functionality for the photosensitization process, which can significantly minimize the above-mentioned side effects.3–6 To attain this objective, organoplatinum(II)-based photosensitizers are regarded as one of the promising candidates.7–15 Owing to the square-planar geometry, they display a strong tendency to form intermolecular Pt(II)⋯Pt(II) interactions, accompanied by the presence of metal–metal-to-ligand charge transfer (MMLCT) optical signals in the visible-NIR region.16,17 By manipulating the distance and strength of Pt(II)⋯Pt(II) forces at the molecular level, the MMLCT spectroscopic signals can be elaborately modulated, which lays the basis for precise tempo-control over photo-sensitization/-catalytic efficiencies.Our research group is interested in constructing organoplatinum(II)-based molecular tweezers such as 1a (Scheme 1) as the supramolecular receptor.18–27 It tends to encapsulate isocyanideplatinum(II) diphenylpyridine 2a (Scheme 1) as the complementary guest, leading to the formation of intermolecular Pt(II)⋯Pt(II) interactions for the non-covalent tweezering system. In the current study, we sought to incorporate dynamic switching elements into the molecular tweezer receptor, which triggers reversible guest encapsulation/release in response to external stimuli,28–32 and thereby regulates the emergence/disappearance of Pt(II)⋯Pt(II) MMLCT signals in a highly controllable manner.
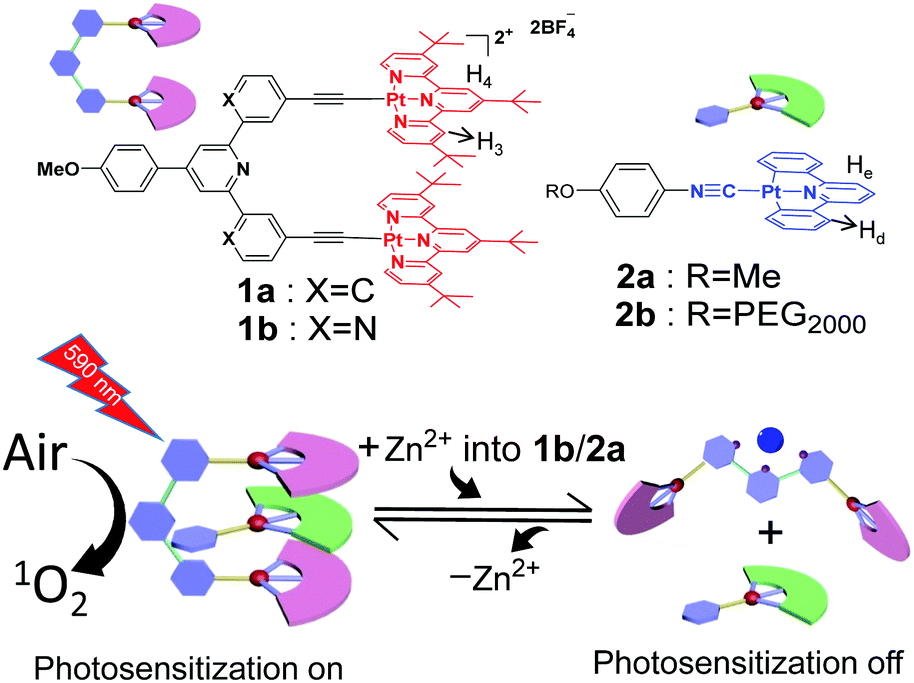 | ||
| Scheme 1 Schematic representation of the construction of supramolecular tweezering systems for “on/off”-switchable photosensitization. | ||
Specifically, molecular tweezer 1b (Scheme 1) has been designed and synthesized (Scheme S1 in the ESI†), in which 2,2′:6′,2′′-terpyridine serves as the rigid spacer unit. As expected, 1b prefers the “U”-shaped conformation (Fig. S1, ESI†),33 with the adoption of co-facial conformation for two electron-deficient alkynylplatinum(II) terpyridine pincers. It is prone to sandwich neutral organoplatinum guest 2a into its cavity. The presence of Pt(II)⋯Pt(II) interactions for the resulting tweezering complexes would lead to the generation of reactive oxygen species upon visible-light irradiation (Scheme 1), which can be further utilized for photo-catalyzed organic transformation. Upon addition of Zn(OTf)2, 1b is expected to undergo mechanical motion due to metal–ligand coordination between the Zn2+ ion and 2,2′:6′,2′′-terpyridine spacer, resulting in the transition from “U”- to “W”-shaped conformation.34,35 Consequently, non-covalent tweezering structures are destroyed, leading to the vanishing of Pt(II)⋯Pt(II) interacting signals. Zn2+ ion-responsiveness for the supramolecular tweezering system provides a novel avenue toward intelligent photosensitization and photocatalytic materials.
Results and discussion
Spectroscopic measurements for molecular tweezer 1b were first performed and compared with those of the counterpart molecular tweezer 1a. For the dilute CHCl3/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution of 1b, the maximum absorption and emission bands are located at 410 and 527 nm, respectively (Fig. 1a and b). According to the previous literatures, these bands are tentatively assigned to the admixture of dπ(Pt) → π*(t-Bu3tpy) metal-to-ligand charge transfer (MLCT) and π(C
:1, v/v) solution of 1b, the maximum absorption and emission bands are located at 410 and 527 nm, respectively (Fig. 1a and b). According to the previous literatures, these bands are tentatively assigned to the admixture of dπ(Pt) → π*(t-Bu3tpy) metal-to-ligand charge transfer (MLCT) and π(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR) → π*(t-Bu3tpy) ligand-to-ligand charge transfer (LLCT) bands.18 Notably, the MLCT/LLCT absorption and emission bands of 1b are blue-shifted compared to those of 1a (440 nm and 593 nm for the maximum absorption and emission bands, Fig. S2, ESI†).24 Considering that 1a–b feature the same platinum(II) center and t-Bu3tpy main ligand, the relatively higher energy levels for MLCT/LLCT transition of 1a can be ascribed to the structural variation on the ancillary dialkynyl ligand. Briefly, 1b features a more electron-withdrawing ancillary ligand than that of 1a (terpyridine versus diphenylpyridine), which renders the metal center more electron-deficient. As a result, it reduces the energy of the highest occupied molecular orbital (HOMO) for the platinum(II) center.36
CR) → π*(t-Bu3tpy) ligand-to-ligand charge transfer (LLCT) bands.18 Notably, the MLCT/LLCT absorption and emission bands of 1b are blue-shifted compared to those of 1a (440 nm and 593 nm for the maximum absorption and emission bands, Fig. S2, ESI†).24 Considering that 1a–b feature the same platinum(II) center and t-Bu3tpy main ligand, the relatively higher energy levels for MLCT/LLCT transition of 1a can be ascribed to the structural variation on the ancillary dialkynyl ligand. Briefly, 1b features a more electron-withdrawing ancillary ligand than that of 1a (terpyridine versus diphenylpyridine), which renders the metal center more electron-deficient. As a result, it reduces the energy of the highest occupied molecular orbital (HOMO) for the platinum(II) center.36
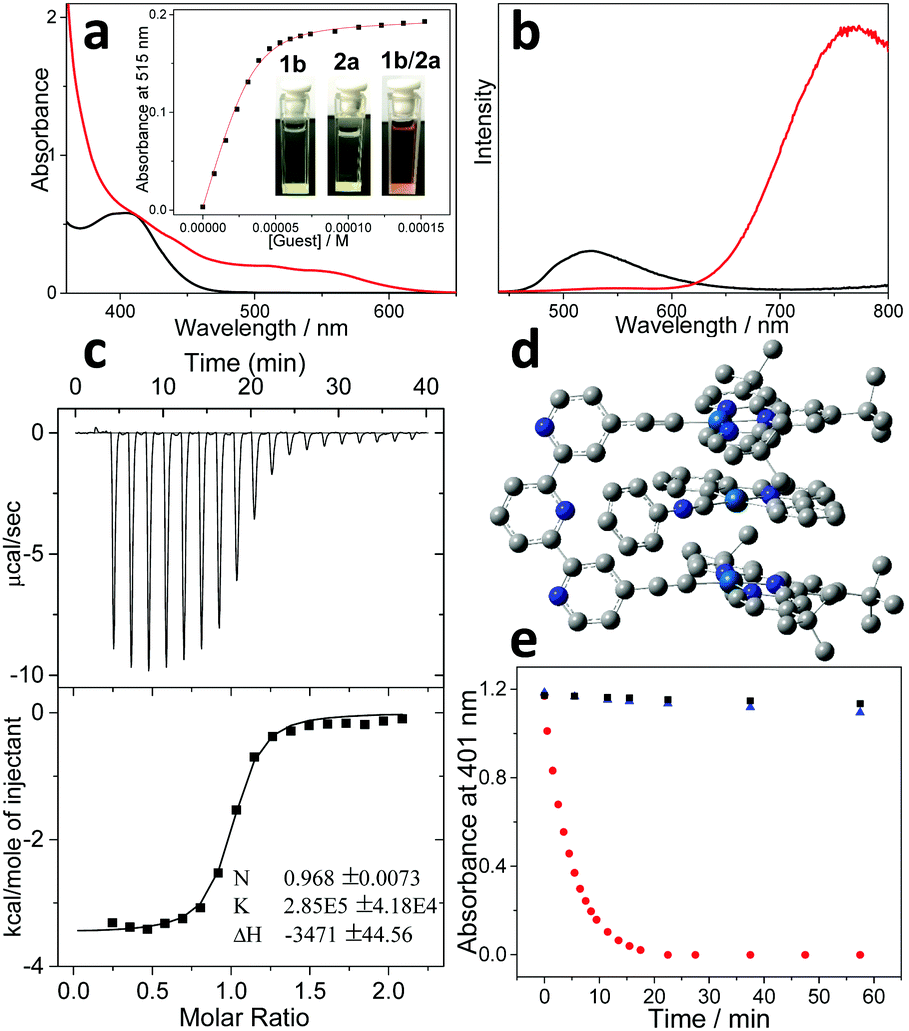 | ||
Fig. 1 (a) UV/Vis absorbance and (b) fluorescence changes upon addition of 2a into 1b (CHCl3/CH3CN = 1:1, 0.05 mM). Inset for (a): intensity changes at λ = 515 nm and the nonlinear curve fitting (red line). (c) ITC data for titrating 2a (8.00 mM) into the solution (CHCl3/CH3CN = 1:1) of 1b (0.40 mM). (d) Optimized structure of complex 1b/2avia the DFT method. (e) Photosensitization (OLED lamp, 12 w, 590 nm) efficiency of 1b/2a ( ), 2a (■), and 1b ( ), 2a (■), and 1b ( ), by monitoring time-dependent absorbance of DMA at 401 nm. ), by monitoring time-dependent absorbance of DMA at 401 nm. | ||
On this basis, non-covalent complexation was investigated between 1b and the organoplatinum(II) guest 2a. Upon adding an equimolar amount of 2a (colorless) into 1b (light yellow color), the mixed solution promptly becomes deep-red (Fig. 1a, inset). Simultaneously, a considerably bathochromically shifted band (λmax = 515 nm) emerges in the UV-Vis spectrum (Fig. 1a). In addition, a new emission band appears in the near infrared region (λmax = 780 nm), which shows enhanced intensity compared to the original MLCT/LLCT band (Fig. 1b). The newly formed bathochromically shifted signals can be assigned to the MMLCT (metal–metal-to-ligand charge transfer) transition,16,17 suggesting the proximity of the Pt(II) atoms upon complexation between 1b and 2a.
The binding thermodynamics for the resulting complex 1b/2a can be further elucidated. Based on UV-Vis Job's plots, the MMLCT absorbance intensity reaches the maximum value at the equivalent ratio, validating 1:1 binding stoichiometry between 1b and 2a (Fig. S4, ESI†). Nonlinear curve-fitting of the collected MMLCT absorbance data at 550 nm provides the Ka value of (2.53 ± 0.29) × 105 M−1 for complex 1b/2a (Fig. 1a, inset). A similar binding affinity can be acquired via isothermal titration calorimetry (ITC) measurements (Ka = (2.85 ± 0.42) × 105 M−1) (Fig. 1c). It is worthy to note that complex 1b/2a displays approximately five times enhancement for the binding strength compared to that of 1a/2a (Ka,UV = (4.69 ± 0.13) × 104 M−1, Fig. S8, ESI†).
The exact binding mode for complex 1b/2a was further clarified by density functional theory (DFT) calculation. As can be seen in Fig. 1d, the distances for the proximity Pt(II) atoms are determined to be 3.48 Å and 3.53 Å, with the Pt⋯Pt⋯Pt angle of 161.9°. Meanwhile, inter-planar π-distances between the electron-rich diphenylpyridine unit on 2a and two electron-deficient terpyridine pincers on 1b are calculated to be approximately 3.63 Å. The presence of electron donor–acceptor interactions between 1b and 2a is further validated by 1H NMR measurements, which display remarkable upfield shifts for the aromatic protons on both 1b and 2a (−0.22, −0.30, −0.58 and −0.71 ppm for H3, H4, Hd, and He, respectively, Fig. S3, ESI†). Accordingly, it can be concluded that non-covalent complexation between 1b and 2a is co-driven by Pt(II)⋯Pt(II) metal–metal and electron donor–acceptor interactions.
In view of the fact that complex 1b/2a features bathochromically shifted MMLCT absorbance, its visible-light photosensitization capability was then investigated. When photo-exciting the mixture of 1b/2a and 9,10-dimethylanthracene (DMA) with an OLED lamp (12 w, 590 nm), the absorbance of DMA at 401 nm decays and levels off after 20 min (Fig. 1e). Hence, it validates the generation of singlet oxygen (1O2) in situ, which is captured by DMA to form 9,10-dimethylanthracene-9,10-endoperoxide. In sharp contrast, DMA absorbance hardly shows any changes for the individual species (1b or 2a) (Fig. 1e), because of the absence of MMLCT transition signals. In addition, photosensitization capability can be influenced by the non-covalent complexation strength, as evidenced by the higher 1O2 generation efficiency of complex 1b/2a than that of 1a/2a (725 min−1 M−1vs. 322 min−1 M−1, Fig. S11, ESI†).37
Furthermore, the photo-catalytic efficiency of complex 1b/2a was examined, by performing the oxidative cyanation reaction for N-phenyl-1,2,3,4-tetrahydroisoquinoline 3 (Table 1). In detail, upon irradiating (LED lamp, >590 nm, 100 W) the reactants and 1b/2a (1.0–0.2 mol%), 89–94% product yields can be achieved (entries 1–3 in Table 1). In stark contrast, almost no conversion can be detected for the individual species even with prolonged irradiation time (either 1b or 2a, entries 7–8 in Table 1). Besides, the turnover (TON) value of 1b/2a is 1.37 times higher than that of 1a/2a (entries 1 and 4 in Table 1), which is consistent with the stronger 1O2 generation capability of the former complex. The photocatalytic mechanism can be explained as follows: due to the spectral overlap between photo-irradiation wavelength and the MMLCT absorbance of 1b/2a, 1O2 can be generated in situ. The reactive oxygen species interacts with 3 to afford the unstable iminium ion intermediate,38 which further reacts with trimethylsilyl cyanide (TMSCN) to give the cyanation product 4. No conversion can be detected with the absence of oxygen or light (entries 5 and 6 in Table 1), validating that 1O2 is the active oxidant during the organic transformation process.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Photocatalyst | Time [h] | Conversion [%] | Yield [%] | TON [h−1] |
|
a Compound 3 (10.00 mmol L−1), photocatalyst (0.4 mol%), and TMSCN (15.00 mmol L−1) are dissolved in CHCl3:CH3CN (1:1, 10 mL), with O2 bubbling for 10 seconds. After the photo-catalyzed reaction, the solvent was evaporated and the residue was subjected to 1H NMR analysis. Conversion was determined by crude 1H NMR analysis using 4,4′-dimethyl-2,2′-bipyridine as the internal standard. Yield was calculated based on the starting amount of substrate. The similar reaction conditions described in entry a were also used for entries b–h, except where otherwise specified.
b Catalyst loading was 1 mol%.
c Catalyst loading was 0.2 mol%.
d Complex 1a/2a was employed as the catalyst.
e N2 instead of O2 was purged into the solution for 30 seconds.
f The solution mixture was kept in the dark.
g
1b was employed as the catalyst.
h
2a was employed as the catalyst. n.d. = not determined.
|
|||||
| 1 | 1b/2a | 1.25 | 100 | 92 | 184.0 |
| 2b | 1b/2a | 0.42 | 100 | 94 | 223.8 |
| 3c | 1b/2a | 4 | 98 | 89 | 111.3 |
| 4d | 1a/2a | 1.67 | 100 | 90 | 134.7 |
| 5e | 1b/2a | 24 | 4 | n.d. | — |
| 6f | 1b/2a | 24 | 2 | n.d. | — |
| 7g | 1b | 24 | 7 | n.d. | — |
| 8h | 2a | 24 | 4 | n.d. | — |
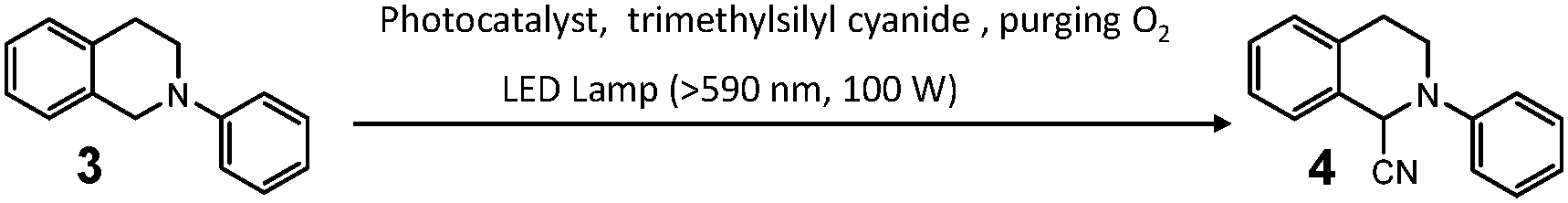
Considering that water is a green and environmentally friendly medium, it is keenly pursued to perform photo-catalytic organic transformation in an aqueous solution. To guarantee sufficient water-solubility of the supramolecular photocatalyst, poly(ethylene glycol) (PEG2000) was attached to the guest structure (guest 2b, see Scheme 1). For the resulting complex 1b/2b, color change together with the appearance of MMLCT transition signals takes place in water (Fig. 2b), which are similar to those of complex 1b/2a in an organic medium. Notably, the presence of a PEG chain exerts minor impact on the non-covalent binding affinity, since both complexes 1b/2a and 1b/2b show comparable Ka values (Ka,UV = (2.30 ± 0.22) × 105 M−1 and Ka,ITC = (3.90 ± 0.17) × 105 M−1 for 1b/2b in CHCl3:CH3CN (1:1, v/v), Fig. S6 and S7, ESI†). On this basis, photo-triggered cleavage of coumarin derivative 5 in an aqueous solution was performed, by employing 1b/2b as the catalyst (Fig. 2a). As previously reported by You and others,39,40 the fluorescence of 5 is significantly quenched due to the presence of the amino-acrylate linker. Upon photo-irradiation (590 nm, 12 w), the linker can be cleavaged by 1O2, leading to the formation of highly fluorescent product 6. In the current system, the emission signal at 450 nm gradually increases and reaches a plateau after 40 min (Fig. 2c). 1H NMR experiments provide additional evidence for the quantitative conversion of 5 to 6, which shows upfield shifting of the aromatic protons after the photo-cleavage process (Fig. S13, ESI†). Under the same conditions, no emission intensity varies for complex 1b/2a (Fig. 2c). Hence, attachment of a PEG solubilizing group on the supramolecular tweezering structure represents an efficient approach for photo-catalytic reactions in aqueous solution.
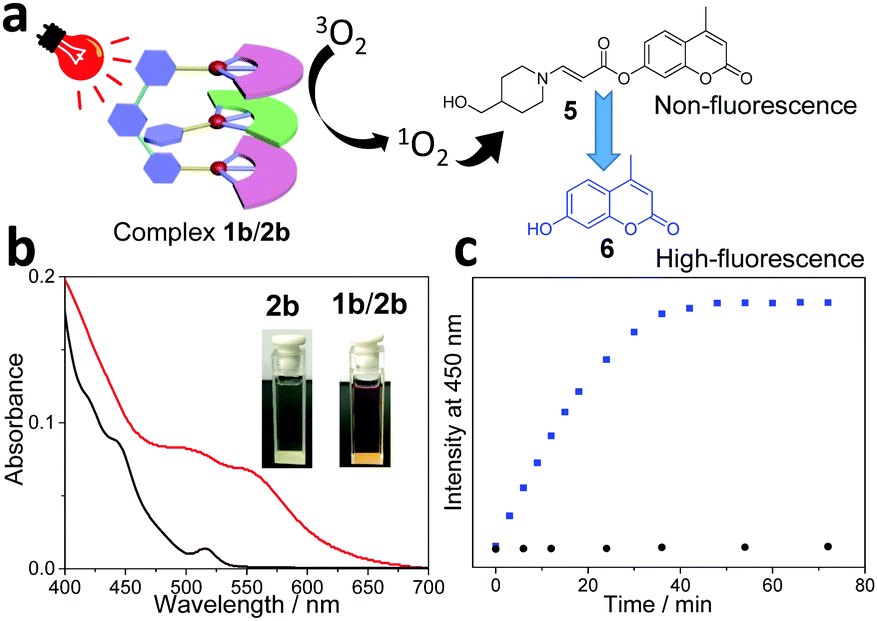 | ||
Fig. 2 (a) Schematic representation for the photo-cleavage of coumarin derivative 5 with complex 1b/2b as the catalyst. (b) UV/Vis absorbance of 2b and complex 1b/2b (0.05 mM, 95% water/5% DMF). Inset: The solution colors of 2b and 1b/2b. (c) Time-dependent fluorescence changes of 5 (20 μM in 95% water/5% DMF) upon visible-light irradiation (OLED lamp, 12 w, 590 nm), with the employment of 1b/2b ( ) and 1b/2a (●) as the photocatalysts. ) and 1b/2a (●) as the photocatalysts. | ||
Stimuli-responsiveness of the non-covalent tweezering complexes toward Zn2+ ions was then exploited. As an initial step, we exploited cation-triggered nanomechanical motion of 1b. Upon gradually titrating Zn(OTf)2 into the CHCl3:CH3CN (1:1, v/v) solution of 1b, the MLCT/LLCT absorbance located at 410 nm exhibits a hypsochromic shift, accompanied by an isosbestic point at 427 nm (Fig. S15, ESI†). Blue-shifting of the MLCT/LLCT band primarily arises from the decreased electron-donating capability of the ancillary ligand, denoting metal–ligand complexation between Zn2+ ions and the rigid terpyridine spacer on 1b. Simultaneously, “U”- to “W”-shaped conformational transition takes place for 1b, which further affects the non-covalent binding affinity toward the complementary guest 2a. It can be directly reflected by the disappearance of MMLCT absorbance and emission bands (Fig. 3a and Fig. S16, ESI†). Further evidence for the decomplexation between 1b and 2a came from ITC experiments, as evidenced by the negligible heat exchange upon titrating 2a into the mixture solution of 1b/Zn(OTf)2 (Fig. S17a, ESI†). In sharp contrast, the presence of Zn2+ ions hardly influences the isothermal curve between 1a and 2a (Fig. S17b, ESI†), validating the importance of dynamic switching elements for cation-responsive molecular tweezer/guest complexation behaviors.
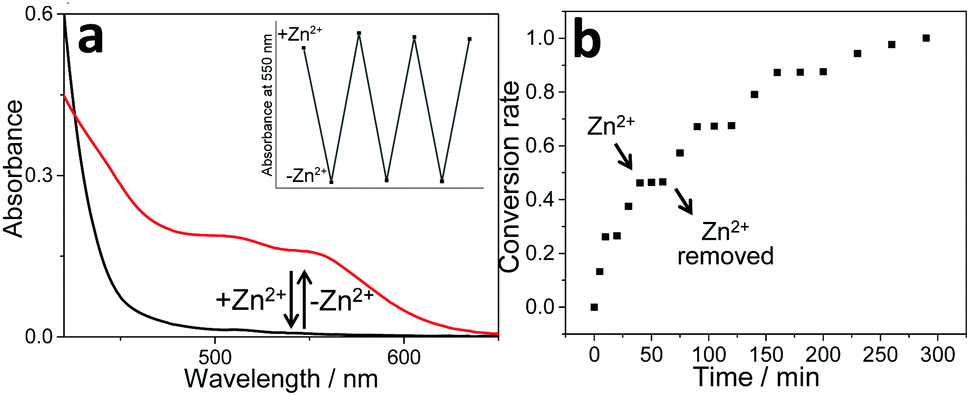 | ||
| Fig. 3 (a) UV-Vis spectral changes of complex 1b/2a in CHCl3/CH3CN (1:1, v/v) upon sequential addition of Zn(OTf)2 and cyclen as the competitive ligand to trap Zn2+ ions. The inset shows the changes in the absorption intensity at 550 nm. (b) Photocatalytic conversion of 3 to 4 upon sequential addition of Zn(OTf)2 and unsubstituted terpyridine (as the competitive ligand to trap Zn2+ ion)41 to the photo-catalyst 1b/2a in CHCl3/CH3CN (1:1, v/v). | ||
Notably, the reversible “on/off” switching of the Pt(II)⋯Pt(II) absorbance signal for 1b/2a (Fig. 3a) provides extra control over their photo-catalytic efficiency, which can be demonstrated by monitoring the conversion from 3 to 4 upon addition/removal of Zn(OTf)2. In detail, when the Zn2+ ion is added to complex 1b/2a, it breaks up the photo-catalytic capability (Fig. S18, ESI†). The successive addition of unsubstituted terpyridine as the competitive ligand41 traps Zn2+ ions, and thereby leads to the recovery of photo-catalytic efficiency (Fig. S18, ESI†). Noteworthily, with the sequential addition of Zn(OTf)2 and unsubstituted terpyridine, “on-demand” revival and loss of photo-oxidation capability for 3 can be achieved for several repeated cycles (Fig. 3b).
Conclusions
In summary, herein we have successfully developed a novel supramolecular tweezering system with the involvement of Pt(II)⋯Pt(II) metal–metal interactions. For the non-covalent molecular tweezer/guest complexes 1b/2a and 1b/2b, MMLCT absorbance signals emerge in the visible-light region, which can be sensitized by O2. As a result, they can be utilized for photo-oxidative cyanation and photo-cleavage reactions in organic and aqueous media. More importantly, temporally “on-demand” photo-catalytic efficiency can be achieved, by taking advantage of the Zn2+-responsive conformational switch of the molecular tweezer receptor. Hence, the current study opens up new avenues toward intelligent photocatalytic materials via elaborate supramolecular design.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21274139), the Fundamental Research Funds for the Central Universities (WK3450000001), and CAS Youth Innovation Promotion Association (2015365).Notes and references
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285 CrossRef CAS PubMed.
- L.-L. Hou, X.-Y. Zhang, T. C. Pijper, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 910 CrossRef CAS PubMed.
- Y. Jiao, K. Liu, G. Wang, Y. Wang and X. Zhang, Chem. Sci., 2015, 6, 3975 RSC.
- X.-Q. Wang, Q. Lei, J.-Y. Zhu, W.-J. Wang, Q. Cheng, F. Gao, Y.-X. Sun and X.-Z. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 22892 CAS.
- K. Feng, R.-Y. Zhang, L.-Z. Wu, B. Tu, M.-L. Peng, L.-P. Zhang, D. Zhao and C.-H. Tung, J. Am. Chem. Soc., 2006, 128, 14685 CrossRef CAS PubMed.
- J.-J. Zhong, Q.-Y. Meng, G.-X. Wang, Q. Liu, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2013, 19, 6443 CrossRef CAS PubMed.
- Q.-Y. Meng, T. Lei, L.-M. Zhao, C.-J. Wu, J.-J. Zhong, X.-W. Gao, C.-H. Tung and L.-Z. Wu, Org. Lett., 2014, 16, 5968 CrossRef CAS PubMed.
- P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 127, 2112 CrossRef.
- J.-J. Zhong, C. Yang, X.-Y. Chang, C. Zou, W. Lu and C.-M. Che, Chem. Commun., 2017, 53, 8948 RSC.
- K. Mori, K. Watanabe, M. Kawashima, M. Che and H. Yamashita, J. Phys. Chem. C, 2011, 115, 1044 CAS.
- K. Mori, K. Watanabe, K. Fuku and H. Yamashita, Chem. – Eur. J., 2012, 18, 415 CrossRef CAS PubMed.
- K. Mori, K. Watanabe, Y. Terai, Y. Fujiwara and H. Yamashita, Chem. – Eur. J., 2012, 18, 11371 CrossRef CAS PubMed.
- K. Mori and H. Yamashita, Chem. – Eur. J., 2016, 22, 11122 CrossRef CAS PubMed.
- K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef CAS PubMed.
- S. Y.-L. Leung, A. Y.-Y. Tam, C.-H. Tao, H. S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2012, 134, 1047 CrossRef CAS PubMed.
- Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Chem. Sci., 2012, 3, 1185 RSC.
- Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Chem. – Eur. J., 2013, 19, 390 CrossRef CAS PubMed.
- Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 125, 14367 CrossRef.
- A. K. W. Chan, W. H. Lam, Y. Tanaka, K. M. C. Wong and V. W. W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 690 CrossRef CAS PubMed.
- T. Haino, T. Fujii, A. Watanabe and U. Takayanagi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10477 CrossRef CAS PubMed.
- Y.-J. He, T.-H. Tu, M.-K. Su, C.-W. Yang, K. V. Kong and Y.-T. Chan, J. Am. Chem. Soc., 2017, 139, 4218 CrossRef CAS PubMed.
- Y.-K. Tian, Y.-G. Shi, Z.-S. Yang and F. Wang, Angew. Chem., Int. Ed., 2014, 53, 6090 CrossRef CAS PubMed.
- Z. Gao, Y.-F. Han, J.-J. Chen, X. Wang and F. Wang, Chem. – Asian J., 2016, 11, 1775 CrossRef CAS PubMed.
- Z.-J. Li, Y.-F. Han, F. Jin, Z.-C. Gao, Z. Gao, L. Ao and F. Wang, Dalton Trans., 2016, 45, 17290 RSC.
- Z.-J. Li, Y.-F. Han, Z.-C. Gao and F. Wang, ACS Catal., 2017, 7, 4676 CrossRef CAS.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342 CrossRef CAS PubMed.
- M. Zhang, M. L. Saha, M. Wang, Z. Zhou, B. Song, C. Lu, X. Yan, X. Li, F. Huang, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2015, 139, 5067 CrossRef PubMed.
- X. Yan, M. Wang, T. R. Cook, M. Zhang, M. L. Saha, Z. Zhou, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580 CrossRef CAS PubMed.
- S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596 CrossRef CAS PubMed.
- B. Jiang, J. Zhang, J.-Q. Ma, W. Zheng, L.-J. Chen, B. Sun, C. Li, B.-W. Hu, H. Tan., X. Li and H.-B. Yang, J. Am. Chem. Soc., 2016, 138, 738 CrossRef CAS PubMed.
- B. Doistau, A. Tron, S. A. Denisov, G. Jonusauskas, N. D. McClenaghan, G. Gontard, V. Marvaud, B. Hasenknopf and G. Vives, Chem. – Eur. J., 2014, 20, 15799 CrossRef CAS PubMed.
- A. Petitjean, R. G. Khoury, N. Kyritsakas and J.-M. Lehn, J. Am. Chem. Soc., 2004, 126, 6637 CrossRef CAS PubMed.
- B. Doistau, L. Benda, J.-L. Cantin, L.-M. Chamoreau, E. Ruiz, V. Marvaud, B. Hasenknopf and G. Vives, J. Am. Chem. Soc., 2017, 139, 9213 CrossRef CAS PubMed.
- F. Guo, W. Sun, Y. Liu and K. Schanze, Inorg. Chem., 2005, 44, 4055 CrossRef CAS PubMed.
- Y. Liu and J. Zhang, Chem. Commun., 2012, 48, 3751 RSC.
- W.-P. Wong, G. S.-M. Tong, W. Lu, C. Ma, J. Liu, A. L.-F. Chow and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 2654 CrossRef PubMed.
- A. M. L. Hossion, M. Bio, G. Nkepang, S. G. Awuah and Y. You, ACS Med. Chem. Lett., 2013, 4, 124 CrossRef CAS PubMed.
- Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. A. Palmans and E. W. Meijier, J. Am. Chem. Soc., 2015, 137, 13096 CrossRef CAS PubMed.
- For the temporally “on-demand” photo-catalytic experiments, we initially employed cyclen to trap Zn2+ ions. However, the amine units on cyclen might influence the photosensitization efficiency of complex 1b/2a, leading to the irreversible deactivation of the photo-catalytic efficiency. To avoid this issue, unsubstituted terpyridine was chosen as the competitive ligand instead of cyclen.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, and other materials. See DOI: 10.1039/c7qm00424a |
| This journal is © the Partner Organisations 2018 |